

Supplemental Digital Content is available in the text.
Key Words: head and neck, TCR, bifunctional, cell therapy, optimization, cytotoxicity, Jurkat
Abstract
Next-generation T-cell therapies will likely continue to utilize T-cell receptors (TCRs) and chimeric antigen receptors (CARs) because each receptor type has advantages. TCRs often possess exceptional properties even when tested unmodified from patients’ T cells. CARs are generally less sensitive, possibly because their ligand-binding domains are grafted from antibodies selected for binding affinity or avidity and not broadly optimized for a functional response. Because of the disconnect between binding and function among these receptor types, the ultimate potential of CARs optimized for sensitivity and selectivity is not clear. Here, we focus on a thoroughly studied immuno-oncology target, the HLA-A*02/HPV-E629–38 complex, and show that CARs can be optimized by a combination of high-throughput binding screens and low-throughput functional assays to have comparable activity to clinical TCRs in acute assays in vitro. These results provide a case study for the challenges and opportunities of optimizing high-performing CARs, especially in the context of targets utilized naturally by TCRs.
T-cell receptors (TCRs) are well known to achieve extraordinary levels of sensitivity and selectivity.1,2 These features and their ready adaptation to engineered T-cell therapy has prompted their use in multiple clinical trials, especially in cancer settings.3–6 TCRs that target tumor-associated antigens have been identified over the years and tested in the clinic with mixed results. In some cases, their utility has been limited by on-target toxicity (eg, carcinoembryonic antigen),4 off-target toxicity (eg, MAGE-A3),5,7 sporadic, unpredictable efficacy, or acquired resistance [eg, NY-ESO-1, human papillomavirus (HPV)].8,9 Nonetheless, the virtues of the engineered T-cell modality remain attractive, given the capacity of T cells for potent, specific killing, migration, and proliferation. Moreover, T cells are endowed with natural reactivity to major histocompatibility complexes that display cytoplasmic antigens, the fraction of the proteome that is both larger than the secreted/surface-antigen fraction and inaccessible to large-molecule therapeutics.
Antibodies offer an independent route to potent peptide-loaded major histocompatibility complex (pMHC)-directed cell therapeutics. Their ligand-binding structures can be grafted onto chimeric antigen receptors (CARs) to trigger cytotoxicity by engineered T cells.10–28 In principle, CARs may have advantages over TCRs that include: (i) compatibility with added vector payloads based on the smaller size, a benefit when vector cargo limitations are in play; (ii) modularity and tolerance to structural change, permitting additional molecular engineering, for example, bifunctional receptors29,30; (iii) simpler single-subunit compositions that do not comingle with native TCR/CD3 subunits; and, (iv) possible freedom from normal TCR regulatory mechanisms, including costimulation.13,28,31 Given the near inevitability of acquired resistance to therapeutics directed at single targets, some of these features may prove useful in next-generation cell therapies.32 However, compared with TCRs, CAR sensitivity and selectivity as a receptor class has been called into question.25 Whereas TCRs isolated from human blood are frequently able to respond to—even kill—target cells that express a few molecules/cell, CARs have rarely been shown to achieve sensitivities below ∼1000 molecules/cell and do not fair well in head-to-head comparisons with TCRs against the same targets.21,33,34 A few studies have demonstrated high-sensitivity killing of individual CAR-Ts; for example, against the OTS8 glycoprotein (∼300 molecules/cell), CD20 (∼200 molecules/cell), and CD19 (<100 molecules/cell).35–37 However, others have claimed an upper limit of senstivity for CARs, well above the threshold needed for TCRs.21
HPV is a serious human health problem, causing over 600,000 cancers each year worldwide,38 and is an attractive indication for cell therapy. Originally linked to cervical cancer, it is now clear that subsets of a variety of tumor types are associated with HPV. Almost 2 decades ago, highly effective prophylactic vaccines were developed.39 Despite their success, diagnoses of HPV-positive cancers continue to increase, and they are expected to continue to do so.40 In addition, therapeutic HPV vaccines have so far proved disappointing, so it is likely that new therapies directed at HPV-positive tumors will be needed.41 Cell therapy targeting HPV antigens offers a route to such treatments.
The mechanism by which HPV causes cancer involves 2 genes carried by the virus: E6 and E7. Both are classic viral oncogenes that inactivate host cancer defenses: E6 binds the tumor suppressor p53, and E7 binds Rb.42 The expression of both genes is required for continued survival and growth of tumor cells.43 E6 and E7 have little homology to human proteins, so they are especially attractive targets for drug discovery, as they afford absolute discrimination from normal cells. However, they reside in the cytoplasm, inaccessible to antibodies and other large molecules. In addition, neither is considered conventionally druggable; that is, inhibitable by small molecules. However, various E6 and E7 peptides are known to elicit an immune response by display on the cell surface, bound to specific HLA class I molecules.44,45 Because of their association with solid tumor malignancy, accounting for about three fourths of HPV-positive cancer, serotypes 16 and 18 have attracted special attention. For these reasons, both E6 and E7 have been pursued in the tumor-infiltrating lymphocyte and TCR-T fields.46,47 Tumor-infiltrating lymphocytes have demonstrated broad activity against HPV-positive cancers,46 and one of the high-quality TCRs developed by C. Hinrichs and colleagues is currently in the clinic. In the most recent trial, an HLA-A*02-restricted TCR selective for HPV E711–19 was given to 12 patients, 6 of whom experienced partial responses.3,9 These encouraging data nonetheless demonstrate the need for further improvement of the cell therapy platform for this tumor type.
We set out to test the limits of CAR pMHC screening and design, using HPV clinical TCRs as a comparator. Here, we provide a flow schema for making high-quality pMHC CARs. We use HPV as an example because of available benchmarks and other well-characterized reagents. Highly optimized CARs, roughly equal to TCRs in sensitivity and selectivity, were generated and examined in Jurkat and primary T-cell assays alongside the clinical TCRs. These CARs provide proof of concept that CARs can be engineered that are comparable to patient-derived TCRs in potency and other preclinical attributes but also provide insight into the challenges of achieving ultra-high sensitivity for CARs.
METHODS
Cell Lines and Peptides
T2 (CTL-1992), CaSki (CRL-1550), and A375 firefly luciferase (CRL-1619-LUC2) cells were purchased from ATCC. Jurkat NFAT-firefly luciferase cells were purchased from BPS Bioscience (#60621). When needed, green fluorescent protein (GFP) was stably expressed in A375 and CaSki cells using lentivirus vectors. All cell lines were cultured in media as recommended by the vendors. Penicillin-streptomycin of 100 U/mL (Gibco; 15140163) (1× P/S) was used in all media. Suspension cells were maintained below a density of 1 E6/mL. Adherent cells lines were passaged at ∼80% confluency.
All peptides were purchased from GenScript by custom order.
pMHC AlphaScreen
Streptavidin-conjugated donor beads were purchased from PerkinElmer (6760002B). Acceptor beads (6762002; PerkinElmer) were conjugated in-house with 1 mg/mL W6/32 (BE0079; Bioxell). The reaction was performed in 15 uL refolding buffer (50 mM Tris-maleate pH 6.6 and 0.03% pluronic acid) containing purified alpha subunit (see below) and beta-2-microglobulin (B2M) (0877095-CF; MP Biomedicals) and varying concentration of peptides (4 points tested per peptide: 50, 5, 0.05, or 0.005 μM; custom order from Genscript). Optimized concentrations of each purified alpha subunit in combination with B2M were used on a per allele basis as follows: 0.3 μM A*01:01+0.6 μM B2M, 1 μM A*02:01+2 μM B2M, 0.1 μM A*03:01+0.2 μM B2M, 0.1 μM A*11:01+0.2 μM B2M, 0.1 μM C*07:01+0.6 μM B2M, or 0.1 μM C*07:02+0.6 μM B2M. The refolding reaction mixture was incubated overnight at 37°C for 18 hours before being transferred to a 384-well Proxiplate (6008289; PerkinElmer). Then, 5 μL mixture of donor beads and acceptor beads in a 1:1 ratio was added at a final total bead concentration of 20 μg/mL. The plates were incubated at room temperature for 1 hour before the signal was measured via EnSpire 2300 (PerkinElmer).
pMHC Probe Generation
HLA-A*02:01 and HLA-A*11:01 were expressed and purified from Escherichia coli as previously described.48,49 Briefly, the extracellular domains of HLA-A*02:01 or HLA-A*11:01 were expressed in strain BL21 (DE3) pLysS cells (Invitrogen; C606003). IPTG at 1 mM for 4 hours at 42°C was added to induce expression. Cells were pelleted and lysed in 50 mM Tris-HCl, pH 8.0, 5 mM MgCl2, 1% (v/v) Triton-X 100, protease inhibitor (Thermo Scientific; 78439) and benzonase. Inclusion bodies were collected by centrifugation, resuspended, and washed in inclusion-body wash buffer (50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.1% (w/v) sodium azide and 0.5% (v/v) Triton-X 100). The centrifugation/wash step was repeated 6–8×; the final wash step used inclusion-body wash buffer without 0.5% (v/v) Triton-X 100. The inclusion bodies were solubilized with 25 mM MES, pH 6.0, 8 M urea, and 10 mM EDTA, followed by centrifugation to deplete debris. To generate pMHC complexes, peptides were dissolved in dimethyl sulfoxide (DMSO); then, B2M (purified in-house or from MP Biomedicals; 08770953), and the alpha chain of HLA class I were added dropwise to fresh refolding buffer (100 mM Tris-HCl, pH 8, 0.5 M l-arginine, 2 mM EDTA, 5 mM reduced glutathione, 0.5 mM oxidized glutathione, and 1:100 diluted protease inhibitor) in sequence to reach final concentration 1, 2, 3 μM, respectively. The mixture was stirred at 10°C for at least 6–10 hours between each addition. The supernatant was collected and concentrated before purification using an ion-exchange Hi-Trap Q column (GE; 3 17115401), followed by a size-exclusion HiLoad 16/600 Superdex 00 column. Post purification, protein concentration and biotinylation efficiency were determined by absorbance at 280 nm and HABA assays (Invitrogen; 28005), respectively.
