

Abstract
Recent advances in high-resolution structural studies of protein amyloids have revealed parallel in-register cross-β-sheets with periodic arrays of closely spaced identical residues. What do these structures tell us about the mechanisms of action of common amyloid-promoting factors such as heparan sulfate, nucleic acids, polyphosphates, anionic phospholipids, and acidic pH?
Keywords: Amyloid structure, nucleation and growth; Parallel in-register cross-β-sheet; Periodic polyanions as templates for secondary nucleation; Heparan sulfate and heparin; Cooperative electrostatic interactions
Amyloidoses, including Alzheimer’s, Parkinson’s, type-2 diabetes and other life-threatening diseases, involve pathologic deposition of normally soluble proteins or peptides as insoluble amyloid fibrils. Amyloid deposition in vital organs, including the brain, kidney, liver, and heart, causes organ damage [1, 2]. These incurable disorders afflict hundreds of millions worldwide. Therapeutic targeting of amyloidoses requires understanding their molecular basis. How do native structures of unrelated proteins convert into cross-β-sheets characteristic of amyloids? What factors influence the rate-limiting steps of this conversion?
Detailed structural studies of amyloids have been hindered by the transient nature of the pre-fibrillar intermediates and the large size and heterogeneity of fibrils. A breakthrough came circa 2000 when the first atomic-resolution structures of amyloid peptides were determined by x-ray crystallography and solid-state NMR. Both parallel and antiparallel cross-β-sheets were observed in various packing arrangements [3]. The next breakthrough came with the atomic structures of protein amyloids determined thanks to major advances in cryo-electron microscopy (cryo-EM, 2017 Nobel Prize in ChemistryI). Rapidly emerging new structures (Table I, Fig 1A), paralleled by advanced understanding of protein misfolding intermediates [1, 4], provide unprecedented insights into amyloid formation, function, and modulation by cofactors, which is the focus of this article.
Table I.
Human amyloid fibrils whose atomic structures have been determined by cryo-electron microscopy (cryoEM) or solid-state nuclear magnetic resonance (ssNMR).
| Protein / peptide | Pathology / Function | Method | PDB ID | Year |
|---|---|---|---|---|
| Neurodegenerative Disease | ||||
| Amyloid-β | Alzheimer’s | ssNMR | 2LMN, 2LMO, 2LMP, 2LMQ | 2008 |
| Amyloid-β | Alzheimer’s | ssNMR | 2M4J | 2013 |
| Amyloid-β | Alzheimer’s | ssNMR | 2MPZ | 2015 |
| Amyloid-β | Alzheimer’s | cryoEM | 5OQV | 2017 |
| Amyloid-β | Alzheimer’s | cryoEM | 6SHS | 2019 |
| Amyloid-β | Alzheimer’s | ssNMR | 6TI5, 6TI6 | 2020 |
| α-synuclein | Parkinson’s | ssNMR | 2N0A | 2016 |
| α-synuclein | Parkinson’s | cryoEM | 6CU7*, 6CU8* | 2018 |
| α-synuclein | Parkinson’s | cryoEM | 6FLT*, 6H6B* | 2018 |
| α-synuclein | Parkinson’s | cryoEM | 6RT0*, 6SSX*, 6RTB*, 6SST* | 2019 |
| Tau | Alzheimer’s | cryoEM | 5O3L*, 5O3T* | 2017 |
| Tau | Alzheimer’s | cryoEM | 6HRE*, 6HRF* | 2018 |
| Tau | Pick’s | cryoEM | 6GX5* | 2018 |
| Tau | CTEa | cryoEM | 6NWP*, 6NWQ* | 2019 |
| Tau | CBDb | cryoEM | 6TJX* [6] | 2020 |
| Other Amyloid Diseases | ||||
| Amylin | T2 diabetes | cryoEM | 6VW2 | 2020 |
| Amylin | T2 diabetes | cryoEM | 6Y1A | 2020 |
| β2 microglobulin | Dialysis-related | cryoEM | 6GK3 | 2018 |
| Igc light chain | AALd | cryoEM | 6HUD* | 2018 |
| Ig light chain | AALd | cryoEM | 6IC3 | 2019 |
| Serum amyloid A | AAe | cryoEM | 6MST | 2018 |
| Prion | TSEf | cryoEM | 6LNI | 2020 |
| Transthyretin V30M | ATTRg | cryoEM | 6SDZ* | 2019 |
| Functional Amyloids | ||||
| β-endorphin | Storage | cryoEM | 6TUB [5] | 2020 |
| Glucagon | Storage | cryoEM | 6NZN | 2019 |
| RIPK1/RIPK3 | Signaling | ssNMR | 5V7Z | 2018 |
CTE – chronic traumatic encephalopathy
CBD – corticobasal degeneration
Ig – immunoglobulin
AAL – antibody light chain
AA – amyloid-A
TSE – transmissible spongiform encephalopathy
ATTR – amyloid transthyretin
Amyloid structures containing additional features that probably represent a bound cofactor
Figure 1.
