

In this study, Yoon et al. set out to study translesion synthesis (TLS) mechanisms in normal versus cancer cells. Using reporter assays and knockdown cells, the authors report that in normal cells, TLS through cisplatin intrastrand cross-links is promoted by Polη- or Polι- dependent pathways, both of which require Rev1 as a scaffolding component. In contrast, cancer cells require Rev1-Polζ. The authors also show that the Rev1 inhibitor JH-RE-06 abrogates Rev1's ability to function with Y family Pols, leading to loss of TLS in various contexts and increased sensitivity to cisplatin in normal cells.
Keywords: DNA repair, nucleotide excision repair, translesion synthesis, Y family DNA polymerases, cisplatin intrastrand cross-links, Rev1
Abstract
Chemotherapy with cisplatin becomes limiting due to toxicity and secondary malignancies. In principle, therapeutics could be improved by targeting translesion synthesis (TLS) polymerases (Pols) that promote replication through intrastrand cross-links, the major cisplatin-induced DNA adduct. However, to specifically target malignancies with minimal adverse effects on normal cells, a good understanding of TLS mechanisms in normal versus cancer cells is paramount. We show that in normal cells, TLS through cisplatin intrastrand cross-links is promoted by Polη- or Polι-dependent pathways, both of which require Rev1 as a scaffolding component. In contrast, cancer cells require Rev1-Polζ. Our findings that a recently identified Rev1 inhibitor, JH-RE-06, purported to specifically disrupt Rev1 interaction with Polζ to block TLS through cisplatin adducts in cancer cells, abrogates Rev1's ability to function with Y family Pols as well, implying that by inactivating Rev1-dependent TLS in normal cells, this inhibitor will exacerbate the toxicity and tumorigenicity of chemotherapeutics with cisplatin.
Although cisplatin is widely used to treat a variety of cancers, including ovarian, breast, and non-small cell lung cancer (Wang and Lippard 2005; Kelland 2007; Goss et al. 2010; Wheate et al. 2010), this therapy becomes limiting due to toxicity and the advent of secondary malignancies. Since cisplatin-induced DNA damage is largely responsible for its cytotoxic properties, an understanding of the mechanisms of cisplatin resistance in normal cells and in cancer cells is essential for developing strategies that specifically target tumors and have minimal impact on normal cell toxicity and tumorigenicity.
The platinum atom of cisplatin binds covalently to the N7 of guanines and, to a lesser extent, the N7 of adenines, forming GG intrastrand cross-links (∼65%–70%), AG intrastrand cross-links (∼25%), GNG intrastrand cross-links (∼3%), monoadducts (∼2%), and G-G interstrand cross-links formed between guanines on opposite DNA strands (∼2%) (Chaney et al. 2005; Wang and Lippard 2005; Noll et al. 2006; Kelland 2007). X-ray crystal structures of DNA containing a cisplatin-GG intrastrand cross-link have indicated that the helix is bent by ∼50° toward the major groove and the DNA is kinked at the platinum site (Jamieson and Lippard 1999). In contrast, the cisplatin interstrand cross-link confers a more significant distortion of the DNA helix at the site of the cross-link; the cisplatin moiety lies in the minor groove, and the DNA helix is bent ∼45° toward the minor groove and is unwound by ∼80°(Noll et al. 2006). Cisplatin interstrand and intrastrand cross-links are removed by nucleotide excision repair (NER). Because of their low frequency of formation and highly efficient recognition by the NER machinery, interstrand cross-links (ICLs) will be effectively removed from DNA (Miller et al. 1982; Wang et al. 2001; Sarkar et al. 2006); in contrast, the high abundance and less efficient recognition of intrastrand cross-links by NER would make them less vulnerable to removal. In fact, a recent detailed analysis of the kinetics of removal of cisplatin GG intrastrand cross-links from mouse liver has shown that while these adducts are effectively eliminated from the transcribed strand within 48 h, the damage in the nontranscribed strand persists for weeks (Yang et al. 2019). The unrepaired intrastrand cross-links would present a block to replication in highly proliferating normal tissues such as skin, gastrointestinal tract, and bone marrow; however, replication through the cross-links can be restored by translesion synthesis (TLS) DNA polymerases (Pols). By promoting proficient replication through cisplatin intrastrand cross-links in normal cells, TLS Pols will prevent fork collapse and the consequent increase in chromosome aberrations and tumorigenicity (Yoon et al. 2019b). Thus, while TLS mechanisms will protect normal cells from cisplatin toxicity during chemotherapy, they will also limit the toxicity of cisplatin in tumor cells, impeding the effectiveness of therapy.
In this study, we identify the TLS Pols that promote replication through cisplatin intrastrand cross-links in normal cells, determine whether they act in an error-free or error-prone manner, and analyze their role in conferring resistance to cisplatin-induced toxicity. We provide evidence that replication through cisplatin intrastrand cross-links is promoted by a Polη-dependent error-free TLS pathway and by an alternate Polι/Polθ-dependent error-prone pathway. In addition, we show that Rev1's role as a scaffolding component of Y family Pols (Yoon et al. 2015, 2017, 2018, 2019a) is indispensable for TLS by both the Polη- and Polι-dependent pathways. Importantly, we found no evidence for the requirement of Polζ in TLS opposite cisplatin-induced intrastrand cross-links in normal cells.
In striking contrast to the lack of requirement of Polζ for TLS through cisplatin intrastrand cross-links in normal cells, depletion of Rev1 or Polζ sensitizes cancer cells to cisplatin and other DNA damaging agents (Lin et al. 2006; Doles et al. 2010; Hicks et al. 2010; Xie et al. 2010) and inhibits DNA damage-induced mutagenesis (Lin et al. 2006; Doles et al. 2010). Hence, to increase the efficacy of chemotherapy with cisplatin, recently a small molecule inhibitor, JH-RE-06, has been identified that blocks Rev1 interaction with the Rev7 subunit of Polζ and improves tumor response to cisplatin in a xenograft mouse model (Wojtaszek et al. 2019). However, we found that this inhibitor lacks specificity for only blocking Rev1 interaction with Polζ; it also blocks Rev1 interaction with Y family Pols, and thereby it inhibits TLS through cisplatin intrastrand cross-links as well as other DNA lesions in normal human cells. We discuss how this JH-RE-06 effect on TLS in normal cells would exacerbate the toxicity and tumorigenicity of chemotherapeutics with cisplatin.
Results
TLS Pols required for replication through the cisplatin-GG intrastrand cross-link in normal cells
Since cisplatin (Pt)-GG intrastrand cross-links constitute the most abundant cisplatin-induced DNA adduct, we first carried out studies to identify the TLS Pols that promote replication through this adduct. The Pt-GG intrastrand cross-link was incorporated in the lagging strand template of SV40-based duplex plasmid in which bidirectional replication initiates from a replication origin and TLS through the DNA adduct generates Kan+ blue colonies (Supplemental Fig. S1). The frequency of Kan+ blue colonies among total Kan+ colonies gives a highly reliable and repeatable estimate of TLS frequency, and we have shown previously that studies with this duplex plasmid are highly informative of the TLS mechanisms that operate during replication fork progression through lesions in genomic DNA (Yoon et al. 2019b).
To identify the TLS Pols required for replication through the Pt-GG intrastrand cross-link, we first analyzed the effects of depletions of TLS Pols in XPA human fibroblasts (HFs), defective in NER. As shown in Table 1, in XPA HFs treated with control siRNA (NC), TLS occurs with a frequency of ∼42%, and TLS frequency remains the same in cells depleted for Polκ, indicating that Polκ has no apparent role in TLS opposite this cross-link. However, depletion of either Polη, Polι, or Polθ reduces TLS frequency to ∼20%. To determine whether these Pols function together in one TLS pathway or whether they act independently, we examined the effects of their depletion in various combinations. The results that codepletion of Polη with Polι or with Polθ reduces TLS frequency to ∼9%, whereas codepletion of Polι with Polθ confers no significant change in TLS frequency from that observed upon their individual depletion, indicates that Polη functions independently of Polι and Polθ and that Polι and Polθ function together in one TLS pathway.
Table 1.