HuTARG Sort
HuTARG primary libraries were obtained from Innovative Targeting Solutions Inc. An in vitro V(D)J repertoire with >1 billion diversity was generated by the expression of RAG-1 and TdT in the host cells as described previously.50 Using a flow sorter device (Aria II; BD Biosciences), the library was enriched for cells that expressed antibodies that bind specifically to target pMHC probes but not to off-target pMHCs. Multiple enrichment rounds were performed to increase on-target and decrease off-target binding. In the final round, on-target and off-target binding cells were isolated and subjected to RNA extraction and reverse transcription. Polymerase chain reaction fragments containing the complementarity-determining region (CDR) regions were produced from the cDNA template, followed by targeted next-generation sequencing (NGS) to determine the frequency of each unique CDR region. The degree of enrichment/depletion was determined by comparing the output and input NGS counts.
Target-specific binders from the primary libraries were used to generate optimization libraries to further improve on-target sensitivity and/or reduce off-target cross-reactivity. Optimization libraries were constructed by diversification of CDR1, CDR2, or CDR3 of the light or heavy chains of parental binders via in vitro RAG-mediated V(D)J recombination. The optimization library was enriched for on-target activity and depleted for off-target activity, followed by NGS, as described for the primary library.
Molecular Cloning
All CAR constructs were created from fusions of single-chain variable fragment (scFv), hinge, transmembrane, and intracellular signaling domains. The hinge was derived from CD8, the transmembrane domain from CD28, and the signaling domain from CD28, 4-1BB, and CD3, unless otherwise specified. Gene segments were fused using the Golden Gate cloning systems and inserted downstream of a human EF1α promoter in a lentivirus expression plasmid. These expression plasmids were used for transient expression and lentivirus packaging.
A2 Scan
Sets of overlapping 9-mer and 10-mer peptides with an offset of a single residue were extracted from the human proteome (20,400 reviewed sequences). Peptides of 11,190,659 10-mer and 11,211,081 9-mer were scored using 3 different methods: (1) identity scoring; (2) anchor scoring; and (3) incompatibility scoring. Identity score compared the 9-mer and 10-mers to the target peptide; an identical amino acid to the target peptide at each position was scored +1. Anchor score took into account the anchor positions and residue preferences for each major histocompatibility complex class I allele [Source: IEDB (Immune Epitope Database and Analysis Resource): www.iedb.org]. Preferred amino acids at anchor positions were scored as +1, tolerated amino as +0.5, and unfavorable amino acids as −0.5. Incompatibility scores penalized differences in the physicochemical properties of amino acids at each position of the peptides when compared with the target peptide. Cutoffs used for the selection of potential cross-reactive peptides were based on the titin (TTN) peptide which was proposed as the most likely cause of the off-target toxicity for a MAGE-A3 pMHC target.51
Jurkat:T2 Cell Assay
TCR and CAR expression constructs in the lentiviral vector were transfected on day 1 into Jurkat NFAT-firefly luciferase cells using standard protocols for the Lonza 4D Nucleofector (AAF-1002B). T2 cells were loaded with peptides listed in Tables 1–3 and Supplementary Tables 2 and 3 (Supplemental Digital Content 1, http://links.lww.com/JIT/A623). After resuspension in DMSO, peptides were diluted 20 times serially 3× per step. Serially diluted peptide solutions were added to T2 cells resuspended in peptide-loading media (RPMI1640+1% BSA+1× P/S), yielding peptide-loaded T2 cells at ∼1 E6/mL, with peptide concentrations ranging from ∼10 fM to 100 μM, including control at 0 μM. Peptide-loaded T2 cells were incubated overnight at 37°C in 384-well plates (Thermo Scientific; AB0781). On day 2, peptide-loaded T2 cells (10,000 cells/well) were added to CAR/TCR-transfected Jurkat-NFAT-firefly luciferase cells (12,000 cells/well) to a final volume of 20 μL in a 384-well plate (Corning; 3570). Luminescence intensity was measured on a Tecan Infinite M1000 with the One-Step Luciferase assay system (firefly luciferase, 60690; BPS Bioscience) after a 6-hour incubation at 37°C.
TABLE 1.
Peptide-loaded Major Histocompatibility Complex Targets of Chimeric Antigen Receptors Screened in This Study
| Peptide Name | Peptide Position | HLA | Sequences | Clinical Trial |
|---|---|---|---|---|
| HPV E629–38 52,53 | 29–38 | A*02 | TIHDIILECV | NCT02280811 |
| HPV E711–19 53–55 | 11–19 | A*02 | YMLDLQPET | NCT02858310 |
| HPV E782–90 54–56 | 82–90 | A*02 | LLMGTLGIV | — |
| HPV E77–15 54,55 | 7–15 | A*02 | TLHEYMLDL | — |
| HPV E659–67 55 | 59–67 | A*11 | IVYRDGNPY | — |
| HPV E693–101 55 | 93–101 | A*11 | TTLEQQYNK | — |
HPV indicates human papillomavirus.
TABLE 3.
Summary of On-target and Near Off-target Activity of HPV E629–38 TCR and Examples of Light-chain and Heavy-chain Optimized CARs in Jurkat:T2 and Acute Primary T-Cell Assays
| Jurkat:T2 Assay | Primary T-Cell Assay | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Constructs | Construct Format/Source | Test Peptide Name | Test Peptide Sequence | On/Off Target | EC50 (nM) | ECminimum (nM) | Emaximum (RLU) | Selectivity | Primary T-Cell EC 50 (nM) |
| C882 | TCR | HPV E711–19 | YMLDLQPET | On | 0.01 | 0.007 | 11,000 | NA | 0.05 |
| C881 | TCR | HPV E629–38 | TIHDIILECV | On | 0.6 | 0.05 | 18,000 | NA | 0.4 |
| PRR5 | NLVDQILESV | Off | >100,000 | NA | NA | >167 | ND | ||
| PTPRD | FIHDALLEAV | Off | >100,000 | NA | NA | >167 | ND | ||
| C1044 | Primary CAR | HPV E629–38 | TIHDIILECV | On | 2200 | NA | 17,315 | NA | NA |
| C1053 | Primary CAR | HPV E629–38 | TIHDIILECV | On | 1500 | NA | 30,000 | NA | NA |
| C1068 | Primary CAR | HPV E629–38 | TIHDIILECV | On | 930 | NA | 34,000 | NA | NA |
| C1072 | Primary CAR | HPV E629–38 | TIHDIILECV | On | 1100 | NA | 7500 | NA | NA |
| C1073 | Primary CAR | HPV E629–38 | TIHDIILECV | On | 3400 | NA | 9200 | NA | NA |
| C1078 | Primary CAR | HPV E629–38 | TIHDIILECV | On | 550 | NA | 12,500 | NA | NA |
| C1987 | Light-chain optimized CAR (parent of CT503) | HPV E629–38 | TIHDIILECV | On | 28 | 0.56 | 50,000 | NA | 5235 |
| PRR5 | NLVDQILESV | Off | >250 | NA | NA | >9 | ND | ||
| PTPRD | FIHDAILEA | Off | 800 | ND | 38,000 | 29 | ND | ||
| C2388 | Light-chain optimized CAR | HPV E629–38 | TIHDIILECV | On | 7.7 | 0.28 | 71,000 | NA | 600 |
| PRR5 | NLVDQILESV | Off | 88 | ND | 57,000 | 11 | ND | ||
| PTPRD | FIHDAILEA | Off | >100,000 | NA | NA | >12,000 | ND | ||
| C1992 | Light-chain optimized CAR (parent of CT512) | HPV E629–38 | TIHDIILECV | On | 5 | 0.02 | 114,000 | NA | 108 |
| PRR5 | NLVDQILESV | Off | 930 | ND | 55,000 | 186 | ND | ||
| PTPRD | FIHDALLEAV | Off | >100,000 | NA | NA | >20,000 | ND | ||
| CT503 | Light-chain and heavy-chain optimized CAR | HPV E629–38 | TIHDIILECV | On | 0.5 | 0.02 | 30,000 | NA | 6.5 |
| PRR5 | NLVDQILESV | Off | 121 | ND | 28,000 | 242 | ND | ||
| PTPRD | FIHDALLEAV | Off | 34 | ND | 30,000 | 68 | ND | ||
| CT512 | Light-chain and heavy-chain optimized CAR | HPV E629–38 | TIHDIILECV | On | 0.2 | 0.06 | 30,000 | NA | 5.2 |
| PRR5 | NLVDQILESV | Off | 3.5 | ND | 27,000 | 18 | ND | ||
| PTPRD | FIHDALLEAV | Off | 280 | ND | 5200 | 1400 | ND | ||
CAR indicates chimeric antigen receptor; HPV, human papillomavirus; NA, not applicable; ND, not determined; RLU, relative luminescence unit; TCR, T-cell receptor.
