

Abstract
Thyroid-associated ophthalmopathy (TAO), the ocular manifestation of Graves’ disease, is a process in which orbital connective tissues and extraocular muscles undergo inflammation and remodeling. It seems to result from autoimmune responses to antigens shared by the thyroid and orbit. The thyrotropin receptor (TSHR), expressed at low levels in orbital tissues, is a leading candidate antigen. Recent evidence suggests that another protein, the insulin-like growth factor-I receptor (IGF-IR), is over-expressed in TAO, and antibodies against IGF-IR have been detected in patients with the disease. Further, TSHR and IGF-IR form a physical and functional complex, and signaling initiated at TSHR requires IGF-IR activity. Identification of therapy for this rare disease has proven challenging and currently relies on nonspecific and inadequate agents and thus represents an important unmet need. A recently completed therapeutic trial suggests that inhibiting IGF-IR activity with a monoclonal antibody may be an effective and safe treatment for active TAO.
Introduction:
Thyroid-associated ophthalmopathy (TAO), an orphan disease without effective therapy
Graves’ disease (GD) is an autoimmune syndrome affecting multiple, anatomically dispersed tissues (1)(Figure 1). Dominant among the phenotypic attributes associated with the disease is a typically over-active thyroid gland that frequently results in thyrotoxicosis. The majority of patients with GD develop hyperthyroidism sometime during the course of their disease. Among those developing GD, a smaller group also manifests clinically important inflammation and tissue remodeling of the orbit and upper face, an orphan disease process known as TAO (aka Graves’ orbitopathy or thyroid eye disease) (2). Patients with TAO are generally considered to represent those having more serious underlying autoimmunity. GD occurring in the thyroid gland and orbit appears to differ in notable respects. Among the most glaring are the frequently divergent clinical courses of hyperthyroidism and TAO. Hyperthyroidism in GD rarely remits permanently without definitive therapy (either surgical thyroidectomy or radioactive iodine ablation). In contrast, active TAO typically runs a course of 1–3 years in a pattern following Rundle’s curve (3). The disease then enters into the stable stage during which signs and symptoms, including tissue congestion, inflammation, and expanding orbital contents, cease progressing. TAO rarely becomes reactivated; these infrequent instances are usually associated with radioactive iodine treatment of hyperthyroidism or following remedial ocular surgery. Reactivated and particularly severe TAO are commonly found in cigarette smokers (4). Efforts to unambiguously identify the genetic, epigenetic, and environmental factors that might uniquely underpin TAO but not hyperthyroidism have thus far been unsuccessful (5). This suggests that causative factors of the two disease manifestations are very similar. Further, the naturally occurring determinants of susceptibility, disease initiation, progression, and resolution uniquely underpinning TAO have eluded definition. A major obstacle to better understanding TAO is the rarity of the disease. Another is the absence of suitable animal models possessing high-degree fidelity with the human disease has undoubtedly impeded progress toward more complete understanding of TAO (6). This, in turn, has resulted in the lack of specific, effective, and well-tolerated therapies that have been proven effective and safe in large, well-controlled prospective trials. Thus, no currently available therapies for active TAO have achieved registration from the US Food and Drug Administration.
Figure 1.
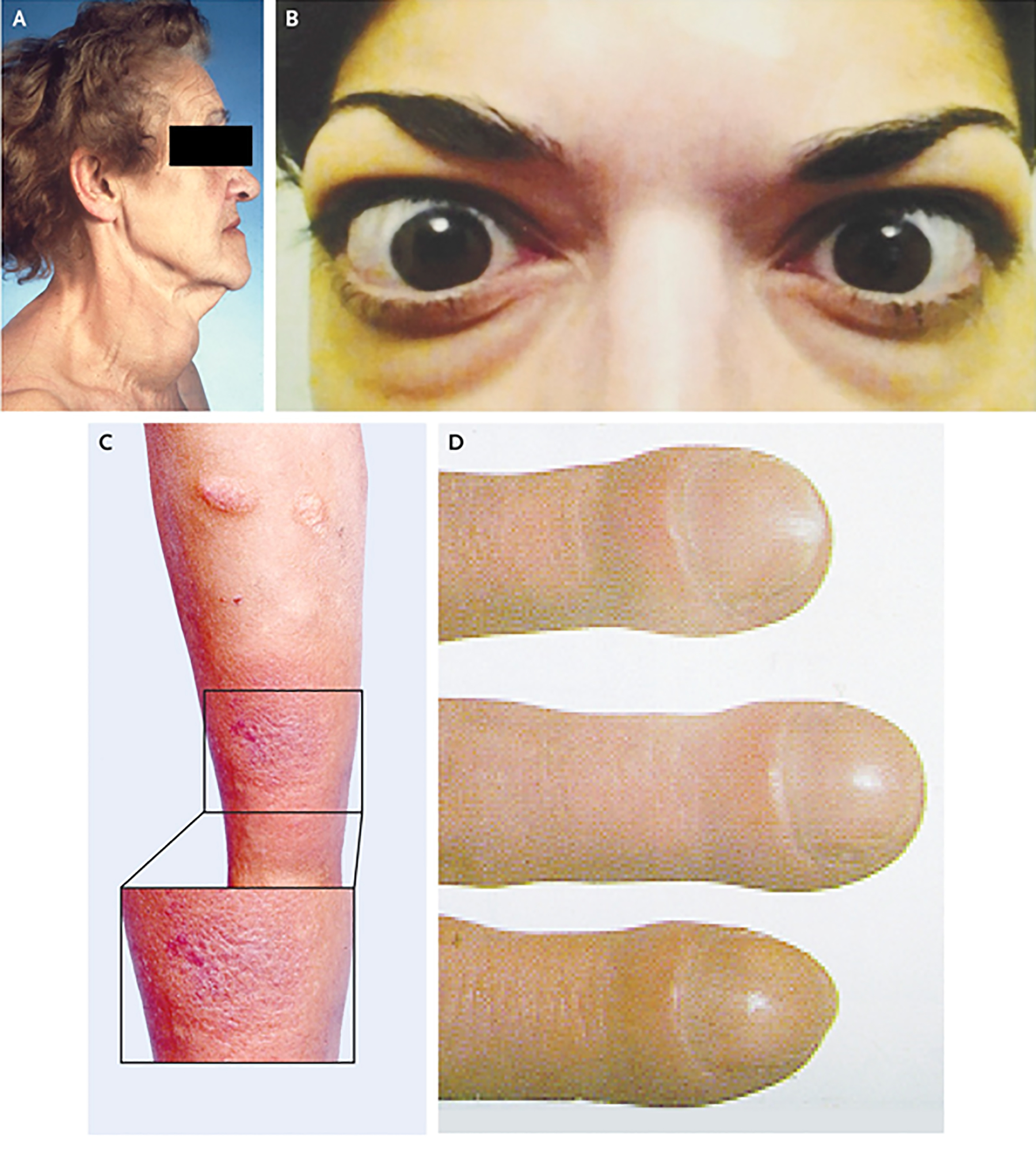
Clinical appearance of Graves’ disease. Panel A demonstrates goiter (thyroid enlargement) frequently developing in the disease. Panel B shows moderate to severe thyroid-associated ophthalmopathy, including bilateral proptosis, lid retraction and periorbital edema. Panel C contains the image of plaque pretibial myxedema. Panel D demosntrates thyroid acropachy. From N. Engl. J. Med, Smith T.J. and Hegedus L., Graves’ Disease, 375; 1552–1565. Copyright © (2016) Massachusetts Medical Society. Reprinted with permission.
Clinical course of TAO
Navigating the mechanistic rationale for developing newer, more targeted treatment of TAO requires familiarity with the disease’s natural course and typical outcomes. The time-frame for evolution and resolution of TAO activity was described in detail more than 70 years ago (3). The disease frequently develops in individuals who have already manifested hyperthyroidism or who will be diagnosed with thyroid dysfunction within months to several years (1). TAO characteristically presents with subtle signs and symptoms, including dry eye, lid retraction, and modest ocular prominence (2). Onset of any of these manifestations, either alone or in combination, whether relatively mild or progressively more aggressive, can herald the onset of active TAO. Disease activity typically continues for 1–3 years but may persist for considerably longer. Changes in the physical signs and symptoms eventually lessen as the disease transitions to the stable disease. The effectiveness of essentially all medical treatments currently available (largely glucocorticoids alone or combined with external beam radiotherapy and B cell depletion) are limited to active disease. Notable exceptions include those locally administered agents that ameliorate ocular surface disease caused by eye exposure (7). These topical drugs can prove beneficial throughout the disease course and in some cases are sight-preserving. Once disease activity ceases, surgical remediation becomes the most effective option. Procedures are usually staged to optimize outcomes.
Pathogenesis of TAO
Details concerning the mechanisms underlying TAO continue to emerge and are depicted in Figure 2. Loss of immunologic tolerance to the thyrotropin receptor (TSHR) appears to underlie the development of GD (8, 9). It remains uncertain how autoreactive T cells and autoantibodies targeting TSHR are involved in the pathogenesis of TAO. The factor(s) overarching involvement of the thyroid gland and orbit in GD remains uncertain. Among the leading candidates are thyroid autoantigens that are also expressed in orbital tissues, albeit at considerably lower levels than those in the thyroid gland. These include TSHR (10) and thyroglobulin (Tg) (11). Anti-TSHR antibodies can be detected in the majority of patients with GD and at especially high levels in those with TAO. Anti-TSHR antibodies that activate the receptor, known as thyroid-stimulating immunoglobulins (TSI), are both specific for GD and are pathogenic (12). TSIs activate cytokine production in orbital fibroblasts (13). It appears that levels of TSI generally correlate with the severity and activity of TAO (14, 15). Anti-Tg and anti-thyroperoxidase antibodies are also commonly detected in GD but are neither disease-specific nor pathogenic (16, 17). Rather, they circulate frequently in other autoimmune thyroid diseases besides GD, such as Hashimoto’s thyroiditis. The absence of detectable TSIs in a small proportion of patients with TAO (18), especially those who fail to develop hyperthyroidism during the course of their GD, suggests that alternate self-antigens might also play roles in the development of TAO.