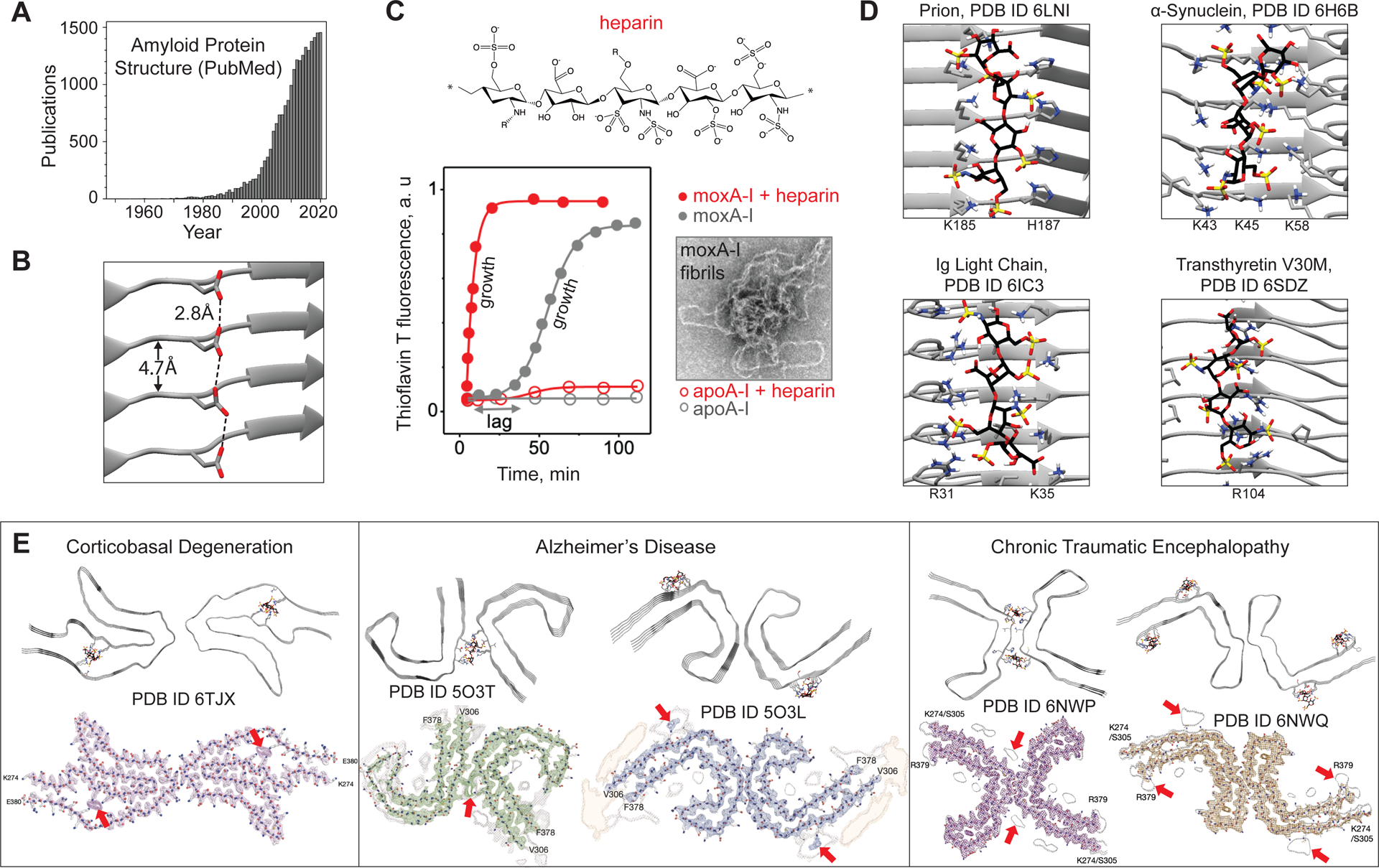
Periodic side-chain arrays in amyloid bind stabilizing counterions. (A) Publications citing “amyloid protein structure”.
