

Abstract
Background and Aims
Zanthoxylum is the only pantropical genus within Rutaceae, with a few species native to temperate eastern Asia and North America. Efforts using Sanger sequencing failed to resolve the backbone phylogeny of Zanthoxylum. In this study, we employed target-enrichment high-throughput sequencing to improve resolution. Gene trees were examined for concordance and sectional classifications of Zanthoxylum were evaluated. Off-target reads were investigated to identify putative single-copy markers for bait refinement, and low-copy markers for evidence of putative hybridization events.
Methods
A custom bait set targeting 354 genes, with a median of 321 bp, was designed for Zanthoxylum and applied to 44 Zanthoxylum species and one Tetradium species as the outgroup. Illumina reads were processed via the HybPhyloMaker pipeline. Phylogenetic inferences were conducted using coalescent and maximum likelihood methods based on concatenated datasets. Concordance was assessed using quartet sampling. Additional phylogenetic analyses were performed on putative single and low-copy genes extracted from off-target reads.
Key Results
Four major clades are supported within Zanthoxylum: the African clade, the Z. asiaticum clade, the Asian–Pacific–Australian clade and the American–eastern Asian clade. While overall support has improved, regions of conflict are similar to those previously observed. Gene tree discordances indicate a hybridization event in the ancestor of the Hawaiian lineage, and incomplete lineage sorting in the American backbone. Off-target putative single-copy genes largely confirm on-target results, and putative low-copy genes provide additional evidence for hybridization in the Hawaiian lineage. Only two of the five sections of Zanthoxylum are resolved as monophyletic.
Conclusions
Target enrichment is suitable for assessing phylogenetic relationships in Zanthoxylum. Our phylogenetic analyses reveal that current sectional classifications need revision. Quartet tree concordance indicates several instances of reticulate evolution. Off-target reads are proven useful to identify additional phylogenetically informative regions for bait refinement or gene tree based approaches.
Keywords: Fagara, gene tree concordance, off-target reads, quartet sampling, Rutaceae, target enrichment, Toddalia, Zanthoxyloideae, Zanthoxylum
INTRODUCTION
With the advances of next-generation sequencing (NGS) approaches in systematics, hitherto recalcitrant phylogenetic relationships, i.e. rapid radiations (Welch et al., 2016) or deep divergences (Zeng et al., 2014), can be tackled with increasingly large datasets at steadily decreasing cost (Straub et al., 2012). Most NGS approaches in systematics aim to achieve a reduced representation of the genome to exclude regions with low phylogenetic signal and reduce computational complexity (Albert et al., 2007; Gnirke et al., 2009; Hörandl and Appelhans, 2015; Zimmer and Wen, 2015). Different methods have emerged, varying in applicability at different taxonomic levels and with regard to sample conservation. For target-enrichment methods, regions of interest are captured and isolated via biotinylated RNA baits designed using reference data (Lemmon et al., 2012; Weitemier et al., 2014). One major advantage of target-enrichment methods is the applicability to herbarium and silica-gel-preserved material as well as fresh material (Villaverde et al., 2018). However, the greatest challenge is often to obtain and analyse high-quality genomic or transcriptomic sequence data from the target or closely related species to identify orthologous and phylogenetically informative regions a priori (Twyford and Ennos, 2012). This disadvantage is mediated by the increasing availability of transcriptomic and genomic data across the angiosperm tree of life.
In addition to the regions of interest, target enrichment also delivers sequence information of a varying percentage from off-target regions. Mapping rates of reads to baits are often in the range of 60–80 %, but rates of ≤20 % have also been reported (Schmickl et al., 2016; Soto Gomez et al., 2019; Tomasello et al., 2020). Thus, off-target reads may serve as a useful resource in target-enrichment approaches. While a fraction of the off-target reads has been frequently utilized to assemble plastid genes or genomes as a ‘by-product’ (e.g. Weitemier et al., 2014; Ma et al., 2021), the remaining off-target reads may be utilized further. They might be used to assemble additional, un-targeted single or low-copy regions, which in turn might be used to expand the existing dataset, refine the bait set for further approaches, and investigate reticulate evolution, or evolution of gene families amongst other purposes.
Zanthoxylum (prickly ash, yellowwood) belongs to subfamily Zanthoxyloideae (Appelhans et al., 2021) and represents the second largest genus within Rutaceae, with about 225 currently accepted species (Kubitzki et al., 2011). It is distributed in all continents except Europe and Antarctica with biodiversity hotspots in the (sub-)tropics. A few species are adapted to a colder climate and are native to North America and temperate eastern and South Asia (Reynel, 2017), where they have been widely used as spices (e.g. Sichuan pepper, sanshō pepper, timur) or herbal medicines (e.g. Lu et al. 2020). Most Zanthoxylum species can be easily recognized by thorny bosses on the trunk and branches, and prickles may be found at a pseudo-stipular position (Weberling, 1970) and/or along the rachis of leaves or leaflets (Zhang et al., 2008). Zanthoxylum has an alternate phyllotaxis with punctate, estipulate and usually pinnate leaves. The plants are usually dioecious and the perianth may be homo- or heterochlamydeous. Seeds stay attached to the opening fruits (follicles) (Hartley, 1966; Kubitzki et al., 2011) and may be dispersed by birds (Silva et al., 2008; Guerrero and Tye, 2009), mammals (Muller-Landau et al., 2008) and ants (Maschwitz et al., 1992; Reynel, 1995) or fish (Reys et al., 2009). Most species reproduce sexually (Kamiya et al., 2008; Costa et al., 2013), but apomixis via nucellar embryony has also been reported (Liu et al., 1987; Naumova, 1993). Due to a significant variation in the flower morphology of Zanthoxylum, Linnaeus (1753, 1759) differentiated between Zanthoxylum s.str. with homochlamydeous flowers, and Fagara L. with heterochlamydeous flowers. Brizicky (1962) hypothesized that the simple perianth in Zanthoxylum s.str. is a secondary condition derived from the double perianth of Fagara. Today, both Zanthoxylum s.str. and Fagara are united as the morphologically diverse Zanthoxylum s.l., since Zanthoxylum s.str. is deeply nested within Fagara (Appelhans et al., 2018). The most recent taxonomic treatment based on morphological traits was published by Reynel (2017) and will be used as the taxonomic framework herein. According to Reynel (2017), Zanthoxylum species formerly accounted to Fagara are ascribed to the pantropical section Macqueria and the American sections Tobinia and Pterota. Members of Zanthoxylum s.str. are divided into an American section Zanthoxylum and an Asian section Sinensis.
Phytochemical (Waterman, 2007) and DNA sequence data (Poon et al., 2007; Appelhans et al., 2018) have confirmed that Zanthoxylum is most closely related to Tetradium and Phellodendron from Asia and Fagaropsis from Africa and Madagascar. The monotypic Toddalia was recently merged with Zanthoxylum (Appelhans et al., 2018) and shows a broad distributional range from tropical Africa and Madagascar to eastern and south-eastern Asia. A rich fossil record is evident in Eocene Europe for all these genera except Fagaropsis (Chandler, 1961; Gregor, 1989; Collinson et al., 2012). Zanthoxylum has been absent from Europe since the late Miocene to early Pliocene (Geissert et al., 1990) but spread over all other continents except Antarctica (i.e. Graham and Larzen, 1969; Jacobs and Kabuye, 1987; Tiffney, 1994; Kershaw and Bretherton, 2007). Recently, we conducted a first worldwide phylogenetic study of Zanthoxylum with 99 specimens comprising 54 species (Appelhans et al., 2018). However, based on only two nuclear and two plastid markers, several nodes in the backbone phylogeny remained unresolved, especially regarding the American and Pacific lineages. The Pacific Zanthoxylum lineage was resolved as monophyletic in the plastid dataset but polyphyletic in the nuclear dataset, possibly related to a previous hybridization event.
Here, target enrichment is applied in Zanthoxylum. We first design a bait set based on newly generated transcriptome data and test its suitability for phylogenetic reconstructions in the genus. The main goal of this study is to improve phylogenetic resolution regarding the main clades within the genus (Appelhans et al., 2018). The large quantity of sequence data will help evaluate whether the low resolution in previous Sanger sequencing studies (Appelhans et al., 2018) was due to a lack of informative characters or cases of reticulate evolution or incomplete lineage sorting (ILS). An additional goal is to test the most recent sectional classification by Reynel (2017) using the phylogenetic framework. Finally, we aim to explore whether off-target reads can be used to identify additional informative regions that can be used in phylogenetic analyses and to improve and/or enlarge bait sets for future studies.
MATERIALS AND METHODS
RNA-seq and bait design
We employed a ‘made-to-measure’ design strategy (Kadlec et al., 2017) to increase bait specificity. Transcriptomic data of four Zanthoxylum L. accessions (representing three species) and three closely related outgroups served as foundation for bait design (Supplementary Data Table S1). Three transcriptomes were publicly available via the NCBI SRA archive, and four additional transcriptomes were generated in the course of this study (Supplementary Data Table S1). Young leaves of plants cultivated at Goettingen Botanical Garden were frozen in liquid nitrogen for RNA preservation. Total RNA was extracted using the RNeasy® Plant Mini Kit (Qiagen) as per the manufacturer’s instructions. Library preparation for Illumina sequencing was performed at the Transcriptome and Genome Analysis Laboratory Goettingen (TAL) using the TruSeq RNA Library Prep Kit v2 (Illumina, San Diego, CA, USA). Pooled libraries were run on an Illumina HiSeq 4000 to produce 50-bp single-end reads. Raw sequence data were trimmed using cutadapt v1.1.6 (Martin, 2011), removing adapter sequences with a minimum overlap of 10 bp and trimming read ends with a PHRED score <30. Trimmed reads with a remaining length of <35 bp were discarded. Trinity v2.5.1 (Grabherr et al., 2011; Haas et al., 2013) was used for de novo assembly of trimmed reads using default options with the exception of max_memory, which was set to 50 GB. Identification of single-copy orthologous loci was conducted as described in Tomasello et al. (2020) using a combination of MarkerMiner (Chamala et al., 2015) and custom python scripts (https://github.com/ClaudiaPaetzold/MarkerMinerFilter). Exons shorter than 120 bp were discarded. The variability between only Zanthoxylum sequence data was assessed and regions showing <0.5 % or >15 % variability were discarded. The obtained 745 exon sequences spanning 354 genes were further processed by Arbor Biosciences (myBaits®, Ann Arbor, MI, USA), which included masking, to produce a set of 20 000 80-mer baits.
