

Abstract
Currently, different sequencing platforms are used to generate plant genomes and no workflow has been properly developed to optimize time, cost, and assembly quality. We present LeafGo, a complete de novo plant genome workflow, that starts from tissue and produces genomes with modest laboratory and bioinformatic resources in approximately 7 days and using one long-read sequencing technology. LeafGo is optimized with ten different plant species, three of which are used to generate high-quality chromosome-level assemblies without any scaffolding technologies. Finally, we report the diploid genomes of Eucalyptus rudis and E. camaldulensis and the allotetraploid genome of Arachis hypogaea.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13059-021-02475-z.
Keywords: Long-read sequencing, Chromosome-level draft genome, High molecular weight DNA extraction, Eucalyptus, Peanut, Arachis, Genomic standardized workflow
Background
Plants represent the dominant kingdom of life in terms of Earth biomass [1], and through colonization of terrestrial and aquatic habitats, are responsible for maintaining ecological and atmospheric balance. Despite being globally distributed, climate change and anthropogenic activities are massively impacting current plant diversity with repercussions for ecophysiology, distribution, and interactions with other organisms [2, 3]. Sequencing present-day plant genomes to better understand genomic diversity is an important requirement for gauging plants’ susceptibility to climate change.
In the last decade, rapid advances in short-read sequencing technology have resulted in the availability of over 300 plant species genomes, of differing quality [4]. Recently, long-read sequencing methods (Pacific Biosciences, PacBio, and Oxford Nanopore Technology, ONT) are becoming more accessible while technological advances have led to increases in the base accuracy and the sequencing length, as well as a significant reduction in cost per base of sequence [5]. The main benefit of long-read sequencing technologies for genomics, compared to the more dominant short-read/Illumina sequencing, is the ability to assemble genomes relatively easily by linking reads that span across repetitive genomic regions. This property when combined with ultra-long reads, highly accurate sequencing, and complementary scaffolding technologies has thereby enabled highly accurate telomere to telomere assemblies [6–8]. The many benefits of long-read sequencing have driven demand for high-quality high molecular weight (HMW) DNA, and led to advances in sequencing technologies and genome assembly tools.
In the case of plants, genomes can be large and highly repetitive, making long-read sequencing ideal for genome assembly [9–11]. However, it is often difficult to extract HMW DNA suitable for long-read sequencing from plants [12–14]. Plants have tough cell walls and contain high levels of metabolic contaminants, such as polyphenols and polysaccharides [15–17] which are difficult to eliminate and impact sequencing quality. Furthermore, a plethora of new initiatives have emerged to sequence millions of species, including those from the plant kingdom [18, 19].
To address the need to sequence new plant genomes, and to achieve the best operating conditions for accomplishing high-quality de novo genomes with one technology, we present and discuss the development of LeafGo. This is a complete workflow designed to generate de novo genomes from plant tissue with relatively modest resources within 7 days for genomes less than approximately one gigabase (Gb) and increasing incrementally for larger or more complex genomes (Fig. 1).
Fig. 1.
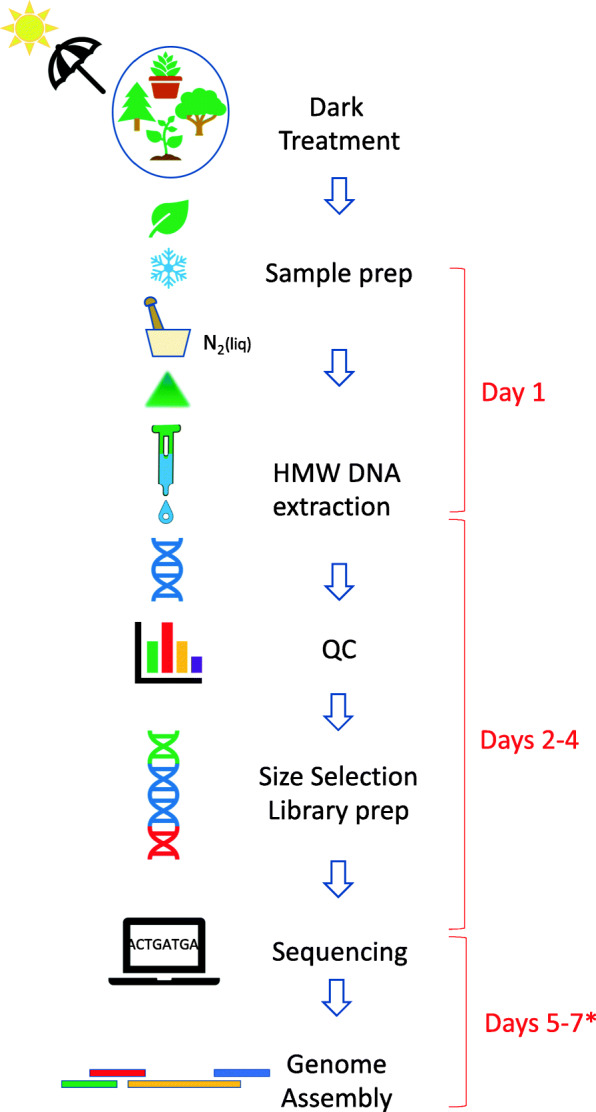
Plant long-read sequencing workflow. Asterisk indicates sequencing time depends on genome size and ploidy. Seven days completion time is based on a diploid organism with a haploid genome < 0.6–1 gigabases
To develop LeafGo, we selected ten plant species (Arachis hypogaea subsp. fastigiata, Brassica rapa subsp. chinensis, Distichlis palmeri, Eucalyptus rudis subsp. rudis, E. camaldulensis subsp. obtusa, Pennisetum glaucum, Salicornia bigelovii, Salvadora persica, Solanum melongena, and Zea mays; Additional file 1: Table S1) from seven diverse plant taxonomic families for HMW DNA extraction and long-read sequencing. Maize (Z. mays), pearl millet (P. glaucum), and peanut (A. hypogaea) are well-studied, globally important crops responsible for feeding millions of people with large, highly repetitive genomes [10, 11, 20–25]. Similarly, bok choy (B. rapa) and eggplant (S. melongena) are high production crops important for human nutrition with published genomes [26–28]. Nipa grass (D. palmeri), dwarf saltwort (S. bigelovii), and toothbrush tree (S. persica) are lesser-researched plants without published genomes that are, or could be developed into, agriculturally/pharmacologically important crops [29–32]. Finally, the flooded gum (E. rudis) and river red gum (E. camaldulensis) trees were selected. Eucalypts are the most commonly planted hardwood trees in the world due to their fast growth, environmental adaptability, and many commercial uses [33]. However, good-quality HMW DNA is relatively difficult to extract from eucalypts due to their high phenolic and polysaccharide content [12, 17, 34]. Moreover, of the > 800 eucalypt species, only a few high-quality genomes have been published [33, 35, 36].
This paper provides the rationale for the adoption of LeafGo, a workflow that combines different resources to generate de novo genomes of different plants. LeafGo consists of a robust HMW DNA extraction method, library preparation and sequencing approach, and genome assembly suggestions. This work also provides the community with three high-quality de novo genomes of E. camaldulensis, E. rudis, and A. hypogaea.
Results
One of the most challenging aspects of long-read sequencing is applying stringent quality controls at every step of the laboratory workflow in order to obtain good-quality sequencing results. We outline the best conditions to extract HMW DNA and to process it into long-read sequencing libraries for the PacBio and ONT platforms. In particular, we prepared PacBio continuous long read (CLR) and high fidelity (HiFi, also known as circular consensus sequencing, CCS) libraries and sequenced them on the latest PacBio platforms, Sequel I and Sequel II. For the Eucalyptus species, libraries were produced and sequenced with the GridION platform demonstrating the suitability of the laboratory component of LeafGo for ONT sequencing [34, 37, 38] (see Additional file 1: Oxford Nanopore Technology Sequencing; Tables S2 and S3; Fig. S1). Finally, we compared CLR and HiFi data using the latest tools for genome assembly and assembled the two Eucalyptus species and A. hypogaea into high-quality draft genomes.