Figure 2.
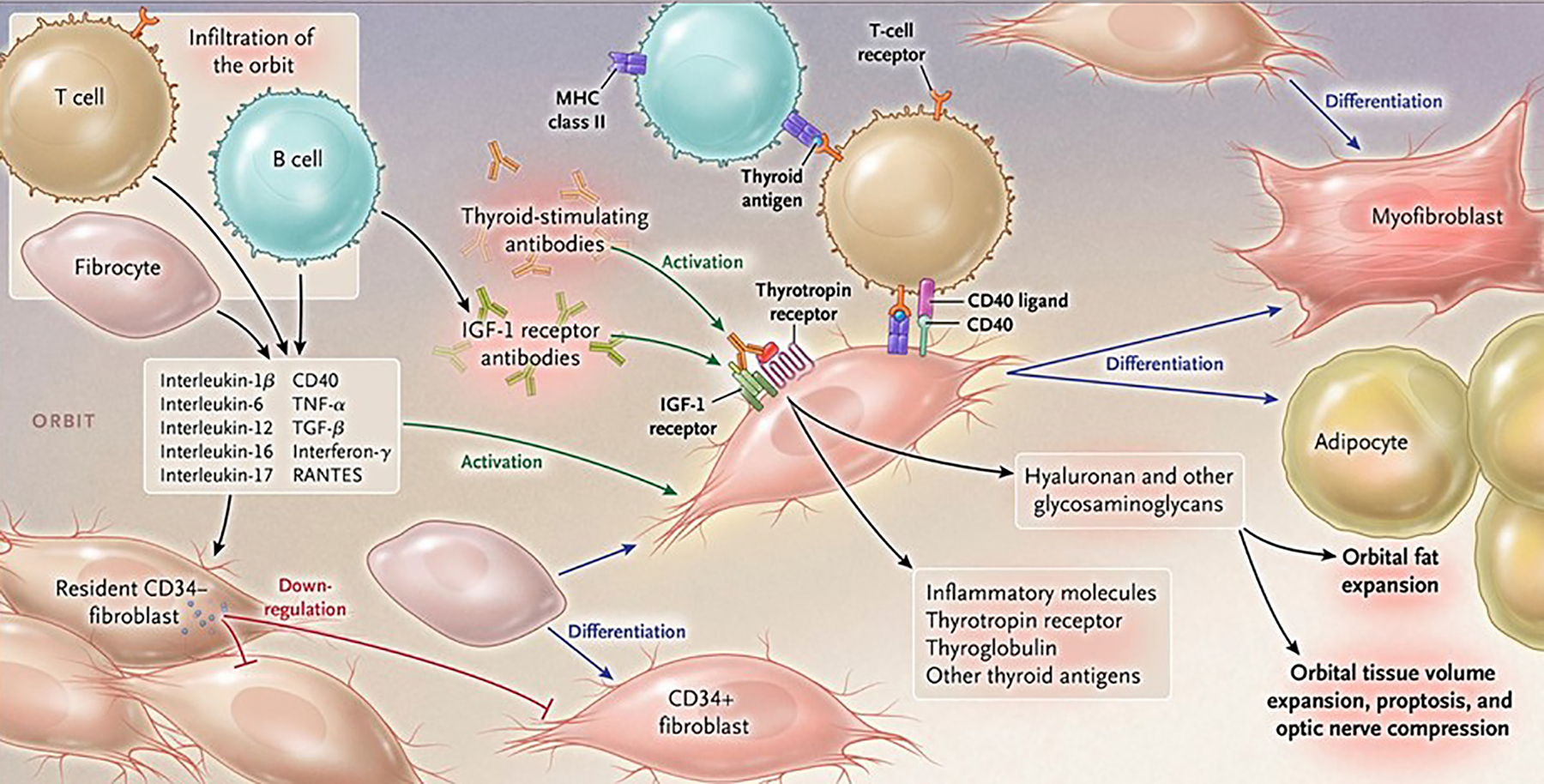
Pathogenesis of thyroid-associated ophthalmopathy. Orbital tissues infiltrated by B and T cells and CD34+ fibrocytes become activated in the disease process by virtue of cell-cell cross talk. Fibrocytes trafficked from bone marrow promiscuously express “thyroid-specific” proteins, including the thyrotropin receptor, thyroglobulin, and other inflammatory genes. They undergo differentiation into myofibroblasts or adipocytes, a consequence of their molecular environment. CD34+ fibroblasts, derived from the fibrocytes take up residence with “garden variety” CD34− fibroblasts. In aggregate, these fibroblasts generate interleukins 1β, 6, 8, 10, 16, IL-1 receptor antagonists, tumor necrosis factor α, RANTES, and CD40 ligand. Thyroid-stimulating immunoglobulins and potentially other autoantibodies directed specifically at the insulin-like growth factor-I receptor, activate the thyrotropin/insulin-like growth factor receptor-1 complex. This results in activation of signaling pathways and target genes participating in disease pathogenesis. Hyaluronan is synthesized following fibroblast activation that culminates in orbital tissue volume expansion which can result in proptosis and optic nerve compression. Orbital fat can also expand following de novo adipogenesis. From N. Engl. J. Med, Smith T.J. and Hegedus L., Graves’ Disease, 375; 1552–1565. Copyright © (2016) Massachusetts Medical Society. Reprinted with permission.
The initiating processes promoting TAO remain uncertain. The distinctive tissue remodeling occurring in active disease results in expansion of the orbital contents and anterior displacement of the globe. Two discrete processes appear to mediate increases in tissue volume in TAO, namely de novo adipogenesis (19) and accumulation of the hydrophilic glycosaminoglycan, hyaluronan (20). Factors that drive differentiation of orbital fibroblasts into triglyceride-accumulating cells appear responsible for the accumulation of orbital fat (21). These include PPAR γ agonists (22–24). Increased hyaluronan deposition in soft orbital tissues results in excessive water content (20). It remains uncertain whether increased synthesis or diminished macromolecular degradation is primarily responsible for this accumulation (20). Both fatty connective tissue and extraocular muscles can expand in TAO (2). Cytokines such as IL-1β, IL-6, IL-17, IL-23 and CD40 ligand appear to play roles in orbital inflammation and remodeling through characteristic patterns of orbital fibroblast responses (24, 25). Cytokines such as IL-1β and CD40 ligand can drive the synthesis of hyaluronan in these cells (26, 27). Orbit-infiltrating lymphocytes secrete a wide array of these cytokines as do residential fibroblasts (28, 29).
Putatively underlying the immune reactivity found uniquely in the TAO orbit are infiltrating CD34+ CXCR4+ Col I+ monocyte lineage progenitor cells, known as fibrocytes (30). These cells derive from the bone marrow and circulate in extremely low abundance in states of health but in GD their numbers increase substantially (31). In experimental models of lung fibrosis, they accumulate in a chemokine-dependent manner in tissues undergoing pathological remodeling (32). They infiltrate the TAO orbit where they become CD34+ orbital fibroblasts (CD34+ OF). They coexist with residential CD34− OF (33). Notable among their phenotypic attributes, fibrocytes express high constitutive levels of MHC class II (34, 35) autoimmune regulator protein (AIRE) (36, 37), and several proteins, the expression of which had been considered thyroid-specific. (36). These include TSHR, functional Tg, sodium iodide symporter, and thyroperoxidase. Within the orbit space, CD34+ OF possess the capacity to undergo terminal differentiation into either adipocytes or myofibroblasts (21, 31). Their fate in differentiation depends on the molecular cues they receive from their microenvironment.
Evidence supporting Insulin-like growth factor-1 receptor involvement in TAO
Insulin-like growth factor-I (IGF-I) plays diverse roles in the regulation of mammalian development, growth, and metabolism (38). IGF-I receptor (IGF-IR) is a more recently identified protein potentially involved in the pathogenesis of TAO (39, 40). IGF-IR is expressed ubiquitously in tissues and cell types throughout the mammalian body. This membrane-spanning tyrosine kinase receptor shares substantial structural identity with the insulin receptor (41). This accounts for the overlap in actions of insulin and IGF-I (42). IGF-IR comprises two α subunits containing the ligand binding domains and two β subunits in which the transmembrane and catalytic domains are located. IGF-IR and the insulin receptor can form heterodimers, a protein complex that exhibits unique signaling properties (43). High-affinity binding of IGF-I to IGF-IR results in receptor auto-phosphorylation occurring at tyrosine residues 1131, 1135, and 1136. The current model for IGF-IR activation involves ligand binding followed by internalization and degradation mediated by proteasomal and lysosomal pathways. Proteins associated with IGF-IR activation and signaling include β-arrestins (44), acting as adaptors for the receptor and the E3 ubiquitin ligase, Mdm2 (45). IGF-IR activation provokes the tyrosine phosphorylation of two post-receptor adaptor proteins, IRS and Shc. This, in turn, activates multiple signaling pathways leading to the altered expression of IGF-I target genes. Among these are genes encoding proteins associated with cell survival and anti-apoptosis (46, 47). Greater than 99% of circulating IGF-I is bound to IGF-I binding proteins of which 6 have been identified (48). The most abundant binding protein, IGFBP3, represents 80% of circulating IGF-I binding capacity. IGF-I binding affinity for these proteins is similar to that for IGF-IR.