Taxon sampling and DNA extraction
We sampled a total of 47 Zanthoxylum specimens representing 44 different species and one specimen of Tetradium (Table 1) as outgroup. All major distributional areas and sections according to Reynel (2017) are covered. Total DNA was extracted from herbarium or silica-dried material using a variation of the CTAB protocol by Doyle and Doyle (1987) or the DNeasy Plant® Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions.
Table 1.
Specimens sampled for target enrichment and phylogenetic analysis including voucher information, date of collection and geographic region. Z., Zanthoxylum
| Species name | Voucher | Collection Date | Origin |
|---|---|---|---|
| Tetradium austrosinense | Wen 13625 (US) | 2016 | China, Guangdong |
| Z. acuminatum ssp. juniperinum | Ortíz 1735 (US) | 1971 | Guatemala |
| Z. acuminatum ssp. juniperinum | Torres-Diaz 1033 (–) | – | Mexico |
| Z. ailanthoides | Konta 18323 (L) | 1997 | Japan, Honshu |
| Z. americanum | Appelhans MA 542 (GOET) | 2015 | Germany, Göttingen (cult.) |
| Z. armatum | Wen 12410 (US) | 2013 | Indonesia, Bali |
| Z. asiaticum | Wen 13271 (US) | 2016 | China, Guangdong |
| Z. brachyacanthum | Forster PIF28159 (L) | 2002 | Australia, Queensland |
| Z. bungeanum | Wen et al. 1541 (US) | 2007 | China, Yunnan |
| Z. caribaeum ssp. caribaeum | Gabriel Flores F. 5332 (MO) | 2003 | Mexico |
| Z. chalybeum 1 | Mhoro 6225 (US) | - | Tanzania |
| Z. chalybeum 2 | Seegeler 2231 (MO) | 1972 | Ethiopia |
| Z. clava-herculis | Wen 12771 (US) | 2014 | USA, Florida |
| Z. coco | Nee & Wen 53858 (US) | 2008 | Bolivia |
| Z. coreanum | Appelhans MA 710 (GOET) | 2017 | Germany, Göttingen (cult.) |
| Z. dimorphophyllum | Tsiang 6852 (US) | 1930 | China, Guizhou |
| Z. dipetalum | Trauernicht 750 (US) | 2009 | USA, Kauai |
| Z. dissitum | Wen 12840 (US) | 2015 | China, Hubei |
| Z. echinocarpum | Wen 13309 (US) | 2016 | China, Guangdong |
| Z. esquirolii | Wen 12813 (US) | 2015 | China, Yunnan |
| Z. fagara ssp. culantrillo | Sánchez 1310 (US) | 2014 | Mexico |
| Z. fagara ssp. fagara | Jestrow 2015-FTG-55 (US) | 2015 | USA, Florida (cult.) |
| Z. foliolosum | Zarate-Marcos 124 (MO) | 2006 | Mexico |
| Z. gilletii | Hamill 1079 (MO) | 1977 | Uganda |
| Z. hawaiiense | Wood 12463 (US) | 2007 | USA, Kauai |
| Z. heterophyllum | Loreno 2607 (MO) | 1979 | Mauritius |
| Z. holtzianum | Rulangaranga 199 (US) | – | Tanzania |
| Z. kauaense | Wood 15131 (US) | 2012 | USA, Kauai |
| Z. madagascariense | Capuron 28595-SF (US) | – | Madagascar |
| Z. mayu | Skottsberg 78 (US) | 1955 | Chile, Juan Fernández Islands |
| Z. mollissimum | Reyes-Garcia 5972 (MO) | 2003 | Mexico |
| Z. nadeaudii | Meyer 1038 (US) | 2002 | French Polynesia, Austral Islands |
| Z. nitidum | Wen 13280 (US) | 2016 | China, Guangdong |
| Z. ovalifolium | Schodde 2967 (US) | 1962 | New Guinea |
| Z. ovatifoliolatum | Swanepoel SWA3/76 (US) | 2004 | Namibia |
| Z. oxyphyllum | Wen et al. 2916 (US) | 2009 | China, Xizang (Tibet) |
| Z. paniculatum | Magdalena 001 (MO) | 2007 | Mauritius, Rodrigues Island |
| Z. pinnatum | Drake 282 (US) | 1995 | Tonga, Vava‘u group |
| Z. poggei | Harris & Fay 1030 (MO) | 1988 | Central African Republic |
| Z. rhetsa | Wen 12411 (US) | 2013 | Indonesia, Bali |
| Z. rhodoxylon | Wen 11907 (US) | 2011 | Jamaica |
| Z. rhoifolium | Stevens 33275 (MO) | 2012 | Nicaragua |
| Z. sapindoides ssp. sapindoides | Jestrow 2015-FTG-63 (US) | 2015 | USA, Florida (cult.) |
| Z. scandens | Wen 13279 (US) | 2016 | China, Guangdong |
| Z. schinifolium | Wen 12055 (US) | 2011 | China, Jiangxi |
| Z. tragodes | Liogier 12644 (US) | 1968 | Dominican Republic |
| Z. viride | Jongkind & Bilivegui 11383 (MO) | 2012 | Guinea |
| Z. zanthoxyloides | Jongkind (US) | 1993 | Ghana |
Library preparation for target enrichment and sequencing
For each sample we used a Q800R sonicator (Qsonica, Newtown, CT, USA) to shear 800 ng of DNA to an approximate fragment size of 350 bp. Library preparation was conducted using the NEBNext® Ultra™ II DNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, MA, USA) with NEBNext Multiplex Oligos for Illumina® (Dual Index Primers Set 1) and AMPure XP magnetic beads (Beckman Coulter, Brea, CA, USA). DNA content and quality of indexed libraries were examined with a Qubit 4.0 using the high-sensitivity kit (ThermoFisher Scientific, Waltham, MA, USA) and a 1 % agarose gel. Samples were pooled in pairs of two or four based on their respective quality, resulting in pools containing a total of 500 ng equimolar DNA. Hybridization of baits to pooled libraries was conducted according to the myBaits® - Hybridization Capture for Targeted NGS Manual v4.01 (April 2018; Arbor Biosciences) with the exception of hybridization time, which was extended to 40 h. Captured DNA libraries were amplified using the KAPA HiFi HotStart Ready Mix (Hoffmann-La Roche, Basel, Switzerland) with 14 cycles. Enriched libraries were purified with AMPure XP magnetic beads and checked for quality with qPCR, using i5 and i7 Illumina TruSeq primers, and Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Samples were sequenced on an Illumina HiSeq 4000 producing 2 × 150-bp paired-end reads (Illumina, San Diego, CA, USA) at Novogene (Sacramento, CA, USA).
Data analysis of targeted reads
The HybPhyloMaker pipeline (Fér and Schmickl, 2018) provided the bash scripts for all the steps from initial quality trimming of reads to species tree reconstruction. Trimmomatic v0.33 (Bolger et al., 2014) was used for adapter and quality trimming with thresholds set to a minimum of 65 bp read length and a minimum PHRED score of 30. Duplicated reads were removed with FastUniq v1.1 (Xu et al., 2012). Reads were mapped to the bait reference using bowtie2 v2.2.9 (Langmead and Salzberg, 2012), applying options --local and --very-sensitive. Kindel v0.1.4 (Constantinides and Robertson, 2017) was utilized to create consensus sequences with a minimum coverage of ×5. Consensus sequences were split into single-exon contigs, which were then compared with the original bait sequences with BLAT (Kent, 2002) using a minimum identity threshold of 85 %. Exons were aligned and concatenated using MAFFT v7.304 (Katoh and Standley, 2013) and catfasta2phyml.pl (Nylander, 2016). There is evidence for polyploidy in Zanthoxylum (Guerra, 1984; Stace et al., 1993; Kiehn and Lorence, 1996) and thus greater risk of paralogues in the dataset. HybPhyloMaker cannot filter against paralogues directly but considers the most abundant sequence to be the orthologue (Fér and Schmickl, 2018). SAMtools v1.8 and BCFtools v1.8 (Li et al., 2009) were used to filter for paralogous sequences, and the setting for the number of heterozygous sites was increased to eight due to the presence of polyploid samples with a potentially higher number of alleles of a gene in the dataset. Loci with >70 % missing data or >25 % missing taxa were removed from further analyses, leaving 258 of the targeted 354 genes after filtering. Phylogenetic inference was conducted with two different approaches, on the concatenated dataset and by coalescent analysis of gene trees. For the concatenated dataset a maximum likelihood (ML) analysis was conducted with ExaML v3.0 (Kozlov et al., 2015) as implemented in the HybPhyloMaker pipeline. Starting trees for ExaML including 100 bootstrap replicates were created with RAxML v8.2.12 (Stamatakis, 2014) under the GTR + G substitution model. For the coalescent analysis gene trees were estimated with RAxML v8.2.12 (Stamatakis, 2014) applying the GTR + G substitution model and 100 bootstrap replicates. Gene trees were combined into a single multi-NEWICK file (Junier and Zdobnow, 2010) and rooted using Tetradium as outgroup. ASTRAL-III v5.6.1 (Mirarab et al., 2014; Zhang et al., 2018) was employed for species-tree reconstruction. Phylogenetic trees were visualized in FigTree v1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
We analysed species-tree discord using quartet sampling (Pease et al., 2018), with a partitioned alignment and the ExaML phylogeny as input. For our approach, the minimum likelihood differential between the best and the second-best likelihood quartet tree was set to 2, and 300 quartet replicates were performed for every branch. For each node, quartet concordance (QC), quartet differential (QD) and quartet informativeness (QI) were computed. In addition, quartet fidelity (QF) values were computed per sample. QC values measure quartet concordances; QD values assess the ratio of the two possible discordance topologies. While QI values indicate the informative capacity of the dataset to resolve a respective node, QF values provide information on the amount of concordant topologies resolved when incorporating the specific taxon/specimen.
