

Abstract
Advances in molecular biology, microfluidics and bioinformatics have empowered the study of thousands or even millions of individual cells from malignant tumours at the single-cell level of resolution. This high-dimensional, multi-faceted characterization of the genomic, transcriptomic, epigenomic and proteomic features of the tumour and/or the associated immune and stromal cells enables the dissection of tumour heterogeneity, the complex interactions between tumour cells and their microenvironment, and the details of the evolutionary trajectory of each tumour. Single-cell transcriptomics, the ability to track individual T cell clones through paired sequencing of the T cell receptor genes and high-dimensional single-cell spatial analysis are all areas of particular relevance to immuno-oncology. Multidimensional biomarker signatures will increasingly be crucial to guiding clinical decision-making in each patient with cancer. High-dimensional single-cell technologies are likely to provide the resolution and richness of data required to generate such clinically relevant signatures in immuno-oncology. In this Perspective, we describe advances made using transformative single-cell analysis technologies, especially in relation to clinical response and resistance to immunotherapy, and discuss the growing utility of single-cell approaches for answering important research questions.
Tumours are a complex mixture of malignant, immune and stromal cells, which often have substantial levels of intratumour and intertumour heterogeneity. The tumour microenvironment (TME) comprises an amalgam of tumour-promoting and anti-tumour signals that are able to modulate tumour growth and influence tumour evolution. Bulk genomic and transcriptomic analyses have provided valuable insights into these processes, but the averaging of signals from large numbers of cells rendered by these methods often obscures specific subpopulations and/or cellular states. Indeed, rare and unique cellular subtypes or states that might be instrumental in disease biology, such as potential ‘cancer stem cells’ or immune cells crucial for a therapeutic response, might not be detected in bulk analyses derived by averaging data from millions of cells. For cancer immunotherapy, in particular, single-cell analyses can provide unique insights into the tumour-intrinsic and tumour-extrinsic mechanisms that drive responses, as well as primary or acquired resistance to novel immunotherapeutics1,2.
The genomic, transcriptomic, epigenomic and/or proteomic examination of individual cells overcomes many of the limitations of bulk analysis, enabling a more refined dissection of the cellular and molecular basis of cancer. The tremendous power of single-cell analysis has been recognized for decades, as shown by the fact that immunohistochemistry, in situ hybridization and flow cytometry have all been indispensable tools in investigating the differences between non-malignant cells and cancer cells, both in the laboratory and in the clinic. These classic methods, however, often only detect a limited number of analytes in any given experiment or test, and this limitation reduces the power to distinguish the diversity of cellular subtypes and states present in the TME.
Breakthroughs in sequencing technologies, cell-isolating microfluidics and analytical bioinformatics have led to a rapid proliferation of single-cell analysis methodologies, and have been reviewed extensively elsewhere3–6. These techniques enable the detection of many analytes at single-cell resolution in a large number of cells (initially a few hundred, but now thousands to hundreds of thousands of cells) in an increasingly cost-effective manner. The concomitant blossoming of the availability of bioinformatic tools has enabled researchers to interrogate the massive datasets provided by such methods in order to exploit the covariate power at the heart of single-cell data, visualize the diversity of cellular subtypes and elucidate the interactions between cells. Continual improvements in single-cell methodological workflows and analytics, including the ascertainment of single cells, reduction of high-dimensional data, unsupervised clustering, phylogeny modelling, harmonization of multiple datasets, trajectory inference, RNA velocity, lineage tracing and ligand–receptor interaction mapping, have been crucial to the success of single-cell approaches and have been reviewed in detail elsewhere7–11. Single-cell approaches are uniquely poised to help answer many of the questions that persist in immuno-oncology, including: the role of tumour and immune-cell heterogeneity (including heterogeneity in expression of the antigen-presenting machinery); the relationship between T cell clonotype and phenotype; the network of immune-checkpoint interactions present in the TME; and the spatiofunctional relationships between tumour cells and immune cells, as well as between different immune cell types. Herein, we focus on how high-dimensional single-cell analysis has been leveraged to investigate the interplay between tumour cells and their microenvironment, and especially how this relates to response to anti-cancer immunotherapy.
In this Perspective, we first examine how single-cell transcriptomic, proteomic, epigenomic and genomic technologies have transformed our understanding of responsiveness and resistance to anti-cancer immunotherapy, before exploring analytical approaches that integrate multiple modalities. Next, we consider the critical importance of single-cell T cell receptor (TCR) analysis. We then survey emerging technologies enabling single-cell spatial analysis, with a focus on their applicability to immuno-oncology. Finally, we discuss the future directions of single-cell technologies and their possible prospective roles in advancing the field of immuno-oncology.
Single-cell omics
Transcriptomics
Single-cell RNA sequencing (scRNA-seq) refers to a collection of techniques that enable the untargeted quantification of the transcripts present in individual cells. Svensson et al.12 have reviewed the remarkable technological advances over the past decade that culminated in the Human Cell Atlas project adopting scRNA-seq as the definitive method for classifying cell types and states13. As an untargeted approach to surveying the transcriptome, scRNA-seq can identify novel cell types (such as the pulmonary ionocyte fundamentally responsible for the disease biology of cystic fibrosis14), interrogate rare cell populations, and define a spectrum of cellular states and phylogenies (such as the reclassification of human dendritic cells (DCs) and monocytes in the peripheral blood15) (FIG. 1). After dissociating a tumour into individual cells, scRNA-seq can be used to analyse the single-cell suspension either without enrichment or after fractionation into defined subsets of cells. In the following subsections, a few illustrative examples of the value of scRNA-seq are discussed in detail. Additional examples highlight the key insights derived from scRNA-seq — especially those that are relevant to the involvement of immune cells in cancer in general and in the response to immunotherapy in particular (TABLE 1).
Fig. 1 |. Workflow for single-cell analysis in immuno-oncology.
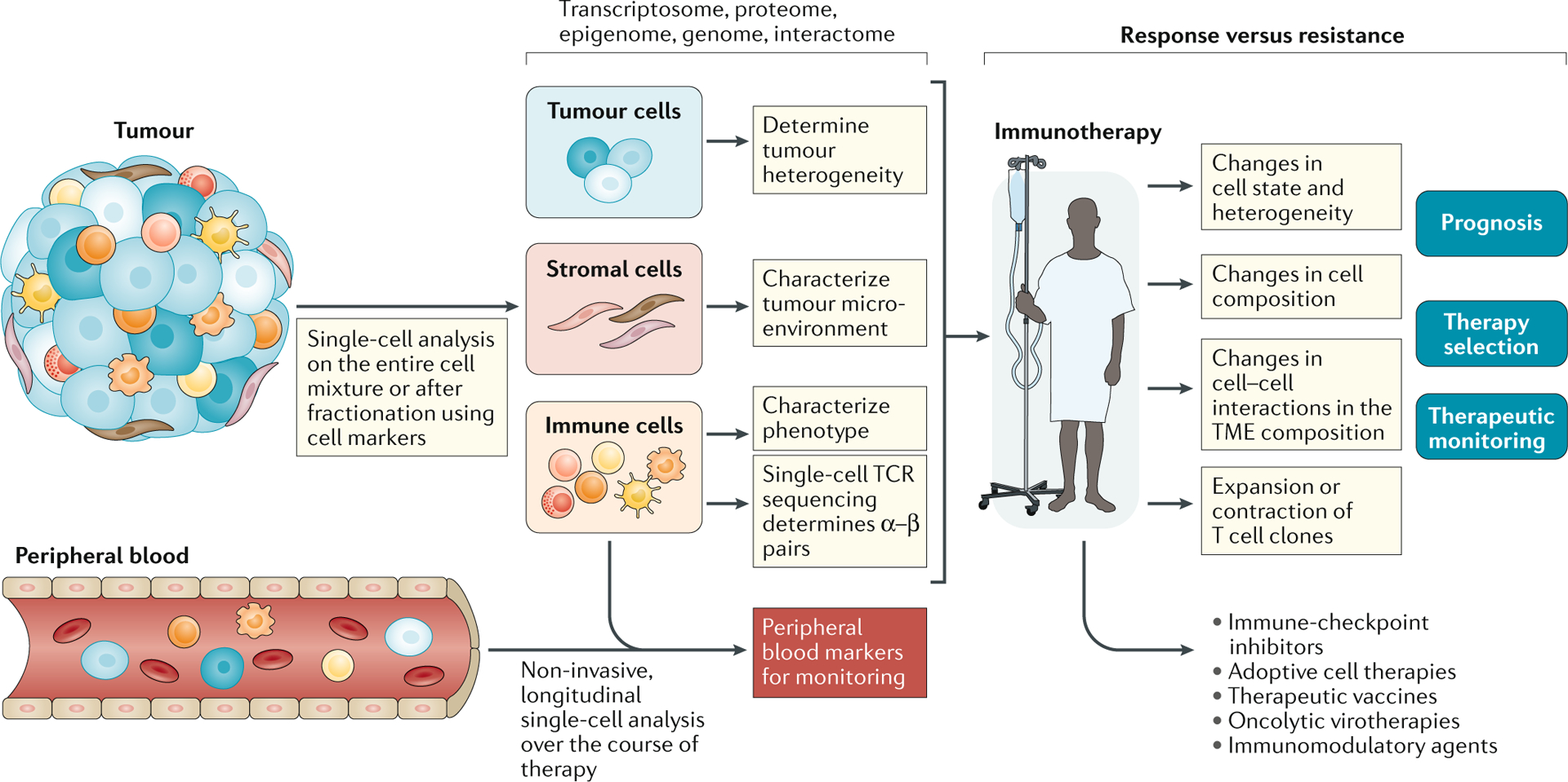
The tumour microenvironment (TME) is often complex and contains myriad cell types and states that can be disaggregated, with varying degrees of success, into a single-cell suspension. Cells can then be processed without further manipulation, or specific cell subsets can be isolated for downstream analysis. Single-cell analysis — most commonly by single-cell RNA sequencing (scRNA-seq) — enables the assessment of tumour, immune and/or stromal cells to yield high-dimensional information on tumour heterogeneity and an atlas of the immune and/or stromal microenvironment in relation to clinical stage, disease subtype and tumour location. Sampling before and after immunotherapy, especially if patients can be stratified based on their response, facilitates a detailed dissection of the mechanisms underlying response and resistance. The ability to track immune cell populations, specifically T cells through paired T cell receptor (TCR) α and β subunit sequencing, enables the detailed characterization of T cell clones involved in mediating response. These findings can be applied to a wider selection of patients, with the goals of improving prognostication and optimal treatment selection in the face of an ever-expanding range of therapeutic options, and enabling therapeutic monitoring through the identification of cellular populations or markers that can be monitored using high-throughput conventional technologies. Findings from within the TME might also be linked to changes in the peripheral circulation, which are more amenable to regular monitoring.