Lack of progress in elucidating the mechanisms involved in the pathogenesis of TAO and the absence of safe and effective therapies for TAO has invigorated “out of the box” thinking and consideration of unorthodox drug targets. Among the new wave of drug candidates are molecules that interrupt the IGF-IR pathway. The rationale for determining the safety and efficacy of inhibitory anti-IGF-IR antibodies in autoimmune diseases such as GD rests on substantial evidence that this pathway is involved in their pathogenesis (39). A central role for IGF-I and IGF-IR in the physiological regulation of immunity is implied by the already known alterations of host defense, inflammation and immunity found with deficient function of the pathway. With regard to TAO, several pieces of evidence suggest that IGF-IR may be a target antigen for pathogenic antibody production in GD (49–51). Potential involvement of the IGF-I pathway in TAO was first suggested by the observation of Weightman et al that IgGs isolated from the sera of patients with GD (GD-IgG) could displace radiolabeled IGF-I from binding sites on TAO orbital fibroblasts (49). Those binding sites remained unidentified until subsequent studies revealed IGF-IR as the target antigen (50). Over-expression of IGF-IR was detected in several cell types in GD, including TAO-derived orbital fibroblasts (50), T cells (52) and B cells (53). IGF-IR density on orbital fibroblasts and memory CD45RO+ T cells was elevated in GD while the abundance of circulating IGF-IR+ B cells was increased. Follow-up studies also demonstrated that GD-IgGs could initiate signaling in TAO orbital fibroblasts but not in those cells cultured from healthy individuals (50, 51). GD-IgGs as well as IGF-I induced the expression of cytokines such as IL-8 and regulated on activation, normal T cell expressed and secreted (RANTES). 1H7, an inhibitory monoclonal antibody targeting IGF-IR or transfecting cells with a dominant negative IGF-IR attenuated the induction (50). The epitope(s) recognized by stimulatory GD-IgG has not been mapped but is presumed to be located either at the ligand-binding site or in its close proximity. Similar findings reported subsequently regarding anti-IGF-IR antibodies involved their detection in patients with rheumatoid arthritis (54). These pieces of evidence derive entirely from studies conducted in vitro. Recent experiments conducted in a mouse model of GD (55, 56) revealed the generation of anti-IGF-IR antibodies that have remained uncharacterized. They were detected in animals immunized with TSHR A subunit encoding DNA. It is currently uncertain whether these antibodies exhibit a capacity to activate IGF-IR signaling. Further, the biological implications for their generation and their relationship to anti-TSHR antibodies remain uncertain. Their generation does provide additional plausibility for the concept that anti-IGF-IR antibodies are produced in GD.
The issues of whether anti-IGF-IR antibodies exist, are more abundant in GD, and might play a role in TAO remain controversial. Studies addressing their presence and activities have come from a small cohort of laboratory groups using a variety of experimental in vitro models, response parameters, and cellular targets (57, 58). One group succeeded in identifying a subset of patients in whom functional anti-IGF-IR antibodies could be detected (59).
Another component of the proposed interactions between the TSHR and IGF-IR pathways in TAO involves the identification of a physical and functional complex formed between the two receptor proteins (60). Recognition of a potential relationship between the two pathways can be attributed to the work of Ingbar and colleagues 30 years ago (61, 62). Those early studies disclosed an enhancement of TSH and TSI actions on thyroid epithelial cells by IGF-I. The mechanism underlying that amplification by IGF-I remained uncertain for decades until Tsui et al (60) reported the physical complex of the two receptor proteins. They co-localize in situ in orbital fibroblasts, thyroid epithelial cells, and in orbital fat in situ as determined by confocal microscopy (60). The localization studies accompanied others involving co-immunoprecipitation experiments. Those studies unambiguously demonstrated physical contact between the two proteins. That report contained strong evidence that signaling initiated directly from TSHR was dependent on IGF-IR activity to allow activation of the downstream kinase, Erk 1/2 (60)(Figure 3). Using the IGF-IR inhibiting antibody 1H7, the authors reported that anti-IGF-IR blocking antibodies could attenuate signaling provoked by IGF-I, rhTSH, and GD-IgG. These findings strongly suggest IGF-IR transactivation by TSHR (60). Thus, IGF-IR appears to play a critical role in mediating at least a subset of actions initiated through TSHR. In aggregate, these observations provided a rationale for considering interruption of the IGF-IR pathway as a potential therapy for GD and TAO.
Figure 3.
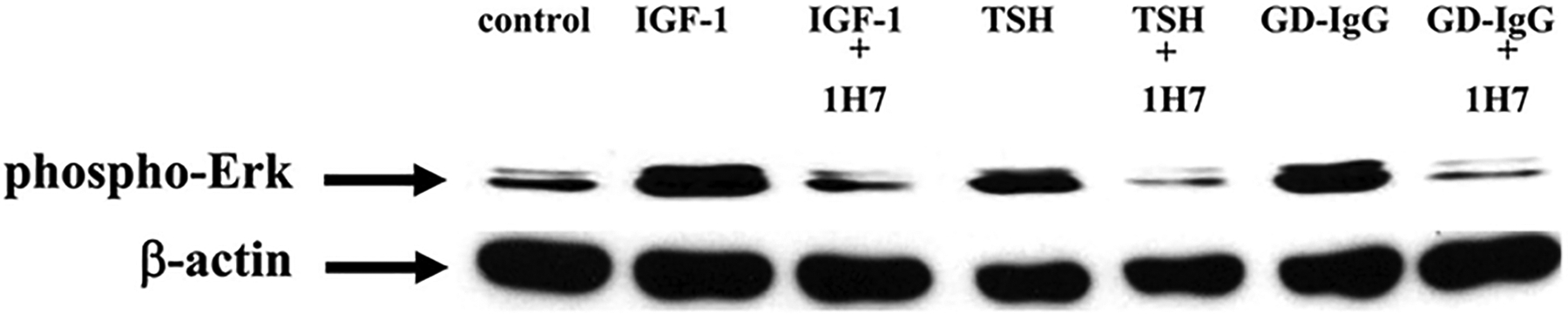
Western blot analysis of ERK activation in cultures of primary human thyrocytes treated with IGF-1 (10 nM), rhTSH (1 mU/ml), or GD-IgG (15 g/ml) alone or in combination with 1H7, an anti-IGF-1R mAb (5 g/ml). Cultures were treated for 15 min, monolayers collected and proteins subjected to Western blot for phospho-ERK 42/44 kDa. Loading equivalence was confirmed by blotting with anti- actin. From J Immunol, Tsui S, Naik V, Hoa N, Hwang CJ, Afifiyan NF, Sinha Hikim A, Gianoukakis AG, Douglas RS, and Smith TJ., Evidence for an association between thyroid-stimulating hormone and insulin-like growth factor 1 receptors: a tale of two antigens implicated in Graves’ disease, 181; 4397–4405.
History of pharmacotherapy for active TAO
Despite the potential for TAO to severely disfigure and threaten sight, no currently available medical therapy has thus far achieved registration by the United States Food and Drug Administration or analogous agencies in other countries. Further, none appears to alter the natural course of the disease, to modify its ultimate outcome, or to decrease the need for rehabilitative surgery. Virtually every current treatment for TAO targets pathways found to be involved in other autoimmune and inflammatory processes. Existing therapy relies upon non-specific, repurposed medical agents, external beam radiotherapy, and surgical strategies that remain unevaluated in adequately powered, well-controlled and prospective therapeutic trials. Treatment has not advanced appreciably in recent decades, despite the substantial gains made in treating other autoimmune diseases such as rheumatoid arthritis (63). Those diseases have benefited greatly from increasingly clarified disease pathogenesis. In the case of TAO, commercial interest in disease-specific developmental programs has not been sufficient, resulting in part from it being an orphan disease. Preclinical animal models in which to test candidate therapies have not been widely available, although recent reports suggest that these may become accessible in the future.
Complicating treatment of TAO is substantial case heterogeneity. It is not yet possible to reliably stratify patients with early disease into those destined for a mild, self-limited course compared with individuals who will ultimately develop sight-threatening, life-altering disease. Other challenges to developing therapy concern barriers to standardizing clinical assessment and identifying response endpoints for use in therapeutic trials. Defining how far along Rundle’s curve a given patient may be is often difficult. Accurate and reproducible measures of disease severity remain to be optimized. Further, there are no specific systemic biomarkers available.
Current therapy for mild active TAO
Selection of treatment for TAO depends on disease stage and severity. The majority of individuals with GD have no signs or symptoms of TAO or they are very mild. If carefully questioned, subjected to detailed physical examination, and studied with high-resolution orbital imaging, subtle evidence of TAO can usually be detected. Frequent complaints of foreign body sensations, dry eye, and intolerance to wind and bright light can dominate the clinical presentation. Over half of those patients manifesting TAO never worsen beyond these mild symptoms. Mild disease is frequently managed effectively with local supportive measures, including protective eyewear and topical ocular solutions to enhance the tear film. For those patients with more symptomatic disease, short courses of oral glucocorticoids alone or in combination with non-steroidal anti-inflammatory agents might reduce orbital inflammation and lessen discomfort.
Moderate to severe, active TAO
Glucocorticoids
For active TAO, non-specific anti-inflammatory agents are commonly used. None of these has thus far been proven safe and effective in moderate to severe TAO. Among them, systemic corticosteroids are frequently administered in relatively high dosages. These became widely used in the 1950s (64) and remain therapy mainstays (65). Despite their widespread use and the popular assumption of their effectiveness, the benefits of glucocorticoids in TAO remain without rigorous validation. Many of the most recent studies of these agents have centered on identifying the optimal dosage and route of administration. A sizable group of investigators favor high-dose pulse glucocorticoid infusions as opposed to more frequently administered oral agents. This preference is based on perceived greater efficacy and reduced side effect profiles although liver toxicity represents a significant concern (66–68). Substantial debate currently exists as to whether oral prednisone should be used routinely in mild, active TAO. Some believe that systemic steroids should be reserved for symptomatic patients, especially those whose vision is threatened. But many patients with moderate to severe disease fail to respond to glucocorticoids while others cannot tolerate these drugs. Worsening diabetes mellitus, impaired glucose tolerance, hypertension, osteoporosis, psychiatric problems, renal disease, and infections are among the most common adverse effects associated with long-term exposure to glucocorticoids. Experts at a limited number of specialized centers advocate external beam radiotherapy (69) delivered either alone or in combination with steroids (70). Radiation therapy for TAO was pioneered in the United States by Kriss and colleagues (71). It is generally accepted that the subset of patients benefiting from steroids are more likely to also respond positively to radiotherapy (72). Despite symptomatic improvement in a substantial proportion of patients treated with these modalities, no convincing evidence exists that they alter the ultimate outcome of the disease or reduce the likelihood of necessary remedial surgeries once the stable phase of TAO has been reached.