Analysis of off-target reads
To explore the possible utility of the by-catch, unmapped reads were extracted from the individual *.sam files resulting from HybPhyloMaker (Fér and Schmickl, 2018) using Samtools v1.9 (Li et al., 2009; Fig. 1). Unmapped reads were then assembled de novo using the software SPAdes v3.13.2 (Bankevich et al., 2012) and k values 21, 33, 55 and 77. Contigs were annotated using BLAST (Altschul et al., 1990) and the January 2020 release of UniProt (The UniProt Consortium, 2019) as the reference database with an e-value cut-off of 10 e−3 and reporting only the best hit. Blast hits were filtered to retain only entries with alignment lengths >120 bp, in order to facilitate the possible design of baits during subsequent bait refinement and to remove spurious hits. Trinotate v3.2.1 (Bryant et al., 2017) was used to create annotation reports for each sample. A custom python script (https://github.com/ClaudiaPaetzold/off-target-reads.git, v1.0.0) was used to compare annotation reports across samples. In a first step, contigs with blast hits to non-spermatophyte gene sequences were excluded as putative contaminations. We catalogued the origin of contigs representing putative contaminations into categories bacteria, fungi, insects, rodents, humans and non-spermatophyte embryophytes. For the remaining contigs annotated as spermatophyte genes, coverage across samples was assessed and filtered to a minimum of 24 (50 % of all samples). For these contigs, blast hits per gene per sample were assessed, and if more than eight in any sample the gene was marked as a putatively repetitive region and excluded. We elected to not filter to one blast hit per gene per sample, in order not to exclude multiple non-overlapping fragments for the same gene. For the remaining genes, sample-specific sequences were collected into per-gene *.fasta files and aligned using MAFFT v7.304 (Katoh and Standley, 2013) with a maximum of 100 iterations and the localpair option. Alignments were checked visually for sequences not overlapping with the remaining alignment (these were deleted) and sorted according to copy number status. Alignments in which each sample was represented by only one sequence were regarded as putative single-copy genes (pSCGs) in Zanthoxylum, the remainder as putative low-copy genes (pLCGs). A custom python script (https://github.com/ClaudiaPaetzold/off-target-reads.git) was used to remove columns containing only gaps and trim alignment ends to a sequence coverage of 75 %. Trimmed pSCG alignments were concatenated using the AMAS (Borowiec, 2016) python suite. RAxML v8.2.8 (Stamatakis, 2014) was used for ML-based tree inference on the concatenated pSCG alignment with the substitution model GTR + G. Statistical support was assessed with 1000 bootstrap replicates. In addition, gene trees of pSCG alignments were estimated with RAxML v8.2.8 and summarized in a coalescence framework using ASTRAL III v5.6.1. The resulting species trees were also subjected to quartet sampling (Pease et al., 2018) to further assess support and results were compared with those from on-target reads. Alignments of pLCGs were also visually checked and edited as necessary. Gene trees resulting from pLCGs were screened for taxon composition and the number of duplicated taxa. We differentiated between pLCG alignments in which only one or few specimens were represented by more than one sequence (pLCG_few), and putative gene families with multiple copies in all or nearly all samples (pLCG_most). As a case study, their potential to provide additional information for the Hawaiian Zanthoxylum species was assessed. The entire workflow (Fig. 1) and all custom python scripts are available on github (https://github.com/ClaudiaPaetzold/off-target-reads.git).
Fig. 1.

Flow chart of the analysis pipeline for the on- and off-target sequence reads. Intermediate results including file formats are in white boxes and final alignments in grey boxes. Intermediate steps in the pipeline including software or scripts used are depicted on connectors. Solid lines, off-target pipeline; dashed lines, on-target pipeline; grey arrows, subsequent phylogenetic analyses.
RESULTS
Raw data and processing of on-target reads
The Illumina HiSeq run resulted in an average of 11 343 488 raw reads (3 851 028–24 070 974; Supplementary Data Table S2). After quality trimming and deduplication 62·11 % of the reads remained. Of these, an average of 49·1 % (29·43–65·89 %) per sample mapped to the bait reference. Out of the 354 genes, 96 did not meet the thresholds of <70 % missing data and <25 % missing taxa, resulting in a final dataset of 258 genes. Of these 258 gene alignments, 231 included all 48 taxa and 11 included 47 taxa. The remaining 16 gene alignments contained between 36 and 46 taxa. The individual alignments ranged from 118 to 3294 bp in length with a median of 587·5 bp, and the concatenated alignment of all 258 genes comprised 187 686 bp. The percentage of missing data in the individual alignments ranged from 0 to 69·1 % with an average of 10·8 %. The individual alignments contained between 10 and 1030 variable sites each, of which 6–498 were parsimony-informative. The concatenated alignment contained 49 993 variable sites, 24 030 of which were parsimony-informative.
Phylogenetic analyses
The three major Zanthoxylum clades identified recently (Appelhans et al., 2018) are confirmed based on the analyses of the concatenated (Fig. 2) and coalescent datasets (Supplementary Data Fig. S1), and a fourth clade has emerged comprising Z. asiaticum only. Clade 1 comprises all sampled Zanthoxylum species endemic to continental Africa, Madagascar and Mauritius (100 % bootstrap support [BS, concatenated analysis, Fig. 2]/1·00 local posterior probability [lPP, coalescent analysis, Supplementary Data Fig. S1]). It is resolved as sister to the remainder of the genus (100 % BS/1·00 lPP), which consists of the major Clades 3 and 4, with Z. asiaticum (Clade 2) as sister to Clade 3 and Clade 4. The split between Clade 3 and Clade 4 is resolved with moderate support (79 % BS/0·68 lPP). Clade 3 comprises species from continental Asia, Malesia and Australia, with a monophyletic Pacific group embedded within (100 % BS/0·67 lPP). Clade 4 comprises species distributed across the Americas, with a second monophyletic Asian lineage and a species from the Juan Fernandéz Islands (Chile, South Pacific) embedded within (100 % BS/1·00 lPP). Several backbone nodes of Clade 4 are not well supported in the coalescent species tree (Supplementary Data Fig. S1) but all except one node received BS values of >90 in the concatenated analysis (Fig. 2). Support of more recent nodes is strong in either analysis. The species tree topologies from the concatenated and coalescent datasets are congruent except for three cases. Within Clade 1, Z. ovatifoliolatum is nested within a paraphyletic Z. chalybeum with low support in the concatenated analysis (Fig. 2; 58 % BS). Both species are resolved as sisters in the coalescent analysis with maximum support (Supplementary Data Fig. S1; 1.00 lPP). In the concatenated analysis, Z. dissitum and Z. scandens form a clade (Fig. 2; 75 % BS) that is sister to Z. echinocarpum. In the coalescent analysis, Z. dissitum and Z. echinocarpum are resolved as sisters, with low support (Supplementary Data Fig. S1; 0.56 lPP). Zanthoxylum caribaeum ssp. caribaeum is resolved as sister to the American clade that contains all species from sections Pterota and Tobinia within Clade 4 in the concatenated analysis (Fig. 2; 93 % BS). In the coalescent analysis it is not resolved as sister to sections Pterota and Tobinia, but as an early-branching lineage of Clade 4 in a part of the tree with low support (Supplementary Data Fig. S1; 0·76 lPP). None of these cases represent a hard conflict, since at least one reconstruction method did not succeed in resolving the respective branch with high support. Concerning the deeper nodes, the phylogeny based on the concatenated dataset (Fig. 2) lacks support only at the ancestral node of Clade 3 and Clade 4 (79 % BS) and at the ancestral node of American and Asian Zanthoxylum species within Clade 4 (85 % BS). In contrast, the coalescent species tree (Supplementary Data Fig. S1) shows a largely unsupported backbone regarding Clades 3 and 4. Both, however, show generally strongly supported younger nodes.
Fig. 2.

ExaML phylogenetic tree of Zanthoxylum based on the concatenated alignment of 258 targeted nuclear genes. Bootstrap values are shown at each branch. Branch lengths are proportional to the number of substitutions per position. Abbreviations after species names refer to their current sectional classification according to Reynel (2017): MAC, Macqueria; PTE, Pterota; SIN, Sinensis; TOB, Tobinia; ZAN, Zanthoxylum. World map source: https://pngkey.com.
Sectional relationships
Only two or three of the Zanthoxylum sections according to Reynel (2017) are resolved as monophyletic here. Sections Pterota and Tobinia each form a monophyletic group with high support in both concatenated and coalescent analyses (Fig. 2, Supplementary Data Fig. S1). Section Sinensis is only monophyletic if Z. dimorphophyllum is excluded. This species produces both homo- and heterochlamydeous flowers (Zhang et al., 2008). It has not been assigned to a section yet, but floral morphology places it in either section Sinensis (homochlamydeous) or Macqueria (heterochlamydeous). Section Zanthoxylum is polyphyletic. Of the two sampled species, Z. americanum is sister to a large part of Clade 4, while Z. mollissimum is resolved as sister to Z. clava-herculis of section Macqueria. Section Macqueria is polyphyletic and its members are scattered in all main clades. Zanthoxylum s.str. (= sections Sinensis and Zanthoxylum) is deeply nested within Fagara (= all other sections) and it is not monophyletic due to the placement of Z. americanum, which makes Zanthoxylum s.str. paraphyletic with respect to sections Pterota and Tobinia as well as Z. caribaeum.