Table 1 |.
Selected immuno-oncology insights from scRNA-seq-based analyses
| Cancer type | Key findings | Single-cell technologies used | Cell types characterized | Ref. |
|---|---|---|---|---|
| Oligodendroglioma and astrocytoma | Microglia/macrophages are the predominant non-malignant cells in the TME with increased infiltration of these cells in IDH-mutant astrocytomas compared with IDH-mutant oligodendrogliomas | Smart-seq2 (plate-based) | Tumour cells, microglia and TAMs | 22 |
| Ovarian ascites | Smart-seq2, 10× Genomics (droplet-based) | Tumour cells, CAFs and immune cells | 20 | |
| Elements of the JAK/STAT signalling pathway are highly expressed in tumour cells and CAFs | ||||
| Breast cancer | The presence of gene-expression signatures of three myofibroblastic CAF subpopulations at diagnosis is associated with resistance to anti-PD-1 antibodies | 10× Genomics | CAFs | 126 |
| Pancreatic cancer | Used a published scRNA-seq dataset to characterize CAFs expressing LRRC15, then correlated the LRRC15+ CAF signature with a poor response to anti-PD-L1 antibodies | 10× Genomics | CAFs | 127 |
| Breast cancer | inDrop (droplet-based), 10× Genomics | CD45+ immune cells | 28 | |
| Trajectory analysis revealed a continuum of T cell states along axes of activation, hypoxia and terminal differentiation | ||||
| NSCLC | Based on CD8+ T cell phenotypes, higher ratios of progenitor exhausted cells to terminally exhausted cells were associated with a better prognosis | Smart-seq2 | T cells | 128 |
| HCC | Increased expression of LAYN associated with exhausted tumour-infiltrating CD8+ T cells and a poor prognosis | Smart-seq2, 10× Genomics | T cells | 129 |
| Colon cancer | Smart-seq2, 10× Genomics | TAMs and dendritic cells | 130 | |
| High SPP1+/low C1QC+ TAM signature associated with a poor prognosis | ||||
| B-ALL | Characterized remodelling of the myeloid compartment at diagnosis including an increase in non-classic CD16− monocyte subpopulations associated with unfavourable survival | 10× Genomics | Myeloid cells (all immune cells included in sequencing) | 131 |
| Melanoma, HNSCC, NSCLC | Published data (Smart-seq2, 10× Genomics, inDrop) | TAMs | 132 | |
| Implicated macrophages with elevated levels of CXCL10 and CXCL11 at baseline with response to immune-checkpoint inhibitors in patients with melanoma using published scRNA-seq datasets |
The capabilities of single-cell RNA sequencing (scRNA-seq) technologies that use end counting (such as 10× Genomics) versus whole-transcript sequencing (such as Smart-seq2) have been reviewed in detail elsewhere3,12,133. B-ALL, B cell acute lymphoblastic leukaemia; CAFs, cancer-associated fibroblasts; HCC, hepatocellular carcinoma; HNSCC, head and neck squamous cell carcinoma; NK, natural killer; NSCLC, non-small-cell lung cancer; TAMs, tumour-associated macrophages; TCGA, The Cancer Genome Atlas; TME, tumour microenvironment.
Tumour cells and immunotherapy response.
The heterogeneous nature of cancer, both between and within individual tumours, is the predominant feature highlighted by scRNA-seq analysis of tumour cells. When clustering malignant cells from multiple patients, authors have noted that transcriptional profiles of individual cells group primarily by patient of origin in those with glioma16, melanoma17,18, head and neck squamous cell carcinoma19 and ovarian cancer20, among others. This observation is perhaps not surprising and attests to the fact that each tumour has a unique evolutionary trajectory. This patient-to-patient heterogeneity can complicate the use of scRNA-seq analysis of single tumour cells to study response to therapy. Consequently, single-cell analysis of immune and stromal cells has been more fruitful when applied to the identification of cell types and states associated with immunotherapy response that are shared among patients.
Despite this heterogeneity, identifying malignant cells with specific transcriptional states that are shared across patients is possible. Studies of the transcriptomes of single tumour cells have refined the classification of tumour types and their cellular hierarchies16,19,21–24, and have also enabled the identification of transcriptome-wide signatures associated with therapeutic response or resistance. The powerful potential of this capability is illustrated by data from an scRNA-seq-based analysis of samples from 31 patients with melanoma receiving immune-checkpoint inhibitors (ICIs), in which the authors defined a transcriptional programme expressed in malignant cells that was associated with T cell exclusion and intrinsic resistance to therapy18. This programme resulted in increased expression of CDK4 with concomitant reductions in the expression of genes encoding proteins involved in antigen processing and presentation (such as B2M, HLA-A, HLA-B and HLA-E), interferon signalling or components of the complement system. Iterative multiplex immunofluorescence using tissue-based cyclic immunofluorescence (t-CyCIF) (BOX 1) confirmed the association of this programme with T cell exclusion in situ. By applying single-cell insights to bulk RNA-seq data, the researchers showed that the resistance programme was expressed prior to therapy in a longitudinal cohort of 26 patients with metastatic melanoma, was associated with inferior survival in 473 patients with melanoma included in The Cancer Genome Atlas and was predictive of poor response to anti-PD-1 in an independent cohort of 112 patients. These observations collectively showcase the potential of single-cell analyses of a small cohort to aid in the deconvolution of bulk data from larger cohorts and to identify cellular subpopulations that are amenable to therapy. Testing of these single-cell insights in a mouse model18 suggested that CDK4/6 inhibition might be a tractable target to reverse the resistance programme and sensitize melanomas to immune-checkpoint inhibition.
Box 1 |. Methods of high-dimensional spatial analysis.
Immunohistochemistry and in situ hybridization have long been used to localize one or a few proteins and RNAs, respectively, in tissue sections. Multiplex methods that have emerged over the past few years have dramatically increased the number of targets that can be detected.
Protein detection
- Iterative multiplexed immunofluorescence. The use of multiple rounds of labelling with fluorescent antibodies to detect 50–100 targeted proteins in tissue sections at subcellular resolution. The widespread availability of fluorescent antibodies and microscopy cores, and the use of routine formalin-fixed paraffin-embedded (FFPE) pathological specimens make these approaches relatively inexpensive and straightforward to apply in most immunotherapy studies. Examples include:
- Mass cytometry. Use of metal-tagged antibodies to detect 30–100 target proteins in FFPE tissue sections. Single-cell detection is possible, although resolution is typically lower than with fluorescence-based methods. These approaches require specialized core facilities and expertise, but enable simultaneous measurement across all targets. Examples include:
- Oligonucleotide barcoding. Use of oligonucleotide-tagged antibodies to detect up to 50 targeted proteins in fresh frozen sections at subcellular levels of resolution. Examples include:
- co-detection by indexing (CODEX)145
RNA Detection
- Iterative multiplexed in situ hybridization. Use of barcoded oligonucleotide probes to greatly expand the capabilities of conventional in situ hybridization in order to detect thousands of transcripts with subcellular resolution in FFPE tissue sections. These approaches require specialized analytics and expertise in order to localize individual mRNA transcripts below the optical diffraction limit. Examples include:
- Spatial barcoding with hybrid capture of mRNA. Use of barcoded reverse transcriptase primers attached to slides or beads to enable untargeted whole-transcriptomic sequencing with the resulting resolution determined by spot or bead size. Unlike spatial protein detection methods, these early incarnations of spatial RNA methods are limited by transcript drop-out, overlapping capture of transcripts from neighbouring cells and the need for frozen, non-fixed tissue. Largely amenable to processing by existing single-cell cores with relatively short turnaround times, but do require specialized analytics and expertise. Examples include:
- Untargeted in situ sequencing. Use of tailed random hexamer reverse transcriptase primers followed by rolling-circle amplification of transcript sequences and in situ sequencing in frozen, non-fixed tissue sections. Capable of single-cell detection. Examples include:
- Targeted in situ sequencing. Use of specific probes to initiate rolling-circle amplification of target transcripts followed by in situ sequencing in frozen, non-fixed tissue sections. Capable of single-cell detection. Examples include:
- Protein and/or RNA detection. This targeted method uses antibodies labelled with oligonucleotide barcodes and barcoded probes for RNA followed by location-specific release of the barcodes. Detection and quantification of the barcodes is conducted using a separate instrument. Although the barcode-releasing mechanism is capable of single-cell detection, current system sensitivity limits practical resolution to regions of interest at the level of 10–100 cells. Examples include:
Immune and/or stromal cells and immunotherapy response.