Stable TAO
Active TAO eventually transitions to stable disease, a period when inflammation abates and the periocular tissue remodeling ceases. At this point, in theory, any deficits in eye motility, proptosis, and eyelid dysfunction remain static. This affords the orbital surgeon opportunity to remediate the disease and its negative impact on visual function and facial appearance. In the absence of active disease, systemic medical therapies and orbital radiation cease to exhibit benefit. Some patients with mild disease may require strabismus surgery and eyelid repair for persistent diplopia and anterior surface exposure, respectively (73). Others may desire cosmetic procedures to correct defects in the upper face and eyelid abnormalities. Those patients with moderate to severe disease are more likely to require remedial surgery, including orbital decompression to correct proptosis, (74–76) extraocular muscle adjustment, (77) and eyelid repair (78).
History of IGF-IR as a potential therapeutic target in human disease
Fundamental insights that IGF-IR might be involved in disease processes emerged during early studies following the protein’s identification, molecular cloning, and characterization. Its apparent participation in development of certain cancers had prompted focus on the IGF-I pathway as a potential drug target (79–83). Several pharmaceutical companies launched independent developmental programs focusing on therapeutic monoclonal antibodies and small molecules that could inhibit IGF-IR. In fact, the vast majority of experience with monoclonal antibody antagonists targeting IGF-IR in human beings has involved their administration to patients with various forms of cancer who participated in drug trials.
The results of those studies, involving a variety of solid tumors, were sufficiently discouraging to result in the curtailment of most efforts in drug development. Qu et al (84) recently reviewed the fate of those clinical trial programs supporting development of anti-IGF-IR inhibitory antibodies in solid tumors. Those authors concluded that these antibodies have uniformly failed to improve prognosis in solid malignancies. They identified evidence for an overall negative impact of these drugs in subsets of tumor types. Their meta-analysis included 17 clinical studies involving 6 different types of cancer and 5 different anti-IGF-IR monoclonal antibodies.
Thus, most if not all of the commercial efforts for the development of agents targeting IGF-IR in cancer have now ceased. Many investigators in the field perceive a major obstacle as relating to the heterogeneity of the tumors targeted in these trials. These experts contend that revisiting the question of IGF-IR targeting for solid tumors must be accompanied by better discriminators for allowing stratification of cases to identify a priori those most likely to respond. One exception is the apparent continuation of the Ganitumab (Nanthealth) program. This antibody was designated by the US FDA as an orphan drug for treatment of metastatic Ewings sarcoma. It is currently listed as an active phase III trial in clinical trials.gov (NCTO2306161) as a combination treatment with multiple chemotherapeutic agents.
Teprotumumab, a repurposed IGF-IR inhibitor as a candidate in TAO
While it appears that the initial rationale for developing inhibitory anti-IGF-IR antibodies in solid tumors has not led to useful treatments, the availability of these agents for repurposing for other diseases represents a great and unforeseen opportunity. Teprotumumab (RV001, R1507) became available for potential testing in TAO. The rationale for this line of inquiry rests entirely on observations made in vitro. These included the demonstration of IGF-IR overexpression in multiple patient-derived cell types (85), detection of activating antibodies of the IgG class that targeted IGF-IR in patients with GD (50), and the identification of physical and functional TSHR/IGF-IR complex formation in human tissues (60). In particular, examination of these receptor protein complexes revealed that signaling initiated at either protein could be blocked by IGF-IR inhibitory antibodies (60, 86). The initial studies were conducted with 1H7, a mouse monoclonal antibody with similar inhibitory properties to those of teprotumumab (60). Preliminary studies in vitro revealed further that teprotumumab might be ideal for testing in human trial studies of active TAO (86).
Teprotumumab is a fully human inhibitory antibody directed against IGF-IR that lacks any detectable agonistic activity toward either canonical IGF-IR signaling or toward the insulin receptor. It was developed as an anti-neoplastic agent and underwent several clinical trials in multiple cancers with outcomes that were inadequate for program continuation (84). The antibody appears to have absolute specificity for IGF-IR. The variable region of teprotumumab, an IgG1 subclass molecule, was derived from a transgenic HCo7 mouse that had been immunized with recombinant IGF-1R protein. Each mw ~150 kDa molecule comprises two heterodimers consisting of a heavy and a light chain. A single carbohydrate chain is located in the constant region of both heavy chains. The antibody was initially generated in a stably transfected murine cell line, Sp2/0Ag14; however stable CHO-DG44 cells have been used to produce material destined for clinical evaluation. Teprotumumab binds to the cysteine-rich region of IGF-IR extracellular domain with high-affinity and specificity. This blocks the receptor’s binding pocket and thus precludes binding of both IGF-I and IGF-II. Once bound by teprotumumab, IGF-IR is internalized and subsequently the receptor/antibody complexes enter degradation pathways. In cancer cells in vitro, the antibody attenuates post-receptor signaling and cell proliferation. Teprotumumab was found to induce apoptosis in a variety of tumor cell lines while halting proliferation in others. A subset of actions of the antibody were enhanced with addition of other agents including erlotinib in cultured non-small cell cancer cells and the combination of erlotinib and gemcitabine in pancreatic cancer cells.
Effectiveness of teprotumumab in moderate to severe TAO
A randomized, multicenter, double-masked, prospective therapeutic trial was organized, predicated entirely on the then-existing evidence for IGF-IR playing a role in TAO (Figure 4). That study began enrolment at 15 performance sites throughout North America and Europe between 2 July, 2013 and 23 September, 2015. It examined the efficacy of teprotumumab, an inhibitory, fully human monoclonal antibody in 88 patients with active (clinical activity score (CAS) ≥ 4 on a 7 point scale), moderate to severe TAO who were randomly assigned to receive either placebo (saline infusion) or the active drug. Major inclusion criteria included participants between the ages of 18 and 75 years, who had developed TAO within 9 months of study entrance. Both treatment groups were administered 8 infusions each, at 3 weekly intervals over a 24-week span. The initial (reduced) drug dosage was 10 mg/kg. If tolerated, the full dosage of 20 mg/kg was then administered for the duration of the treatment period. The primary response end-point was an aggregate of achieving reduction in CAS ≥ 2 points AND reduction of ≥ 2 mm of proptosis at the 24 week assessment in the more severely affected (study) eye in the absence of a similar worsening in the contralateral (fellow) eye. Responders were further analyzed according to the magnitude of their responses. Secondary endpoints included assessment of changes in CAS and proptosis measured as continuous variables over time, changes in quality of life as assessed by a validated patient survey comprising 2 subscales (GO-Qol) (87, 88)(Figure 5), and improvement of subjective diplopia. Patients were assessed at 6, 12, 18 and 24 weeks following their initial infusion. With regard to the primary outcome, 20% (9/45) of the patients receiving placebo and 69% (29/42) of those receiving the drug were responders (p<0.001) in the intention to treat cohort. In the per protocol population, 22% (8/36) of those in the placebo arm and 78% (26/33) receiving teprotumumab were responders (adjusted odds ratio 12.73, p<0.001). Time to first response was considerably shorter in the group receiving active drug (p<0.001). Therapeutic effects of teprotumumab were rapid. 18/42 patients in the teprotumumab group (43%) versus 2/45 patients receiving placebo (4%) had a response at week 6 following the initial infusion (P<0.001). The proportion of patients termed “high responders” (CAS improvement ≥3 points and proptosis reduction ≥3 mm) was substantially greater in the teprotumumab population (p<0.001). GO-Qol and strabismus also improved significantly at 24 weeks. Thus, teprotumumab proved to be an effective therapy for this select cohort of patients with active, moderate to severe TAO. These study patients are now participating in long-term follow-up, which should reveal the durability of treatment response and any as yet unrecognized, late-developing, untoward consequences of IGF-IR inhibition over time. On the basis of the encouraging results from this trial, the U.S. FDA has designated teprotumumab with “breakthrough” status for the treatment of active, moderate to severe TAO.
Figure 4.