Concordance analysis
A quartet concordance analysis was conducted for the main dataset with the alignment partitioned by genes and the concatenated ML tree as input (Pease et al., 2018; Fig. 3). Nodes with lower QC values, implying a significant degree of discordant quartets computed, are predominantly found in the backbone regions. A consistently high QI is inferred (0·78–1·0) over the entire topology, indicating that most of the quartets computed for a given branch were informative for the respective branch in question. Taxon-specific QF scores are in a similar range (0·75–1·0), hence suggesting all taxon placements within the concatenated ML tree are consistent. The split of Z. asiaticum as well as the divergences of Clade 3 and Clade 4 are associated with a medium QC and low QD, an indication that one discordant topology was predominantly inferred. In contrast, the ancestor to Clades 3 and Clade 4 is characterized by a combination of low QC and high QD values. Here, none of the discordant quartets is inferred significantly more often compared with the other. The most recent common ancestor of the Pacific lineage shows a low QC in combination with a QD value of 0, indicating strong discordance and a single alternative topology. The backbone of the American–Asian lineage within Clade 4 shows several nodes with low QC values combined with medium to low QD values and short branch lengths. In two cases of topological incongruences between the concatenated and the coalescent trees, the concatenated tree showed low bootstrap support (see Results section Phylogenetic analyses; placement of Z. dissitum, Z. echinocarpum and Z. scandens; placement of Z. chalybeum and Z. ovatifoliolatum). The respective nodes show low or medium QC values and mixed QD values.
Fig. 3.

Zanthoxylum quartet sampling scores (300 replicates) on the basis of the ExaML tree topology. On each node quartet concordance (QC)/quartet differential (QD)/quartet informativeness (QI) scores are displayed. Quartet fidelity (QF) scores are shown next to the species names. Quartet sampling scores in grey represent nodes with no or nearly no discord. World map source: https://pngkey.com. NA = not applicable.
Off-target reads
Target-enrichment sequencing resulted in an average of 3 524 882 off-target reads (1 153 183–8 409 705; Supplementary Data Table S2) per sample. On average, 16·95 % of the off-target reads (6·50–54·66 %) were identified as putative contaminations of the plant material (Supplementary Data Table S3, Fig. S2). The most prominent taxonomic group among the putative contaminants was fungi (531 contigs on average), followed by insects (501 contigs on average). Specimens showed major variation in their putative contamination patterns (Supplementary Data Table S3, Fig. S2). For example, fungi represent the largest percentage of putative contaminations in the Z. mayu specimen, while the highest number of contigs representing putative contaminations in the Z. chalybeum and Z. viride specimens are insects and bacteria, respectively. The Z. dimorphophyllum and Z. mayu samples were taken from herbarium specimens collected in 1930 and 1955, respectively. The percentage of putative contamination in the Z. dimorphophyllum sample ranks among the lowest in the whole dataset, and the composition of taxonomic groups among the putative contaminants is highly similar to the average. In contrast, the Z. mayu sample is the only sample that shows >50 % putative contaminants among the off-target contigs, and 74·5 % of them are of fungal origin (Supplementary Data Table S3, Fig. S2).
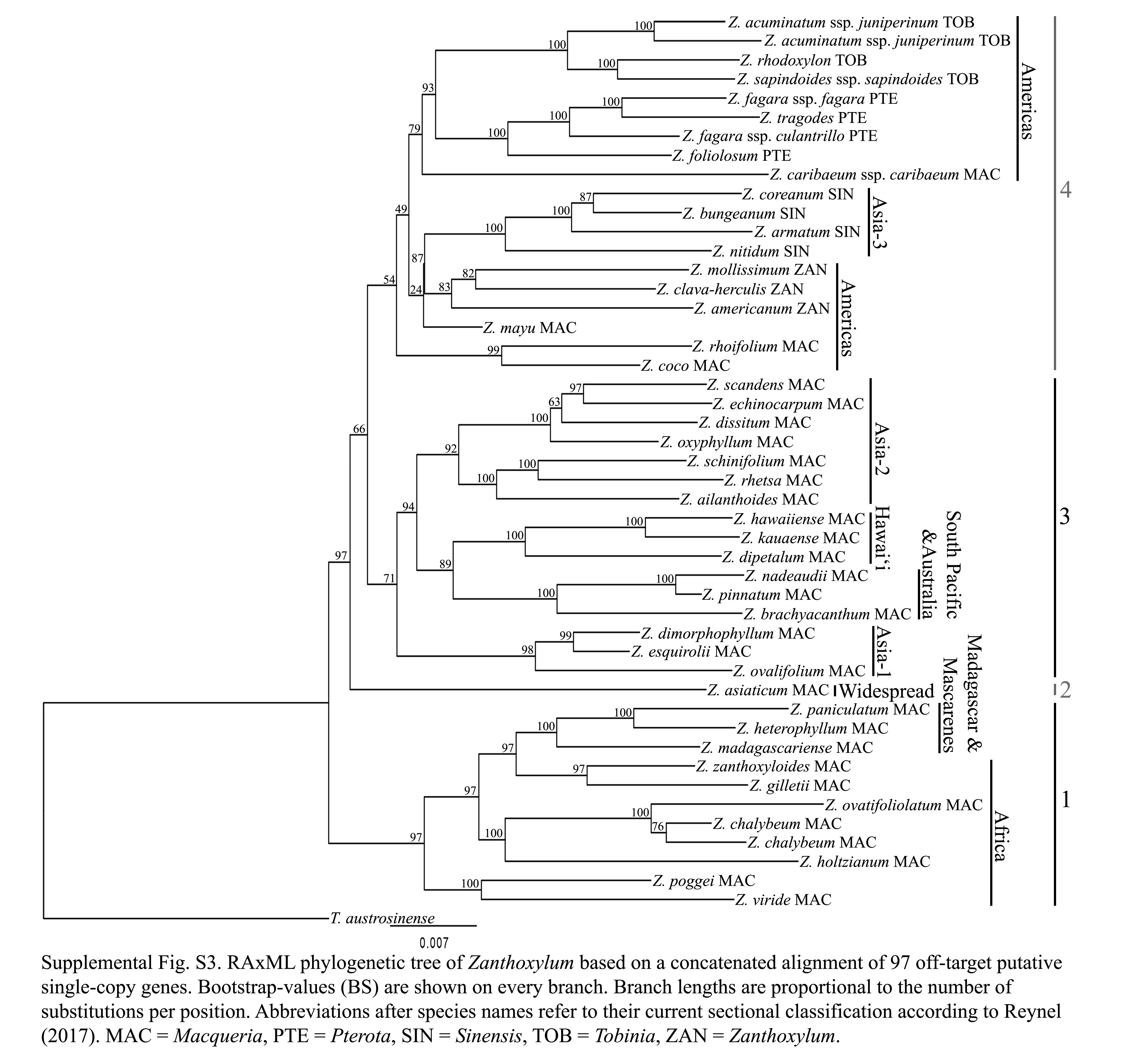
In total, 97 pSCGs alignments with a total length of 60 521 bp could be assembled, with four of these from the chloroplast genome and another four from the mitochondrial genome (Supplementary Data Table S4). The concatenated ML tree (Supplementary Data Fig. S3) based on the 97 pSCGs is largely congruent with the phylogenies based on targeted loci. However, node support is overall slightly lower (Fig. 2, Supplementary Data Figs S2 and S3). Like in the coalescent analysis of the on-target reads, the coalescent pSCG tree (Supplementary Data Fig. S4) is characterized by lower lPP values. It shows one major deviation from both on-target phylogenetic trees (Fig. 2, Supplementary Data Fig. S1) and the concatenated pSCGs (Supplementary Data Fig. S3) since it does not resolve the Hawaiian Zanthoxylum species as sister to the South Pacific and Australian lineage. In contrast to the on-target trees, Z. chalybeum is monophyletic in the concatenated pSCG tree (76 % BS) but polyphyletic in the coalescent pSCG tree, albeit with low support (0·46 lPP). Both the concatenated and coalescent pSCG topologies diverge from on-target trees with Z. echinocarpum and Z. scandens as sister to each other (97 % BS; 0·82 lPP), and Z. dissitum at the base to these (0·63 % BS; 0·29 lPP). Zanthoxylum mayu is resolved as sister to Z. coco and Z. rhoifolium with high support (100 % BS; 1·00 lPP) in the on-target trees but resolved as sister to the clade of sections Pterota and Tobinia in the coalescent pSCG tree (0·85 lPP). In the concatenated pSCGs tree its placement next to a clade with sections Sinensis and Zanthoxylum is not supported (0·24 % BS). The concatenated pSCG tree diverges from all other trees as it resolves Z. americanum as direct sister to Z. clava-herculis and Z. mollissimum (0·83 % BS; Supplementary Data Fig. S3). Partitioned quartet sampling of the coalescent pSCG gene tree results in overall lower QC and mixed QD values (not shown) in comparison with the quartet sampling analysis of targeted data. Likewise, QI (0·11–0·73) and QF values (0·18–0·41) are generally low in the coalescent pSCG tree.