Effects of siRNA knockdowns of TLS Pols on replicative bypass of a Pt-GG intrastrand cross-link carried on the lagging strand DNA template in human fibroblasts (HFs)
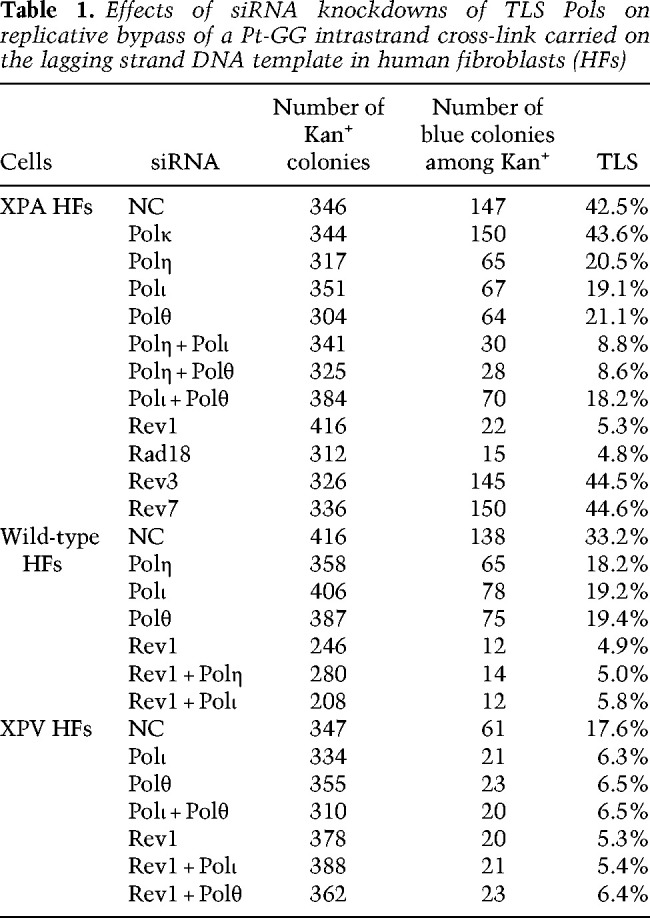
We also examined the effect of depletions of TLS Pols in NER-proficient WT HFs and in XPV HFs. In control (NC) WT HFs, TLS opposite the Pt-GG cross-link occurs with a frequency of ∼33% and TLS frequency declines to ∼18% in cells depleted for Polη, Polι, or Polθ (Table 1). Accordingly, TLS in XPV cells, defective in Polη, occurs with a frequency of ∼18% and, as expected, TLS in these cells declines to ∼6% upon depletion of either Polι or Polθ or upon their codepletion (Table 1). Altogether, these results support the conclusion that TLS through the Pt-GG intrastrand cross-link is mediated by two alternative pathways, dependent on Polη and Polι/Polθ, respectively (Supplemental Fig. S2).
Rad6-Rad18-mediated PCNA ubiquitination plays an essential role in TLS (Yoon et al. 2012). As expected from the requirement of Rad6-Rad18-mediated PCNA ubiquitination, the frequency of TLS through the Pt-GG intrastrand cross-link was reduced to a residual level of ∼5% in Rad18-depleted XPA cells (Table 1).
Genetic control of error-free and mutagenic TLS through cisplatin GG and AG intrastrand cross-links in normal cells
Sequence analyses of TLS products in control (NC) siRNA-treated XPA HFs indicated that, in ∼2% of TLS products, an A is inserted opposite the 3′ G of the Pt-GG cross-link rather than a C (Supplemental Table S1). We did not observe any mutational changes at the 5′ G of the cross-link or at any other subsequent 5′ template residues. Out of 282 TLS products sequenced, we observed only one non-GG cross-link-derived mutation in which the C residue on the 3′ side of the cross-link was converted to a G. In XPA HFs depleted for Polη, or in XPV HFs that lack functional Polη, the frequency of mutational TLS products increased to ∼4%, but the pattern of mutational change at the 3′ G residue of the cross-link remained the same as in control (NC) siRNA-treated XPA HFs (Supplemental Table S1). In contrast, mutational TLS products were absent in cells depleted for Polι, Polθ, or depleted for both Pols (Supplemental Table S1). These data suggested that Polη promotes error-free TLS through the Pt-GG cross-link and that Polι/Polθ-dependent TLS operates in an error-prone manner in which an A is inserted opposite the 3′ G of the GG cross-link.
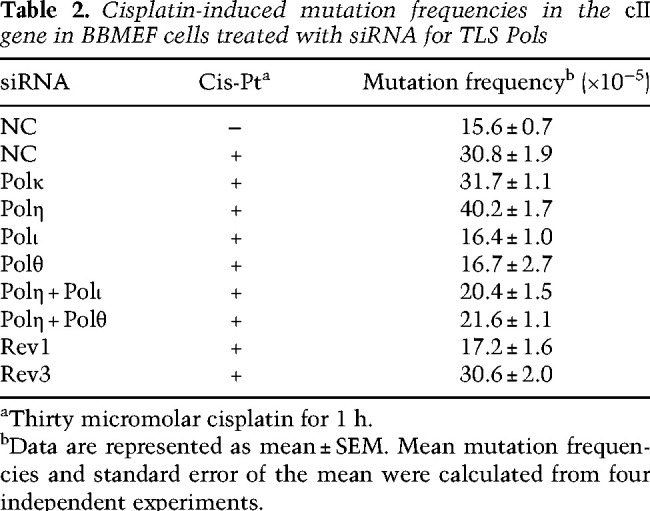
Because mutagenic TLS opposite the defined Pt-GG cross-link in the duplex plasmid occurs at a low frequency (∼2%), and because in addition to GG cross-links, cisplatin treatment generates AG intrastrand cross-links at a considerable frequency, to evaluate the relative contribution of GG and AG cross-links to mutagenesis and to decipher the targets and the spectra of mutations generated by the Polι/Polθ error-prone pathway at the Pt-GG and Pt-AG cross-links, we determined the effects of depletion of TLS Pols on the frequency and pattern of mutations induced by cisplatin in the cII gene that has been integrated into the genome of big blue mouse embryonic fibroblasts (BBMEFs). A large number of studies have established that, in response to DNA damaging agents, the mutational pattern in the cII gene resembles that in native chromosomal genes (You and Pfeifer 2001; You et al. 2001; Besaratinia and Pfeifer 2006; Yoon et al. 2019b). In undamaged cells treated with NC siRNA, spontaneous mutations in the cII gene occur at a frequency of ∼16 × 10−5, and this frequency rises to ∼31 × 10−5 in cisplatin-treated cells (Table 2). Consistent with the lack of an effect of Polκ on TLS through the Pt-GG cross-link in the duplex plasmid assay (Table 1), its depletion has no effect on the frequency of cisplatin-induced mutations in the cII gene. Depletion of Polη, however, raises the cisplatin-induced cII mutation frequency to ∼40 × 10−5, and depletion of Polι or Polθ reduces cisplatin-induced mutation frequency to the same level (∼16 × 10−5) as in untreated cells (Table 2). Moreover, the elevated frequency of cisplatin-induced mutations in Polη-depleted cells was largely eliminated upon codepletion with Polι or Polθ. These results provide confirmatory evidence for a role of Polη in primarily error-free TLS and of Polι and Polθ in error-prone TLS as was inferred from mutational analyses of TLS products generated from replication through a Pt-GG intrastrand cross-link carried on the duplex plasmid in human cells (Supplemental Table S1).
Table 2.
Cisplatin-induced mutation frequencies in the cII gene in BBMEF cells treated with siRNA for TLS Pols
Sequence analyses of cisplatin-induced mutations in the cII gene revealed a high degree of targeting to GG and AG sequences. In BBMEFs treated with NC siRNA, cisplatin-induced mutations in the cII gene are largely clustered at nine hotspot locations (Fig. 1A). Three of the hotspots occur at GG sequences (hotspots 3, 5, and 7), and six of the hotspots occur at AG sequences (hotspots 1, 2, 4, 6, 8, and 9). At each of the GG hot spots, 3′ G > T was the primary mutational change, consistent with the insertion of an A opposite the 3′ G of the Pt-GG cross-link as was seen in the SV40-based plasmid system (Supplemental Table S1). At AG hotspots, the pattern of mutational change was more varied and differed among hotspots. For instance, at hotspot 1, mutagenic TLS would have occurred by the insertion of either a T, a G, or an A opposite the 3′ G of the AG sequence in the opposite strand. At hotspot 2, mutations would have occurred primarily by the insertion of a G opposite the 5′ A and less frequently by the insertion of an A or a G opposite the 3′ G of the AG sequence. At hotspot 4, the 3′ G > T or 3′ G > C would have occurred by the insertion of an A or a G opposite the 3′ G of the AG sequence. At hotspot 6, the pattern of mutations at the AG sequence (in the opposite strand) is very similar to that at hotspot 2. At hotspot 8, the conversion of G > T in the AG sequence resembles that at hotspot 4. Hotspot 9 represents yet another AG site at which mutagenic TLS would have occurred by the insertion of primarily an A opposite the 5′ A and an A opposite the 3′ G. Thus, depending on the sequence context of the AG cisplatin cross-link, a G or an A is inserted opposite the 5′ A, while the insertion of an A and less frequently of a G occurs opposite the 3′ G.
Figure 1.

Cisplatin-induced mutational spectra in the cII gene in BBMEFs. (A) Mutational spectra of cisplatin-induced mutations in control siRNA (NC)-treated cells are shown above the sequence, and mutational spectra in cells treated with Polη siRNA are shown below the sequence. (B) Mutational spectra in Polι-depleted cells are shown above the sequence and in Polθ-depleted cells are shown below the sequence. (C) Mutational spectra in cells not treated with cisplatin are shown above the sequence and mutational spectra in cells codepleted for Polη and Polθ are shown below the sequence. The designations for other mutational changes are X for deletion and + for addition. Underlines indicate a mutational change in flanking bases.