Overall, a comprehensive understanding of immune cell composition and state is crucial to understanding responsiveness and resistance to current immunotherapies, and to the rational design of novel immune-modulating therapies and combinations. Initial efforts in this area were focused on creating detailed immune atlases of the cells that reside within non-malignant tissues and then applying those atlases and methods to the neoplastic setting. For example, Szabo et al.25 generated a reference map of non-malignant human T cell activation by analysing >50,000 resting and activated T cells from the lungs, lymph nodes, bone marrow and blood. The authors then projected published scRNA-seq profiles of tumour-associated T cells from patients with non-small-cell lung cancer26, colorectal cancer27, breast cancer28 or melanoma29 onto their map of non-malignant T cell activation states, and thereby demonstrated that tumours harbour activated CD8+ T cells, but not functionally activated CD4+ T cells25. Furthermore, intratumour CD8+ T cells expressing exhaustion markers also expressed genes associated with conventional CD8+ T cell effector functions and ongoing proliferation.
Extending single-cell transcriptional mapping to the TME across a range of tumour types has revealed several intriguing insights into the heterogeneity and diversity of immune responses in cancer (TABLE 1). An important finding is that clusters based on untargeted transcriptional assessments of cellular states often do not cleanly segregate into conventional immune cell subsets based on cell-surface protein expression, thus leading to a more nuanced understanding of the roles of immune cells in the TME.
Applying these approaches to the immune and stromal cells present in the TME in the context of immunotherapy has enabled the elucidation of the transcriptional states that underlie response and resistance to immunotherapies, such as ICIs. In a prototypical example, Sade-Feldman et al.29 analysed 16,291 immune cells from 48 well-annotated tumour biopsy samples from 32 patients with metastatic melanoma treated with ICIs and identified 11 immune cell clusters. Focusing on CD8+ T cells revealed two broadly defined cellular states that correlated with response and resistance. Features of the cellular state enriched in responders included increased expression of genes associated with memory, activation and cellular survival (including TCF7) and reduced expression of genes associated with co-inhibitory effects. By contrast, the cellular state enriched in non-responders included increased expression of genes associated with T cell exhaustion (such as ENTPD1, BATF, HAVCR2, PDCD1, LAG3 and CTLA4). Notably, this distinction between responders and non-responders was observed in tumour-infiltrating lymphocytes (TILs) from both pretreatment and post-treatment samples, suggesting that these signatures have predictive potential. Indeed, conventional multiplex immunofluorescence analysis of tissue sections from an independent cohort of 33 patients confirmed the association of TCF7 expression with a response. Additionally, the finding that CD8+ TILs in non-responders are enriched with cells that co-express CD39 and TIM3 prompted the testing of targeted inhibitors of these proteins in a mouse model of melanoma29. Enhanced levels of tumour control were observed when inhibiting CD39 in combination with antibodies targeting TIM3, PD-1, or PD-1 and CTLA-4, thus illustrating the potential of single-cell transcriptomics, when combined with protein-level confirmation, to identify new co-expressed targets for novel immunotherapy combinations.
Importantly, analyses designed to map the immune landscapes of a diverse range of tumour types have identified a nascent, but growing portfolio of transcriptional states associated with responsiveness to immunotherapy across lymphocyte, DC, monocyte, macrophage and fibroblast compartments, many of which are now being studied as putative biomarkers (TABLE 1). Having gained a better understanding of the transcriptional landscapes of tumours and their microenvironmental constituents, investigators can now examine how the tumour and immune system co-evolve in concert over the course of treatment and relapse, at the single-cell level of resolution.
Interaction analysis.
Many studies have explored the roles of either tumour cells or immune and/or stromal cells in isolation, despite complex interactions between and within these cell types having a crucial role in cancer biology. Information on these important cell–cell interactions can be inferred from scRNA-seq data by correlating the expression of ligand–receptor pairs between different cell types30,31. For example, assessments of the interactome by Zhang et al.32 have provided insights into the interplay between tumour-associated DCs and peripheral T cells. In this study involving 16 treatment-naive patients with hepatocellular carcinoma, leukocytes from the tumour, adjacent liver, hepatic lymph nodes, blood and ascites were analysed using scRNA-seq. Lineage analysis revealed a novel class of LAMP3+ DCs capable of migrating from tumours to lymph nodes. These LAMP3+ DCs expressed many putative inhibitory ligand–receptor pairs and were associated with multiple T cell subsets, suggesting a role in promoting the dysfunction of peripheral T cells. Novel technologies are also being developed that enable the exploration of physical cell–cell interactions — including spatially resolved scRNA-seq methods (BOX 1) — in an attempt to better understand the intricately choreographed interactions between tumour cells and those of the TME.
Limitations of scRNA-seq.
scRNA-seq has revolutionized the delineation of cell types and cellular states, although this technique also has several notable drawbacks. First, scRNA-seq data are intrinsically noisy because eukaryotic transcription does not take place at a consistent basal rate; it occurs in pulses33,34. For example, monoallelic expression in single cells is predominantly the result of burst-like stochastic transcription35,36. Thus, a failure to detect a transcript from a particular gene in a cell at a single specific point in time is an ambiguous result: it could mean that the gene is inactive in that cell, that the gene is active, but the transcript was not detected owing to burst kinetics and the timing of sampling, or that technical deficiencies meant that expression of the gene was not detected. This challenge in substantiating negative findings is particularly relevant for genes with lower levels of expression. Analytical approaches that consider the entirety of the cellular transcriptome (such as those involving clustering by differentially expressed genes) have helped to overcome this limitation of scRNA-seq. Consequently, the interpretation of results should focus on pathway analysis and gene-set enrichment, rather than the expression or effects of any single gene.
Second, the use of transcriptome data alone to establish correlations between genotype and phenotype remains challenging. For example, scRNA-seq data have been used to infer somatic copy number variants (commonly referred to as sCNVs)37,38, although this only applies to relatively large deletions or amplifications. The larger number of somatic single-nucleotide variants (sSNVs) present in tumour cells enables the assessment of their genetic architecture at a higher level of resolution than sCNVs; nonetheless, the ability to confidently detect sSNVs within scRNA-seq data remains limited. For example, sSNVs in transcripts with low levels of expression are detected only in a minority of cells owing to the combined challenges of transcriptional bursting and technical dropout. Additionally, because the highest-throughput methods involve end counting (by sequencing just the 3′ or 5′ end of the RNA molecule, rather than the full transcript), sSNVs located away from the end of a transcript are often poorly captured and thus under-represented in the data. Several new approaches, including TARGET-seq39, genotyping of transcriptomes40 and the approach described by van Galen et al.41, have incorporated primers for known mutations into otherwise untargeted scRNA-seq protocols in an attempt to address this limitation, but these approaches require a priori knowledge of the variants to target. As a result, scRNA-seq approaches are a suboptimal method of evaluating the implications of somatic mutations in individual tumour cells for therapeutic response or resistance.
In addition to these inherent limitations of scRNA-seq, specific details relating to sample procurement and processing can also confound results, with important implications for the clinical applicability of these technologies. Indeed, scRNA-seq has relied upon viable single-cell suspensions, which are readily available from peripheral blood and/or lymph nodes, but are often more difficult to obtain from solid tumour tissues. Differences in dissociation methods and cryopreservation conditions can also induce considerable transcriptome-wide changes42. Even with blood samples, the amount of time blood draws are kept at room temperature before cryopreservation substantially alters the transcriptional profile of individual cells, including a pronounced downregulation of immune cell type-specific genes, in the absence of a loss of RNA fidelity after storage for >2 hours at room temperature43. In an attempt to address these issues, a large group of researchers with an interest in single-cell genomic analysis of tumours has begun developing protocols designed to standardize tissue dissociation and the preparation of single-cell suspensions44. Investigators studying tissue development have found that single-nucleus RNA sequencing (snRNA-seq) enables a more streamlined workflow that is applicable to many different tissue types, is compatible with frozen samples and can sometimes provide less-biased cellular coverage compared with scRNA-seq45–48. This method is beginning to be applied to tumour samples and should substantially improve the clinical utility of single-cell approaches44.
Finally, batch effects can occur owing to the processing of samples on different days with even slight technical variations, as often happens when comparing samples obtained over a time course. Multiple methods of correcting such batch effects have been developed49, although the use of these corrections must be balanced with the risk of obscuring true biological differences.
Proteomics
Mass cytometry.
Untargeted detection of proteins using mass spectrometry lacks sufficient sensitivity to be relevant to single-cell analysis50. Therefore, single-cell proteomics studies rely predominantly on selective target detection using antibodies or similar affinity reagents (such as affibodies or aptamers)51–55. Flow cytometry has been the longstanding workhorse for investigators seeking to assess protein expression at the single-cell level of resolution, although the number of epitopes that can be detected concurrently is limited by spectral overlap between fluorophores. The advent of mass cytometry56, which involves the use of metal isotopes to label antibodies, has greatly expanded the number of markers that can be analysed simultaneously, thereby enabling the high-dimensional detection of proteins expressed on single cells. For example, Wagner et al.57 used a 35-marker immune cell-centric panel and a 38-marker tumour cell-centric panel to generate a comprehensive single-cell atlas of breast cancers.
In general, the analytical precision of single-cell proteomics methods is greater than that of current scRNA-seq methods. Furthermore, the longer half-life of proteins compared with RNA means that protein measurements are less prone to the fluctuations often observed in scRNA-seq data. Antibody-targeted approaches also provide additional detection capabilities, such as the detection of post-translational modifications58 or the assessment of morphological features via cell morphometry59. Similar to flow cytometry, however, the targeted nature of mass cytometry analysis requires the prospective selection of analytes, which can bias the identification of cellular groups compared with the untargeted simultaneous detection of hundreds to thousands of RNA transcripts using scRNA-seq.
