

Abstract
Background Childhood ataxia with central nervous system hypomyelination (CACH) is a recently described childhood inherited white matter disease, caused by mutations in any of the five genes encoding eukaryotic translation initiation factor ( eIF2B ).
Methods Retrospective review of the charts of children with CACH was performed from January 2014 to March 2020 at tertiary care center from Southern India. Diagnosis was based on magnetic resonance imaging (MRI) criteria or genetic testing.
Results Total number of children with CACH enrolled were 18. Male/female ratio was 10:8. Mean age of presentation was 37.11 months (range = 6–144 months). Affected siblings were seen in five (28%) cases. All children had spasticity, ataxia, and diffuse white matter changes with similar signal as cerebrospinal fluid on all pulse sequences on MRI brain. Of the 18 children, only nine are alive. Duration of illness among deceased children was 9.6667 months (range = 2–16 months). Waxing and waning of symptoms were seen in seven cases. Genetic analysis of EIF2B gene was performed in five cases, among which three mutations were novel.
Conclusion A diagnosis of childhood ataxia with central nervous system hypomyelination should be considered in patients presenting with acute onset neuroregression following infection or trauma with associated neuroimaging showing classical white matter findings.
Keywords: EIF2B, childhood ataxia with central hypomyelination, vanishing white matter disease
Introduction
Childhood ataxia with diffuse central nervous system hypomyelination (CACH), also called as vanishing white matter disease (VWMD; MIM 603896), is a major white matter disease characterized by ataxia, spasticity, and neuroregression with variable progression. 1 2 3 4 5 6 The disease is caused by a gene encoding human eukaryotic translation initiation factor ( EIF2B ). 7 8 EIF2B has five subunits, EIF2B1 , EIF2B2 , EIF2B3 , EIF2B4 , and EIF2B5 . The phenotypic range includes a prenatal/congenital form, a subacute infantile form (onset age < 1 year), an early childhood-onset form (onset age 1 to < 4 years), a late childhood-/juvenile-onset form (onset age 4 to < 18 years), and an adult-onset form (onset ≥ 18 years). 9 The diagnostic criteria include normal initial psychomotor development, deterioration following infection or trauma, and typical clinical findings of ataxia and spasticity. Magnetic resonance imaging (MRI) brain shows diffuse white matter signal changes with signal intensity of cerebrospinal fluid (CSF) on all pulse sequences. 10 11 12 Until now 250 cases and 150 mutations of EIF2B1, five have been reported in the literature. 6 Most commonly reported among Caucasians, the precise incidence in other populations are not known. In Asia, incidence has only been reported in China and Japan. 13 Only a few cases have been reported in India to date. 14 15 16 Hence, we aimed to study and describe clinical, neuroimaging and molecular findings of Indian children with CACH.
Methods
This is a retrospective chart review of CACH patients from a single center tertiary care referral center from Southern India. The medical records of children attending the pediatric neurology clinic and those who were admitted in pediatric neurology and pediatric ward from January 2016 to March 2020 were analyzed. Among them, only children who were confirmed to have a diagnosis of CACH either by MRI criteria or genetic studies were included and formed the study group. Those case records were retrieved separately and analyzed in detail. The data were extracted as per predesigned proforma. The diagnostic criteria for CACH proposed by van der Knaap et al 1 was followed in this study. All the following four criteria should be satisfied:
Psychomotor development is normal or near normal initially.
Onset of neurological deterioration is episodic with chronic progressive course and occurs in childhood.
-
Neurological signs typically include:
Cerebellar ataxia
Spasticity
Optic atrophy (not always)
Epilepsy (not always)
Motor function disproportionately affected
Imaging (MRI) bilateral and symmetric cerebral hemispheric signal white matter intensities like CSF. 1
Details of history including family history, examination including fundus, investigations like complete hemogram, liver function, renal function, blood glucose, serum electrolytes, serum ammonia, serum lactate, arterial blood gas, urine ketones, tandem mass spectrometry (TMS), and MRI brain were collected. Special investigations if performed on a case to case basis like disease specific enzyme assays, nerve conductions studies, and urinary gas chromatography mass spectrometry were also included.