Next, we analyzed the effects of depletion of Polη, Polι, or Polθ on cisplatin-induced hotspot mutations in the cII gene. In Polη-depleted BBMEFs, the pattern of hotspot mutations is almost identical to that in control cells (Fig. 1A). Conversely, these mutational hotspots are absent in BBMEFs depleted for Polι or Polθ (Fig. 1B) or codepleted for Pols η and θ (Fig. 1C). From these mutational data, we conclude that TLS through the GG or AG intrastrand cross-links is mediated via two major pathways dependent on Polη, which functions in an error-free manner, and upon Polι and Polθ, which act in an error-prone manner (Supplemental Fig. S2).
Replication fork (RF) progression through cisplatin-induced intrastrand cross-links in primary Polη−/−, Polθ−/−, and Polη−/− Polθ−/− MEFs
Since TLS through cisplatin-induced GG and AG intrastrand cross-links occurs via Polη- and Polι/Polθ-dependent pathways and these intrastrand cross-links account for over 90% of cisplatin-induced DNA adducts, we surmised that DNA replication through cisplatin-induced DNA lesions will be strongly inhibited in the absence of Polη, Polι, or Polθ. To verify this, we monitored RF progression on single DNA fibers in WT, Polη−/−, Polθ−/−, and Polη−/− Polθ−/− primary MEFs treated with cisplatin. Whereas no significant impairment of RF progression occurs in these mutant primary MEFs not treated with cisplatin (Fig. 2A), RF progression in cisplatin-treated cells was reduced by ∼40% in Polη−/−, ∼30% in Polθ−/−, and ∼60% in Polη−/− Polθ−/− MEFs compared with that in WT MEFs (Fig. 2B). These results validate the involvement of Polη and Polθ in the two alternative pathways for replicating through the cisplatin-induced DNA intrastrand cross-links and, more importantly, attest to the crucial role TLS plays in maintaining RF progression through cisplatin-induced DNA damage in normal cells.
Figure 2.

Analysis of RF progression through cisplatin-induced intrastrand cross-links in wild-type (WT), Rev1−/−, Polη−/−, Polθ−/−, and Polη−/− Polθ−/− primary MEFs. (A, left) Schematic of DNA fiber assay and images of stretched DNA fibers in untreated primary WT, Rev1−/−, Polη−/−, Polθ−/−, and Polη−/− Polθ−/− MEFs. (Right) Quantification of RF progression (mean CldU:IdU ratio) in these MEFs. (B) Analyses of RF progression through cisplatin-induced intrastrand cross-links in primary WT, Rev1−/−, Polη−/−, Polθ−/−, and Polη−/− Polθ−/− MEFs. (Top left) Schematic of DNA fiber assay and representative images of stretched DNA fibers. (Top right) Quantification of RF progression through cisplatin-induced cross-links (mean CldU:IdU ratio). In A and B, quantification was done on ∼400 DNA fibers from four independent experiments. Error bars indicate the standard deviation of results of four independent experiments. Student's two-tailed t-test P-values,(**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. (Bottom left) The percentage of replication tracks and the Cldu:IdU ratio measured in fibers from cisplatin-treated primary MEFs. (C) Defects in TLS through intrastrand cross-links reduce survival in cisplatin-treated primary MEFs. Error bars indicate the standard deviation from four independent experiments. Student's two-tailed t-test P-values, (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001.
TLS through intrastrand cross-links attenuates cisplatin toxicity
The requirement of Polη, Polι, and Polθ for replication through cisplatin-induced intrastrand cross-links implies a role for these Pols in reducing the toxicity of normal cells to cisplatin. To ascertain this, we examined the survival of Polη−/−, Polθ−/−, and Polη−/− Polθ−/− primary MEFs in response to cisplatin treatment (Fig. 2C). Congruent with their roles in alternate TLS pathways and similar to their effect on RF progression through cisplatin adducts, survival is reduced to a greater extent in Polη−/−MEFs than in Polθ−/−MEFs, and a further reduction in survival occurs in Polη−/− Polθ−/− MEFs than in Polη−/− MEFs (Fig. 2C). Conversely, WT HFs depleted for Polι, Polθ, or both Pols simultaneously and treated with cisplatin (Supplemental Fig. S3A) exhibit a similar reduction in survival. Similarly, survival of cisplatin-treated XPV HFs is reduced to the same level upon depletion of Polι or Polθ or upon codepletion of both these Pols (Supplemental Fig. S3B). Altogether, these results support the inference that TLS by the Polι/Polθ and Polη pathways limits the toxicity of cisplatin.
Biochemical analysis of roles of Polι and Polθ in TLS through the Pt-GG intrastrand cross-link
Biochemical and structural studies have shown that Polη replicates through both the guanines of the Pt-GG cross-link and that both the Gs in the cross-link form a Watson-Crick base pair with the incoming dCTP in the active site (Ummat et al. 2012; Zhao et al. 2012). The requirement of Polι and Polθ in the Polη independent pathway for TLS through the Pt-GG cross-link implicates a role for these Pols in replicating through the cross-link at either the nucleotide insertion or the subsequent extension step of TLS, respectively. Polι replicates DNA by forcing template purines into the syn conformation and uses the Hoogsteen edge for base-pairing with the incoming nucleotide (Nair et al. 2005). As such, dNTP incorporation opposite the unadducted template G by Polι exhibits a typical pattern of C and T incorporation, owing to the syn conformation of the template G (Supplemental Fig. S4A). Polι also incorporates opposite the next 5′ G as well and, in the presence of all four dNTPs, Polι continues synthesis to the third template nucleotide (Supplemental Fig. S4A). On the Pt-GG cross-linked DNA, Polι is effective at inserting a C opposite the 3′ G of the cross-link; however, unlike incorporation opposite unadducted template G, little, if any, T is incorporated (Supplemental Fig. S4A). In the presence of all four dNTPs, Polι is unable to incorporate a nucleotide opposite the 5′ G of the cross-link (Supplemental Fig. S4A,B).
The inability of Polι to insert a nucleotide opposite the 5′ G of the cross-link (Supplemental Fig. S4A,B) and the involvement of Polθ in Polι-dependent TLS opposite cisplatin intrastrand cross-links in normal cells suggested that Polθ might perform this step of TLS opposite the Pt-GG cross-link. Therefore, we examined the proficiency of Polθ for inserting a nucleotide opposite the 5′ G of the cross-link. As shown in Supplemental Figure S4C, on undamaged DNA, Polθ is highly error-prone, as it inserts a G, T, A, or C opposite the G residue (corresponding to the 5′ G of the cisplatin cross-link), and it also exhibits untemplated A insertion. Polθ is very highly error-prone at inserting nucleotides opposite the 5′ G of the cross-link and it exhibits a pattern of untemplated insertion of an A, as well as a G. Importantly, in the presence of all four dNTPs, Polθ incorporates an A opposite the 5′ G of the cisplatin adduct and it continues synthesis past the cross-link nearly as well as on the nonadducted GG template. Polθ, however, exhibits high error-proneness in extending synthesis opposite from the 5′ G of the cross-link; for example, opposite the next 5′ T template residue, it inserts an A as efficiently as a C. Overall, purified Polθ exhibits an extreme high error-proneness in inserting a nucleotide opposite the 5′ G of the cross-link and in extending synthesis therefrom.
Indispensability of Rev1 for TLS through cisplatin intrastrand cross-links in normal cells
For TLS that occurs during DNA replication and promotes RF progression through DNA lesions in normal human cells, Rev1 functions as an indispensable scaffolding component of Y family Pols η, ι, and κ (Yoon et al. 2015, 2017, 2018, 2019a). Hence, we carried out studies to determine whether Rev1 was similarly required for mediating Polη- and Polι-dependent TLS through cisplatin intrastrand cross-links. We found that (1) TLS through the Pt-GG intrastrand cross-link in the duplex plasmid is reduced in Rev1-depleted XPA HFs (∼5%) to the same level as in Rad18-depleted cells, indicating that TLS by both the Polη- and Polι-dependent pathways is inactivated (Table 1); (2) Rev1 exhibits epistasis with Polη and Polι for TLS through the Pt-GG intrastrand cross-link (Table 1); (3) the frequency of cisplatin-induced mutations in the cII gene in Rev1-depleted BBMEFs is reduced to levels near to that in undamaged cells (Table 2); (4) cisplatin-induced mutational hotspots in the cII gene are absent in Rev1-depleted BBMEFs (Fig. 3A); (5) RF progression in cisplatin-treated primary Rev1−/− MEFs is drastically reduced to a level nearly identical to that seen in Polη−/− Polθ−/− primary MEFs (Fig. 2B); and (6) cisplatin treatment reduces survival of primary Rev1−/− MEFs to nearly the same level as of Polη−/− Polθ−/− primary MEFs (Fig. 2C), and Rev1 exhibits epistasis over Polη and Polι for survival in cisplatin-treated WT or XPV HFs (Fig. 3B,C). Thus, similar to its role in TLS opposite other DNA lesions, Rev1 functions as an essential scaffolding component in both the Polη error-free and Polι/Polθ error-prone pathways of TLS through the major cisplatin-induced DNA adducts: intrastrand cross-links.
Figure 3.