Mass cytometry is a particularly useful method of characterizing the specific phenotypes of cells involved in responses to immunotherapy at the single-cell level, as illustrated by the results of two investigations of responses to ICIs in patients with melanoma. In the first study60, investigators used mass cytometry to demonstrate that a high frequency of circulating classic monocytes before treatment is a strong predictor of a response to anti-PD-1 antibodies, which was confirmed by analysing samples from a validation cohort of 31 patients using targeted flow cytometry. In the second study, Gide et al.61 analysed melanoma biopsy samples obtained from a subset of patients who received either anti-PD-1 antibodies as monotherapy or in combination with anti-CTLA-4 antibodies. Dimensionality reduction analysis of a 43-marker mass cytometry panel identified 14 immune cell clusters, including three distinct T cell clusters. Key markers of activation, differentiation and exhaustion were found to co-cluster with a CD45RO+EOMES+ T cell population, including markers of recent activation and tissue residency (CD69), tissue-resident memory (CD103) and effector memory (T-BET, HLA-DR and low CCR7). This subpopulation expressed the inhibitory receptors PD-1 and TIGIT, but had only low levels of CD57 expression, suggesting they are not terminally exhausted. Responders to combination therapy had a higher proportion of this CD45RO+EOMES+ subpopulation among both the CD4+ and CD8+ cells compared with non-responders, but no difference was observed among patients receiving monotherapy. When the insights gained from mass cytometry were used to examine a larger cohort of 140 samples from 73 patients with melanoma using bulk RNA-seq analysis, a memory T cell phenotype featuring high levels of CD4 or CD8, EOMES, CD69 and CD45RO expression corresponded with responsiveness to both ICI regimens and was predictive of a longer progression-free survival duration in those receiving anti-PD-1 antibodies as monotherapy. These observations underscore the importance of evaluating immune cell phenotypes at the single-cell level.
As noted previously, the ability to detect post-translational modifications is a distinct advantage of antibody-targeted approaches, such as mass cytometry. This capability was utilized in a study analysing chimeric antigen receptor (CAR) T cells using a mass cytometry panel that focused on the detection of phosphoproteins with roles in maintaining T cell function53. The single-cell resolution provided by mass cytometry enabled a more sophisticated analysis of network dynamics. Analysis of second-generation CD19–28ζ CAR cells indicated that tonic signalling, resulting in antigen-independent CAR activation, is predominantly associated with the CD3ζ, but not the CD28, endodomain. Tonic signalling can affect the efficacy of therapy by inducing CAR T cell exhaustion. On the basis of these single-cell findings, the authors explored an alternative CAR design that circumvents tonic signalling by using γδT cells expressing a chimeric costimulatory receptor. By separating the CD3ζ domain in the γδTCR from the CD28 domain in the chimeric costimulatory receptor, this modified CAR design enabled efficient cytotoxic responses to CD19-positive leukaemic cells without evidence of activation of tonic signalling. The single-cell analysis of post-translational modifications will continue to be of particular benefit to understanding the signalling pathways associated with effectiveness and/or resistance of adoptive cellular therapies and native immune cells in the context of immunotherapy.
Emerging technologies for single-cell proteome analysis.
Next-generation spectral analysers promise to extend the capabilities of fluorescence-based flow cytometry to a level that rivals that of mass cytometry. An advantage of these systems over mass cytometry is the ability to utilize the vast number of fluorescence-labelled antibodies that are already available and, because they do not destroy cells like mass cytometry does, enable the sorting and isolation of cells for subsequent experiments. As another emerging technology, tagging antibodies with barcode oligonucleotides, instead of fluorophores or heavy metal ions, obviates the constraints placed on the number of simultaneously resolvable targets.
Additional antibody-targeted single-cell strategies include tissue-based cytometry (BOX 1), which adds an improved level of spatial resolution, and microfluidic cytometry, which is exemplified by chipcytometry62,63 wherein cells are immobilized on a microfluidic chip instead of being analysed in a flow stream. Chipcytometry enables iterative staining; therefore, scores of markers (up to 110) can be detected and quantified on the same sample of cells. Another innovation based on single-cell barcode chip technology64,65 is the IsoLight system66, which enables the detection of secreted proteins from single cells. The system utilizes microfluidic chips to capture information from 200 to 2,000 single live cells and sandwich-ELISA-like assays to measure >30 cytokines, chemokines, growth factors and other secreted ligands over a multi-hour incubation period. This system also has the flexibility to incorporate additional analytes, such as metabolites. The preliminary utility of the IsoLight system has been demonstrated in a phase 1 trial in which patients with advanced-stage melanoma received adoptive cell transfer supported by pegylated IL-2, which resulted in enhanced levels of cytokine polyfunctionality among circulating T cells and natural killer cells67. These new platforms of spectral analysers and microfluidic systems are still in the early stages of development and need to be validated in additional clinical settings. Nonetheless, the early successes thus far suggest that they will increasingly have a key role in immuno-oncology.
Genomics
Methods enabling whole-genome and whole-exome sequencing of single cells have been developed (reviewed extensively elsewhere by Gawad et al.68), although these methods have not yet made the transition to high-dimensional analysis, largely because the high costs of such sequencing preclude scaling to sizeable numbers of cells per patient. A more efficient approach is to first use deep sequencing of bulk tumour DNA samples to identify sSNVs, then use targeted sequencing of DNA from single cells to detect putative driver mutations or mutations implicated in clonal structure. By combining microfluidics with the ability to merge droplets, the Tapestri system uses a two-step process whereby robust lysis and liberation of DNA occurs in a first droplet followed by merging with a second droplet to perform PCR amplification and cell-specific barcoding of target genomic loci69. Although not yet applied in the context of immunotherapy, this system has been used to analyse hundreds of thousands of single cells to track specific mutations through diagnosis, remission and relapse in patients with acute myeloid leukaemia70–74 — thus demonstrating the promise of this technology.
Epigenomics
Epigenomic analysis methods, such as the assay for transposase-accessible chromatin using sequencing (ATAC-seq, which enables the detection of open segments of chromatin), bisulfite-based DNA methylation sequencing, chromatin immunoprecipitation sequencing (ChIP-seq, which detects DNA–protein interactions) and chromosome conformation capture (such as 3C and Hi-C), have all been adapted for single-cell analysis (reviewed in detail elsewhere75–77). Of these methods, single-cell (sc)ATAC-seq is currently the only one with sufficiently high throughput and is thus widely adopted for this purpose. For example, application of this method to pretreatment and post-treatment samples obtained from patients with advanced-stage basal cell carcinoma revealed expanded populations of exhausted CD8+ T cells and CD4+ T follicular helper cells after treatment with anti-PD-1 antibodies78. In these cells, the PDCD1 locus contained a +5 kb cis-element with specific accessibility, suggesting that post-treatment CD8+ T cells and T follicular helper cells have a shared enhancer that drives persistent PD-1 expression. Single-cell epigenomics is a rapidly developing field that, particularly when integrated with single-cell transcriptomics79,80, is likely to be one of the next major advances in single-cell analysis in oncology. In this regard, single-cell reduced representation bisulfite sequencing81 for DNA methylation analysis and CUT&Tag82 for single-cell ChIP-seq, are particularly notable, and these methods should further expand the ability to analyse epigenetic changes at the level of individual cells.
Multimodal analysis
Interest in integrating distinct modalities of single-cell analysis into consolidated technologies is increasing83,84. The application of such multimodal approaches to investigating responses to immunotherapy has been limited thus far, although several promising methods with broad applicability are briefly reviewed here.
RNA or DNA plus protein.
Techniques incorporating single-cell detection of proteins into scRNA-seq protocols (such as CITE-seq85 and REAP-seq86) use antibodies conjugated with barcode oligonucleotides to enable the detection of protein-based markers via the cDNA sequencing steps of scRNA-seq, thereby enabling protein detection to proceed without the constraints imposed by spectral overlap or the number of resolvable metal ions. For example, the proximity extension assay87 that uses barcoded antibodies has enabled the development of commercial 96-plex panels for the detection of proteins present in serum and plasma samples. The ability to add the antibody-based detection of tens to hundreds of proteins to untargeted transcriptome analysis will substantially improve the ability to assess the TME by defining transcriptional clusters within the framework of traditional immunology and diminishing ambiguities caused by transcript dropout. The use of oligonucleotide-tagged antibodies has also been incorporated into the Tapestri workflow for single-cell DNA sequencing, providing a direct method of linking genotype and phenotype88.
Lineage tracing.
scATAC-seq data contain sequencing reads from the mitochondrial genome. Although these signals were originally treated as an experimental nuisance, this feature enables the detection of mitochondrial mutations that can be used to perform clonal tracing89,90. Thus, scATAC-seq enables the simultaneous measurement of cell lineage (via the mitochondrial genome) and cell fate (via the nuclear epigenome). Mitochondrial mutations can also be detected in scRNA-seq data84, although not as robustly as in scATAC-seq data owing to lower and less consistent coverage of the mitochondrial genome. Such lineage tracing analysis could be applied in the future to better understand which tumour cell clones expand or contract in response to a specific treatment, and thus which clones might be linked to therapeutic resistance.