In total, 260 alignments based on the off-target reads were of good quality, but had at least one sample represented by more than one sequence (Supplementary Data Fig. S4). The gene trees inferred from these alignments were generally not well resolved. In 41 cases, only one or a few specimens were represented by more than one sequence (pLCG_few), while 219 alignments constituted putative gene families with multiple copies in all samples (pLCG_most). In the 41 pLCG_few alignments, 21–43 (average 31·4; s.d. 6·3) of the 48 samples were included and 1–15 specimens (average 3·8; s.d. 3·4) were represented by two to four sequences. Some specimens were nearly always represented in the 41 pLCG_few alignments (Z. dipetalum, Z. hawaiiense [38 alignments each], Z. acuminatum ssp. juniperinum 2, Z. rhetsa, Z. rhoifolium [37 each]), while three samples were present only in <10 alignments (Z. poggei [8], Tetradium austrosinense [7], Z. mayu [1x]). Nine specimens never showed any duplicated sequences and another 12 specimens were duplicated in only one or two alignments. Only two specimens had duplicated sequences in more than ten alignments (Z. asiaticum, Z. rhoifolium [12 each]). As a case study for the informative value of these gene trees, we focused on the Hawaiian lineage. In 11 of the 41 pLCG_few alignments, one or more Hawaiian species were represented by more than one sequence and in four of these alignments only Hawaiian species had duplicated sequences. In 18 out of the 41 alignments, the gene trees resolved the Hawaiian lineage as polyphyletic with low support. In some cases, the relationships of the polyphyletic Hawaiian groups could not be determined due to low resolution of the gene trees. In the case of reasonable resolution of gene trees, in all cases of a polyphyletic Hawaiian group, one copy resolved Hawaiian species as closely related to South Pacific and Australian species (Z. brachyacanthum, Z. pinnatum, Z. nadeaudii), while the other copy was most closely related to Asian species, most frequently to the clade of Z. ailanthoides, Z. rhetsa and Z. schinifolium (Supplementary Data Fig. S4). This latter relationship of the Hawaiian lineage was also found in the coalescent analysis of the pSCGs (Supplementary Data Fig. S4).
Discussion
Phylogenetic relationships
We present the first phylogenomic study for the genus Zanthoxylum using a target-enrichment high-throughput sequencing approach. Concatenated and coalescent analyses of both the on-target as well as the off-target pSCG dataset resulted in phylogenetic trees largely congruent with the recently published Zanthoxylum phylogeny based on four genes only (Appelhans et al., 2018). However, phylogenetic resolution and support in several clades have greatly improved in this study. Herein, the genus Zanthoxylum is divided into four major clades.
The African clade (Clade 1). Zanthoxylum species endemic to the African continent, Madagascar and the Mascarene Islands represent a monophyletic lineage that is sister to the remainder of the genus. Within this clade, the accessions from Madagascar and the Mascarene Islands are resolved as monophyletic (Clade 1; Fig. 2, Supplementary Data Fig. S1). Zanthoxylum heterophyllum and Z. paniculatum are the only Zanthoxylum species from the Mascarenes and Z. paniculatum is an extremely rare endemic to the island of Rodrigues (Bone, 2004). The two species are resolved as sisters in our study and they might have evolved from a common Malagasy ancestor.
The Zanthoxylum asiaticum clade (Clade 2). Zanthoxylum asiaticum, formerly recognized as Toddalia asiatica, represents a separate lineage and is sister to the major Clades 3 and 4 in the analyses of both the on-target and off-target datasets. In a previous study the identical Z. asiaticum individual was resolved as sister to the African species (Clade 1 in the present study; Appelhans et al., 2018). Appelhans et al. (2018) sampled several outgroup taxa, including most species of Fagaropsis, Phellodendron and Tetradium, as well as the more distantly related Acronychia and Melicope. Thus, the different placements of Z. asiaticum might be affected by the large difference in outgroup sampling.
The Asian–Pacific–Australian clade (Clade 3). Clade 3 consists of four subclades: the Asia-1 subclade, the South Pacific–Australian subclade, the Hawaiian subclade and the Asia-2 subclade. Phylogenetic support values are, with a few exceptions, generally high. Both concatenated and coalescent analyses show high support for the Asia-1 subclade (comprising Z. dimorphophyllum, Z. esquirolii and Z. ovalifolium; 100 % BS, 1·00 lPP; Fig. 2, Supplementary Data Fig. S1) and there are no conflicting quartet topologies (Fig. 3). Zanthoxylum esquirolii and Z. dimorphophyllum, both common in South China, are resolved as sister to each other. However, Z. dimorphophyllum is also native to other regions from Central China to Thailand and Vietnam (Zhang et al., 2008). Zanthoxylum ovalifolium is absent from China (Zhang et al., 2008; mistakenly synonymized with Z. dimorphophyllum previously) but shows a broad distributional range from the Himalayas and India to Australia (Hartley, 2013).
Regarding the Hawaiian subclade, three out of the four Hawaiian species are sampled here, and the herein missing Z. oahuense was sampled by Appelhans et al. (2014). It showed a close relationship with Z. hawaiiense and Z. kauaense, so the Hawaiian species are most likely monophyletic. Zanthoxylum dipetalum was confirmed as the earliest-diverging lineage and sister to the remaining Hawaiian species (Fig. 2, Supplementary Data Fig. S1; Appelhans et al., 2014), which is supported by distinct morphological features (Hillebrand, 1888; Wagner et al., 1999) such as (usually) two petals, the lowest pair of leaflets reduced in size, and larger fruits with a beaked apex.
The sister group of the Hawaiian clade comprises the Australian Z. brachyacanthum and two Pacific species, Z. nadeaudii, endemic to the Society Islands, and Z. pinnatum, which is more widespread in the South Pacific (Lord Howe Island to the Austral Islands; Butaud and Meyer, 2004). These two small lineages are sister to an Asian lineage (Asia-2 subclade) with species ranging from South-East Asia to China (Fig. 2, Supplementary Data Fig. S1; Clade 3).
The American–eastern Asian clade (Clade 4). Clade 4 includes all American species and a subclade of species from eastern Asia (Asia-3 subclade or section Sinensis). This biogeographically disjunct clade (also see Valcárcel and Wen, 2019) is morphologically diverse and includes taxa that represent all five sections recognized by Reynel (2017).
Our analyses reveal that Z. clava-herculis, distributed in the southern USA and northern Mexico, and the Central American Z. mollissimum are the closest relatives of the Asian lineage of Clade 4, although strong support for this placement is only apparent in the concatenated analysis of the on-target reads. A close relationship to more temperate or subtropical American Zanthoxylum species is also indicated by the ecology of the embedded Asian lineage (Asia-3 subclade; section Sinensis) as most of their members are well adapted to a temperate and subtropical climate in Asia, and only Z. nitidum is also present in tropical Asia (Zhang et al., 2008). The Asian subclade of Z. bungeanum, Z. coreanum, Z. armatum and Z. nitidum, all of economic importance as spices or medicine, is apparently nested within a New World grade (Fig. 2, Supplementary Data Fig. S1).
Only 15 of the several dozen members of South and Central American and Caribbean species are sampled, so detailed hypotheses about their relationships will not be made here. Two subspecies of Z. fagara (ssp. fagara and ssp. culantrillo) have been sampled and they are resolved as paraphyletic with respect to Z. tragodes. Zanthoxylum fagara is a widely distributed and morphologically diverse species. The subspecies culantrillo differs remarkably from the typical form by its spinulose fruit. Reynel (2017) described that intermediates between the two subspecies exist in regions where they co-occur. Our study suggests that Z. fagara ssp. culantrillo should be regarded as a separate species from Z. fagara and the intermediate specimens might represent interspecific hybrids of the two species.
Zanthoxylum mayu is one of two Zanthoxylum species that occur on the Juan Fernández Islands, Chile. It is endemic to Robinson Crusoe Island (Masatierra), while the second species, Z. externum, is endemic to Alejandro Selkirk Island (Masafuera; Penneckamp, 2019). Spatial/geographic isolation (between islands) has been identified as the primary driver of speciation on the Juan Fernández Islands (Stuessy et al., 1998; Stuessy, 2020), which results in species pairs with one species endemic to Robinson Crusoe Island and one species endemic to Alejandro Selkirk Island, as is the case in Zanthoxylum. Engler (1896) placed Z. mayu in the monotypic section Mayu (Z. externum had not been described then), which is morphologically only differentiated from its closest Central and South American relatives (section Macqueria series Paniculatae sensuEngler, 1896) by its inflorescence type, which is an axillary raceme in section Mayu versus panicles in section Macqueria series Paniculatae (Engler, 1896). Reynel (2017) did not recognize section Mayu and included it in section Macqueria, which is supported by our results.
Reticulate evolution and incomplete lineage sorting
Phylogenetic resolution and support in several clades have greatly improved in this study compared with our previous analysis based on Sanger sequencing (Appelhans et al., 2018). Nevertheless, similar regions of conflict, especially regarding the backbone phylogeny and the Pacific radiation, remained. This observation indicates that low support in our previous study (Appelhans et al., 2018) is not only due to the limited size of the Sanger dataset but also to conflicts within the dataset itself that may be attributed to reticulate evolution or ILS (Figs 2 and 3).
The African clade (Clade 1). With the exception of Z. chalybeum and Z. ovatifoliolatum, the relationships in Clade 1 are well resolved and quartet sampling revealed no signals of reticulate evolution in this clade. The on-target concatenated analysis (Fig. 2) and off-target coalescent pSCG tree (Supplementary Data Fig. S4) both resolved Z. chalybeum as paraphyletic with respect to Z. ovatifoliolatum, but with low bootstrap support (58 % BS; 0·46 lPP) and the quartet sampling showed a low QC value and a QD value of 0 (Fig. 3). Since there is only one dominant alternative topology, a hybridization event is a likely cause for the conflict. However, the on-target coalescent tree (Supplementary Data Fig. S1) and concatenated pSCG tree (Supplementary Data Fig. S3) resolved the two specimens of Z. chalybeum as monophyletic and were sister to Z. ovatifoliolatum with medium to high support.
