

Summary:
Environmental Enteric Dysfunction (EED), is a gastrointestinal inflammatory disease caused by malnutrition and chronic infection. EED is associated with stunting in children and reduced efficacy of oral vaccines. To study the mechanisms of oral vaccine failure during EED, we developed a microbiota- and diet-dependent mouse EED model. Analysis of E. coli-Labile Toxin vaccine-specific CD4+ T cells in these mice revealed impaired CD4+ T cell responses in the small intestine and but not the lymph nodes. EED mice exhibited increased frequencies of small intestine-resident RORγT+FOXP3+ regulatory T (Treg) cells. Targeted deletion of RORγT from Treg cells restored small intestinal vaccine-specific CD4 T cell responses and vaccine-mediated protection upon challenge. However, ablation of RORγT+FOXP3+ Treg cells made mice more susceptible to EED-induced stunting. Our findings provide insight into the poor efficacy of oral vaccines in EED and highlight how RORγT+FOXP3+ Treg cells can regulate intestinal immunity while leaving systemic responses intact.
eTOC Blurb
Environmental Enteric Dysfunction (EED) is caused by chronic malnutrition and infection, and is associated with stunting and reduced oral vaccine efficacy in children. Bhattacharjee et al. develop a mouse model of EED and reveal both oral vaccine failure and stunting is controlled by a localized increase in intestinal RORγT+FOXP3+ regulatory T cells.
Graphical Abstract
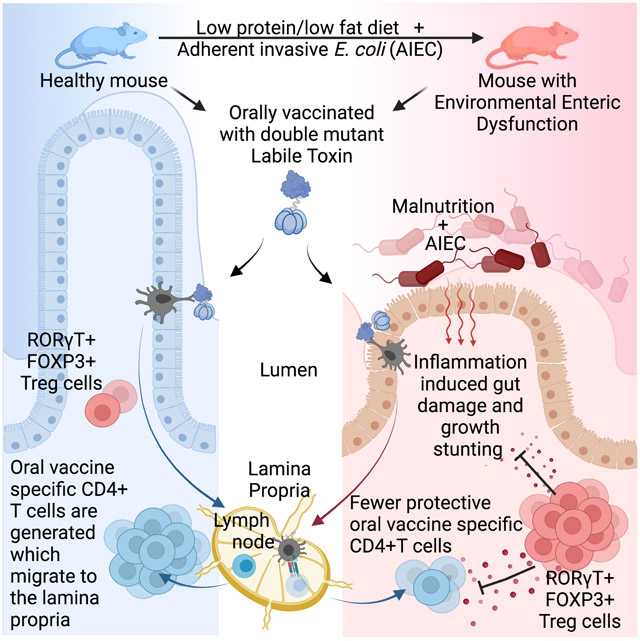
Introduction:
Oral vaccines are an effective mechanism to provide immunity against infection in the gastrointestinal tract and are used to prevent the spread of enteric viral and bacterial infections (Vela Ramirez et al., 2017). Bacterial enterotoxins, such as the E. coli heat labile toxin (LT), have the potential to be excellent oral vaccines because they possess intrinsic adjuvant activity. Attenuated enterotoxins are currently in trials as both adjuvants and vaccines against various enteric infections (Clements and Norton, 2018; Harro et al., 2019; Norton et al., 2011; Shaikh et al., 2020). Oral vaccines can induce long-lived intestinal-resident CD4+ T cells and IgA+ plasma B cells, which mediate protection at the site of infection (Bemark et al., 2016; Clements and Norton, 2018; Pasetti et al., 2011). How intestinal-resident T and B cells develop and are regulated after oral vaccination and the role of the local intestinal microbiota in their biology is not completely understood.
Intestinal immunity, requires a balance between restricting infectious microorganisms and avoiding persistent activation by the gut microbiota to prevent autoinflammatory disorders such as Inflammatory Bowel Disease. This equilibrium is complicated by the ever-changing environment of the intestine, where the host-microbiota relationship can be modified by shifts in diet and infection (Carmody et al., 2015; Cotillard et al., 2013; Hand, 2016). Diet-driven microbial shifts are associated with a variety of chronic diseases, and therapies that restore health to the microbiota can influence effective treatments (Blanton et al., 2016; Hand et al., 2016; Ridaura et al., 2013; Turnbaugh et al., 2006). Regardless of the trigger, microbial dysbiosis affects the immune response both locally, at barrier sites, and systemically, contributing to inflammatory disease (Henao-Mejia et al., 2012; Honda and Littman, 2016).
Intestinal regulatory T (Treg) cells play an important role in shaping immune responses to mucosally-encountered antigens, such as those derived from oral vaccines. The role of Treg cells in oral vaccination is complex, because in contrast to their regulation of T cell responses Treg cells support the generation of IgA-producing B cells, particularly within Peyer’s Patches (Cong et al., 2009; Gribonika et al., 2019; Kawamoto et al., 2014; Tsuji et al., 2009). Intestinal Treg cells can be categorized by markers that differ according to the source of the antigens that they are specific to, where Treg cells responding to self-antigens are Neuropilin+, RAR–related orphan receptor γ 2−( NRP1+RORγT−), those responding to microbiome-derived antigens are NRP1− RORγT+ and those responding to food are negative for both molecules (Kim et al., 2016; Ohnmacht et al., 2015; Sefik et al., 2015; Weiss et al., 2012; Xu et al., 2018). The balance of Treg and T helper cells in the intestine is important because inflammatory diseases have been associated with an increased frequencies of Th1 and Th17 cells and decreased Treg cells (Leung et al., 2014; Lochner et al., 2008). Whether Treg or T helper cell differentiation dominates the immune response to intestinal bacteria is dependent both upon the biology of the bacteria and extrinsic environmental factors and is likely important to efficacy of oral vaccination (Ansaldo et al., 2019; Chai et al., 2017; Chiaranunt et al., 2018; Hand et al., 2012; Xu et al., 2018; Zhao et al., 2017).
Environmental Enteric Dysfunction (EED) is a gastrointestinal inflammatory disease most commonly seen in children in resource-poor settings that contributes to developmental stunting (Keusch et al., 2014; Korpe and Petri, 2012). While EED is rare in resource-rich countries, approximately 150 million children are at-risk worldwide. EED is caused by a combination of malnutrition and chronic intestinal infection and is characterized by microbial dysbiosis, lymphocytic infiltration and a flattening of villi in the small intestine, which causes malabsorption due to reduced surface area (Korpe and Petri, 2012). Areas where EED is endemic are also associated with a reduced efficacy of oral vaccines (Naylor et al., 2015). There are several factors that may contribute to oral vaccine failure in resource-poor countries including EED-induced disruption of intestinal immunity (Bhattacharjee and Hand, 2018; Di Luccia et al., 2020; Naylor et al., 2015).
To identify how EED disrupts intestinal immune responses and reduces the efficacy of oral vaccines, we developed a mouse model of EED dependent upon malnutrition and intestinal colonization with adherent invasive E.coli. This model recapitulated multiple aspects of human EED, including growth stunting, intestinal villi shortening and reduced intestinal barrier function. Upon oral vaccination, EED-affected mice exhibited a near complete block in the accumulation of intestinal oral vaccine-specific CD4+ T cells while responses in secondary lymphoid tissue were intact. This failure of vaccine-specific CD4+T cell responses in the intestine was accompanied by a coincident increase in the number and function of RORγT+ Treg cells in the intestine, and ablation of these cells restored oral vaccine efficacy. However, depletion of RORγT+ Treg cells increased the severity of EED-induced stunting, indicating the importance of these cells in regulating chronic intestinal inflammation. Taken together, our data supports the idea that oral vaccine failure in EED is a consequence of a localized intestinal immune regulation and uncovers a modular structure for tissue and systemic immune responses.
Results
Murine EED is associated with weight loss, growth stunting, and increased intestinal permeability.
We developed an animal model of EED to replicate the intestinal inflammation, barrier defects and growth stunting associated with the disease in children. Existing mouse models require changes to both the diet and microbiota to induce EED-like symptoms and affected animals show an enrichment for Enterobacteriaceae in the mucus layer of the small intestine (Brown et al., 2015; Kau et al., 2015). We hypothesized that a combination of low protein/fat diet and colonization with an adherent-invasive E. coli isolate that intrinsically associated with the small intestinal epithelium would be sufficient to induce EED (Rolhion and Darfeuille-Michaud, 2007). The E. coli isolate ‘CUMT8’ was chosen because it is derived from mice, adheres to the small intestinal epithelium and ‘blooms’ under inflammatory conditions (Craven et al., 2012; Dogan et al., 2014). To control for diet and microbiota, our experiments had four groups: i) Isocaloric controls (ISO) fed a diet with sufficient fat and protein, ii) Malnourishment controls (MAL) fed a low protein/fat chow, iii) CUMT8 E. coli colonized controls (MT8) and iv) mice which receive both low protein/fat chow and CUMT8 E. coli which could develop EED-like disease(EED) (Fig. 1A). Mice were initiated onto their respective diets at 3 weeks of age, colonized with CUMT8 E. coli 16, 18 and 20 days later. All mice were fed an equal amount of food (by calories) to prevent the mice fed the low fat/protein diet from over-eating. As expected, mice fed with the malnourished diet (MAL and EED) gained weight more slowly than mice fed the isocaloric control diet (Fig. 1B). Independent of diet, administration of CUMT8 E. coli temporarily slowed the growth of mice (MT8 and EED), and the combination of low protein/fat diet and CUMT8 E. coli led to a reduction in growth of EED mice compared to controls (Fig. 1B and Supplemental Fig. 1A). EED mice also exhibited other signs of growth stunting, as evidenced by reduced tail lengths (Fig. 1C). In humans, EED is characterized by a reduction in the length of the intestinal villi (‘blunting’), increased immune cell infiltration, elevated intestinal inflammation and reduced intestinal barrier function (Korpe and Petri, 2012). Histological analysis of the terminal ileum revealed that EED mice, but not any of the controls, exhibited villous blunting and lymphocytic infiltration (Fig. 1D and E). The small intestine of mice on the EED protocol was more inflamed, as indicated by increased expression inflammatory cytokines (IFNγ and TNFα) and the anti-bacterial gene Lcn2 (Supplemental Fig. 1B and C) (Chen et al., 2020; Haberman et al., 2021; Watanabe and Petri, 2016). EED mice also exhibited decreased intestinal barrier function as measured by the detection of orally-administered FITC-dextran particles in the blood (Fig. 1F) (Keusch et al., 2014). To test whether colonization with an adherent/invasive strain of E. coli was required for the development of EED-like disease, we substituted CUMT8 E. coli for either a sessile commensal E. coli ‘ECMB’ or a human adherent-invasive E. coli ‘2A’ that can attach to the mouse intestine (Molloy et al., 2013; Viladomiu et al., 2021). These experiments showed that the non-adherent ECMB E. coli isolate, while colonizing in similar amounts compared to CUMT8 E. coli did not induce stunting, while colonization with the adherent 2A isolate reduced growth to a comparable degree as CUMT8 (Supplemental Fig 1D -F ). Conversely, gavage of CUMT8 E. coli alone had no obvious long-term effects on the mice fed a diet with standard amounts of fat and protein (Fig. 1B-F). We hypothesized that this might be due to differences in the colonization efficiency of CUMT8 E. coli under different diets (Brown et al., 2015). Quantification of CUMT8 E. coli from ileal and fecal samples of mice fed different diets revealed that malnourishment was necessary for colonization, as mice on the high protein/high fat diet (MT8) displayed only low level CUMT8 colonization that dissipated quickly (Fig. 1G and Supplemental Fig. 1G). In contrast, mice on the malnourished diet (EED) had > 10-fold more CUMT8 E. coli compared to controls two days after CUMT8 gavage and many animals maintained colonized status for a week or more (Fig. 1G and Supplemental Fig. 1G). Therefore, a diet low in protein and fat sets the stage for EED-like disease by directly limiting host growth and allowing for greater levels of colonization with adherent/invasive E. coli and thereby intestinal inflammation.