Single-cell T cell receptor analysis
T cells are the main arm of the adaptive immune system responsible for anti-tumour immunity and are thus the primary focus of most FDA-approved cancer immunotherapies (such as ICIs, adoptive cell therapies, peptide or RNA vaccines, cytokine-based therapies and oncolytic virotherapies). Owing to this focus, the analysis of individual T cell clonotypes is currently of increasing research interest. The TCR is a heterodimer comprising two chains, for which genetic recombination creates a diverse TCR repertoire. The majority of T cells (95%) carry individual TCR α and β subunits, which together recognize a cognate peptide–MHC antigen91. On the basis of this TCR–peptide–MHC interaction, naturally occurring TILs in immunogenic tumours have the potential to eliminate tumours92. Determining whether a T cell is capable of targeting a tumour cell requires single-cell paired TCR α and β subunit sequences for TCR reconstruction and specificity testing. Furthermore, in order for immunotherapies to generate an effective antigen-specific anti-tumour immune response, tumour antigens must ultimately be presented as antigen–MHC complexes and recognized by cognate TCRs present on tumour-infiltrating T cells. A growing body of evidence suggests that many tumour-infiltrating T cells are simply ‘bystanders’ that are incapable of recognizing and attacking tumour cells93. Therefore, a tumour might appear to be highly infiltrated by immune effector cells, but nonetheless be ‘qualitatively cold’ owing to a lack of tumour-specific T cells capable of anti-tumour activity94. Beyond quantifying cellular composition and characterizing transcriptional states, single-cell approaches also enable the identification of paired α and β TCR subunits, which is a crucial step in determining the specificity of infiltrating T cells. Such information might not only help explain resistance to current immunotherapies (such as a lack of tumour specificity), but also facilitate the discovery of high-avidity anti-tumour TCRs that can be utilized in novel engineered TCR-based cellular therapies.
Bulk TCR sequencing cannot assign paired TCR subunits to individual T cells; therefore, this information was historically obtained by isolating TILs using in vitro culture methods, then subcloning cells through a low-throughput, labour-intensive process of creating homogeneous populations of T cells that could then be sequenced to determine individual TCR αβ subunit combinations. In the subsequent years, numerous PCR-based techniques were developed to enable sequencing of individual T cell TCR–complementarity determining region (CDR3) sequences in order to unambiguously assign TCR αβ subunits to individual T cells in a tumour95–98. Paired αβ sequences can now be obtained at higher throughput using droplet-based methods. Moreover, the identified TCR sequences can be reconstituted in native T cells or reporter cell lines using viral transfection to deduce antigen specificity99–101. Combining scRNA-seq with single-cell TCR sequencing (scTCR-seq) links T cell phenotypes, such as activation, memory and exhaustion, with the specific TCR clonotypes of individual T cells. The ability to unambiguously assign TCR sequences to single T cells has led to the development of microfluidic and flow cytometry-based methods designed to directly identify antigen-specific T cells from TIL, tissue and peripheral blood samples102–104. Together, the integration of scTCR-seq, antigen specificity determination and the single-cell analysis approaches described previously have enabled more detailed investigations of several important topics in immuno-oncology, such as the roles of clonal replacement versus clonal expansion and of T cell clonotypic phenotypes in immunotherapy responses.
scTCR-seq of mRNA had a key role in the analysis of samples obtained in a phase 1 trial involving personalized vaccination with neoantigen peptides to treat patients with glioblastoma105. In one patient, scTCR-seq enabled the identification of clonotypes shared between TILs and peripheral blood T cells. Cloning and expression of the shared TCRs demonstrated the presence of TILs within the tumour that were specific for a vaccinating peptide, thus highlighting the power of these techniques to track and evaluate individual T cell clonotypes during an immunotherapy response. Tumour material from other patients in this study was not analysed in this manner owing to difficulties in obtaining TILs as single-cell suspensions from clinical samples, which is a persistent issue in applying scTCR-seq to the management of patients with solid tumours.
Integrated scRNA-seq and scTCR-seq provides a particularly effective method of analysing the phenotypic and functional characteristics of immune cells over the course of treatment with immunotherapy. For example, analysis of pretreatment and post-treatment biopsy samples from patients with advanced-stage basal cell carcinoma (n = 11) or squamous cell carcinoma (n = 4) receiving treatment with an anti-PD-1 antibody revealed an increase in the number of both activated and chronically activated (exhausted) CD8+ TILs following treatment106. Notably, the greatest degree of TCR clonality (implying the lowest level of diversity) was observed in post-treatment exhausted CD8+ TILs, and the overwhelming majority of these clonotypes consisted of expanded novel clones that were not detected prior to anti-PD-1 therapy. Exhausted TIL clones that were present before treatment did not expand upon treatment nor did they transition to non-exhausted phenotypes. Bulk repertoire analysis of pretreatment peripheral blood samples revealed that 11.8% of the post-treatment TILs were novel exhausted T cell clonotypes, even though these clonotypes were not detected in pretreatment TILs. These results suggest that pre-existing tumour-specific TILs have only a limited ability to be reactivated and that ICIs can induce the influx and expansion of novel T cell clones (clonal replacement). Additionally, in an analysis of samples from eight primary uveal melanomas and three metastases107, scTCR-seq revealed expanded populations of specific T cell clonotypes, and scRNA-seq indicated that these expanded T cell clones were largely exhausted. The expression of genes encoding exhaustion-associated immune checkpoints was strongest for LAG3, with minimal expression of PDCD1 and CTLA4, which might contribute to the observed ineffectiveness of anti-PD-1 and anti-CTLA4 antibodies in this setting. The results of single-cell studies like these suggest that responsiveness to ICIs might depend on non-exhausted T cells and T cells recruited from the periphery (clonal replacement) rather than only on the reinvigoration of exhausted T cells in the tumour (clonal expansion).
By complementing single-cell phenotypic profiling of T cells (such as via scRNA-seq) with the identification of their respective TCR clonotypes, these integrated approaches add crucial information on T cell antigen specificity to the analyses of immune cells, thereby enabling finer dissection of the roles of antigen-specific T cells in immunotherapy responses. Furthermore, the non-invasive identification of expanded TCR clones in blood samples collected longitudinally over the course of treatment with immunotherapy might provide insights into the identity and functionality of clinically relevant TCRs in tumours. The development of a cost-effective method of identifying paired TCR αβ subunit clonotypes from peripheral blood108 will make the longitudinal analysis of blood samples obtained from patients receiving immunotherapy in clinical trials even more powerful.
High-dimensional spatial analysis
Spatial variability in the tumour immune microenvironment is increasingly becoming recognized as contributing to the heterogeneous nature and variable therapeutic responses seen with most cancer types. Time-honoured spatial methods dating back to Coons et al.109 (selective immunostaining of proteins by immunohistochemistry and immunofluorescence) and Gall and Pardue110 (in situ hybridization of nucleic acid sequences) have been indispensable for cancer diagnosis, classification, treatment selection and prognostication. Contemporary applications include the quantification of PD-L1 expression using immunohistochemistry and HER2 amplification using in situ hybridization, both of which are required by the FDA as companion diagnostics for certain anti-PD-(L)1 checkpoint inhibitors and HER2-targeted monoclonal antibodies, respectively, in defined clinical indications. Over the past few years, these more traditional methods have been substantially expanded, multiplexed and coupled with advances in bioinformatics to enable the simultaneous visualization of multiple proteins, DNA and/or RNA molecules. Several approaches that combine spatial resolution with next generation sequencing are also being developed (BOX 1). Most published data relating to these new approaches are limited to preliminary studies designed to demonstrate proof-of-concept.
Multiplexing of immunohistochemistry and immunofluorescence has already demonstrated preliminary clinical utility in a few immunotherapy studies. A meta-analysis of data from 8,135 patients across ten different cancer types treated with anti-PD-1 or anti-PD-L1 antibodies demonstrated that multiplexed immunohistochemistry plus immunofluorescence strategies outperform tumour mutational burden, gene expression profiling and PD-L1 immunohistochemistry expression biomarkers in predicting response to immunotherapy111. The diagnostic benefits of multiplexed imaging were not dependent on a large number of analytes — improved prediction was observed with as few as two or three markers — but from the incorporation of information on the spatial relationships and protein co-expression at the single-cell level of resolution. Indeed, in an investigation of tumour biopsy samples from patients with metastatic melanoma who received anti-PD-1 antibodies, tumours with both increased spatial interactions between PD-L1+ cells (both tumour and myeloid) and PD-1+ non-tumour cells (mostly immune) as well as those featuring higher proportions of tumour cells co-expressing HLA-DR and IDO-1 were most likely to respond to immunotherapy and were associated with improved survival outcomes112. These single-cell level spatial findings reinforce the hypothesis that ample and proximal expression of both PD-1 in T cells and PD-L1 in tumour cells is needed to derive therapeutic benefit from anti-PD-1 or anti-PD-L1 antibodies.
High-dimensional protein detection methods have the potential to increase the multiplex capabilities of spatial analysis to a level of 20–100 markers, thus expanding the number of cell types and cell states that can be characterized (BOX 1). This increased dimensionality enables an unprecedented depth of analysis of the complex spatial interactions between networks of single-cell phenotypes, thereby spatially defining ‘communities’ of cells in the tumour and/or their microenvironment that might be better prognostic biomarkers than quantification of single-cell phenotypes alone113,114. Cytometry by time of flight (CyTOF) and scRNA-seq-based analyses implicated intratumoural B cell receptor diversity and memory B cell phenotypes in responses to ICIs in patients with melanoma, with the addition of spatial methods indicating the importance of B cell localization to tertiary lymphoid structures (TLSs)115. By using the GeoMx DSP platform to quantify both the expression and location of 60 proteins in B cells, T cells and tumour cells in melanoma biopsy samples, Cabrita et al.116 found that B cells interact closely with CD4+ T cells in TLSs and are associated with increases in the numbers of TCF7+-naive and memory T cells. Conversely, TILs present in tumours that lack TLSs were more likely to have molecular signatures associated with immunological dysfunction and/or exhaustion. By integrating these key spatial findings with scRNA-seq data, the authors derived a TLS gene signature that predicted an improved response to ICIs116.