The Asian–Pacific–Australian clade (Clade 3). Resolution, support and quartet sampling values are high in Clade 3 except for two nodes regarding the Pacific lineages and the relationships among Z. dissitum, Z. echinocarpum and Z. scandens (Figs 2 and 3, Supplementary Data Figs S3 and S4). In the most recent Zanthoxylum phylogeny (Appelhans et al., 2018), the Pacific group, comprising species from Australia up to Hawaii, was resolved as monophyletic based on data on the plastid genes trnL-trnF and rps16 but polyphyletic based on nuclear ITS and ETS sequences. Thus, the authors hypothesized a hybridization event prior to the colonization of the islands. In the study presented here, quartet sampling (Pease et al., 2018) indicates a strong discord for the ancestral Pacific node (QC = 0·16; Fig. 3) with one dominant discordant topology (QD = 0), supporting the hypothesis of a previous hybridization event. Furthermore, it is striking that 18 out of the 41 off-target pLCG_few alignments resolved the Hawaiian lineage as polyphyletic (Supplementary Data Fig. S4). In nearly all of these cases, one Hawaiian gene lineage was resolved as sister to South Pacific and Australian species, while the other gene lineage was closely related to an Asian clade (Asia-2) and the close relationship to the Asia-2 clade was also found in the coalescent pSCG tree (Supplementary Data Fig. S4). Chromosome numbers are known only for one of the four Hawaiian species, Z. hawaiiense, where a chromosome count of 136–144 suggests an octoploid cytotype (Kiehn and Lorence, 1996). The majority of Zanthoxylum species appear to be tetraploids with 68–72 chromosomes (e.g. Guerra, 1984; Zhang et al., 2008), hence the Hawaiian lineage might be the result of an allopolyploidization event prior to the colonization of the Hawaiian Islands. Further cytological data of Hawaiian, South Pacific and Asian taxa will be crucial in providing insights into the ploidy levels and evolutionary relationships within these lineages.
Instead of another case of putative hybridization, the conflict among Z. dissitum, Z. echinocarpum and Z. scandens from the Asia-2 subclade points towards cases of ILS. These species are resolved incongruently across our different analyses with high to moderate support (Figs 2 and 3, Supplementary Data Figs S3 and S4). In Appelhans et al. (2018), the topology was identical with the concatenated analysis of the on-target reads in the present study. The QC values are low or even negative for the respective nodes (Fig. 3), yet the QD values are relatively high (0·81–0·89), indicating ILS as a source of the incongruence.
The American–eastern Asian clade (Clade 4). Of the majority of clades within Clade 4 with lower support, the quartet sampling results indicate non-reticulate evolution and/or ILS. The concatenated analysis of the on-target reads resolved the backbone phylogeny of Clade 4 well (one node with <90 % BS; Fig. 2), but branch lengths are generally short. The coalescent tree and the off-target pSCGs results show lower support, similar to the results of Appelhans et al. (2018; Supplementary Data Figs S1, S3 and S4). Quartet sampling reveals strong quartet discordance (low QC values) in combination with high QD values, with one exception (QD = 0·1) (Fig. 3). Thus, none of the alternative topologies is favoured among the quartets that have been sampled. Combined with the consistently short branches in the backbone phylogeny of Clade 4, this pattern gives an indication for ILS during periods of rapid diversification in the past. In contrast, the node separating the American Z. mollissimum and Z. clava-herculis from the eastern Asian species shows a medium QC (0·46) coupled with a strong preference for one discordant topology (QD = 0). Thus, a hybridization event prior to the dispersal to Asia might have occurred.
In summary, we have identified a number of nodes that are putatively associated with past hybridization or ILS events. Among these, the putative hybridization events prior to the colonization of the Hawaiian Islands and the colonization of temperate Asia by a North American ancestor are particularly interesting. The Hawaiian Islands are among the areas with the highest percentage of polyploid plants in the world and most of the polyploidization events are inferred to have taken place prior to the immigration (Paetzold et al., 2018). The success of (allo)polyploids as colonizers of oceanic islands and as long-distance dispersers in general has often been associated with the smaller effect of inbreeding depression of allopolyploids (Lindner & Barker, 2014; Pannell, 2015). Hawaiian Zanthoxylum are a good example of this. Some, but not all, of the temperate Asian Zanthoxylum species are octaploids (e.g. Z. armatum and Z. simulans; Desai, 1960; Guerra, 1984), so that the putative hybridization at the base of this lineage probably did not result in a polyploidization event. Instead, at least two polyploidization events occurred within the temperate Asian Zanthoxylum lineage. As far as we know, all of the temperate Asian species are apomicts (Liu et al., 1986; Naumova, 1993). The formation of apomicts is often strongly correlated to hybridization (Hojsgaard and Hörandl, 2019). Apomictic species often have a wider distribution compared with their sexual relatives and often occur in more extreme habitats (‘geographical parthenogenesis’; Cosendai et al., 2013; Kirchheimer et al., 2018). Apomixis in Zanthoxylum might thus represent a case of geographical parthenogenesis, where the evolution of apomictic reproduction facilitated the colonization of temperate areas of Eastern Asia.
Sectional classifications
Our results reveal that several of the morphological sections recently described or newly circumscribed by Reynel (2017) are polyphyletic. The mainly Caribbean section Tobinia, characterized by 3-merous flowers (rarely 4-merous; Reynel, 2017), and the Neotropical section Pterota, which is distinguished by its winged leaf rachis and 4-merous flowers (Reynel, 2017), are both monophyletic. The temperate Asian section Sinensis, which has homochlamydeous flowers, might be monophyletic. Section Zanthoxylum is resolved as polyphyletic due to the placement of Z. americanum and Z. clava-herculis. The position of Z. americanum could not be clarified in this study. Appelhans et al. (2018) resolved it as sister to the remainder of section Zanthoxylum and section Sinensis. If a wider taxon sampling in future studies confirms this, section Zanthoxylum would form a grade with section Sinensis nested within it. Zanthoxylum clava-herculis was placed in section Zanthoxylum (Engler, 1896), and Reynel (2017) moved the species to section Macqueria, but highlighted that this was provisional. Like Z. dimorphophyllum, Z. clava-herculis exhibits characters that are transitional between the two sections. Heterochlamydeous flowers are typical for section Macqueria, whereas a deciduous perianth in fruiting carpels, a conspicuous dorsal gland in the ovules as well as a globose stigma are characteristic of section Zanthoxylum (Reynel, 2017). Section Zanthoxylum sensuReynel (2017) consists of only three species and the only species missing in our study is Z. ciliatum. This species shares clear morphological similarities with Z. mollissimum (tepals apically pubescent, base of staminal filament pubescent), and their close relationship is very likely. The Asian section Sinensis is monophyletic only if Z. dimorphophyllum, a species comprising both homo- and heterochlamydeous flowers, is excluded from it. The species was originally described as a Zanthoxylum species, at a time when Fagara was still recognized as a separate genus (Hemsley, 1895; Hartley, 1966). Accordingly, it could be considered to be part of Reynel’s (2017) section Sinensis. However, Engler (1896) placed the species in Fagara section Macqueria. Our study places the species in a clade with species from section Macqueria. Section Macqueria is highly polyphyletic and its members are found in all main clades, and its polyphyly has already been documented by Appelhans et al. (2018). Section Macqueria needs to be split up into at least four sections in order to establish a classification of monophyletic sections (Fig. 2). The type species of section Macqueria is Z. heterophyllum from the Mascarene Islands (Reynel, 2017). This species is part of Clade 1 in our analyses and the name Macqueria should therefore be applied to the African, Malagasy and Mascarene species of Zanthoxylum only. A formal proposal of a new sectional or subgeneric classification is premature at this stage, and the taxon sampling, especially regarding Central and South American and Chinese species, needs to be increased significantly in future studies. Nevertheless, the four major clades recognized in our study set the foundation for a subgeneric classification of Zanthoxylum.
Off-target reads as a source of additional information
In previous studies, off-target read information has mainly been utilized for the assembly of partial or complete plastid or mitochondrial genomes (e.g. Weitemier et al., 2014; Ma et al., 2021). Here, we identified 89 off-target nuclear pSCGs in addition to four mitochondrial and four plastid pSCGs (Supplementary Data Table S4), and 260 pLCGs are recovered in at least half of all samples. The Zanthoxylum phylogeny inferred from the concatenated pSCGs phylogenetic tree (Supplementary Data Fig. S3) is largely congruent with the topologies from on-target phylogenies (Fig. 2, Supplementary Data Fig. S1). Thus, the newly identified pSCGs represent a useful resource to complement the targeted dataset and may also be employed to improve the bait set for future studies. The pLCGs, on the other hand, can be utilized to evaluate if a locus represents a gene family or if it only duplicated in one or several taxa (Supplementary Data Fig. S4). The reasons why a locus is duplicated in few taxa are manifold and duplicated loci might represent the parental lineages of a hybrid taxon. In this study, such information from pLCGs is shown to support the hypothesis of a past hybridization event prior to the colonization of the Hawaiian Islands based on the quartet concordances of the targeted data. The reasons why a specimen is missing in the alignments of a pLCG are also manifold, and a likely explanation is the low coverage of sequence reads. Zanthoxylum mayu is only included in a single pLCG_few alignment. It is the sample from an old herbarium specimen with the second lowest number of reads that mapped to the baits and the sample with the highest percentage of putative contamination (Supplementary Data Tables S2 and S3). Hence, information from off-target pSCG and pLCG loci should be corroborated by subsequent targeted sequencing to conclusively capture sequences across samples and copy numbers. Putative contaminations identified for the off-target reads (Supplementary Data Table S3, Fig. S2) may indicate the minimum sequencing coverage necessary for sufficient data when working with leaf material for which sampling or subsequent conservation treatments are unknown.
