

Abstract
A Silver syndrome is a rare autosomal dominant spastic paraparesis in which spasticity of the lower limbs is accompanied by amyotrophy of the small hand muscles. The causative gene is the Berardinelli-Seip congenital lipodystrophy 2 ( BSCL2) , which is related to a spectrum of neurological phenotypes. In the current study, we presented a 14-year-old male with a slowly progressive spastic paraparesis with urinary incontinence that later on exhibited atrophy and weakness in the thenar and dorsal interosseous muscles. Magnetic resonance imaging (MRI) revealed discrete atrophy of the corpus callosum isthmus and an extended next-generation sequencing panel identified a de novo heterozygous mutation in BSCL2 gene, c.269C > T p.(S90L). Various clinical expression and incomplete penetrance of BSCL2 gene mutations complicate the establishment of a genetic etiology for these cases. Therefore, Silver syndrome should be included in the differential diagnosis if the initial presentation is a spastic paraparesis by urinary involvement with childhood-onset, even with MRI atypical findings. This report described the first Iberian Silver syndrome case carrying a de novo c.269C > T p. (S90L) BSCL2 gene mutation.
Keywords: BSCL2, Silver syndrome, S90L
Introduction
Mutations of Berardinelli-Seip congenital lipodystrophy 2 ( BSCL2 ) gene, mapped to chromosome region 11q12-q14, cause a variety of clinically heterogeneous phenotypes: a congenital severe generalized lipodystrophy of autosomal recessive transmission, and isolated neurological manifestations of motor neuron disease in a heterozygous state, including hereditary spastic paraparesis (HSP) type 17 (SPG17), Silver syndrome (SS), distal hereditary motor neuropathy type-V (dHMN-V) and Charcot-Marie-Tooth disease(CMT)-type 2. 1 2 3 4 5 6 The exact pathophysiology of these mutations and an explanation for different clinical expressions remain to be elucidated. 6
SS is a rare form of autosomal dominant HSP, originally characterized by an initially spastic paraparesis phenotype with marked weakness and atrophy of thenar, first dorsal interosseous muscles of the hand and, to a lesser extent, foot muscles. 1 5 6 7 8 9 10 11 The phenotypical variability in SS is being highly recognized. 7 12 Urinary incontinence has demonstrated in one case with childhood onset 13 and pes cavus is also rarely reported. 10 13 Age at onset ranges from 8 to 70 years 13 14 and the progression of disease is rather slow and leads to a relatively mild impact on daily live functioning. 7 13 14 Due to the incomplete and variable penetrance, BSCL2 mutations can manifest as a sporadic disease. 1 3 9 Clinically, the carriers can range from asymptomatic to severely affected patients. We hereby report the first Iberian patient with SS carrying a de novo BSCL2 gene c.269C > T p.(S90L) mutation, presenting urinary dysfunction and atypical findings on MRI, and discuss literature review.
Case Report
We reported a 14-year-old Caucasian male patient with right foot deformity and associated complaints of pain and urinary incontinence with onset at the age of 6. He is the only child of a nonconsanguineous healthy couple, with no family history for neurological diseases. Pregnancy and birth were uneventful, psychomotor development was adequate and past medical history was unremarkable. The first examination report when he was 6 years old revealed weakness of the ankle dorsiflexors [right/left side grade ¾ Medical Research Council (MRC) scale], lower limb hyperreflexia, mild lower limb distal spasticity, and bilateral pes cavus , worse on the right side. No other abnormalities on examination were found, namely cognitive impairment. He did not present any walking difficulties, falls, or compromise in his daily activities.
During the next 6 years, symptoms slowly progressed and the weakness in lower limbs worsened. He was progressively having difficulties in climbing stairs and was unable to run at school. Urinary complaints were stable. At neurological examination, he presented a scissoring gait pattern, possible without support, and symmetric lower limb weakness (grade 4 proximal and grade 2 in ankle dorsiflexors, MRCs). He maintained spasticity and hyperreflexia, pes cavus with equinus deformity and bilateral claw toes.
At the age of 11, the patient performed a neuroaxis MRI: brain MRI showed a discrete atrophy of isthmus of the corpus callosum ( Fig. 1 ). The spinal cord image was normal. Needle electromyography showed chronic neurogenic motor unit potentials restricted to the anterior tibial muscles, with normal motor and sensory nerve conduction studies. At the same time, invasive urodynamic testing suggested a terminal overactive bladder. Genetic testing included a normal array-comparative genomic hybridization and a negative molecular single gene analysis of SPAST , ATL1, and SPG11 genes.