DNA extraction, quality controls, library, and long-read sequencing
DNA extraction
The extraction protocol implemented in LeafGo generated large amounts of HMW DNA with high purity within a day and using minimal resources and effort. The protocol yielded high-quality HMW DNA in 27 separate extractions from ten different plant species over different days by different technicians. The yield (per 1 g wet weight of leaf) ranged from 10 to 278 μg (average ± standard deviation [SD], 79.6 ± 71.6) with high variability in the Eucalyptus species, compared to the other species (Fig. 2A). All the extracted HMW DNA had high purity and integrity, despite the different composition, including potential contaminants, between species [12, 29, 32, 34, 36]. Notably, the Eucalyptus samples showed signs of oxidation during lysis, such as dark coloration of the solution; however, the quality of DNA was not compromised, without the need for added antioxidants [12, 34]. The absorbance ratios indicated low levels of contaminants, such as protein, carbohydrates and phenolics, and high purity (average A260/280 = 1.83 ± 0.05 SD and A260/230 = 2.21 ± 0.13 SD; Fig. 2B) [9, 34, 36, 39]. The integrity of the extracted genomic DNA (gDNA) was assessed by pulsed-field gel electrophoresis and capillary electrophoresis. The extracted samples typically showed a DNA smear ranging from approximately > 15 to < 150 Kb and a mode of over 80 Kb for all samples (Additional file 1: Fig. S2 and S3). Our results are comparable to, or improve on, similar studies in terms of DNA yield, fragment length, and purity but with a simpler and less toxic extraction protocol [12, 17, 34] that produces HMW gDNA suitable for long-read sequencing in 1 day.
Fig. 2.

DNA extraction and long-read sequencing output from study plant species. Yield (A) and absorbance ratios (B) of extracted HMW DNA from ten study plant species. Subread N50 of CLR libraries on Sequel I and Sequel II for seven of the ten study plant species (C). The total throughput (D), subread N50 length (E), and Q20 yield (F) for HiFi libraries sequenced on Sequel II for five study plant species. The average for all data is plotted along the margin. CLR sequencing was not completed for A. hypogaea, B. rapa, and S. melongena
PacBio CLR and HiFi sequencing
The extracted HMW gDNA from eight of the species (Arachis hypogaea, Distichlis palmeri, Eucalyptus rudis, E. camaldulensis, Pennisetum glaucum, Salicornia bigelovii, Salvadora persica, and Zea mays), was processed into 26 CLR and 7 HiFi libraries prior to sequencing with the PacBio Sequel I (22: 1M SMRT cells) and Sequel II (17: 8M SMRT cells). SMRTLink analysis (Additional file 1: Table S4) showed that sequence statistics for both CLR and HiFi libraries were above the PacBio recommended specifications [40] with optimal internal controls metrics indicating that no inhibition of the sequencing reaction was observed.
The CLR libraries (Additional file 1: Fig. S4A to S4D) showed a mode > 30 kb with few shorter fragments. The sequencing results confirmed that the extracted gDNA was of good purity and of high molecular weight. The average CLR throughput per Sequel I SMRT cell was 11.5 Gb (± 4.7 Gb [SD]; 3.3–18.1 Gb [min–max]) while the Sequel II yielded 167.6 Gb/SMRT cell (± 26.1 Gb; 140.8–195.4 Gb) (Additional file 1: Fig. S4E and S4F). The average N50 subread length was 36.6 kilobases (Kb) (± 5.5 Kb; 21.5–44.7 Kb) for the 22 Sequel I CLR libraries, and 36.4 Kb (± 3.2 Kb; 32.4–39.8 Kb) for the four Sequel II CLR libraries (Fig. 2C). Sheared DNA was tested for CLR library preparation; however, there was no obvious improvement in yield or N50, as previously shown with ONT libraries [34], and unsheared DNA was used for all other libraries. To test whether library loading affected the subread N50 length, some SMRT cells were intentionally underloaded. There was a borderline significant correlation (Additional file 1: Table S5 and Fig. S5) between ZMW (zero mode waveguide) occupancy or library underloading (high P0%) and subread N50 (Spearman’s rho = 0.42, p = 0.0499) suggesting a small benefit in subread N50 at the expense of throughput yield; unsurprisingly, library loading (determined from P0% and P1%) was significant correlated with throughput yield (Spearman’s rho = − 0.84 and 0.86, p-value < 0.00005). Although not presented here, initial testing with higher size-selected CLR libraries (35 Kb and 40 Kb) yielded diminished sequencing results with inconsistent throughput and lower N50 sizes. For this reason, we exclusively used 30-Kb size-selected CLR libraries. The reason for the suboptimal results with higher size-selected libraries is not clear and might require follow up studies.
The HiFi libraries showed a mode of approximately 20 Kb (Additional file 1: Fig. S6) and, when sequenced with Sequel II, yielded an average total throughput of 322.9 Gb/SMRT cell (± 87.6 Gb; 113.9–477.3 Gb), with the Q20 yield of 23.3 Gb (± 5.4 Gb; 7.6–29.4 Gb) and subread N50 of 20.1 Kb (± 1.3 Kb; 17.6–22.2 Kb) as shown in Fig. 2D–F. The total throughput and Q20 yield were high for all the samples except for one D. palmeri HiFi library that was underloaded (P1% = 13%); subsequent resequencing of the library with optimal loading yielded more typical results (356.3 Gb total throughput; 23 Gb Q20 yield).
De novo genome assembly of Eucalyptus and Arachis species
Assembling a de novo genome requires a combination of coverage, read length, base quality, and computational resources. An accurate reconstruction, preferably in a short time frame, is indeed crucial, as both the continuity and base accuracy of an assembly can affect the quality of the genome. LeafGo bioinformatics recommendations are based on testing existing and recently developed tools to optimize both genome quality and computational time, a commonly limited resource. To evaluate the best approach, both HiFi and CLR sequencing data of two diploid eucalypts (2n = 2x = 20) and HiFi data for the allotetraploid peanut (2n = 4x = 40) were assessed and assembled.
Quality control of raw sequencing data
The first bioinformatics step is the quality control (QC) of the raw sequencing data. Platform-specific metrics represent the first informative statistics for the overall quality of the sequencing run (Additional file 1: Table S4). However, a more comprehensive QC step was performed using LongQC [41]. LongQC, or similar software, allows rapid and in depth cross-platform QC of the raw sequencing data. LongQC results for the Eucalyptus species and A. hypogaea sequencing data are shown in Additional file 1: Fig. S7 and S8.
All HiFi data for the three species are of good quality as indicated by the high scores for per read base calling accuracy (Additional file 1: Fig. S7A, S7F and S7M) and by a normal distribution of per read coverage, except for the slight bimodality in E. rudis (Additional file 1: Fig. S7C, S7H and S7O). Both Eucalyptus species showed a similar GC content and sequence complexity (Additional file 1: Fig. S7B, S7G and S7D, S7I). The GC content for all the Eucalyptus reads shows a sharp unimodal distribution around a mean of 0.39 (± 0.03 [SD]), but with an upper sub-mode outlier near 0.55. Closer inspection of this higher GC content peak revealed the presence of telomere repeats.
