

Abstract
Enzymes selectively hydrolyze the carbohydrate fractions of lignocellulosic biomass into corresponding sugars, but these processes are limited by low yields and slow catalytic turnovers. Under certain conditions, the rates and yields of enzymatic sugar production can be increased by pretreating biomass using solvents, heat and dilute acid catalysts. However, the mechanistic details underlying this behavior are not fully elucidated, and designing effective pretreatment strategies remains an empirical challenge. Herein, using a combination of solid-state and high-resolution magic-angle-spinning NMR, infrared spectroscopy and X-ray diffractometry, we show that the extent to which cellulase enzymes are able to hydrolyze solvent-pretreated biomass can be understood in terms of the ability of the solvent to break the chemical linkages between cellulose and non-cellulosic materials in the cell wall. This finding is of general significance to enzymatic biomass conversion research, and implications for designing improved biomass conversion strategies are discussed. These findings demonstrate the utility of solid-state NMR as a tool to elucidate the key chemical and physical changes that occur during the liquid-phase conversion of real biomass.
Keywords: biomass, pretreatment, solid-state NMR, cellulose, enzymatic conversion
Graphical Abstract
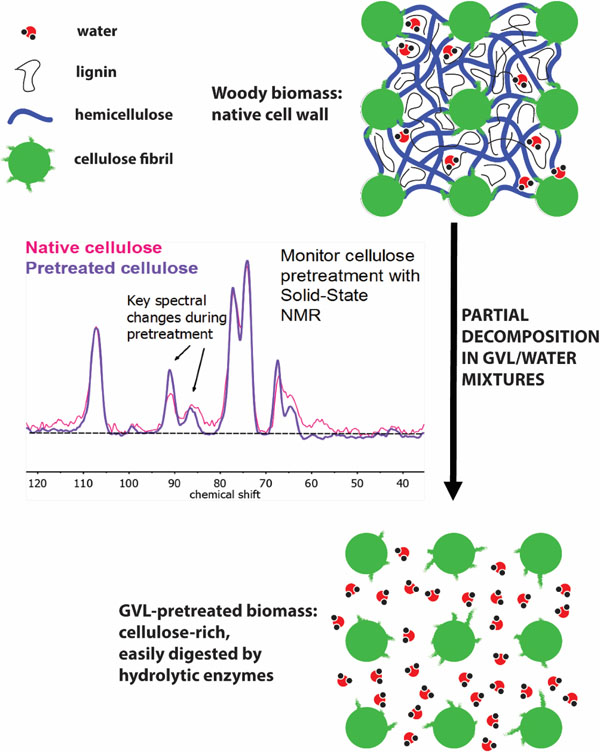
Introduction
Lignocellulosic plant matter (biomass) is a heterogeneous, polymeric material composed mainly of xylan (hemicellulose), glucan (cellulose) and lignin, along with entrained water, minerals, proteins and extractives.1–3 Of these constituent fractions, cellulose is present in amorphous phases and relatively crystalline phases4 with characteristic lateral dimensions (microfibrils).5–6 The microfibrils are bound to the less-ordered (amorphous) cellulose, hemicellulose and lignin via covalent and non-covalent interactions to form the cell wall.7–8 These structures are represented schematically in Figure 1.
Figure 1.
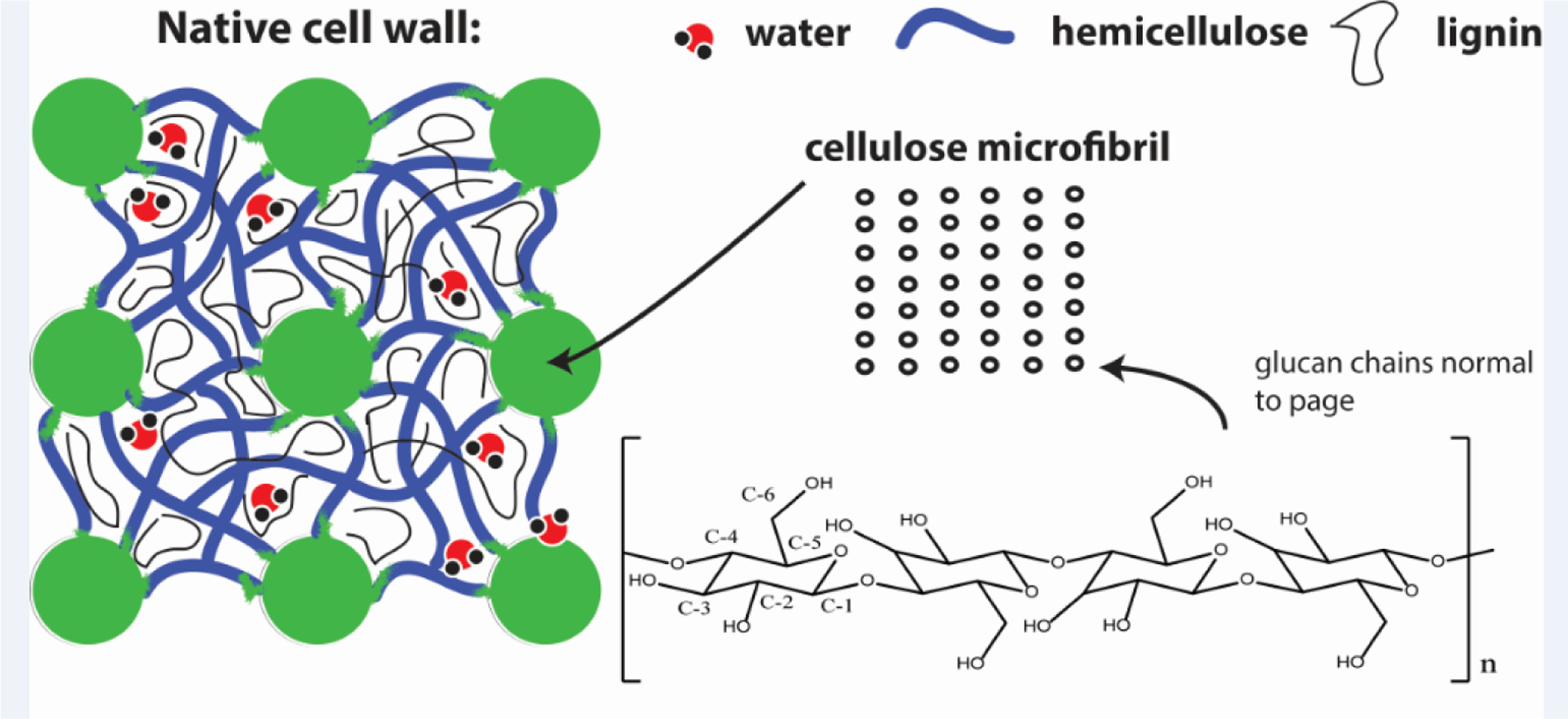
Schematic representation of the cellulose-, hemicellulose- and lignin-containing structures present in the cell wall of woody biomass. Cellulose microfibrils are semi-crystalline in nature, and are constituted by parallel chains of glucan arrayed in ordered planes (represented here by an arbitrary m × n matrix, with ellipses denoting single chains whose long axes are oriented normal to the page). The six carbon centers present in the repeating units of cellulose are labeled according to IUPAC convention.
As an abundant and naturally occurring source of carbon, deconstruction of biomass to yield platform chemicals is a strategy to partially displace global demand for fossil resources.9–11 To this end, several technologies have been proposed, including enzymatic conversion12–13 and solvent-based methods.14–15 Of these two examples, solvent-assisted conversion of biomass over acid catalysts is characterized by rapid catalytic turnovers, but suffers from poor selectivity and often requires elevated temperatures (>= 160°C).9, 16 Hydrolytic enzymes selectively hydrolyze cellulose and hemicellulose into corresponding sugars at mild temperatures,17 but these processes are limited by high operating costs and slow catalytic turnovers.18–19
One strategy to address these challenges is to combine enzymatic hydrolysis of biomass with a solvent-mediated pretreatment step,20 whereby the solvent (usually containing a dilute acid catalyst) first dissolves the hemicellulose and lignin fractions, leaving behind a cellulose-rich solid that is more easily hydrolyzed by enzymes than the native cell wall.21 However, the details connecting the physical and chemical changes effected by the solvent to the improved yield of the enzymatic hydrolysis step are not fully elucidated. For example, it has been proposed that the solvent alters the cellulose pore structure,22 and/or the crystallinity of cellulose;23 these changes hypothetically improve the accessibility of the glyosidic linkages to enzymes, or the affinity of enzymes for the surface accessible facets of the cellulose, respectively. However, attempts to verify these hypotheses have often proven to be inconclusive,24 or yielded contradictory results.25–26 As such, few fundamental approaches exist to understand the effectiveness of biomass pretreatment strategies, as determined by improved sugar yields in the enzymatic hydrolysis step.
Herein, we present a strategy to probe the physical and chemical properties of crystalline cellulose present in raw and solvent-pretreated biomass using a combination of high resolution magic-angle-spinning (HR/MAS) and solid-state cross-polarization (CP)/MAS nuclear magnetic resonance spectroscopy (NMR), along with infrared spectroscopy (FTIR) and X-ray diffraction (XRD) techniques. Proton spin-relaxation-edited (PSRE) CP/MAS NMR is used to differentiate 13C signals originating from the crystalline and amorphous portions of cellulose, which overlap in non-PSRE NMR spectra. We show that as hemicellulose and lignin are removed by the solvent (observable by NMR and FTIR), the characteristic dimensions of the remaining crystalline cellulose microfibrils (observable by XRD) are unchanged. In contrast, as the lignin and hemicellulose are removed, the magnetic environment at the exterior of the crystalline cellulose microfibrils (observable by 13C PSRE CP/MAS NMR) is quantifiably altered. The latter result indicates that the cohesive forces in the interior of the crystalline cellulose microfibrils are comparable to those bonding the microfibril exteriors to hemicellulose and lignin in the plant cell wall. Accordingly, the presence of hemicellulose and lignin likely prevents cellulose-specific enzymes from binding to the surface-accessible glycosidic linkages on the microfibril exterior, inhibiting cellulose hydrolysis. 1H HR/MAS NMR reveals that as the residual cellulose from pretreated-biomass is dried, distinct hydration environments form within the pore structure, and water binds more strongly with the surface accessible facets of the pore walls. This behavior likely slows the diffusion of cellulases within the cellulose pore structure, further hindering enzymatic conversion.
We combine these characterization results with enzymatic reactivity studies to show that the NMR-observable reductions in cellulose-xylan interactions and increased water binding strength in solvent-pretreated biomass can be correlated with the yields of sugars afforded by hydrolysis of the residual cellulose by engineered cellulases. To our knowledge, this study represents the first use of solid-state and HR/MAS NMR to establish this relationship. This finding is of general significance, as the correlation between reduced cellulose-xylan interactions and increased enzymatic sugar production from cellulose is of general importance between different biomass types (corn stover, hard woods, etc.) and solvents (tetrahydrofuran, dimethyl sulfoxide, etc.). Therefore, any strategy to reduce cellulose-xylan interactions in the cell wall while preserving the native cellulose pore structure should produce a corresponding increase in the enzymatic digestibility of the resulting cellulose. Furthermore, we believe that this study demonstrates the use of solid-state NMR to study the liquid-phase conversion of real biomass, affording insights that complement techniques such as solution-phase NMR, X-ray scattering and FTIR.
Results
Solvent-mediated pretreatment of biomass over sulfuric acid
Figure 2 displays a three-step pretreatment process based on prior work by our own group and others.10, 14, 27–29 Generally, this process consists of:
Figure 2.