Fig. 1.
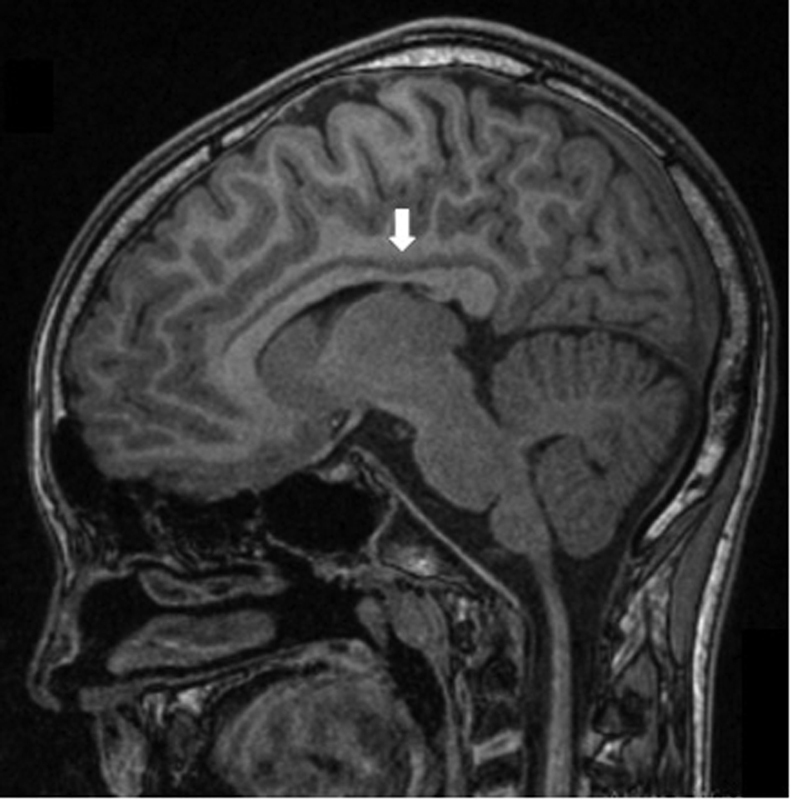
MRI of the brain, showing a discrete atrophy of isthmus of the corpus callosum ( arrow ) (FLAIR image). FLAIR, fluid-attenuated inversion recovery; MRI, magnetic resonance imaging.
At the age of 13, the patient noted symmetrical wasting and weakness of both his hands. His recent neurological examination revealed bilateral atrophy of thenar and first dorsal interosseous muscles of the hand, which resulted in split-hands , accompanied with weakness of finger abduction (grade 4 MRCs), a predominantly distal lower limb symmetric weakness (hip flexion/extension grade 4 MRCs, ankle dorsiflexion grade 2 MRCs), lower limb spasticity, generalized hyperreflexia, and bilateral pes cavus deformity ( Fig. 2 ). He had a scissoring gait pattern with bilateral steppage , which got slower, needing bilateral support for greater distances. Furthermore, no other abnormalities were observed. A recent neurophysiological study revealed normal, sensitive nerve conduction in the upper and lower limbs and an absence of response in peroneal and tibial motor nerve conduction. Needle electromyography showed severe signs of chronic neurogenic change in combination with abnormal spontaneous activity, including positive sharp waves and fibrillations, with a proximal-distal gradient ( Table 1 ). The patient and the parents were consulted in a Genetic Unit, confirming a biological paternity, and at this time an extended next-generation sequencing panel of 6110 genes (focused exome, Agilent) identified as a heterozygous mutation of BSCL2 gene, c.269C > T p.(S90L). His parents were confirmed as noncarriers for the mutation, establishing this genetic alteration as de novo. All the family received genetic counseling.
Fig. 2.
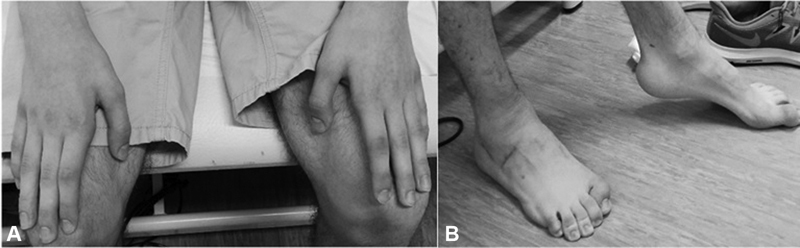
( A ) The hands of the patient had predominant atrophy and weakness in the thenar and first dorsal interosseous muscles (s plit-hands) . ( B ) Pes cavus deformity and claw toes bilaterally.