SUPPLEMENTARY DATA
Supplementary data are available online at https://academic.oup.com/aob and consist of the following. Table S1: accessions of Zanthoxylum and closely related genera used for bait design. Table S2: Illumina sequencing output information. Table S3: contigs of assembled off-target reads considered as putative contamination. Table S4: functional annotation of 97 off-target pSCGs. Figure S1: ASTRAL species tree of Zanthoxylum based on 258 targeted genes. Figure S2: pie charts displaying putative contaminants in the off-target reads. Figure S3: RAxML phylogenetic tree of Zanthoxylum based on a concatenated alignment of 97 off-target pSCGs. Figure S4: ASTRAL species tree of Zanthoxylum based on 97 off-target pSCGs. Figure S5: four representative pLCG_few trees that show gene families and an example of a polyphyletic Hawaiian lineage.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We thank the curators of the herbaria L (Leiden University, Netherlands) and MO (Missouri Botanical Garden, St Louis, MO, USA) for additional samples, and two anonymous reviewers for their constructive and helpful comments on the manuscript. Special thanks also to Gabriel Johnson for his assistance. Raw reads for all specimens are available at the NCBI Sequence Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra) under BioProject Number PRJNA733013. The bait set used herein, alignments and tree files are available on Dryad (https://doi.org/10.5061/dryad.hqbzkh1gh). Python scripts used for analysing off-target reads are available on github at https://github.com/ClaudiaPaetzold/off-target-reads.git.
FUNDING
This study was supported by the Smithsonian Institution Fellowship Program and PROMOS (DAAD).
LITERATURE CITED
- Albert TJ, Molla MN, Muzny DM, et al. 2007. Direct selection of human genomic loci by microarray hybridization. Nature Methods 4: 903–905. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. Journal of Molecular Biology 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Appelhans MS, Wen J, Wood KR, Allan GJ, Zimmer EA, Wagner WL. 2014. Molecular phylogenetic analysis of Hawaiian Rutaceae (Melicope, Platydesma and Zanthoxylum) and their different colonization patterns. Botanical Journal of the Linnean Society 174: 425–448. [Google Scholar]
- Appelhans MS, Reichelt N, Groppo M, Pätzold C, Wen J. 2018. Phylogeny and biogeography of the pantropical genus Zanthoxylum and its closest relatives in the proto-Rutaceae group (Rutaceae). Molecular Phylogenetics and Evolution 126: 32–44. [DOI] [PubMed] [Google Scholar]
- Appelhans MS, Bayly M, Heslewood MM, et al. 2021. . A new subfamily classification of the Citrus family (Rutaceae) based on six nuclear and plastid markers. Taxon (in press). doi: 10.1002/tax.12543 [DOI] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, et al. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology 19: 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone R. 2004. A proposal for rare plant rescue: Zanthoxylum paniculatum Balf. fil.(Rutaceae), endemic to Rodrigues.https://www.dendrology.org/publications/dendrology/a-proposal-for-rare-plant-rescue-zanthoxylum-paniculatum-balf-fil-rutaceae-endemic-to-rodrigues/ (13 March 2021 date last accessed).
- Borowiec ML. 2016. AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 4: e1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizicky GK. 1962. Taxonomic and nomenclatural notes on Zanthoxylum and Glycosmis (Rutaceae). Journal of the Arnold Arboretum 43: 80–93. [Google Scholar]
- Bryant DM, Johnson K, DiTommaso T, et al. 2017. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Reports 18: 762–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butaud JF, Meyer JY. 2004. Plans de conservation pour des plantes menacées et/ou protégées en Polynésie française. Contribution à la biodiversité de Polynésie française No. 11. Papeete: Service du Développement Rural/Délégation à la Recherche, 1–51. [Google Scholar]
- Chamala S, García N, Godden GT, et al. 2015. MakerMiner 1.0: a new application for phylogenetic marker development using angiosperm transcriptomes. Applications in Plant Sciences 3: 1400115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler MEJ. 1961. The lower tertiary floras of Southern England. In: Chandler MEJ, ed. I. Palaeocene floras, London Clay flora (Supplement). London: British Museum, 1–101. [Google Scholar]
- Collinson ME, Manchester SR, Wilde V. 2012. Fossil fruits and seeds of the Middle Eocene Messel biota, Germany. Abhandlungen der Senckenbergischen Gesellschaft für Naturforschung 570: 1–251. [Google Scholar]
- Constantinides B, Robertson D. 2017. Kindel: indel-aware consensus for nucleotide sequence alignments. Journal of Open Source Software 2: 282. [Google Scholar]
- Cosendai AC, Wagner J, Ladinig U, Rosche C, Hörandl E. 2013. Geographical parthenogenesis and population genetic structure in the alpine species Ranunculus kuepferi (Ranunculaceae). Heredity 110: 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa MC, Vergara-Roig VA, Kivatinitz SC. 2013. A melissopalynological study of artisanal honey produced in Catamarca (Argentina). Grana 52: 229–237. [Google Scholar]
- Desai S. 1960. Cytology of Rutaceae and Simaroubaceae. Cytologia 25: 28–35. [Google Scholar]
- Doyle JJ, Doyle JL. 1987. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 19: 11–15. [Google Scholar]
- Engler A. 1896. Rutaceae. In: Engler A, ed. Die natürlichen Pflanzenfamilien, III. Teil 4. Abteilung. Leipzig: Wilhelm Engelmann, 95–201. [Google Scholar]
- Fér T, Schmickl RE. 2018. HybPhyloMaker: target enrichment data analysis from raw reads to species trees. Evolutionary Bioinformatics 14: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissert F, Gregor HJ, Mai DH. 1990. Die „Saugbaggerflora“, eine Frucht- und Samenflora aus dem Grenzbereich Miozän-Pliozän von Sessenheim im Elsaß (Frankreich). Documenta Naturae 57: 1–207. [Google Scholar]
- Gnirke A, Melnikov A, Maguire J, et al. 2009. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology 27: 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham A, Larzen DL. 1969. Studies in neotropical paleobotany. I. The Oligocene communities of Puerto Rico. Annals of the Missouri Botanical Garden 56: 308–357. [Google Scholar]
- Gregor HJ. 1989. Aspects of the fossil record and phylogeny of the family Rutaceae (Zanthoxyleae, Toddalioideae). Plant Systematics and Evolution 162: 251–265. [Google Scholar]
- Guerra MDS. 1984. New chromosome numbers in Rutaceae. Plant Systematics and Evolution 146: 13–30. [Google Scholar]
- Guerrero AM, Tye A. 2009. Darwin’s finches as seed predators and dispersers. Wilson Journal of Ornithology 121: 752–764. [Google Scholar]
- Haas BJ, Papanicolaou A, Yassour M, et al. 2013. . De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 8: 1494–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley TG. 1966. A revision of the Malesian species of Zanthoxylum (Rutaceae). Journal of the Arnold Arboretum 47: 171–221. [Google Scholar]
- Hartley TG. 2013. Rutaceae. In: Wilson A, Kuchlmayr B, McCusker A, Zhang X, eds. Flora of Australia, Volume 26, Melicaceae, Rutaceae, Zygophyllaceae. Melbourne: CSIRO Publishing, 43–510. [Google Scholar]
- Hemsley WB. 1895. Descriptions of some new plants from Eastern Asia, chiefly from the island of Formosa, presented by Dr. Augustine Henry, F.L.S., to the Herbarium, Royal Gardens, Kew. Annals of Botany. 9: 143–160. [Google Scholar]
- Hillebrand W. 1888. Flora of the Hawaiian Islands: a description of their phanerogams and vascular cryptogams. New York: B. Westermann. [Google Scholar]
- Hojsgaard D, Hörandl E. 2019. The rise of apomixis in natural plant populations. Frontiers in Plant Science 10: 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hörandl E, Appelhans MS. 2015. Introduction to chapters and methodological overview. In: Hörandl E, Appelhans MS, eds. Next-generation sequencing in plant systematics. Regnum Vegetabile 158. Königsstein: Koeltz Scientific Books, 1–8. [Google Scholar]
- Jacobs BF, Kabuye CH. 1987. A Middle Miocene (12.2 my old) forest in the East African rift valley. Kenya. Journal of Human Evolution 16: 147–155. [Google Scholar]
- Junier T, Zdobnow EM. 2010. The Newick Utilities: high-throughput phylogenetic tree processing in the UNIX shell. Bioinformatics 26: 1669–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlec M, Bellstedt DU, Le Maitre NC, Pirie MD. 2017. Targeted NGS for species level phylogenomics: “made to measure” or “one size fits all”? PeerJ 5: e3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya K, Moritsuka E, Yoshia T, Yahara T, Tachida H. 2008. High population differentiation and unusual haplotype structure in a shade-intolerant pioneer tree species, Zanthoxylum ailanthoides (Rutaceae) revealed by analysis of DNA polymorphism at four nuclear loci. Molecular Ecology 17: 2329–2338. [DOI] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Resources 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershaw AP, Bretherton SC. 2007. A complete pollen record of the last 230 ka from Lynch’s Crater, North-Eastern Australia. Palaeogeography, Palaeoclimatology, Palaeoecology 251: 23–45. [Google Scholar]
- Kiehn M, Lorence DH. 1996. Chromosome counts on angiosperms cultivated at the National Tropical Botanical Garden, Kaua’i, Hawai’i. Pacific Science 50: 317–323. [Google Scholar]