We did targeted gene sequencing by next generation sequencing. DNA extracted from blood was used to perform targeted gene capture using a custom capture kit. The libraries were sequenced to mean > 80 to 100 × coverage on Illumina sequencing platform as per the manufacturer protocol. Sequences obtained were aligned to human reference genome (GRCh37/hg19), QC, data mapping, variant calling, and annotation of variants with external and internal data sources were achieved with a customized genome analysis toolkit framework. Gene/variant annotation was achieved using variant effect predictor program against the ensemble release 91 human gene model. With each transcript listed, the analyzed region includes coding exons and ± 10 base pair of flanking intronic region on both sides of each exon. Clinically relevant mutations were annotated using published variants in literature and a set of diseases databases: ClinVar, OMIM (updated on November 21, 2018), genome-wide association study, human gene mutation database (v2018.3), and SwissVar. Common variants are filtered based on allele frequency in 1000Genome Phase 3, ExAC (v1.0), gnomAD (v2.1), exome variant server, single nucleotide polymorphism database (v151), 1000 Japanese Genome, and Indian population database. Nonsynonymous variants effect was calculated using multiple algorithms such as polymorphism phenotyping v2 (PolyPhen-2), sorting intolerant from tolerant (SIFT), MutationTaster2, and LRT. Only nonsynonymous and splice site variants found in the targeted gene capture were used for clinical interpretation and synonymous, and deep intronic variants not previously reported were not considered for the analysis. QIAGEN Ingenuity Variant Analysis was used to identify variants which are relevant to the clinical indication and were classified as per the American College of Medical Genetics and Genomics (ACMG) guidelines. The variants were classified based on the standard guidelines of ACMG-Association for Molecular Pathology, and scored based on the evidences and strength of each criteria. Simple descriptive statistics were used to analyze the data. Ethical clearance was obtained from institutional ethical committee.
Results
Among 383 children with well characterized genetic white matter diseases from our records, there were totally 18 cases of CACH (4.6%). Male:female ratio was 10:8. The mean age of presentation was 37.11 months with a range from 6 to 144 months. Table 1 shows clinical and MRI findings of individual cases. The phenotypic range included no prenatal/congenital form, six (33%) subacute infantile form, nine (50%) early childhood-onset form (onset age 1 to < 4 years) and three (17%) late childhood-/juvenile-onset form. Consanguinity was seen in 17 (94%) with family history of sibling deaths in five (28%). Two pairs of affected siblings were observed.
Table 1. Clinical and radiological features of childhood ataxia with central nervous system hypomyelination cases.
| Details/patient number | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gender | M | F | F | F | M | F | M | M | F | M | F | M | M | M | F | M | M | F |
| Affected siblings | Y | Y | Y | Y | Y | N | N | N | N | N | N | N | N | N | N | N | N | N |
| Age at onset (mo) | 9 | 10 | 24 | 9 | 12 | 6 | 84 | 42 | 24 | 36 | 9 | 42 | 84 | 42 | 30 | 9 | 52 | 144 |
| Initial motor delay | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N |
| Course | SP | W | W | RP | RP | W | SP | RP | W | SP | RP | SP | W | W | RP | RP | W | SP |
| Precipitating factors | Inf | NK | F | Inf | Inf | NK | NK | F | Inf | Inf | Inf | NK | NK | Inf | Inf | Inf | Inf | NK |
| Spasticity | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| Epilepsy | Y | Y | N | Y | Y | Y | Y | N | Y | Y | N | Y | Y | Y | Y | Y | Y | Y |