Cisplatin-induced mutation spectra in the cII gene in Rev1-depleted BBMEFs and cisplatin survival in Rev1-depleted HFs. (A) Mutational spectra of cisplatin-induced mutations in control siRNA (NC)-treated cells are shown above the sequence and mutational spectra in cells treated with Rev1 siRNA are shown below the sequence. (B) Effects of Rev1 depletion alone or together with Polη or Polι depletion on cisplatin survival in wild-type HFs. (C) Effects of Rev1 depletion alone or together with Polι depletion on cisplatin survival in XPV HFs. In B and C, error bars indicate the standard deviation from four independent experiments. Student's two-tailed t-test P-values, (ns) not significant, (****) P < 0.0001.
Lack of Polζ requirement for TLS through cisplatin intrastrand cross-links in normal cells
A number of studies have indicated that, in cancer cells, Polζ performs a critical role in TLS opposite cisplatin-induced DNA adducts and is required for cisplatin-induced mutations (Lin et al. 2006; Doles et al. 2010; Hicks et al. 2010). For that reason, we inquired whether Polζ was also required for TLS opposite cisplatin adducts in normal cells. As shown in Table 1, the TLS frequency opposite the Pt-GG intrastrand cross-link in the duplex plasmid in XPA HFs depleted for either the Rev3 (catalytic) or Rev7 (accessory) subunit of Polζ remains the same as that in control siRNA-treated cells, thus indicating that Polζ plays no role in TLS opposite this cisplatin adduct in normal cells. Moreover, the evidence that the frequency of cisplatin-induced mutations (Table 2) and the pattern of cisplatin-induced mutational hotspots at position 1–9 in the cII gene remain the same in Rev3-depleted BBMEFs as in control (NC) siRNA-treated cells (Supplemental Fig. S5) adds further support for the lack of any involvement of Polζ in TLS through cisplatin-induced intrastrand cross-links in normal cells. Thus, in contrast to the requirement of Polζ for TLS through UV and other DNA lesions in normal cells (Yoon et al. 2019a,b), Polζ is not required for TLS through cisplatin intrastrand cross-links.
Abrogation of TLS through cisplatin intrastrand cross-links in normal cells by JH-RE-06
Our findings that in normal cells, Rev1 functions as an indispensable scaffolding component of Pols η and ι, which provide the two alternative pathways for replicating through cisplatin intrastrand cross-links, and that Polζ is not required, taken together with the evidence that cancer cells require Rev1 and Polζ for replicating through cisplatin adducts and other DNA lesions (Lin et al. 2006; Doles et al. 2010; Hicks et al. 2010; Xie et al. 2010), implicate Polζ inhibition as an effective strategy for improving cisplatin cancer therapy. The very high specificity ascribed to JH-RE-06 for inhibiting Rev1 interaction with the Rev7 subunit of Polζ, the evidence that JH-RE-06 enhances cisplatin toxicity of cancer cells, and that it improves tumor response to cisplatin in an A375 xenograft mouse model of human melanoma (Wojtaszek et al. 2019), strongly suggested that this inhibitor would improve chemotherapeutics with cisplatin with minimal adverse effects on normal tissues.
To ascertain the lack of any significant effect of JH-RE-06 in normal cells, we first analyzed the effects of this inhibitor on TLS through the Pt-GG intrastrand cross-link in WT HFs. As shown in Figure 4A, TLS in control HFs occurs with a frequency of ∼31%; however, TLS in cells treated with JH-RE-06 drops to ∼ 5%, a frequency similar to that in Rev1-depleted cells. The similar reduction in TLS frequency to ∼ 4%–5% in cells treated with JH-RE-06, depleted for Rev1, or depleted for Rev1 and treated with JH-RE-06 (Fig. 4A) indicated that JH-RE-06 inhibits Rev1′s role as a scaffolding component of Pols η and ι in normal cells. To elaborate upon this JH-RE-06 role further, next we examined its effects on cisplatin-induced mutations in the cII gene in Rev1+/+ and Rev1−/− primary BBMEFs (Yoon et al. 2015). Our results that the high frequency of cisplatin-induced mutations in the cII gene is reduced in JH-RE-06-treated Rev1+/+ MEFs near to the level in undamaged cells, that a similar reduction in cisplatin-induced mutation frequency occurs in Rev1−/− BBMEFs, and that this frequency stays about the same in these MEFs additionally treated with JH-RE-06 (Fig. 4B) add further to the evidence that JH-RE-06 inhibits Rev1's TLS function in normal cells.
Figure 4.

Effects of JH-RE-06 on TLS through cisplatin intrastrand cross-links in HFs and primary MEFs. (A) TLS through Pt-GG intrastrand cross-link in JH-RE-06-treated WT HFs. (B) Cisplatin-induced mutation frequencies in the cII gene in JH-RE-06-treated primary wild-type or Rev1−/− MEFs. (C) Schematic of DNA fiber assay and images of stretched DNA fibers in JH-RE-06-treated primary WT or Rev1−/− MEFs not treated with cisplatin and quantification of RF progression (mean CldU:IdU ratio) in these MEFs. Student's two-tailed t-test P-value, (ns) not significant. (D) Analyses of RF progression through cisplatin-induced intrastrand cross-links in JH-RE-06-treated primary WT or Rev1−/− MEFs. (Left) Schematic of DNA fiber assay and representative images of stretched DNA fibers. (Right) Quantification of RF progression through cisplatin-induced cross-links (mean CldU:IdU ratio). Quantification in C and D were done on ∼300 DNA fibers from three independent experiments. Error bars indicate the standard deviation of results of three independent experiments. Student's two-tailed t-test P-values, (ns) not significant, (****) P < 0.0001. (E) Effects of JH-RE-06 on survival of cisplatin-treated WT or Rev1−/− primary MEFs. Error bars indicate the SD results from three independent experiments. Student's two-tailed t-test P-values, (****) P < 0.0001. (F) Effects of JH-RE-06 on survival of cisplatin-treated (30 µM for 2 h) GM637 HFs. Error bars indicate the SD results from three independent experiments. Student's two-tailed t-test P-values, (ns) not significant, (****) P < 0.0001.
The strong inhibitory effect of JH-RE-06 on TLS through Pt-GG intrastrand cross-link (Fig. 4A) and on cisplatin-induced mutations in the cII gene (Fig. 4B) suggested that it would severely impair DNA replication in normal cells treated with cisplatin. Although JH-RE-06 has no significant effect on RF progression in undamaged primary WT (Rev1+/+) or Rev1−/− MEFs (Fig. 4C), JH-RE-06 is strongly inhibitory to RF progression in cisplatin-treated Rev1+/+ primary MEFs, the level of inhibition being nearly identical to that in cisplatin-treated Rev1−/− primary MEFs, and JH-RE-06 causes no further reduction in RF progression in cisplatin-treated Rev1−/− MEFs (Fig. 4D). Thus, by inhibiting Rev1's scaffolding role with Pols η and ι, JH-RE-06 abrogates the proficiency of normal cells to replicate through cisplatin intrastrand cross-links. Consequently, JH-RE-06 reduces survival of cisplatin-treated Rev1+/+ primary MEFs near to that of cisplatin-treated Rev1−/− MEFs, and JH-RE-06 has no effect on survival of cisplatin-treated Rev1−/− MEFs (Fig. 4E). Treatment with JH-RE-06 also reduced the survival of cisplatin-treated WT HFs similar to that in cisplatin-treated Rev1-depleted HFs (Fig. 4F).
Ablation of TLS through other DNA lesions in normal cells by JH-RE-06
To ascertain that the inhibitory effects of JH-RE-06 on Rev1's interaction with Y family Pols extend to other DNA lesions as well, we analyzed its effect on TLS opposite UV lesions and opposite an N2-dG minor groove DNA adduct (r)-γ-HOPdG. We have shown previously that TLS through UV-induced cyclobutane pyrimidine dimers (CPDs) in normal human and mouse cells operates via a Polη-dependent error-free pathway and via a Polθ-dependent error-prone pathway in which, following nucleotide insertion opposite the 3′ pyrimidine of the CPD by Polθ, Polκ or Polζ extend synthesis (Yoon et al. 2019b), and Rev1 is required as a scaffolding component for Polη- and Polκ-dependent TLS opposite CPDs (Yoon et al. 2015). As shown in Figure 5A, in WT HFs, TLS opposite a cis-syn TT dimer occurs with a frequency of ∼25%. JH-RE-06 treatment reduces TLS frequency to ∼ 10%, a level similar to that in Rev1-depleted cells, and TLS frequency remains the same in Rev1-depleted cells additionally treated with JH-RE-06. Thus, JH-RE-06 inhibits Rev1-dependent TLS by Polη and Polκ opposite CPDs.
Figure 5.

Effects of JH-RE-06 on TLS through UV lesions and an N2-dG minor groove adduct. (A) TLS through a cis-syn TT dimer or a (6-4) TT photoproduct in JH-RE-06-treated WT HFs. (B) Effects of JH-RE-06 on UV survival of GM637 HFs. Error bars indicate the SD results from three independent experiments. Student's two-tailed t-test P-values, (***) P < 0.001. (C) Effects of JH-RE-06 on UV survival of primary wild-type or Rev1−/− MEFs. Error bars indicate the SD results from three independent experiments. Student's two-tailed t-test P-values, (****) P < 0.0001. (D) TLS through (r) γ-HOPdG in JH-RE-06-treated HFs.