Figure 1. Murine EED is associated with weight loss, growth stunting and increased intestinal permeability.

A) Schematic of the EED protocol. ISO (blue) and MT8 (violet) mice received control diets while MAL (green) and EED (red) mice were provided low protein/fat diets. EED and MT8 mice were colonized with CUMT8 E. coli.
B) Percent weight gain during EED protocol. Dotted vertical lines represent days when CUMT8 E. coli was orally gavaged. Data shown are representative of 3 independent experiments (n=4). Data points represent mean +/−SEM. Statistics calculated from weights at day 28.
C-G show analyses from day 28 of the EED protocol. Data points represent a single mouse
C) Tail lengths. Data shown are one representative example of 3 independent experiments
D) Representative Hematoxylin and Eosin staining of ileal sections. Representative of 2 separate experiments.
E) Histological scoring of D). Pooled from 2 independent experiments (n=3/experiment)
F) Measurement of intestinal permeability by quantification of serum FITC Dextran (4kDA) concentration 4 hrs. after oral gavage. Data are pooled from 2 independent experiments (n=3-4/experiment).
G) Level of CUMT8 E. coli in ileal samples from day 23 and day 28 of the EED protocol. Genomic DNA was isolated from ileal contents and the abundance of CUMT8 was measured by qPCR and compared against a standard curve. Dotted lines represent limit of detection. Data are pooled from 2 separate experiments (n=3-4/experiment). Graphs show the mean ±SEM. Statistics calculated by one-way ANOVA (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001).
EED induces tissue-specific failure of oral vaccination
In humans, EED is hypothesized to suppress the efficacy of oral vaccines (Bhattacharjee and Hand, 2018; Naylor et al., 2015). To evaluate oral vaccine responses in the murine EED model, we vaccinated mice with an attenuated Heat Labile Toxin (LT) derived from enterotoxigenic E. coli (ETEC). This attenuated LT, called double mutant Labile Toxin (dmLT) carries two mutations that prevent diarrhea induction by inhibiting host protease-mediated cleavage of the toxin from the plasma membrane and by reducing activation of adenylate cyclase (Clements and Norton, 2018). Despite reduced toxicity, dmLT maintains its immunogenicity as an oral vaccine and generates a robust mixed Th1/Th17 CD4+ T cell and anti-LT IgA B cell response that is dependent upon immune cell activation in the draining mesenteric lymph nodes (MLNs) (Fonseca et al., 2015; Leach et al., 2012). Critically, vaccination of mice with dmLT leads to a CD4+ T cell-dependent protection against intestinal colonization with LT-expressing ETEC (Supplemental Fig. 2A) (Roy et al., 2011).
To test oral vaccine efficacy in our EED model, , mice were primed and boosted with dmLT via oral gavage at the end of the EED protocol (Fig. 2A) (Hall et al., 2011a). MHC class II tetramers loaded with an epitope from the A-subunit of LT (LTA166-176; LT-specific) were used to magnetically isolate and analyze antigen-specific CD4+ T cells from the small intestinal lamina propria (siLP), Peyer’s patches (PPs) and MLNs (Fig. 2B, C and supplemental Fig 2B) (Moon et al., 2009; Pepper et al., 2009). While LT-specific CD4+ T cell responses did not vary across groups in the MLNs and PPs, siLP anti-LT CD4+ T cell responses were almost undetectable in EED mice (>18 fold lower compared to ISO controls), indicating that the vaccine-specific T-cells accumulated in the secondary lymphoid tissue but not at the gut mucosa (Fig 2B, 2C and Supplemental Fig. 2B). EED mice also had lower levels of intestinally-secreted dmLT-specific IgA compared to controls, indicating that intestinal IgA+ B cell responses to oral vaccination were also affected (Fig 2D). Vaccinated EED mice were also incapable of controlling ETEC colonization compared to controls (Fig. 2E). In addition, mice fed a low protein/fat MAL diet but orally gavaged with the sessile E. coli isolate ECMB (that is insufficient to induce EED) had normal siLP LT-specific CD4+ T cell responses (Supplemental Fig. 1E, F and Supplemental Fig. 2C). Taken together, in a model of EED we have shown that oral vaccines were capable of priming CD4+ T cell responses in the lymph nodes but vaccine-specific CD4+ T cells fail to accumulate in the small intestine. This suggests that oral vaccine failure in EED might be due to inhibition of intestine-resident T and B cell responses.
Figure 2. EED induces tissue-specific oral vaccine failure.

A) Schematic of the EED protocol and oral vaccination regime.
B) Representative flow cytometric plots showing LT166-176:I-Ab specific CD4+ T cells from the siLP and MLN, 14 days after primary vaccination with dmLT (day 42). Cells gated: Live, Lineage−, CD90+, CD3+, CD8b−, CD4+. Numbers on plots denote mean and SEM, respectively. Data are representative examples of 3 independent experiments.
C) Quantification B. Data are pooled from 3 independent experiments (n=2-3/experiment).
D) Concentration of LT-specific IgA in the small intestine (from PBS lavage) of mice from C.
E) Mice in the EED protocol were vaccinated with dmLT as shown in A. After vaccination (day 42 or 45) mice were colonized with ETEC H10407 (KanaR) and a day later numbers of bacterial CFU counted on LB/Kanamycin plates. Data are pooled from 2 separate experiments (n=3/experiment).
(C-E) Data points represent a single mouse. Graphs show the mean ±SEM. Statistics calculated by one-way ANOVA (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001).
EED-associated shifts in the intestinal microbiota are necessary for oral vaccine failure.
The intestinal microbiota of children suffering from EED is characterized by overgrowth by Enterobacteriaceae and an overrepresentation of bacteria typical to the oropharyngeal cavity (Blanton et al., 2016; Chen et al., 2020; Kau et al., 2015; Vonaesch et al., 2018). In a healthy gut, Enterobacteriaceae colonization is controlled by the resident microbiota (Buffie and Pamer, 2013). We wanted to measure whether the increased efficiency of CUMT8 E.coli colonization in mice fed a low protein/fat diet was associated with shifts in the intestinal microbiota. To equalize the initial microbiota, mice in these experiments were purchased from a single source and assorted into singly housed cages to avoid ‘cage-mate’ effects on the microbiota. Longitudinal analysis of the fecal microbiota (16S rRNA amplicon sequencing) from our four cohorts revealed that the primary shift in was driven by diet, as mice on the low protein/fat diet (MAL:green and EED:red) largely clustered away from mice fed the isocaloric diet (ISO:blue and MT8:purple), highlighting the effect of this diet on the microbiota (Supplemental Fig. 3A and B). As mice progressed through the protocol, principal coordinates analysis (PCoA) of the fecal microbiome did not differentiate later fecal sampling timepoints (experimental day 23 and day 28), indicating the dominance of diet for the overall structure of the colonic microbiome in these experiments (Supplemental Fig. 3A and B). Intestinal inflammation in EED is centered on the ileum, and despite heterogeneity associated with singly-housed mice, comparison of the ileal microbiota of ISO and EED mice at day 28 revealed larger differences than for the colonic microbiome (Supplemental Fig. 3C-F). In the ileum, the microbiome did not indicate groupings according to diet as MAL and EED were not associated by PCoA, perhaps indicating a dominant role for EED-driven inflammation at this site (Supplemental Fig. 3C-E). In concert with human and animal model studies on EED, we observed an increase in Enterobacteriaceae and bacteria more commonly associated with the mouth microbiome (Moraxacellaceae, Veillonellaceae) amongst mice with EED (Supplemental Fig. 3F)(Vonaesch et al., 2018). Therefore, we observe that induction of EED in our murine model is associated with diet and inflammation-dependent shifts in the intestinal microbiota.
We next sought to determine if EED-associated changes in the intestinal microbiota were necessary for the reduction in oral vaccine efficacy. To test this question, we treated the EED mice or ISO mice with a broad-spectrum antibiotic cocktail (Metronidazole, Ampicillin, Neomycin and Vancomycin; MANV) to ablate the intestinal microbiota after disease initiation (Fig. 3A) (Rakoff-Nahoum et al., 2004). To limit the size of these experiments, we tested only the ISO and EED groups, which show the largest difference in oral vaccine responses. We hypothesized that ablation of the microbiota might reset local immunity and the response to oral vaccination with dmLT. Comparison of the anti-dmLT CD4+ T cell response after three weeks of treatment with antibiotics revealed no difference between mice with EED and ISO controls due to an increase in dmLT-specific T cell responses in antibiotic-treated EED mice (Fig. 3B). In these experiments, anti-dmLT CD4+ T cell responses in non-antibiotic treated EED mice were not as strongly inhibited, perhaps indicating that the mice began to recover approximately one month post-colonization with CUMT8 E. coli (Fig. 3B). Thus, our EED model exhibits shifts in the composition of the microbiota characteristic of humans with EED and these shifts are required to maintain EED-associated deficits in the CD4+ T cell response to oral vaccination (Vonaesch et al., 2018).
Figure 3. EED-associated shifts in the intestinal microbiome are necessary for oral vaccine failure.

A) Schematic of EED protocol, antibiotic treatment and vaccination regime. Mice on ISO or EED protocols were treated with antibiotics for 3 weeks, ending on day 49 when they are orally vaccinated.
B) Numbers of LTA166-176:I-Ab specific CD4+ T cells isolated from the siLP and MLN of mice vaccinated after 3 weeks of treatment with (shaded bars) or without (clear bars) antibiotics. Data points represent a single mouse. Data are pooled from 2 independent experiments (n=3/experiment). Graphs show the mean ±SEM. Statistics calculated by one-way ANOVA (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001).
EED-induced Treg cells are necessary for intestinal oral vaccine failure
The intestine-specific block in oral vaccine responses in the EED mice suggested that the intestinal immune microenvironment may be altered. Therefore, we evaluated the immune cells of the siLP in EED mice and controls. We have previously demonstrated that infection-induced damage to the lymphatics inhibited the traffic of intestinal CD103+ Dendritic cells (DCs) to the draining MLNs and induced large shifts in intestinal immune cell populations (Fonseca et al., 2015). In contrast, here we saw no statistically significant differences in the relative abundance of any of the three DC subsets in the MLN , which was consistent with normal activation of LT-specific CD4+ T cells in the MLNs . However, we did observe an increase of CD11B+ CD103+ DCs in the siLP (Supplemental Fig. 4A-C). Since the defect in vaccine-specific CD4+ T cells was focused on the small intestine, we next examined major immune populations of the siLP and saw no differences in the proportions of major innate immune cells, B cells or T cells (Supplemental Fig. 4D-I). Children with EED have increased levels of CD3+CD25+ T cells in their intestine, consistent with an increased number of FOXP3+ Treg cells (Campbell et al., 2003). In accord with human EED phenotypes, mice with EED were found to have an increased number and frequency of Treg cells in their siLP, compared to ISO controls (Fig. 4A). In contrast, MLN Treg cell percentages and numbers were invariant across all groups (Fig. 4A and B). Thus, the abundance of Treg cells at the time of oral vaccination inversely correlated with the accumulation of LT-specific CD4+ T cells at that site. To test the hypothesis that EED-induced Treg cells inhibited oral vaccine responses, EED was induced in Foxp3DTR-GFP mice, where injection with diphtheria toxin (DT) specifically and transiently ablates FOXP3+ Treg cells (Fig. 4C and Supplemental Fig. 4J) (Kim et al., 2007). To target EED-induced Treg cells, DT was administered only for the week post-CUMT8 colonization but prior to vaccination with dmLT (Fig. 4C). Treg cell depletion in EED mice completely restored LT-specific CD4+ T cell responses and anti-dmLT IgA responses, while PBS-treated Foxp3DTR-GFP mice still maintained an impairment in these responses (Fig. 4D-F). Therefore, oral vaccine failure in our EED mouse model requires a local increase in small intestinal Treg cells.