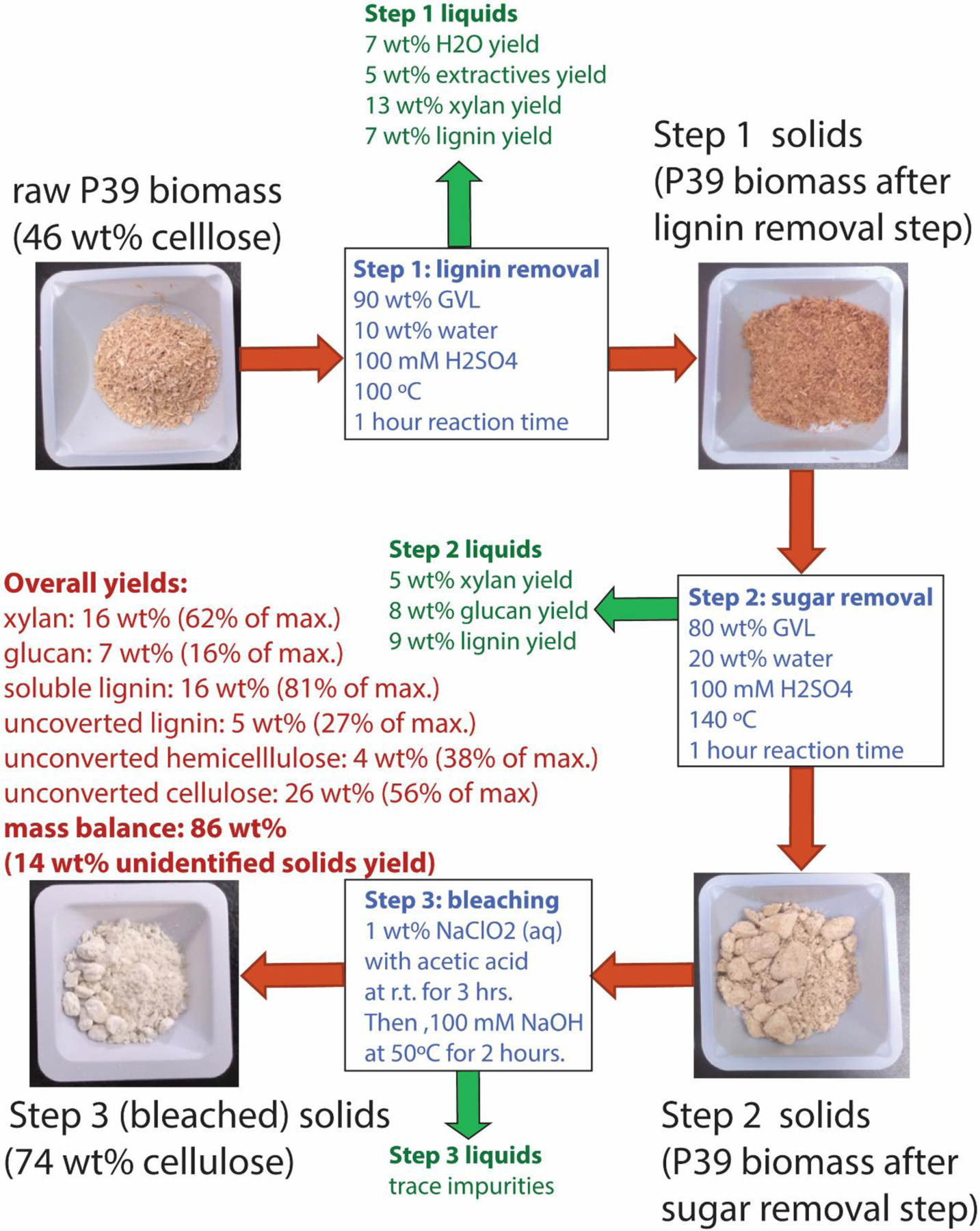
An exemplary solvent-assisted biomass pretreatment sequence based on the general two-step lignin- and then sugar-removal process described above, followed by an additional bleaching step. Reaction conditions: γ-valerolactone (GVL) solvent with HPLC-grade water as a cosolvent. Sulfuric acid (H2SO4) is used as catalyst. Each step was carried out at 9/1 wt/wt liquids to solids in pressure-sealed glass reactors, with mixing facilitated by a magnetic stir bar agitated at 500 rpm. See ESI for further details.
A low temperature (60–120°C) lignin removal step, wherein raw biomass is treated in a solvent system consisting of mostly of an organic solvent, with some water and a dilute mineral acid catalyst.
A higher temperature (100–160°C) sugar removal step, wherein the residual solids from Step (1) are treated in a solvent system, again consisting of an organic solvent and dilute acid catalyst, but with more water than Step (1), and;
A bleaching step, wherein trace impurities are removed from the remaining solids to yield a pristine cellulose product.
The purpose of the first step is to remove as large a portion of the lignin as possible in a form that closely resembles the native lignin in the cell wall; higher temperatures and solvents systems containing more water alter the chemical nature of the extracted lignin.30–32 The first step also removes part of the hemicellulose in the form of soluble sugar monomers and oligomers (xylan), along with the entrained water and extractives in the biomass. The purpose of the second step is to remove additional amounts of hemicellulose and lignin, but a portion of the cellulose may also be removed in this step in the form of soluble six-carbon sugar monomers and oligomers (glucan). The bleaching step, if used, removes trace lignin and degraded sugars (humins),33 which have been shown to inhibit the activity of some hydrolytic enzymes.34
In the example displayed in Figure 2, the organic solvent used is γ-valerolactone (GVL), a non-toxic and bio-renewable solvent35 that has the demonstrated abilities to solubilize all fractions of woody biomass,14 and to facilitate selective liquid-phase biomass conversion reactions over acid catalysts.27, 36 Other organic solvents may be used, so long as they are miscible with water (which is required to solubilize the carbohydrate fractions of biomass),37 and are stable at the desired pretreatment temperatures. Other solvents used in this study were: tetrahydrofuran (THF), acetonitrile (MeCN); N-methyl-pyrrolidinone (NMP); and dimethyl sulfoxide (DMSO).
The biomass type shown in Figure 2 is P39 poplar, an engineered deciduous energy crop optimize for growth on marginal lands (i.e.. lands that are not useable for growing food).38 Three other high yield but non-food-competitive biomass feedstocks were studied in this work: corn stover, switchgrass, sorghum and an additional engineered poplar (NM6).39 The catalyst (H2SO4) used in Figure 2 is a standard reagent used in demonstrated biomass pretreatment/conversion strategies.40 The bleaching step is based on industry-standard protocols, using water a solvent.41
In Figure 2, representative reaction conditions are shown, with corresponding yields of soluble products in each liquid fraction. Note that the mass balance for the overall process is less than 100%, which results from the formation of unaccountable soluble and insoluble polymeric species (humins), which are ubiquitous in biomass conversion processes.42 The yields shown in Figure 2 are typical for those reported under similar conditions.14, 27 Table 1, below, summarizes all pretreatment conditions investigated in this study.
Table 1.
Reaction conditions for solvent-assisted pretreatment of various biomass, and corresponding enzymatic sugar yields and the NMR-derived observable XNMR. The temperature in the lignin removal step was 100°C for each entry. The catalyst was 100 mM H2SO4 for all lignin- and sugar-removal steps. Bleaching was carried out as described in the methods above. See ESI for full details and corresponding liquid-phase product assays. Enzymatic hydrolysis was performed with 10 wt% CelR cellulase (see methods) and 0.5 mg/mL pretreated material in a total volume of 1 mL for 24 hours with shaking.
| Entry | Biomass type | Organic solvent (wt%) – lignin removal step | Organic solvent (wt%) – sugar removal step | Temp. – sugar removal step / °C | Bleached? (Y/N) |
Dried? (Y/N) |
XNMR | Enzymatic sugar yield from residual cellulose (wt% of theor. maximum) |
|---|---|---|---|---|---|---|---|---|
| 1 | P39 | GVL (90) | -- | -- | N | N | 0.504 | 0.0 |
| 2 | P39 | GVL (90) | -- | -- | N | Y | 0.485 | 0.0 |
| 3 | P39 | GVL (90) | -- | -- | Y | N | 0.530 | 0.0 |
| 4 | P39 | GVL (90) | -- | -- | Y | Y | 0.507 | 0.0 |
| 5 | P39 | GVL (90) | GVL (80) | 120 | N | N | 0.545 | 0.3 |
| 6 | P39 | GVL (90) | GVL (80) | 120 | N | Y | 0.522 | 0.0 |
| 7 | P39 | GVL (90) | GVL (80) | 120 | Y | N | 0.608 | 3.7 |
| 8 | P39 | GVL (90) | GVL (80) | 120 | Y | Y | 0.580 | 1.8 |
| 9 | P39 | GVL (90) | GVL (80) | 140 | N | N | 0.745 | 31.8 |
| 10 | P39 | GVL (90) | GVL (80) | 140 | N | Y | 0.710 | 7.4 |
| 11 | P39 | GVL (90) | GVL (80) | 140 | Y | N | 0.775 | 38.0 |
| 12 | P39 | GVL (90) | GVL (80) | 140 | Y | Y | 0.747 | 11.7 |
| 13 | P39 | THF (90) | THF (80) | 140 | N | N | 0.779 | 32.3 |
| 14 | P39 | MeCN (90) | MeCN (80) | 140 | N | N | 0.684 | 14.1 |
| 15 | P39 | NMP (90) | NMP (80) | 140 | N | N | 0.609 | 6.4 |
| 16 | P39 | DMSO (90) | DMSO (80) | 140 | N | N | 0.633 | 6.9 |
| 17 | NM6 | GVL (90) | GVL (80) | 140 | N | N | 0.711 | 28.1 |
| 18 | Sorghum | GVL (90) | GVL (80) | 140 | N | N | 0.582 | 7.9 |
| 19 | switchgrass | GVL (90) | GVL (80) | 140 | N | N | 0.545 | 10.4 |
| 20 | Corn stover | GVL (90) | GVL (80) | 140 | N | N | 0.698 | 20.1 |
13C solid-state cross-polarization magic-angle-spinning (CP/MAS) NMR
13C solid-state CP/MAS NMR is a technique whereby magnetization is transferred from the protons to their attached carbon nuclei in a solid sample for the purpose of enhancing the 13C signal and improving resolution for this low natural-abundance nucleus. The sample is spun at high frequency (~1–10 kHz) to mimic the solution-phase behaviors that average out chemical shift anisotropies in liquid samples and produce sharp, well-separated features in the NMR spectrum. This technique has seen increasing use in biomass conversion research as late, as it is a non-destructive technique that allows for the in-situ characterization of whole biomass.43–45 See methods and ESI for details.
Figure 3 displays 13C CP/MAS NMR spectra for three of the representative biomass samples shown in Figure 2: raw P39 biomass, the same biomass pretreated in GVL-water mixtures to remove lignin (Figure 2, Step 1 solids as described in Entry 1 of Table 1), and then hemicellulose (Figure 2, Step 2 solids as described in Entry 9 of Table 1). Key resonances are delineated in Figure 3, corresponding to the six carbon centers in cellulose,46 as well as the acetate groups in hemicellulose, and the methoxy groups present in lignin.47 These latter two resonances decrease in intensity by about 74 and 65%, respectively, in the pretreated samples as compared to the raw P39, indicating a corresponding reduction in the lignin and hemicellulose content, as expected. See ESI for quantitative, NMR-enabled assays of lignin, hemicellulose cellulose contents in raw and solvent-pretreated biomass samples. This behavior is confirmed by Fourier Transform Infrared Spectroscopy (FTIR), also shown the ESI.
Figure 3.
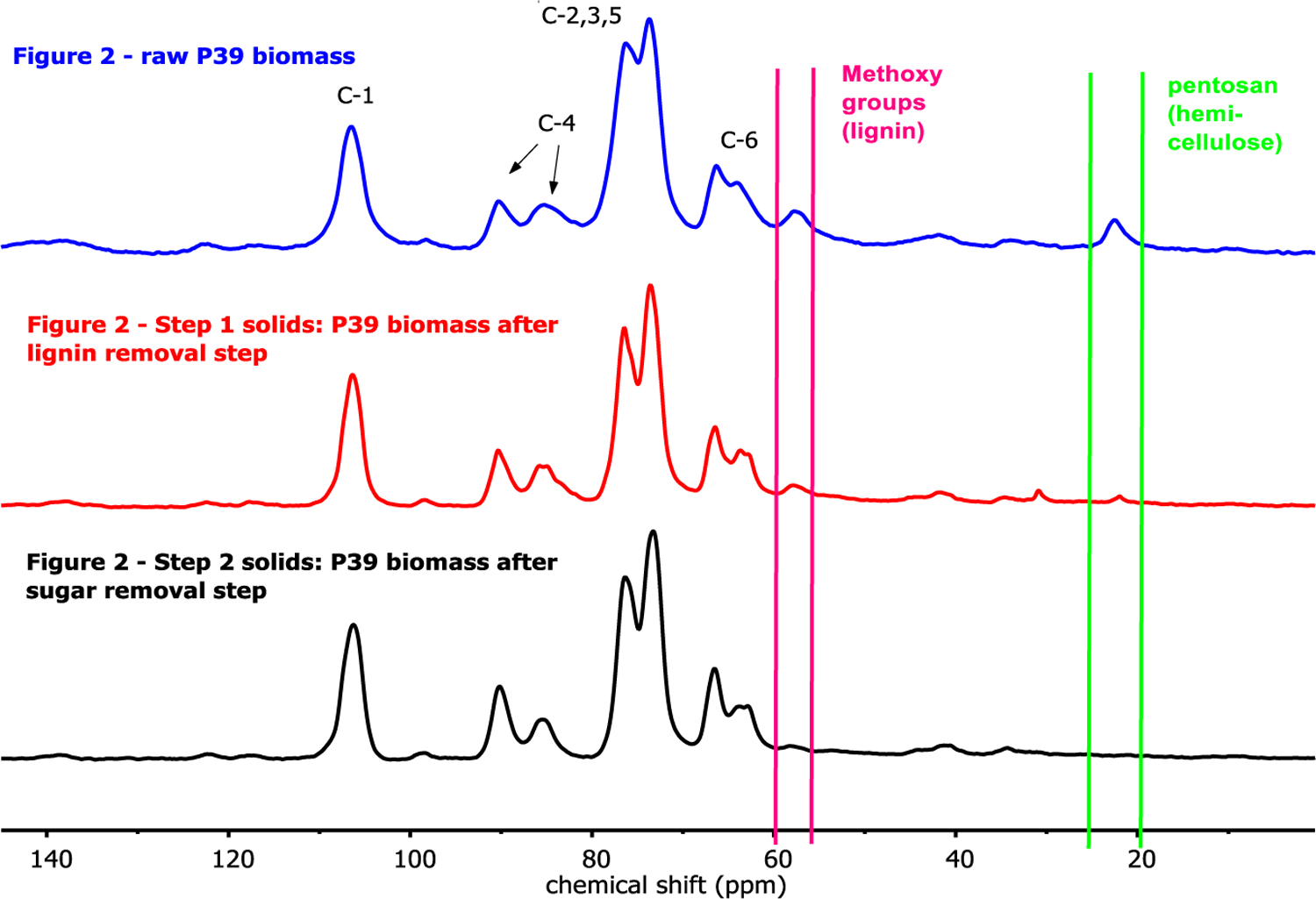
13C MAS-NMR spectra corresponding to native P39 biomass, and P39 biomass pretreated to remove lignin and hemicellulose. Conditions for the pretreatment steps associated with each spectrum are noted in the Figure 2.