TLS through (6-4) pyrimidine-pyrimidone photoproducts is conducted by two alternative Polη/Polθ- or Polι/Polθ-dependent pathways in which, following nucleotide insertion opposite the 3′ pyrimidine by Polη or Polι, Polθ extends synthesis, and Rev1 functions as a scaffolding component of Pols η and ι (Yoon et al. 2015, 2019b). In an alternative pathway, Polζ would extend synthesis from a nucleotide inserted opposite the 3′ pyrimidine by another Pol (Yoon et al. 2019b). Our results that JH-RE-06 inhibits TLS frequency opposite (6-4) TT photoproduct by ∼ 50%, a level similar to that in cells depleted for Rev1 and untreated or treated with JH-RE-06 (Fig. 5A), add further evidence that JH-RE-06 inhibits Rev1-dependent TLS by Pols η and ι opposite (6-4) photoproduct.
Consistent with inhibition of Rev1 function in TLS opposite UV lesions, treatment with JH-RE-06 reduced survival of UV-irradiated WT HFs similar to that in UV-irradiated Rev1-depleted cells, and treatment with JH-RE-06 did not further reduce the survival of UV-irradiated Rev1-depleted cells (Fig. 5B). We also analyzed the effects of JH-RE-06 on survival of UV-irradiated Rev1+/+ and Rev1−/− primary MEFs. Again, JH-RE-06 reduced survival of UV-irradiated Rev1+/+ MEFs to the same level as in UV-irradiated Rev1−/− MEFs, and additional treatment with JH-RE-06 caused no reduction in survival of UV-irradiated Rev1−/− MEFs (Fig. 5C).
Next, we analyzed the effects of JH-RE-06 on TLS opposite an N2-dG minor groove DNA adduct. The reaction of acrolein, an α,β unsaturated aldehyde generated in vivo as the end product of lipid peroxidation, with the N2 of guanine in DNA leads to the formation of γ-hydroxy-1-N2-propano-2′-deoxyguanosine (γ-HOPdG), which can exist in DNA in a ring-closed or ring-opened form. We have shown previously that TLS through the permanently ring-opened reduced form of γ-HOPdG [(r)-γ-HOPdG] is conducted by Rev1/Polη-, Polι/Polκ-, and Polθ-dependent pathways, that Rev1's role as a scaffolding component is required for TLS by all the Y family Pols, and that TLS opposite this lesion operates in a predominantly error-free manner (Yoon et al. 2018). As shown in Figure 5D, TLS opposite (r)-γ-HOPdG occurs with a frequency of ∼ 34% in WT HFs, whereas in cells treated with JH-RE-06, TLS is reduced to ∼12%, and a similar reduction in TLS frequency occurs in cells depleted for Rev1, or depleted for Rev1 and treated with JH-RE-06. Thus, JH-RE-06 is inhibitory to Rev1's functional interaction with Y family pols required for TLS through (r)-γ-HOPdG. From our extensive TLS and related data opposite a number of DNA lesions, we conclude that JH-RE-06 inhibits Rev1's ability to interact with the Y family Pols η, ι, and κ; thereby, this inhibitor would abrogate all Rev1-dependent TLS in normal cells, including TLS through cisplatin intrastrand cross-links and TLS through other DNA lesions.
Discussion
Role of TLS in protection from the toxicity and tumorigenicity of cisplatin cancer therapy
GG and AG intrastrand cross-links account for >90% of cisplatin-induced DNA adducts, and they are highly blocking to DNA replication. Here, we show that TLS through cisplatin intrastrand cross-links in normal cells is mediated by two alternative pathways dependent on Polη and Polι/Polθ, wherein Polη conducts error-free TLS and Polι in conjunction with Polθ promotes error-prone TLS. In the absence of these Pols, RF progression through the cisplatin adducts is severely impaired and cell survival is greatly reduced. Thus, by promoting replication through cisplatin intrastrand DNA cross-links, these TLS Pols would protect normal cells from the toxicity of cisplatin chemotherapy.
Furthermore, RF progression through cisplatin cross-links mediated by Polη or Polι/Polθ would protect the replication fork from collapse, thereby preventing the ensuing formation of double-strand breaks and chromosomal aberrations. By preventing chromosomal instability, TLS through the intrastrand cross-links would protect normal cells from the tumorigenic impact of cisplatin, in a way similar to the role of error-free TLS by Polη and error-prone TLS by Polθ opposite UV lesions in protection against skin cancers (Yoon et al. 2019b).
The high fidelity of TLS opposite cisplatin intrastrand cross-links in human cells
In striking contrast to the ability of purified Polη or Polι to insert a C opposite the 3′ G of the Pt-GG cross-link, purified Polθ is highly error-prone for insertion opposite the 5′ G of the cross-link and during extension of synthesis (Supplemental Fig. S4). Hence, the lack of mutations at or near the 5′ G of the Pt-GG cross-link carried on the duplex plasmid in HFs (Supplemental Table S1), as well as the scarcity of mutations at the 5′ G or thereafter at the hotspots formed at the GG sequences in the cII gene in cisplatin-treated BBMEFs (Fig. 1) is highly discordant with the extremely error-prone TLS by purified Polθ (Supplemental Fig. S4C). The observed highly error-free TLS opposite the 5′ G of the Pt-GG cross-link and opposite the subsequent 5′ residues carried out by Polθ can only be explained by the assumption that, in human and mouse cells, the fidelity of Polθ is actively regulated within the multiprotein ensemble that comprises it (Yoon et al. 2019a).
Addiction of cancer cells to DNA polymerase ζ
In contrast to the requirement of Polη for error-free TLS and of Polι and Polθ for error-prone TLS through cisplatin intrastrand cross-links in normal human or mouse cells, we found that depletion of the Rev3 catalytic subunit of Polζ has no effect on TLS opposite the Pt-GG cross-link in HFs (Table 1) or on cisplatin-induced mutations at the Pt-GG or Pt-AG cross-links in HFs or BBMEFs (Table 2; Supplemental Fig. S5). However, a number of studies have indicated that, for TLS and very likely for other DNA repair processes as well, cancer cells become highly dependent on DNA polymerase ζ. Depletion of Rev3 was shown to reduce cisplatin-induced mutagenesis in human colon carcinoma (HCT116) cells (Lin et al. 2006), and our studies have indicated that cisplatin-induced mutations do not occur in Rev3- or Rev7-depleted MCF7 breast cancer cells (Supplemental Fig. S6). Similarly, studies with a mouse model of a lung adenocarcinoma cell line have shown that Rev3-deficient cells exhibit a large reduction in cisplatin-induced mutations, and that, in aggressive late stage lung cancers, a reduction in Rev3 levels caused enhanced cisplatin sensitivity in tumors (Doles et al. 2010). Furthermore, the sensitivity of HeLa cervical cancer cells and of Burkitt's lymphoma (BL2) cells to cisplatin, satraplatin, oxaliplatin, and picoplatin was greatly enhanced upon Rev3 depletion, whereas depletion of Polη or Polι had little effect (Sharma et al. 2012).
Additionally, analysis of expression levels of the Rev7 Polζ subunit in 137 epithelial ovarian carcinoma (EOC) tissue samples has indicated a strong association between Rev7 expression and chemoresistance to cisplatin (Niimi et al. 2014). Moreover, inhibition of Rev3 expression specifically reduces survival of lung (A549 and Calu-3), breast (MCF7 and MD-M-231), mesothelioma (IL45 and ZL55), and colon (HCT116) cancer cell lines, but not of normal cell lines (Knobel et al. 2011). Altogether, all the evidence in cancer cells indicates that Polζ takes over the roles of Y family Pols and other Pols in TLS and possibly in other DNA repair processes and that Polζ functions in these processes in an error-prone manner.
Role of Rev1 in TLS through cisplatin adducts in normal vs. cancer cells
In a number of previous TLS studies, we have provided evidence for an indispensable role of Rev1 as a scaffolding component of Y family Pols η, ι, and κ (Yoon et al. 2015, 2017, 2018). In this study, we provide evidence for the indispensability of Rev1 for both the Polη- and Polι-dependent pathways for TLS through the Pt-GG cross-link in normal HFs (Table 1), for RF progression through cisplatin-induced DNA adducts in primary MEFs (Fig. 2B), and for cisplatin-induced mutations at the GG and AG sequences in the cII gene in BBMEFs (Table 2; Fig. 3A). A large reduction in survival occurs in cisplatin-treated Rev1−/− MEFs (Fig. 2C), and Rev1 exhibits epistasis over both Polη and Polι for survival in cisplatin-treated HFs (Fig. 3B,C).