Table 1. Neurophysiological study.
| Activity | Spontaneous activity | Voluntary activity | Recruitment | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Muscle | Insertion | Fibrillations | Positive waves | Fasciculations | Polyphasia | MUP amp. | Configuration | MUP pattern | MUP duration | Max. amp. | Recruitment | Max. effort |
| R. first dorsal interosseous | N | +1 | +1 | Absent | Absent | N | N | Freq | Inc + | N | Red | Max |
| R. tibial anterior | N | +2 | +2 | Absent | Absent | N | N | Freq | Inc + | N | S | Max |
| L. tibial anterior | N | +1 | +1 | Absent | Absent | N | N | Freq | Inc + | N | S | Max |
| R. medial gastrocnemius | N | +2 | +2 | Absent | Absent | |||||||
| L. medial gastrocnemius | N | +2 | +2 | Absent | Absent | N | N | Freq | Inc + | N | S | Max |
| R. vastus medialis | N | Absent | Absent | Absent | Absent | N | N | Freq | Inc + | N | S | Max |
| L. vastus medialis | N | Absent | Absent | Absent | Absent | N | N | N | N | N | N | Max |
Abbreviations: amp., amplitude; freq, frequent; inc, increased; L., left; max., maximal; MUP, motor unit potential; N, normal; R., right; red, reduced; S, simple.
Supportive management was given to the patient, with a physiotherapy program since an early age, currently twice per week, and nocturnal ankle splinters. Spasticity management also included botulinum toxin injections in the lower limbs and medication with baclofen 5 mg/d. A surgical correction of the right foot deformity was done at the age of 13 (percutaneous lengthening of the Achilles tendon, tibial posterior transfer through the interosseous membrane to the second cuneiform, peroneus longus tendon tenodesis to peroneus brevis , tarsectomy and first metatarsal proximal extension chevron-type osteotomy), with clinical improvement of pain and improving foot posture and gait. A surgical intervention to his left foot was performed at the age of 15 (percutaneous lengthening of the Achilles tendon, tibial posterior transfer through the interosseous membrane to the second cuneiform, peroneus longus tendon tenodesis to peroneus brevis , valgus-Dwyer-like osteotomy of the calcaneus and first metatarsal proximal extension chevron-type osteotomy). Urinary dysfunction was managed with lifestyle measures.
Discussion
Our patient was an example of the clinical heterogeneity of BSCL2 gene mutations, as a cause of upper and lower motor neuron involvement. Although initially presenting as an HSP with neurogenic bladder, resembling a pure SPG17, his symptoms progressed to a clinically relevant motor-neuron disease with upper limb involvement, as described in SS. The initial differential diagnoses could include a complicated HSP or an atypical CMT-like presentation, as he also showed lower neuron involvement in lower limbs ( pes cavus , atrophy, and steppage gait). In cases of dHMN-V, hand weakness usually precedes spastic paraparesis, 4 5 8 which was not the case. Therefore, the presence of upper motor neuron signs directed the initial diagnosis. We would like to point out two different aspects of this case. First, urinary involvement is rarely described in SS. Neurogenic bladder is described in HSP spectrum, although its frequency might not be routinely addressed and hence ignored as a common complaint. 15 As urinary incontinence is not so easily overlooked, it can account for the clinical remark in the other reported case. We remind that thorough inquest about bladder complaints is clinically important in complex neurological cases. Second, we are unaware of any previous reports of SS with abnormal brain MRI—our case presented a thin corpus callosum isthmus, and not all of its length as is characteristically found in other complicated HSP (mainly SPG11 gene mutations). This finding can reveal the heterogeneity of these disorders and the overlap between different mutations and clinical manifestations. 16
In our case, molecular characterization allowed identification of an already known causative mutation, albeit being de novo. Cases reported of SS are mainly caused by c.269C > T p.(S90L) and c.263A > G p.(N88S) mutations. 1 2 3 4 5 6 7 Cen et al 4 reported that c.269C > T p.(S90L) mutation is predominantly associated with SS (87.5% of the patients). Moreover, the most recent review about the variability phenotype of so-far published cases/families with BSCL2 mutations described only 14 patients with SS caused by c.269C > T p.(S90L). 1 This phenotype may be more severe than that of c.263A > G p.(N88S). 4 15 A novel mutation—c.269C > G p.(S90W)—has been recently described in a CMT-type 2 phenotype in a Korean family, which demonstrates that the same protein can cause involvement in upper and lower motor neurons. 1 10 Furthermore, in the last few weeks, a Japanese group reported two more novel mutations with specific and interesting clinical findings: c.263A > C p.(N88T) causing an earlier onset phenotype of tetraparesis with vocal cord paresis and respiratory dysfunction; and c.421T > G p.(S141A) mutation carriers, with electrophysiological changes resembling CMT-type 2. 17 Other studies expanded the neurological involvement of BSCL2 gene mutations, with a severe epileptic encephalopathy described in two patients with de novo mutations in exon 4 of the gene. 18 Previous studies suggest that the mutations in exon 3 are probably only two associated with SS and dHMN-V and therefore, the analysis for these disorders may be restricted to this exon. 3 8 Remarkably, the neurological phenotypical spectrum of BSCL2 gene mutations is quite variable, and further genotype–phenotype relations might arise.