Proton spin-relaxation edited (PSRE) CP/MAS NMR
Proton spin-relaxation editing (PSRE) is a solid-state NMR technique whereby differences in the rotating-frame 1H relaxation behavior for crystalline materials (such as cellulose) and amorphous materials (such as hemicellulose or lignin) are exploited to differentiate their corresponding NMR signals when they have similar chemical shifts. Briefly, a key time delay in the CP/MAS pulse sequence (spin-lock time) is modulated; increasing spin-lock time results in less magnetization being transferred to the carbon nuclei, and therefore decreased signal in the CP/MAS spectrum. Importantly, the extent to which the NMR signal for different carbon nuclei in a solid sample is reduced at a fixed spin-lock time is a function of the chemical environment, allowing for signals for different materials or solid phases to be differentiated, even if their NMR signals overlap in the chemical shift domain. Herein, we followed the method developed by Newman et al.48 to filter out the solid-state NMR signals corresponding to amorphous cellulose, hemicellulose and lignin in biomass, allowing for the NMR signal corresponding to crystalline cellulose to be isolated and analyzed. See the methods section for more details, and the ESI for a more complete description of this technique.
Figure 4 displays the total 13C CP/MAS NMR spectra for raw P39 biomass, along with the PSRE sub-spectrum corresponding only to the crystalline cellulose in the same sample. Note that several broad spectral features (corresponding to amorphous cellulose, lignin and hemicellulose) are mostly removed in the PSRE spectrum, resulting in better-separated peaks and a flat baseline as compared to the normal CP/MAS sprectum.48 The resonances corresponding to the C-4 and C-6 carbon centers in the PSRE and the unedited CP/MAS NMR spectra are split into two non-equivalent features. This behavior has been attributed to differences in the chemical environment between cellulose chains located on the outside vs. the inside of the microfibrils.49 In particular, the C-4 carbon resonances that correspond to these two environments are well separated in the PSRE spectrum.
Figure 4.
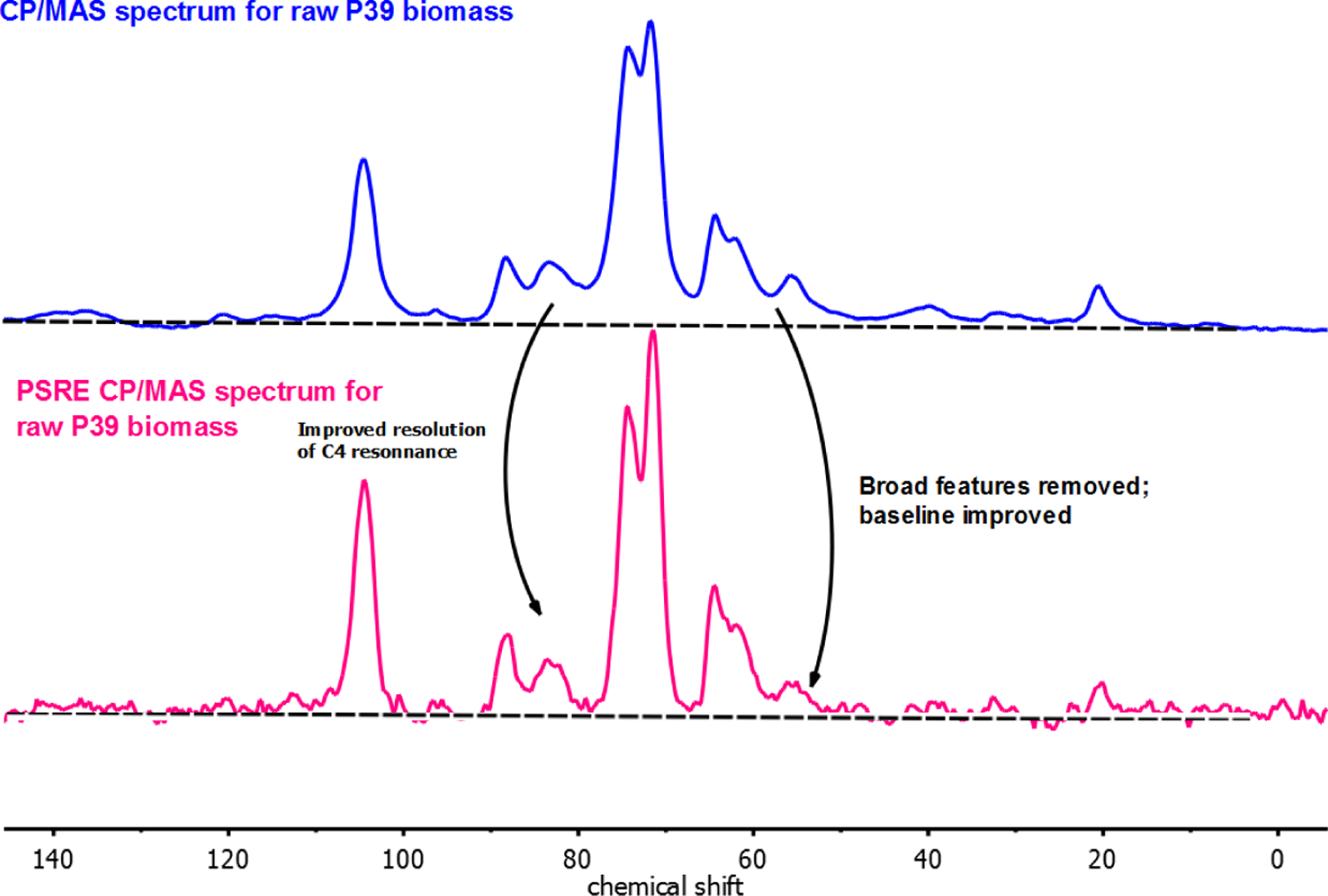
CP/MAS-NMR spectra for native P39 biomass. The top display is the unedited spectrum produced by averaging 2056 scans with a 10 sec recycle delay time and 0.03 ms spin-lock mixing time. The bottom display is the proton spin-relaxation edited (PSRE) spectrum corresponding to the crystalline cellulose fraction of native P39.
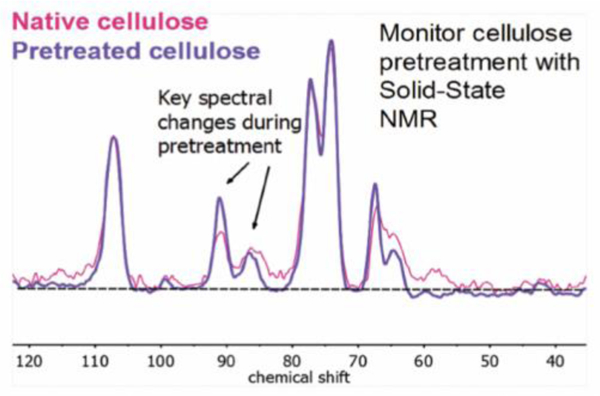
Following these insights, PSRE cellulose spectra corresponding to the native and pretreated biomass investigated in this study were collected, and compared on the basis of the relative peak areas for the downfield C-4 resonance at ~90 ppm and the upfield C-4 resonance at ~86 ppm, denoted C90 and C86, respectively. Figure 5 displays this procedure. When comparing the PSRE cellulose spectra corresponding to raw and pretreated P39 biomass (with peak areas normalized to the C-1 resonance), note that the combined area for the upfield and downfield C-4 peaks remains constant. In contrast, the relative C-4 peak areas, are expressed by the relationship:
| (1) |
reveals an enrichment of the downfield C-4 peak area going from the raw to the pretreated P39 cellulose. Together, these results indicate a redistribution of the C-4 carbons from one chemical environment to the other, rather than a selective depletion of one phase during the GVL pretreatment process. This behavior has been attributed to a reduction in xylan-cellulose interactions at the surface of the cellulose microfibrils.50–52 Furthermore, the average size of the crystalline cellulose microfibrils (observable by XRD; see ESI) did not decrease during pretreatment, indicating that changes in XNMR during biomass pretreatment could not be attributed to an increase in the fraction of glucan chains on the exterior of the microfibrils, as reported elsewhere.48 Accordingly, we attribute increases in XNMR resulting from solvent-mediated pretreatment of biomass to the destruction of cellulose-hemicellulose linkages at the surface of the cellulose microfibrils.
Figure 5.
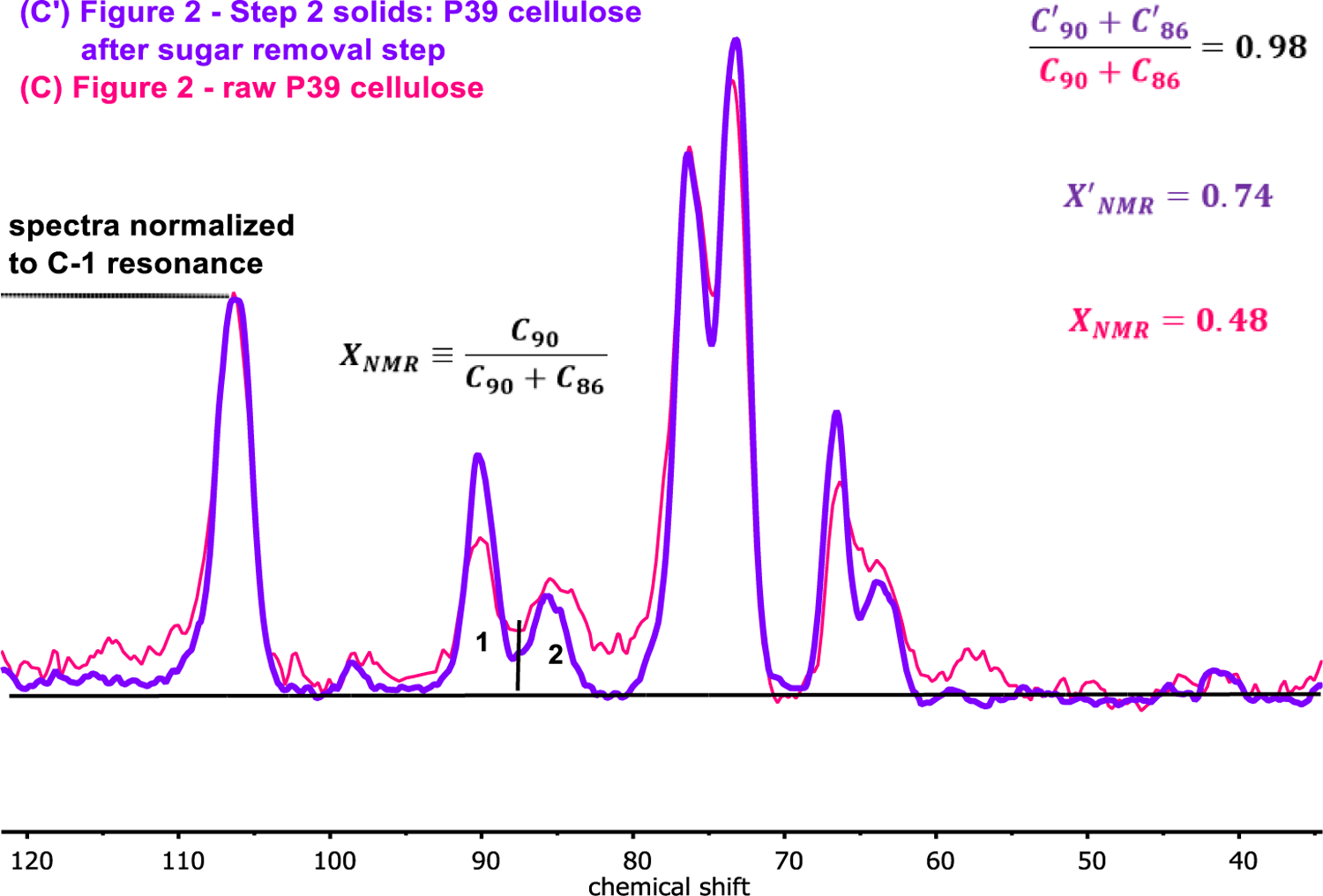
Proton spin-lock edited (PSRE) CP/MAS NMR spectra corresponding to the crystalline cellulose fraction of native P39 biomass (pink display), and P39 biomass pretreated in GVL/water systems to remove lignin and hemicellulose (purple display).