| Optic atrophy | N | N | Y | Y | Y | Y | N | Y | Y | N | N | Y | N | Y | Y | N | N | N |
| Macrocephaly | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N | N |
| Ataxia | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| Disease duration (mo) | 24 | 14 | 14 | 12 | 9 | 3 | 48 | 12 | 9 | 40 | 16 | 6 | 48 | 78 | 10 | 2 | 4 | 12 |
| MRI of brain | ||||||||||||||||||
| Diffuse white matter involvement | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| Medulla oblongata | Y | Y | N | Y | Y | N | Y | Y | Y | Y | N | Y | Y | Y | N | Y | Y | Y |
| Central tegmental tract | Y | Y | N | Y | Y | N | Y | Y | N | Y | Y | N | Y | Y | N | Y | N | Y |
| Basal ganglia and thalamus | Y | N | Y | N | Y | Y | N | N | Y | N | Y | N | Y | N | Y | N | Y | Y |
| Cerebellar white matter | Y | N | Y | N | Y | Y | N | N | Y | N | Y | N | Y | N | Y | N | Y | Y |
| Mid brain | Y | N | Y | Y | N | Y | N | N | Y | Y | N | N | Y | N | Y | N | Y | Y |
| Internal capsule | Y | N | N | Y | Y | N | Y | N | N | Y | N | N | Y | N | Y | N | Y | N |
| Pons | N | Y | N | Y | Y | Y | N | N | N | Y | N | N | Y | N | Y | N | Y | N |
| Corpus callosum | N | N | N | Y | Y | N | Y | N | N | Y | N | N | Y | N | N | N | Y | N |
| Outcome | A | A | E | E | E | E | A | E | E | A | E | A | A | A | E | E | A | A |
Abbreviations: A, alive; C, cerebellum; CC, carpus callosum; CTT, central tegmental tracts; D, diffuse; DG, deep gray matter lentiform nucleus, caudate nucleus, and thalamus; E, expired; F, fall; IC, internal capsule; Inf, infections; M, medulla; MB, midbrain; N, no; NA, not available; NK, not known; P, pons; RP, rapidly progressive; SP, slowly progressive; W, waxing and waning; Y, yes.
The initial clinical presentation of all 18 (100%) individuals predominantly included motor regression, spasticity, and ataxia. Epilepsy was observed in 15 (83.33%) and optic atrophy in nine (50%). Among the 15 epilepsy cases, 10 had generalized tonic, four had focal, and one had myoclonic seizures. Initial precipitating factors were identified in 12 (67%). Initial precipitating factors in our study included falls in three, infection/febrile illnesses in 10, and in five; no triggers were identified. Developmental delay was not observed prior to illness in any of our cases.
Baseline investigations including complete hemogram, TMS, serum ammonia, serum lactate, and arterial blood gas were normal in all. Brain magnetic resonance imaging was done in all children. It revealed white matter signal changes same as CSF intensity on all pulse sequences in all (100%) and involvement of all lobes were observed in all children (100%). Fig. 1 shows a summary of MRI brain findings and structures affected in this series. Fig. 2 shows T2WI displaying diffuse periventricular and lobar white matter hyperintensity with a somewhat striated appearance and fluid attenuation inversion recovery (FLAIR) image showing partial inversion of the frontal and parietal white matter lesions. Genetic testing was done in five cases ( Table 2 ) in which two previously reported pathogenic variants, and three novel pathogenic variants were observed. The three novel variants were p.R195H on EIF2B5 gene, p.R15Y on EIF2B3 , and p.V117A on EIF2B3 gene. The clinical course of the children was rapidly progressive in seven (39%), slowly progressive in four (22%), and waxing and waning in seven (39%). Eight (44%) children succumbed and 10 (56%) children are on follow-up with predominant motor impairment and neurological problems as described above with 40% of surviving children having swallowing disturbances.
Fig. 1.
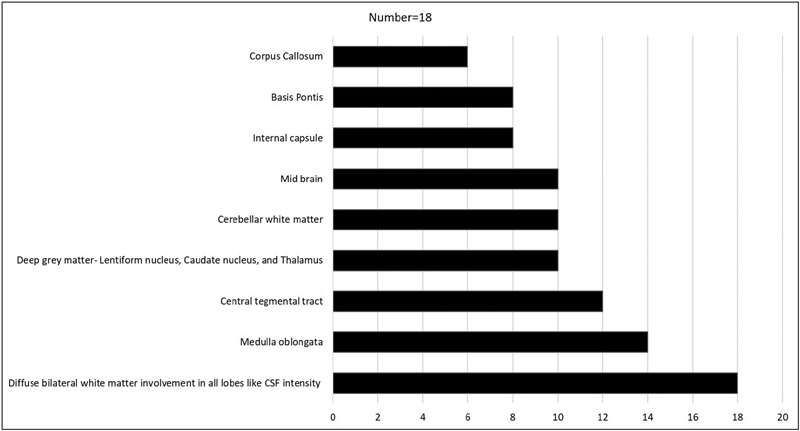
Showing involvement of various structures in magnetic resonance imaging of brain in childhood ataxia with central nervous system hypomyelination.