- Kirchheimer B, Wessely J, Gattringer A, et al. 2018. Reconstructing geographical parthenogenesis: relative effects of niche differentiation and reproductive mode during the Holocene range expansion of an alpine plant. Ecology Letters 21: 392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov AM, Aberer AJ, Stamatakis A. 2015. ExaML version 3: a tool for phylogenomic analyses on supercomputers. Bioinformatics 31: 2577–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubitzki K, Kallunki JA, Duretto M, Wilson PG. 2011. Rutaceae. In: Kubitzki K, ed. The families and genera of vascular plants. Berlin: Springer, 276–356. [Google Scholar]
- Langmead B, Salzberg S. 2012. Fast gapped-read alignment with Bowtie 2. Nature Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon AR, Emme SA, Lemmon EM. 2012. Anchored hybrid enrichment for massively high throughput phylogenomics. Systematic Biology 61: 727–744. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner HP, Barker NP. 2004. Does polyploidy facilitate long-distance dispersal? Annals of Botany 113: 1175–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnaeus C. 1753. Zanthoxylum. In: Linnaeus C, ed., Species plantarum, Vol. 1. Stockholm: Laurentii Salvii, 270. [Google Scholar]
- Linnaeus C. 1759. Fagara. In: Linnaeus C, ed., Systema Naturae, Vol. 10. Stockholm: Laurentii Salvii, 897. [Google Scholar]
- Liu Y. Wang F.Qian N.1986. Apomixis in Zanthoxylum bungeanum and Z. simulans. Acta Genetica Sinica 14: 107–113. [Google Scholar]
- Liu Y, Wang F, Qian NF. 1987. Apomixis in Zanthoxylum bungeanum and Z. simulans. Journal of Genetics & Genomics 14: 107–113. [Google Scholar]
- Lu Q, Ma R, Yang Y, Mo Z, Pu X, Li C. 2020. Zanthoxylum nitidum (Roxb.) DC: traditional uses, phytochemistry, pharmacological activities and toxicology. Journal of Ethnopharmacology 260: 112946. [DOI] [PubMed] [Google Scholar]
- Ma Z-Y, Nie ZL, Ren C, Liu X-Q, Zimmer EA, Wen J. 2021. Phylogenomic relationships and character evolution of the grape family (Vitaceae). Molecular Phylogenetics and Evolution 154: 106948. [DOI] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12. [Google Scholar]
- Maschwitz U, Fiala B, Linsenmair KE. 1992. A new ant-tree from SE Asia: Zanthoxylum myriacanthum (Rutaceae), the thorny ivy-rue. Malayan Nature Journal 46: 101–109. [Google Scholar]
- Mirarab S, Reaz R, Bayzid MS, Zimmermann T, Swenson MS, Warnow T. 2014. ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30: i541–i548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Landau HC, Wright SJ, Calderón O, Condit R, Hubbell SP. 2008. Interspecific variation in primary seed dispersal in a tropical forest. Journal of Ecology 96: 653–667. [Google Scholar]
- Naumova TN. 1993. Occurrence of nucellar and integumentary embryony and its evolutionary significance. In: Naumova TN, ed. Apomixis in angiosperms. Boca Raton: CRC Press, 49–56. [Google Scholar]
- Nylander J. 2016. catfasta2phyml.https://github.com/nylander/catfasta2phyml.
- Paetzold C, Kiehn M, Wood KR, Wagner WL, Appelhans MS. 2018. The odd one out or a hidden generalist: Hawaiian Melicope (Rutaceae) do not share traits associated with successful island colonization. Journal of Systematics and Evolution 56: 621–636. [Google Scholar]
- Pannell JR. 2015. Evolution of the mating system in colonizing plants. Molecular Ecology 24: 2018–2037. [DOI] [PubMed] [Google Scholar]
- Pease JB, Brown JW, Hinchliff CE, Smith SA. 2018. Quartet sampling distinguishes lack of support from conflicting support in the green plant tree of life. American Journal of Botany 105: 385–403. [DOI] [PubMed] [Google Scholar]
- Penneckamp D. 2019. Suplemento a la flora vascular silvestre del Archipiélago Juan Fernández (Primera Edición). Capítulo adicional. 724–750. https://www.chlorischile.cl/Suplemento_Flora_JF_Penneckamp_2019.pdf (26 February 2021, date last accessed).
- Poon WS, Shaw PC, Simmons MP, But PPH. 2007. Congruence of molecular, morphological, and biochemical profiles in Rutaceae: a cladistic analysis of the subfamilies Rutoideae and Toddalioideae. Systematic Botany 32: 837–846. [Google Scholar]
- Reynel C. 1995. New Andean Zanthoxylum (Rutaceae) with distinctive vegetative characters. Novon 5: 362–367. [Google Scholar]
- Reynel C. 2017. Zanthoxylum (Rutaceae), Flora Neotropica Monograph 117. New York: New York Botanical Garden Press. [Google Scholar]
- Reys P, Sabion J, Galetti M. 2009. Frugivory by the fish Brycon hilarii (Characidae) in western Brazil. Acta Oecologica 35: 136–141. [Google Scholar]
- Schmickl R, Liston A, Zeisek V, et al. 2016. Phylogenetic marker development for target enrichment from transcriptome and genome skim data: the pipeline and its application in southern African Oxalis (Oxalidaceae). Molecular Biology Resources 16: 1124–1135. [DOI] [PubMed] [Google Scholar]
- Silva IA, Antônio de Figueiredo R, da Silva Matos, DM. 2008. Feeding visit time of fruit eating birds in Cerrado plants: revisiting the predation risk model. Revista Brasileira de Zoologia 25: 682–688. [Google Scholar]
- Soto Gomez M, Pokorny L, Kantar MB, et al. 2019. A customized nuclear target enrichment approach for developing a phylogenomic baseline for Dioscorea yams (Dioscoreaceae). Applications in Plant Sciences 7: e11254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stace HM, Armstrong JA, James SH. 1993. Cytoevolutionary patterns in Rutaceae. Plant Systematics and Evolution 187: 1–28. [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub SC, Parks M, Weitemier K, Fishbein M, Cronn R, Liston A. 2012. Navigating the tip of the genomic iceberg: next-generation sequencing for plant systematics. American Journal of Botany 99: 349–364. [DOI] [PubMed] [Google Scholar]
- Stuessy TF. 2020. The importance of historical ecology for interpreting evolutionary processes in plants of oceanic islands. Journal of Systematics and Evolution 58: 751–766. [Google Scholar]
- Stuessy TF, Crawford DJ, Marticorena C, Silva M. 1998. Isolating mechanisms and modes of speciation in endemic angiosperms of Juan Fernández Islands. In: Stuessy TF, Ono M, eds. Evolution and speciation of island plants. Cambridge: Cambridge University Press, 79–96. [Google Scholar]
- The UniProt Consortium . 2019. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Research 47: D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffney BH. 1994. Re-evaluation of the age of the Brandon lignite (Vermont, USA) based on plant megafossils. Review of Palaeobotany and Palynology 82: 299–315. [Google Scholar]
- Tomasello S, Karbstein K, Hodač L, Paetzold C, Hoerandl E. 2020. Phylogenomics unravels Quaternary vicariance and allopatric speciation patterns in temperate-montane plant species: a case study on the Ranunculus auricomus species complex. Molecular Ecology 29: 2031–2049. [DOI] [PubMed] [Google Scholar]
- Twyford AD, Ennos RA. 2012. Next-generation hybridization and introgression. Heredity 108: 179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcárcel V, Wen J. 2019. Chloroplast phylogenomic data support Eocene amphi-Pacific early radiation for the Asian Palmate core Araliaceae. Journal of Systematics and Evolution 57: 547–560. [Google Scholar]
- Villaverde T, Pokorny L, Olsson S, et al. 2018. Bridging the micro- and macroevolutionary levels in phylogenomics: Hyb-Seq solves relationships from populations to species and above. New Phytologist 220: 636–650. [DOI] [PubMed] [Google Scholar]
- Wagner WL, Herbst DR, Sohmer SH. 1999. Rutaceae. In: Stone BC, Wagner WL, Herbst DR, eds. Manual of the flowering plants of Hawai’i. Revised Edition. Vol. 2. Bishop Museum Special Publication 83. Honolulu: Bishop Museum Press, 1174–1216. [Google Scholar]
- Waterman PG. 2007. The current status of chemical systematics. Phytochemistry 68: 2896–2903. [DOI] [PubMed] [Google Scholar]
- Weberling F. 1970. Die vermeintlichen Stipulardornen bei Zanthoxylum L. und Fagara L. (Rutaceae) sowie bei Acanthopanax (Araliaceae). Bericht der Oberhessischen Gesellschaft für Natur- und Heilkunde zu Gießen, Neue Folge, Naturwissenschaftliche Abteilung 37: 141–147. [Google Scholar]
- Weitemier K, Straub SC, Cronn RC, et al. 2014. Hyb-Seq: combining target enrichment and genome skimming for plant phylogenomics. Applications in Plant Sciences 2: 1400042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch AJ, Collins K, Ratan A, Drautz-Moses DI, Schuster SC, Lindqvist C. 2016. The quest to resolve recent radiations: plastid phylogenomics of extinct and endangered Hawaiian endemic mints (Lamiaceae). Molecular Phylogenetics and Evolution 99: 16–33. [DOI] [PubMed] [Google Scholar]
- Xu H, Luo X, Qian J, et al. 2012. FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS ONE 7: e52249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Qiang Z, Sun R, Kong H, Zhang N, Ma H. 2014. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nature Communications 5: 4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Rabiee M, Sayyari E, Mirarab S. 2018. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19: 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Hartley TG, Mabberley DJ. 2008. Zanthoxylum. In: Zhang D, Hartley TG, eds. Flora of China, Vol. 11, 53–66. http://www.efloras.org (4 February2015, date last accessed). [Google Scholar]
- Zimmer EA, Wen J. 2015. Using nuclear gene data for plant phylogenetics: Progress and prospects II. Next-gen approaches. Journal of Systematics and Evolution 53: 371–379. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.