Primary T-Cell Transduction
For human peripheral blood mononuclear cells (PBMCs), collection protocols and donor informed-consent were approved by an Institutional Review Board at Allcells. Allcells followed HIPAA compliance and approved protocols (www.allcells.com/cell-tissue-procurement/donor-facilities/). PBMCs were purified from Leukopaks purchased from Allcells according to the method described by Garcia et al.57 Unless otherwise specified, all LymphoONE media (Takara; WK552) was supplemented with 1% human AB Serum (GeminiBio; 100-512). Lentivirus CAR and TCR constructs were packaged at Alstem (Richmond, CA) using the expression constructs in the lentiviral vector. Human PBMCs were grown in LymphoONE and supplemented with TransAct (Miltenyi; 130-111-160) following the manufacturers guidelines (1:100 dilution) for 24 hours before being transduced with lentivirus encoding a CAR or TCR. Additional LymphoONE supplemented with interleukin 2 (IL-2) (300 IU/mL) was added 24 hours after transduction to transduced cells and cultured for 3 days before transfer to a 24-well G-Rex plate (Wilson Wolf; 80192M). Fresh IL-2 (300 IU/mL) was added every 48 hours with a media change every 7 days during expansion in G-Rex plates. Expression and antigen binding of transduced CARs or TCRs in primary T cells was confirmed by flow cytometry as described below. CAR-expressing or TCR-expressing cells were labeled with protein l-biotin/streptavidin-PE or mTCR-PE, followed by anti-PE microbeads (Miltenyi; 130-048-801) according to the manufacturer’s protocol, and subsequently enriched using AutoMACS Pro Separator (Miltenyi). Enriched cells were grown in G-Rex plates until harvest.
Cytotoxicity Measurements
For peptide-loading assays, A375 firefly luciferase cells were loaded with peptides as described above in the Jurkat:T2 section. For cytotoxicity on cell lines with or without endogenous HPV expression (ie, CaSki and A375, respectively), cells were plated in LymphoONE supplemented with 1% human serum and 1× P/S before coculture with T cells. To determine the number of target cells seeded per well, 24 hours after peptide loading or target cell seeding, a calibration curve was generated using CellTiter-Glo (Promega; G7570) readout. T cells were mixed with target cells at 3:1 effector cell to target cell ratio (E:T) (for peptide-loaded target cells) or 9:1 (for target cells without peptide loading), according to the target cell number determined by the calibration curve. Cytotoxicity of primary T cells was quantified by the bioluminescent signal using One-Step Luciferase assay system firefly (BPS Bioscience; 60690), or Renilla Luciferase Assay System (Promega; E2810) on Tecan Infinite M1000 after 48-hour coculture at 37°C. Loss of GFP signal in Caski or A375 cells was monitored using IncuCyte S3 to determine cytotoxicity.
Interferon γ (IFN-γ) Secretion Assay
T cells engineered with a TCR or scFv CAR were cocultured with 10,000 HPV E6 or E7 peptide-loaded A375 cells per well at a 1:1 ratio in 96-well flat-bottom plates. After 16 hours concentration of IFN-γ was measured in the supernatant using BD CBA Flex Set and BD human soluble protein master buffer kits according to the manufacturer’s instructions (BD Biosciences). Briefly, 50 µL of the provided standard or the cell culture supernatant was incubated together with 50 µL of capture beads in the dark for 1 hour at room temperature. Then, 50 µL of the detection reagent was added, the samples were incubated and mixed for 2 hours, followed by analysis by flow cytometry on a BD FACS Canto II. Concentrations were estimated from interpolation of median fluorescence intensity on a 10-point dilution standard curve.
Repeated Antigen-challenge Assay
A375 GFP+ target cells were loaded with target peptide (custom ordered from Genescript) in LymphoONE supplemented with 1% human serum and 1× P/S as described above. Peptide-loaded A375 target cells were seeded in either 48-well (for “cycle” 1) or 96-well (for subsequent cycles) plates at 27,000/well or 9000/well, respectively. Twenty-four hours after peptide loading, T cells were mixed with target cells at 3:1 E:T ratio. Plates were placed in the IncuCyte S3 Platform (Sartorius) and imaged every 2 hours for 48 or 72 hours for cycle 1 and subsequent cycles, respectively. At the end of each cycle, cells in suspension were separated from adherent target cells and counted before added to the subsequent cycle of coculture with equivalently peptide-loaded target A375 cells and incubated for 72 hours. This process was repeated until T cells no longer killed peptide-loaded target cells (<10% loss of GFP signal observed in a given round). Data was reported as percent of control of the GFP signal relative to A375+ DMSO-only controls.
RESULTS
Identification of Potent, Selective scFvs That Bind HPV E629–38 and E711–19 pMHCs
As a first step, we assembled a set of 454 overlapping 9-mer and 10-mer peptides, each offset by 1 amino acid, spanning the E6 (1–148 amino acids) and E7 (1–98 amino acids) sequences of HPV16 (E6: 139 9-mers and 138 10-mers tested; E7: 89 9-mers and 88 10-mers tested, Supplementary Fig. 1, Supplemental Digital Content 1, http://links.lww.com/JIT/A623). The HPV16 serotype accounts for over half of HPV-positive malignancies (Supplementary Table 1, Supplemental Digital Content 1, http://links.lww.com/JIT/A623).58 The HPV peptides were mixed individually with 6 purified common HLA class I proteins and assayed via AlphaScreen, a standard method for quantifying biochemical interactions.59 Positives were retested and plotted as percent control. A total of 317 unique pMHC complexes were identified, showing >5 percent of control binding to at least 1 of the 6 HLA class I proteins tested (Figs. 1A, B). Sixteen different peptides, the top hits from the Alphascreen, were selected, synthesized, and tested for pMHC probe assembly. Eight of the 16 peptides formed stable pMHC complexes. All 16 of these pMHCs are part of the sequence set encompassed by 10 longer, nonoverlapping peptides described in a previous report that demonstrated immunoreactivity of HPV-positive patients’ T cells against E6/E7 pMHCs54 (Fig. 1C). Our analysis determined HLA association for all 30 peptides. In addition, we also defined 3 peptides that bound to 2 different HLA allelic forms tested.
FIGURE 1.
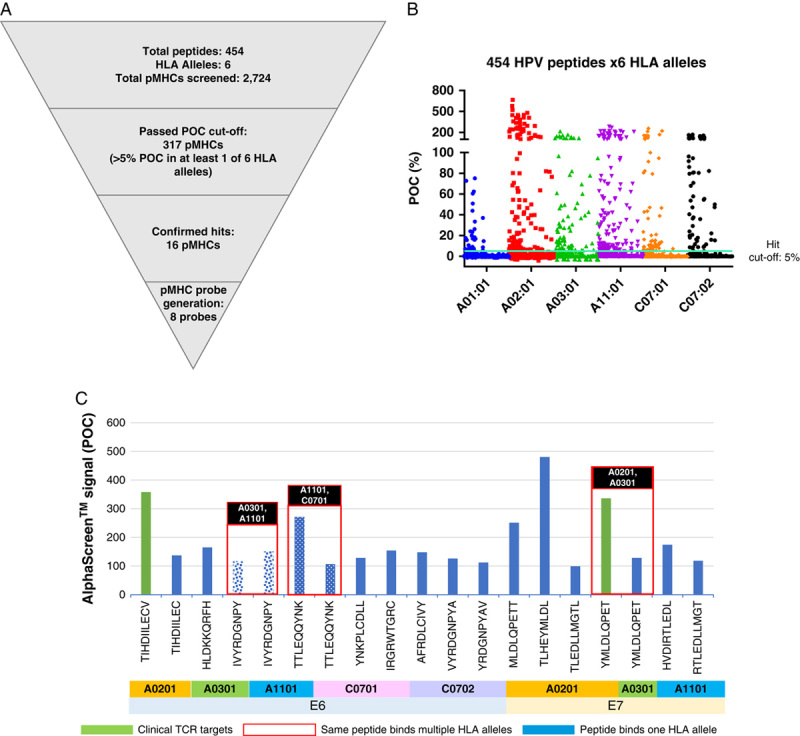
HPV pMHC target identification. A, Diagram of filters used for the Alphascreen of HPV peptides. B, Hit selection for AlphaScreen; hit cutoff: >5 POC in at least 1 of 6 HLA alleles; (3× SD <5% POC). C, Sixteen different peptides were identified and confirmed in Alphascreen; 8 formed stable complexes and were used in binder isolation (see the Methods section). Peptides that can bind to 2 HLA alleles are shown in the red boxes. All peptides shown are derived from larger sequences identified by Ramos et al.54 HPV indicates human papillomavirus; pMHC, peptide-loaded major histocompatibility complex; POC, percent of control; TCR, T-cell receptor.
We selected 6 pMHCs (Table 1, Supplementary Fig. 1, Supplemental Digital Content 1, http://links.lww.com/JIT/A623), all HLA-A*02-restricted, or A*11-restricted, for further study and screened for antibody binders against all of these complexes using a mammalian display system called HuTARG.50 This system allows the generation of an entire heavy-chain antibody repertoire paired with a single light chain or scFV libraries with custom heavy-chain and light-chain combinations, with diversity in the range of 1 billion idiotypes.30 By linking the pMHCs to biotin during expression in E. coli, streptavidin triggered the formation of tetramers that could be conjugated to a fluorochrome. These labeled probes were used to screen the library with a fluorescence-activated cell sorter (FACS; Figs. 2A, B). The primary screens yielded binders that were converted to scFvs and tested in Jurkat:T2 functional assays for sensitivity and selectivity (Supplementary Figs. 2, 3, Supplemental Digital Content 1, http://links.lww.com/JIT/A623). Four of 6 pMHCs yielded functional CARs with EC 50 in or below the range of 1 μM (see Fig. 3, Table 2, Supplementary Fig. 4, Supplementary Table 3, Supplemental Digital Content 1, http://links.lww.com/JIT/A623, for examples of primary binders for all 4 pMHCs). Two pMHCs, HPV E629–38, and HPV E711–19, were selected for further study because they produced primary binders with better sensitivity and had TCR benchmarks available for comparison (see Figs. 3A, 4A, Supplementary Fig. 4A, Supplemental Digital Content 1, http://links.lww.com/JIT/A623, for examples of E6 and E7 primary binder dose-response curves; Tables 2, 3, for summary of activity).