(B) An unstable acidic residue ladder may be stabilized upon protonation. Hypothetical H-bonds connect adjacent carboxyl oxygens spaced at 2.8±0.2Å (dashed lines).
(C) Heparin influences amyloid formation kinetics monitored by thioflavine T fluorescence. Human apolipoprotein A-I (apoA-I) exemplifies the effect. Native apoA-I does not bind heparin and does not form amyloid. Methionine-oxidized apoA-I (moxA-I) forms amyloid in vivo [2] and in vitro at 37°C, pH7.5, showing sigmoidal “nucleation-growth” kinetics; electron micrograph shows amyloid fibrils. Heparin eliminates the nucleation (lag) phase and accelerates the growth.
(D) Flexible docking using ClusProii of heparin tetrasaccharide onto human amyloid structures suggests glucosaminoglycan binding sites. All sites contain arrays of solvent-accessible basic residues forming salt-bridge networks with heparin sulfates and carboxylates. Heparin-coordinating residues are shown; N (blue). Heparin S (yellow), O (red), C (black). Fibril axis is vertical.
(E) Cryo-EM structures of patient-extracted fibrils of pathogenic tau strains contain unidentified contiguous density running along the fibril axis (bottom figures reused with permission from [6]; Fitzpatrick, A.W.P. et al. (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 547(7662), 185–190; Falcon B. et al. (2019) Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 568, 420–423]). Heparin docking by ClusPro (top) matches some of this density (arrows), suggesting that HS bound via basic residue arrays is an obligatory physiologic cofactor of tau amyloid. View down the fibril axis.
Parallel in-register β-sheets and their likely bottlenecks
Unlike short peptides, all currently available amyloid protein structures contain parallel in-register cross-β-sheets, which explains protein self-recognition and template-based amyloid self-assembly. In this conformation, protein molecules pack in stacks running along the fibril axis, wherein ladders of identical polar residues, such as Asn or Tyr, stabilize the assembly [3]. However, similar arrays of uncompensated charged residues spaced at ~4.7Å are expected to be destabilizing (Fig. 1B). Typically, charged residue arrays buried in the amyloid core are compensated by the adjacent oppositely-charged residues. One exception is functional amyloid of β-endorphin, whose dissolution is modulated by titration of an uncompensated buried glutamate [5]. Another exception is patient-derived amyloid of tau protein, whose buried basic residues coordinate a nonproteinaceous polyanion [6].
Unlike buried charges, solvent-exposed charges in amyloid structures are not necessarily compensated. We hypothesize that arrays of such uncompensated charged residues decelerate amyloid formation via electrostatic repulsion. Conversely, counterion binding to these residues is expected to accelerate amyloid nucleation and elongation. In particular, periodic arrays of anionic counterions with ~5Å spacing were proposed to catalyze amyloid formation via scaffolding [3, 7, 8], although atomic details were unclear.