In cancer cells, however, Rev1 functions as a scaffolding component of Polζ (Doles et al. 2010; Hicks et al. 2010; Xie et al. 2010), similar to that in yeast (Prakash et al. 2005). Consequently, cisplatin sensitivity in HeLa and Burkitt's lymphoma (BL2) cells is similarly enhanced upon depletion of Polζ or Rev1, and suppression of Rev1 sensitizes B-cell lymphoma to cisplatin and inhibits cisplatin-induced mutagenesis (Hicks et al. 2010). We found that cisplatin-induced mutations are reduced to near spontaneous levels in Rev1-depleted MCF7 breast cancer cells, similar to the effect of Rev3 depletion (Supplemental Fig. S6). Thus, in normal human cells, Rev1 mediates TLS through cisplatin intrastrand cross-links and a variety of other DNA lesions via interaction with Y family Pols, whereas in cancer cells, Rev1 functions in TLS in conjunction with Polζ.
JH-RE-06 inhibits Rev1 interaction with Y family pols—essential for TLS through cisplatin intrastrand cross-links and other DNA lesions in normal cells
JH-RE-06 was identified in a screen for inhibitors of Rev1 interaction with the Rev7 subunit of Polζ. JH-RE-06 induces dimerization of the Rev1-C-terminal domain (CTD) such that the compound is accommodated into a deep pocket inside the Rev1 CTD dimer (Wojtaszek et al. 2019), thereby disrupting Rev1-Rev7 interaction. A very high specificity for disrupting Rev1 interaction with Polζ was ascribed to this inhibitor. This compound enhances the cisplatin toxicity of cancer cells and improves tumor response of A375 human melanoma xenografts to cisplatin treatment. This study also observed that JH-RE-06 reduced cisplatin-induced mutations in normal Rev1+/+ MEFs, and this was presumed to result from inhibition of Rev1 interaction with Polζ. Based on the presumption of high specificity of JH-RE-06 for the disruption of Rev1-Polζ-dependent mutagenic TLS, it was suggested that, by disrupting Rev1-Polζ TLS in normal cells, this inhibitor would reduce the advent of secondary malignancies caused by cisplatin-induced mutagenesis. However, our study clearly establishes that, for TLS through the predominant cisplatin-induced intrastrand cross-links in normal cells, Polζ plays no role and that Rev1 is essential for both error-free Polη-dependent and error-prone Polι-dependent TLS pathways. Since it was unknown whether the dimerization of Rev1 induced by JH-RE-06 could interfere with its function in TLS dependent on Y family Pols, it was highly important to determine the effects of JH-RE-06 on TLS opposite cisplatin DNA adducts and other DNA lesions as well in normal cells.
Our results show that JH-RE-06 abrogates Rev1-dependent TLS by Y family Pols opposite cisplatin intrastrand cross-links as well as opposite UV lesions and the N2-dG adduct (r) γ-HOPdG in normal cells, and that treatment of normal cells with this compound sensitizes them to cisplatin and UV treatment. Thus, we have provided strong evidence that this inhibitor lacks the specificity for inhibiting only Polζ-dependent TLS via disruption of Rev1-Rev7 interaction and conclude that all the effects of JH-RE-06 pursuant to TLS inhibition in normal cells emanate from inhibition of Rev1 interaction with Y family Pols.
Implications of inhibition of TLS in normal cells by JH-RE-06 for cisplatin-based cancer therapy
Since treatment with cisplatin causes numerous side effects (Waseem et al. 2015; Dugbartey et al. 2016) and increases the risk of secondary malignancies unrelated to the original cancer (Ratain et al. 1987; Kushner et al. 1998; Travis et al. 2005), it becomes consequential that the means adopted for improving the efficacy of cisplatin treatment specifically target the malignancies with minimal effect on normal tissues. Thus, even though JH-RE-06 would increase the cisplatin toxicity of cancer cells, by inactivating Rev1 interaction with Y family Pols, this inhibitor would annihilate the proficiency of normal cells for replicating through cisplatin-induced intrastrand cross-links, thereby greatly increasing the cisplatin toxicity of highly proliferating tissues such as gastrointestinal tract and bone marrow (Supplemental Fig. S7).
Additionally, since suppression of TLS through the cisplatin adducts by the JH-RE-06 inhibitor would cause extensive RF stalling in cisplatin-treated normal cells, the ensuing RF collapse would lead to the formation of double-strand breaks and, consequently, to an increase in large chromosomal alterations (Yoon et al. 2019b). Such chromosomal instability would be causal for tumorigenesis similar to that which occurs upon inhibition of error-free or error-prone TLS opposite UV lesions (Yoon et al. 2019b). Hence, inhibition of TLS by JH-RE-06 in normal cells during cisplatin therapy would lead to a rise in secondary malignancies, particularly in rapidly proliferating tissues (Supplemental Fig. S7). Moreover, inhibition of TLS by Y family Pols through UV and other DNA lesions would increase susceptibility to sunlight-induced skin cancers and other cancers.
Materials and methods
Protein expression in yeast
Full-length GST-tagged human Polι was expressed from plasmid pPOL114 as described (Washington et al. 2004). The human Polθ (1708–2590) protein harboring the catalytically active C-terminal DNA polymerase domain was expressed as a fusion with glutathione S-transferase from plasmid pPOL507 as described (Yoon et al. 2014). Proteins were expressed in yeast strain YRP654 and affinity-purified using glutathione Sepharose as described (Johnson et al. 2006). The GST fusion tags were removed from Polι and Polθ (1708–2590) proteins by treatment with prescission protease. Proteins were quantified by densitometry of Coomassie-stained protein samples separated by 11% SDS-PAGE using Imagequant software.
DNA polymerase assays
The DNA substrates consisted of a 5′ 32P-labeled DNA primer annealed to a 78-mer template with the sequence 5′-AGCAAGTCACCAATGTCTAAGAGTTTCTTGGTCTCCTCCTACACTGGAGTACCGGAGCATCGTCGTGACTGGGAAAAC-3′, wherein the underlined GG was either undamaged or consisted of a GG cisplatin intrastrand cross-link. The cisplatin adduct was introduced into a 13-mer oligonucleotide (5′-TTCTTGGTCTCCT-3′) as described (Ummat et al. 2012) and was subsequently ligated to flanking 25-mer and 40-mer oligonucleotides by use of a scaffold to generate the 78-mer. The 78-mer was isolated by TBE-PAGE containing 8 M urea. To assay incorporation opposite the 3′ G of either undamaged G or the cisplatin adduct, the 30-mer oligonucleotide 5′-GATGCTCCGGTACTCCAGTGTAGGAGGAGA-3′ was used as primer. To assay incorporation opposite the 5′ G of the cisplatin adduct, or the undamaged counterpart, a 31-mer oligo with the identical sequence as the 30-mer but harboring an additional C at the 3′ end was used. Each substrate was generated by annealing the 5′ 32P-labeled primer to the DNA substrate in a 1:1.5 ratio by heating to 95°C and slow cooling to room temperature. The standard DNA polymerase assay (5 µL) contained 25 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM DTT, 10% glycerol, and 0.1 mg/mL bovine serum albumin. Reactions contained a single dGTP, dATP, dCTP, or dTTP at 25 µM or all four dNTPs at 25 µM each. All reactions were carried out for 5 min at 37°C. Protein concentrations are indicated in Figure 4. Reactions were assembled on ice and were initiated by the addition of 1 µL of DNA polymerase in 5× reaction buffer (125 mM Tris-HCl at pH 7.5, 5 mM DTT, 0.5 mg/ml BSA) and terminated by the addition of 6 vol of 95% formamide loading buffer containing 0.06% xylene cyanol/0.06% bromophenol blue. Reaction products were separated by 16% TBE/8M urea-PAGE. Gels were fixed in 10% methanol:10% acetic acid for 10 min and dried, and products were visualized by phosphorimaging on a Typhoon FLA7000 (GE Biotech).
Oligonucleotide synthesis containing a cisplatin GG cross-link and construction of plasmid vectors containing a cisplatin GG
The 16-mer oligonucleotide 5′-CTTCCTCGGCTCTTCC-3′ containing a GG-Pt cross-link (underlined) was synthesized as described previously (Gelasco and Lippard 1998; Ummat et al. 2012). The heteroduplex vector containing a GG-Pt cross-link on the lagging strand template was constructed as described (Yoon et al. 2009, 2010).
Cell lines
Human cell lines used were normal fibroblast (GM00637), XPA fibroblast (GM04429), and XPV fibroblast (GM03617). Polη−/−, Polθ−/−, and Polη−/− Polθ−/− MEFs have been described previously (Yoon et al. 2019b), and Rev1−/− MEFs were derived as described (Yoon et al. 2015).
Translesion synthesis assays in human cells
For siRNA knockdown of TLS Pols, HPLC-purified duplex siRNA for human and mouse genes were purchased from Ambion. The sense sequence of siRNA target sequence and the siRNA knockdown efficiency of TLS Pols as well as the detailed methods for TLS assays and for mutational analyses of TLS products have been described previously (Yoon et al. 2009, 2015, 2019b).
Cisplatin survival assays
Cisplatin (Sigma) stock was prepared in 0.1 M NaCl. Cells were transfected with siRNAs, and 48 h after siRNA transfection, cells were treated with cisplatin. For primary MEFs, cells were seeded on duplicated six-well plates and incubated overnight. For cisplatin treatment, cells were washed with PBS buffer, MEFs were treated with 0–60 µM cisplatin for 2 h (Fig. 2C), and HFs were treated with 30 µM cisplatin for 2 h (Fig. 3B,C; Supplemental Fig. S3A,B) in the presence of PBS buffer. After cisplatin treatment, fresh growth DMEM medium (GenDEPOT) was added and cells were incubated for an additional 48 h. Cytotoxicity was determined by the MTS assay (Promega). Briefly, 100 µL of MTS assay solutions were added to each well and incubated for 30 min. Cell viability was determined by measuring OD at 490 nM, and four independent experiments were carried out.