The GC content of Arachis (Additional file 1: Fig. S7N) presents a sharp unimodal distribution around a mean of 0.36 (± 0.05 [SD]), but two sub-modes are observed: an upper sub-mode outlier near 0.55 like the eucalypts, and one lower sub-mode outlier near 0.15. A closer investigation of the lower sub-mode showed the presence of centromere repeats [42]. No artificial sequence adapters are present in the flanking region for either datasets (Additional file 1: Fig. S7E, S7L and S7Q).
In contrast, CLR data for the two eucalypts does not achieve the same level of quality shown by HiFi data. The Phred scores are not provided in CLR mode and thus read base calling accuracy cannot be directly assessed. The sequence complexity of CLR is lower (sequences with low complexity, 0–40% and 0–20% in CLR and HiFi, respectively), and the flanking regions seem to extend over a hundred bases pointing to the possibility of artificial sequences (Additional file 1: Fig. S8B, S8F and S8D, S8H). Both Eucalyptus species’ CLR sequences have a similar GC content to that for HiFi data. The GC content for all the reads has a sharp normal distribution around a mean of 40% (± 4%), but the upper sub-mode peak, indicative of telomere repeats and present in HiFi data, is not found within the CLR distribution. Surprisingly, CLR sequencing may be less sensitive at detecting telomere repeats than HiFi.
Genome assemblies
Prior to assembly, k-mer counting can provide insights into the genome size of an unknown genome. A k-mer assessment on HiFi data using GenomeScope 2.0 [36] estimated the genome size of E. rudis, E. camaldulensis, and A. hypogaea as 506 Mb, 510 Mb, and 2.54 Gb, respectively (Additional file 1: Fig. S9). The heterozygosity level was also estimated using the same k-mer approach and was relatively high in both eucalypts (ab: E. camaldulensis, 2.19%; E. rudis, 1.57%) which is expected to affect genome assembly [37] and estimated genome size [38]. The patterns of nucleotide heterozygosity rates shown by A. hypogaea follow the expected distinct patterns for allotetraploid genomes [43] (Additional file 1: Fig. S9C).
The three genomes were assembled with tools optimized for long-read sequences: Canu [44] for CLR data (only the two eucalypts) and hifiasm [45] for HiFi data. A comparison among different assemblers has shown Canu is an efficient assembler for CLR PacBio data [46]. For HiFi generated data, we carried out a comparison among different assemblers (hifiasm v0.8 [45], HiCanu v2 [44], Flye v2.8.1 [47], Wtdbg2 v2.5 [48]) and found that hifiasm outperformed the other assemblers (Additional file 1: Tables S6 and S7).
Eucalyptus diploid genomes
The overall genome assembly statistics are shown in Table 1. For the two eucalypts, the assembled genome size based on the HiFi and on the CLR data is similar for E. rudis and E. camaldulensis. The HiFi assembly is superior to the CLR assembly for both Eucalyptus species, which is in agreement with HiFi-based assemblies for other species [8, 44, 49]. The contig N50/N90 and L50/L90 show noticeably higher contiguity in the HiFi assemblies compared to the CLR assemblies. Furthermore, the HiFi assemblies consistently produced the longest contigs (E. rudis, 61.8 Mb, 33.7 Mb; E. camaldulensis, 69.1 Mb, 58.1 Mb; for HiFi and CLR, respectively).
Table 1.
Genome assembly statistics for two Eucalyptus species and A. hypogaea. We calculated the assembly statistics using Quast. CLR-based assemblies were 3-cycle polished as detailed in “Methods”. Results are based on the purged assemblies (see “Methods”).
| Plants | Type | Sized (Mb), ≥ 1 Mb|total | No. contigsd, ≥ 1 Mb|total | N50 (Mb)/L50 | N90 (Mb)/L90 | Longest contig (Mb) | Alternative size (Mb) |
|---|---|---|---|---|---|---|---|
| E. rudisa | HiFi (~ 40×) | 531|549 | 26|331 | 36.0/7 | 7.3/15 | 61.8 | 425 |
| CLR (~ 50×) | 506|518 | 44|138 | 16.3/11 | 5.2/30 | 33.7 | 399 | |
| E. camaldulensisb | HiFi (~ 51×) | 525|532 | 14|149 | 41.4/5 | 23.2/12 | 69.1 | 520 |
| CLR (~ 230×) | 516|523 | 28|77 | 29.3/7 | 8.5/19 | 58.1 | 570 | |
| A. hypogaeac | HiFi (~ 74×) | 2,564|2,623 | 114|1417 | 42.3/22 | 10.37/69 | 90.3 | 51 |
aE. rudis unknown genome size. Coverage estimated based on assembly size
bE. camaldulensis reference genome size, 558.6 Mb (AC: GCA_014182705.1)
cA. hypogaea reference genome size, 2557 Mb (AC: GCF_003086295.2)
N50 the smallest length contig at which the cumulative contig lengths equal to 50% of the assembled size, L50 N50 contig count, N90 the smallest length contig at which the cumulative contig lengths equal to 90% of the assembled size, L90 N90 contig count
dMetric calculated based on (1) minimum contig length cut-off of 1Mb or (2) no cut-off
The assembly haploid genome sizes were estimated by separating haplotypes and purging haplotigs, and the completeness was assessed using BUSCO scores (Additional file 1: Table S8). Based on the primary haplo-purged assembly, the haploid genome size of E. rudis varies between 518 Mb (CLR) and 549 Mb (HiFi) and produced an estimated ploidy of 1.77N. The high complete BUSCO scores (> 96%) for the primary contigs and a relatively high score for the alternative contigs (CLR, 73.4%; HiFi, 87.2%) indicate a reasonable separation of the haplotypes. In addition, the alternative contig set for the CLR assembly shows a lower degree of completeness which further demonstrates the superiority of the HiFi assembly. The haploid genome size for E. camaldulensis is about 523 and 532 Mb based respectively on CLR and HiFi primary assemblies. Both the primary and the alternative contig sets show high BUSCO scores (primary, > 97%; alternative, > 93%) in both CLR and HiFi assemblies. The estimated ploidy of the assembly after haplo-purging is over 1.98 N which, taken together with the BUSCO scores, suggests that the assembly is comprehensive. Alignment to the E. grandis genome [33] shows that our HiFi E. camaldulensis assembly has 9 full chromosomes with the remaining two chromosomes spanned almost fully by two contigs each (Fig. 3). Similarly, the E. rudis HiFi assembly has five full chromosomes and the remaining chromosomes spanned almost fully by two to three contigs each. In terms of computational time, assembling the draft genome with HiFi data is 65 times quicker than using CLR for similar coverage (40× HiFi, 53 CPU hrs; 50× CLR, 3444 CPU hrs; E. rudis). This gap surges to 895 times when the coverage of CLR over HiFi increases about 4 times (51× HiFi, 81 CPU hrs; 230× CLR, 72491 CPU hrs; E. camaldulensis). The two Eucalyptus genomes show the same assembly complexity and similar computational time is expected. The two Eucalyptus species were further identified by phenotypic and in silico inspection [50, 51] (see Additional file 1: Phenotypic and in silico identification of the Eucalyptus species; Fig. S10).
Fig. 3.

Chord diagrams of E. camaldulensis, E. rudis, and A. hypogaea de novo assemblies mapped against a reference genome. Alignment of E. camaldulensis (A), E. rudis (B), and A. hypogaea (C) HiFi assemblies against E. grandis (A and B) and A. hypogaea (C) reference genomes
The recent assembly of E. pauciflora, a genome of similar size and complexity, offers a possible comparison with our assemblies. A hybrid assembly strategy, with ONT long-read scaffolding contigs generated by Illumina short reads, has been adopted to generate the E. pauciflora genome [36]. The LeafGo approach is simpler and less labor intensive by using only one long-read technology, and produced genomes with twelve times more contiguity (contig N50 36/41 Mb vs 3.2 Mb, E. rudis/camaldulensis, E. pauciflora respectively) with a hundred times less computational resources (see Additional file 1: Genome assembly: computational resources).