FIGURE 2.
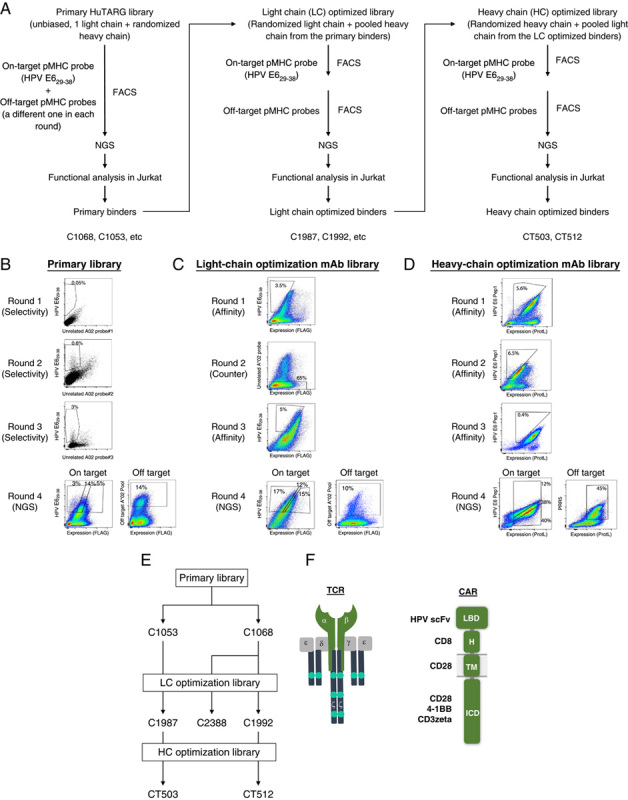
Overview of sorting strategy and representative scatter plots. HPV E629–38 is used as an example. A, Flowchart of the HuTARG library, FACS, and optimization process. B, An unbiased HuTARG library was screened against HPV E629–38 pMHC mixed with an unrelated pMHC probe to serve as counterscreen in each round. Cells that bound to HPV E629–38 pMHC were enriched over 3 rounds of FACS, while nonspecific binders were depleted by gating against cells binding the counterscreen probe. Subsequently, cells with high, medium, and low on-target probe binding and off-target probes binding were collected separately. cDNAs were generated from these cells and pooled with barcodes. Complementarity-determining regions of binders were determined by NGS using pooled cDNA samples. C, Thirty-two HPV E629–38 binders from the primary HuTARG library (primary binders) were selected based on their functional activity in Jurkat assays as parents for light-chain optimization. A HuTARG library was built containing a mixture of binders with light-chain variations paired with the heavy chains from the 32 parents. The optimization library was screened against HPV E629–38 using the same strategy as the primary library, except that the on-target and off-target counterscreen were done in separate rounds by fluorescence-activated cell sorter. D, Thirty-five binders from the light-chain optimization library were selected based on their functional activity in Jurkat assays as parents for heavy-chain optimization. A HuTARG library was built containing a mixture of binders with heavy-chain variations paired with the light chains from the 35 parents. The optimization library was screened against HPV E629–38 using the same strategy as the light-chain optimization library. E, Diagram of relationships of the HPV E6 binders tested. F, Structural diagram of CARs and TCRs tested. CAR indicates chimeric antigen receptor; FACS, fluorescence-activated cell sorter; HPV, human papillomavirus; ICD, intracellular domain; LBD, ligand-binding domain; mAb, monoclonal antibody; NGS, next-generation sequencing; pMHC, peptide-loaded major histocompatibility complex; TCR, T-cell receptor.
FIGURE 3.
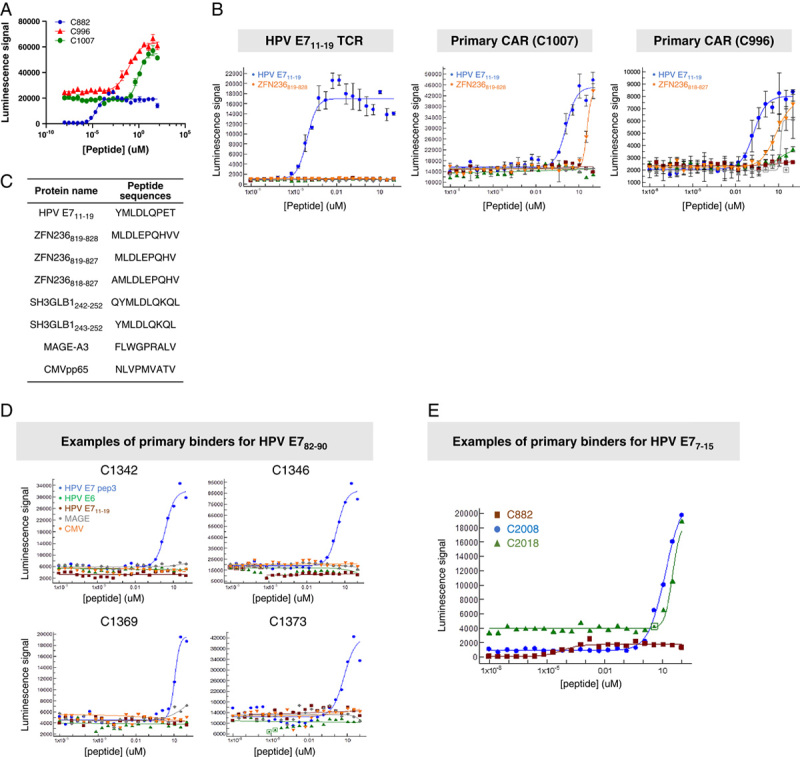
Sensitive and selective HPV E711–19 primary binders (C1007 and C996; examples of the best binders in peptide-loading Jurkat:T2 assay). A, Comparison of the primary binders to the TCR (C882). B, Selectivity of the best primary binder compared with the TCR. Blue lines are the on-target dose-response curves. Other colors are near off-target curves. C, Near off-target peptide sequences for HPV E711–19 are listed in the inserted table. Primary binder: binders identified from primary HuTARG library. D, Examples of on-target and far off-target activity of primary binders against HPV E782–90. E, Examples of on-target activity of primary binders against HPV E77–15. CAR indicates chimeric antigen receptor; CMV, cytomegalovirus; HPV, human papillomavirus; TCR, T-cell receptor.
TABLE 2.
Summary of On-target and Near Off-target Activity of HPV E711–19 TCR and Examples of Best Primary CARs in Jurkat:T2 Assays
| Jurkat:T2 Assay | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Constructs | Construct Format or Source | Test Peptide Name | Test Peptide Sequence | On/Off Target | EC50 (μM) | ECminimum (μM) | Eminimum (RLU) | Emaximum (RLU) | Selectivity |
| C882 | TCR | HPV E711–19 | YMLDLQPET | On | 0.00005 | 0.000008 | 900 | 14,000 | NA |
| ZFN236819–828 | MLDLEPQHVV | Off | >100 | NA | 740 | NA | >2,000,000 | ||
| ZFN236819–827 | MLDLEPQHV | Off | >100 | NA | 900 | NA | >2,000,000 | ||
| SH3GLB1242–252 | QYMLDLQKQL | Off | >100 | NA | 900 | NA | >2,000,000 | ||
| SH3GLB1243–252 | YMLDLQKQL | Off | >100 | NA | 900 | NA | >2,000,000 | ||
| C1007 | Primary binder CAR | HPV E711–19 | YMLDLQPET | On | 1.7 | 0.137 | 16,000 | 52,000 | NA |
| ZFN236819–828 | MLDLEPQHVV | Off | 35 | ND | 17,000 | 39,000 | 21 | ||
| ZFN236819–827 | MLDLEPQHV | Off | >100 | NA | NA | NA | >59 | ||
| SH3GLB1242–252 | QYMLDLQKQL | Off | >100 | NA | NA | NA | >59 | ||
| SH3GLB1243–252 | YMLDLQKQL | Off | >100 | NA | NA | NA | >59 | ||
| C996 | Primary binder CAR | HPV E711–19 | YMLDLQPET | On | 0.07 | 0.005 | 6806 | 27,604 | NA |
| ZFN236819–828 | MLDLEPQHVV | Off | >100 | NA | NA | NA | >1428 | ||
| ZFN236819–827 | MLDLEPQHV | Off | >100 | NA | NA | NA | >1428 | ||
| ZFN236818–827 | AMLDLEPQHV | Off | 13.2 | ND | 2365 | 3705 | 189 | ||
| SH3GLB1242–252 | QYMLDLQKQL | Off | 13.7 | ND | 1994 | 6108 | 196 | ||
| SH3GLB1243–252 | YMLDLQKQL | Off | 6.5 | ND | 2245 | 7111 | 93 | ||
C996 is the parent of C1800 and C1806 shown in Supplementary Figure 4A and Supplementary Table 3 (Supplemental Digital Content 1, http://links.lww.com/JIT/A623).
CAR indicates chimeric antigen receptor; HPV, human papillomavirus; NA, not applicable; ND, not determined; RLU, relative luminescence unit; TCR, T-cell receptor.
FIGURE 4.