Fig. 2.
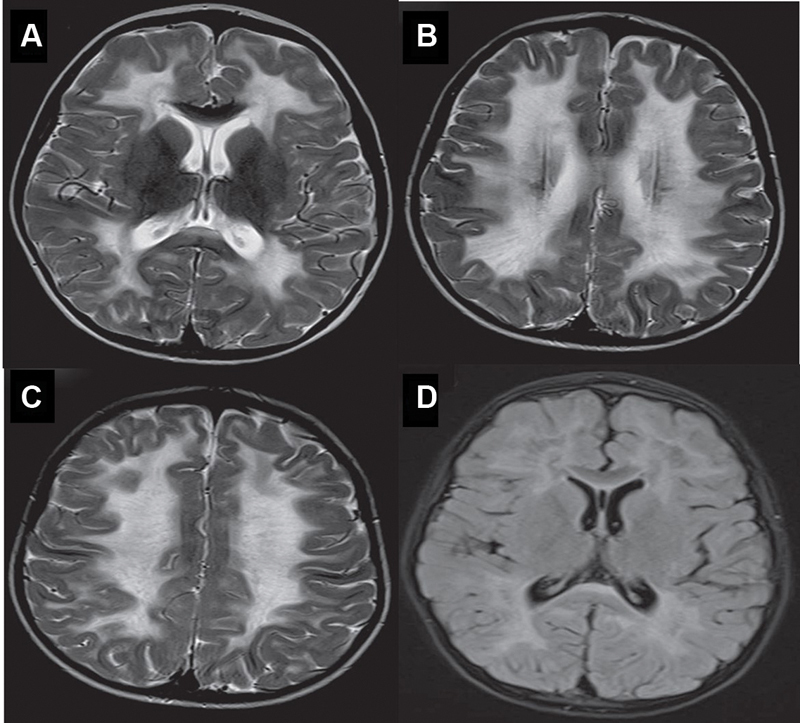
( A–C ) Axial T2WI showing diffuse periventricular and lobar white matter hyperintensity with a somewhat striated appearance and Axial FLAIR image showing partial inversion of the frontal and parietal white matter lesions noted in ( D ).
Table 2. Genetic analysis in five children with childhood ataxia with central nervous system hypomyelination.
| Case number | Gene | Ex | Zygosity | Variant | HGVS annotation | Amino acid position | ACMG | HGMD | CLinVAr | dbSNP | Mutation taster | SIFT | LRT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8 and 18 | EIF2B3 | 11 | HOM | c.1270 T > G/ p.C424G | NM_020365.5:c.1270T > G | p.Cys424Gly | VUS | DC | DC | DE | DE | ||
| 10 | EIF2B3 | 4 | HOM | c.350T > C/ p.V117A | NM_020365.5:c.350T > C | p.Val117Ala | VUS | VUS | rs1423018308 | DC | DE | DE | |
| 11 | EIF2B5 | 4 | HOM | c.584G > A/ p.R195H | NM_003907.3:c.584G > A | p.Arg195His | LP | DC | Pathogenic | rs113994054 | DC | DE | DE |
| 17 | EIF2B3 | 2 | HOM | c.43C > T/ p.R15Y | NM_020365.5:c.43C > T | p.Arg15Tyr | VUS | VUS | rs200409938 | DC | DE | DE |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; DC, disease causing; DE, deleterious; Ex, exon; HGMD, human genetic variation database; HOM, homozygous; LP, likely pathogenic; VUS, variants of uncertain significance.