Arachis hypogaea allotetraploid genome
The assembly of the allotetraploid genome of A. hypogaea was carried out only with HiFi data. This resulted in a 2623-Mb genome size with 42.3 Mb and 10.37 Mb contigs N50 and N90 respectively. Notably, the A. hypogaea genome is a large genome rich in repetitive content [25] and a large quantity of long reads are needed to help bridge difficult regions in the genome. Our assembly proves to be more contiguous than a previously reported assembly generated only by older PacBio technology showing a lower N50 of 1.5 Mb and N90 of 0.34 Mb [25]. Alignment to the A. hypogaea reference genome (GCA_003086295.2) shows that our HiFi A. hypogaea assembly is comprehensive where the majority of the 20 chromosomes are spanned by a few contigs each. A closer inspection of the alignment revealed central and peripheral chromosomal regions (likely telomeres and/or centromeres) not fully assembling into the larger contigs and therefore falling short of chromosome-level assembly (Fig. 3C). The assembly, indeed, faces the challenge of the repetitive nature of the peanut genome [24], richer in repetitive content than the eucalypt ones (E. rudis, 37.4%; E. camaldulensis, 38.3%; A. hypogaea, 82.9%; Additional file 1: Fig. S9). The high BUSCO score of 97.5% supports the high quality and completeness of our HiFi assembly, although the computational time to assemble the large peanut allotetraploid genome with HiFi data is longer compared to the smaller diploid eucalypt genomes (74× HiFi, 1081 CPU hrs, A. hypogaea).
Time estimates
The estimated total time from raw reads to HiFi/CCS reads to the assembly of a high-quality contiguous draft for a diploid genome of 0.6 to 1.0 Gb is less than one day which increases to approximately 2 days for a tetraploid genome of 2.5 Gb (see Additional file 1: Genome assembly: computational resources). When combined with time estimates of HMW DNA extraction (1 day), HiFi library preparation and sequencing (5 days) and assembly, a high-quality draft genome of 0.6–1.0 Gb can be prepared from plant samples within a minimum of 7 days, depending on available compute resources. This time increases for a polyploid (2.5 Gb) genome in which sequencing will require approximately 9 additional days bringing the time up to 16 days (Additional file 1: Fig. S11). All time estimates are based on sequencing using a single Sequel II machine and will vary according to coverage requirements.
Discussion
The current era of genomics is very promising with ambitious projects to sequence much of life on Earth [18, 19, 52]. The ideal goal for these projects would be to produce high-quality error-free, gapless or near-gapless haplotype-resolved genome assemblies. Very few gapless chromosome-level reference genomes today exist and typically require two or more different technologies like PacBio/ONT long reads, Illumina short and 10X linked reads sequencing, optical mapping, and Hi-C [5, 9, 11, 53]. In particular, plants prove to have some of the most challenging genomes to assemble as they are rich in repetitive content, with high levels of heterozygosity and complex polyploidy [54]. Assembling a high-quality plant genome with complete or near complete chromosomes therefore requires high coverage, long-read length, and high-quality sequences with a low error rate. Recently, PacBio has improved the circular consensus sequencing approach to generate long (≥ 15 Kb) HiFi reads with base accuracy upwards of 99.8% [8], and we took advantage of this progress and developed LeafGo, a streamlined workflow able to produce a high-quality draft plant genome from plant tissue within 7 days.
LeafGo is designed to be used as a rapid one-pass approach for assembling a high-quality draft genome that generates data suitable for most plant species. Our workflow targets a crucial problem: optimally assembling high-quality de novo genomes using only one technology. Our goal was achieved by considering the overall procedure, and modifying and optimizing each single step for consistent results. On this basis, we provide a robust HMW DNA extraction method, guidance for library preparation and sequencing, and recommendations for genome assembly.
The use of one sequencing technology such as PacBio is a distinctive advantage of LeafGo, although compatibility of the extracted HMW DNA with long-read ONT sequencing is also demonstrated. The adoption of other sequencing technologies might improve the contiguity of a genome assembly, but a significant investment of additional resources may be needed, such as ultra-high molecular weight DNA extractions, increased wet-lab labor and time, additional equipment and reagents, various software packages, and extensive bioinformatics analysis with manual intervention [5, 9, 11, 52, 53].
LeafGo uses a modified column-based gDNA extraction protocol delivering high molecular weight DNA of excellent quality. All the extracted gDNA samples had a mode length of over 80 Kb, with optical density ratios indicating absence of organic contamination. We then benchmarked the effectiveness and utility of two different methods of PacBio library preparation and sequencing, HiFi, and CLR. The HiFi protocol proved to generate high base-level accuracy long reads, while the CLR protocol produced subreads of considerable length. The CLR subread N50 is further evidence of the quality of the extracted HMW DNA. The averaged CLR N50 subread length was greater than 36 Kb on both Sequel I and Sequel II platforms, exceeding therefore the N50 of many recent studies [11, 49, 52, 55], and we introduced the N50 subread length parameter as a quality control for assessing HMW DNA extractions. It should be highlighted that although we extracted high-quality HMW DNA from ten different species, our extraction method might not work for all plant species.
Although the eucalypt genomes assembled in this manuscript with both CLR and HiFi data are very contiguous, the higher base-level accuracy given by HiFi improves the assembly considerably thus removing the need for polishing with short-read sequencing [44]. Our results emphasize several strengths of the HiFi technology. Compared to the CLR assemblies, HiFi assemblies demanded less computational requirements, had higher BUSCO scores, showed several fold improvement of contig N50/N90 and L50/L90, and generated more complete genome assemblies, as has been previously published [8, 45, 49]. In fact, HiFi sequencing data assembled with hifiasm produced near-chromosome-level haploid draft genomes.
The two Eucalyptus species genomes assembled with the LeafGo workflow will improve our genomic knowledge of eucalypts, which at the moment is relatively sparse, and will assist with conservation issues and commercial uses. There are more than 800 eucalypt species, but only three genomes have been published: E. grandis [33], E. pauciflora [36], and E. camaldulensis [35]. The high-quality draft genome of E. camaldulensis generated with LeafGo improves upon the published and highly fragmented genome produced by short-read sequencing [35], and the unpublished reference-based and less contiguous assembly recently produced from ONT long-reads [56] as shown by the assembly metrics (LeafGo de novo assembly contig N50 of 41.4 Mb; ONT reference-based assembly contig N50 of 2.5 Mb). The first genome assembly of E. rudis is generated of mostly complete chromosomes and shows a primary haploid genome size of 549 Mb (HiFi assembly) which is similar to other eucalypts [33, 35, 36, 56].
Polyploidy plays an important role in plant evolution affecting phenotypic diversification, ecological tolerance, and species richness. The A. hypogaea genome is a large polyploid genome, about four times larger than the above eucalypt species. It carries two sets of chromosome pairs, originating from a hybridization event of two distinct ancestral Arachis species 9400 years ago (A. ipaensis, genome A; A. duranensis, genome B) which generated the allotetraploid genome of A. hypogaea (AABB-type genome; 2n = 4× = 40 chromosomes; genome size of ~ 2.5 Gb) [57].
Recently, different allotetraploid plant genomes were assembled with PacBio and other sequencing technologies [58]; here, we provide a highly contiguous and complete Arachis hypogaea genome assembly using only PacBio sequencing. Our assembly is comparable to the already published peanut genome [24, 25] and other polyploid plant species based on HiFi reads [45] consolidating the strength of our workflow. A careful examination of the alignment of our assembly against the published reference reveals that the source of fragmentation can be directly attributed to the central and peripheral regions (possibly centromeric and/or telomeric). Considering the high repeat content in the peanut genome (~ 82.9%), the contiguity of our assembly is impressive. Presently, for large and repetitive allotetraploid genomes such as the peanut, a scaffolding technology can improve the contiguity of the assembly [24] but it will take more time and add further costs. The generation of the A. hypogaea genome took 9 days longer than for a eucalypt species, due to the necessity of sequencing the larger genome at sufficient coverage with a consequential increase in platform sequencing time; however, significant reductions in sequencing time could be achieved by parallel sequencing on multiple machines.