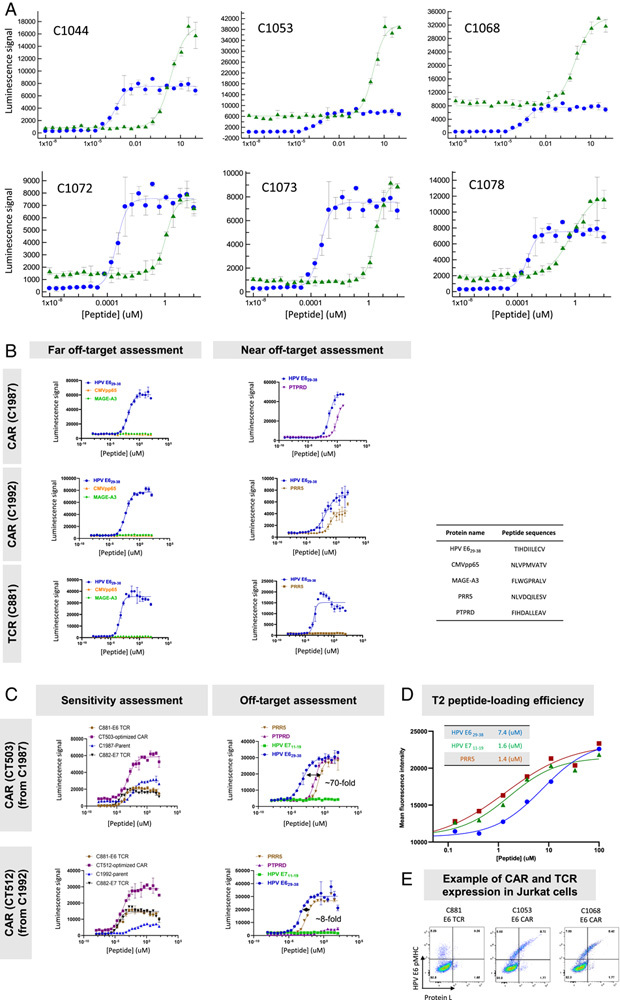
Sensitive and selective HPV E629–38 primary, light-chain optimized, and heavy-chain optimized binders identified by sorting (examples of the best binders in peptide-loading Jurkat:T2 assay). Each data point is an average of 3 replicates, and all experiments were run at least twice, except where noted. An example of the repeated experiments is shown in this figure. A, Examples of dose-response curves of on-target activity of HPV E629–38 binders from primary HuTARG screen (green) compared with the HPV E629–38 TCR (blue). Their EC 50 and E maximum are summarized in Table 3. B, Examples of HPV E629–38 light-chain optimized binders: sensitivity, near and far off-target selectivity. C, Examples of HPV E629–38 heavy-chain optimized binders: sensitivity, near and far off-target selectivity. D, Peptide-loading efficiency comparison of HPV E629–38, HPV E711–19, and PRR5. This experiment was run once. MFI was measured after staining with W6/32 antibody (see the Results section). E, Flow plot of CAR and TCR expression in Jurkat cells. The sequences of peptides used in this figure are listed in the inserted table. Peptide-loading efficiencies are summarized in the inserted table in (C). CAR indicates chimeric antigen receptor; CMV, cytomegalovirus; HPV, human papillomavirus; TCR, T-cell receptor.
To drive further sensitivity, optimization libraries for binders of HPV E629–38 and HPV E711–19 were constructed (see the Methods section, Supplementary Figs. 2, 3, Supplemental Digital Content 1, http://links.lww.com/JIT/A623). These libraries involved pooling the top primary binder heavy-chain CDR3s and using the HuTARG platform to generate diversity in the CDRs of the light chain (see the sorting strategy in Fig. 2A). The CDR-optimization libraries were screened via FACS to select higher avidity binders. The best-optimized binders were identified based on DNA sequence frequencies (see the Methods section; enrichment in on-target sorted cells; depletion in off-target sorted cells). Functional analysis of scFv-formatted binders in CARs revealed a range of sensitivities (Fig. 4A, Table 3, Supplementary Table 3, Supplemental Digital Content 1, http://links.lww.com/JIT/A623). For unknown reasons, the selected HPV E711–19 binders all had high backgrounds; that is, target-peptide-independent signaling in Jurkat cells (2 examples are shown in Fig. 3A). In addition, light-chain binder optimization did not significantly improve sensitivity and selectivity for HPV E711–19 primary binders (Table 2, Supplementary Fig. 4, Supplementary Table 3, Supplemental Digital Content 1, http://links.lww.com/JIT/A623). Thus, HPV E711–19 pMHC binders were dropped from further investigation to focus on E629–38 binders. However, the HLA-A*02-restricted TCR selective for HPV E711–19 was retained as a benchmark given the encouraging clinical experience with this engineered T-cell product.
To further optimize HPV E629–38 binders, we selected 32 primary binders with different heavy chains and a common light chain for additional light-chain optimization (Fig. 2C). Following binder enrichment by FACS and NGS analysis to identify the most enriched on-target binders, the top-ranked binders (C1987 and C1992 as 2 examples) were further studied to measure potential off-target cross-reactivity against unrelated peptides and “near off-target” functional activity against 47 closely related peptides in Jurkat-cell assays (Fig. 4B, Table 3, Supplementary Fig. 5, Supplementary Table 2, Supplemental Digital Content 1, http://links.lww.com/JIT/A623). These 47 peptides, predicted from the human proteome as most similar to HPV E629–38, were identified by NCBI BLAST and a simple matching algorithm (see A2 Scan section in the Methods section). All of these peptides derive from human proteins with transcript levels between 1 and 40 TPM in multiple tissues (GTEx Web site, www.gtexportal.org/home). Thirty-five light-chain optimized binders with unique light-heavy-chain combinations and minimal near off-target activity were selected for heavy-chain optimization (Fig. 2D). The 2 most sensitive heavy-chain optimized binders, CT503 and CT512, were derived from 2 different light-chain parents, C1987 and C1992, respectively (Fig. 2E).
Characterization of Fully Optimized HPV E629–38 CARs in Jurkat Cell Assays
The most enriched, fully optimized (light chain and heavy chain) E6 binders were studied to determine sensitivity and selectivity. In Jurkat-cell assays, the 2 most sensitive binders (CT503 and CT512) had EC 50 <1 nM (Fig. 4C, Table 3). The improvement over their parental (light-chain optimized only) sequences was ∼10× (Figs. 4B, C; Table 3). CT503 and CT512 matched the benchmark E6 TCR but were ∼20× less potent than the benchmark E7 TCR. Some of this shortfall was explained by the loading efficiency of HPV E711–19 compared with HPV E629–38, where the observed EC 50 for loading in T2 cells was 5× right-shifted for HPV E629–38 (Fig. 4D). E maximum was higher for both CARs than for the TCR and baseline (tonic-signaling) slightly higher. Selectivity was high. However, the fully optimized E6 scFv CT503 retained cross-reactivity to the PTPRD peptide from its parent (C1987) and acquired cross-reactivity to the PRR5 peptide (Figs. 4B, C). The other selected fully optimized E6 scFv C512 retained reactivity from its parent (C1992) to the PRR5 peptide. This suggests that boosting potency did not, of itself, improve relative selectivity, and heavy-chain optimization may increase cross-reactivity. We noted that E maximum values in the Jurkat:T2 assay were up to 2–3× higher for the E629–38 CARs (CT503, CT512) compared with the E7 TCR. Previous results suggest that E maximum correlates with total functional receptor levels, and in this case, the CARs appeared to express at ∼3× higher levels compared with the TCR, based on probe binding16 (Fig. 4E).
We next explored the potential role of different CAR signaling formats in acute sensitivity of the HPV E629–38 CARs (Fig. 5). We tested the CT503 and CT512 CAR ligand-binding domains in a Gen2 construct (hinge/transmembrane/intracellular domain=CD8/CD28/4-1BB−CD3ζ) and 4 variants of a Gen3 construct (=CD8/CD28/CD28−4-1BB−CD3ζ): heavy chain variable domain (VH)/light chain variable domain (VL) scFv; VL/VH scFv; bivalent scFv; and tandem CD3ζ [CD3ζ(×2)] (Fig. 5). Even though EC 50 varied ∼15–50× among these variants, none of the variants showed higher sensitivity than the original CT503 or CT512 Gen3 constructs. In addition, E maximum was affected in some cases (Supplementary Table 4, Supplemental Digital Content 1, http://links.lww.com/JIT/A623). Thus, simple alteration of format did not significantly improve the acute sensitivity of the CARs in Jurkat cells.
FIGURE 5.
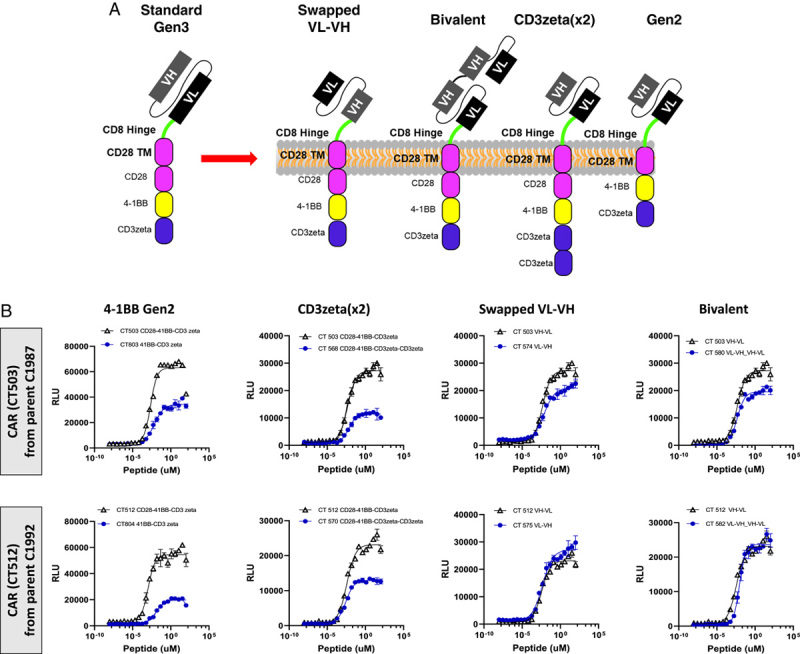
Effect of intracellular domain and binder format on the activity of the HPV E629–38 CARs in Jurkat:T2 assays. Two heavy-chain optimized binders, CT503 and CT512, with the highest activity were further optimized by varying the intracellular signaling domains and the scFv format. A, Diagram of constructs. B, Intracellular domain and ligand-binding domain variants. These changes did not show substantial improvement in EC 50 compared with CT503 or CT512 in Jurkat:T2 assays. The data are summarized in Supplementary Table 4 (Supplemental Digital Content 1, http://links.lww.com/JIT/A623). CAR indicates chimeric antigen receptor; RLU, relative luminescence unit; VH, heavy chain variable domain; VL, light chain variable domain.