Although still in their technological infancy, spatially-resolved multiplexed profiling approaches are reshaping the ability to interrogate both the intercellular interactions and the architectural relationships between tumour cells and cells of the TME that govern tumour immunology and responsiveness to immunotherapy in patients.
Future directions
scRNA-seq and its various multimodal incarnations are revolutionary research tools that are advancing both our understanding of human cancer immunology and the mechanisms driving responsiveness and resistance to immunotherapies in patients. Although immunotherapies have transformed the management of many advanced-stage malignancies, the majority of patients either do not respond to these agents or will ultimately acquire resistance1. Thousands of clinical trials exploring combination immunotherapy approaches, with other immunotherapy agents, with non-immunotherapy agents or both, have either been completed or are currently underway117; nonetheless, the design of rational immunotherapy-containing combinations designed to overcome primary and/or acquired resistance will require the comprehensive characterization of the tumour immune microenvironment of each cancer type, and a detailed understanding of the determinants of effective anti-tumour immunity in the context of therapy. Single-cell technologies enable comprehensive profiling of the TME and are therefore especially well-suited to studying both tumour and immune cell heterogeneity and how such differences in tumour biology contribute to therapeutic resistance118,119.
In order to effectively leverage single-cell technologies to improve our understanding of mechanisms of immunotherapy response and resistance and to aid the development of novel therapeutics, clinical trials will have to be carefully designed to incorporate single-cell approaches in a feasible way. Many of the current technologies are costly and require the isolation of viable single cells from disaggregated tumour tissues, thus posing substantial financial and logistical challenges that preclude their routine use in clinical care. If clinical studies are carefully designed in collaboration with translational scientists, however, incorporating single-cell analysis is feasible in a subset of patients and could yield informative results that can be applied to larger cohorts. Careful consideration must be given to what types (among others, tumour tissue, non-malignant adjacent tissues and/or peripheral blood) and formats (including fresh, snap-frozen and/or formalin-fixed paraffin-embedded) of samples should be collected, the optimal timing of collection (pretreatment, on-treatment and/or post-treatment) and how sample material will be rapidly processed for immediate single-cell analysis (or cryopreserved for later analysis). While challenging, the insights gleaned from an in-depth investigation of a relatively small number of patients (5–50) can inform the analysis of a larger sample set using a more cost-effective and logistically more straightforward method (FIG. 2). As indicated in a number of studies described in this Perspective, an emerging approach is to exploit published scRNA-seq data rather than to generate these data de novo. This type of resource has now been formalized in the establishment of the Human Tumor Atlas Network120.
Fig. 2 |. General biospecimen framework for incorporating single-cell analyses in clinical trials involving immunotherapies.
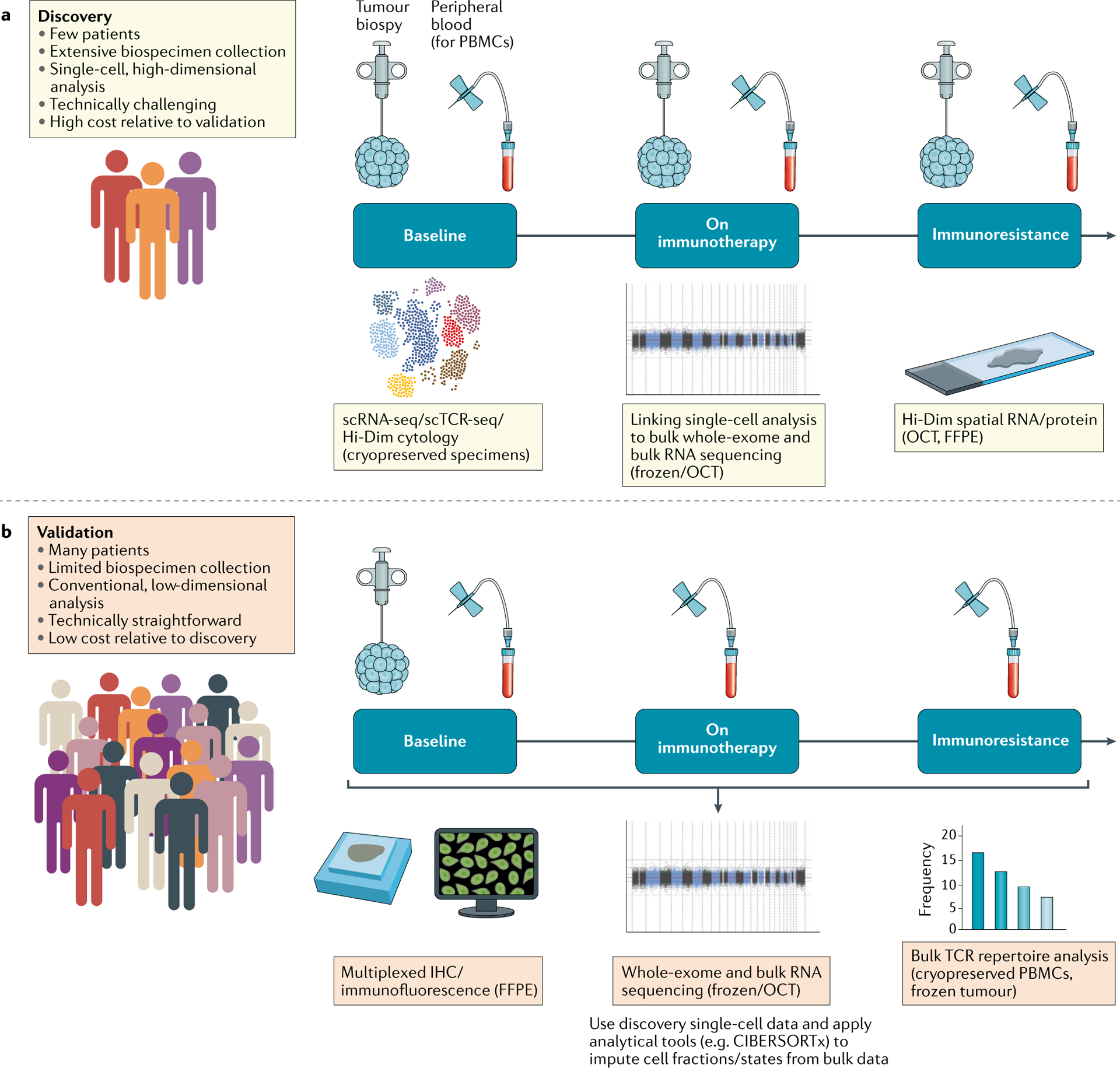
a | Discovery efforts for a limited subset of patients. In-depth characterization of pre-treatment, on-treatment and post-treatment (resistance) tumour biopsies and peripheral immune cells using single-cell and bulk approaches enables the discovery of biological determinants of therapeutic response and resistance. These analyses are technically challenging and expensive, and so would only be performed in a limited number of patients. b | Validation using conventional technologies for a larger set of patients. For a larger set of clinical trial patients, more conventional sample collection (such as frozen or formalin-fixed paraffin-embedded (FFPE) tumour tissue at baseline, peripheral blood cells before and during treatment) is feasible. These samples can be used to validate proposed biological determinants of immunotherapy response and resistance (from the ‘discovery’ efforts depicted in part a) using standardized assays. PBMCs, peripheral blood mononuclear cells; Hi-Dim, high-dimensional; IHC, immunohistochemistry; OCT, optimal cutting temperature compound; scRNA-seq, single-cell RNA sequencing; TCR, T cell receptor.
Mining high-dimensional single-cell data to define low-dimensional panels of genes and/or proteins that show promise as predictive biomarkers provides a clear path towards clinical utility. Another avenue for exploiting data-rich single-cell information is provided by the use of bioinformatic algorithms to map bulk RNA-seq data onto the cell types defined by scRNA-seq and related techniques. The use of these in silico dissection approaches is less subject to the compromises inherent in using low-dimensional panels alone. A good example of this approach is provided by CIBERSORTx121, a machine learning-based method that takes a signature matrix generated from scRNA-seq data and applies it to bulk tissue RNA-seq profiles in order to estimate the composition and abundance of cell types in each tissue sample. Thus, highly refined cell types defined by whole-transcriptome data can be readily quantified in routine clinical samples that might be of limited quality, fixed and/or difficult to disaggregate into intact single cells — thereby helping to substantially reduce the barriers to implementation in future immuno-oncology studies.
High-dimensional single-cell techniques that could be more readily applied clinically include those that expand the information content of traditional methods, such as immunohistochemistry and flow cytometry. For samples from haematological malignancies and others that can be easily dispersed into single cells, mass cytometry and flow cytometry panels incorporating increased numbers of fluorescent markers (such as 20–50) have already begun to be successfully utilized in clinical trials (NCT03734198, NCT03915379 and NCT04181827). For solid tumours, spatially resolved multiplexed technologies (BOX 1) have the ability to add genotypic and phenotypic dimensions to our understanding of how cells interact in the tumour immune microenvironment, and constitute the next frontier in elucidating the mechanisms underlying resistance to immunotherapies. Devices that automate these technologies and are compatible with existing clinical laboratory workflows, such as the in situ sequencing microfluidic tissue processor122, indicate potential ways forward to the type of advances that will ease the adoption of these techniques.