Cisplatin-induced cII mutational assays in siRNA-treated BBMEF cells
Forty-eight hours after siRNA knockdown, cells were washed with PBS buffer (GenDEPOT) and treated with 30 µM cisplatin for 1 h. Fresh growth DMEM medium (GenDEPOT) was added and cells were incubated for 24 h. After the 24-h incubation period, the second siRNA transfection was carried out to maintain the siRNA knockdown of the target gene(s). Cells were incubated for an additional 4 d to allow for mutation fixation. Mouse genomic DNA was isolated using the genomic DNA isolation kit (Qiagen). The LIZ shuttle vector was rescued from the genomic DNA by mixing DNA aliquots and transpack packaging extract (Agilent), and the cII assay was carried out as previously described (Yoon et al. 2009, 2010). The mutation frequency was calculated by dividing the number of mutant plaques by the number of total plaques. For mutation analysis, the sequences of PCR products of the cII gene from the mutant plaques were analyzed as described previously (Yoon et al. 2009, 2010).
DNA fiber assays
Primary MEFs were isolated from embryos as described previously (Tommasi et al. 2005; Yoon et al. 2015, 2019b). Cells were pulse-labeled with 25 µM IdU (Sigma) for 20 min and subsequently washed twice with PBS buffer, followed by treatment with cisplatin (60 µM) and 250 µM CldU (Sigma) for 30 min. DNA fibers were spread on glass slides, and slides were incubated in 2.5 M HCl for 90 min and then washed with PBS buffer. The slides were incubated in blocking buffer (5% BSA in 1× PBS) for 2 h. Primary antibodies—rat anti-BrdU antibody (Abcam) and mouse anti-BrdU antibody (BD Bioscience)—were diluted in blocking buffer and incubated for 1 h followed by extensive washing with PBS buffer. Secondary antibodies—goat anti-rat Alexa 594 (Thermo Scientific) and goat anti-mouse Alexa 488 (Thermo Scientific)—were applied for 30 min and slides were mounted with antifade gold mounting medium (Invitrogen). DNA fibers were analyzed using a Nikon eclipse fluorescence microscope and quantified using NLS-Elements AR software.
Cisplatin-induced supF mutation assay in MCF7 cells
MCF7 cells were transfected with siRNAs for 48 h. Cells were cotransfected with siRNA and with cisplatin-treated (600 µM for 2 h) pSP189 shuttle vector. Plasmid DNA was rescued after 48 h of incubation and treated with DpnI to remove any unreplicated plasmid DNA. The rescued plasmids were transformed into MB7070 bacterial cells. The transformed bacterial cells were grown on LB plates containing ampicillin, IPTG, and X-gal, and the frequency of cisplatin-induced mutations in the SupF gene was determined.
Translesion synthesis assays in JH-RE-06-treated HFs
JH-RE-06 stock (3 mM) was prepared in DMSO. GM637 cells were treated with siRNAs for 24 h, following which cells were treated with 1.5 µM JH-RE-06 for 24 h in fresh growth medium. After 24 h of incubation, cells were cotransfected with siRNA and with pSB or pBS vector containing a DNA lesion. Three hours after transfection, fresh growth medium containing 1.5 µM JH-RE-06 was added to cells and cells were incubated for 24 h. Plasmid DNA was then rescued and treated with DpnI to remove the unreplicated plasmid DNA. TLS assays were carried out as previously described (Yoon et al. 2009, 2018, 2019b).
Cisplatin survival assays in JH-RE-06-treated cells
Primary MEFs were seeded on six-well plates, incubated overnight, and then treated with 1.5 µM JH-RE-06 for 24 h in fresh growth medium. GM637 HFs were treated with siRNAs for 24 h, followed by the addition of 1.5 µM JH-RE-06 for 24 h. MEFs were then incubated with 0–60 µM cisplatin for 2 h and HFs were treated with 30 µM cisplatin for 2 h in PBS buffer. After cisplatin treatment, cells were incubated for 48 h in fresh growth DMEM medium (GenDEPOT) containing 1.5 µM JH-RE-06. The cytotoxicity was determined by MTS assay (Promega).
UV survival assays in JH-RE-06-treated cells
Following treatment of primary MEFs and GM637 HFs with JH-RE-06 as described above, cells were washed with PBS buffer and irradiated with 0–20 J/m2 of UVC light in PBS buffer. After UV irradiation, cells were incubated for 48 h in fresh growth DMEM medium (GenDEPOT) containing 1.5 µM JH-RE-06. Cytotoxicity was determined by MTS assay (Promega).
Cisplatin-induced cII mutational assays in JH-RE-06-treated primary WT or Rev1−/− MEFs
Twenty-four hours after treatment with 1.5 µM JH-RE-06, primary MEFs were washed with PBS buffer (GenDEPOT) and incubated with 30 µM cisplatin for 1 h. After washing with PBS buffer, fresh growth DMEM medium (GenDEPOT) containing 1.5 µM JH-RE-06 was added to cells for a 24-h incubation period, after which medium containing JH-RE-06 was replaced by fresh growth medium. Cells were incubated for an additional 4 d to allow for mutation fixation. The mouse genomic DNA was isolated using the genomic DNA isolation kit (Qiagen).
DNA fiber assays in JH-RE-06-treated cells
Primary MEFs were treated with 1.5 µM JH-RE-06 for 24 h in fresh growth medium. Cells were then pulse-labeled with 25 µM IdU (Sigma) for 20 min. After that, cells were washed with PBS buffer twice and treated with 60 µM cisplatin and 250 µM CldU (Sigma) for 30 min.
Data availability
All of the study data are included in the tables and figures here and in the Supplemental Material.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grant R01 GM126087. We are most grateful to Dr. Pei Zhou and Dr. Jiyong Hong for providing JH-RE-06 for these studies.
Author contributions: J.-H.Y. designed and performed the genetic and cellular studies in HFs and MEFs. R.E.J. designed and performed DNA synthesis assays. S.P. and L.P. designed and coordinated the study. S.P., J.-H.Y., R.E.J., and L.P. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.348662.121.
Competing interest statement
The authors declare no competing interests.