Figure 4. EED-induced Treg cells are necessary for intestinal oral vaccine failure.

(A and B) Mice were placed on the ISO, EED, MAL or MT8 protocols as shown in Fig 1A and sacrificed on day 28.
A) Representative flow cytometric plots showing percent FOXP3+ CD4+ T cells from the siLP and MLN, as indicated. Gated: Live, CD90+, TCRb+, CD8b−, CD4+. Numbers on plots denote mean and SEM, respectively. Data are representative of 3 independent experiments (n=3/experiment).
B) Percent (left) and numbers (right) of CD4+FOXP3+ T cells from siLP and MLNs from (A). Data are pooled from 3 experiments.
C) Schematic of mouse Treg cell depletion experiments using the ISO or EED protocol, Foxp3DTR-GFP mice and treatment with Diphtheria toxin (C-F).
D) Representative flow cytometric plots of LTA166-176:I-Ab specific CD4+ T cells following oral vaccination of Foxp3DTR-GFP mice with dmLT as in Fig. 2. Mice were treated with DT as indicated. Numbers on plots denote mean and SEM, respectively. Data shown are representative of 2 independent experiments (n=3/experiment)
E) Number of LTA166-176:I-Ab specific CD4+ T cells calculated from D). Shown are 2 pooled experiments.
F) Concentration of LT-specific IgA in the small intestine (from PBS lavage) from (E).
Data points represent a single mouse. Graphs show the mean ±SEM. Statistics calculated by one-way ANOVA (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001).
EED induces intestinal RORγT-expressing Treg cells that potently suppress CD4+ T cell responses
Given that siLP Treg cells were necessary for the EED-mediated inhibition of oral vaccination, we wanted to determine whether EED-associated intestinal Treg cells were functionally distinct. We found that Treg cells from EED mice expressed higher levels of the proliferation marker Ki67 than ISO mice, potentially accounting for their increased number (Fig. 5A and B). A critical function of Treg cells is suppression of the clonal expansion of helper T cells. We quantified the suppression capacities of intestinal Treg cells from mice with EED using a microsuppression assay. Treg cells isolated from the siLP of EED mice had greater suppressive potential than the same cells isolated from ISO controls (Fig. 5C). Despite their increased functionality, phenotypic characterization of siLP Treg cells from EED mice revealed no statistically significant differences in characteristic Treg proteins such as FOXP3, CD25, CTLA-4 and IL-10 (Supplemental Figure 5A-D). However, flow cytometric analysis indicated that siLP Treg cells from EED mice had increased expression of CD39 and CD73 (Supplemental Fig. 5E), a pair of ectonucleotidases that dephosphorylate extracellular ATP sequentially to adenosine and are associated with increased suppressive function in Treg cells (Deaglio et al., 2007).
Figure 5. EED induces intestinal RORγT-expressing Treg cells that potently suppress CD4+ T cell responses.

Mice were placed on the EED protocol as shown in Fig 1A and sacrificed on day 28
A) Flow cytometric histograms showing percent Ki67 expression in FOXP3+CD4+ Treg cells from the siLP. Numbers on plots denote mean and SEM, respectively. Data shown is representative of 2 separate experiments (n=3/experiment)
B) Mean Fluorescence Intensity (MFI) of Ki67 expression from A). Data pooled from 2 experiments
C) Percent suppression of naïve CD4+ T cell proliferation by siLP Treg cells from EED mice or ISO controls (n=5). Top is line graph showing relative suppression at different cellular concentrations. Bottom is representative histograms of Tconv CFSE dilution. Black histograms represent suppression by splenic Treg cells from ISO mice. Data points and error bars represent mean and SEM respectively (**p % 0.01; 2-way ANOVA). Data shown are representative of 2 independent experiments where siLP Treg cells were sorted from 4 EED and 4 ISO mice.
D) Representative flow cytometric contour plots of RORγT expression on siLP (top) and MLN (bottom) FOXP3+ CD4+ Treg cells. Numbers on plots denote mean and SEM, respectively. Data shown are representative of 3 independent experiments (n=3/experiment).
E) Percent and F) numbers of FOXP3+ CD4+ Treg cells from D) that express RORγT. Data is pooled from 3 separate experiments.
B, E-F) Data points represent a single mouse. Graphs show the mean ±SEM. Statistics for B,E and F were calculated by one-way ANOVA (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001).
The microbiota and in particular, intestinal bacteria that adhere to the intestinal epithelium are associated with the induction of intestinal Treg cells that express the transcription factor RORγT (Kim et al., 2016; Ohnmacht et al., 2015; Sefik et al., 2015; Xu et al., 2018). Analysis of EED mice and controls revealed that siLP Treg cells from EED mice showed a distinct increase in RORγT+ Treg cells that did not extend to Treg cells isolated from the MLNs (Fig. 5D-F). The development of RORγT+ Treg cells in the colon requires the presence of specific types of intestinal bacteria (Abdel-Gadir et al., 2019; Sefik et al., 2015; Song et al., 2020; Xu et al., 2018). Therefore, we sought to analyze the effects of the EED microbiota on siLP Treg cells. Antibiotic depletion of the microbiota for three weeks eliminated the EED-induced increase RORγT+ Treg cells in the small intestine (Supplemental Fig. 5F) which corresponds well with the finding that ablation of the microbiota can largely restore the ability of mice with EED to induce intestinal immune responses to oral vaccination (Fig. 3B). Intestinal RORγT+ Treg cells are also supported by the expression of inflammatory cytokines, such as IL-6 and IL-23, and the presence of the Vitamin A metabolite, retinoic acid (Ohnmacht et al., 2015; Yan et al., 2021). Analysis of the cytokine milieu of the small intestine of EED mice and ISO controls did not reveal differences in IL-6 and IL-23, but did show increases IL-2 and IL-10, two cytokines important for Treg cell homeostasis in the intestine (Murai et al., 2009; Zhou et al., 2019) (Supplemental Fig. 5G). The production of retinoic acid (RA) from retinaldehyde is catalyzed by an aldehyde dehydrogenase: RALDH2, which is expressed in migratory dendritic cells found in the siLP and MLN (Hall et al., 2011b). Flow cytometric analysis showed that aldehyde dehydrogenase (ALDH) enzymatic activity was specifically increased amongst dendritic cells in the siLP but not DCs isolated from the MLNs (Supplemental Fig. 5H and I). Most ALDH activity was found amongst CD103+ DCs, though an increase in activity was found in both intestinal CD103+ and CD103− DCs (Supplemental Fig. 5H and I). Taken altogether, we have shown that EED is associated with the specific expansion of small intestinal RORγT+CD39+CD73+ Treg cells endowed with increased regulatory function. Further, the accumulation of RORγT+ Treg cells in the small intestine requires the presence of the intestinal microbiota and is associated with the distinct up-regulation of retinoic acid production capability of intestinal DCs.
RORγT-expression in Treg cells is necessary to inhibit vaccine-specific CD4+ T cell responses and EED-associated stunting
RORγT+ Treg cells can suppress inflammation in animal models of colonic pathology (Ohnmacht et al., 2015; Sefik et al., 2015), but their role in the small intestine and specifically in oral vaccine responses is not known. Therefore, we bred Foxp3Cre ERT2 Rorcfl/fl mice (FOXP3RORγT+/−) where the expression of RORγT can be specifically extinguished in FOXP3+ Treg cells via the administration of tamoxifen (FOXP3RORγT/−)(Fig. 6A and Supplemental Fig. 6A) (Rubtsov et al., 2010). We gavaged mice with tamoxifen to excise the Rorc gene in developing Treg cells starting from five days before CUMT8 E. coli gavage to the end of the EED protocol. When immunized with dmLT, tamoxifen-treated FOXP3RORγT− mice showed completely restored LT-specific CD4+T cell populations in the siLP and had normal production of anti-dmLT IgA, while vehicle-gavaged controls (FOXP3RORγt+) still showed reduced responses (Fig. 6B and C). Critically, deletion of the Rorc gene from Treg cells also restored the ability of mice on the EED protocol to develop oral vaccine-mediated protective immunity, as vaccinated FOXP3RORγT− mice showed a reduction in ETEC colonization compared to vehicle-treated EED mice (Fig. 6D). Treg-specific ablation of RORγT had an effect on the total number and frequency of siLP Treg cells in EED mice, indicating that, RORγT may play a role in the accumulation and survival of FOXP3+Treg cells in the intestine(Supplemental Fig. 6B and C).
Figure 6. Blocking RORγT induction in EED induced Treg cells restores oral vaccine responses.

A) Schematic of ISO or EED protocols, tamoxifen (TX) or vehicle(Veh; corn oil) treatment for RORγT deletion and oral vaccination and in Foxp3Cre ERT2 Rorcfl/fl mice (FOXP3RORgT+/−).
(B-E) Data from mice which were treated with tamoxifen(FOXP3RORgT−) are represented in open circles while data from vehicle treated controls (FOXP3RORgT+) are in closed circles.
B) Numbers of LT166-176:I-Ab specific CD4+ T cells isolated from the siLP and MLNs following oral vaccination with dmLT (as in Fig. 2A) of Foxp3Cre ERT2 Rorcfl/fl with and without tamoxifen treatment. Data is pooled from 2 separate experiments (n=3/experiment).
C) Concentration of LT-specific IgA in the small intestine (from PBS lavage) of Foxp3Cre ERT2 Rorcfl/fl mice from mice from B.
D)Foxp3Cre ERT2 Rorcfl/fl mice in the EED protocol were vaccinated with dmLT, with and without tamoxifen treatment as shown in 6A. After vaccination (day 42 or 45) mice were colonized with ETEC H10407 (KanaR) and a day later numbers of bacterial CFU counted on LB/Kanamycin plates. Data are pooled from 2 separate experiments (n=3/experiment).
E) Percent weight gain during EED establishment in FOXP3Cre ERT2 Rorc fl/fl mice with and without tamoxifen treatment. Dotted lined represent days when CUMT8 E.coli was orally gavaged. Data shown is representative of 2 separate experiments (n=3-4/experiment). Statistics calculated from weights at day 28.
Graphs show the mean ±SEM. Data points represent a single mouse. Statistics calculated by one-way ANOVA (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001)
Since we treated mice with tamoxifen during EED induction, we were able to determine if RORγT+ Treg cells are important for controlling any of the symptoms of EED. Interestingly, FOXP3RORγt− EED mice were more stunted than RORγT+ Treg sufficient EED controls (FOXP3RORγt+) under the same protocol and gained minimal weight after colonization with CUMT8 E.coli (Fig. 6E and Supplemental Fig. 6D). Thus, RORγT expression in Treg cells is not only necessary to inhibit the accumulation of vaccine-specific T cells in the small intestine but also important for reducing stunting associated with EED. We hypothesize that the accumulation of RORγT+ Treg cells in the intestine of EED may result from an effort to alleviate chronic intestinal inflammation in the small intestine and that the localized failure of vaccine-specific T cell responses at this site develops as a side effect of a tissue-specific immunoregulation.