The molecular effects of these mutations have been further explored. The structure of human protein Seipin, coded by the BSCL2 gene, has been already studied; it is an integral membrane protein of the endoplasmic reticulum (ER) that helps to maintain the phospholipid homeostasis and the surface tension. 19 It plays a critical role in the assembly and/or expansion of lipid droplets formation and adipocyte differentiation, although the molecular function remains controversial. 19 20 21 Loss-of-function mutations of Seipin are associated with the most severe form of congenital lipodystrophy, the BSCL2 , an autosomal recessive disorder characterized by an almost complete loss of adipose tissue, insulin resistance, fatty liver, and hypertriglyceridemia. 6 20 21 Specifically, c.269C > T p.(S90L) mutation is a gain-of-function mutation, affecting a predicted N -glycosylation site in the encoded protein, leading to an accumulation and aggregation of the unfolded protein in the ER, neuronal toxicity, and cell death. 22
Treatment of this neurodegenerative condition is only symptomatic, including physiotherapy, orthopaedic correction surgeries for foot deformities and also drug therapy for spasticity, with limited benefits. 5 14 Genetic counselling of these patients is essential and requires a multidisciplinary team.
In conclusion, the variable clinical expression of BSCL2 gene mutations and incomplete penetrance potentially complicate the molecular diagnostic strategy. Although the absence of family history and the slow progression of our patient resembled an autosomal recessive transmission pattern, it is important, to consider BSCL2 mutations as a disorder when the clinical syndrome is alike to SS. Also, as hand involvement in SS presents later, these mutations related to SS should be included in the differential diagnosis of HSP, especially in pediatric-onset cases and even if urinary dysfunction is present, as the full clinical picture may still be emerging. In the present report, we highlight the value of the new techniques of genetic sequencing for an accurate molecular diagnosis in phenotypically challenging cases.
Funding Statement
Funding None.
Footnotes
Conflict of Interest None declared.
References
- 1.Musacchio T, Zaum A K, Üçeyler N. ALS and MMN mimics in patients with BSCL2 mutations: the expanding clinical spectrum of SPG17 hereditary spastic paraplegia. J Neurol. 2017;264(01):11–20. doi: 10.1007/s00415-016-8301-2. [DOI] [PubMed] [Google Scholar]
- 2.Rakocević-Stojanović V, Milić-Rasić V, Perić S. N88S mutation in the BSCL2 gene in a Serbian family with distal hereditary motor neuropathy type V or Silver syndrome J Neurol Sci 2010296(1-2):107–109. [DOI] [PubMed] [Google Scholar]
- 3.Brugman F, Scheffer H, Schelhaas H J. Seipin/BSCL2 mutation screening in sporadic adult-onset upper motor neuron syndromes. J Neurol. 2009;256(05):824–826. doi: 10.1007/s00415-009-5009-6. [DOI] [PubMed] [Google Scholar]
- 4.Cen Z, Lu X, Wang Z, Ouyang Z, Xie F, Luo W. BSCL2 S90L mutation in a Chinese family with Silver syndrome with a review of the literature. J Clin Neurosci. 2015;22(02):429–430. doi: 10.1016/j.jocn.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Rowland L P, Bird T D. Silver syndrome: the complexity of complicated hereditary spastic paraplegia. Neurology. 2008;70(21):1948–1949. doi: 10.1212/01.wnl.0000312519.62351.5b. [DOI] [PubMed] [Google Scholar]
- 6.Windpassinger C, Auer-Grumbach M, Irobi J. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat Genet. 2004;36(03):271–276. doi: 10.1038/ng1313. [DOI] [PubMed] [Google Scholar]