Periodic polyanions influence amyloid formation
Besides fibrils, patient-derived amyloid deposits contain non-proteinaceous moieties such as heparan sulfate (HS), RNA, DNA, polyphosphate, and lipids, particularly anionic lipids [9, 10]. These anionic molecules often accelerate formation of amyloid and influence its structure, infectivity and toxicity. Although the exact effects are protein-specific and depend on the polyanion charge distribution and molecular length, common trends have emerged. For example, HS, polyphosphate, and nucleic acids accelerate amyloid nucleation and/or elongation in tau and other proteins; anionic phospholipid arrays in neural membranes catalyze amyloid formation for α-synuclein and Aβ peptide; and phosphatidylethanolamine and single-stranded nucleic acids are obligatory cofactors in prion fibrillation ([9] and references therein). The common effects of these cofactors on proteins with unrelated sequences and native structures suggest that the cofactors stabilize common structural motifs in metastable misfolding intermediates such as amyloid oligomers. Such oligomers likely contain fibril-like structural motifs [4], including parallel in-register β-strands. This compels us to postulate that periodic polyanions augment amyloid formation by helping to assemble and stabilize linear arrays of basic residues in amyloid oligomers.
Consider HS, a particularly potent amyloid agonist. HS is found in all patient-derived amyloid deposits and explains their starch-like properties (“amyloid” means “starch” in Greek) [2, 10]. HS contributes to the disease pathogenesis in inflammation-linked amyloidosis [2]. HS and its highly sulfated mimetic, heparin, accelerate amyloid formation in vitro for various proteins including tau, Aβ, serum amyloid A, immunoglobulin light chain, transthyretin, α-synuclein, etc. [10]. This effect depends neither on the net charge of the protein nor on its native-state binding to HS, suggesting specific HS-amyloid interactions. HS and heparin accelerate amyloid nucleation (shorten the lag phase) and can speed up the growth (Fig. 1C) [8]. HS can catalyze amyloid formation via transient interactions resembling surface catalysis, or it can be an integral part of amyloid, as in tau fibrils ([11] and references therein). Moreover, HS can influence amyloid structure in various proteins [10, 11]. These effects exemplify secondary nucleation wherein periodic molecular surfaces such as HS, anionic liposomes, or the fibril side provide templates for amyloid growth [1, 3].
To illustrate such interactions, we docked heparin tetrasaccharide onto cryo-EM fibril structures of various proteins. All heparin-binding sites identified by flexible dockingII contained linear arrays of uncompensated basic residues forming salt-bridge networks with heparin’s sulfates and carboxylates (Fig. 1D). Notably, heparin/HS chains, which are coordinated by basic residues running along the fibril axis in our models, match unidentified nonproteinaceous density found in cryo-EM structures of tau fibrils, which were extracted from patients with various neurodegenerative diseases (Table I, Fig. 1E). This shows how HS can be incorporated into various strains of tau fibrils, supporting its role as a physiologic amyloid cofactor [11].
Our docking concurs with experimental evidence that Coulombic attraction drives amyloid-heparin/HS interactions, and with structural studies suggesting heparin complementarity to Aβ amyloid [12]. Such binding resembles interactions between regularly-spaced anionic moieties of amyloid-stabilizing drugs with complementary basic residue arrays in prions[13]. These interactions exemplifiy how HS/heparin and other periodic anionic arrays can provide docking platforms to nucleate amyloid formation and modulate its structure and stability.
Acidic side-chain arrays in amyloid
Like basic residues, arrays of uncompensated acidic residues are also expected to counteract amyloid formation. Perhaps such periodic arrays are stabilized at acidic pH by cooperative binding of protons or hydronium ions (Fig. 1B), similar to the “Caspar carboxylate pairing” that regulates the assembly of viral coat proteins. This could explain why acidic pH is a common amyloid-promoting factor that not only destabilizes the native protein conformation, but also potentially stabilizes amyloid oligomers. Furthermore, acidic side-chain arrays in amyloid can perhaps cooperatively bind divalent metal ions, which often, albeit not always, act as amyloid cofactors [4, 9].
Summary and future directions
This paper tells a brief story that omits details. Since protein interactions with amyloid modulators influence both the native and the amyloid states, the same modulator can act as either agonist or antagonist for different protein amyloids. Nevertheless, common trends observed in the effects of diverse cofactors on amyloid formation by unrelated proteins must stem from the structural periodicity common to amyloids. This begs a question: can periodic arrays of identical residues in amyloid catalyze the assembly of complementary arrays in other molecules, act as scaffolds for DNA repair, as activators of pattern-recognition receptors in immune response, as relays for signal transmission, or perform other highly cooperative functions? One example is the unusual electronic conductivity via Tyr stacks [14]; others, such as signaling or storage, are emerging from studies of functional amyloids [3, 5] (Table I).