Discussion
We have developed a murine model of EED that mimics several aspects of human disease, including a reduction in immunity conferred by oral vaccination. Areas of the world where EED is endemic exhibit a lack of efficacy of oral vaccines, which is tragic since these communities would probably benefit most from effective mucosal immune protection (Grassly et al., 2009; Levine, 2010). There are a variety of factors other than EED that contribute to the failure of oral vaccines in resource-poor countries, including extended breast feeding, which may contain vaccine inhibitory IgA and milk oligosaccharides, increased exposure to pollution, micronutrient deficiency and pathogen heterogeneity (Burr et al., 2020; Moon et al., 2010; Santos and Hoshino, 2005). We found that independent of other factors, EED is sufficient to inhibit siLP CD4+ T cell responses and IgA production, indicating that EED is an important contributor to failure of oral vaccines in resource poor countries(Di Luccia et al., 2020).
Adherent-Invasive E. coli were first described in patients with Crohn’s Disease where they contribute to pathology, but are not sufficient to induce disease since they are common in the general population (Barnich and Darfeuille-Michaud, 2007). It is likely that our ancestors were commonly colonized with bacteria that attach to and irritate the intestinal epithelium and engendered CD4+ T cell responses. We propose that EED is inducing local immune suppression, mediated by RORγt+ Treg cells, to limit inflammatory intestinal damage. In this model, systemic immunity is left intact to guard against more invasive organisms. However, a side effect of this EED-induced intestinal immune shutdown is the loss of efficient tissue responses to oral vaccines. We hypothesize that this represents an evolutionary mechanism to prevent chronic inflammation in the small intestine, such as Inflammatory Bowel Disease in the face of constant colonization with intestinally-adherent bacteria.
Most research on oral vaccines has focused on antibodies. However, there is a growing interest in the potential of intestinal-resident CD4+ T cells in mediating protection via the rapid production of cytokines such as IL-17A and IL-22, that can activate defense mechanisms in the intestinal epithelium (Cao et al., 2012; Omenetti et al., 2019; Schreiber et al., 2015). A recent trial using oral dmLT in combination with live-attenuated enterotoxigenic E. coli vaccine revealed significant protection that was not correlated to antigen-specific IgA or IgG, leaving the possibility of a T cell-mediated mechanism (Clements and Norton, 2018; Harro et al., 2019). CD4+ T cells also support the differentiation and development of IgA+ plasma B cells in Peyer’s Patches (PP) (Bemark et al., 2016; Lycke and Bemark, 2017), but, surprisingly, we saw very few LT-specific T cells in the PP in our studies and they did not vary according to nutritional/EED status. After activation in the PP, IgA+ plasma cells traffic to siLP where they will reside long-term (Lycke and Bemark, 2017). Perhaps siLP CD4+ T cells assist B cells in the small intestinal lamina propria or alternatively, the inflammatory and nutrient-deficient environment of the EED intestine may inhibit IgA-producing B cells independently of T cell help. Future studies on LT-specific B cells in EED will be necessary to elucidate this point.
In our model of EED, we correlated oral vaccine failure with an increased presence of RORγT+ Treg cells, within the small intestine but not in the draining MLN tissue. This is consistent with human studies where children suffering from EED have increased CD3+CD25+ cells in their intestine (Campbell et al., 2003). Most previous descriptions of RORγT+ Treg cells have focused on the colon, where these cells are dependent upon the production of bile acid metabolites and specific members of the microbiota (Sefik et al., 2015; Song et al., 2020; Xu et al., 2018). The majority of bile acids are recycled by the ileum so they could also be important in inducing RORγT+ Treg cells in the small intestine(Ridlon et al., 2006). The intestinal metabolite profile of the small intestine is substantially different between EED mice and controls with bile acid metabolism/synthesis amongst the largest differences (Brown et al., 2015). Alternatively, RORγT+ Treg cells are also observed in lung tissue, where bile acids are not present at high levels, implying that there may be multiple pathways that drive this subset (Lochner et al., 2008). Retinoic acid production, presumably mediated by intestine-derived DCs, is also required for colonic RORγT+ Treg cells and here we found an intestine-specific increase in ALDH activity amongst those DCs,. While induction of Treg cells specific to intestinally-derived antigens occurs in the gut-draining mesenteric lymph nodes, RORγT+ Treg cells are rare at this site, implying that like RORγT-expressing Th17 cells, further differentiation of these cells occurs upon arrival in the intestine (Sano et al., 2015). Indeed, IL-6 expressed by enteric nerves is critical for the development of colonic RORγT+ Treg cells and we hypothesize that the increase in intestinal RA production has a similar effect, acting on Treg cells after they arrive in the intestine (Yan et al., 2021). Antigen-specific RORγT+ Treg cells are also induced by colonization of the cecum and colon with Helicobacter hepaticus (Xu et al., 2018). It is not clear whether the RORγT+ Treg cells induced by EED are specific to CUMT8 E. coli but both H. hepaticus and CUMT8 E. coli share the ability to adhere to the surface of the intestine (Dogan et al., 2014; Fox et al., 1994). Adherence to the gut lining is likely critical for the induction of RORγY+ Treg cells (Kim et al., 2018), as seen by the lack of oral vaccine failure in malnourished mice that were given ECMB, a non-adherent strain of E.coli. Further experimentation will be needed to identify whether epithelium-adherent bacteria are required to activate RORγT+ Treg cells in the small intestine.
The molecular mechanism by which RORγT+ Treg cells limit the accumulation of LT-specific CD4+ T cells is not clear. Our hypothesis is that RORγT+ Treg cells are blocking local antigen presentation and cytokine signals that are necessary for secondary proliferation once LT-specific T cells arrive in the intestine (Sano et al., 2015). The function that RORγT performs as a transcription factor in Treg cells is also not clear at this point, but our results showed that RORγT expression may be important for Treg proliferation and survival in the intestine. Loss of RORγT expression from Treg cells does not lead to spontaneous intestinal inflammation but, instead, is critical to limiting inflammation once it is induced (Boehm et al., 2012; Ohnmacht et al., 2015; Sefik et al., 2015). Accordingly, RORγT+ Treg cells were superior to RORγT− Treg cells at suppressing inflammation in a model of T cell transfer colitis (Yang et al., 2016). In concert, our results show that RORγt+ Treg cells reduce EED-associated stunting. Since the primary function of the gut is to absorb nutrients, we hypothesize that RORγt+ Treg cells prioritize epithelial health and absorption and that the block in immune responses is a by-product of the necessity to regulate damaging inflammation. Patients suffering from EED can take years to re-establish full intestinal functionality, and it will be important to determine whether, once established, intestinal resident immune cells maintain the disease state long after the inciting conditions have subsided (Gerson et al., 1971; Lindenbaum et al., 1972). In the future, as we develop a deeper knowledge of the intestinal immune defects in EED, we hope that this information can be incorporated into treatment plans and oral vaccine formulations so we can break the chain of malnutrition and infection that underlies this disease.
Limitations of the study
Our study tests an animal model of a complex human disease. While our model recapitulates many aspects of human EED, there are undoubtedly many ways that mouse physiology differs from humans, some of which may be important to this model. While we feel that our EED model will be useful for the research community to test hypotheses mechanistically, it will be important to determine these differences. For example, our model relies on adherent/invasive E. coli (AIEC) and while E. coli is increased in children with EED, AIEC may be irrelevant to human disease. Our study uncovered a failure of vaccine-specific CD4+ T cells to accumulate in the intestine due to local increases in Treg cells, however, limitations on how many tissues we could analyze per experiment prevented us from determining whether EED was driving vaccine-specific T cells to a different tissue. In addition, we relied on flow cytometric measurements of antigen-specific CD4+ T cells isolated from enzymatically-degraded tissue. A more comprehensive view of the oral vaccine-specific CD4+ T cell response would be provided by microscopy, but this will necessitate a shift away from tetramer-based identification of T cells, which is technically challenging in fixed tissue. Finally, while we saw a drop in LT-specific IgA, we did not measure a difference in total IgA or IgA-bound bacteria in the small intestine. We have hypothesized that EED is primarily affecting T cell dependent IgA+ B cell differentiation and that the level of T independent IgA remains intact. A more in depth study of LT-specific B cells and potentially T-B interaction in the Peyer’s Patches of EED mice will be required to answer these questions.
STAR methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Timothy Hand (timothy.hand@chp.edu)
Materials Availability Statement
The LT specific MHCII tetramer generated for this study will be available by contacting the Lead Contact.
The data generated by 16s sequencing for this study has been deposited to the BioProject database.
Data and code availability statement
16S rDNA amplicon sequencing data from stool samples and ileal contents from mice are publicly available through NCBI under the accession number PRJNA736663.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
3 week old wild type C57BL/6Tac mice (B6 MPF; Taconic) were used for EED establishment and characterization unless otherwise noted. Foxp3DTR-GFP (Foxp3tm3(DTR/GFP)Ayr) mice, were the kind gift of John Williams (UPMC Children’s Hospital of Pittsburgh) (Kim et al., 2007). Foxp3eGFP-cre-ERT2 (Foxp3tm9(EGFP/cre/ERT2)Ayr) mice were the kind gift of Dario Vignali (University of Pittsburgh) and bred with Rorcfl/fl mice (B6(Cg)-Rorctm3Litt) from Jackson Laboratories (Choi et al., 2016; Rubtsov et al., 2010). Both male and female age-matched mice (3 weeks) were used for all experiments. Littermates of the same sex were randomly assigned to experimental groups. All experiments were performed in an American Association for the Accreditation of Laboratory Animal Care-accredited animal facility at the University of Pittsburgh. Mice were weaned onto special diets and kept in specific pathogen-free conditions and housed in accordance with the procedures outlined in the Guide for the Care and Use of Laboratory Animals under an animal study proposal approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. For 16s sequencing experiments, all animals were singly housed to prevent cage effects of the microbiota.
Cell culture and Microbial strains
Drosophila s2 cells were used for tetramer production and were cultured in Schneider's Medium at 28°C before transfection. CUMT8, an autochthonous mouse adherent invasive E. coli (Dogan et al., 2014), was provided by Kenneth Simpson (Cornell University, Ithaca, NY; transferred to Univ. of Pittsburgh by MTA). In some experiments, a mouse commensal E. coli (ECMB) was used in place of CUMT8 (Molloy et al., 2013) to study the effect of adherence as ECMB lacks a flagellar operon and is non-motile. E coli 2A (Viladomiu et al., 2021; transferred to Univ. of Pittsburgh by MTA), an AIEC isolated from a patient with Crohn’s disease was provided by from Randy Longman (NYU School of Medicine, New York,NY). ETEC jf876, a Kanamycin resistant strain derived from ETEC H10407 (Roy et al., 2011) was obtained from James Fleckenstein (Washington University School of Medicine in St. Louis). All bacteria were grown in Luria Broth in 125 ml conical flasks in a 37°C shaker incubator.
METHOD DETAILS
EED Induction
Immediately following arrival, mice are placed on one of 2 special diets (Research Diets); a malnourished low protein/low fat diet (7% protein/7% fat; D14071001)), or a control isocaloric diet (20% protein/15% fat; D09051102) (Brown et al., 2015). Chow was added bi-weekly to ensure that malnourished mice did not compensate for protein/fat deficiencies in their diet by increasing intake and all mice were provided the same amount of calories each week. Some groups of mice (MT8 and EED) were orally gavaged with 1x109 CUMT8 E. coli on experimental days 16, 18 and 20. Mice were weighed biweekly. After 28 days on diets, mice were sacrificed and evaluated for signs of EED. Alternatively, mice were then treated or immunized at experimental day 28.