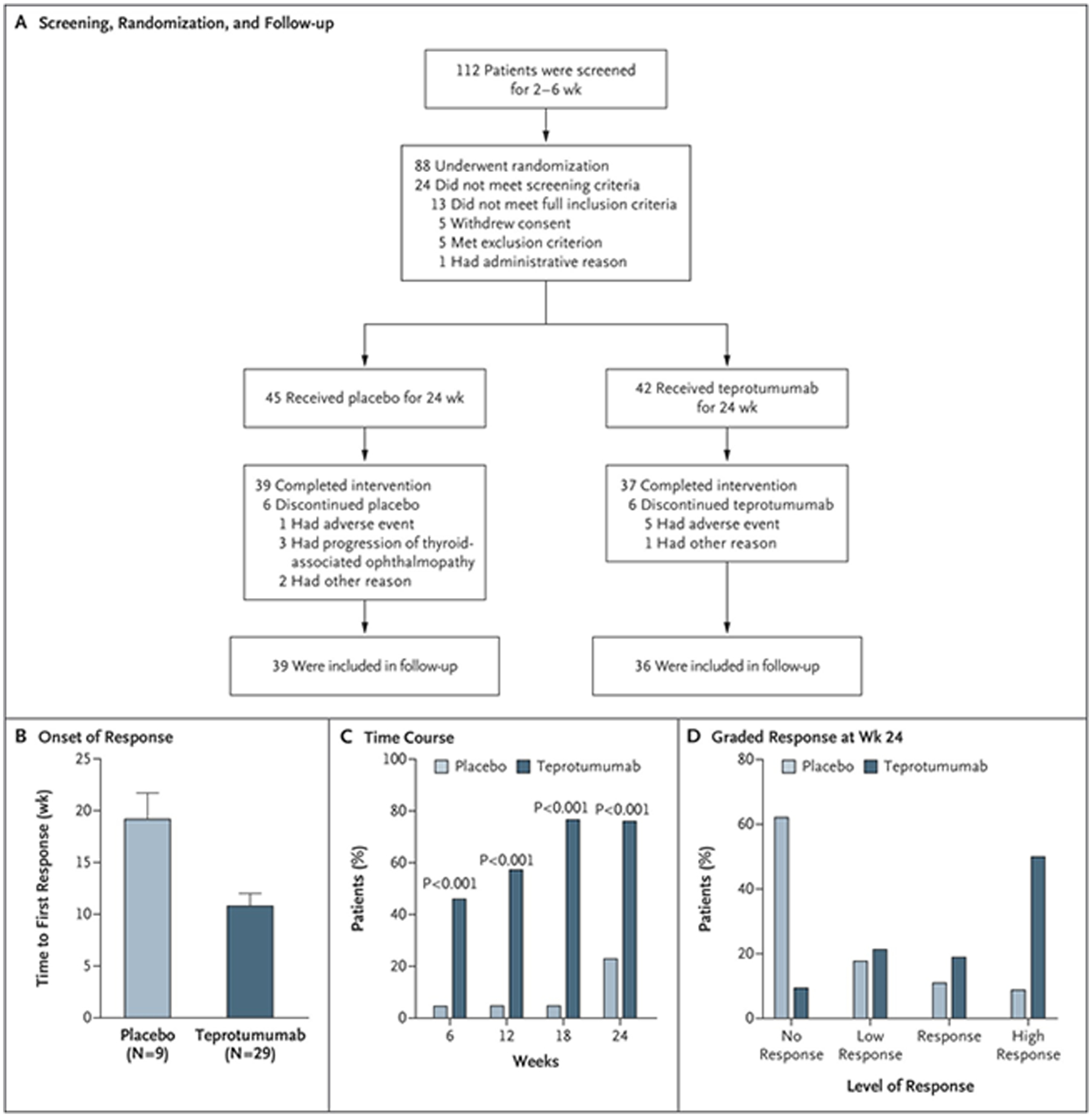
Panel A. Strategy for screening patients, their randomization into the two treatment arms, and follow-up. Panel B. Analysis to first response (a reduction of ≥ 2 mm in proptosis AND an improvement of ≥ 2 points on a 7-point scale in clinical activity score). Panel C. Time course in patients meeting response criteria. Panel D. Grading of the response at week 24. From N. Engl. J. Med, Smith T.J., Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, et al, Teprotumumab for Thyroid-Associated Ophthalmopathy, 376:1748–61. Copyright © (2017) Massachusetts Medical Society. Reprinted with permission.
Figure 5.

Secondary efficacy endpoints. Panel A. Change in proptosis from baseline. Panel B. Changes in clinical activity score from baseline. Panel C. Percentage of patients with a clinical activity score of 0 or 1 at week 24. Panel D. Change from baseline of visual functioning subscale in the thyroid-associated ophthalmopathy-specific quality of life scale (GO-QOL). Panel E. Change from baseline in GD-QOL appearance subscale. Panel F. Response in subjective diplopia. From N. Engl. J. Med, Smith T.J., Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, et al, Teprotumumab for Thyroid-Associated Ophthalmopathy, 376:1748–61. Copyright © (2017) Massachusetts Medical Society. Reprinted with permission.
Assessment of teprotumumab toxicity in TAO
With regard to safety, teprotumumab had previously demonstrated good tolerability in multiple cancer studies y at a dosage of 9 mg/kg weekly or 16 mg/kg every 3 weeks (89). In that study, the AUC increased with dose escalation. Antibody exposure resulted in increased serum levels of IGF-I. In cynomolgus monkeys, teprotumumab depleted thymic lymphocytes and resulted in the hypocellularity and involution of that tissue which was dose-independent. Thymic involution appeared to be partially reversible. There was no increased incidence of opportunistic infection.
The safety profile emerging from the recent teprotumumab clinical trial for TAO appears to be encouraging. Hyperglycemia, especially in patients with preexistent diabetes mellitus occurs more frequently in the group receiving the drug. This adverse event was the only one unambiguously identified as drug-related (90). In non-diabetics, it was effectively managed with frequent assessment of blood glucose and was uniformly grade 1. A small group of patients receiving teprotumumab who were already diabetic at their study entrance developed grade 2 or grade 3 hyperglycemia, requiring increased anti-diabetic medications. Importantly, the glycemic control in these individuals uniformly returned to baseline following their completion of the treatment phase of the study. The majority of adverse events were mild and did not require medical treatment. None of the other side effects reported in earlier clinical studies of teprotumumab and similar drugs for cancer manifested themselves in the TAO trial (90). These included thrombocytopenia and systemic hypertension and were not seen in the TAO trial. These adverse events can probably be attributed to the other drugs, some cytotoxic, also being administered to fragile patients with various forms of malignancy.
Pharmacokinetics of teprotumumab
In cynomolgus monkeys, administration of teprotumumab resulted in a relatively long T1/2 and low systemic clearance. Animals administered either 3 mg/kg or 15 mg/kg exhibited non-linear kinetics while linear kinetics were found at higher dosages, up to 150 mg/kg. This profile suggests that both receptor-mediated (saturable) and non-receptor mediated (non-saturable) clearance was involved in the elimination of teprotumumab. Using a weekly IV infusion schedule, drug accumulation was detected at all dosages. The PK characteristics of teprotumumab in human subjects resemble those of other antibodies directed at a variety of antigenic targets. The antibody is slowly cleared, with a relatively prolonged T1/2. In patients receiving multiple doses of 16–20 mg/kg IV every 3 weeks, the T1/2 was 20 days. The PK parameters are consistent with two clearance pathways, one saturable at low drug concentrations and the other probably mediated by the reticuloendothelial system.
The road ahead for teprotumumab therapy in TAO
A phase III trial of teprotumumab in moderate to severe TAO has been initiated involving several performance sites in North America and Europe. Many of these sites participated in the initial phase II study (90). Patients enrolled in the follow-up study must meet the identical inclusion criteria as those in the earlier trial. This study will include a cross-over phase where non-responders at 24 weeks will be eligible for treatment with teprotumumab. The primary endpoint of the study involves a single component of proptosis reduction in the study eye of ≥ 2 mm in the absence of similar worsening of proptosis in the fellow eye at 24 weeks. This follow-up study also involves 8 infusions of placebo or drug at 3 weekly intervals.
Other emerging therapeutic targets in TAO
Rituximab
B cell depleting agents such as rituximab (anti-CD20) were originally developed for the treatment of non-Hodgkin’s B cell lymphoma (91). They have proven effective in several autoimmune diseases, including rheumatoid arthritis, systemic lupus erythematosus, neuromyelitis optica, and ulcerative colitis (92). Early advocates of anti-CD20 use in GD focused on hyperthyroidism (93). More recently, the drug’s potential in active, moderate to severe TAO has been examined in two small, simultaneously reported clinical trials (94, 95). Both trials were prospective, double-masked, and controlled and each conducted at a single performance site. One compared the drug to placebo (94). In that study, 25 patients were enrolled in the trial with the primary endpoint, > 2 point improvement in the clinical activity score, was assessed at 24 weeks. No differences emerged between the two treatment groups (94). In the other study, 32 patients were randomized to receive either rituximab or intravenous methylprednisolone (7.5 grams). The study found that rituximab improved clinical activity score more than did IV methylprednisolone (95). Neither rituximab nor methylprednisolone improved proptosis in either study. The dramatically different results from the two trials have raised importance of patient selection and the potential for experimental bias in studies that are not placebo-controlled. Factors such as disease duration prior to study enrolment, patient age, and levels of anti-TSHR antibodies may account for these differences in study outcome. In any event, the question of B cell depletion (anti-CD20 or anti-CD19) in TAO remains largely unanswered and will require additional study. Ultimately, demonstrating that this drug alters the course and ultimate outcome of the disease, such as avoidance of surgical remediation, may be necessary to justify its high costs and significant side effects.
Anti-cytokine and antimetabolite agents
A small number of anti-cytokine agents have thus far been examined in small, frequently uncontrolled, open-label clinical studies. Like most of the biological agents mentioned in this article, these were developed for other diseases but the increasing awareness of mechanistic similarities between the diseases has invited consideration for use in TAO. A good example is tociluzimab, which interrupts IL-6 signaling by blocking the IL-6 receptor (96). The drug was developed for rheumatoid arthritis and has been evaluated in several other autoimmune diseases including systemic lupus erythematosis, giant cell arteritis, Sjӧgren syndrome, Crohn’s disease, and neuromyelitis optica (97). The largest clinical study of tociluzimab examined its impact in an uncontrolled, open-label study of 18 patients with active TAO (98). The results are difficult to interpret and illuminate the importance of properly controlled and adequately powered studies. A very recent report suggests that mycophenolate mofetil may offer benefit in active moderate to severe TAO compared to corticosteroids (99). That drug inhibits the rate-limiting enzyme in de novo synthesis of guanosine nucleotides, inosine monophosphate dehydrogenase (100). Lymphocytes are particularly vulnerable to the inhibition of this enzyme. Analysis of these and other candidate agents must necessarily be expanded to include large prospective (ideally placebo controlled) trials.
TSHR as a therapeutic target
The widely held view that TSHR represents the central autoimmune target in the TAO orbit aligns with its critical role in hyperthyroidism (12). That concept serves as the rationale for developing therapeutic agents that interrupt TSHR signaling in both hyperthyroidism and TAO. Agents that inhibit TSH/TSI binding and signal initiation through direct interactions with TSHR include both monoclonal antibodies and small molecules. Several antibodies, including those isolated from patients with thyroid autoimmunity exhibit blocking activities (101–103). Among the anti-TSHR antibodies are those acting as inverse agonists, so designated because they inhibit constitutive activities of the receptor (104). Several of the blocking antibodies exhibit affinities for TSHR of approximately 100 pM. Small molecules have also been developed to specifically inhibit TSHR activity. Some lack specificity, including Org41841 which inhibits both TSHR and leutinizing hormone receptor (105). Small molecule inhibitors can block actions of TSIs, both in vitro and in small animals (106, 107). None of the molecules thus far developed has been subjected to study in human therapeutic trials. As a developmental direction, these agents may offer clinical benefit and target a receptor protein with relatively limited anatomical expression.