Characterization of Optimized CARs in Primary T-Cell Assays
Having achieved acute functional sensitivity in Jurkat cells matching the E6 TCR and within 10× of the E7 TCR when corrected for peptide loading, we tested the fully optimized E6 CARs in primary T-cell assays. First, we explored acute sensitivity in a highly controlled, peptide-loading context using A375 (HPV-negative) target cells, with cytotoxicity as the readout. Acute HPV E629–38 CAR (CT512) sensitivity was ∼10× lower than the E6 TCR and ∼100× lower than the E7 TCR, regardless of IL-2 addition (Fig. 6A, Table 4). These findings are consistent with previous observations that primary T-cell cytotoxicity assays increase resolution and amplify differences in acute sensitivity measured in Jurkat:T2 assays.16 The cytotoxicity response was mirrored by IFN-γ secretion (Fig. 6C).60–62 In addition, we confirmed the E6 CAR (CT512), E6 TCR, and E7 TCR killed the HPV-positive tumor cell line CaSki similarly at multiple E:T ratios, while they had little effect on the HPV-negative cell line A375 (Fig. 6B). The E7 TCR killed better than the others. Expression of both CAR and TCR constructs was similar to one another in these experiments and comparable to what we typically observed (Fig. 6D, Supplementary Fig. 6, Supplemental Digital Content 1, http://links.lww.com/JIT/A623).
FIGURE 6.
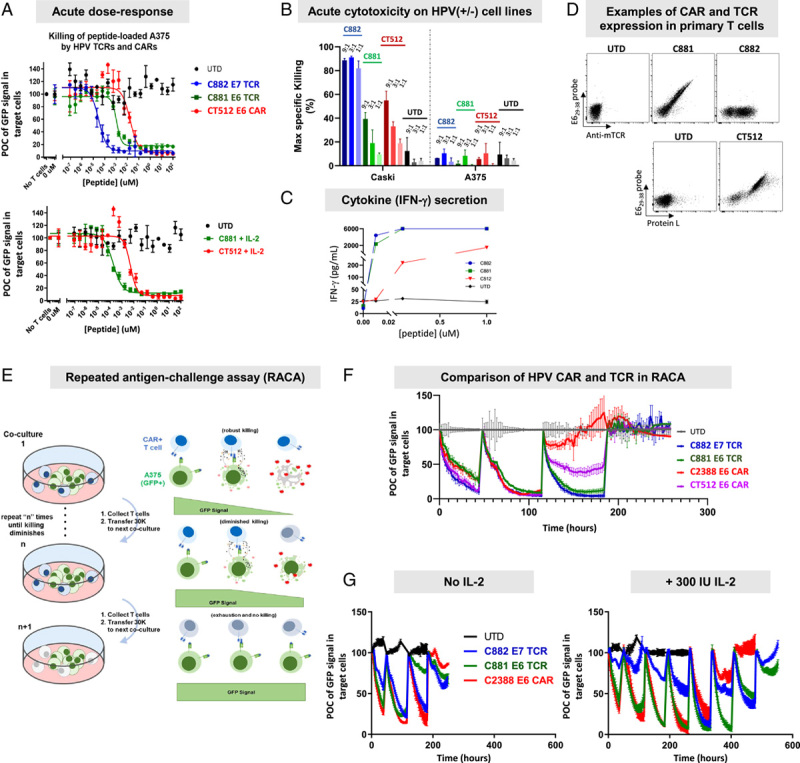
Cytotoxicity in acute assay and RACA of optimized HPV binders. A, Acute sensitivity by peptide loading. HPV E629–38 was loaded on A375 target cells expressing firefly luciferase at serially diluted concentrations. T cells expressing the TCRs or the CAR were cocultured with peptide-loaded A375 for 48 hours. Luciferase signal was determined as an indication of live cell number. B, Acute cytotoxicity on HPV(+/−) cell lines. T cells expressing the E7 TCR (C882), E6 TCR (C881), or the CAR (CT512) were cocultured with the HPV(+) cell line, CaSki, or HPV(−) cell line, A375, at 9:1, 3:1, and 1:1 E:T ratios, and cytotoxicity was monitored by imaging for 70 hours on the Incucyte. The maximum specific killing was calculated and plotted. C, IFN-γ secretion in acute cytotoxicity assay at 1:1 E:T ratio after 16 hours cocultured with peptide-loaded A375 cells. D, Flow cytometry plots of CAR and TCR expression in primary T cells. E, Diagram of RACA. F, Comparison of cytotoxicity of TCRs, a light-chain optimized CAR (C2388), and a heavy-chain optimized CAR (CT512) in RACA at 3:1 E:T ratio. No IL-2 was present in this assay. The process is described in (E). G, Antigen-dependent exhaustion of TCRs and light-chain optimized CAR (C2388) in RACA with and without IL-2. The results show that the addition of 300 IU IL-2 in the assay significantly extended the cytotoxicity of TCRs and the CARs. CAR indicates chimeric antigen receptor; E:T, effector cell to target cell ratio; GFP, green fluorescent protein; HPV, human papillomavirus; IFN-γ, interferon γ; IL-2, interleukin 2; POC, percent of control; TCR, T-cell receptor; UTD, untransduced T cells.
TABLE 4.
Summary of Antigen-dependent Cytotoxicity and Exhaustion of HPV E629–38 and HPV E711–19 TCRs and Examples of Light-chain and Heavy-chain Optimized HPV E629–38 CARs in RACA
| Constructs | Construct Format/Source | Test Peptide Sequence | Primary T-Cell Acute EC 50 (nM) | Peptide Concentration Used in RACA (nM) | Cycles (No IL-2) | Cycles (+IL-2) |
|---|---|---|---|---|---|---|
| C882 | HPV E711–19 TCR | YMLDLQPET | 0.05 | 25 | 4–5 | 8 |
| C881 | HPV E629–38 TCR | TIHDIILECV | 0.4 | 500 | 4 | 8 |
| C2388 | HPV E629–38 CAR (light chain optimized) | TIHDIILECV | 600 | 25,000 | 3 | 6 |
| CT512 | HPV E629–38 CAR (light and heavy chain optimized) | TIHDIILECV | 5.2 | 5000 | 4 | ND |
CAR indicates chimeric antigen receptor; HPV, human papillomavirus; IL-2, interleukin 2; ND, not determined; RACA, repeated antigen-challenge assay; TCR, T-cell receptor.
Next, we studied long-term antigen-dependent cytotoxicity in serial cocultures with peptide-loaded A375 target cells (HPV-negative, GFP-positive), with loss of GFP signal relative to target-negative controls as the readout (Fig. 6E). We used both the fully optimized CT512 CAR and the partially optimized (light-chain only) C2388 CAR. While both CARs killed target-positive cells throughout 3 cycles of serial coculture, they did not display an advantage over either TCR (Fig. 6F, Table 4). Instead, the E7 TCR maintained target-dependent killing for at least 1 additional coculture cycle in comparison to E6 CARs. We also characterized the C2388 CAR together with E6 and E7 TCRs in the same assay in the presence and absence of IL-2. IL-2 increased the number of active cycles (adding ∼3 cycles) for all constructs similarly (Fig. 6G). Together, these results demonstrated that the optimized HPV E629–38 CARs were potent and selective after heavy-chain and/or light-chain optimization, behaving similarly in many respects to the clinical TCRs. However, the E7 TCR exhibited exceptional potency, eclipsing the E6 TCR and CARs (Table 3).