Conclusions
Single-cell RNA sequencing and related modalities are increasingly becoming critical research tools for characterizing response and resistance to immunotherapy. Additionally, single-cell TCR analysis is required to determine T cell antigen specificity and provides crucial information on the role of the adaptive immune system in tumour formation, growth and treatment. The next frontier in single-cell analysis is the combination of high-dimensional content with precise tissue localization. The clinical utility of high-dimensional single-cell analysis includes identifying high-dimensional biomarkers of immunotherapy response, robustly informing the design of low-dimensional biomarkers, enabling refined analysis of bulk sequencing data and understanding the complex cell–cell spatial interactions in the TME that drive responses to immunotherapy. Given the growing matrix of possible immunotherapies coupled with the personalized nature of tumour heterogeneity and therapeutic resistance, the future of cancer treatment is combination therapy123–125. As a result, multidimensional biomarker signatures will be crucial to making the optimal management choices for each patient with cancer. High-dimensional single-cell technologies will provide the resolution and richness of data required to generate these signatures in immuno-oncology.
Acknowledgements
The work of S.H.G. is supported by a Kay Kendall Leukaemia Fund Fellowship. The work of J.B.I. is supported by the NCI (K12CA090354) and Conquer Cancer Foundation-Sontag Foundation Young Investigator Award. The work of D.A.B. is supported by the DF/HCC Kidney Cancer SPORE Career Enhancement Program (P50CA101942-15), DOD CDMRP (KC170216, KC190130) and the DOD Academy of Kidney Cancer Investigators (KC190128). The work of D.B.K. is supported by the NIH/NCI (R21 CA216772-01A1 and NCI-SPORE-2P50CA101942-11A1). The work of K.J.L. is supported by the NIH/NCI (U24 CA224331 R01CA229261-01, NIH/NCI P01CA229092) and the NIH/NIAID (U19 AI082630).
Competing interests
D.A.B. has received non-financial support from Bristol Myers Squibb, honoraria from LM Education/Exchange Services and personal fees from Adept Field Solutions, Blueprint Partnerships, Charles River Associates, Dedham Group, Defined Health, Insight Strategy, Octane Global, Slingshot Insights and Trinity Group. D.B.K. has acted as an advisor of and has received consulting fees from Neon Therapeutics and owns equity in Aduro Biotech, Agenus, Armata Pharmaceuticals, Breakbio, BioMarin Pharmaceutical, Bristol Myers Squibb, Celldex Therapeutics, Editas Medicine, Exelixis, Gilead Sciences, IMV, Lexicon Pharmaceuticals, Moderna, Regeneron Pharmaceuticals and Stemline Therapeutics. The other authors declare no competing interests.
References
- 1.Sharma P, Hu-Lieskovan S, Wargo JA & Ribas A Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iorgulescu JB, Braun D, Oliveira G, Keskin DB & Wu CJ Acquired mechanisms of immune escape in cancer following immunotherapy. Genome Med. 10, 87 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang L, Livak KJ & Wu CJ High-dimension single-cell analysis applied to cancer. Mol. Aspects Med 59, 70–84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finotello F & Eduati F Multi-omics profiling of the tumor microenvironment: paving the way to precision immuno-oncology. Front. Oncol 8, 430 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomes T, Teichmann SA & Talavera-López C Immunology driven by large-scale single-cell sequencing. Trends Immunol. 40, 1011–1021 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Tang X, Huang Y, Lei J, Luo H & Zhu X The single-cell sequencing: new developments and medical applications. Cell Biosci. 9, 53 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen G, Ning B & Shi T Single-cell RNA-Seq technologies and related computational data analysis. Front. Genet 10, 317 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qi R, Ma A, Ma Q & Zou Q Clustering and classification methods for single-cell RNA-sequencing data. Brief. Bioinform 21, 1196–1208 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luecken MD & Theis FJ Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol 15, e8746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saelens W, Cannoodt R, Todorov H & Saeys Y A comparison of single-cell trajectory inference methods. Nat. Biotechnol 37, 547–554 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Lähnemann D et al. Eleven grand challenges in single-cell data science. Genome Biol. 21, 31 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Svensson V, Vento-Tormo R & Teichmann SA Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc 13, 599–604 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Regev A et al. The human cell atlas. eLife 6, e27041 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plasschaert LW et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villani A-C et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Filbin MG et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 360, 331–335 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jerby-Arnon L et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997.e24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puram SV et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 171, 1611–1624.e24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Izar B et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med 26, 1271–1279 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tirosh I et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venteicher AS et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neftel C et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178, 835–849.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Young MD et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 361, 594–599 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szabo PA et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun 10, 4706 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lambrechts D et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med 24, 1277–1289 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Zhang L et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Azizi E et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308.e36 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sade-Feldman M et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013. e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Efremova M, Vento-Tormo M, Teichmann SA & Vento-Tormo R CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc 15, 1484–1506 (2020). [DOI] [PubMed] [Google Scholar]
- 31.Cabello-Aguilar S et al. SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 48, e55 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Q et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell 179, 829–845.e20 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Chubb JR, Trcek T, Shenoy SM & Singer RH Transcriptional pulsing of a developmental gene. Curr. Biol 16, 1018–1025 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dar RD et al. Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc. Natl Acad. Sci. USA 109, 17454–17459 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng Q, Ramsköld D, Reinius B & Sandberg R Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343, 193–196 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Reinius B & Sandberg R Random monoallelic expression of autosomal genes: stochastic transcription and allele-level regulation. Nat. Rev. Genet 16, 653–664 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Patel AP et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Müller S et al. Single-cell sequencing maps gene expression to mutational phylogenies in PDGF-and EGF-driven gliomas. Mol. Syst. Biol 12, 889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriguez-Meira A et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel RNA sequencing. Mol. Cell 73, 1292–1305.e8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nam AS et al. Somatic mutations and cell identity linked by genotyping of transcriptomes. Nature 571, 355–360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Galen P et al. Single-cell RNA-Seq reveals AML hierarchies relevant to disease progression and immunity. Cell 176, 1265–1281.e24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van den Brink SC et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 14, 935–936 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Massoni-Badosa R et al. Sampling time-dependent artifacts in single-cell genomics studies. Genome Biol. 21, 112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slyper M et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med 26, 792–802 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Habib N et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bakken TE et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE 13, e0209648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu H, Kirita Y, Donnelly EL & Humphreys BD Advantages of single-nucleus over single-cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J. Am. Soc. Nephrol 30, 23–32 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang Q et al. Single-nuclei RNA-seq on human retinal tissue provides improved transcriptome profiling. Nat. Commun 10, 5743 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tran HTN et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L & Vertes A Single-cell mass spectrometry approaches to explore cellular heterogeneity. Angew. Chem. Int. Ed. Engl 57, 4466–4477 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Löfblom J, Frejd FY & Ståhl S Non-immunoglobulin based protein scaffolds. Curr. Opin. Biotechnol 22, 843–848 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Banta S, Dooley K & Shur O Replacing antibodies: engineering new binding proteins. Annu. Rev. Biomed. Eng 15, 93–113 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Reverdatto S, Burz DS & Shekhtman A Peptide aptamers: development and applications. Curr. Top. Med. Chem 15, 1082–1101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ståhl S et al. Affibody molecules in biotechnological and medical applications. Trends Biotechnol. 35, 691–712 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Zhou Z, Liu M & Jiang J The potential of aptamers for cancer research. Anal. Biochem 549, 91–95 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Spitzer MH & Nolan GP Mass cytometry: single cells, many features. Cell 165, 780–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wagner J et al. A single-cell atlas of the tumor and immune ecosystem of human breast cancer. Cell 177, 1330–1345.e18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fisher J et al. Engineering γδT cells limits tonic signaling associated with chimeric antigen receptors. Sci. Signal 12, eaax1872 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsai AG et al. Multiplexed single-cell morphometry for hematopathology diagnostics. Nat. Med 26, 408–417 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krieg C et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med 24, 144–153 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Gide TN et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell 35, 238–255.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Hennig C, Adams N & Hansen G A versatile platform for comprehensive chip-based explorative cytometry. Cytometry A 75, 362–370 (2009). [DOI] [PubMed] [Google Scholar]
- 63.Teo J et al. A preliminary study for the assessment of PD-L1 and PD-L2 on circulating tumor cells by microfluidic-based chipcytometry. Future Sci. OA 3, FSO244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shi Q et al. Single-cell proteomic chip for profiling intracellular signaling pathways in single tumor cells. Proc. Natl Acad. Sci. USA 109, 419–424 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Su Y et al. Multi-omic single-cell snapshots reveal multiple independent trajectories to drug tolerance in a melanoma cell line. Nat. Commun 11, 2345 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu D, Paczkowski P, Mackay S, Ng C & Zhou J Single-cell multiplexed proteomics on the IsoLight resolves cellular functional heterogeneity to reveal clinical responses of cancer patients to immunotherapies. Methods Mol. Biol 2055, 413–431 (2020). [DOI] [PubMed] [Google Scholar]