Conclusions
Despite the absence of proven-effective medical therapies currently available, the immediate horizon for therapeutic development targeting TAO appears encouraging. These prospects are enabled by expanding insights into the pathogenesis of the disease. The direction appearing most likely to yield an accessible treatment is the monoclonal antibody, teprotumumab, which inhibits IGF-IR. This agent is currently in phase III clinical trial and has been designated “break-through” therapy status by the US FDA after it demonstrated substantial effectiveness and an encouraging safety profile in a recently completed phase II trial. Evaluation of other attractive targets, including TSHR and cytokines networks, is less advanced and will require considerably more investigation if they are to come to fruition. In any event, care of the patient with moderate to severe TAO is likely to be far better served in the near future. Progress made in treating TAO could provide important guidance to improving outcomes of other orphan diseases.
Acknowledgements
The author thanks Ms. Darla Kroft for her expert help in manuscript preparation. This work was supported in part by National Institutes of Health grants EY008976, EY11708, DK063121, and 5UM1AI110557, Center for Vision core grant EY007002 from the National Eye Institute, an unrestricted grant from Research to Prevent Blindness, and by the Bell Charitable Foundation.
Footnotes
Conflicts
The author has been issued patents related to the detection of antibody-mediated inflammatory auto-immune disorders (US 6936426), the diagnosis and therapy of antibody-mediated inflammatory autoimmune disorders (US 7998681 and US 8153121), and diagnostic methods related to Graves’ disease and other autoimmune disorders (US 8178304). He was previously a paid consultant for River Vision.
References
- 1.Smith TJ, Hegedus L. 2016. Graves’ Disease. N. Engl. J. Med 375:1552–65 [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Smith TJ. 2014. Current concepts in the molecular pathogenesis of thyroid-associated ophthalmopathy. Invest. Ophthalmol. Vis. Sci 55:1735–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rundle FF, Wilson CW. 1945. Development and course of exophthalmos and ophthalmoplegia in Graves’ disease with special reference to the effect of thyroidectomy. Clin. Sci 5:177–94 [PubMed] [Google Scholar]
- 4.Bartalena L, Piantanida E. 2016. Cigarette smoking: number one enemy for Graves ophthalmopathy. Pol. Arch. Med. Wewn 126:725–6 [DOI] [PubMed] [Google Scholar]
- 5.Chu X, Pan CM, Zhao SX, Liang J, Gao GQ, et al. 2011. A genome-wide association study identifies two new risk loci for Graves’ disease. Nat. Genet 43:897–901 [DOI] [PubMed] [Google Scholar]
- 6.Ungerer M, Fassbender J, Li Z, Munch G, Holthoff HP. 2017. Review of Mouse Models of Graves’ Disease and Orbitopathy-Novel Treatment by Induction of Tolerance. Clin. Rev. Allergy. Immunol 52:182–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Novaes P, Diniz Grisolia AB, Smith TJ. 2016. Update on thyroid-associated Ophthalmopathy with a special emphasis on the ocular surface. Clin. Diabetes Endocrinol 2:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen CR, Pichurin P, Nagayama Y, Latrofa F, Rapoport B, McLachlan SM. 2003. The thyrotropin receptor autoantigen in Graves disease is the culprit as well as the victim. J. Clin. Invest 111:1897–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morshed SA, Davies TF. 2015. Graves’ Disease Mechanisms: The Role of Stimulating, Blocking, and Cleavage Region TSH Receptor Antibodies. Horm. Metab. Res 47:727–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feliciello A, Porcellini A, Ciullo I, Bonavolonta G, Avvedimento EV, Fenzi G. 1993. Expression of thyrotropin-receptor mRNA in healthy and Graves’ disease retro-orbital tissue. Lancet 342:337–8 [DOI] [PubMed] [Google Scholar]
- 11.Kriss JP. 1970. Radioisotopic thyroidolymphography in patients with Graves’ disease. J. Clin. Endocrinol. Met 31:315–23 [DOI] [PubMed] [Google Scholar]
- 12.Adams DD, Purves HD, Sirett NE. 1961. The response of hypophysectomized mice to injections of human serum containing long-acting thyroid stimulator. Endocrinology 68:154–5 [DOI] [PubMed] [Google Scholar]
- 13.Raychaudhuri N, Fernando R, Smith TJ. 2013. Thyrotropin regulates IL-6 expression in CD34+ fibrocytes: clear delineation of its cAMP-independent actions. PLoS One 8:e75100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang SY, Shin DY, Lee EJ, Lee SY, Yoon JS. 2013. Relevance of TSH-receptor antibody levels in predicting disease course in Graves’ orbitopathy: comparison of the third-generation TBII assay and Mc4-TSI bioassay. Eye 27:964–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jang SY, Shin DY, Lee EJ, Yoon JS. 2014. Clinical characteristics of Graves’ orbitopathy in patients showing discrepancy between levels from TBII assays and TSI bioassay. Clinic. Endocrinol 80:591–7 [DOI] [PubMed] [Google Scholar]
- 16.Sikorska HM. 1986. Anti-thyroglobulin anti-idiotypic antibodies in sera of patients with Hashimoto’s thyroiditis and Graves’ disease. J.Immuno 137:3786–95 [PubMed] [Google Scholar]
- 17.Chardes T, Chapal N, Bresson D, Bes C, Giudicelli V, et al. 2002. The human anti-thyroid peroxidase autoantibody repertoire in Graves’ and Hashimoto’s autoimmune thyroid diseases. Immunogenetics 54:141–57 [DOI] [PubMed] [Google Scholar]
- 18.Tabasum A, Khan I, Taylor P, Das G, Okosieme OE. 2016. Thyroid antibody-negative euthyroid Graves’ ophthalmopathy. Endocrinol. Diabetes Metab. Case Rep 2016:160008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar S, Coenen MJ, Scherer PE, Bahn RS. 2004. Evidence for enhanced adipogenesis in the orbits of patients with Graves’ ophthalmopathy. J. Clin. Endocrinol. Metab 89:930–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith TJ, Bahn RS, Gorman CA. 1989. Connective tissue, glycosaminoglycans, and diseases of the thyroid. Endocr. Rev 10:366–91 [DOI] [PubMed] [Google Scholar]
- 21.Sorisky A, Pardasani D, Gagnon A, Smith TJ. 1996. Evidence of adipocyte differentiation in human orbital fibroblasts in primary culture. J. Clin Endocrinol. Metab 81:3428–31 [DOI] [PubMed] [Google Scholar]
- 22.Smith TJ. 2002. Role of orbital fat in thyroid-associated ophthalmopathy. Graves’ Associated Orbital Disease. In Thyroid Eye Disease : Diagnosis and Treatment, ed. Dutton JJ, Haik BG:215–21.New York, NY: Marcel Dekker, Inc. Publishers. Number of215–21 pp. [Google Scholar]
- 23.Smith TJ. 2002. Fibroblast biology in thyroid diseases. Curr. Opin. Endocrinol. Diabetes Obes 9:393–400 [Google Scholar]
- 24.Smith TJ, Koumas L, Gagnon A, Bell A, Sempowski GD, et al. 2002. Orbital fibroblast heterogeneity may determine the clinical presentation of thyroid-associated ophthalmopathy. J. Clin. Endocrinol. Metab 87:385–92 [DOI] [PubMed] [Google Scholar]
- 25.Fang S, Huang Y, Wang S, Zhang Y, Luo X, et al. 2016. IL-17A Exacerbates Fibrosis by Promoting the Proinflammatory and Profibrotic Function of Orbital Fibroblasts in TAO. J. Clin. Endocrinol. Metab 101:2955–65 [DOI] [PubMed] [Google Scholar]
- 26.Kaback LA, Smith TJ. 1999. Expression of hyaluronan synthase messenger ribonucleic acids and their induction by interleukin-1beta in human orbital fibroblasts: potential insight into the molecular pathogenesis of thyroid-associated ophthalmopathy. J. Clin. Endocrinol. Metab 84:4079–84 [DOI] [PubMed] [Google Scholar]
- 27.Cao HJ, Wang HS, Zhang Y, Lin HY, Phipps RP, Smith TJ. 1998. Activation of human orbital fibroblasts through CD40 engagement results in a dramatic induction of hyaluronan synthesis and prostaglandin endoperoxide H synthase-2 expression. Insights into potential pathogenic mechanisms of thyroid-associated ophthalmopathy. J. Biol. Chem 273:29615–25 [DOI] [PubMed] [Google Scholar]
- 28.de Carli M, D’Elios MM, Mariotti S, Marcocci C, Pinchera A, et al. 1993. Cytolytic T cells with Th1-like cytokine profile predominate in retroorbital lymphocytic infiltrates of Graves’ ophthalmopathy. J. Clin. Endocrinol. Metab 77:1120–4 [DOI] [PubMed] [Google Scholar]
- 29.McLachlan SM, Prummel MF, Rapoport B. 1994. Cell-mediated or humoral immunity in Graves’ ophthalmopathy? Profiles of T-cell cytokines amplified by polymerase chain reaction from orbital tissue. J. Clin. Endocrinol. Metab 78:1070–4 [DOI] [PubMed] [Google Scholar]
- 30.Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A. 1994. Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol. Med 1:71–81 [PMC free article] [PubMed] [Google Scholar]
- 31.Douglas RS, Afifiyan NF, Hwang CJ, Chong K, Haider U, et al. 2010. Increased generation of fibrocytes in thyroid-associated ophthalmopathy. J. Clin. Endocrinol. Metab 95:430–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hong KM, Belperio JA, Keane MP, Burdick MD, Strieter RM. 2007. Differentiation of human circulating fibrocytes as mediated by transforming growth factor-beta and peroxisome proliferator-activated receptor gamma. J. Biol. Chem 282:22910–20 [DOI] [PubMed] [Google Scholar]