Enzymatic hydrolysis of residual cellulose
The GVL-pretreated solids derived from each step in Figure 2 were collected, washed with water, filtered to produce a solid product of about 70 wt% moisture content, and hydrolyzed in the presence of engineered cellulases (see ESI for details). Similar experiments were also conducted for the different biomass types (corn stover, switchgrass, sorghum and NM6-poplar), and solvent systems (THF, DMSO, NMP and MeCN) used in this study. Bleaching steps were applied to some of the GVL-pretreated samples prior to enzymatic hydrolysis to investigate the effects of this procedure on the digestibility of the residual cellulose. Finally, some of the bleached and unbleached GVL-pretreated celluloses were dried in a vacuum oven at 85°C overnight, and then subjected to enzymatic hydrolysis as well. The yields of soluble sugars derived from this enzymatic hydrolysis step were then compared to assess the effect of each pretreatment step on the enzymatic reactivity of the residual cellulose, whereby higher yields of sugars from the enzymatic hydrolysis step indicate a more effective pretreatment process. These results are displayed in Table 1, along with the values of XNMR.
1H High-Resolution (HR)/MAS NMR spectroscopy
1H High-Resolution (HR)/MAS NMR is a technique whereby solution-phase protons in liquid/solid mixtures can be characterized. Radiofrequency pulse strengths in HR/MAS NMR experiments are set to probe the liquid-phase protons in the same manner as a solution NMR experiment, but high frequency sample spinning is still required to average out chemical shift anisotropies resulting from intermolecular interactions at the liquid-solid interface. See ESI for details.
Figure 6 displays representative HR/MAS spectra corresponding to P39 biomass pretreated in GVL-water solvents to remove hemicellulose and lignin for never-dried and dried materials (Entries 9 and 10 in Table 1). The dried material has been re-wetted by soaking in water (7/3 water to dry solids) for 72 hours (see methods and ESI). HR/MAS spectra were collected with 100 single-rotor-cycle interpulse delays in a Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence for a total of 50 ms transverse relaxation time. Note that each of the spectra within Figure 6 display resonances corresponding to the solution-phase protons in the wetted cellulose; i.e., to liquid water only. Without any magnification, only one resonance is visible: a large multiplet at about 9.5 ppm corresponding to water wetting the surface-accessible facets of the cellulose. The inset of Figure 6 shows the same spectra magnified 100 times. In these magnified images, we see the singlet at 4.7 ppm corresponding to bulk water. Notably, there is a small feature present in the spectra corresponding to the re-wetted material that is not present in the never-dried material. The appearance of this feature indicates the formation of distinct hydration environment upon drying and re-wetting, which we attribute to a change in the cellulose pore structure upon drying. This behavior has been attributed to water becoming trapped in segregated hydration environments upon rapid changes in the pore structure of semi-crystalline polymeric materials.53 Accordingly, assign the minor feature at 8.9 ppm to a partially “collapsed” cellulose pore structure that forms upon drying.
Figure 6.
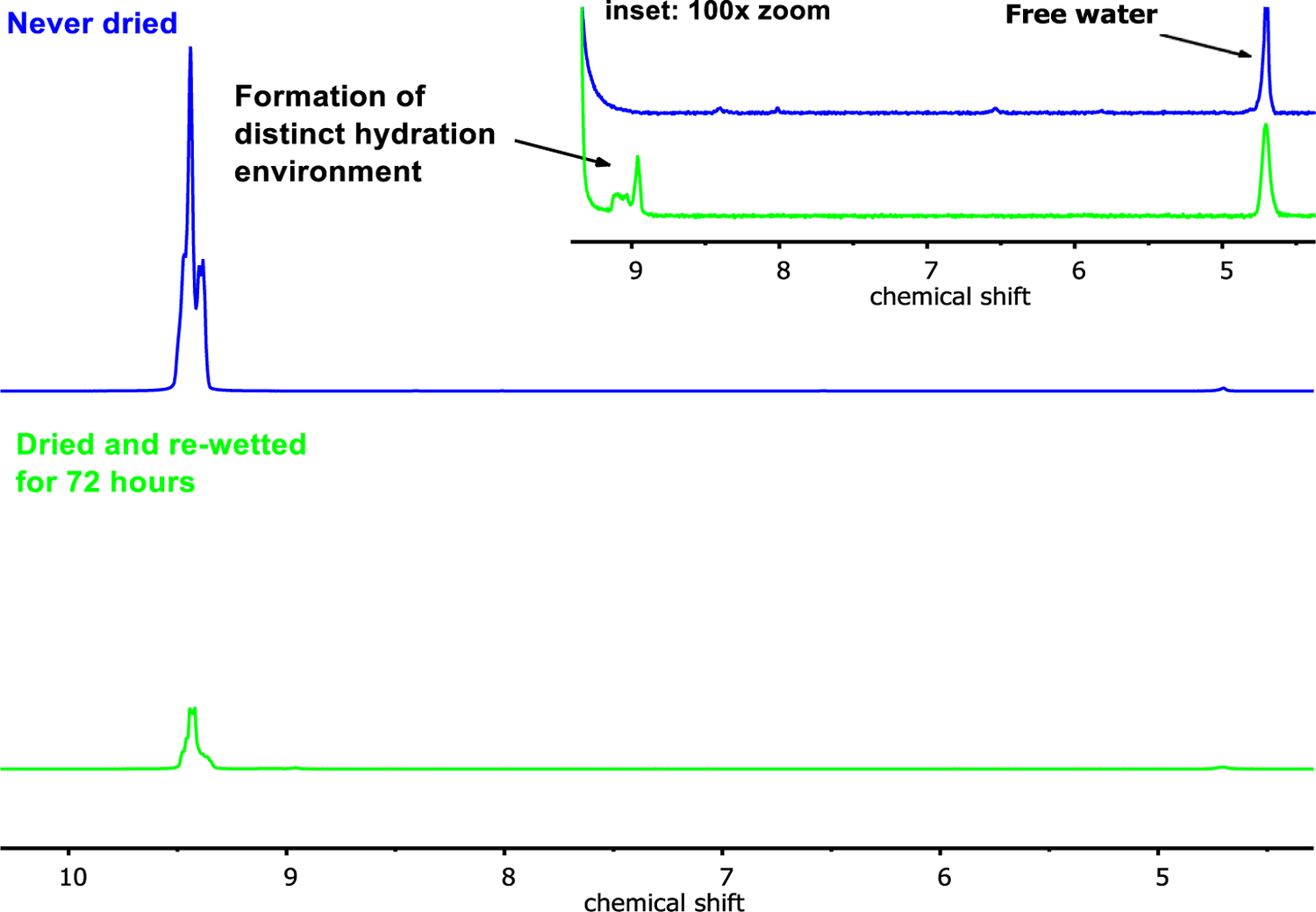
HR/MAS NMR spectra with a 50 ms Hahn-echo delay applied (100 single-rotor-cycle interpulse delays in a Carr–Purcell–Meiboom–Gill (CPMG) pulse sequence) for cellulose from P39 biomass pretreated in 90 wt% GVL at 100ºC for one hour, then 80 wt% GVL at 140ºC for one hour (Table 1, Entries 9 and 10). Top spectrum corresponds to cellulose that was never dried, and analyzed directly after collection from the pretreatment steps. Bottom spectrum corresponds to the same material after drying in a vacuum oven overnight, then re-wetting by soaking in water for 72 hours.
Discussion
To understand the effects of different solvent-mediated biomass pretreatment strategies on the enzymatic digestibility of the residual cellulose, we first analyze the GVL-pretreated P39 biomass samples and corresponding enzymatic sugar yields to probe the effects of the lignin removal step, the sugar removal step, and the bleaching and drying steps. In doing so, we establish that the enzymatic sugar yields from GVL-pretreated P39 cellulose can be correlated with respect to XNMR, and we propose a physical basis for this behavior. We then establish that this relationship can be extended across all pretreated cellulose samples investigated in this study, regardless of the native biomass type or pretreatment solvent used.
Effects of GVL-pretreatment, bleaching and drying on the enzymatic digestibility of cellulose derived from P39 biomass
If increases in XNMR correspond to a reduction in cellulosic material that is bonded to hemicellulose and lignin, and if these extracellular materials occlude enzymes’ access to the cellulose microfibrils25, then the value of XNMR might serve as a predictor of the enzymatic digestibility of the residual cellulose derived from GVL-pretreated biomass. Following this hypothesis, Figure 7 displays the yields of enzymatic sugars from the hydrolysis of GVL-pretreated P39 (both with and without additional bleaching) as function of XNMR. Two curves are shown: one corresponding to the never-dried samples, and the same samples after drying in a vacuum oven at −15 in. Hg for 24 hours (open symbols). In general, for both the never-dried and dried GVL-pretreated P39 celluloses, the yield of enzymatic sugars after a 24 reaction time increases monotonically with XNMR.
Figure 7.
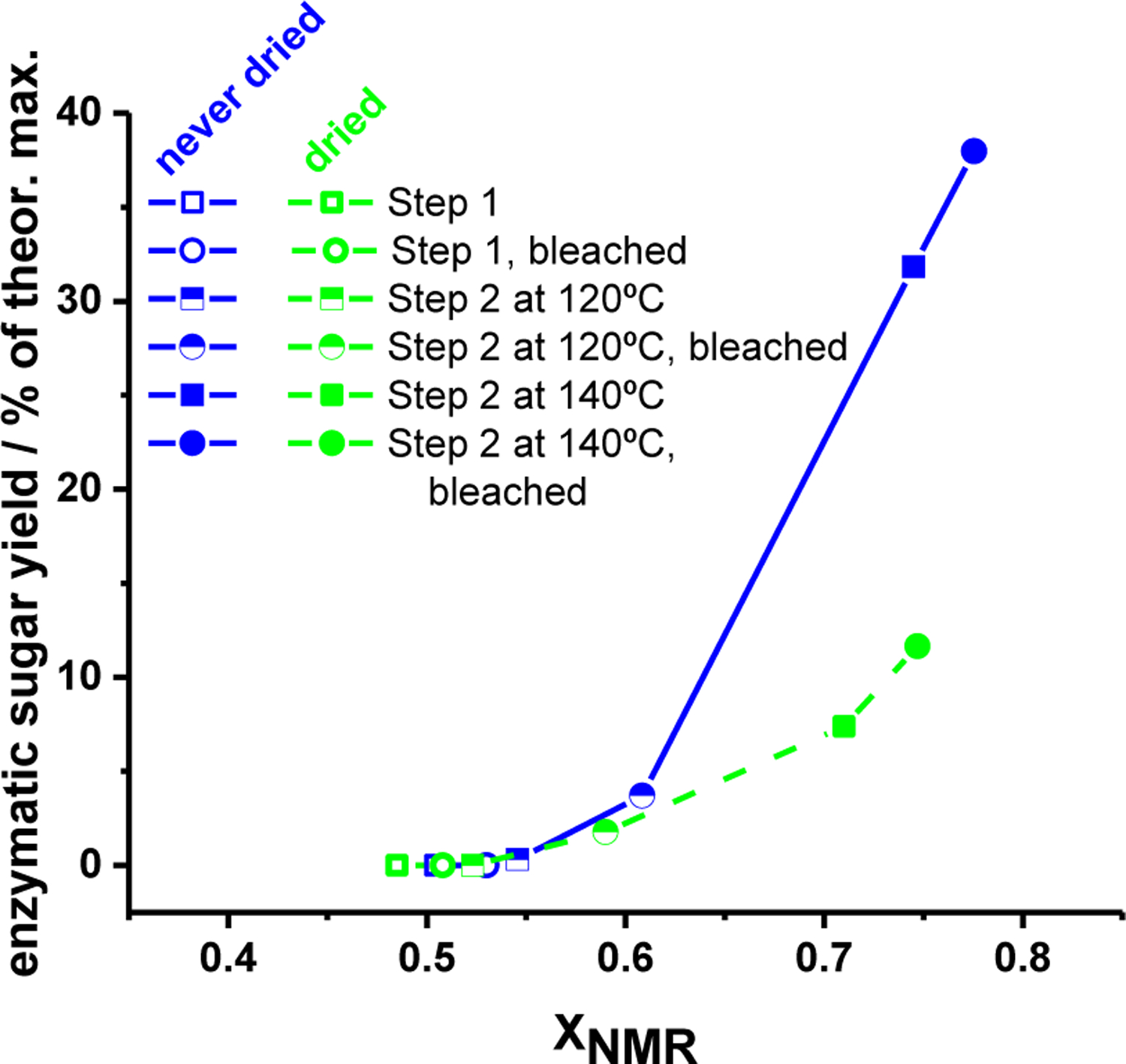
Enzymatic sugar yields from P39 biomass pretreated with GVL-water solvent systems under various conditions, both with and without bleaching steps (entries 1 through 12 in Table 1).
We next compare enzymatic sugar yields as a function of XNMR for the different biomass types and solvent systems studied, where there were no bleaching steps or drying, and the sugar removal step was fixed at 140°C (Figure 8). As demonstrated by the linear regression analyses displayed in Figure 8, enzymatic reactivity generalizes universally with XNMR across all solvent systems and biomass types at a fixed set of reaction times and temperatures for the lignin removal and sugar removal steps.
Figure 8.