Our HiFi assemblies of two diploid Eucalyptus genomes and the allotetraploid genome of Arachis hypogaea are therefore comparable to single technology assemblies based on high coverage CLR and HiFi assemblies of homozygous maize strains and, more relevant, to heterozygous plant species based on HiFi reads [45].
Conclusions
The global initiatives to sequence and assemble genomes for thousands of eukaryotic life forms, including plants, do not yet have a published standardized workflow [18, 19, 52, 59, 60]. Pursuing this goal, LeafGo is a valuable tool as it provides the foundation to produce high-quality genome assemblies within 7 days. The timeline and the resulting genome contiguity therefore might be further improved by future bioinformatics and sequencing technology developments. However, currently LeafGo will be an extremely valuable resource for those scientists aiming for a fast and cost-effective genome assembly workflow. We envisage that the simplified and efficient LeafGo will be useful for plant researchers as well as specialized genome sequencing centers.
Methods
Sample collection and HMW DNA extraction
We chose ten plants from seven taxonomic families that are economically relevant and represent typical species that may require high-quality draft genomes (Additional file 1: Table S1). Leaves were collected after at least 48 h of dark treatment. If possible, an individual plant was used for leaf collection and HMW DNA extraction, as was the case with the species used for genome assembly (A. hypogaea, E. rudis, and E. camaldulensis). To dark treat an entire plant or a branch from a large tree, the target was covered with light-opaque black plastic sheets with a few holes that allowed air flow. The leaves were sprayed with ethanol and wiped to remove contaminating organisms. Leaves were removed and weighed before flash-freezing in liquid nitrogen.
HMW DNA was extracted by modifying a Qiagen Genomic protocol (Qiagen, Hilden, Germany). Briefly, frozen leaves were ground to a fine powder in a mortar and pestle under liquid nitrogen and stored at − 80°C. For extraction, 1 g of ground leaf powder was resuspended in lysis buffer with 1 mg/ml of proteinase K (19133, Qiagen) and 190 μg/ml of RNase A (19101, Qiagen). The lysis solution was incubated at 50 °C for at least 3.5 h with gentle rocking. Following centrifugation for 15 min at room temperature at 3220×g the supernatant was purified with genomic tip columns according to the manufacturer’s recommendations. After elution from the column, the DNA was precipitated with 0.7 volumes of isopropanol and inverted slowly until the appearance of a floating DNA mass, or “jellyfish”. The DNA was hooked out with a plastic loop and washed in fresh 80% ethanol for 1 min three times. Repeated washing in ethanol, a major modification in the protocol, has been shown to improve DNA purity without a decrease in yield [13]. The DNA pellet was resuspended in EB buffer overnight at room temperature. The yield and quality of the DNA was assessed with a Broad Range dsDNA Qubit assay (Q32853, ThermoFisher, Waltham, MA, USA) and NanoDrop 8000 (ThermoFisher). In the event that absorbance ratios were not optimal [9, 61], potentially indicating the presence of contaminants, a repeat bead clean was performed. To visualize the DNA smear, pulse field gel electrophoresis (PFGE) was run with a 1% agarose TBE gel over 24 h (Initial switch 1 s, final switch 25 s, 6 volt/cm, 120° included angle, Chef III, Bio-Rad, Hercules, CA, USA) with MidRange PFG and Lambda PFG markers (N0342S, N0341S, NEB, Ipswich, MA, USA). Alternatively, HMW DNA was also imaged with a gDNA 165 Kb kit (FP-1002-0275) on the Femto Pulse system (Agilent, Santa Clara, CA, USA). A detailed procedure is deposited in protocols.io (dx.doi.org/10.17504/protocols.io.bafmibk6).
PacBio and Oxford Nanopore sequencing
CLR library preparation sequencing with PacBio Sequel I and II
SMRTbell libraries were constructed with SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences, Menlo Park, CA, USA, 100-938-900). The input DNA, with a size distribution mode predominantly above 80 Kb, was processed for SMRTbell library construction without any shearing (except for one library; Additional file 1: Table S2). Initially, 15–20 μg of gDNA was cleaned-up and concentrated with 0.45x AMPure PB (Pacific Biosciences, 100-265-900), then 10 μg of gDNA was used in the first enzymatic reactions to remove single-strand overhangs followed by the DNA damage repair, end-repair/A-tailing reaction and finally, adapter ligation. The SMRTbell libraries were then purified with 0.45x AMPure PB beads before size selection on the BluePippin system (Sage Science, Beverley, MA, USA) with a 30-Kb cut-off using a 0.75% agarose cassette with U1 ladder. Following size selection, the libraries were given a final 1x AMPure PB bead clean-up and eluted in 10 μl of EB (Pacific Bioscience, 101-633-500). The concentration and size of SMRTbell were assessed with the Qubit dsDNA assay kit (Thermo Fisher Scientific, Waltham, MA, USA; Q32854), and the Genomic DNA 165 Kb Kit on the FEMTO Pulse, respectively. For Sequel I, SMRTbell libraries were prepared for sequencing by annealing to Sequencing Primer v4 and Polymerase 3.0 with Sequel Sequencing kit 3.0 (Pacific Biosciences, 101-597-900) and Sequel Binding and Internal Control Kit 3.0 (Pacific Biosciences, 101-626-600), and SMRT Cell 1M v3 LR (Pacific Biosciences, 101-531-001) according to SMRT link Sample Setup v.7.0 instructions and sequencing for 10 or 20 h without any pre-extension time. For Sequel II, SMRTbells were annealed with primer v4 and polymerase with Sequel II Binding Kit 2.0 and Internal Control Kit 1.0 (Pacific Biosciences,101-842-900), SMRT Cell 8M (Pacific Biosciences, 101-389-001), and according to SMRT link Sample Setup v.8.0 and sequence for 15 or 30 h without any pre-extension time.
HiFi library preparation and sequencing with PacBio Sequel II
Fifteen to 20 μg of gDNA was diluted in EB and was sheared using g-TUBE or Megaruptor 2. Samples were loaded into g-TUBEs (Covaris, 520079) and sheared with an Eppendorf 5424 (Eppendorf, Hamburg, Germany) for 2 min each spin. A repeat spin was implemented to make sure the entire gDNA had passed through the orifice. Alternatively, samples were sheared using the Megaruptor 2 (Diagenode, Denville, USA) with Long Hydropores (E07010002) and Hydrotubes (C30010018). For shearing, small scale test shears were performed to make sure that the mode of the fragments was in the 15–20-Kb size range (size checked with FEMTO Pulse). A minimum 10 μg of sheared and 0.45x AMPure purified gDNA was carried into SMRTbell construction by using Express Template Prep Kit 2.0 + Enzyme Clean Up (101-843-100).
Additional step of nuclease treatment of the HiFi library after the ligation step was done to remove any non-intact SMRTbell templates. Following nuclease treatment, the SMRTbell library was size-selected with 3.1x of diluted 35% v/v AMPure PB beads or the BluePippin system. The concentration and size of HiFi SMRTbell were assessed with Qubit dsDNA assay kit and gDNA 165 Kb kit of FEMTO Pulse, respectively. The SMRTbell libraries were annealed and bound with sequencing primer v2 (101-847-900), and Sequel II DNA polymerase 2.0 from Sequel II Binding kit 2.0, 101-842-900, respectively, using conditions specified in SMRT Link Sample Setup v.8.0. The final sample bound complex was sequenced with Sequel II Sequencing Kit 2.0 (101-820-200), and SMRT cell 8M Tray (101-389-001), and ran for 30 h with 2 or 4 h of pre-extension.