DISCUSSION
We have explored the potential of CARs in one of the most challenging areas of target biology, pMHCs, with HPV E6 as the focal point. pMHC targets pose challenges for CARs for at least 2 reasons: (i) the target density is low compared with cases where CARs have demonstrated unequivocal success in the clinic46; and, (ii) TCRs evolved to target pMHCs, and they can be selected in the body for high potency and specificity, often requiring little further optimization. The E6 and E7 TCRs characterized by Hinrichs and colleagues offer good comparators that exemplify the high standard that CARs must meet to compete with good TCRs.9,47 Both TCRs are highly potent and selective. Nonetheless, we were able to optimize a CAR that nearly matches the E6 TCR while falling short of the E7 TCR in sensitivity. This shortfall, especially notable in primary T-cell cytotoxicity assays, may be partly because of differences in peptide display and/or loading.63
The peptide-loading assay offers a convenient measure of sensitivity, the EC 50 at which T-cell response is half-maximal. This value is correlated with pMHC molecules/cell, which can be estimated directly using potent, selective binding reagents and a combination of single-molecule microscopy and cell fluorescence, providing a standard curve.64 If these data are applied to HPV binders and corrected for loading efficiency measured by surface-stabilization of HLA-A*02, we estimate that at its Jurkat:T2 EC 50 the E6 CAR (CT512) reacted to cells bearing an average of ∼10–20 target pMHCs/cell; the E7 TCR, ∼5 pMHCs/cell (Supplementary Fig. 7, Supplemental Digital Content 1, http://links.lww.com/JIT/A623). For comparison, HPV E711–19 is reported to be displayed on HPV-positive CaSki cells at ∼25 copies/cell.45 The sensitivity of the E6 CAR is impressive and may be as high as any CAR ever reported. Many groups have developed monoclonal antibodies (mAbs) that bind pMHCs, and some have explicitly optimized them for affinity and/or avidity.21 Where reported, the absolute sensitivities typically fall in the range of 100–1000 molecules/cell, though one study found that in vitro treatment with CD19 CAR-T cells could kill target CD19(+) myeloma cells with <100 molecules/cell.35,37 It is also generally thought that high-affinity CARs have poor sensitivity, though we have not observed such a correlation.65
We chose especially potent TCRs with which to compare our CARs. Though these HPV TCRs are not outliers, there are many characterized patient-derived TCRs relevant to immuno-oncology with a sensitivity well below the best HPV TCRs. Good examples are mutant KRAS-directed TCRs. Despite enormous effort, published TCRs that discriminate between the mutant and wild-type KRAS forms often include relatively weak TCRs based on in vitro cytotoxicity assays.66,67 Therefore, it appears likely that, at least for certain antigens, potency optimization will also be required for TCRs, potentially providing another entré for CARs that must be optimized in vitro to gain acceptable potency.
We do not understand certain aspects of the primary binding and optimization process; for example, why certain pMHC probes yield reasonable binders after primary screening while others do not. It is possible other library formats or larger numbers of variants might improve the initial output from the binding screen. However, we have not noticed large differences in results when different types of library construction are used (eg, mAb, scFv, heavy-chain vs. light-chain optimization, CDR3 combination, etc.). The HPV E711–19 pMHC probe appeared to enrich more rapidly during the primary binding campaign, and it is possible this probe is less specific than, for instance, the HPV E629–38 probe. This problem might also explain the poor results from the optimization of HPV E711–19 binders. We also noted here a disconnect between binding and function that we have observed generally.16 Though binding is necessary, and further optimization rounds yielded improved function, the relationship between binding and function is apparently not simple. Finally, in the case of HPV E629–38, we completed 2 optimization cycles on top of the initial primary binder selection. Further or different types of optimization may produce additional increments in sensitivity.
With regard to specificity, the HPV CARs described here are equally selective, or nearly so, compared with the highly selective HPV TCRs. This finding is not consistent with the view that high-potency CARs directed at low-density antigens such as pMHCs cannot be selective.65 In addition, it is relatively easy to identify and study-related peptides that could cross-react with scFv binders that, unlike many TCRs, are not derived from the human blood. This in turn, suggests an easy way to flag and de-risk such possible safety concerns in vitro based on quantitative assays and comparisons with expression levels of proteins that encode potentially cross-reacting peptides in human tissues.
As cell therapy evolves to include more complex engineering, it seems likely that some of the innovation that has characterized mAb therapeutic development will apply. A good example is multifunctionality, illustrated by bispecific T-cell engagers and dual-targeted checkpoint inhibitors.68–70 Such antibody-based molecules either furnish unique functional capability that cannot be achieved by combining individual mAbs, or offer additive benefits for convenience; or, in the case of immuno-oncology, mitigation of preexisting and acquired resistance.31 In the context of HPV-induced cancers, we imagine that improved cell therapies will incorporate, at a minimum, binding elements that permit multiple targeting of different epitopes, perhaps including additional viral serotypes and HLA allelic forms.
CONCLUSIONS
A common view holds that CARs cannot be optimized to reach the level of sensitivity and selectivity observed in natural TCRs.65 This feature may not be an absolute difference but rather a result of their divergent origins. TCRs typically come from the body optimized for signaling function. CAR ligand-binding domains, on the other hand, are selected principally from binding screens. Here we show that sensitive, selective pMHC-directed CARs can be recovered. However, our experiments also illustrate the effort required to achieve these features using current technology and suggest the possible value of high-throughput functional screens.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.
ACKNOWLEDGMENTS
The authors thank Tanya Kim and Dr Jee-Young Mock for generating pMHC probes, Bella Lee for molecular cloning, Dr Agnes Hamburger for supervising various activities and helpful discussions, and Dr Armen Mardiros for comments on the manuscript.
Conflicts of Interest/Financial Disclosures
All authors are employees of A2Biotherapeutics Inc. or Innovative Targeting Solutions.
Footnotes
Present address: Kathleen R. Negri, Onchilles Pharma, 9880 Campus Point Drive #420, San Diego, CA.
Present address: Ming-Lun Wu, Mantra Bio, 600 Gateway Boulevard, South San Francisco, CA.
X.W. and M.L.S. contributed equally.
X.W., M.L.S., A.D.M., D.P.N., J.O., H.X., and A.K.: helped in project conceptualization, planning, and experimental design. X.W., M.L.S., A.D.M., K.R.N., G.B.G., D.P.N., M.L.W., M.E.M., W.H.L., J.O., F.C., C.Y., J.F., M.D., F.C., C.Y., and J.F.: executed the experiment. X.W., M.L.S., A.D.M., K.R.N., G.B.G., D.P.N., D.T.W., J.O., C.P., and H.X.: carried out data analysis. A.K., H.X., A.D.M., M.L.S., and X.W.: drafted the manuscript.
Contributor Information
Xueyin Wang, Email: xwang@a2bio.com.
Mark L. Sandberg, Email: msandberg@a2bio.com.
Aaron D. Martin, Email: amartin@a2bio.com.
Kathleen R. Negri, Email: Knegri3178@gmail.com.
Grant B. Gabrelow, Email: ggabrelow@a2bio.com.
Daniel P. Nampe, Email: dnampe@a2bio.com.
Ming-Lun Wu, Email: minglwu427@gmail.com.
Michele E. McElvain, Email: mmcelvain@a2bio.com.
Dora Toledo Warshaviak, Email: dwarshaviak@a2bio.com.
Wen-Hua Lee, Email: Wlee@a2bio.com.
Julyun Oh, Email: Joh@a2bio.com.
Mark E. Daris, Email: mdaris@a2bio.com.
Falene Chai, Email: fchai@innovativetargeting.com.
Christine Yao, Email: cyao@innovativetargeting.com.
James Furney, Email: jfurney@innovativetargeting.com.
Craig Pigott, Email: cpigott@innovativetargeting.com.
Alexander Kamb, Email: akamb@a2biotherapeutics.com.
Han Xu, Email: hxu@a2biotherapeutics.com.
REFERENCES
- 1.Irvine DJ, Purbhoo MA, Krogsgaard M, et al. Direct observation of ligand recognition by T cells. Nature. 2002;419:845–849. [DOI] [PubMed] [Google Scholar]
- 2.Chakraborty AK, Weiss A. Insights into the initiation of TCR signaling. Nat Immunol. 2014;15:798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagarsheth NB, Norberg SM, Sinkoe AL, et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat Med. 2021;27:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19:620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan RA, Chinnasamy N, Abate-Daga D, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Angelo SP, Melchiori L, Merchant MS, et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 (c259)T cells in synovial sarcoma. Cancer Discov. 2018;8:944–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cameron BJ, Gerry AB, Dukes J, et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5:197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramachandran I, Lowther DE, Dryer-Minnerly R, et al. Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J Immunother Cancer. 2019;7:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Norberg S, Nagarsheth N, Sinkoe A, et al. Safety and clinical activity of gene-engineered T-cell therapy targeting HPV-16 E7 for epithelial cancers. J Clin Oncol. 2020;38:101. [Google Scholar]
- 10.Ataie N, Xiang J, Cheng N, et al. Structure of a TCR-mimic antibody with target predicts pharmacogenetics. J Mol Biol. 2016;428:194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang AY, Dao T, Gejman RS, et al. A therapeutic T cell receptor mimic antibody targets tumor-associated PRAME peptide/HLA-I antigens. J Clin Invest. 2017;127:2705–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubrovsky L, Dao T, Gejman RS, et al. T cell receptor mimic antibodies for cancer therapy. Oncoimmunology. 2016;5:e1049803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris DT, Hager MV, Smith SN, et al. Comparison of T cell activities mediated by human TCRs and CARs that use the same recognition domains. J Immunol. 2018;200:1088–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lev A, Denkberg G, Cohen CJ, et al. Isolation and characterization of human recombinant antibodies endowed with the antigen-specific, major histocompatibility complex-restricted specificity of T cells directed toward the widely expressed tumor T-cell epitopes of the telomerase catalytic subunit. Cancer Res. 2002;62:3184–3194. [PubMed] [Google Scholar]
- 15.Liu H, Xu Y, Xiang J, et al. Targeting alpha-fetoprotein (AFP)-MHC complex with CAR T-cell therapy for liver cancer. Clin Cancer Res. 2017;23:478–488. [DOI] [PubMed] [Google Scholar]