Histological analysis of terminal ileums
Terminal ileum samples were fixed in formalin, dehydrated and paraffin embedded. Sections of ileum were stained with hematoxylin and eosin stains for morphological analysis and slides were scored using the modified Marsh-Oberhuber classification (Dickson et al., 2006). Scoring was carried out by a student who was blinded to the experimental groups Outline of scoring system: 0=Pre - infiltrative, 1=Infiltrative, 2=Infiltrative - hyperplastic, 3a=Flat destructive with mild villous atrophy, 3b= Flat destructive with moderate villous atrophy, 3c= Flat destructive with total villous atrophy and 4=Atrophic - hypoplastic.
FITC–dextran assay
For evaluation of gut permeability, mice were orally gavaged with 4 kDa FITC–dextran (Sigma-Aldrich) after fasting for 4 hrs. FITC–dextran (100 mg/ml) was dissolved in PBS and gavaged at 44mg/100g body weight. Four hours post gavage, mice were euthanized and blood was collected immediately via cardiac puncture. Serum was isolated and was diluted with an equal volume of PBS, of which 100μl which was added to a 96-well microplate in duplicate. FITC in blood sera was quantified using florescence spectrometer. The plate was read at an excitation of 485 nm (20 nm band width) and an emission wavelength of 528 nm, and serially diluted FITC-dextran was used to generate a standard curve to calculate serum concentrations. A naïve mouse gavaged with PBS for the same duration was used to determine background fluorescence.
LT-specific tetramer Production
MHC class II (I-Ab) LTA166-176 tetramers (RYYRNLNIAPA) were produced in our laboratory as previously described (Moon et al., 2007). Peptide:I-Ab molecules were expressed in Drosophila S2 cells using the Drosophila Expression System kit (Invitrogen). Cells were co-transfected via calcium phosphate with plasmids encoding the I-Ab alpha chain, the I-Ab beta chain, a birA gene, and a blasticidin resistance gene at a molar ratio of 9:9:9:1. Transfected cells were selected in blasticidin-containing media for two weeks at 28° C, passaged into serum-free media, and scaled up to one liter cultures in three liter spinner flasks. When cell densities exceeded 107 /ml, expression was induced by the addition of 0.8 mM copper sulfate. Peptide:I-Ab heterodimers were purified from supernatants 8 days later using” a His-Bind Purification Kit (Millipore Sigma) followed by a Monomeric Avidin Column (Thermo Scientific). The peptide:I-Ab ‘monomer’ was then separated from the free biotin used to elute from the latter column by multiple washes with 1X PBS over a 30 kDa molecular weight cut-off centrifugal filter unit (Millipore Sigma). Monomer concentration was determined by absorbance at 280nm via a Nanodrop 2000 (Thermo Scientific, extinction coefficient = 0.066 cm-1 uM-1). Monomers created this way contain an 11 amino acid sequence encoding the antigenic peptides LTA166-176 (including the immunogenic nonamer LTA168-176 plus two N terminal amino acids) was fused to the N-terminus of the beta chain via a flexible polyglycine linker (Kozono et al., 1994). The streptavidin binding efficacy of each monomer is determined by comparing monomer alone and monomer and streptavidin mixed in a molar ratio of 4 monomer: 1 PE-conjugated streptavidin run on a native polyacrylamide gel (Bio-Rad) stained with Coomassie Blue R-250. Tetramers were made by mixing a molar ratio of 4.5 monomer: 1 APC-conjugated streptavidin (Prozyme).
Oral Vaccination and isolation of vaccine specific cells
Double mutant E. coli heat labile toxin (R192G/L211A) (dmLT), was produced from E. coli clones expressing recombinant protein as previously described (Norton et al., 2011). Mice were immunized twice, 7 days apart by oral gavage with 20μg of dmLT on completion of the EED protocol and vaccine responses were assayed 2 weeks after primary gavage as described before (Hall et al., 2011a). In some experiments mice were treated for with antibiotics for 3 weeks prior to vaccination. Staining of tetramer positive T cells was carried out after magnetic isolation of the cells as described (Moon et al., 2009). Briefly, at the completion of the vaccination regime, the small intestines of differently treated mice were harvested, cleaned ,flayed open and digested to a single cell suspension. Cells were then incubated with the APC-LT166 tetramer at room temperature for an hour to allow binding of dmLT-specific cells, and then subjected to MACS enrichment using the EasySep™ APC Positive Selection Kit II. Cells were stained as fixed as described in the flow cytometry section.
In vivo ETEC challenge
Mice were challenged with ETEC H10407 KanRes (ETEC jf876) as shown previously (Roy et al., 2011), with a few modifications. All mice were given streptomycin (5g/liter) in their drinking water 48 hours prior to ETEC challenge to eradicate normal resident bacterial flora in the intestinal tract (Wadolkowski et al., 1988). 12 hours prior to inoculation, food was withdrawn and antibiotic-treated water was replaced with regular water. 20 minutes prior to ETEC challenge, mice were gavaged with bicarbonate solution to reduce the effect of stomach acidity on the bacteria. Each mouse was orally inoculated with a suspension of 1x104 ETEC in a final volume of 200μl via a gavage needle. For isolating mucosa-associated ETEC, a 3cm segment of the terminal ileum, beginning from the ileocecal junction and extending proximally, was removed, opened longitudinally and placed in 2mL of sterile PBS with 0.05% Tween 20. The sample was vortexed for 5 seconds, incubated for 10 minutes at room temperature, and vortexed again for 5 seconds. 100μL of the resulting suspension was then plated in duplicate onto Kanamycin/LB agar plates and incubated at 37°C overnight prior to counting of colonies. CUMT8 E. coli (gavaged for EED induction prior to ETEC challenge) was susceptible to Kanamycin and did not confound the analysis of the ETEC colonization/protection assay.
In some experiments mice were depleted of CD4 T cells after immunization via IP injection of 200μg an anti-CD4 antibody (GK1.5 clone) at day 8 and day 11 of the prime/boost oral vaccination protocol.
Antibiotic treatment
The antibiotic cocktail used comprised of Metronidazole (1mg/mL), Ampicillin (1mg/mL), Neomycin (1mg/mL) and Vancomycin (0.5mg/mL) (MANV). Antibiotics were provided in the drinking water and were given to mice for 3 weeks after EED establishment (days 28 to 49 on diets). To reduce the bitter taste of metronidazole and prevent dehydration, antibiotic water was supplemented with 0.5 g/L of saccharin.
qRT-PCR analysis
Real-time RT-PCR (qPCR) was performed using SYBR Green supermix (Bio-RAD) on a CFX Connect Real-time PCR Detection Machine (Bio-RAD). Please see the Key resources table for primer sequences .For evaluation of bacterial abundance, DNA from stool and ileal contents were isolated using QIAamp Fast DNA Stool Mini Kit (Qiagen). CUMT8 E. coli was measured using primers that amplify the FliC gene and number of bacteria calculated against a standard curve prepared from known quantities of CUMT8 E. coli. ECMB E. coli was identified by PCR identification of a distinct sequence in the OmpL gene. For LCN2 expression, terminal ileums (3cm from ileocecal junction) from ISO and EED mice were homogenized in Trizol buffer (Life Technologies). RNA was transcribed into cDNA using iScript reagent (Bio-RAD) according to manufacturer’s instructions. LCN2 expression was normalized to Gapdh and expression level determined by the delta-delta Ct method. Reaction: 95°C for 3 minutes, 49 cycles at 95°C for 10 s (s) and 60°C for 30 s.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Anti-mouse CD45.2 (104), APC-eFluor 780 | Invitrogen | Cat#47-0454-82; RRID: AB_1272175; Clone: 104 |
| Rat Anti-Mouse CD90.2 (30-H12), Brilliant Violet 605 | BD Biosciences | Cat#740334; RRID: AB_2740067; Clone: 30-H12 |
| Rat Anti-mouse CD3 molecular complex (17A2), PerCP-Cyanine5.5 | BD Biosciences | Cat#560527; RRID: AB_1727463; Clone: 17A2 |
| Hamster Anti-mouse TCR beta (H57-597), APC-eFluor 780 | Invitrogen | Cat# 47-5961-82; RRID: AB_1272173; H57-597 |
| Rat Anti-Mouse CD4 (RM4-5), Brilliant Violet 786 | BD Biosciences | Cat#563727; RRID: AB_2728707; Clone: RM4-5 |
| Rat Anti-mouse CD8b (H35-17.2), PE-Cyanine7 | Invitrogen | Cat#25-0083-82; RRID: AB_11218494; Clone H35-17.2 |
| Rat Anti-mouse CD44 (IM7), FITC | BD Biosciences | Cat#553133; RRID: AB_2076224; Clone: IM7 |
| Rat Anti-mouse CD44 (IM7), PerCP-Cyanine5.5 | Invitrogen | Cat#45-0441-82; RRID: AB_925746; Clone: IM7 |
| Rat Anti-mouse FOXP3 (FJK-16s), Alexa Fluor 488 | Invitrogen | Cat#53-5773-82; RRID: AB_763537; Clone: FJK-16s |
| Rat Anti-mouse FOXP3 (FJK-16s), eFluor 450 | Invitrogen | Cat#48-5773-82; RRID: AB_1518812; Clone: FJK-16s |
| Rat Anti-mouse CD304 (Neuropilin-1) (3E12), PE-Cyanine7 | Biolegend | Cat#145212; RRID: AB_2562360; Clone: 3E12 |
| Rat Anti-mouse ROR gamma (t) (B2D), PE | Invitrogen | Cat#12-6981-82; RRID: AB_10807092; Clone: B2D |
| Rat Anti-mouse Ki67 (16A8), FITC | Biolegend | Cat#652410; RRID: AB_2562141; Clone: 16A8 |
| Rat Anti-mouse CD39 (Duha59), PE-Dazzle™ 594 | Biolegend | Cat#143812; RRID: AB_2750322; Clone: Duha59 |
| Rat Anti-mouse CD73 (TY/11.8), Alexa Fluor 700 | Biolegend | Cat#127230; RRID: AB_2800625; Clone: TY/11.8 |
| Rat Anti-mouse CD11b (M1/70), PE-Cyanine7 | Invitrogen | Cat#25-0112-82; RRID: AB_469588; Clone: M1/70 |
| Hamster Anti-mouse CD11c (HL3), PerCP-Cyanine5.5 | BD Biosciences | Cat# 560584; RRID: AB_1727422; Clone: HL3 |
| Rat Anti-mouse Ly-6C (HK1.4), eFluor 450 | Invitrogen | Cat#48-5932-82; RRID: AB_10805519; Clone: HK1.4 |
| Rat Anti-mouse I-A/I-E (M5/114.15.2), Brilliant Violet 786 | BD Biosciences | Cat#742894; RRID: AB_2734759; Clone: M5/114.15.2 |
| Mouse Anti-mouse Cd64 (FcγRI) (X54-5/7.1), Brilliant Violet 605 | Biolegend | Cat#139323; RRID: AB_2629778; Clone: X54-5/7.1 |
| Rat Anti-mouse CD170 (Siglec F) (1RNM44N), eFluor 660 | Invitrogen | Cat#50-1702-82; RRID: AB_2574187; Clone: 1RNM44N |
| Rat Anti-mouse Ly-6G (1A8), PE-CF594 | BD Biosciences | Cat#562700; RRID: AB_2737730; Clone: 1A8 |