Oxford Nanopore sequencing
Oxford Nanopore sequencing was performed on the GridION sequencer only for the two Eucalyptus species (E. camaldulensis and E. rudis). Prior to library preparation, short fragments were depleted from the extracted HMW DNA using BluePippin with a 30-Kb cut-off or the Short Read Eliminator XL kit (SRE XL, Circulomics, Baltimore, MD, USA) using the manufacturer’s instructions. The size-selected DNA was cleaned-up with AMPure XP beads and then processed into libraries using genomic DNA by ligation protocol (SQK-LSK109, Oxford Nanopore, Oxford, UK). Briefly, DNA (E. camaldulensis, 1.3 μg; E. rudis, 4 μg) was repaired and end-prepped prior to bead clean-up and adapter ligation. The ligated product is bead cleaned with Long Fragment Buffer (LFB) and eluted. The prepared library (E. camaldulensis, 8 fmol; E. rudis, 50 fmol) is then loaded onto GridION (FLO-MIN106D).
Bioinformatics and genome assembly
Sequencing data analysis
For PacBio SMRTLink v8.0/v9.0 was used for designing and monitoring sequencing runs and analyzing and managing sequence data. For ONT MinKNOW Core 3.6.0 was used for data acquisition and real-time analysis. Reads were base called using Guppy 3.2.8 from FAST5 files to produce FASTQ files. Statistical analyses were performed using R 3.6.1 [62].
The quality metrics of base called reads were also calculated using LongQC version 1.2 [41]. We applied pb-hifi, pb-sequel, and ont-ligation profiles for PacBio HiFi, PacBio CLR, and Oxford Nanopore datasets, respectively.
Genome size estimation
GenomeScope 2.0 [43] was used to estimate, in silico, the genome sizes of both Eucalyptus species. The software was run with the following parameters: [k-mer length = 21, Ploidy = 2, Max k-mer coverage = − 1, average k-mer coverage for polyploid genome = − 1]. We calculated the k-mer distribution, which we then fed to GenomeScope, using JellyFish [63] with the following parameters: [jellyfish count -C -m 21 -s 1000000000]. For the peanut genome, we run the k-mer analysis using kmc v3.1.2 [64] [-k21 -m500 -ci1 -cs 100000000], then kmc_tools [-cx100000000] then GenomeScope2 with the following [ploidy = 4, kmer = 21].
Genome assemblies
We assembled the HiFi data for both Eucalyptus species using the newly released assembler hifiasm v0.8 - r279 [45]. We run hifiasm with default settings (-r3 -a4 -k51 -w51 -f37 -D5.0 -N100 -z0 -m10000000 -p 100000 -n3 -x0.8 -y0.2). We also separated haplotigs using the purge_dups module in hifiasm using default settings (-l2 -s0.75 -O1). Output from hifiasm is two GFA graph files: one for the primary contigs and another for the alternative haplotigs. We converted the GFA files to the FASTA format using gfatools v0.4-r179-dirty [65]. To produce the total assembly statistics, we remerged the primary and alternative haplotigs using seqkit [66].
For PacBio CLR we used the Canu assembler v2 (github r9818) [44]. To force Canu to keep haplotigs separate, we set the following parameters: batOptions=-dg 3 -db 3 -dr 1 -ca 500 -cp 50. For speed gains, we run Canu in cluster mode. Where deep coverage was available, we increased the amount of data used in the assembly beyond the default 40x coverage.
Polishing of genome assemblies
We polished the CLR-based assemblies using the Arrow algorithm in the gcpp tool from PacBio’s SMRT Link v8.0 stack. First, we aligned the raw CLR data against the initial assembly using pbmm2 v1.1.0, which is a version of Minimap2 [67] adapted to PacBio’s native format. The alignment is then used for consensus calling and polishing using gcpp v1.0.0. We repeated the process for two additional polishing cycles whereby we feed the polished assembly from the previous cycle as the alignment reference in the next cycle. The HiFi-based assemblies do not require additional polishing to the highly accurate starting CCS sequences [44].
Genome assemblies’ assessment
We generated comprehensive assembly statistics using QUAST-LG v5.0.2 [68]. To assess the biological integrity of the assemblies, we used BUSCO v3 [69] as a proxy of genome completeness.
Haplotig purging
We removed haplotigs for the CLR-based assemblies using purge_dups [70] using default settings. We manually inspected read depth to adjust coverage cut-offs where necessary for best performance. The hifiasm pipeline, on the other hand, is able to separate haplotigs for the HiFi-based assemblies without additional tools (Additional file 1: Fig. S12). We validated the success of this step for all assemblies using BUSCO v3 [69].
Supplementary Information
Additional file 1. Supplementary materials. Includes supplementary results and all supplementary tables (S1-S8) and figures (S1-S12).
Acknowledgements
We thank Dr. Boubacar Kountche and Prof. Salim AlBabili for insightful discussions on the wet-lab work and their constructive support for BCL, and the KAUST Supercomputing Core Lab for providing support for the computing resources. We thank Dr. Dean Nicolle for the phenotypic identification of the two Eucalyptus species and the three anonymous reviewers for their constructive criticism. We thank the KAUST Core Labs management for supporting this study. In fond memory of our talented colleague Dr. Kamel Jabbari.
Review history
The review history is available as Additional file 2.
Peer review information
Barbara Cheifet was the primary editor of this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Authors’ contributions
P.D, K.C., and A.P performed laboratory work, PacBio and ONT sequencing, and interpreted the results. S.B. carried out the genome assemblies and all bioinformatics analyses with support from Y.F. K.J. estimated size and the heterozygosity level on the unassembled HiFi sequencing data. M.P. and R.S. provided plant samples for sequencing. MS.C. supported the study and provided managerial guidance. Y.F. performed LongQC analyses, advised on bioinformatic analysis. L.E. conceived and planned this study, interpreted the results, and supervised all aspects of this work. L.E., P.D., and S.B. wrote the manuscript with input from K.C and A.P. The authors read and approved the final manuscript.
Authors’ information
Twitter handle: @patrick_driguez (Patrick Driguez), @mrdoods (Salim Bougouffa), @KAUSTBCL (Ming Sin Cheung)
Funding
This study used resources provided by the KAUST Core Labs.
Availability of data and materials
The assembled genomes and relevant raw data are available under NCBI BioProject ID PRJNA674723 [71], PRJNA739547 [72], and PRJNA737587 [73] and WGS accessions JAHSVA000000000, JAHSVB000000000, JAHMTN000000000, JAIGYT000000000, and JAIGYU000000000.
Declarations
Ethics approval and consent to participate
Not applicable
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Kamel Jabbari is deceased.
Patrick Driguez, Salim Bougouffa, Karen Carty and Alexander Putra contributed equally to this work.
References
- 1.Burgess MG, Gaines SD. The scale of life and its lessons for humanity. Proc Natl Acad Sci. 2018;115:6328–6330. doi: 10.1073/pnas.1807019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shukla PR, Skea J, Buendia EC, Masson-Delmotte V, Pörtner H-O, Roberts DC, et al. IPCC, 2019: Summary for Policymakers. In: Climate change and land: an IPCC special report on climate change, desertification, land degradation, sustainable land management, food security, and greenhouse gas fluxes in terrestrial ecosystems. 2019. https://www.ipcc.ch/srccl/chapter/summary-for-policymakers.
