

Abstract
Mutations in the DHDDS gene (MIM: 617836), encoding a subunit of dehydrodolichyl diphosphate synthase complex, have been recently implicated in very rare neurodevelopmental diseases. In total, five individuals carrying two de novo mutations in DHDDS have been reported so far, but genotype–phenotype correlations remain elusive. We reported a boy with a de novo mutation in DHDDS (NM_205861.3: c.G632A; p.Arg211Gln) featuring a complex neurological phenotype, including mild intellectual disability, impaired speech, complex hyperkinetic movements, and refractory epilepsy. We defined the electroclinical and movement disorder phenotype associated with the monoallelic form of the DHDDS -related neurodevelopmental disease and possible underlying dominant-negative mechanisms.
Keywords: DHDDS, neurodevelopmental disorder, epilepsy
An 11-year-old boy, born to unrelated healthy parents of Italian descent ( Fig. 1A ), presented with hypotonia and small head circumference (<third centile). Pregnancy and delivery were uncomplicated and growth parameters at birth were all within normal limits (weight 3,470 g, length 48 cm, occipitofrontal circumference 34 cm). There was no family history of neurological disorders. Abnormal motor coordination and walking difficulties became evident following the first year of life. At the age of 8 months, he could not get to the sitting position on his own nor remain seated. He learned to sit unaided at the age of 18 months and started to walk at the age of 26 months. He currently has a broad-based gait. He experienced two tonic-clonic seizures during an intercurrent illness at the age of 10 months evolving into short weekly febrile and afebrile tonic-clonic seizures at the age of 14 months. Moreover, the boy did suffer from daily drops of the head or falls to the ground, refractory to multiple antiseizure drugs, including valproic acid, topiramate, felbamate, rufinamide, and clobazam. From the age of 15 months, he presented with a movement disorder, that is, nonepileptic hyperkinetic behavior, including stereotyped hand movements, and myoclonus associated with choreoathetosis (see Video 1 ). He did show distinctive craniofacial features, namely, short and sloping forehead, laterally prominent eyebrows, periorbital fullness, low-set columella, large nostrils, droopy lower lip, tooth dysgenesis (malocclusion and enamel abnormalities), and open mouth appearance with drooling ( Fig. 1B ). Some skeletal anomalies, including hyperlaxity of the knees, flat feet, genua valga, and thoracic (dorsal and lumbar) dextroconvex scoliosis were also present. The speech was absent at the age of 5 years. His hearing and vision were unremarkable. Metabolic workup (e.g., urinary organic acids, blood amino acids, sialotransferrine profile) was also reported as normal. His electroencephalography (EEG) recordings showed sharp-wave and slow-wave complexes more evident on the posterior and frontal regions, in the context of a dysregulated background activity. Hyperventilation and photic stimulation were ineffective. Magnetic resonance imaging of the brain at age 5 and 10 years was unremarkable. Genetic studies, including karyotype, array comparative genome hybridization, SCN1A and GLUT1 genes sequencing, and targeted resequencing of genes ( n = 52) associated with developmental epileptic encephalopathies were all reported as normal.
Fig. 1.
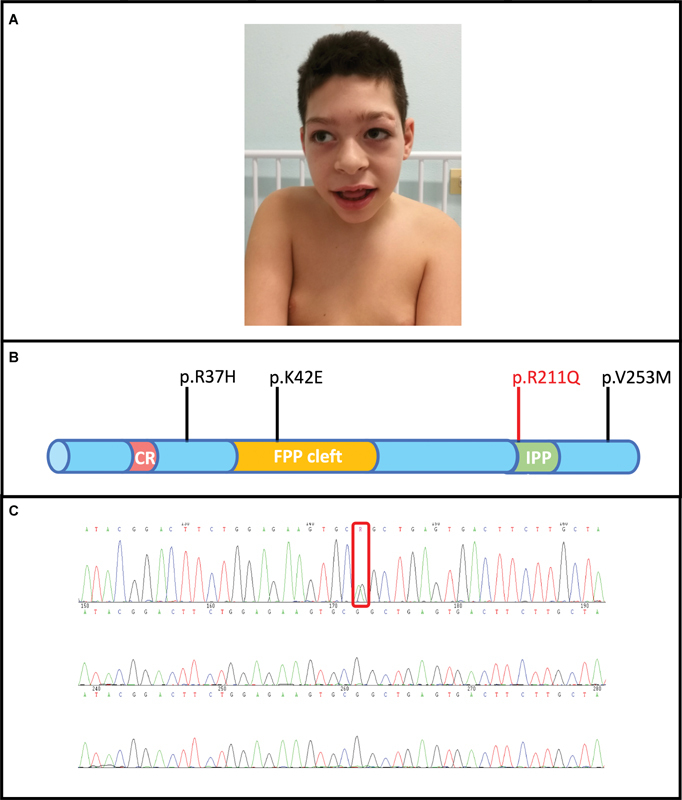
( A ) Distinctive facial features: short and sloping forehead, laterally prominent eyebrows, large nostrils, and an open mouth appearance with drooling. ( B ) A schematic illustration of the main functional domains of DHDDS protein: catalytic residue (CR)-farnesyl pyrophosphate (FPP) binding site; FPP cleft; isopentenyl pyrophosphate (IPP) binding site. All other previously reported DHDDS mutations are indicated in black, while our mutated de novo missense version of DHDDS protein is highlighted in red. ( C ) Sanger sequencing showing the proband carrying the de novo missense variant in DHDDS (c.G632A; p.Arg211Gln).