- 33.Fernando R, Atkins S, Raychaudhuri N, Lu Y, Li B, et al. 2012. Human fibrocytes coexpress thyroglobulin and thyrotropin receptor. Proc. Natl. Acad. Sci. U.S.A 109:7427–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chesney J, Bacher M, Bender A, Bucala R. 1997. The peripheral blood fibrocyte is a potent antigen-presenting cell capable of priming naive T cells in situ. Proc. Natl. Acad. Sci. U.S.A 94:6307–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grab DJ, Lanners H, Martin LN, Chesney J, Cai C, et al. 1999. Interaction of Borrelia burgdorferi with peripheral blood fibrocytes, antigen-presenting cells with the potential for connective tissue targeting. Mol. Med 5:46–54 [PMC free article] [PubMed] [Google Scholar]
- 36.Fernando R, Lu Y, Atkins SJ, Mester T, Branham K, Smith TJ. 2014. Expression of thyrotropin receptor, thyroglobulin, sodium-iodide symporter, and thyroperoxidase by fibrocytes depends on AIRE. J. Clin. Endocrinol. Metab 99:E1236–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pitkanen J, Peterson P. 2003. Autoimmune regulator: from loss of function to autoimmunity. Genes Immun. 4:12–21 [DOI] [PubMed] [Google Scholar]
- 38.Gallagher EJ, LeRoith D. 2011. Minireview: IGF, Insulin, and Cancer. Endocrinology 152:2546–51 [DOI] [PubMed] [Google Scholar]
- 39.Smith TJ. 2010. Insulin-like growth factor-I regulation of immune function: a potential therapeutic target in autoimmune diseases? Pharmacol. Rev 62:199–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith TJ. 2013. Is IGF-I receptor a target for autoantibody generation in Graves’ disease? J. Clin. Endocrin. Metab 98:515–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Meyts P, Whittaker J. 2002. Structural biology of insulin and IGF1 receptors: implications for drug design. Nat. Rev. Drug Discov 1:769–83 [DOI] [PubMed] [Google Scholar]
- 42.Mastick CC, Kato H, Roberts CT Jr., LeRoith D, Saltiel AR. 1994. Insulin and insulin-like growth factor-I receptors similarly stimulate deoxyribonucleic acid synthesis despite differences in cellular protein tyrosine phosphorylation. Endocrinology 135:214–22 [DOI] [PubMed] [Google Scholar]
- 43.Soos MA, Whittaker J, Lammers R, Ullrich A, Siddle K. 1990. Receptors for insulin and insulin-like growth factor-I can form hybrid dimers. Characterisation of hybrid receptors in transfected cells. Biochem. J 270:383–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng H, Shen H, Oprea I, Worrall C, Stefanescu R, et al. 2012. beta-Arrestin-biased agonism as the central mechanism of action for insulin-like growth factor 1 receptor-targeting antibodies in Ewing’s sarcoma. Proc. Natl. Acad. Sci. U.S.A 109:20620–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Girnita L, Girnita A, Larsson O. 2003. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc. Natl. Acad. Sci. U.S.A 100:8247–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams TE, Epa VC, Garrett TP, Ward CW. 2000. Structure and function of the type 1 insulin-like growth factor receptor. Cell. Mol. Life Sci 57:1050–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dupont J, Fernandez AM, Glackin CA, Helman L, LeRoith D. 2001. Insulin-like growth factor 1 (IGF-1)-induced twist expression is involved in the anti-apoptotic effects of the IGF-1 receptor. J. Biol. Chem 276:26699–707 [DOI] [PubMed] [Google Scholar]
- 48.Kim MS, Lee DY. 2015. Insulin-like growth factor (IGF)-I and IGF binding proteins axis in diabetes mellitus. Ann. Pediatr. Endocrinol. Metab 20:69–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weightman DR, Perros P, Sherif IH, Kendall-Taylor P. 1993. Autoantibodies to IGF-1 binding sites in thyroid associated ophthalmopathy. Autoimmunity 16:251–7 [DOI] [PubMed] [Google Scholar]
- 50.Pritchard J, Han R, Horst N, Cruikshank WW, Smith TJ. 2003. Immunoglobulin activation of T cell chemoattractant expression in fibroblasts from patients with Graves’ disease is mediated through the insulin-like growth factor I receptor pathway. J. Immunol 170:6348–54 [DOI] [PubMed] [Google Scholar]
- 51.Smith TJ, Hoa N. 2004. Immunoglobulins from patients with Graves’ disease induce hyaluronan synthesis in their orbital fibroblasts through the self-antigen, insulin-like growth factor-I receptor. J. Clin. Endocrin. Metab 89:5076–80 [DOI] [PubMed] [Google Scholar]
- 52.Douglas RS, Smith TJ. 2007. Thyroid Related Orbitopathy: New Immunologic Concepts and Future Implications. In Oculoplastics and Orbit, ed. Guthoff R, Katowitz J:123–41.Berlin: Springer-Verlag. Number of123–41 pp. [Google Scholar]
- 53.Douglas RS, Naik V, Hwang CJ, Afifiyan NF, Gianoukakis AG, et al. 2008. B cells from patients with Graves’ disease aberrantly express the IGF-1 receptor: implications for disease pathogenesis. J. Immunol 181:5768–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pritchard J, Tsui S, Horst N, Cruikshank WW, Smith TJ. 2004. Synovial fibroblasts from patients with rheumatoid arthritis, like fibroblasts from Graves’ disease, express high levels of IL-16 when treated with Igs against insulin-like growth factor-1 receptor. J. Immunol 173:3564–9 [DOI] [PubMed] [Google Scholar]
- 55.Zhao SX, Tsui S, Cheung A, Douglas RS, Smith TJ, Banga JP. 2011. Orbital fibrosis in a mouse model of Graves’ disease induced by genetic immunization of thyrotropin receptor cDNA. J. Endocrinol 210:369–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berchner-Pfannschmidt U, Moshkelgosha S, Diaz-Cano S, Edelmann B, Gortz GE, et al. 2016. Comparative Assessment of Female Mouse Model of Graves’ Orbitopathy Under Different Environments, Accompanied by Proinflammatory Cytokine and T-Cell Responses to Thyrotropin Hormone Receptor Antigen. Endocrinology 157:1673–82 [DOI] [PubMed] [Google Scholar]
- 57.Minich WB, Dehina N, Welsink T, Schwiebert C, Morgenthaler NG, et al. 2013. Autoantibodies to the IGF1 receptor in Graves’ orbitopathy. J. Clin. Endocrin. Metab 98:752–60 [DOI] [PubMed] [Google Scholar]
- 58.Krieger CC, Place RF, Bevilacqua C, Marcus-Samuels B, Abel BS, et al. 2016. TSH/IGF-1 Receptor Cross Talk in Graves’ Ophthalmopathy Pathogenesis. J. Clin. Endocrin. Metab 101:2340–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Varewijck AJ, Boelen A, Lamberts SW, Fliers E, Hofland LJ, et al. 2013. Circulating IgGs may modulate IGF-I receptor stimulating activity in a subset of patients with Graves’ ophthalmopathy. J. Clin. Endocrin. Metab 98:769–76 [DOI] [PubMed] [Google Scholar]
- 60.Tsui S, Naik V, Hoa N, Hwang CJ, Afifiyan NF, et al. 2008. Evidence for an association between thyroid-stimulating hormone and insulin-like growth factor 1 receptors: a tale of two antigens implicated in Graves’ disease. J. Immunol 181:4397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tramontano D, Cushing GW, Moses AC, Ingbar SH. 1986. Insulin-like growth factor-I stimulates the growth of rat thyroid cells in culture and synergizes the stimulation of DNA synthesis induced by TSH and Graves’-IgG. Endocrinology 119:940–2 [DOI] [PubMed] [Google Scholar]
- 62.Tramontano D, Moses AC, Veneziani BM, Ingbar SH. 1988. Adenosine 3’,5’-monophosphate mediates both the mitogenic effect of thyrotropin and its ability to amplify the response to insulin-like growth factor I in FRTL5 cells. Endocrinology 122:127–32 [DOI] [PubMed] [Google Scholar]
- 63.Alfonso-Cristancho R, Armstrong N, Arjunji R, Riemsma R, Worthy G, et al. 2017. Comparative effectiveness of biologics for the management of rheumatoid arthritis: systematic review and network meta-analysis. Clin. Rheumatol 36:25–34 [DOI] [PubMed] [Google Scholar]
- 64.Chandler GN, Hartfall SJ. 1952. Cortisone and ACTH in exophthalmic ophthalmoplegia. Lancet 1:847–51 [DOI] [PubMed] [Google Scholar]
- 65.Bartalena L, Krassas GE, Wiersinga W, Marcocci C, Salvi M, et al. 2012. Efficacy and safety of three different cumulative doses of intravenous methylprednisolone for moderate to severe and active Graves’ orbitopathy. J. Clin. Endocrin. Metab 97:4454–63 [DOI] [PubMed] [Google Scholar]
- 66.Sisti E, Coco B, Menconi F, Leo M, Rocchi R, et al. 2015. Intravenous glucocorticoid therapy for Graves’ ophthalmopathy and acute liver damage: an epidemiological study. Eur. J. Endocrinol 172:269–76 [DOI] [PubMed] [Google Scholar]
- 67.Sisti E, Coco B, Menconi F, Leo M, Rocchi R, et al. 2015. Age and Dose Are Major Risk Factors for Liver Damage Associated with Intravenous Glucocorticoid Pulse Therapy for Graves’ Orbitopathy. Thyroid 25:846–50 [DOI] [PubMed] [Google Scholar]
- 68.Marino M, Morabito E, Altea MA, Ambrogini E, Oliveri F, et al. 2005. Autoimmune hepatitis during intravenous glucocorticoid pulse therapy for Graves’ ophthalmopathy treated successfully with glucocorticoids themselves. J. Endocrinol. Invest 28:280–4 [DOI] [PubMed] [Google Scholar]
- 69.Kim JW, Han SH, Son BJ, Rim TH, Keum KC, Yoon JS. 2016. Efficacy of combined orbital radiation and systemic steroids in the management of Graves’ orbitopathy. Graefes Arch. Clin. Exp. Ophthalmol 254:991–8 [DOI] [PubMed] [Google Scholar]