- 3.Sala OE, Chapin FS, Armesto JJ, Berlow E, Bloomfield J, et al. Global biodiversity scenarios for the year 2100. Science. 2000;287:1770–1774. [DOI] [PubMed]
- 4.Surfing the genomic new wave. Nat Plants. 2018;4:393–3.https://www.nature.com/articles/s41477-018-0209-7. [DOI] [PubMed]
- 5.Logsdon GA, Vollger MR, Eichler EE. Long-read human genome sequencing and its applications. Nat Rev Genet. 2020;21:597–614. doi: 10.1038/s41576-020-0236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miga KH, Koren S, Rhie A, Vollger MR, Gershman A, Bzikadze A, et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature. 2020;585:79–84. doi: 10.1038/s41586-020-2547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36:338–345. doi: 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT, et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat Biotechnol. 2019;37:1155–1162. doi: 10.1038/s41587-019-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung H, Winefield C, Bombarely A, Prentis P, Waterhouse P. Tools and strategies for long-read sequencing and de novo assembly of plant genomes. Trends Plant Sci. 2019;24:700–724. doi: 10.1016/j.tplants.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 10.Ou S, Liu J, Chougule KM, Fungtammasan A, Seetharam AS, Stein JC, et al. Effect of sequence depth and length in long-read assembly of the maize inbred NC358. Nat Commun. 2020;11:2288. doi: 10.1038/s41467-020-16037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Seetharam AS, Chougule K, Ou S, Swentowsky KW, Gent JI, et al. Gapless assembly of maize chromosomes using long-read technologies. Genome Biol. 2020;21:121. doi: 10.1186/s13059-020-02029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inglis PW, de Pappas MCR, Resende LV, Grattapaglia D. Fast and inexpensive protocols for consistent extraction of high quality DNA and RNA from challenging plant and fungal samples for high-throughput SNP genotyping and sequencing applications. PLOS ONE. 2018;13:e0206085. doi: 10.1371/journal.pone.0206085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mayjonade B, Gouzy J, Donnadieu C, Pouilly N, Marande W, Callot C, et al. Extraction of high-molecular-weight genomic DNA for long-read sequencing of single molecules. BioTechniques. 2016;61:203–205. doi: 10.2144/000114460. [DOI] [PubMed] [Google Scholar]
- 14.Vaillancourt B, Buell CR. High molecular weight DNA isolation method from diverse plant species for use with Oxford Nanopore sequencing. 2019; Available from: http://biorxiv.org/lookup/doi/10.1101/783159.
- 15.Varma A, Padh H, Shrivastava N. Plant genomic DNA isolation: an art or a science. Biotechnol J. 2007;2:386–392. doi: 10.1002/biot.200600195. [DOI] [PubMed] [Google Scholar]
- 16.Zhang M, Zhang Y, Scheuring CF, Wu C-C, Dong JJ, Zhang H-B. Preparation of megabase-sized DNA from a variety of organisms using the nuclei method for advanced genomics research. Nat Protoc. 2012;7:467–478. doi: 10.1038/nprot.2011.455. [DOI] [PubMed] [Google Scholar]
- 17.Healey A, Furtado A, Cooper T, Henry RJ. Protocol: a simple method for extracting next-generation sequencing quality genomic DNA from recalcitrant plant species. Plant Methods. 2014;10:21. doi: 10.1186/1746-4811-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lewin HA, Robinson GE, Kress WJ, Baker WJ, Coddington J, Crandall KA, et al. Earth BioGenome project: sequencing life for the future of life. Proc Natl Acad Sci. 2018;115:4325–4333. doi: 10.1073/pnas.1720115115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng S, Melkonian M, Smith SA, Brockington S, Archibald JM, Delaux P-M, et al. 10KP: a phylodiverse genome sequencing plan. GigaScience. 2018;7:1–9. doi: 10.1093/gigascience/giy013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Debieu M, Kanfany G, Laplaze L. Pearl millet genome: lessons from a tough crop. Trends Plant Sci. 2017;22:911–913. doi: 10.1016/j.tplants.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Jiao Y, Peluso P, Shi J, Liang T, Stitzer MC, Wang B, et al. Improved maize reference genome with single-molecule technologies. Nature. 2017;546:524–527. doi: 10.1038/nature22971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- 23.Varshney RK, Shi C, Thudi M, Mariac C, Wallace J, Qi P, et al. Pearl millet genome sequence provides a resource to improve agronomic traits in arid environments. Nat Biotechnol. 2017;35:969–976. doi: 10.1038/nbt.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhuang W, Chen H, Yang M, Wang J, Pandey MK, Zhang C, et al. The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nat Genet. 2019;51:865–876. doi: 10.1038/s41588-019-0402-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertioli DJ, Jenkins J, Clevenger J, Dudchenko O, Gao D, Seijo G, et al. The genome sequence of segmental allotetraploid peanut Arachis hypogaea. Nat Genet. 2019;51:877–884. doi: 10.1038/s41588-019-0405-z. [DOI] [PubMed] [Google Scholar]
- 26.Zhang L, Cai X, Wu J, Liu M, Grob S, Cheng F, et al. Improved Brassica rapa reference genome by single-molecule sequencing and chromosome conformation capture technologies. Hortic Res. 2018;5:1–11. doi: 10.1038/s41438-017-0012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li P, Su T, Zhao X, Wang W, Zhang D, Yu Y, et al. Assembly of the non-heading pak choi genome and comparison with the genomes of heading Chinese cabbage and the oilseed yellow sarson. Plant Biotechnol J. 2021;19:966–976. doi: 10.1111/pbi.13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barchi L, Pietrella M, Venturini L, Minio A, Toppino L, Acquadro A, et al. A chromosome-anchored eggplant genome sequence reveals key events in Solanaceae evolution. Sci Rep. 2019;9:11769. doi: 10.1038/s41598-019-47985-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pearlstein SL, Felger RS, Glenn EP, Harrington J, Al-Ghanem KA, Nelson SG. Nipa (Distichlis palmeri): a perennial grain crop for saltwater irrigation. J Arid Environ. 2012;82:60–70. doi: 10.1016/j.jaridenv.2012.02.009. [DOI] [Google Scholar]
- 30.Glenn EP, Anday T, Chaturvedi R, Martinez-Garcia R, Pearlstein S, Soliz D, et al. Three halophytes for saline-water agriculture: an oilseed, a forage and a grain crop. Environ Exp Bot. 2013;92:110–121. doi: 10.1016/j.envexpbot.2012.05.002. [DOI] [Google Scholar]
- 31.Reddy MP, Shah MT, Patolia JS. Salvadora persica, a potential species for industrial oil production in semiarid saline and alkali soils. Ind Crops Prod. 2008;28:273–278. doi: 10.1016/j.indcrop.2008.03.001. [DOI] [Google Scholar]
- 32.Monfared MA, Samsampour D, Sharifi-Sirchi GR, Sadeghi F. Assessment of genetic diversity in Salvadora persica L. based on inter simple sequence repeat (ISSR) genetic marker. J Genet Eng Biotechnol. 2018;16:661–667. doi: 10.1016/j.jgeb.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, et al. The genome of Eucalyptus grandis. Nature. 2014;510:356–362. doi: 10.1038/nature13308. [DOI] [PubMed] [Google Scholar]
- 34.Schalamun M, Nagar R, Kainer D, Beavan E, Eccles D, Rathjen JP, et al. Harnessing the MinION: an example of how to establish long-read sequencing in a laboratory using challenging plant tissue from Eucalyptus pauciflora. Mol Ecol Resour. 2019;19:77–89. doi: 10.1111/1755-0998.12938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirakawa H, Nakamura Y, Kaneko T, Isobe S, Sakai H, Kato T, et al. Survey of the genetic information carried in the genome of Eucalyptus camaldulensis. Plant Biotechnol. 2011;28:471–480. doi: 10.5511/plantbiotechnology.11.1027b. [DOI] [Google Scholar]
- 36.Wang W, Das A, Kainer D, Schalamun M, Morales-Suarez A, Schwessinger B, et al. The draft nuclear genome assembly of Eucalyptus pauciflora: a pipeline for comparing de novo assemblies. GigaScience. 2020;9:1–12. doi: 10.1093/gigascience/giz160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi JY, Lye ZN, Groen SC, Dai X, Rughani P, Zaaijer S, et al. Nanopore sequencing-based genome assembly and evolutionary genomics of circum-basmati rice. Genome Biol. 2020;21:21. doi: 10.1186/s13059-020-1938-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Belser C, Istace B, Denis E, Dubarry M, Baurens F-C, Falentin C, et al. Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat Plants. 2018;4:879–887. doi: 10.1038/s41477-018-0289-4. [DOI] [PubMed] [Google Scholar]
- 39.Simbolo M, Gottardi M, Corbo V, Fassan M, Mafficini A, Malpeli G, et al. DNA qualification workflow for next generation sequencing of histopathological samples. PLOS ONE. 2013;8:e62692. doi: 10.1371/journal.pone.0062692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pacific Biosciences. Guide - step-by-step run performance evaluation. 2020. Available from: https://www.pacb.com/documentation/guide-step-by-step-run-performance-evaluation/.