Video 1 Video of the boy showing hyperkinetic movements including choreoathetosis and myoclonus.
To investigate the molecular etiology of the phenotype in the affected individual, we then performed whole-exome sequencing (WES) of the trio as previously described. 1 2 In our analysis, we prioritized autosomal or X-linked recessive (homozygous, compound heterozygous, hemizygous) or heterozygous de novo variants that were plausible candidates for the complex clinical phenotype of the affected individual, presenting with intellectual disability, speech impairment, hyperkinetic movements, and epilepsy. As part of our filtering strategy, we focused on exonic and donor/acceptor splicing variants absent in the general population or present only at a very low frequency (<0.01% in public databases, including gnomAD v2.1.1; https://gnomad.broadinstitute.org/ ) and affecting genes previously implicated in neurodevelopmental diseases, epilepsy and movement disorders. We identified a de novo missense variant in DHDDS (NM_205861.3: c.G632A; p.Arg211Gln) that has been already reported as a cause of neurodevelopmental disease 3 variably associated with global developmental delay, intellectual disability, impaired (or absent) speech, and seizures/EEG abnormalities. No additional candidate variants emerged as plausible from the analysis of the WES data.
The dehydrodolichyl diphosphate synthase encoded by DHDDS belongs to a complex (the so-called human cis-prenyltransferase complex) 4 which plays a crucial role in protein glycosylation. 5 Protein N-glycosylation is an essential posttranslational modification crucial to protein folding and oligomerization, as well as sorting and transport of proteins. 6 Variations in glycosylation processes may markedly affect protein structure and function, potentially resulting in a wide range of disease phenotypes. 7 8 Biallelic mutations in DHDDS were initially shown to be implicated in autosomal recessive retinitis pigmentosa and severe developmental epileptic encephalopathies (MIM: 613861). 9 Recently, recurrent monoallelic de novo missense variants in DHDDS (c.G110A, p.Arg37His and c.G632A, p.Arg211Gln, MIM: 617836) have been reported in five patients with a milder glycosylation disorder presenting with impaired neurodevelopment and variable seizures and abnormal movements. 10 Interestingly, the allelic heterogeneity associated with the DHDDS -related neurodevelopmental disease suggests a possible dominant-negative mechanism underlying the monoallelic form of the condition. 3
In conclusion, we describe the electroclinical phenotype associated with monoallelic DHDDS deficiency, and in addition, we show that hyperkinetic movements can be an early feature in infancy. Further descriptions of patients with de novo DHDDS mutations will define the genotype–phenotype correlations in this rare genetic neurodevelopmental disorder.
Funding Statement
Funding This work was developed within the framework of the DINOGMI Department of Excellence of MIUR 2018–2022 (Legge 232/2016).
Conflict of Interest None declared.
Authors' Contributions
G.P. performed study design and experimental data acquisition, analysis, and interpretation of data for this study. E.A., M.S.V., and V.G. conceptualized the study, helped acquire clinical data, contributed to the manuscript, analyzed and interpreted phenotype data. M.I. contributed to the manuscript, and helped in analysis and interpretation of genetic data. F.M.V.S. performed critical revision of the manuscript document. P.S. and F.Z. supervised the study and obtained funding.
Consent for Publication
Written consent for recording and publishing photographs and videos of the patient were obtained from his parents.
References
- 1.Salpietro V, Malintan N T, Llano-Rivas I. Mutations in the neuronal vesicular SNARE VAMP2 affect synaptic membrane fusion and impair human neurodevelopment. Am J Hum Genet. 2019;104(04):721–730. doi: 10.1016/j.ajhg.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salpietro V, Dixon C L, Guo H. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat Commun. 2019;10(01):3094. doi: 10.1038/s41467-019-10910-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sabry S, Vuillaumier-Barrot S, Mintet E. A case of fatal Type I congenital disorders of glycosylation (CDG I) associated with low dehydrodolichol diphosphate synthase (DHDDS) activity. Orphanet J Rare Dis. 2016;11(01):84. doi: 10.1186/s13023-016-0468-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lisnyansky Bar-El M, Lee S Y, Ki A Y. Structural characterization of full-length human dehydrodolichyl diphosphate synthase using an integrative computational and experimental approach. Biomolecules. 2019;9(11):660. doi: 10.3390/biom9110660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grabińska K A, Park E J, Sessa W C. CIS-prenyltransferase: new insights into protein glycosylation, rubber synthesis, and human diseases. J Biol Chem. 2016;291(35):18582–18590. doi: 10.1074/jbc.R116.739490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Helenius A, Aebi M.Intracellular functions of N-linked glycans Science 2001291(5512):2364–2369. [DOI] [PubMed] [Google Scholar]
- 7.Ng B G, Freeze H H. Perspectives on glycosylation and its congenital disorders. Trends Genet. 2018;34(06):466–476. doi: 10.1016/j.tig.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fiumara A, Barone R, Del Campo G, Striano P, Jaeken J. Electroclinical features of early-onset epileptic encephalopathies in congenital disorders of glycosylation (CDGs) JIMD Rep. 2016;27:93–99. doi: 10.1007/8904_2015_497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freeze H H. Understanding human glycosylation disorders: biochemistry leads the charge. J Biol Chem. 2013;288(10):6936–6945. doi: 10.1074/jbc.R112.429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamdan F F, Myers C T, Cossette P. High rate of recurrent de novo mutations in developmental and epileptic encephalopathies . Am J Hum Genet. 2017;101(05):664–685. doi: 10.1016/j.ajhg.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]