| Rat Anti-mouse CD19 (1D3), PE-CF594 | BD Biosciences | Cat#562291; RRID: AB_11154223; Clone: 1D3 |
| Rat Anti-mouse TER-119 (TER-119), PE-eFluor 610 | Invitrogen | Cat#61-5921-80; RRID: AB_2574637; Clone: TER-119 |
| Rat Anti-mouse CD11b (M1/70), PE-CF594 | BD Biosciences | Cat#562317; RRID: AB_11154422; Clone: M1/70 |
| Hamster Anti-mouse CD11c (HL3), PE-CF594 | BD Biosciences | Cat#562454; RRID: AB_2737617; Clone: HL3 |
| InVivoMAb anti-mouse CD4 (GK1.5) | BioXCell | Cat#BE0003-1; RRID: AB_1107636; Clone: GK1.5 |
| Goat Anti-Mouse IgA-HRP | SouthernBiotech | Cat#1040-05; RRID: AB_2714213 |
| Rat Anti-mouse CD16/32 (93) | Invitrogen | Cat#14-0161-86; RRID: AB_467135; Clone: 93 |
| Bacterial and virus strains | ||
| CUMT8 bacteria | (Craven et al., 2012; Dogan et al., 2014) | N/A |
| ECMB bacteria | (Molloy et al., 2013) | N/A |
| E coli 2A | Viladomiu et al., 2021) | N/A |
| ETEC KanaRes(jf876) | (Roy et al., 2011) | N/A |
| Biological samples | ||
| Stool samples and ileal contents from EED mice | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Double mutant Labile Toxin (dmLT) from E. coli | Elizabeth Norton (Tulane) (Norton et al. 2011) | |
| Ionomycin | Millipore Sigma | Cat#I0634; CAS: 56092-82-1 |
| Phorbol 12-myristate 13-acetate | Millipore Sigma | Cat#P8139; CAS: 16561-29-8 |
| Brefeldin A | Invitrogen | Cat#00-514506-51 |
| Fluorescein isothiocyanate–dextran | Millipore Sigma | Cat#FD4; CAS: 60842-46-8 |
| LB Agar Plates, Kanamycin-50 | Teknova | Cat#L1025 |
| Metronidazole | Millipore Sigma | Cat#M1547; CAS: 443-48-1 |
| Ampicillin sodium salt | Millipore Sigma | Cat#A0166; CAS: 69-52-3 |
| Neomycin trisulfate salt hydrate | Millipore Sigma | Cat#N6386; CAS: 1405-10-3 |
| Vancomycin hydrochloride | Millipore Sigma | Cat#V1130; CAS: 1404-93-9 |
| Diphtheria Toxin | Millipore Sigma | Cat#D0564; MDL: MFCD00163490 |
| Tamoxifen | Millipore Sigma | Cat#T5648; CAS: 10540-29-1 |
| Corn oil | Millipore Sigma | CAS: 8001-30-7 |
| Fixable Live/Dead Aqua | Invitrogen | Cat#L34966 |
| Saccharin | Acros Organics | Cat#AC149005000 |
| Monomeric Avidin UltraLink− Resin | Thermo Scientific | Cat#53146 |
| Streptavidin-APC | Prozyme/Agilent | Cat#PJ27S-1 |
| Streptavidin-PE | Prozyme/Agilent | Cat#PJRS25-1 |
| Blasticidin | Invivogen | Cat#ant-bl-1 |
| Critical commercial assays | ||
| FoxP3/Transcription Factor Staining Buffer Set | Invitrogen | Cat#:00-5523-00 |
| Intracellular Fixation and and Permeabilization Buffer Set | Thermo Fisher Scientific | Cat#: 88-8824-00 |
| EasySep− APC Positive Selection Kit II | Stemcell Technologies | Cat#17681 |
| QIAamp Fast DNA Stool Mini Kit | Qiagen | Cat#51604 |
| MILLIPLEX MAP Mouse TH17 Magnetic Bead Panel - Immunology Multiplex Assay | Millipore Sigma | Cat#MTH17MAG-47K |
| SsoAdvanced Universal SYBR® Green Supermix | Bio-Rad | Cat#1725270 |
| Calcium Phosphate Transfection Kit | Invitrogen | Cat# K278001 |
| His-Tag Purification Kit | Millipore Sigma | Cat#70239-3 |
| Ultra-15 30kDa MWCO Centrifugal Filter Units | Millipore Sigma | Cat#UFC903024 |
| Deposited data | ||
| 16S rDNA amplicon sequencing of Stool samples and ileal contents from EED and control mice | This manuscript | PRJNA736663 |
| Experimental models: Cell lines | ||
| Drosophila S2 Cells in Schneider's Medium | Gibco | Cat#R69007 |
| Experimental models: Organisms/strains | ||
| Specific Pathogen Free C57BL/6NTac mice | Taconic Farms | MGI:5658006 |
| Foxp3DTR mice | Jackson Labs (Kim et al., 2007) | JAX stock #016958 |
| Foxp3eGFP-Cre-ERT2 mice | Jackson Labs Rubtsov et al., 2010). | JAX stock #016961 |
| Rorγtfl mice | Jackson Labs (Choi et al., 2016; . | JAX stock #008771 |
| Oligonucleotides | ||
| 16S primer Fwd 5'-ACTCCTACGGGAGGCAGCAGT-3' | IDT | N/A |
| 16S primer Rev 5'-ATTACCGCGGCTGCTGGC-3'. | IDT | N/A |
| ECMB OmpL Fwd 5’-TTCGGGTGGTGCAGTGTATC-3’ | IDT | N/A |
| ECMB OmpL Rev 5’-TGCTGCTATTAAAGTCATTTACGC-3’ | IDT | N/A |
| CUMT8 FliC Fwd 5’-CGACAGTGCCGGTATTTGTA-3’ | IDT | N/A |
| CUMT8 FliC Rev 5’-TGAGTACTGCGGATGGTTCA-3’ | IDT | N/A |
| LCN2 | Qiagen | PPMO3770A |
| Gapdh | Qiagen | PPM41731A |
| Recombinant DNA | ||
| Mouse I-Ab alpha chain on pRMHa-3 backbone | Jenkins lab, Moon et al | N/A |
| Mouse I-Ab beta chain with LT166-176 peptide DNA sequence on pRMHa-3 backbone | Jenkins lab, Moon et al | N/A |
| Software and algorithms | ||
| FlowJo X | BD Biosciences | N/A |
| FACSDiva | BD Biosciences | N/A |
| Prism 9 | GraphPad | 9.02 |
| R Studio | R Studio | N/A |
| Phyloseq | https://joey711.github.io/phyloseq/ | N/A |
| QIIME 2 2020.2 | (Bolyen et al., 2019). | N/A |
| DADA2 | (Callahan et al., 2016) | N/A |
| mafft | (Katoh et al., 2002) | N/A |
| fasttree2 | (Price et al., 2010) | N/A |
| Greengenes 13_8 99% OTUs reference sequences | (McDonald et al., 2012) | N/A |
| MicrobeR | J Bisanz, 2018 https://www.biorxiv.org/content/10.1101/304840v1 | N/A |
| LEfSe | https://huttenhower.sph.harvard.edu/galaxy/root | N/A |
| Other | ||
| Low protein/low fat diet (7% protein/7% fat) | Research Diets Brown et al., 2015 | D14071001 |
| Isocaloric diet (20% protein/15% fat) | Research Diets Brown et al., 2015 | D09051102 |
16S rRNA amplicon analysis of relative bacterial abundance
DNA was isolated using the QIAamp Fast DNA Stool Mini Kit (Qiagen) and the extracted DNA was stored at −20 °C prior to 16S amplicon PCR and sequencing. Analysis of small subunit ribosomal RNA gene (16S rRNA) expression was done using following primers; 16S primer Fwd: 5'-ACTCCTACGGGAGGCAGCAGT-3', Rev: 5'-ATTACCGCGGCTGCTGGC-3'. Sequencing was carried out by BGI Genomics. The dataset analyzed is available through NCBI under the accession number PRJNA736663. Microbiome informatics were performed using QIIME2 2020.2 (Bolyen et al., 2019). Raw sequences were quality-filtered and denoised with DADA2 (Callahan et al., 2016). Amplicon variant sequences (ASVs) were aligned with MAFFT (Katoh et al., 2002) and used to construct a phylogeny with FastTree2(Price et al., 2010). Alpha diversity metrics (observed OTUs), beta diversity metrics (Bray Curtis dissimilarity) and Principle Coordinate Analysis (PCoA) were estimated after samples were rarefied to 63,000 (subsampled without replacement) sequences per samples. Clustering of fecal and ileal microbiota samples was performed by PCoA based on bacterial community similarity (using the Bray Curtis/Jaccard algorithm). Taxonomy was assigned to ASVs using naive Bayes taxonomy classifier against the Greengenes 18_8 99% OTUs reference sequences (McDonald et al., 2012). All plots were made with publicly available R packages. The linear discriminant analysis (LDA) effect size (LEfSe) method was use to analyze the intergroup differences at phylum, class, order, family and genus level in each group were analyzed using the default settings on the website (https://huttenhower.sph.harvard.edu/galaxy/root). 2-tailed nonparametric Kruskal-Wallis test (determine significance of differences in OTUs between EED v ALL) and unpaired Wilcoxon test (pair-wise test among the 2 groups) are employed by LEfSe followed by LDA to determine effect size of each differentially abundant OTU (Segata et al., 2011). Results are expressed at mean+/−SEM, where significant differences had a p value <0.05 and LDA score (log10)>2.4
Flow cytometry
All antibodies used for flow cytometry were purchased from either ThermoFisher, BD Biosciences, or BioLegend and are listed in the Key resources table. Dead cells were discriminated in all experiments using LIVE/DEAD fixable dead stain (ThermoFisher). All stains were carried out in media containing anti-CD16/32 blocking antibody (clone 93, ThermoFisher). For intracellular staining, cells were fixed and permeabilized using the FoxP3/Transcription factor staining buffer set according to the manufacturer’s directions (ThermoFisher). Intracellular staining for IL-10 production from Treg cells was carried out on siLP cell suspensions treated with PMA/ionomycin/brefeldin A for four hours and fixed using Fix/Perm (BD Biosciences). All flow cytometry was acquired on an LSRFortessa FACS analyzer(BD Biosciences) and cell sorting was carried out on a FACS Aria (BD Biosciences). Cells were isolated from the small intestine, Peyer’s Patches, spleen and lymph nodes for flow cytometry as described previously (Hand et al., 2012)
Suppression assay
Small intestinal Treg cells (CD45+CD90+CD4+GFP+ T cells) from ISO and EED Foxp3DTR-GFP mice were sorted using the BD FACSAria sorter for comparing suppressive efficacy along with splenic non-Treg cells(CD45+CD90+CD4+GFP− T cells) cells and splenic antigen presenting cells (APCs) (CD45+CD90−) from a naïve mouse. Splenic Treg cells (CD45+CD90+CD4+GFP+T cells) were also sorted as a positive control for suppression. The responder cells (splenic non-Treg cells) from naïve Foxp3DTR-GFP mice were labeled with 5μM CellTrace Violet (Life Technology). Responder cells (4×103), APCs (8×103), and different concentrations of Treg cells (1:2-1:64 Treg: Teff ratio, 500-2000 Treg cells) were activated with 2μg/ml anti-CD3 (Biolegend) in a 96-well round bottom plate with 100μL RPMI for 72hrs (Turnis et al., 2016). Suppression was calculated as previously described (McMurchy and Levings, 2012). Briefly, cells were acquired by the BD LSR Fortessa™, and the division index of responder cells was analyzed on the division of CellTrace Violet. Suppression was then calculated with the formula % Suppression = (1-DITreg/DIctrl) × 100% (DITreg stands for the division index of responder cells with Treg cells, and DICtrl stands for the division index of responder cells activated without Treg cells).
Anti-dmLT ELISA
For detection of dmLT-specific IgA antibody from intestinal lavage, 96-well-plates were coated with 1 μg/ml of dmLT protein overnight at 4°C, blocked with 1% BSA for 1 hour followed by incubation with intestinal lavage (cleared of solid particles/bacteria by centrifugation) overnight at 4°C. Next day, after washing, IgA antibody bound to dmLT was detected by HRP-labeled anti-IgA antibody and developed using standard ELISA substrate. A naïve mouse was used as a negative control.