- 41.Fukasawa Y, Ermini L, Wang H, Carty K, Cheung M-S. LongQC: a quality control tool for third generation sequencing long read data. G3 Genes Genomes Genet. 2020;10:1193–1196. doi: 10.1534/g3.119.400864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Yang X, Tian L, Chen L, Yu W. Identification of peanut (Arachis hypogaea) chromosomes using a fluorescence in situ hybridization system reveals multiple hybridization events during tetraploid peanut formation. New Phytol. 2016;211:1424–1439. doi: 10.1111/nph.13999. [DOI] [PubMed] [Google Scholar]
- 43.Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020;11:1432. doi: 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, Grothe R, et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 2020;30:1291–1305. doi: 10.1101/gr.263566.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021;18:170–175. doi: 10.1038/s41592-020-01056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murigneux V, Rai SK, Furtado A, Bruxner TJC, Tian W, Harliwong I, et al. Comparison of long-read methods for sequencing and assembly of a plant genome. GigaScience. 2020;9:1–11. doi: 10.1093/gigascience/giaa146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37:540–546. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- 48.Ruan J, Li H. Fast and accurate long-read assembly with wtdbg2. Nat Methods. 2020;17:155–158. doi: 10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vollger MR, Logsdon GA, Audano PA, Sulovari A, Porubsky D, Peluso P, et al. Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Ann Hum Genet. 2020;84:125–140. doi: 10.1111/ahg.12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Banchi E, Ametrano CG, Greco S, Stanković D, Muggia L, Pallavicini A. PLANiTS: a curated sequence reference dataset for plant ITS DNA metabarcoding. Database. 2020;2020:baz155. [DOI] [PMC free article] [PubMed]
- 51.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 52.Rhie A, McCarthy SA, Fedrigo O, et al. Towards complete and error-free genome assemblies of all vertebrate species. Natur. 2021;592:737–46. [DOI] [PMC free article] [PubMed]
- 53.Logsdon GA, Vollger MR, Hsieh P, Mao Y, Liskovykh MA, Koren S, et al. The structure, function and evolution of a complete human chromosome 8. Nature. 021;593:101–7. [DOI] [PMC free article] [PubMed]
- 54.Mishra DC, Lal SB, Sharma A, Kumar S, Budhlakoti N, Rai A. Strategies and tools for sequencing and assembly of plant genomes. In: Kumar Chakrabarti S, Xie C, Kumar Tiwari J, editors. Potato genome. Cham: Springer International Publishing; 2017. pp. 81–93. [Google Scholar]
- 55.Zhou Y, Chebotarov D, Kudrna D, Llaca V, Lee S, Rajasekar S, et al. A platinum standard pan-genome resource that represents the population structure of Asian rice. Sci Data. 2020;7:113. doi: 10.1038/s41597-020-0438-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eucalyptus camaldulensis (Murray red gum) genome assembly ASM1418270v1: NCBI. Available from: https://www.ncbi.nlm.nih.gov/assembly/GCA_014182705.1
- 57.Bertioli DJ, Cannon SB, Froenicke L, Huang G, Farmer AD, Cannon EKS, et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet. 2016;48:438–446. doi: 10.1038/ng.3517. [DOI] [PubMed] [Google Scholar]
- 58.Kyriakidou M, Tai HH, Anglin NL, Ellis D, Strömvik MV. Current strategies of polyploid plant genome sequence assembly. Front Plant Sci. 2018;9:1–15. doi: 10.3389/fpls.2018.01660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Darwin tree Of life. 2020. Available from: https://www.darwintreeoflife.org/
- 60.Eucalyptus genome project- BioProject ID 509734: NCBI. Available from: https://www.ncbi.nlm.nih.gov/bioproject/509734
- 61.Pacific Biosciences. Technical note: preparing DNA for PacBio HiFi- sequencing extraction and quality control. Prep. DNA PacBio HiFi Seq. — Extr. Qual. Control. 2020. Available from: https://www.pacb.com/wp-content/uploads/Technical-Note-Preparing-DNA-for-PacBio-HiFi-Sequencing-Extraction-and-Quality-Control.pd
- 62.R Core Team . R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2017. [Google Scholar]
- 63.Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kokot M, Długosz M, Deorowicz S. KMC 3: counting and manipulating k-mer statistics. Berger B, editor. Bioinformatics. 2017;33:2759–2761. doi: 10.1093/bioinformatics/btx304. [DOI] [PubMed] [Google Scholar]
- 65.Li H, Feng X, Chu C. The design and construction of reference pangenome graphs with minigraph. Genome Biol. 2020;21:265. doi: 10.1186/s13059-020-02168-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLOS ONE. 2016;11:e0163962. doi: 10.1371/journal.pone.0163962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mikheenko A, Prjibelski A, Saveliev V, Antipov D, Gurevich A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics. 2018;34:i142–i150. doi: 10.1093/bioinformatics/bty266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seppey M, Manni M, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness. In: Kollmar M, editor. Gene Predict. New York: Springer New York; 2019. pp. 227–245. [DOI] [PubMed] [Google Scholar]
- 70.Guan D, McCarthy SA, Wood J, Howe K, Wang Y, Durbin R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 2020;36:2896–2898. doi: 10.1093/bioinformatics/btaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Driguez P, Bougouffa S, Carty K, Putra A, Jabbari K, Reddy MP, et al. LeafGo - Eucalyptus and Peanut genome sequencing - BioProject ID 674723: NCBI; 2021. Available from: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA674723
- 72.Driguez P, Bougouffa S, Carty K, Putra A, Jabbari K, Reddy MP, et al. LeafGo - Eucalyptus and Peanut genome sequencing - BioProject ID 739547: NCBI; 2021. Available from: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA739547
- 73.Driguez P, Bougouffa S, Carty K, Putra A, Jabbari K, Reddy MP, et al. LeafGo - Eucalyptus and Peanut genome sequencing - BioProject ID 737587: NCBI; 2021. Available from: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA737587.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supplementary materials. Includes supplementary results and all supplementary tables (S1-S8) and figures (S1-S12).
Data Availability Statement
The assembled genomes and relevant raw data are available under NCBI BioProject ID PRJNA674723 [71], PRJNA739547 [72], and PRJNA737587 [73] and WGS accessions JAHSVA000000000, JAHSVB000000000, JAHMTN000000000, JAIGYT000000000, and JAIGYU000000000.