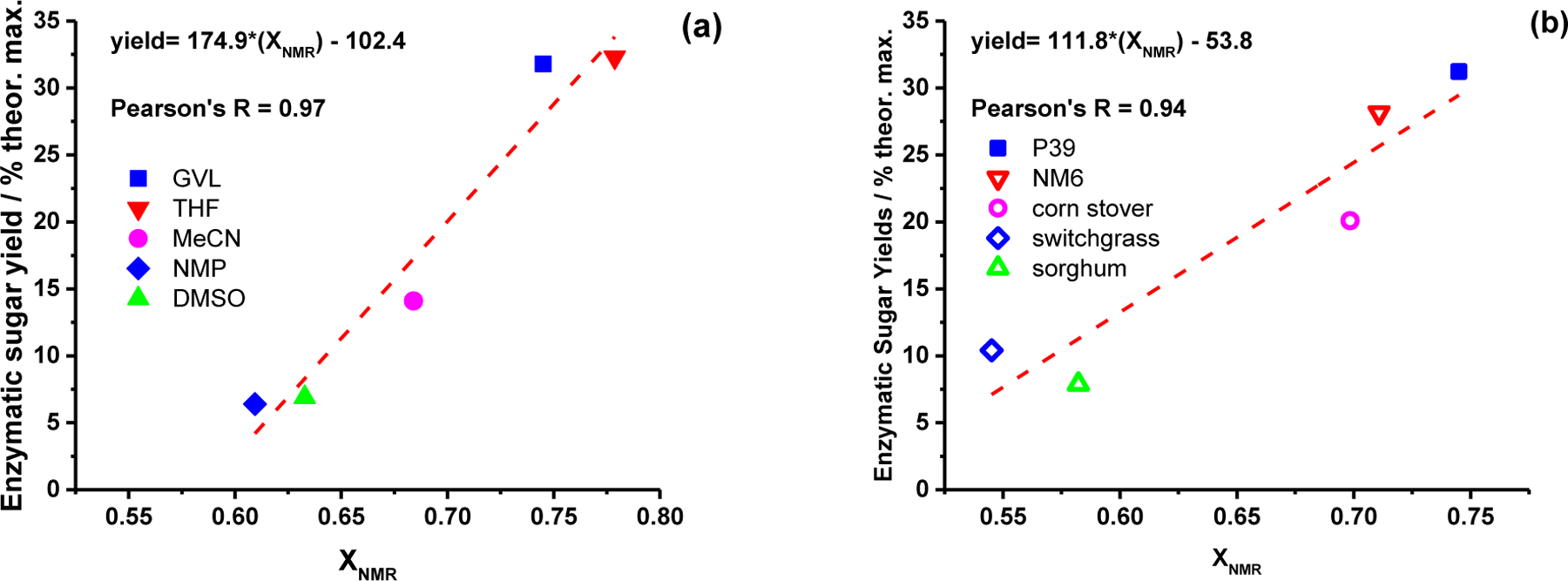
Enzymatic sugar yields versus XNMR for: (a) P39 biomass pretreated in various water/organic-cosolvent mixtures to remove lignin and hemicellulose (entries 11, 15–18 in Table 1), and; (b) different biomass types pretreated in GVL-water solvent mixtures to remove lignin and hemicellulose (entries 11, 19–22 in Table 1). Least squares linear fit to the data are shown, demonstrating a quantiative corrlation between XNMR and enzyamtic sugar yields across various biomass types and pretreatment solvents.
Finally, Figure 9 displays enzyamtic sugar yields as a function of XNMR for all entires in Table 1 corresponding to never-dried celluoses. As demonstrated by the linear regression in Figure 9, there is a quantitative, linear and universal correlation between XNMR and enzyamtic reactivity (as expressed by sugar yields) for all biomass types, solvent systems, and pretreatement temperatures tested in this study. Therefore, the results displayed in Figure 9 indicate that for a fixed set of enzymatic hydroylsis conditions, XNMR serves as a universal predictor of the enzymatic digestibility of cellulose following pretreatment in mixtures of water with sulfuric acid and polar aprotic cosolvents. That said, it is possible that different catalysts (such as bases) or cosolvents (such as proton donors or ionic liquids) would effect different changes, which may or may not improve the enzymatic digestibility of the residual cellulose; only the effects of Brønsted-acid catalysts and polar aprotic cosolvents are studied herein.
Figure 9.
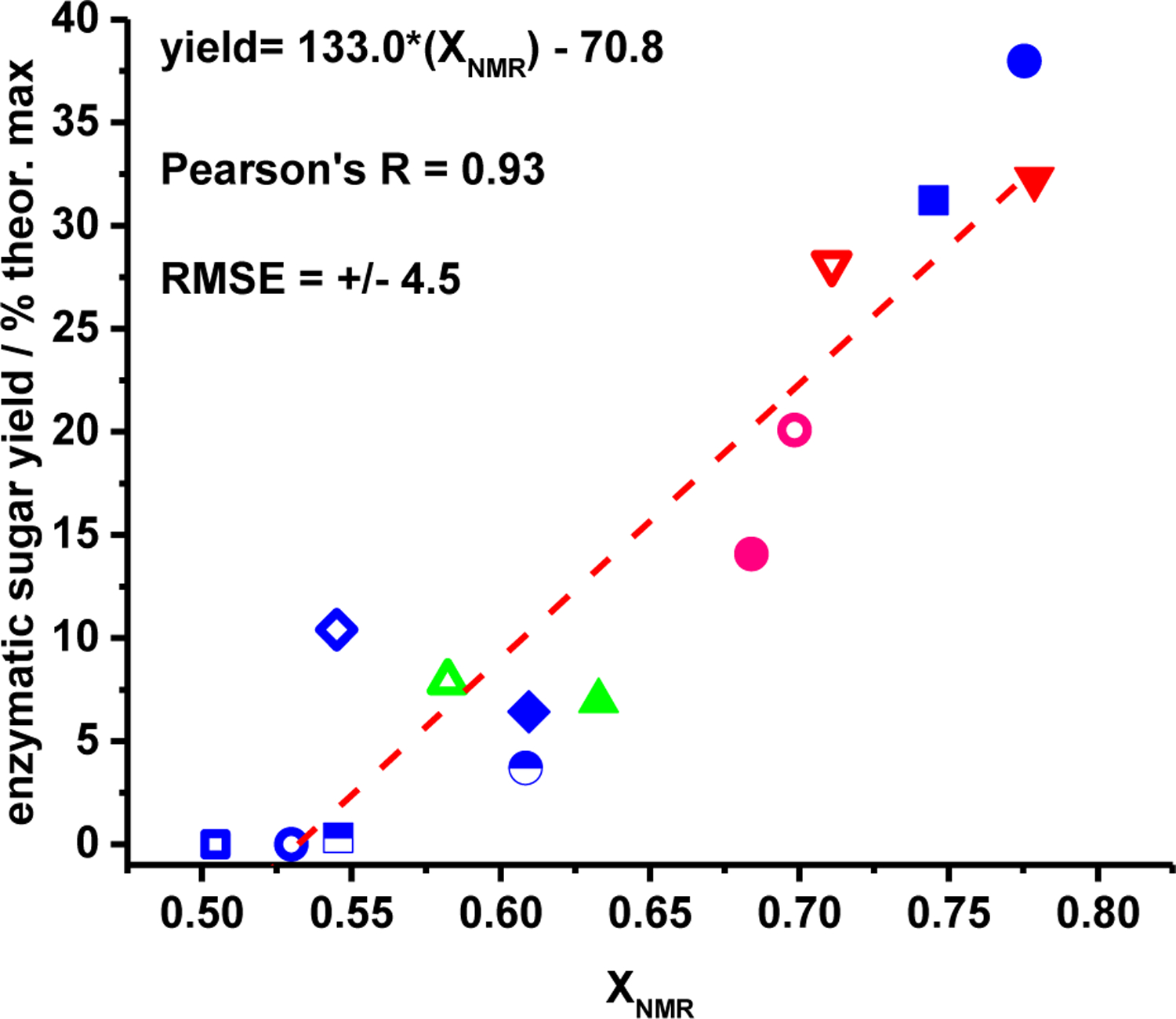
enzymatic sugar yields versus XNMR for all never-dried entries in Table 1. Symbols correspond to those datapoints indicated in the legends in Figures 7 and 8.
The effects of drying on the hydration behavior of GVL-pretreated P39 cellulose
In contrast to the never-dried celluloses, the dried celluloses in Figure 7 exhibit a slight decrease in XNMR, but systematically lower yields of enzymatic sugars at similar values of XNMR as compared to the never dried samples. This result indicates that decreasing the moisture content of pretreated cellulose alters its physical or chemical properties, other than the fraction of microfibril surface chains liberated from the lignin-hemicellulose matrix, and that these changes correspond to a decrease in enzymatic digestibility. Furthermore, the results displayed in Figure 6 indicate that drying GVL-pretreated celluloses alters the pore structure, creating distinct hydration environments as compared to the never-dried material. It has been reported that polymeric materials composed of identical monomer units but synthesized using different methods exhibit similar behavior, whereby different preparation methods produce distinct pore structures, and therefore different hydration environments between materials.53 These differences in hydration environments can be characterized in terms of the binding strengths of water with the surface accessible facets of the pore structure, the diffusivity of water within the pores, and the extent to which water exchanges readily between the pore structure and the bulk fluid. Changes in these characteristics would suggest changes in the ease with which macromolecules, such as cellulases, diffuse from the bulk solution to the interior pores of the cellulose, explaining differences in reactivity between dried and never-dried cellulose materials, and these changes can be probed using 1H HR/MAS NMR.54–55
Figure 10 displays the total integrated area of the 1H HR/MAS resonance at ~9.5 ppm (which we attribute to the main pore structure of wetted cellulose) as a function of the the Hahn Echo delay time in a rotor-synchronized CPMG pulse sequence for GVL-pretreated P39 cellulose. Closed symbols correspond to samples that were never dried, while open symbols corresond to the same samples after being dried in a vacuum oven overnight, and then re-wetted by soaking in DI water at room temperature for 72 hours. Transverse spin-spin relaxation times (T2) were estimated by fitting the data in Figure 8 to a bi-exponential curve (shown in dashed lines). Accordingly, two time constants were assigned to the T2 relaxation process for the never-dried and the dried-and-re-wetted GVL-pretreated cellulose samples: one long relaxation time on the order of 100 ms (T2, long), and one short relaxation time on the order of 10 ms (T2, short). These time constants are also displayed in Figure 10. Going from the never-dried to the dried-and-re-wetted samples, the time constants associated with the spin-spin-relaxation processes for water within the cellulose pore structure roughly double. Short spin-spin relaxation times have been attributed to water that binds more strongly within a polymer matrix.56
Figure 10.
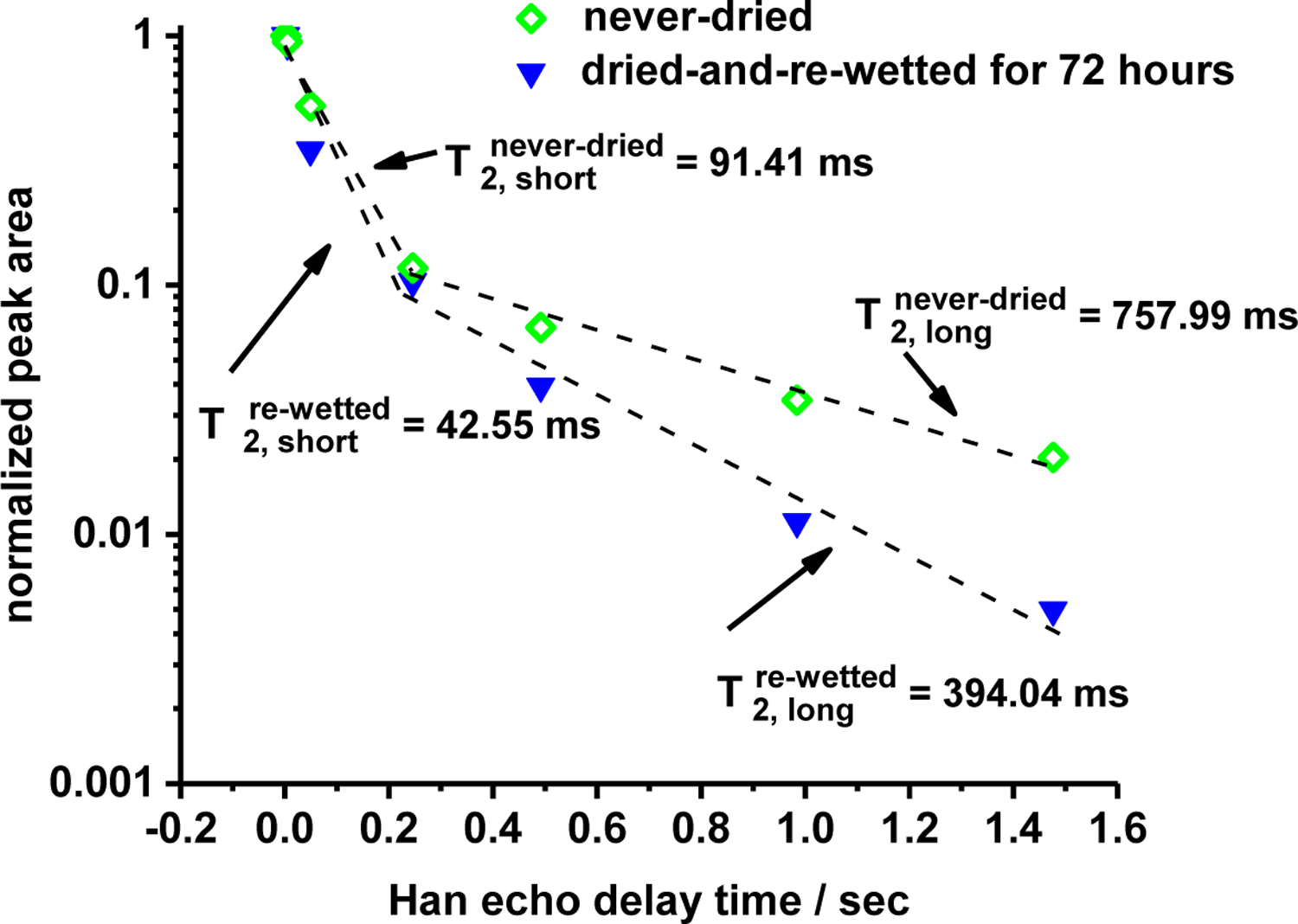
Normalized, integrated areas of the main hydration resonance at ~9.45 ppm in the 1H HR-MAS spectra as a function of the number of CPMG rotor cycle delays or, equivalently, the Hahn echo delay time. Integrals shown are for never-dried, and dried-then-re-wetted P39 biomass that was treated with GVL-water solvents to remove hemicellulsoe and lignin (entries 9 and 10 in Table 1, respectively).