- 70.Marcocci C, Bartalena L, Tanda ML, Manetti L, Dell’Unto E, et al. 2001. Comparison of the effectiveness and tolerability of intravenous or oral glucocorticoids associated with orbital radiotherapy in the management of severe Graves’ ophthalmopathy: results of a prospective, single-blind, randomized study. J. Clin. Endocrin. Metab 86:3562–7 [DOI] [PubMed] [Google Scholar]
- 71.Donaldson SS, Bagshaw MA, Kriss JP. 1973. Supervoltage orbital radiotherapy for Graves’ ophthalmopathy. J. Clin. Endocrin. Metab 37:276–85 [DOI] [PubMed] [Google Scholar]
- 72.Bartalena L 2002. Radioiodine therapy and Graves’ ophthalmopathy. Nucl. Med. Commun 23:1143–5 [DOI] [PubMed] [Google Scholar]
- 73.Del Monte MA. 2002. 2001 an ocular odyssey: lessons learned from 25 years of surgical treatment for graves eye disease. Am. Orthopt. J 52:40–57 [DOI] [PubMed] [Google Scholar]
- 74.Braun TL, Bhadkamkar MA, Jubbal KT, Weber AC, Marx DP. 2017. Orbital Decompression for Thyroid Eye Disease. Semin. Plast. Surg 31:40–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boboridis KG, Uddin J, Mikropoulos DG, Bunce C, Mangouritsas G, et al. 2015. Critical Appraisal on Orbital Decompression for Thyroid Eye Disease: A Systematic Review and Literature Search. Adv. Ther 32:595–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang-Nunes SX, Dang S, Garneau HC, Hwang C, Isaacs D, et al. 2015. Characterization and outcomes of repeat orbital decompression for thyroid-associated orbitopathy. Orbit 34:57–65 [DOI] [PubMed] [Google Scholar]
- 77.Boergen KP. 1989. Surgical repair of motility impairment in Graves’ orbitopathy. Dev. Ophthalmol 20:159–68 [DOI] [PubMed] [Google Scholar]
- 78.Papageorgiou KI, Ang M, Chang SH, Kohn J, Martinez S, Goldberg RA. 2012. Aesthetic considerations in upper eyelid retraction surgery. Ophthal. Plast. Reconstr. Surg 28:419–23 [DOI] [PubMed] [Google Scholar]
- 79.Olmos D, Tan DS, Jones RL, Judson IR. 2010. Biological rationale and current clinical experience with anti-insulin-like growth factor 1 receptor monoclonal antibodies in treating sarcoma: twenty years from the bench to the bedside. Cancer J. 16:183–94 [DOI] [PubMed] [Google Scholar]
- 80.Mahadevan D, Sutton GR, Arteta-Bulos R, Bowden CJ, Miller PJ, et al. 2014. Phase 1b study of safety, tolerability and efficacy of R1507, a monoclonal antibody to IGF-1R in combination with multiple standard oncology regimens in patients with advanced solid malignancies. Cancer Chemother. Pharmacol 73:467–73 [DOI] [PubMed] [Google Scholar]
- 81.Ramalingam SS, Spigel DR, Chen D, Steins MB, Engelman JA, et al. 2011. Randomized phase II study of erlotinib in combination with placebo or R1507, a monoclonal antibody to insulin-like growth factor-1 receptor, for advanced-stage non-small-cell lung cancer. J. Clin. Oncol 29:4574–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fleuren ED, Versleijen-Jonkers YM, Heskamp S, Roeffen MH, Bouwman WH, et al. 2013. The strength of small: improved targeting of insulin-like growth factor-1 receptor (IGF-1R) with F(ab’)(2)-R1507 fragments in Ewing sarcomas. Eur. J. Cancer 49:2851–8 [DOI] [PubMed] [Google Scholar]
- 83.Ferte C, Loriot Y, Clemenson C, Commo F, Gombos A, et al. 2013. IGF-1R targeting increases the antitumor effects of DNA-damaging agents in SCLC model: an opportunity to increase the efficacy of standard therapy. Mol. Cancer Ther 12:1213–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Qu X, Wu Z, Dong W, Zhang T, Wang L, et al. 2017. Update of IGF-1 receptor inhibitor (ganitumab, dalotuzumab, cixutumumab, teprotumumab and figitumumab) effects on cancer therapy. Oncotarget 8:29501–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smith TJ. 2015. TSH-receptor-expressing fibrocytes and thyroid-associated ophthalmopathy. Nat. Rev. Endocrinol 11:171–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen H, Mester T, Raychaudhuri N, Kauh CY, Gupta S, et al. 2014. Teprotumumab, an IGF-1R blocking monoclonal antibody inhibits TSH and IGF-1 action in fibrocytes. J. Clin. Endocrin. Metab 99:E1635–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Terwee CB, Dekker FW, Mourits MP, Gerding MN, Baldeschi L, et al. 2001. Interpretation and validity of changes in scores on the Graves’ ophthalmopathy quality of life questionnaire (GO-QOL) after different treatments. Clinic. Endocrinol 54:391–8 [DOI] [PubMed] [Google Scholar]
- 88.Terwee CB, Gerding MN, Dekker FW, Prummel MF, Wiersinga WM. 1998. Development of a disease specific quality of life questionnaire for patients with Graves’ ophthalmopathy: the GO-QOL. Br. J. Ophthalmol 82:773–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kurzrock R, Patnaik A, Aisner J, Warren T, Leong S, et al. 2010. A phase I study of weekly R1507, a human monoclonal antibody insulin-like growth factor-I receptor antagonist, in patients with advanced solid tumors. Clin. Cancer Res 16:2458–65 [DOI] [PubMed] [Google Scholar]
- 90.Smith TJ, Kahaly GJ, Ezra DG, Fleming JC, Dailey RA, et al. 2017. Teprotumumab for Thyroid-Associated Ophthalmopathy. N. Engl. J. Med 376:1748–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Scott SD. 1998. Rituximab: a new therapeutic monoclonal antibody for non-Hodgkin’s lymphoma. Cancer Pract. 6:195–7 [DOI] [PubMed] [Google Scholar]
- 92.Franks SE, Getahun A, Hogarth PM, Cambier JC. 2016. Targeting B cells in treatment of autoimmunity. Curr. Opin. Immunol 43:39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Salvi M, Vannucchi G, Campi I, Curro N, Dazzi D, et al. 2007. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur. J. Endocrinol 156:33–40 [DOI] [PubMed] [Google Scholar]
- 94.Stan MN, Garrity JA, Carranza Leon BG, Prabin T, Bradley EA, Bahn RS. 2015. Randomized controlled trial of rituximab in patients with Graves’ orbitopathy. J. Clin. Endocrin. Metab 100:432–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Salvi M, Vannucchi G, Curro N, Campi I, Covelli D, et al. 2015. Efficacy of B-cell targeted therapy with rituximab in patients with active moderate to severe Graves’ orbitopathy: a randomized controlled study. J. Clin. Endocrin.Metab 100:422–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nakahara H, Nishimoto N. 2006. Anti-interleukin-6 receptor antibody therapy in rheumatic diseases. Endocr. Metab. Immune. Disord. Drug Targets 6:373–81 [DOI] [PubMed] [Google Scholar]
- 97.Paul-Pletzer K 2006. Tocilizumab: blockade of interleukin-6 signaling pathway as a therapeutic strategy for inflammatory disorders. Drugs Today 42:559–76 [DOI] [PubMed] [Google Scholar]
- 98.Perez-Moreiras JV, Alvarez-Lopez A, Gomez EC. 2014. Treatment of active corticosteroid-resistant graves’ orbitopathy. Ophthal. Plast. Reconstr. Surg 30:162–7 [DOI] [PubMed] [Google Scholar]
- 99.Ye X, Bo X, Hu X, Cui H, Lu B, et al. 2017. Efficacy and safety of mycophenolate mofetil in patients with active moderate-to-severe Graves’ orbitopathy. Clin Endocrinol. 86:247–55 [DOI] [PubMed] [Google Scholar]
- 100.Allison AC, Eugui EM. 2000. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology 47:85–118 [DOI] [PubMed] [Google Scholar]
- 101.Sanders J, Allen F, Jeffreys J, Bolton J, Richards T, et al. 2005. Characteristics of a monoclonal antibody to the thyrotropin receptor that acts as a powerful thyroid-stimulating autoantibody antagonist. Thyroid 15:672–82 [DOI] [PubMed] [Google Scholar]
- 102.Chen CR, McLachlan SM, Rapoport B. 2009. A monoclonal antibody with thyrotropin (TSH) receptor inverse agonist and TSH antagonist activities binds to the receptor hinge region as well as to the leucine-rich domain. Endocrinology 150:3401–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sanders P, Young S, Sanders J, Kabelis K, Baker S, et al. 2011. Crystal structure of the TSH receptor (TSHR) bound to a blocking-type TSHR autoantibody. J. Mol. Endocrinol 46:81–99 [DOI] [PubMed] [Google Scholar]
- 104.Sanders J, Miguel RN, Furmaniak J, Smith BR. 2010. TSH receptor monoclonal antibodies with agonist, antagonist, and inverse agonist activities. Methods Enzymol. 485:393–420 [DOI] [PubMed] [Google Scholar]
- 105.Jaschke H, Neumann S, Moore S, Thomas CJ, Colson AO, et al. 2006. A low molecular weight agonist signals by binding to the transmembrane domain of thyroid-stimulating hormone receptor (TSHR) and luteinizing hormone/chorionic gonadotropin receptor (LHCGR). J. Biol. Chem 281:9841–4 [DOI] [PubMed] [Google Scholar]
- 106.Neumann S, Nir EA, Eliseeva E, Huang W, Marugan J, et al. 2014. A selective TSH receptor antagonist inhibits stimulation of thyroid function in female mice. Endocrinology 155:310–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Neumann S, Eliseeva E, McCoy JG, Napolitano G, Giuliani C, et al. 2011. A new small-molecule antagonist inhibits Graves’ disease antibody activation of the TSH receptor. J. Clin. Endocrin. Metab 96:548–54 [DOI] [PMC free article] [PubMed] [Google Scholar]