Discussion
CACH/VWM is an autosomal recessive leukodystrophy characterized by ataxia, spasticity, and variable optic atrophy. The disease typically manifests itself by progressive neurological deterioration caused by certain triggers like infections and trauma in usually a completely healthy child or rarely in a child with mild developmental delay. 1 2 3 4 5 6 9 In our study, CACH cases account for 18/383 (4.7%) of all leukodystrophies. This incidence is comparatively higher to the 2/80 (2%) previously reported in a separate Indian study by Gulati et al. 14
van der knaap et al described this entity based on autopsies performed on nine cases which showed cystic degeneration of white matter. Some reports from 1960s to 1970s might correspond to this disease. 2 3 4 17 18 Table 3 shows comparison between the current and previously reported studies.
Table 3. Showing comparison of our study with previously reported studies in childhood ataxia with central nervous system hypomyelination.
| Parameter | Gungor et al ( n = 11) Turkey |
Turon-Vinas et al ( n = 21) Spain |
Current study ( n = 18) India |
|---|---|---|---|
| Mean age of onset (mo) | NR | NR | 37 |
| Infantile onset | 34 | NR | 33 |
| Early childhood onset | 64 | 86 | 50 |
| Late childhood onset | NR | 14 | 17 |
| Consanguinity | 27 | 10 | 94 |
| Epilepsy | 18 | 42 | 83 |
| Course | |||
| Rapidly progressive | 24 | 36 | 39 |
| waxing and waning | NR | NR | 39 |
| Slowly progressive | 76 | 64 | 22 |
| Optic atrophy | NR | 4 (19) | 9 (50) |
Abbreviation: NR, not reported.
The mean age of presentation in this study was 37 months with the youngest case having presented at 6 months and the latest age of presentation was 13 years. This is comparable to Gungor et al 19 and Turon-Vinas et al 20 as shown in in Table 3 . Early childhood onset form is the most common type of CACH in this study, corroborating reported literature. 19 The variation in magnitude is possibly due to different age cutoffs used in different studies. We have adapted the standard classification proposed by van der Knapp et al in this study. 1
In the congenital and early infantile forms, the encephalopathy is severe, seizures are often a predominant clinical feature, and decline is rapid and followed quickly by death. In the early and late childhood-onset forms, initial motor and intellectual development is normal or mildly delayed, followed by neurological deterioration with a chronic progressive or subacute course. 9 In early and late childhood-onset forms, motor decline predominates with ataxia and spasticity. Cognitive decline is relatively mild. In adult-onset forms, cognitive decline and personality changes predominate and motor decline comes later in the disease course. 9 The observation in this study was similar to previously described literature. However, epilepsy was found to be a predominant feature in both subacute infantile as well as early childhood onset forms in this study and a similar observation was made by Hamilton et al. 21
Consanguinity was observed in 94% in this study compared with 10% by Turon-Vinas et al 20 and 27% by Gungor et al. 19 We observed two pairs of affected siblings in this study and a similar observation was reported by Turon-Vinas et al. 20 It has to be noted that while one pair of siblings had a similar age of onset and disease course, the other pair of siblings had contrasting presentations. One child had onset of symptoms at 9 months with an acutely rapid course and the other child had onset of symptoms at 6 years of age with slowly progressive course which highlights the extremely variable phenotype of this disorder. The phenotype is so variable that there have been mildly symptomatic cases who had headache, dizziness, migraine like symptoms, and incidental neuroimaging revealed features suggestive of vanishing white matter disease confirming the diagnosis. 22 We also observed one child with only headache and mild ataxia as the presenting complaint with neuroimaging clinching the diagnosis. However, the child subsequently developed the other classical clinical features of the disease.