We next examined the temperature-dependent spin-spin relaxation behavior of the 1H HR/MAS resonances in Figure 6 that we attribute bulk water (4.7 ppm), water in the main pore structure (9.4 ppm) and “collapsed” pore stucture (8.9 ppm) of dried-and-re-wetted cellulose. These data are displayed in Figure 11. Briefly, 1H HR-MAS NMR spectra were collected using a CPMG pulse sequence with a Han-echo delay between 4 and 250 ms, with the temperature varied between 23 and 70°C. The absolute integrals of the aforementioned resonances were analyzed as a function of Hahn echo delay time using a single exponential decay to estimate spin-spin relaxation time constants (T2 values; see ESI).
Figure 11.
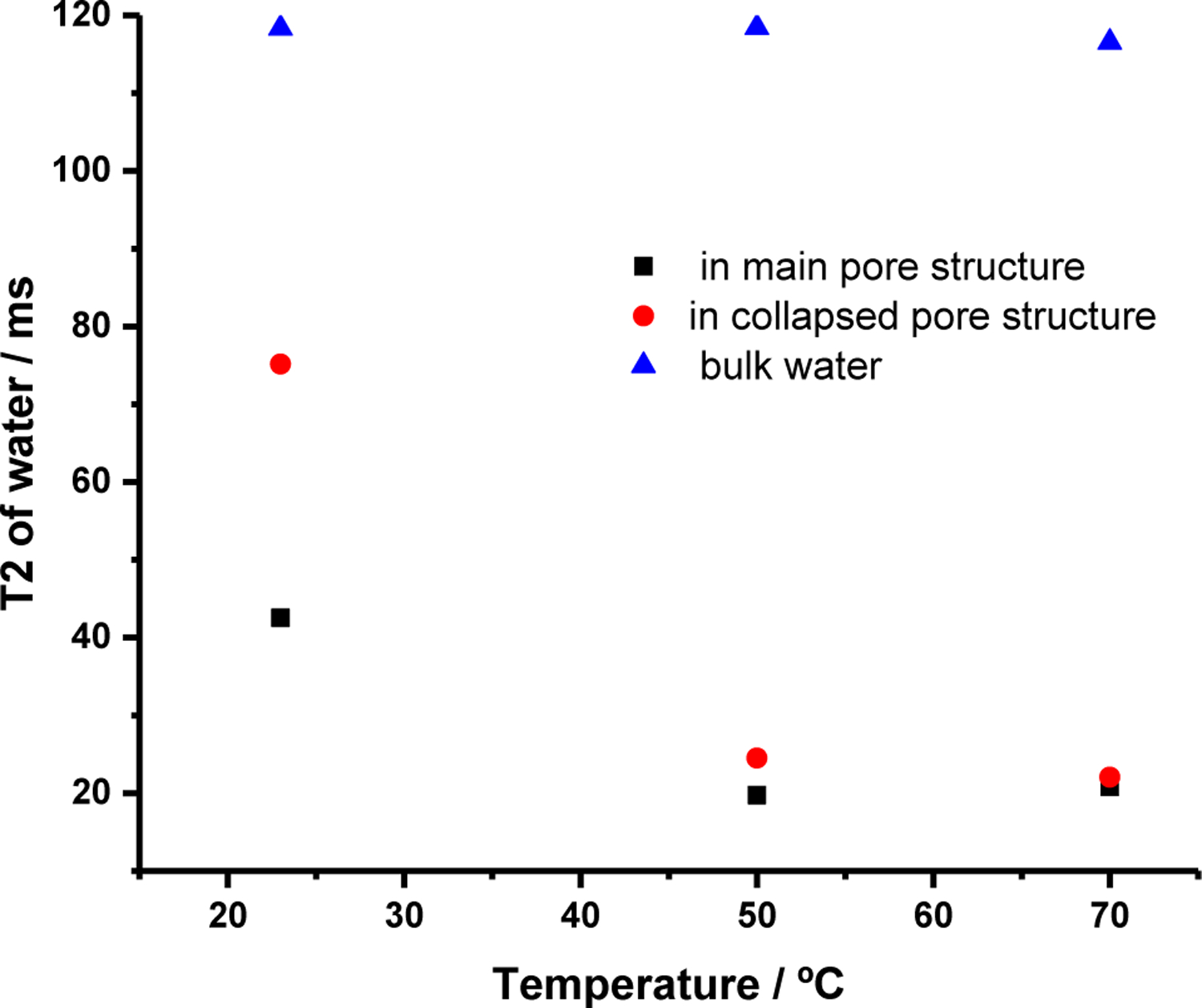
T2 relaxation time contants for water in the main pore structure of cellulose (9..4 ppm in Figure 11) and the “collapsed” pore strucutre of cellulose that forms upon drying (8.9 ppm in Figure 11) as a function of temperature. Spectra were collected with a CPMG filter (Hahn echo delay time) of 2 to 250 ms, and the absolute integrals of the correpsonding resonances were analyzed as a function of Hahn echo delay time using a single exponential decay to estimate spin-spin relaxation time constants (T2 values). The standard error for these six T2 measurements was +/− 4.3%.
At room temperature, the spin-spin relaxation times (T2 values) of bulk water, and water in the main and collapsed pore stucture of previously dried celluose are clearly differentiated. If water were able to exchange readily between the cellulose pore structure and the bulk water, then it would be expected that the T2 values of the corresponding resonances would approach one another with increasing temperature, as the rate of exchange increases.53 However, we find that the T2 values for bulk water is contant with tempearture. In contrast, the T2 values for water in the main and collapsed cellulose pore structures are initially differentiated at 23°C, but decrease and approach one another with increasing temperature, becoming indistinguishable within the error (+/− 4.3% or about 1 ms). This behavior indicates that while water may exchange (diffuse) between the main or secondary hydration environments (main pore stucutre and collapsed pore strucutres) at these temperatures, water does not exchange readily between the cellulose pore structures and the bulk.
All things considered, we interpert the results displayed in Figures 6, 7, 10 and 11 to indicate that upon drying GVL-pretreated cellulose, the cellulose pore structure partially collapses, trapping water in a number of segregated hydration environments wherein water binds more strongly with the surface-accessible facets of cellulose, and does not exchange readily with the bulk water outside the cellulose pores. We expect that these changes in hydration behavior, as compared to the never-dried cellulose, inhibit the diffusion of cellulases into the pore structure, as well as the binding of the cellulases to the surface accessible facets of the cellulose, limiting enzymatic sugar yields. Therefore, it follows that drying biomass may decrease the enzymatic digestibilty of the entrained cellulose in general. This result is of general significance to biomass conversion research, as laboratory samples may be dried prior to enzymatic hydrolysis to determine moisture content, or to increase shelf life.
Physical interpretations and predicting the effectiveness of solvent-assisted biomass pretreatment strategies using NMR
We propose in Figure 12 a physical interpretation for the key changes effected by solvent-mediated pretreatment of woody biomass over acid catalysts. Note that the proposed microfibril dimensions shown in Figure 11 are derived from XRD measurements of P39 poplar, and are in agreement with those proposed elsewhere;48, 57 see ESI for details. As native lignocellulosic biomass is pretreated in semi-aqueous organic solvent systems (for example GVL-water mixtures) over acid catalysts and/or bleached, lignin, hemicellulose and other extracellular materials are removed from the cell wall. At sufficiently low temperatures and short reaction times, however, the cellulose microfibrils are largely untouched, so that their characteristic dimensions do not change. Furthermore, without a drying step, water occupies the interstitial areas between the remaining microfibrils, so that the pore structure formed by these features in the native cell wall is preserved. The result is a mostly cellulose-containing solid through which water can easily diffuse and exchange with the bulk liquid, so that hydrolytic enzymes (cellulases) can readily migrate to the pore structure interior.
Figure 12.
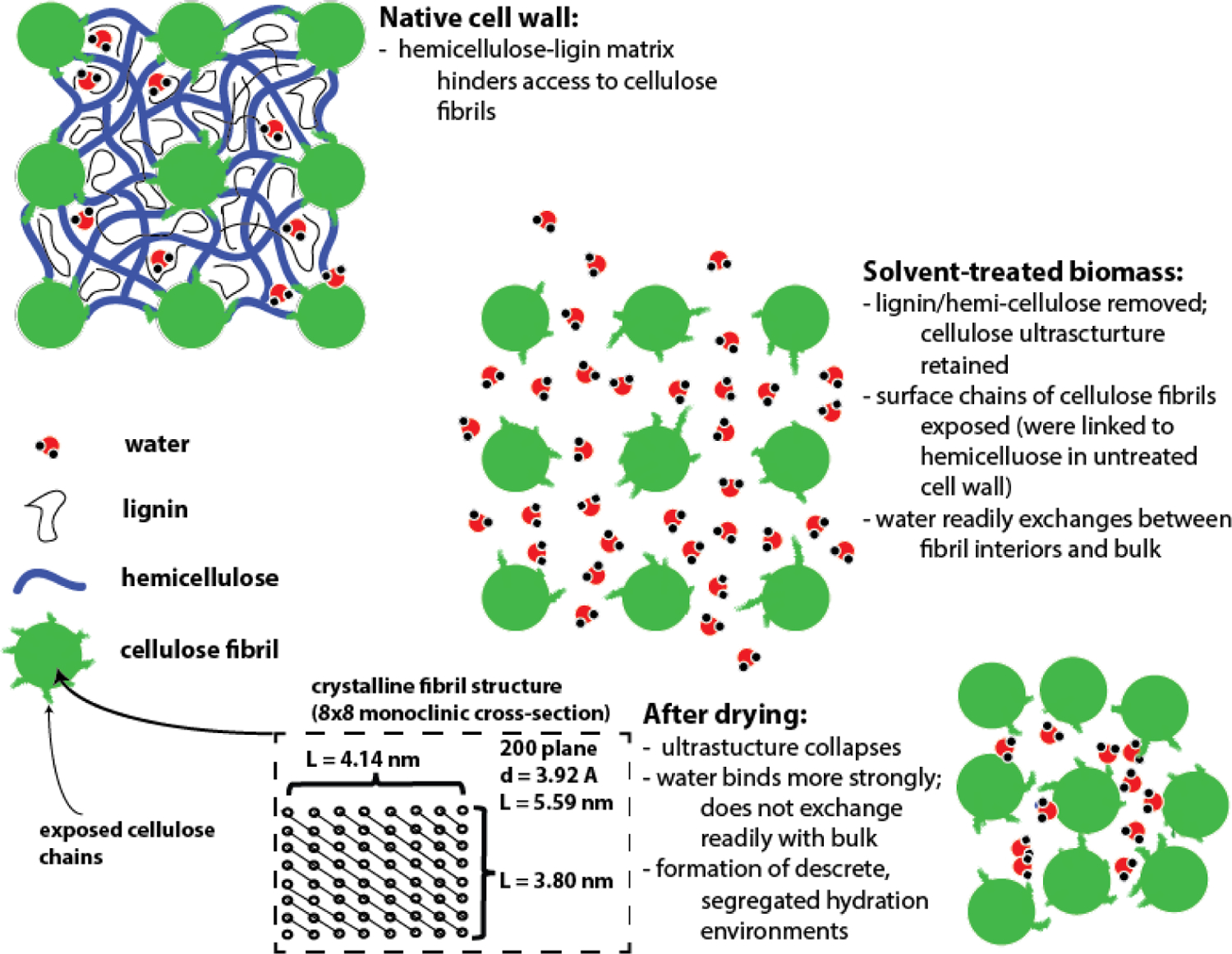
Schematic representation of the structural features of lignocellulosic biomass at various stages of the pretreatment process. Characteristic dimensions of the cellulose microfibrils are for native P39 biomass; values are estimated based on the combined NMR/XRD analysis as described in the ESI.