Intriguingly, several amyloid structures, including tau fibrils extracted from patients with neurologic diseases such as Alzheimer’s, contain unidentified nonproteinaceous densities ([6] and references therein) (Table I), some of which match bound HS/heparin (Fig. 1E). Such fibril-cofactor complexes allow sharp insights into physiologic modulators of amyloid formation, which may help guide therapeutic targeting of amyloidoses.
ACKNOWLEDGEMENTS
We apologize to the authors of many superb publications that cannot be cited here due to journal restriction on the number of references. We are indebted to Dr. John Straub and Harrison Reiter for extremely useful discussions, and to Dr. Sandor Vajda and Amanda Wakefield for help with ClusPro docking. This work was supported by the NIH grants GM067260 and GM135158. The authors would like to dedicate it to the memory of Dr. Robert Kisilevsky, a pioneer of therapeutic targeting of HS-amyloid interactions.
References
- 1.Dobson CM et al. (2020) The amyloid phenomenon and its significance in biology and medicine. Cold Spring Harb. Perspect. Biol 12(2), a033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benson MD et al. (2020) Amyloid nomenclature 2020: update and recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid 27(4), 217–222 [DOI] [PubMed] [Google Scholar]
- 3.Riek R, Eisenberg DS (2016) The activities of amyloids from a structural perspective. Nature 539, 227–235 [DOI] [PubMed] [Google Scholar]
- 4.Nguen PH et al. (2021) Amyloid oligomers: A joint experimental/computational perspective on Alzheimer’s disease, Parkinson’s disease, type II diabetes and amyotrophic lateral sclerosis. Chem. Rev 121(4), 2545–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seuring C et al. (2020) The three-dimensional structure of human β-endorphin amyloid fibrils. Nat. Struct. Mol. Biol 27(12), 1178–1184 [DOI] [PubMed] [Google Scholar]
- 6.Zhang W et al. (2020) Novel tau filament fold in corticobasal degeneration. Nature 580(7802), 283–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motamedi-Shad N et al. (2009) Amyloid formation by the model protein muscle acylphosphatase is accelerated by heparin and heparan sulphate through a scaffolding-based mechanism. J. Biochem 146(6), 805–814 [DOI] [PubMed] [Google Scholar]
- 8.Solomon JP et al. (2011) Heparin binds 8 kDa gelsolin cross-β-sheet oligomers and accelerates amyloidogenesis by hastening fibril extension. Biochemistry 50(13), 2486–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stewart KL, Radford SE (2017) Amyloid plaques beyond Aβ: a survey of the diverse modulators of amyloid aggregation. Biophys. Rev 9, 405–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishitsuji K, Uchimura K (2017) Sulfated glycosaminoglycans in protein aggregation diseases. Glycoconj. J 34(4), 453–466 [DOI] [PubMed] [Google Scholar]
- 11.Fichou Y et al. (2018) Cofactors are essential constituents of stable and seeding-active tau fibrils. Proc. Natl. Acad. Sci. U. S. A 115(52), 13234–13239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stewart KL et al. (2017) Molecular origins of the compatibility between glycosaminoglycans and Aβ40 amyloid fibrils. J. Mol. Biol 429(16), 2449–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herrmann US et al. (2015) Structure-based drug design identifies polythiophenes as antiprion compounds. Sci. Transl. Med 7(299), 299ra123. [DOI] [PubMed] [Google Scholar]
- 14.Shipps C et al. (2021) Intrinsic electronic conductivity of individual atomically resolved amyloid crystals reveals micrometer-long hole hopping via tyrosines. Proc. Natl. Acad. Sci. U. S. A 118(2), e2014139118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Resources
- I.Press release: The Nobel Prize in Chemistry 2017: https://www.nobelprize.org/prizes/chemistry/2017/press-release/
- II.ClusPro server: Kozakov D et al. (2017) The ClusPro web server for protein-protein docking. Nature Protocols 12(2), 255–278; https://cluspro.bu.edu/login.php [DOI] [PMC free article] [PubMed] [Google Scholar]