- 67.Parisi G et al. Persistence of adoptively transferred T cells with a kinetically engineered IL-2 receptor agonist. Nat. Commun 11, 660 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gawad C, Koh W & Quake SR Single-cell genome sequencing: current state of the science. Nat. Rev. Genet 17, 175–188 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Pellegrino M et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 28, 1345–1352 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McMahon CM et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 9, 1050–1063 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Xu L et al. Clonal evolution and changes in two AML patients detected with a novel single-cell DNA sequencing platform. Sci. Rep 9, 11119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ediriwickrema A et al. Single-cell mutational profiling enhances the clinical evaluation of AML MRD. Blood Adv. 4, 943–952 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DiNardo CD et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 135, 791–803 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Choe S et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 4, 1894–1905 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kelsey G, Stegle O & Reik W Single-cell epigenomics: recording the past and predicting the future. Science 358, 69–75 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Shema E, Bernstein BE & Buenrostro JD Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat. Genet 51, 19–25 (2019). [DOI] [PubMed] [Google Scholar]
- 77.Ludwig CH & Bintu L Mapping chromatin modifications at the single cell level. Development 146, dev170217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Satpathy AT et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol 37, 925–936 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cao J et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361, 1380–1385 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reyes M, Billman K, Hacohen N & Blainey PC Simultaneous profiling of gene expression and chromatin accessibility in single cells. Adv. Biosys 3, 1900065 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gaiti F et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature 569, 576–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1930 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stuart T & Satija R Integrative single-cell analysis. Nat. Rev. Genet 20, 257–272 (2019). [DOI] [PubMed] [Google Scholar]
- 84.Peng A, Mao X, Zhong J, Fan S & Hu Y Single-cell multi-omics and its prospective application in cancer biology. Proteomics 20, 1900271 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Stoeckius M et al. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 14, 865–868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Peterson VM et al. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol 35, 936–939 (2017). [DOI] [PubMed] [Google Scholar]
- 87.Assarsson E et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE 9, e95192 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Miles LA et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 10.1038/s41586-020-2864-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ludwig LS et al. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell 176, 1325–1339.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu J et al. Single-cell lineage tracing by endogenous mutations enriched in transposase accessible mitochondrial DNA. eLife 8, e45105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Davis MM T cell receptor gene diversity and selection. Annu. Rev. Biochem 59, 475–496 (1990). [DOI] [PubMed] [Google Scholar]
- 92.Yang JC & Rosenberg SA Adoptive T-cell therapy for cancer. Adv. Immunol 130, 279–294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Simoni Y et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557, 575–579 (2018). [DOI] [PubMed] [Google Scholar]
- 94.Scheper W et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med 25, 89–94 (2019). [DOI] [PubMed] [Google Scholar]
- 95.Wang GC, Dash P, McCullers JA, Doherty PC & Thomas PG T cell receptor αβ diversity inversely correlates with pathogen-specific antibody levels in human cytomegalovirus infection. Sci. Transl Med 4, 128ra42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Han A, Glanville J, Hansmann L & Davis MM Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat. Biotechnol 32, 684–692 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dash P, Wang GC & Thomas PG Single-cell analysis of T-cell receptor αβ repertoire. Methods Mol. Biol 1343, 181–197 (2015). [DOI] [PubMed] [Google Scholar]
- 98.Li S et al. RNase H-dependent PCR-enabled T-cell receptor sequencing for highly specific and efficient targeted sequencing of T-cell receptor mRNA for single-cell and repertoire analysis. Nat. Protoc 14, 2571–2594 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kobayashi E et al. A new cloning and expression system yields and validates TCRs from blood lymphocytes of patients with cancer within 10 days. Nat. Med 19, 1542–1546 (2013). [DOI] [PubMed] [Google Scholar]
- 100.Guo X-ZJ et al. Rapid cloning, expression, and functional characterization of paired αβ and γδ T-cell receptor chains from single-cell analysis. Mol. Ther. Methods Clin. Dev 3, 15054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hu Z et al. A cloning and expression system to probe T-cell receptor specificity and assess functional avidity to neoantigens. Blood 132, 1911–1921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kula T et al. T-Scan: a genome-wide method for the systematic discovery of T cell epitopes. Cell 178, 1016–1028.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang S-Q et al. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat. Biotechnol 36, 1156–1159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Segaliny AI et al. Functional TCR T cell screening using single-cell droplet microfluidics. Lab Chip 18, 3733–3749 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Keskin DB et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 565, 234–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yost KE et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med 25, 1251–1259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Durante MA et al. Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat. Commun 11, 496 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tanno H et al. A facile technology for the high-throughput sequencing of the paired VH:VL and TCRβ:TCRα repertoires. Sci. Adv 6, eaay9093 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Coons AH, Creech HJ & Jones RN Immunological properties of an antibody containing a fluorescent group. Exp. Biol. Med 47, 200–202 (1941). [Google Scholar]
- 110.Gall JG & Pardue ML Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc. Natl Acad. Sci. USA 63, 378–383 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lu S et al. Comparison of biomarker modalities for predicting response to PD-1/PD-L1 checkpoint blockade: a systematic review and meta-analysis. JAMA Oncol. 5, 1195–1204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Johnson DB et al. Quantitative spatial profiling of PD-1/PD-L1 interaction and HLA-DR/IDO-1 predicts improved outcomes of anti-PD-1 therapies in metastatic melanoma. Clin. Cancer Res 24, 5250–5260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jackson HW et al. The single-cell pathology landscape of breast cancer. Nature 578, 615–620 (2020). [DOI] [PubMed] [Google Scholar]
- 114.Bosisio FM et al. Functional heterogeneity of lymphocytic patterns in primary melanoma dissected through single-cell multiplexing. eLife 9, e53008 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Helmink BA et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cabrita R et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565 (2020). [DOI] [PubMed] [Google Scholar]
- 117.Xin Yu J et al. Trends in clinical development for PD-1/PD-L1 inhibitors. Nat. Rev. Drug Discov 19, 163–164 (2020). [DOI] [PubMed] [Google Scholar]
- 118.McGranahan N et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reuben A et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med 2, 10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rozenblatt-Rosen O et al. The Human Tumor Atlas Network: charting tumor transitions across space and time at single-cell resolution. Cell 181, 236–249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Newman AM et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol 37, 773–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maïno N et al. A microfluidic platform towards automated multiplexed in situ sequencing. Sci. Rep 9, 3542 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maus MV et al. Adoptive immunotherapy for cancer or viruses. Annu. Rev. Immunol 32, 189–225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Martin-Liberal J et al. The expanding role of immunotherapy. Cancer Treat. Rev 54, 74–86 (2017). [DOI] [PubMed] [Google Scholar]
- 125.Sanghera C & Sanghera R Immunotherapy – strategies for expanding its role in the treatment of all major tumor sites. Cureus 11, e5938 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kieffer Y et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov. 10, 1330–1351 (2020). [DOI] [PubMed] [Google Scholar]
- 127.Dominguez CX et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 10, 232–253 (2020). [DOI] [PubMed] [Google Scholar]
- 128.Guo X et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med 24, 978–985 (2018). [DOI] [PubMed] [Google Scholar]
- 129.Zheng C et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356.e16 (2017). [DOI] [PubMed] [Google Scholar]
- 130.Zhang L et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 181, 442–459.e29 (2020). [DOI] [PubMed] [Google Scholar]
- 131.Witkowski MT et al. Extensive remodeling of the immune microenvironment in B cell acute lymphoblastic leukemia. Cancer Cell 37, 867–882. e12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.House IG et al. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin. Cancer Res 26, 487–504 (2020). [DOI] [PubMed] [Google Scholar]
- 133.Livak KJ in Field Guidelines for Genetic Experimental Designs in High-Throughput Sequencing (eds Aransay AM & Lavín Trueba JL) 343–365 (Springer, 2016). [Google Scholar]
- 134.Gerdes MJ et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl Acad. Sci. USA 110, 11982–11987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lin J-R et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 7, e31657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bolognesi MM et al. Multiplex staining by sequential immunostaining and antibody removal on routine tissue sections. J. Histochem. Cytochem 65, 431–444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cattoretti G, Bosisio FM, Marcelis L & Bolognesi MM Multiple iterative labeling by antibody neodeposition (MILAN). Protocol Exchange 10.21203/rs.2.1646/v5 (2019). [DOI] [Google Scholar]
- 138.Eng J et al. Cyclic multiplexed-immunofluorescence (cmIF), a highly multiplexed method for single-cell analysis. Methods Mol. Biol 2055, 521–562 (2020). [DOI] [PubMed] [Google Scholar]
- 139.Consentius C et al. In situ detection of CD73+ CD90+ CD105+ lineage: mesenchymal stromal cells in human placenta and bone marrow specimens by chipcytometry. Cytometry A 93, 889–893 (2018). [DOI] [PubMed] [Google Scholar]
- 140.Angelo M et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med 20, 436–442 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Keren L et al. MIBI-TOF: A multiplexed imaging platform relates cellular phenotypes and tissue structure. Sci. Adv 5, eaax5851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Giesen C et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422 (2014). [DOI] [PubMed] [Google Scholar]
- 143.Ramaglia V et al. Multiplexed imaging of immune cells in staged multiple sclerosis lesions by mass cytometry. eLife 8, e48051 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ijsselsteijn ME, van der Breggen R, Farina Sarasqueta A, Koning F & de Miranda NFCCA 40-marker panel for high dimensional characterization of cancer immune microenvironments by imaging mass cytometry. Front. Immunol 10, 2534 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Goltsev Y et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell 174, 968–981.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Eng C-HL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ståhl PL et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 149.Thrane K, Eriksson H, Maaskola J, Hansson J & Lundeberg J Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res. 78, 5970–5979 (2018). [DOI] [PubMed] [Google Scholar]
- 150.Rodriques SG et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vickovic S et al. High-definition spatial transcriptomics for in situ tissue profiling. Nat. Methods 16, 987–990 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lee JH et al. Highly multiplexed subcellular RNA sequencing in situ. Science 343, 1360–1363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lee JH et al. Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat. Protoc 10, 442–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang X et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361, eaat5691 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Decalf J, Albert ML & Ziai J New tools for pathology: a user’s review of a highly multiplexed method for in situ analysis of protein and RNA expression in tissue. J. Pathol 247, 650–661 (2019). [DOI] [PubMed] [Google Scholar]
- 156.Van TM & Blank CU A user’s perspective on GeoMxTM digital spatial profiling. Immunooncol. Technol 1, 11–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]