- 7.van de Warrenburg B P, Scheffer H, van Eijk J J. BSCL2 mutations in two Dutch families with overlapping Silver syndrome-distal hereditary motor neuropathy. Neuromuscul Disord. 2006;16(02):122–125. doi: 10.1016/j.nmd.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Rohkamm B, Reilly M M, Lochmüller H.Further evidence for genetic heterogeneity of distal HMN type V, CMT2 with predominant hand involvement and Silver syndrome J Neurol Sci 2007263(1-2):100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel H, Hart P E, Warner T T. The Silver syndrome variant of hereditary spastic paraplegia maps to chromosome 11q12-q14, with evidence for genetic heterogeneity within this subtype. Am J Hum Genet. 2001;69(01):209–215. doi: 10.1086/321267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monteiro A, Real R, Nadais G, Silveira F, Leão M. BSCL2 N88S mutation in a Portuguese patient with the Silver syndrome. Muscle Nerve. 2015;51(03):456–458. doi: 10.1002/mus.24455. [DOI] [PubMed] [Google Scholar]
- 11.Cho H J, Sung D H, Ki C S. Identification of de novo BSCL2 Ser90Leu mutation in a Korean family with Silver syndrome and distal hereditary motor neuropathy. Muscle Nerve. 2007;36(03):384–386. doi: 10.1002/mus.20792. [DOI] [PubMed] [Google Scholar]
- 12.Irobi J, Van den Bergh P, Merlini L.The phenotype of motor neuropathies associated with BSCL2 mutations is broader than Silver syndrome and distal HMN type V Brain 2004127(Pt 9):2124–2130. [DOI] [PubMed] [Google Scholar]
- 13.Poon M, Nguyen T P. Clinical reasoning: childhood-onset atrophy and spasticity. Neurology. 2016;86(13):e140–e143. doi: 10.1212/WNL.0000000000002519. [DOI] [PubMed] [Google Scholar]
- 14.Ito D. BSCL2-related neurologic disorders/seipinopathy summary genetic counseling GeneReview scope. GeneReviews; 2018:1–12. Accessed February 10, 2020 at:https://www.ncbi.nlm.nih.gov/books/NBK1307/
- 15.Braschinsky M, Zopp I, Kals M, Haldre S, Gross-Paju K. Bladder dysfunction in hereditary spastic paraplegia: what to expect? J Neurol Neurosurg Psychiatry. 2010;81(03):263–266. doi: 10.1136/jnnp.2009.180331. [DOI] [PubMed] [Google Scholar]
- 16.Franco F, Junqueira T, Rezende R. Neuroimaging in hereditary spastic paraplegias. Curr Use Future Perspect. 2019;9:1–1117. doi: 10.3389/fneur.2018.01117. [DOI] [Google Scholar]
- 17.Ishihara S, Okamoto Y, Tanabe H.Clinical features of inherited neuropathy with BSCL2 mutations in Japan J Peripher Nerv Syst 2020(e-pub ahead of print) 10.1111/jns.12369 [DOI] [PubMed] [Google Scholar]
- 18.Fernández-Marmiesse A, Sánchez-Iglesias S, Darling A. A de novo heterozygous missense BSCL2 variant in 2 siblings with intractable developmental and epileptic encephalopathy. Seizure. 2019;71:161–165. doi: 10.1016/j.seizure.2019.07.019. [DOI] [PubMed] [Google Scholar]
- 19.Yan R, Qian H, Lukmantara I. Human SEIPIN binds anionic phospholipids. Dev Cell. 2018;47(02):248–2.56E6. doi: 10.1016/j.devcel.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 20.Cui X, Wang Y, Tang Y. Seipin ablation in mice results in severe generalized lipodystrophy. Hum Mol Genet. 2011;20(15):3022–3030. doi: 10.1093/hmg/ddr205. [DOI] [PubMed] [Google Scholar]
- 21.Gao M, Huang X, Song B L, Yang H. The biogenesis of lipid droplets: lipids take center stage. Prog Lipid Res. 2019;75:100989. doi: 10.1016/j.plipres.2019.100989. [DOI] [PubMed] [Google Scholar]
- 22.Ito D, Fujisawa T, Iida H, Suzuki N. Characterization of seipin/BSCL2, a protein associated with spastic paraplegia 17. Neurobiol Dis. 2008;31(02):266–277. doi: 10.1016/j.nbd.2008.05.004. [DOI] [PubMed] [Google Scholar]