To facilitate cellulose hydrolysis, enzymes must also bind to the glyosidic linkages exposed on the surface-accessible facets of the cellulose microfibrils, and so the proportion of these linkages that are exposed to the cellulases ultimately determines the enzymatic reactivity of the cellulose. This fraction of exposed linkages is determined by the extent to which the solvent is able to remove not just the bulk extracellular material, but to destroy the linkages between the cellulose and the hemicellulose/lignin matrices. As discussed above, this characteristic feature can be probed using CP/MAS NMR, though the observable XNMR. Upon drying, the cellulose pore structure collapses, trapping remaining water into a number of segregated hydration environments. The water in these hydration environments binds more strongly than the water present in the never-dried material, so that it does not diffuse as easily, nor exchange as readily with the bulk liquid. This behavior hinders the ability of macromolecules such as enzymes to diffuse into the cellulose pore structure interior, reducing the reactivity of hydrolytic enzymes, and limiting the yields of enzymatic sugars.
Conclusions
At short reaction times and lower temperatures, pretreatment of lignocellulosic biomass in aqueous mixtures of organic solvents with dilute acid catalysts removes lignin, hemicellulose and extracellular material from the bulk interstitial areas between crystalline cellulose microfibrils in the cell wall. At higher temperatures and longer reaction times, the characteristic dimensions of the crystalline cellulose domains remain unchanged, but the linkages between the exterior of the cellulose microfibrils and the lignin/hemicellulose matrix are broken, exposing surface-accessible cellulose chains on the outsides of the microfibrils. The extent to which these surface-accessible cellulose chains are liberated from the lignin and hemicellulose is observable by solid state NMR, and herein we express these changes in terms of a single, quantitative metric. Changes in this NMR-observable metric as function of biomass pretreatment conditions (reaction time and temperature) universally predict the yields of enzymatic sugars achieved by hydrolysis of the residual cellulose, regardless of biomass type or the organic pretreatment solvent used. Drying the residual cellulose collected from solvent-pretreated biomass alters the pore structure constituted by the cellulose microfibrils, so that water binds more strongly within the pores, and does not exchange readily with the bulk. This behavior limits the diffusion of enzymes into the cellulose pore structure, limiting enzymatic reactivity of the cellulose accordingly.
The results of this study suggest that solvent-mediated pretreatments (and biomass pretreatment methods in general) that are able to liberate the surface-accessible facets of the cellulose microfibrils from the surrounding lignin and hemicellulose render biomass more readily hydrolysable by enzymes, and likewise improve the yields of enzymatic sugars from biomass. The factors which connect the composition and properties of specific solvent systems to their ability to effect these key changes is the subject of future work. Finally, the moisture content of the pretreated cellulose should be carefully controlled prior to enzymatic hydrolysis, as changes in moisture content alter the structure of the residual cellulose and limit enzymatic reactivity.
Together, these results demonstrate the utility of solid-state and HR/MAS NMR as tools to understand the key chemical and physical changes that occur during liquid-phase conversion of real biomass. The most important corollary is that solid-state NMR can resolve details regarding not just the bulk composition of different biomass types, but also the chemical linkages between distinct phases like lignin, hemicellulose and cellulose. As demonstrated herein, these details can complement those afforded by more standard biomass characterization techniques, and lead to new insights. Moreover, details beyond those generated in this study might be obtained by bringing more advanced solid-state NMR techniques to bear, such as 2D-dimensional solid-state NMR. We hope that such efforts will represent a greater fraction of published efforts in biomass conversion research, going forward.
Experimental
Materials
Corn stover, switchgrass, sorghum, and poplars (NM658 and P3959) were obtained from the Great Lakes Bioenergy Research Center (GLBRC) in the form of debarked, 5 mm chips, and used as received. Water (Fisher, HPLC grade), acetone (Sigma, 98%), ethanol (Fisher, HPLC grade, 200 proof), γ-valerolactone (GVL; Sigma, 95%), tetrahydrofuran (THF; Acros, 99%+ anhydrous with 200 ppm BHT inhibitor), acetonitrile (MeCN; Sigma, 98%), dimethyl sulfoxide (DMSO; Sigma, 98%), and N-methyl pyrrolidinone (NMP; Sigma, 99%) were obtained from vendors and used as received. Sulfuric acid (98 wt%), sodium hydroxide, sodium chlorite and acetic acid were obtained from Sigma and used as received. β-D-glucose, xylose, furfural, and 5-hydroxymethylfurfural (Sigma-Aldrich ACS reagent grade) were obtained from vendors, and used as calibration standards.
CelR_cbm3a was created through fusion of the Hungateiclostridium thermocellum CelR enzyme domain (GenBank accession code CAE51308.1 residues 31 – 642, i.e., GH9 and CBM3c modules only) with an additional 153-residue CBM3a from the scaffolding protein CipA, connected via the 48-residue CipA-native linker sequence. The lac operon-controlled expression plasmid was shipped to a third-party vendor, where it was expressed and purified at scale.
Sample prep and solvent-mediated pretreatment of biomass over sulfuric acid
Briefly, solid biomass samples were charged to sealed glass reactors containing a descried amount of an appropriate solvent system (e.g., pure water or 90/10 GVL/water on a mass basis) along with a sulfuric acid catalyst or appropriate bleaching agent (e.g., NaClO2). Reactors were placed in an oil bath at the desired temperature (e.g., 100 °C) and aggerated with a magnetic stir bar. After an appropriate period of time, reactors were quenched in an ice bath, and the liquid and solid contents were separated with filtration, washed (if needed), and characterized using the techniques described elsewhere in this report (e.g., HPLC, NMR, etc.). See ESI for details.
1H/13C Proton spin-relaxation (T1ρ) edited cross-polarization magic-angle-spinning (PSRE CP/MAS) NMR
About 200 mg of solid sample was packed into 4-mm thin-walled silicon nitride rotors and sealed with glass-filled Torlon caps. 1H/13C PSRE CP/MAS NMR spectra were acquired on a Bruker Avance III 500 MHz spectrometer with a proton radio frequency of 500.22 MHz and a 13C radio frequency of 125.76 MHz. Each rotor was spun at 4 kHz in a 4 mm Doty Scientific MAS probe. 1H and 13C 90° pulse lengths of 2.30 and 4.55 μs were used, respectively. Spectra shown represent 2056 signal averages with a 0.3 second acquisition time and an 8 second recycle delay between scans. The spin-lock radiofrequency strength was γBH/(2π) = 46 kHz during spin-locking with a spin-lock time of 3–10000 μs, as described below. Cross-polarization radiofrequency strength was 99.53 kHz (maximum) during cross-polarization with a 70–100% ramp and a contact time of 2000 μs. The decoupler radiofrequency was 100 kHz during acquisition. 13C spectra were secondary referenced to the up field adamantane peak at 28.7 ppm referenced to TMS. See ESI for full details and a more complete description of this technique.
Attenuated total reflectance infrared (ATR-FTIR) spectroscopy
Attenuated total reflection–Fourier transform infrared spectroscopy (ATR–FTIR) was conducted using a Bruker Optics Vertex system with a diamond-germanium ATR single reflection crystal. Untreated and solvent-pretreated biomass samples were dried in a vacuum oven overnight to remove water content prior to analysis, and were pressed uniformly against the diamond surface using a spring-loaded anvil. Sample spectra were obtained in triplicates using an average of 128 scans over the range between 400 cm−1 and 4000 cm−1 with a spectral resolution of 2 cm−1. Air was used as background.
X-ray diffraction characterizations of native and pretreated cellulose
Cellulose microfibrils scatter incident X-rays to produce a diffraction pattern consistent with a monoclinic unit cell.60 Accordingly, X-ray diffractograms were collected for the native P39 and solvent-pretreated biomass samples, using a Bruker D8 Discovery diffractometer with a Cu Ka X-ray source operating at 1000 kV and 100 mA, with a 5-mm aperture and 600 sec exposure time. Biomass samples were analyzed without additional grinding to reduce particle size. A separate sample of boron nitride was analyzed to assess the inherent line broadening of the instrument.
Enzymatic hydrolysis of residual cellulose
Hydrolysis reactions were prepared with 0.5 mg/mL pretreated cellulose and 0.05 mg/mL CelR in 0.1M phosphate buffer, pH 6.0 to a final volume of 1 mL. Reactions were incubated in a Heidolph Titramax 1000, with a Heidolph Inkubator 1000 used to control temperature. Reactions were run for 24 hours at 50 °C with 1050 rpm (0.0185 × g) shaking. After incubation, reactions were centrifuged for 5 minutes at 21,130 × g. The concentration of soluble sugar produced was determined via the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Briefly, 100 μL of working solution and 5 μL of supernatant were heated for 15 minutes at 80 °C. Control experiments used were CelR only, cellulose only, and buffer only samples, and a glucose standard curve was used for converting absorbance units to mg/mL glucose in solution. Four reactions were performed for each condition, with supernatant being sampled in triplicate.
1H High-Resolution (HR)/MAS NMR spectroscopy
1H HR/MAS NMR experiments were performed on a Bruker Avance 500 MHz spectrometer with a proton radiofrequency of 500.22 MHz equipped with a 4 mm Doty Scientific MAS NMR probe. Proton 90° pulses were applied with a pulse length of 2.48 μs with a 0.5 second acquisition time and a 10 second recycle delay. Solvent-pretreated biomass samples of about 70 wt% moisture content were sealed into 30 μL Kel-F HR/MAS rotor inserts (Bruker Biospin Inc.), with the MAS experiments performed at 4 kHz with a ±0.1 K temperature regulation between 296 and 343 K. Dried, solvent-pretreated biomass samples were re-wetted by soaking in an amount of water corresponding to the moisture content of the never-dried samples (typically ~70 wt% water to 30 wt% solids) for 72 hours, and then loaded into 30 μL Kel-F HR/MAS rotor inserts. The sample temperatures under HR/MAS conditions (TMAS) were calibrated using a neat ethylene glycol thermometer. The one-dimensional 1H NMR spectra were obtained using a single pulse Bloch decay sequence with 16 scan averages and 10 seconds recycle delays. Transverse spin–spin relaxation times (T2) were found using a rotor-synchronized CPMG (Carr–Purcell–Meiboom–Gill) pulse sequence with 16 signal averages, and inter-pulse delays varied between 10 and 20000 rotor cycles (corresponding to 0.03 to 8 seconds transverse spin-spin relaxation times). The 1H NMR chemical shifts were referenced to the internal standard of neat water, δ = +4.7 ppm at 298 K with respect to the chemical shift of TMS, δ = 0 ppm.
Supplementary Material
Synopsis:
Solid state NMR reveals key structural changes relating to the increased enzymatic digestibility of biomass following pretreatment in organic solvents.
Acknowledgements
This material is based upon work supported by the Great Lakes Bioenergy Research Center, U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Numbers DE-SC0018409 and DE-FC02–07ER64494.
Footnotes
- - Details of sample preparation, exemplary mass balances and compositional analysis of biomass by NMR
- - additional background and details relating to relevant solid-state and HR/MAS NMR techniques used in the main text
- - FTIR spectra, X-ray diffractograms and additional HR/MAS and solid-state NMR spectra
References
- 1.Bobleter O., Hydrothermal degradation of polymers derived from plants. Progress in Polymer Science 1994, 19 (5), 797–841. [Google Scholar]
- 2.Mosier N; Wyman C; Dale B; Elander R; Lee Y; Holtzapple M; Ladisch M., Features of promising technologies for pretreatment of lignocellulosic biomass. Bioresource technology 2005, 96 (6), 673–686. [DOI] [PubMed] [Google Scholar]
- 3.Jenkins B; Baxter L; Miles T Jr; Miles T., Combustion properties of biomass. Fuel processing technology 1998, 54 (1–3), 17–46. [Google Scholar]
- 4.O’SULLIVAN AC, Cellulose: the structure slowly unravels. Cellulose 1997, 4 (3), 173–207. [Google Scholar]
- 5.Nishiyama Y., Structure and properties of the cellulose microfibril. Journal of Wood Science 2009, 55 (4), 241–249. [Google Scholar]
- 6.Frey-Wyssling A., The Fine Structure of Cellulose Microfibrils. Science 1954, 119 (3081), 80–82. [DOI] [PubMed] [Google Scholar]
- 7.Salmén L., Micromechanical understanding of the cell-wall structure. Comptes Rendus Biologies 2004, 327 (9), 873–880. [DOI] [PubMed] [Google Scholar]
- 8.Harris PJ; Stone BA, Chemistry and molecular organization of plant cell walls. Biomass recalcitrance: deconstructing the plant cell wall for bioenergy 2009, 61–93.