All the cases in this series had motor predominant neuroregression, spasticity, and ataxia which are the cardinal clinical features of this disease which have been described similarly in other studies as well. Epilepsy in our series is higher than ( Table 3 ) that reported by Gungor et al and Turon-Vinas et al. 19 20 The possible reason could be due to the smaller cohort of children in our series who had a chronic slowly progressive course compared with other studies. There is sufficient literature which describes a lower incidence of epilepsy in children with chronic slowly progressive course compared with acutely rapid course. Optic atrophy is variable and manifests rather late in all forms. 9 Similar observation was noted in our series where 55% had optic atrophy, and it was a late manifestation. Some children develop dysmetric tremor or become comatose spontaneously or acutely following mild head trauma or febrile illness. 21 Head circumference is usually normal; however, severe progressive macrocephaly occurring after the age of 2 years has been reported 21 ; microcephaly has also been observed. The peripheral nervous system is usually normal. 9 All our cases had normal development prior to the onset of the disease and normal head circumference. Ovarian failure has been reported in late onset forms of the disease and can manifest as primary/secondary amenorrhea. 21 We have not observed in any of our children in this series. Children and adolescents Ptosis, hemiparesis, dystonia, and chorea are other atypical clinical features reported in literature in CACH. 20 However, they were not observed in this series.
Neurologic deterioration has a chronic progressive or subacute or acutely rapid course. Chronic progressive decline can be exacerbated by rapid deterioration during febrile illnesses or following head trauma or major surgical procedures, or by acute and extreme fright. 9 These exacerbations often produce a waxing and waning course in this disease. The variabilities in clinical courses are shown in in Table 3 . The reason for these differences is that previous studies have incorporated waxing and waning forms caused by acute exacerbations under chronic progressive forms of the disease itself which exaggerates the magnitude of the chronic progressive form of the disease. The other possible reason could be the spectrum of age of onset in the study. This study has a higher proportion of subacute infantile form of the disease which usually have an acutely rapid progressive course. 9
In CACH, brain MRI is characterized by extensive and symmetrical involvement of white matter. There is a homogeneous abnormal signal in the cerebral white matter which is already obvious at the onset of the disease and remains constant all through the course of the disease, though there can be progressively appearing signs of cystic degeneration showing CSF-like signal intensity that causes progressive rarefaction and complete loss of nearly whole cerebral white matter. 9 19 20 Brain stem and cerebellar white matter can also be involved, but cystic degeneration has not been reported. 23 Extensive and symmetrical involvement of white matter involving all lobes with intensity similar to that of CSF on all pulse sequences was observed in all individuals in this series. Brainstem white matter involvement was noted in 78%, and cerebellar white matter involvement was noted in 56% in this series. There is relative preservation of temporal lobes, absence of cysts in cerebellar white matter, and parenchymal contrast enhancement may be noted. 24 Internal capsule, subcortical fibers, and outer parts of the corpus callosum are spared. 24 25 26 However internal capsule involvement was noted in 44% in this series. FLAIR images show markedly hypointense areas in deep white matter regions probably corresponding to cavitation. 26 A band-like pattern may be found in rarified and cystic white matter. 23 Selective involvement in the inner edge of the corpus callosum and symmetrical central tegmental tract hyperintensity may be a clue in the early stages of the disease. 19 26 The tigroid appearance of the white matter can also be seen in CACH apart from other leukodystrophies like metachromatic leukodystrophy. 27 Additionally, variable cerebellar atrophy primarily affecting the vermis may be present and involvement of pontine tegmentum tracts are prominent. 28 Even at the end stage, when almost all white matter becomes cystic, it is noteworthy that cerebral atrophy if present is usually of mild degree only. 20 The neuroimaging findings observed in this series corroborated previously reported studies, except for deep gray matter (lentiform nucleus, caudate nucleus, and thalamus) involvement in 55% and internal capsule involvement in 44% of our cases.