- 16.Xu H, Hamburger AE, Mock JY, et al. Structure-function relationships of chimeric antigen receptors in acute T cell responses to antigen. Mol Immunol. 2020;126:56–64. [DOI] [PubMed] [Google Scholar]
- 17.Martin AD, Wang X, Sandberg ML, et al. Re-examination of MAGE-A3 as a T-cell TherapeuticTarge. J Immunother. 2020;44:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willemsen RA, Debets R, Hart E, et al. A phage display selected fab fragment with MHC class I-restricted specificity for MAGE-A1 allows for retargeting of primary human T lymphocytes. Gene Ther. 2001;8:1601–1608. [DOI] [PubMed] [Google Scholar]
- 19.Stewart-Jones G, Wadle A, Hombach A, et al. Rational development of high-affinity T-cell receptor-like antibodies. Proc Natl Acad Sci USA. 2009;106:5784–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inaguma Y, Akahori Y, Murayama Y, et al. Construction and molecular characterization of a T-cell receptor-like antibody and CAR-T cells specific for minor histocompatibility antigen HA-1H. Gene Ther. 2014;21:575–584. [DOI] [PubMed] [Google Scholar]
- 21.Oren R, Hod-Marco M, Haus-Cohen M, et al. Functional comparison of engineered T cells carrying a native TCR versus TCR-like antibody-based chimeric antigen receptors indicates affinity/avidity thresholds. J Immunol. 2014;193:5733–5743. [DOI] [PubMed] [Google Scholar]
- 22.Zhang G, Wang L, Cui H, et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci Rep. 2014;4:3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Q, Ahmed M, Tassev DV, et al. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia. 2015;29:2238–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rafiq S, Purdon TJ, Daniyan AF, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms tumor 1 antigen. Leukemia. 2017;31:1788–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akahori Y, Wang L, Yoneyama M, et al. Antitumor activity of CAR-T cells targeting the intracellular oncoprotein WT1 can be enhanced by vaccination. Blood. 2018;132:1134–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, Sosinowski T, Cox AR, et al. Chimeric antigen receptor (CAR) T cells targeting a pathogenic MHC class II:peptide complex modulate the progression of autoimmune diabetes. J Autoimmun. 2019;96:50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuberth PC, Jakka G, Jensen SM, et al. Effector memory and central memory NY-ESO-1-specific re-directed T cells for treatment of multiple myeloma. Gene Ther. 2013;20:386–395. [DOI] [PubMed] [Google Scholar]
- 28.Willemsen RA, Ronteltap C, Chames P, et al. T cell retargeting with MHC class I-restricted antibodies: the CD28 costimulatory domain enhances antigen-specific cytotoxicity and cytokine production. J Immunol. 2005;174:7853–7858. [DOI] [PubMed] [Google Scholar]
- 29.Zah E, Lin MY, Silva-Benedict A, et al. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4:498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh J, Warshaviak DT, Mkrtichyan M, et al. Single variable domains from the T cell receptor β chain function as mono- and bifunctional CARs and TCRs. Sci Rep. 2019;9:17291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang ZL, Chen YY. CARs: synthetic immunoreceptors for cancer therapy and beyond. Trends Mol Med. 2017;23:430–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foo J, Michor F. Evolution of acquired resistance to anti-cancer therapy. J Theor Biol. 2014;355:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harris DT, Kranz DM. Adoptive T cell therapies: a comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol Sci. 2016;37:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maus MV, Plotkin J, Jakka G, et al. An MHC-restricted antibody-based chimeric antigen receptor requires TCR-like affinity to maintain antigen specificity. Mol Ther Oncolytics. 2016;3:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stone JD, Aggen DH, Schietinger A, et al. A sensitivity scale for targeting T cells with chimeric antigen receptors (CARs) and bispecific T-cell Engagers (BiTEs). Oncoimmunology. 2012;1:863–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Watanabe K, Terakura S, Martens AC, et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8+ T cells. J Immunol. 2015;194:911–920. [DOI] [PubMed] [Google Scholar]
- 37.Nerreter T, Letschert S, Gotz R, et al. Super-resolution microscopy reveals ultra-low CD19 expression on myeloma cells that triggers elimination by CD19 CAR-T. Nat Commun. 2019;10:3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Martel C, Plummer M, Vignat J, et al. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int J Cancer. 2017;141:664–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schiller JT, Castellsagué X, Garland SM. A review of clinical trials of human papillomavirus prophylactic vaccines. Vaccine. 2012;30(suppl 5):F123–F138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith BD, Smith GL, Hurria A, et al. Future of cancer incidence in the United States: burdens upon an aging, changing nation. J Clin Oncol. 2009;27:2758–2765. [DOI] [PubMed] [Google Scholar]
- 41.Clark KT, Trimble CL. Current status of therapeutic HPV vaccines. Gynecol Oncol. 2020;156:503–510. [DOI] [PubMed] [Google Scholar]
- 42.Scheffner M, Münger K, Byrne JC, et al. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc Natl Acad Sci USA. 1991;88:5523–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeFilippis RA, Goodwin EC, Wu L, et al. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells. J Virol. 2003;77:1551–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Altmann A, Jochmus-Kudielka I, Frank R, et al. Definition of immunogenic determinants of the human papillomavirus type 16 nucleoprotein E7. Eur J Cancer. 1992;28:326–333. [DOI] [PubMed] [Google Scholar]
- 45.Blatnik R, Mohan N, Bonsack M, et al. A targeted LC-MS strategy for low-abundant HLA class-I-presented peptide detection identifies novel human papillomavirus T-cell epitopes. Proteomics. 2018;18:e1700390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stevanović S, Draper LM, Langhan MM, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus-targeted tumor-infiltrating T cells. J Clin Oncol. 2015;33:1543–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doran SL, Stevanović S, Adhikary S, et al. T-cell receptor gene therapy for human papillomavirus-associated epithelial cancers: a first-in-human, phase I/II study. J Clin Oncol. 2019;37:2759–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altman JD, Moss PA, Goulder PJ, et al. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–96. [PubMed] [Google Scholar]
- 49.Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Eschericia coli and complexed with single antigenic peptides. Proc Natl Acad Sci USA. 1992;89:3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gallo M, Kang JS, Pigott CR. Sequence diversity generation in immunoglobulins. Innovative Targeting Solution Inc. (assignee). USA patent US008012714 B2; 2011.
- 51.Raman MC, Rizkallah PJ, Simmons R, et al. Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci Rep. 2016;6:18851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Draper LM, Kwong ML, Gros A, et al. Targeting of HPV-16+ epithelial cancer cells by TCR gene engineered t cells directed against E6. Clin Cancer Res. 2015;21:4431–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jin BY, Campbell TE, Draper LM, et al. Engineered T cells targeting E7 mediate regression of human papillomavirus cancers in a murine model. JCI Insight. 2018;3:e99488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramos CA, Narala N, Vyas GM, et al. Human papillomavirus type 16 E6/E7-specific cytotoxic T lymphocytes for adoptive immunotherapy of HPV-associated malignancies. J Immunother. 2013;36:66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kast WM, Brandt RM, Sidney J, et al. Role of HLA-A motifs in identification of potential CTL epitopes in human papillomavirus type 16 E6 and E7 proteins. J Immunol. 1994;152:3904–3912. [PubMed] [Google Scholar]
- 56.Ressing ME, Sette A, Brandt RM, et al. Human CTL epitopes encoded by human papillomavirus type 16 E6 and E7 identified through in vivo and in vitro immunogenicity studies of HLA-A*0201-binding peptides. J Immunol. 1995;154:5934–5943. [PubMed] [Google Scholar]
- 57.Garcia A, Keinonen S, Sanchez AM, et al. Leukopak PBMC sample processing for preparing quality control material to support proficiency testing programs. J Immunol Methods. 2014;409:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghosh T, VandeHaar MA, Rivera M, et al. High-risk HPV genotype distribution in HPV co-test specimens: study of a predominantly Midwestern population. J Am Soc Cytopathol. 2018;7:99–105. [DOI] [PubMed] [Google Scholar]
- 59.Harndahl M, Justesen S, Lamberth K, et al. Peptide binding to HLA class I molecules: homogenous, high-throughput screening, and affinity assays. J Biomol Screen. 2009;14:173–180. [DOI] [PubMed] [Google Scholar]
- 60.Huang GL, Nampe DP, Yi J, et al. A multivariate, quantitative assay that disentangles key kinetic parameters of primary human T cell function in vitro. PLoS One. 2020;15:e0241421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Au-Yeung BB, Zikherman J, Mueller JL, et al. A sharp T-cell antigen receptor signaling threshold for T-cell proliferation. Proc Natl Acad Sci USA. 2014;111:E3679–E3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Faroudi M, Utzny C, Salio M, et al. Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: manifestation of a dual activation threshold. Proc Natl Acad Sci USA. 2003;100:14145–14150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Riemer AB, Keskin DB, Zhang G, et al. A conserved E7-derived cytotoxic T lymphocyte epitope expressed on human papillomavirus 16-transformed HLA-A2+ epithelial cancers. J Biol Chem. 2010;285:29608–29622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Purbhoo MA, Li Y, Sutton DH, et al. The HLA A*0201-restricted hTERT(540-548) peptide is not detected on tumor cells by a CTL clone or a high-affinity T-cell receptor. Mol Cancer Ther. 2007;6:2081–2091. [DOI] [PubMed] [Google Scholar]
- 65.Akatsuka Y. TCR-like CAR-T cells targeting MHC-bound minor histocompatibility antigens. Front Immunol. 2020;11:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang QJ, Yu Z, Griffith K, et al. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol Res. 2016;4:204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sim MJW, Lu J, Spencer M, et al. High-affinity oligoclonal TCRs define effective adoptive T cell therapy targeting mutant KRAS-G12D. Proc Natl Acad Sci USA. 2020;117:12826–12835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Staerz UD, Kanagawa O, Bevan MJ. Hybrid antibodies can target sites for attack by T cells. Nature. 1985;314:628–631. [DOI] [PubMed] [Google Scholar]
- 69.Mack M, Riethmüller G, Kufer P. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc Natl Acad Sci USA. 1995;92:7021–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dahlén E, Veitonmäki N, Norlén P. Bispecific antibodies in cancer immunotherapy. Ther Adv Vaccines Immunother. 2018;6:3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.