References
- Besaratinia A, Pfeifer GP. 2006. Investigating human cancer etiology by DNA lesion footprinting and mutagenicity analysis. Carcinogenesis 27: 1526–1537. 10.1093/carcin/bgi311 [DOI] [PubMed] [Google Scholar]
- Chaney SG, Campbell SL, Bassett E, Wu Y. 2005. Recognition and processing of cisplatin- and oxaliplatin-DNA adducts. Crit Rev Oncol Hematol 53: 3–11. 10.1016/j.critrevonc.2004.08.008 [DOI] [PubMed] [Google Scholar]
- Doles J, Oliver TG, Cameron ER, Hsu G, Jacks T, Walker GC, Hemann MT. 2010. Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc Natl Acad Sci 107: 20786–20791. 10.1073/pnas.1011409107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugbartey GJ, Peppone LJ, de Graaf IA. 2016. An integrative view of cisplatin-induced renal and cardiac toxicities: molecular mechanisms, current treatment challenges and potential protective measures. Toxicology 371: 58–66. 10.1016/j.tox.2016.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelasco A, Lippard SJ. 1998. NMR solution structure of a DNA dodecamer duplex containing a cis-diammineplatinum(II) d(GpG) intrastrand cross-link, the major adduct of the anticancer drug cisplatin. Biochemistry 37: 9230–9239. 10.1021/bi973176v [DOI] [PubMed] [Google Scholar]
- Goss GD, Arnold A, Shepherd FA, Dediu M, Ciuleanu TE, Fenton D, Zukin M, Walde D, Laberge F, Vincent MD, et al. 2010. Randomized, double-blind trial of carboplatin and paclitaxel with either daily oral cediranib or placebo in advanced non-small-cell lung cancer: NCIC clinical trials group BR24 study. J Clin Oncol 28: 49–55. 10.1200/JCO.2009.22.9427 [DOI] [PubMed] [Google Scholar]
- Hicks JK, Chute CL, Paulsen MT, Ragland RL, Howlett NG, Guéranger Q, Glover TW, Canman CE. 2010. Differential roles for DNA polymerases η, ζ, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol Cell Biol 30: 1217–1230. 10.1128/MCB.00993-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson ER, Lippard SJ. 1999. Structure, recognition, and processing of cisplatin-DNA adducts. Chem Rev 99: 2467–2498. 10.1021/cr980421n [DOI] [PubMed] [Google Scholar]
- Johnson RE, Prakash L, Prakash S. 2006. Yeast and human translesion DNA synthesis polymerases: expression, purification, and biochemical characterization. Methods Enzymol 408: 390–407. 10.1016/S0076-6879(06)08024-4 [DOI] [PubMed] [Google Scholar]
- Kelland L. 2007. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 7: 573–584. 10.1038/nrc2167 [DOI] [PubMed] [Google Scholar]
- Knobel PA, Kotov IN, Felley-Bosco E, Stahel RA, Marti TM. 2011. Inhibition of REV3 expression induces persistent DNA damage and growth arrest in cancer cells. Neoplasia 13: 961–970. 10.1593/neo.11828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner BH, Cheung NK, Kramer K, Heller G, Jhanwar SC. 1998. Neuroblastoma and treatment-related myelodysplasia/leukemia: the Memorial Sloan-Kettering experience and a literature review. J Clin. Oncol 16: 3880–3889. 10.1200/JCO.1998.16.12.3880 [DOI] [PubMed] [Google Scholar]
- Lin X, Trang J, Okuda T, Howell SB. 2006. DNA polymerase ζ accounts for the reduced cytotoxicity and enhanced mutagenicity of cisplatin in human colon carcinoma cells that have lost DNA mismatch repair. Clincal Cancer Research 12: 563–568. 10.1158/1078-0432.CCR-05-1380 [DOI] [PubMed] [Google Scholar]
- Miller RD, Prakash L, Prakash S. 1982. Genetic control of excision of Saccharomyces cerevisiae interstrand DNA cross-links induced by psoralen plus near-UV light. Mol Cell Biol 2: 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. 2005. Human DNA polymerase ι incorporates dCTP opposite template G via a G.C+ Hoogsteen base pair. Structure 13: 1569–1577. 10.1016/j.str.2005.08.010 [DOI] [PubMed] [Google Scholar]
- Niimi K, Murakumo Y, Watanabe N, Kato T, Mii S, Enomoto A, Asai M, Asai N, Yamamoto E, Kajiyama H, et al. 2014. Suppression of REV7 enhances cisplatin sensitivity in ovarian clear cell carcinoma cells. Cancer Sci 105: 545–552. 10.1111/cas.12390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noll DM, Mason TM, Miller PS. 2006. Formation and repair of interstrand cross-links in DNA. Chem Rev 106: 277–301. 10.1021/cr040478b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash S, Johnson RE, Prakash L. 2005. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Ann Rev Biochem 74: 317–353. 10.1146/annurev.biochem.74.082803.133250 [DOI] [PubMed] [Google Scholar]
- Ratain MJ, Kaminer LS, Bitran JD, Larson RA, Le Beau MM, Skosey C, Purl S, Hoffman PC, Wade J, Vardiman JW, et al. 1987. Acute nonlymphocytic leukemia following etoposide and cisplatin combination chemotherapy for advanced non-small-cell carcinoma of the lung. Blood 70: 1412–1417. 10.1182/blood.V70.5.1412.1412 [DOI] [PubMed] [Google Scholar]
- Sarkar S, Davies AA, Ulrich HD, McHugh PJ. 2006. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase ζ. EMBO J 25: 1285–1294. 10.1038/sj.emboj.7600993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Shah NA, Joiner AM, Roberts KH, Canman CE. 2012. DNA polymerase ζ is a major determinant of resistance to platinum-based chemotherapeutic agents. Mol Pharmacol 81: 778–787. 10.1124/mol.111.076828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, Wilczynski SP, Li J, You M, Pfeifer GP. 2005. Tumor susceptibility of Rassf1a knockout mice. Cancer Res 65: 92–98. [PubMed] [Google Scholar]
- Travis LB, Fosså SD, Schonfeld SJ, McMaster ML, Lynch CF, Storm H, Hall P, Holowaty E, Andersen A, Pukkala E, et al. 2005. Second cancers among 40,576 testicular cancer patients: focus on long-term survivors. J Natl Cancer Inst 97: 1354–1365. 10.1093/jnci/dji278 [DOI] [PubMed] [Google Scholar]
- Ummat A, Rechkoblit O, Jain R, Roy Choudhury J, Johnson RE, Silverstein TD, Buku A, Lone S, Prakash L, Prakash S, et al. 2012. Structural basis for cisplatin DNA damage tolerance by human polymerase η during cancer chemotherapy. Nat Struct Mol Biol 19: 628–632. 10.1038/nsmb.2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Lippard SJ. 2005. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 4: 307–320. 10.1038/nrd1691 [DOI] [PubMed] [Google Scholar]
- Wang X, Peterson CA, Zheng H, Nairn RS, Legerski RJ, Li L. 2001. Involvement of nucleotide excision repair in a recombination-independent and error-prone pathway of DNA interstrand cross-link repair. Mol Cell Biol 21: 713–720. 10.1128/MCB.21.3.713-720.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waseem M, Bhardwaj M, Tabassum H, Raisuddin S, Parvez S. 2015. Cisplatin hepatotoxicity mediated by mitochondrial stress. Drug Chem Toxicol 38: 452–459. 10.3109/01480545.2014.992437 [DOI] [PubMed] [Google Scholar]
- Washington MT, Johnson RE, Prakash L, Prakash S. 2004. Human DNA polymerase ι utilizes different nucleotide incorporation mechanisms dependent upon the template base. Mol Cell Biol 24: 936–943. 10.1128/MCB.24.2.936-943.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheate NJ, Walker S, Craig GE, Oun R. 2010. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans 39: 8113–8127. 10.1039/c0dt00292e [DOI] [PubMed] [Google Scholar]
- Wojtaszek JL, Chatterjee N, Najeeb J, Ramos A, Lee M, Bian K, Xue JY, Fenton BA, Park H, Li D, et al. 2019. A small molecule targeting mutagenic translesion synthesis improves chemotherapy. Cell 178: 152–159.e11. 10.1016/j.cell.2019.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie K, Doles J, Hemann MT, Walker GC. 2010. Error-prone translesion synthesis mediates acquired chemoresistance. Proc Natl Acad Sci 107: 20792–20797. 10.1073/pnas.1011412107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu Z, Selby CP, Sancar A. 2019. Long-term, genome-wide kinetic analysis of the effect of the circadian clock and transcription on the repair of cisplatin-DNA adducts in the mouse liver. J Biol Chem 294: 11960–11968. 10.1074/jbc.RA119.009579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J-H, Prakash L, Prakash S. 2009. Highly error-free role of DNA polymerase η in the replicative bypass of UV induced pyrimidine dimers in mouse and human cells. Proc Natl Acad Sci 106: 18219–18224. 10.1073/pnas.0910121106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon J-H, Prakash L, Prakash S. 2010. Error-free replicative bypass of (6-4) photoproducts by DNA polymerase ζ in mouse and human cells. Genes Dev 24: 123–128. 10.1101/gad.1872810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Prakash S, Prakash L. 2012. Requirement of Rad18 protein for replication through DNA lesions in mouse and human cells. Proc Natl Acad Sci 109: 7799–7804. 10.1073/pnas.1204105109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Roy Choudhury J, Park J, Prakash S, Prakash L. 2014. A role for DNA polymerase θ in promoting replication through oxidative DNA lesion, thymine glycol, in human cells. J Biol Chem 289: 13177–13185. 10.1074/jbc.M114.556977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Park J, Conde J, Wakamiya M, Prakash L, Prakash S. 2015. Rev1 promotes replication through UV lesions in conjunction with DNA polymerases η, ι and κ but not DNA polymerase ζ. Genes Dev 29: 2588–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Roy Choudhury J, Park J, Prakash S, Prakash L. 2017. Translesion synthesis DNA polymerases promote error-free replication through the minor-groove DNA adduct 3-deaza-3-methyladenine. J Biol Chem 292: 18682–18688. 10.1074/jbc.M117.808659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Hodge RP, Hackfeld LC, Park J, Roy Choudhury J, Prakash S, Prakash L. 2018. Genetic control of predominantly error-free replication through an acrolein-derived minor-groove DNA adduct. J Biol Chem 293: 2949–2958. 10.1074/jbc.RA117.000962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Johnson RE, Prakash L, Prakash S. 2019a. DNA polymerase θ accomplishes translesion synthesis opposite 1,N6-ethenodeoxyadenosine with a remarkably high fidelity in human cells. Genes Dev 33: 282–287. 10.1101/gad.320531.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, McArthur MJ, Park J, Basu D, Wakamiya M, Prakash L, Prakash S. 2019b. Error-prone replication through UV lesions by DNA polymerase θ protects against skin cancers. Cell 176: 1295–1309.e15. 10.1016/j.cell.2019.01.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Y-H, Pfeifer GP. 2001. Similarities in sunlight-induced mutational spectra of CpG-methylated transgenes and the p53 gene in skin cancer point to an important role of 5-methylcytosine residues in solar UV mutagenesis. J Mol Biol 305: 389–399. 10.1006/jmbi.2000.4322 [DOI] [PubMed] [Google Scholar]
- You Y-H, Lee D-H, Yoon J-H, Nakajima S, Yasui A, Pfeifer GP. 2001. Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells. J Biol Chem 276: 44688–44694. 10.1074/jbc.M107696200 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Biertumpfel C, Gregory MT, Hua YJ, Hanaoka F, Yang W. 2012. Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proc Natl Acad Sci 109: 7269–7274. 10.1073/pnas.1202681109 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the study data are included in the tables and figures here and in the Supplemental Material.