Luminex assays
Cytokines from ileal homogenates were measured by Luminex according to the manufacturer’s instructions using the MILLIPLEX Mouse Th17 Magnetic Bead Panel (Millipore Sigma),
ALDH activity assay
ALDH activity was analyzed using the Aldefluor kit (STEMCELL™ Technologies) according to the manufacturer's protocol. Briefly, 1×106 cells/ml cells were incubated in 15 mL tubes with 5μl/ml Aldefluor reagent for 30 min in a 37°C water bath with or without the DEAB inhibitor (as a control to determine background). All subsequent washes and staining was done in the assay buffer provided in the kit to prevent Aldefluor loss. Flow cytometric analysis was performed to determine the percent of DCs (Live CD45+ CD11c+ MHCII+) that were Aldefluor+.
Diphtheria toxin (DT) and Tamoxifen (TX) administration
Mice were intraperitoneally (I.P.) injected with 25μg/kg diphtheria toxin (DT, Sigma) twice, with 3 days between injections (PBS used as vehicle). Tamoxifen (TX, Sigma-Aldrich), was orally gavaged at 5mg/mouse/day, first for 5 consecutive days to activate the promoter and then once every 3 days for the duration of the deletion. Since TX is poorly soluble in water, the amount needed for a single day was dissolved in EtOH with heating to 37°C and then diluted in corn oil (Sigma) such that 100ul had 5mg of TX. Corn oil was used for vehicle controls.
QUANTIFICATION AND STATISTICAL ANALYSIS
Due to the necessities of diets and microbiota modification, mouse studies were performed in a non-blinded manner. Data are presented as mean ± SEM. Statistical significance was determined using unpaired Student’s t test when comparing 2 groups, and one-way ANOVA with multiple comparisons, when comparing multiple groups. The Treg suppression assay was analyzed with a 2-way ANOVA. Significance was defined as *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001. All statistical analysis was calculated using Prism software (GraphPad). For details on significance, please see figure legends.
Supplementary Material
Highlights.
Mouse model mimics human EED, including stunting and intestinal inflammation.
EED causes an intestine-specific failure in oral vaccine-specific T cell responses.
EED induces intestinal microbiota-dependent RORγT+FOXP3+ Treg T cells.
Depletion of Treg cells restores oral vaccine responses but increases stunting.
Acknowledgments
This work was supported by the National Institutes of Health (R21AI142051; TWH, NIAID division of Intramural Research; YB, FAPESP-JP 2015/25364-0; DMF), the R.K. Mellon Institute for Pediatric Research (TWH), and the UPMC Children’s Hospital of Pittsburgh RAC award (AB). AEO is supported by a Damon Runyon Cancer Fellowship and AHPB is supported by a NIH T32 (T32AI089443). The authors would like to thank J. Michel and A. Styche for assistance with cell sorting, the UPMC Children’s Hospital of Pittsburgh Histology core for the preparation of tissue slides, and the University of Pittsburgh Division of Laboratory Animal Research. We would also like to thank D. Vignali (Foxp3eGFP-Cre-ERT2), K. Simpson (CUMT8 E. coli), R. Longman (2A E. coli), J. Fleckenstein (ETEC H10407) and J. Williams (Foxp3DTR-GFP) for sharing critical reagents and M. Pepper and M. Jenkins for assistance with making and using MHC class II tetramer reagents. We would like to thank the University of Pittsburgh Center for Research Computing for resources and assistance with microbiome sequence analysis. We would like to thank the members of the Hand, Belkaid, Poholek and Canna labs for discussion and critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References:
- Abdel-Gadir A, Stephen-Victor E, Gerber GK, Noval Rivas M, Wang S, Harb H, Wang L, Li N, Crestani E, Spielman S, et al. (2019). Microbiota therapy acts via a regulatory T cell MyD88/RORgammat pathway to suppress food allergy. Nat Med 25, 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansaldo E, Slayden LC, Ching KL, Koch MA, Wolf NK, Plichta DR, Brown EM, Graham DB, Xavier RJ, Moon JJ, et al. (2019). Akkermansia muciniphila induces intestinal adaptive immune responses during homeostasis. Science 364, 1179–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnich N, and Darfeuille-Michaud A (2007). Adherent-invasive Escherichia coli and Crohn's disease. Curr Opin Gastroenterol 23, 16–20. [DOI] [PubMed] [Google Scholar]
- Bemark M, Hazanov H, Stromberg A, Komban R, Holmqvist J, Koster S, Mattsson J, Sikora P, Mehr R, and Lycke NY (2016). Limited clonal relatedness between gut IgA plasma cells and memory B cells after oral immunization. Nat Commun 7, 12698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, and Hand TW (2018). Role of nutrition, infection, and the microbiota in the efficacy of oral vaccines. Clin Sci (Lond) 132, 1169–1177. [DOI] [PubMed] [Google Scholar]
- Blanton LV, Barratt MJ, Charbonneau MR, Ahmed T, and Gordon JI (2016). Childhood undernutrition, the gut microbiota, and microbiota-directed therapeutics. Science 352, 1533. [DOI] [PubMed] [Google Scholar]
- Boehm F, Martin M, Kesselring R, Schiechl G, Geissler EK, Schlitt HJ, and Fichtner-Feigl S (2012). Deletion of Foxp3+ regulatory T cells in genetically targeted mice supports development of intestinal inflammation. BMC Gastroenterol 12, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EM, Wlodarska M, Willing BP, Vonaesch P, Han J, Reynolds LA, Arrieta MC, Uhrig M, Scholz R, Partida O, et al. (2015). Diet and specific microbial exposure trigger features of environmental enteropathy in a novel murine model. Nat Commun 6, 7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffie CG, and Pamer EG (2013). Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol 13, 790–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr AHP, Bhattacharjee A, and Hand TW (2020). Nutritional Modulation of the Microbiome and Immune Response. J Immunol 205, 1479–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DI, Elia M, and Lunn PG (2003). Growth faltering in rural Gambian infants is associated with impaired small intestinal barrier function, leading to endotoxemia and systemic inflammation. J Nutr 133, 1332–1338. [DOI] [PubMed] [Google Scholar]
- Cao AT, Yao S, Gong B, Elson CO, and Cong Y (2012). Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. J Immunol 189, 4666–4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody RN, Gerber GK, Luevano JM Jr., Gatti DM, Somes L, Svenson KL, and Turnbaugh PJ (2015). Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17, 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai JN, Peng Y, Rengarajan S, Solomon BD, Ai TL, Shen Z, Perry JSA, Knoop KA, Tanoue T, Narushima S, et al. (2017). Helicobacter species are potent drivers of colonic T cell responses in homeostasis and inflammation. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RY, Kung VL, Das S, Hossain MS, Hibberd MC, Guruge J, Mahfuz M, Begum S, Rahman MM, Fahim SM, et al. (2020). Duodenal Microbiota in Stunted Undernourished Children with Enteropathy. N Engl J Med 383, 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaranunt P, Tometich JT, Ji J, and Hand TW (2018). T Cell Proliferation and Colitis Are Initiated by Defined Intestinal Microbes. J Immunol 201, 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, and Huh JR (2016). The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351, 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, and Norton EB (2018). The Mucosal Vaccine Adjuvant LT(R192G/L211A) or dmLT. mSphere 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y, Feng T, Fujihashi K, Schoeb TR, and Elson CO (2009). A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci U S A 106, 19256–19261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, Almeida M, Quinquis B, Levenez F, Galleron N, et al. (2013). Dietary intervention impact on gut microbial gene richness. Nature 500, 585–588. [DOI] [PubMed] [Google Scholar]
- Craven M, Egan CE, Dowd SE, McDonough SP, Dogan B, Denkers EY, Bowman D, Scherl EJ, and Simpson KW (2012). Inflammation drives dysbiosis and bacterial invasion in murine models of ileal Crohn's disease. PloS one 7, e41594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, et al. (2007). Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 204, 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Luccia B, Ahern PP, Griffin NW, Cheng J, Guruge JL, Byrne AE, Rodionov DA, Leyn SA, Osterman AL, Ahmed T, et al. (2020). Combined Prebiotic and Microbial Intervention Improves Oral Cholera Vaccination Responses in a Mouse Model of Childhood Undernutrition. Cell Host Microbe 27, 899–908 e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BC, Streutker CJ, and Chetty R (2006). Coeliac disease: an update for pathologists. J Clin Pathol 59, 1008–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan B, Suzuki H, Herlekar D, Sartor RB, Campbell BJ, Roberts CL, Stewart K, Scherl EJ, Araz Y, Bitar PP, et al. (2014). Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm Bowel Dis 20, 1919–1932. [DOI] [PubMed] [Google Scholar]
- Fonseca DM, Hand TW, Han SJ, Gerner MY, Glatman Zaretsky A, Byrd AL, Harrison OJ, Ortiz AM, Quinones M, Trinchieri G, et al. (2015). Microbiota-Dependent Sequelae of Acute Infection Compromise Tissue-Specific Immunity. Cell 163, 354–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox JG, Dewhirst FE, Tully JG, Paster BJ, Yan L, Taylor NS, Collins MJ Jr., Gorelick PL, and Ward JM (1994). Helicobacter hepaticus sp. nov., a microaerophilic bacterium isolated from livers and intestinal mucosal scrapings from mice. J Clin Microbiol 32, 1238–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerson CD, Kent TH, Saha JR, Siddiqi N, and Lindenbaum J (1971). Recovery of small-intestinal structure and function after residence in the tropics. II. Studies in Indians and Pakistanis living in New York City. Ann Intern Med 75, 41–48. [DOI] [PubMed] [Google Scholar]
- Grassly NC, Jafari H, Bahl S, Durrani S, Wenger J, Sutter RW, and Aylward RB (2009). Mucosal immunity after vaccination with monovalent and trivalent oral poliovirus vaccine in India. J Infect Dis 200, 794–801. [DOI] [PubMed] [Google Scholar]
- Gribonika I, Eliasson DG, Chandode RK, Schon K, Stromberg A, Bemark M, and Lycke NY (2019). Class-switch recombination to IgA in the Peyer's patches requires natural thymus-derived Tregs and appears to be antigen independent. Mucosal Immunol 12, 1268–1279. [DOI] [PubMed] [Google Scholar]
- Haberman Y, Iqbal NT, Ghandikota S, Mallawaarachchi I, Tzipi B, Dexheimer PJ, Rahman N, Hadar R, Sadiq K, Ahmad Z, et al. (2021). Mucosal Genomics Implicate Lymphocyte Activation and Lipid Metabolism in Refractory Environmental Enteric Dysfunction. Gastroenterology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, Wohlfert EA, Chou DB, Oldenhove G, Robinson M, et al. (2011a). Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity 34, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Grainger JR, Spencer SP, and Belkaid Y (2011b). The role of retinoic acid in tolerance and immunity. Immunity 35, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand TW (2016). The Role of the Microbiota in Shaping Infectious Immunity. Trends Immunol 37, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand TW, Dos Santos LM, Bouladoux N, Molloy MJ, Pagan AJ, Pepper M, Maynard CL, Elson CO 3rd, and Belkaid Y (2012). Acute gastrointestinal infection induces long-lived microbiota-specific T cell responses. Science 337, 1553–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hand TW, Vujkovic-Cvijin I, Ridaura VK, and Belkaid Y (2016). Linking the Microbiota, Chronic Disease, and the Immune System. Trends Endocrinol Metab 27, 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harro C, Louis Bourgeois A, Sack D, Walker R, DeNearing B, Brubaker J, Maier N, Fix A, Dally L, Chakraborty S, et al. (2019). Live attenuated enterotoxigenic Escherichia coli (ETEC) vaccine with dmLT adjuvant protects human volunteers against virulent experimental ETEC challenge. Vaccine 37, 1978–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. (2012). Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, and Littman DR (2016). The microbiota in adaptive immune homeostasis and disease. Nature 535, 75–84. [DOI] [PubMed] [Google Scholar]
- Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, Umetsu DT, and Rudensky AY (2012). Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, and Miyata T (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30, 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kau AL, Planer JD, Liu J, Rao S, Yatsunenko T, Trehan I, Manary MJ, Liu TC, Stappenbeck TS, Maleta KM, et al. (2015). Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy. Sci Transl Med 7, 276ra224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, Tsutsui Y, Qin H, Honda K, Okada T, et al. (2014). Foxp3(+) T cells regulate immunoglobulin a selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 41, 152–165. [DOI] [PubMed] [Google Scholar]
- Keusch GT, Denno DM, Black RE, Duggan C, Guerrant RL, Lavery JV, Nataro JP, Rosenberg IH, Ryan ET, Tarr PI, et al. (2014). Environmental enteric dysfunction: pathogenesis, diagnosis, and clinical consequences. Clin Infect Dis 59 Suppl 4, S207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, and Rudensky AY (2007). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 8, 191–197. [DOI] [PubMed] [Google Scholar]
- Kim KS, Hong SW, Han D, Yi J, Jung J, Yang BG, Lee JY, Lee M, and Surh CD (2016). Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 351, 858–863. [DOI] [PubMed] [Google Scholar]
- Kim M, Galan C, Hill AA, Wu WJ, Fehlner-Peach H, Song HW, Schady D, Bettini ML, Simpson KW, Longman RS, et al. (2018). Critical Role for the Microbiota in CX3CR1(+) Intestinal Mononuclear Phagocyte Regulation of Intestinal T Cell Responses. Immunity 49, 151–163 e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpe PS, and Petri WA Jr. (2012). Environmental enteropathy: critical implications of a poorly understood condition. Trends Mol Med 18, 328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozono H, White J, Clements J, Marrack P, and Kappler J (1994). Production of soluble MHC class II proteins with covalently bound single peptides. Nature 369, 151–154. [DOI] [PubMed] [Google Scholar]
- Leach S, Clements JD, Kaim J, and Lundgren A (2012). The adjuvant double mutant Escherichia coli heat labile toxin enhances IL-17A production in human T cells specific for bacterial vaccine antigens. PLoS One 7, e51718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung JM, Davenport M, Wolff MJ, Wiens KE, Abidi WM, Poles MA, Cho I, Ullman T, Mayer L, and Loke P (2014). IL-22-producing CD4+ cells are depleted in actively inflamed colitis tissue. Mucosal Immunol 7, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MM (2010). Immunogenicity and efficacy of oral vaccines in developing countries: lessons from a live cholera vaccine. BMC Biol 8, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbaum J, Harmon JW, and Gerson CD (1972). Subclinical malabsorption in developing countries. Am J Clin Nutr 25, 1056–1061. [DOI] [PubMed] [Google Scholar]
- Lochner M, Peduto L, Cherrier M, Sawa S, Langa F, Varona R, Riethmacher D, Si-Tahar M, Di Santo JP, and Eberl G (2008). In vivo equilibrium of proinflammatory IL-17+ and regulatory IL-10+ Foxp3+ RORgamma t+ T cells. J Exp Med 205, 1381–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lycke NY, and Bemark M (2017). The regulation of gut mucosal IgA B-cell responses: recent developments. Mucosal Immunol 10, 1361–1374. [DOI] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, and Hugenholtz P (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurchy AN, and Levings MK (2012). Suppression assays with human T regulatory cells: a technical guide. Eur J Immunol 42, 27–34. [DOI] [PubMed] [Google Scholar]
- Molloy MJ, Grainger JR, Bouladoux N, Hand TW, Koo LY, Naik S, Quinones M, Dzutsev AK, Gao JL, Trinchieri G, et al. (2013). Intraluminal containment of commensal outgrowth in the gut during infection-induced dysbiosis. Cell Host Microbe 14, 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Hataye J, Pagan AJ, Pepper M, McLachlan JB, Zell T, and Jenkins MK (2009). Tracking epitope-specific T cells. Nat Protoc 4, 565–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, and Jenkins MK (2007). Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 27, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SS, Wang Y, Shane AL, Nguyen T, Ray P, Dennehy P, Baek LJ, Parashar U, Glass RI, and Jiang B (2010). Inhibitory effect of breast milk on infectivity of live oral rotavirus vaccines. Pediatr Infect Dis J 29, 919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, and Kronenberg M (2009). Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol 10, 1178–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor C, Lu M, Haque R, Mondal D, Buonomo E, Nayak U, Mychaleckyj JC, Kirkpatrick B, Colgate R, Carmolli M, et al. (2015). Environmental Enteropathy, Oral Vaccine Failure and Growth Faltering in Infants in Bangladesh. EBioMedicine 2, 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton EB, Lawson LB, Freytag LC, and Clements JD (2011). Characterization of a mutant Escherichia coli heat-labile toxin, LT(R192G/L211A), as a safe and effective oral adjuvant. Clin Vaccine Immunol 18, 546–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, Gaboriau-Routhiau V, Marques R, Dulauroy S, Fedoseeva M, et al. (2015). MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORgammat(+) T cells. Science 349, 989–993. [DOI] [PubMed] [Google Scholar]
- Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, Tolaini M, Li Y, Kelly G, Chakravarty P, Shoaie S, et al. (2019). The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity 51, 77–89 e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasetti MF, Simon JK, Sztein MB, and Levine MM (2011). Immunology of gut mucosal vaccines. Immunol Rev 239, 125–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper M, Linehan JL, Pagan AJ, Zell T, Dileepan T, Cleary PP, and Jenkins MK (2009). Different routes of bacterial infection induce long-lived TH1 memory cells and short-lived TH17 cells. Nat Immunol 11, 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, and Medzhitov R (2004). Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241. [DOI] [PubMed] [Google Scholar]
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, et al. (2013). Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341, 1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, and Hylemon PB (2006). Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47, 241–259. [DOI] [PubMed] [Google Scholar]
- Rolhion N, and Darfeuille-Michaud A (2007). Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflamm Bowel Dis 13, 1277–1283. [DOI] [PubMed] [Google Scholar]
- Roy K, Hamilton DJ, Munson GP, and Fleckenstein JM (2011). Outer membrane vesicles induce immune responses to virulence proteins and protect against colonization by enterotoxigenic Escherichia coli. Clin Vaccine Immunol 18, 1803–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, and Rudensky AY (2010). Stability of the regulatory T cell lineage in vivo. Science 329, 1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, Lee JY, Ziel JW, Miraldi ER, Domingos AI, et al. (2015). An IL-23R/IL-22 Circuit Regulates Epithelial Serum Amyloid A to Promote Local Effector Th17 Responses. Cell 163, 381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos N, and Hoshino Y (2005). Global distribution of rotavirus serotypes/genotypes and its implication for the development and implementation of an effective rotavirus vaccine. Rev Med Virol 15, 29–56. [DOI] [PubMed] [Google Scholar]
- Schreiber F, Arasteh JM, and Lawley TD (2015). Pathogen Resistance Mediated by IL-22 Signaling at the Epithelial-Microbiota Interface. J Mol Biol 427, 3676–3682. [DOI] [PubMed] [Google Scholar]
- Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J, et al. (2015). MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science 349, 993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome Biol 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh H, Lynch J, Kim J, and Excler JL (2020). Current and future cholera vaccines. Vaccine 38 Suppl 1, A118–A126. [DOI] [PubMed] [Google Scholar]
- Singh B, Read S, Asseman C, Malmstrom V, Mottet C, Stephens LA, Stepankova R, Tlaskalova H, and Powrie F (2001). Control of intestinal inflammation by regulatory T cells. Immunol Rev 182, 190–200. [DOI] [PubMed] [Google Scholar]
- Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, Geva-Zatorsky N, Jupp R, Mathis D, Benoist C, et al. (2020). Microbial bile acid metabolites modulate gut RORgamma(+) regulatory T cell homeostasis. Nature 577, 410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, Hori S, and Fagarasan S (2009). Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer's patches. Science 323, 1488–1492. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, and Gordon JI (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031. [DOI] [PubMed] [Google Scholar]
- Turnis ME, Sawant DV, Szymczak-Workman AL, Andrews LP, Delgoffe GM, Yano H, Beres AJ, Vogel P, Workman CJ, and Vignali DA (2016). Interleukin-35 Limits Anti-Tumor Immunity. Immunity 44, 316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vela Ramirez JE, Sharpe LA, and Peppas NA (2017). Current state and challenges in developing oral vaccines. Adv Drug Deliv Rev 114, 116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viladomiu M, Metz ML, Lima SF, Jin WB, Chou L, Bank JRILC, Guo CJ, Diehl GE, Simpson KW, Scherl EJ, et al. (2021). Adherent-invasive E. coli metabolism of propanediol in Crohn's disease regulates phagocytes to drive intestinal inflammation. Cell Host Microbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonaesch P, Morien E, Andrianonimiadana L, Sanke H, Mbecko JR, Huus KE, Naharimanananirina T, Gondje BP, Nigatoloum SN, Vondo SS, et al. (2018). Stunted childhood growth is associated with decompartmentalization of the gastrointestinal tract and overgrowth of oropharyngeal taxa. Proc Natl Acad Sci U S A 115, E8489–E8498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadolkowski EA, Laux DC, and Cohen PS (1988). Colonization of the streptomycin-treated mouse large intestine by a human fecal Escherichia coli strain: role of growth in mucus. Infect Immun 56, 1030–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, and Petri WA Jr. (2016). Environmental Enteropathy: Elusive but Significant Subclinical Abnormalities in Developing Countries. EBioMedicine 10, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JM, Bilate AM, Gobert M, Ding Y, Curotto de Lafaille MA, Parkhurst CN, Xiong H, Dolpady J, Frey AB, Ruocco MG, et al. (2012). Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J Exp Med 209, 1723–1742, S1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ, Galan C, Belkaid Y, Bonneau R, and Littman DR (2018). c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Ramanan D, Rozenberg M, McGovern K, Rastelli D, Vijaykumar B, Yaghi O, Voisin T, Mosaheb M, Chiu I, et al. (2021). Interleukin-6 produced by enteric neurons regulates the number and phenotype of microbe-responsive regulatory T cells in the gut. Immunity 54, 499–513 e495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang BH, Hagemann S, Mamareli P, Lauer U, Hoffmann U, Beckstette M, Fohse L, Prinz I, Pezoldt J, Suerbaum S, et al. (2016). Foxp3(+) T cells expressing RORgammat represent a stable regulatory T-cell effector lineage with enhanced suppressive capacity during intestinal inflammation. Mucosal Immunol 9, 444–457. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Harbour SN, Kolde R, Latorre IJ, Tun HM, Schoeb TR, Turner H, Moon JJ, Khafipour E, Xavier RJ, et al. (2017). Selective Induction of Homeostatic Th17 Cells in the Murine Intestine by Cholera Toxin Interacting with the Microbiota. J Immunol 199, 312–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Chu C, Teng F, Bessman NJ, Goc J, Santosa EK, Putzel GG, Kabata H, Kelsen JR, Baldassano RN, et al. (2019). Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 568, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
16S rDNA amplicon sequencing data from stool samples and ileal contents from mice are publicly available through NCBI under the accession number PRJNA736663.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.