- 9.Chheda JN; Huber GW; Dumesic JA, Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angewandte Chemie International Edition 2007, 46 (38), 7164–7183. [DOI] [PubMed] [Google Scholar]
- 10.Wettstein SG; Alonso DM; Gürbüz EI; Dumesic JA, A roadmap for conversion of lignocellulosic biomass to chemicals and fuels. Current Opinion in Chemical Engineering 2012, 1 (3), 218–224. [Google Scholar]
- 11.Gaddy BE; Sivaram V; Jones TB; Wayman L., Venture capital and cleantech: The wrong model for energy innovation. Energy Policy 2017, 102, 385–395. [Google Scholar]
- 12.Jørgensen H; Kristensen JB; Felby C., Enzymatic conversion of lignocellulose into fermentable sugars: challenges and opportunities. Biofuels, Bioproducts and Biorefining 2007, 1 (2), 119–134. [Google Scholar]
- 13.Nguyen Q; Saddler J., An integrated model for the technical and economic evaluation of an enzymatic biomass conversion process. Bioresource Technology 1991, 35 (3), 275–282. [Google Scholar]
- 14.Luterbacher JS; Rand JM; Alonso DM; Han J; Youngquist JT; Maravelias CT; Pfleger BF; Dumesic JA, Nonenzymatic sugar production from biomass using biomass-derived γ-valerolactone. Science 2014, 343 (6168), 277–280. [DOI] [PubMed] [Google Scholar]
- 15.Cai CM; Zhang T; Kumar R; Wyman CE, THF co-solvent enhances hydrocarbon fuel precursor yields from lignocellulosic biomass. Green Chemistry 2013, 15 (11), 3140–3145. [Google Scholar]
- 16.Liu Z; Zhang F-S, Effects of various solvents on the liquefaction of biomass to produce fuels and chemical feedstocks. Energy conversion and management 2008, 49 (12), 3498–3504. [Google Scholar]
- 17.Kumar R; Singh S; Singh OV, Bioconversion of lignocellulosic biomass: biochemical and molecular perspectives. Journal of industrial microbiology & biotechnology 2008, 35 (5), 377–391. [DOI] [PubMed] [Google Scholar]
- 18.Klein-Marcuschamer D; Oleskowicz-Popiel P; Simmons BA; Blanch HW, The challenge of enzyme cost in the production of lignocellulosic biofuels. Biotechnology and bioengineering 2012, 109 (4), 1083–1087. [DOI] [PubMed] [Google Scholar]
- 19.Humbird D; Davis R; Tao L; Kinchin C; Hsu D; Aden A; Schoen P; Lukas J; Olthof B; Worley M Process design and economics for biochemical conversion of lignocellulosic biomass to ethanol: dilute-acid pretreatment and enzymatic hydrolysis of corn stover; National Renewable Energy Lab.(NREL), Golden, CO (United States): 2011. [Google Scholar]
- 20.Zhao X; Cheng K; Liu D., Organosolv pretreatment of lignocellulosic biomass for enzymatic hydrolysis. Applied Microbiology and Biotechnology 2009, 82 (5), 815–827. [DOI] [PubMed] [Google Scholar]
- 21.Hendriks A; Zeeman G., Pretreatments to enhance the digestibility of lignocellulosic biomass. Bioresource technology 2009, 100 (1), 10–18. [DOI] [PubMed] [Google Scholar]
- 22.Mooney CA; Mansfield SD; Beatson RP; Saddler JN, The effect of fiber characteristics on hydrolysis and cellulase accessibility to softwood substrates. Enzyme and Microbial Technology 1999, 25 (8–9), 644–650. [Google Scholar]
- 23.Chundawat SP; Bellesia G; Uppugundla N; da Costa Sousa L; Gao D; Cheh AM; Agarwal UP; Bianchetti CM; Phillips GN Jr; Langan P., Restructuring the crystalline cellulose hydrogen bond network enhances its depolymerization rate. Journal of the American Chemical Society 2011, 133 (29), 11163–11174. [DOI] [PubMed] [Google Scholar]
- 24.Chang VS; Burr B; Holtzapple MT, Lime pretreatment of switchgrass. In Biotechnology for fuels and chemicals, Springer: 1997; pp 3–19. [DOI] [PubMed] [Google Scholar]
- 25.Fan L; Lee YH; Beardmore D., The influence of major structural features of cellulose on rate of enzymatic hydrolysis. Biotechnology and Bioengineering 1981, 23 (2), 419–424. [Google Scholar]
- 26.Grethlein HE, The Effect of Pore Size Distribution on the Rate of Enzymatic Hydrolysis of Cellulosic Substrates. Bio/Technology 1985, 3 (2), 155–160. [Google Scholar]
- 27.Alonso DM; Hakim SH; Zhou S; Won W; Hosseinaei O; Tao J; Garcia-Negron V; Motagamwala AH; Mellmer MA; Huang K., Increasing the revenue from lignocellulosic biomass: Maximizing feedstock utilization. Science advances 2017, 3 (5), e1603301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shuai L; Questell-Santiago YM; Luterbacher JS, A mild biomass pretreatment using γ-valerolactone for concentrated sugar production. Green Chemistry 2016, 18 (4), 937–943. [Google Scholar]
- 29.Motagamwala AH; Won W; Maravelias CT; Dumesic JA, An engineered solvent system for sugar production from lignocellulosic biomass using biomass derived γ-valerolactone. Green Chemistry 2016, 18 (21), 5756–5763. [Google Scholar]
- 30.Luterbacher JS; Azarpira A; Motagamwala AH; Lu F; Ralph J; Dumesic JA, Lignin monomer production integrated into the γ-valerolactone sugar platform. Energy & Environmental Science 2015, 8 (9), 2657–2663. [Google Scholar]
- 31.Yokoyama T., Revisiting the Mechanism of β-O-4 Bond Cleavage During Acidolysis of Lignin. Part 6: A Review. Journal of Wood Chemistry and Technology 2015, 35 (1), 27–42. [Google Scholar]
- 32.Deuss PJ; Scott M; Tran F; Westwood NJ; de Vries JG; Barta K., Aromatic monomers by in situ conversion of reactive intermediates in the acid-catalyzed depolymerization of lignin. Journal of the American Chemical Society 2015, 137 (23), 7456–7467. [DOI] [PubMed] [Google Scholar]
- 33.Johar N; Ahmad I; Dufresne A., Extraction, preparation and characterization of cellulose fibres and nanocrystals from rice husk. Industrial Crops and Products 2012, 37 (1), 93–99. [Google Scholar]
- 34.Yang Q; Pan X., Correlation between lignin physicochemical properties and inhibition to enzymatic hydrolysis of cellulose. Biotechnology and Bioengineering 2016, 113 (6), 1213–1224. [DOI] [PubMed] [Google Scholar]
- 35.Horváth IT; Mehdi H; Fábos V; Boda L; Mika LT, γ-Valerolactone—a sustainable liquid for energy and carbon-based chemicals. Green Chemistry 2008, 10 (2), 238–242. [Google Scholar]
- 36.Mellmer MA; Sener C; Gallo JMR; Luterbacher JS; Alonso DM; Dumesic JA, Solvent effects in acid-catalyzed biomass conversion reactions. Angewandte chemie international edition 2014, 53 (44), 11872–11875. [DOI] [PubMed] [Google Scholar]
- 37.Mellmer MA; Alonso DM; Luterbacher JS; Gallo JMR; Dumesic JA, Effects of γ-valerolactone in hydrolysis of lignocellulosic biomass to monosaccharides. Green Chemistry 2014, 16 (11), 4659–4662. [Google Scholar]
- 38.Unda F; Kim H; Hefer C; Ralph J; Mansfield SD, Altering carbon allocation in hybrid poplar (Populus alba× grandidentata) impacts cell wall growth and development. Plant biotechnology journal 2017, 15 (7), 865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamilton S; Hussain M; Bhardwaj A; Basso B; Robertson G., Comparative water use by maize, perennial crops, restored prairie, and poplar trees in the US Midwest. Environmental Research Letters 2015, 10 (6), 064015. [Google Scholar]
- 40.Lloyd TA; Wyman CE, Combined sugar yields for dilute sulfuric acid pretreatment of corn stover followed by enzymatic hydrolysis of the remaining solids. Bioresource technology 2005, 96 (18), 1967–1977. [DOI] [PubMed] [Google Scholar]
- 41.Morán JI; Alvarez VA; Cyras VP; Vázquez A., Extraction of cellulose and preparation of nanocellulose from sisal fibers. Cellulose 2008, 15 (1), 149–159. [Google Scholar]
- 42.Patil SK; Lund CR, Formation and growth of humins via aldol addition and condensation during acid-catalyzed conversion of 5-hydroxymethylfurfural. Energy & Fuels 2011, 25 (10), 4745–4755. [Google Scholar]
- 43.Falco C; Perez Caballero F; Babonneau F; Gervais C; Laurent G; Titirici M-M; Baccile N., Hydrothermal carbon from biomass: structural differences between hydrothermal and pyrolyzed carbons via 13C solid state NMR. Langmuir 2011, 27 (23), 14460–14471. [DOI] [PubMed] [Google Scholar]
- 44.Fu L; McCallum SA; Miao J; Hart C; Tudryn GJ; Zhang F; Linhardt RJ, Rapid and accurate determination of the lignin content of lignocellulosic biomass by solid-state NMR. Fuel 2015, 141, 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foston M., Advances in solid-state NMR of cellulose. Current opinion in biotechnology 2014, 27, 176–184. [DOI] [PubMed] [Google Scholar]
- 46.Atalla RH; Gast J; Sindorf D; Bartuska V; Maciel G., Carbon-13 NMR spectra of cellulose polymorphs. Journal of the American Chemical Society 1980, 102 (9), 3249–3251. [Google Scholar]
- 47.Gilardi G; Abis L; Cass AE, Carbon-13 CP/MAS solid-state NMR and FT-IR spectroscopy of wood cell wall biodegradation. Enzyme and Microbial Technology 1995, 17 (3), 268–275. [Google Scholar]
- 48.Newman RH, Estimation of the lateral dimensions of cellulose crystallites using 13C NMR signal strengths. Solid State Nuclear Magnetic Resonance 1999, 15 (1), 21–29. [DOI] [PubMed] [Google Scholar]
- 49.Earl WL; VanderHart D., Observations by high-resolution carbon-13 nuclear magnetic resonance of cellulose I related to morphology and crystal structure. Macromolecules 1981, 14 (3), 570–574. [Google Scholar]
- 50.Larsson PT; Hult E-L; Wickholm K; Pettersson E; Iversen T., CP/MAS 13C-NMR spectroscopy applied to structure and interaction studies on cellulose I. Solid State Nuclear Magnetic Resonance 1999, 15 (1), 31–40. [DOI] [PubMed] [Google Scholar]
- 51.Newman R; Hemmingson J; Suckling I., Carbon-13 nuclear magnetic resonance studies of kraft pulping. Holzforschung-International Journal of the Biology, Chemistry, Physics and Technology of Wood 1993, 47 (3), 234–238. [Google Scholar]
- 52.Hult E-L; Larsson PT; Iversen T., A comparative CP/MAS 13C-NMR study of the supermolecular structure of polysaccharides in sulphite and kraft pulps. Holzforschung 2002, 56 (2), 179–184. [Google Scholar]
- 53.Alam TM; Childress KK; Pastoor K; Rice CV, Characterization of free, restricted, and entrapped water environments in poly (N-isopropyl acrylamide) hydrogels via 1 H HRMAS PFG NMR spectroscopy. Journal of Polymer Science Part B: Polymer Physics 2014, 52 (23), 1521–1527. [Google Scholar]
- 54.Zhang C; Li P; Zhang Y; Lu F; Li W; Kang H; Xiang J.-f.; Huang Y; Liu R., Hierarchical porous structures in cellulose: NMR relaxometry approach. Polymer 2016, 98, 237–243. [Google Scholar]
- 55.Corsaro C; Mallamace D; Vasi S; Pietronero L; Mallamace F; Missori M., The role of water in the degradation process of paper using 1 H HR-MAS NMR spectroscopy. Physical Chemistry Chemical Physics 2016, 18 (48), 33335–33343. [DOI] [PubMed] [Google Scholar]
- 56.Selig MJ; Thygesen LG; Felby C., Correlating the ability of lignocellulosic polymers to constrain water with the potential to inhibit cellulose saccharification. Biotechnology for biofuels 2014, 7 (1), 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leppänen K; Andersson S; Torkkeli M; Knaapila M; Kotelnikova N; Serimaa R., Structure of cellulose and microcrystalline cellulose from various wood species, cotton and flax studied by X-ray scattering. Cellulose 2009, 16 (6), 999–1015. [Google Scholar]
- 58.Zalesny RS Jr; Wiese AH; Bauer EO; Riemenschneider DE, Sapflow of hybrid poplar (Populus nigra L.× P. maximowiczii A. Henry ‘NM6’) during phytoremediation of landfill leachate. Biomass and bioenergy 2006, 30 (8–9), 784–793. [Google Scholar]
- 59.Wilkerson C; Mansfield S; Lu F; Withers S; Park J-Y; Karlen S; Gonzales-Vigil E; Padmakshan D; Unda F; Rencoret J., Monolignol ferulate transferase introduces chemically labile linkages into the lignin backbone. Science 2014, 344 (6179), 90–93. [DOI] [PubMed] [Google Scholar]
- 60.Sugiyama J; Vuong R; Chanzy H., Electron diffraction study on the two crystalline phases occurring in native cellulose from an algal cell wall. Macromolecules 1991, 24 (14), 4168–4175. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.