The diagnosis of CACH/VWM can be established in an individual with typical clinical findings, characteristic abnormalities on brain MRI, and identification of biallelic pathogenic variants in one of five genes, which encode the five subunits of the eukaryotic translation initiation factor 2B ( eIF2B ). There are five variants reported in this study all of which are homozygous; three of which are novel and two have been previously reported. Among the total five variants reported in this series, four are in the EIF2B3 gene and one in the EIF2B5 gene. This contrasts with most other studies which report EIF2B5 as the most common gene in their respective case series. We do not know whether this could be attributed to differences in ethnicities due to the small cohort studied which precluded any conclusive findings being drawn. Case 8 and 18 had a homozygous missense variant c.1270T > G; p. Cys424Gly on EIF2B3, (exon-11). This mutant residue is larger in size and less hydrophobic compared with the wild-type residue. This variant was previously reported by Gowda et al. 16 The detected missense variant in case 10, c.350T > C, (p.V117A) on EIF2B3 is a novel variant with multiple lines of computational evidence supporting a deleterious effect on the gene. Case 11 had a novel missense change c.584G > A (p.R195H) in EIF2B5 gene. This change lies in a critical well-established domain (Glyco-tranf-GTA-type) without benign variants in this domain. Case 17 has a homozygous missense variant c.43C > T (p.R15Y) on exon 2 of EIF2B3 gene. This is a novel variant and multiple in silico predictions by PolyPhen-2, SIFT, and LRT.
All surviving children are on physiotherapy and neurorehabilitation in this series. Children with epilepsy are treated with antiepileptic drugs in addition. They have been advised to avoid contact supports with high risk of head injury and avoid emotional, fearful situations, extremes of temperature, and major surgery. We have not attempted immunosuppressive therapy in any of our children as corticosteroids and intravenous gamma globulin are not effective in the treatment of CACH/VWM. 9 However, there is literature supporting corticosteroids usage with inconsistent results in acute situations, including intractable status epilepticus. 9
One of the limitations of our study is that genetic testing was not performed in all cases. Additionally, being a retrospective study, we have not done any functional or disability assessments using standard tools. Despite the limitations, this is the largest series of CACH from the Indian subcontinent which describes the clinical phenotype, neuroimaging and genotype of these children with the identification of three novel mutations. This further enhances our understanding of the disease, particularly in relation to ethnicity.
Conclusion
CACH should be considered in any child with acute onset of regression, or waxing and waning course precipitated by fall or infections with associated MRI findings showing white matter signal changes in all sequences similar to CSF intensity.
Acknowledgments
We thank Dr Sam Balu and Dr Mohan Rao Naveen from Eurofins Clinical Genetics, Bangalore, India for helping to analyze exome sequencing data for our study population.
Funding Statement
Funding None.
Conflict of Interest None declared.
Authors' Contributions
V.K.G. involved in supervision, guidance, and review of the manuscript. V.M.S. and B.N. were dedicated in the management of the child and the preparation of manuscript. M.B. supported in diagnosis and preparation of the manuscript. S.K.S. contributed in the diagnosis and management of the child. N.B. has given valuable inputs in the management of this child.
References
- 1.van der Knaap M S, Barth P G, Gabreëls F JM. A new leukoencephalopathy with vanishing white matter. Neurology. 1997;48(04):845–855. doi: 10.1212/wnl.48.4.845. [DOI] [PubMed] [Google Scholar]
- 2.Schiffmann R, Moller J R, Trapp B D. Childhood ataxia with diffuse central nervous system hypomyelination. Ann Neurol. 1994;35(03):331–340. doi: 10.1002/ana.410350314. [DOI] [PubMed] [Google Scholar]
- 3.Hanefeld F, Holzbach U, Kruse B, Wilichowski E, Christen H J, Frahm J. Diffuse white matter disease in three children: an encephalopathy with unique features on magnetic resonance imaging and proton magnetic resonance spectroscopy. Neuropediatrics. 1993;24(05):244–248. doi: 10.1055/s-2008-1071551. [DOI] [PubMed] [Google Scholar]
- 4.Van der Knaap M S, Kamphorst W, Barth P G, Kraaijeveld C L, Gut E, Valk J. Phenotypic variation in leukoencephalopathy with vanishing white matter. Neurology. 1998;51:540–547. doi: 10.1212/wnl.51.2.540. [DOI] [PubMed] [Google Scholar]
- 5.Vermeulen G, Seidl R, Mercimek-Mahmutoglu S, Rotteveel J J, Scheper G C, van der Knaap M S. Fright is a provoking factor in vanishing white matter disease. Ann Neurol. 2005;57(04):560–563. doi: 10.1002/ana.20418. [DOI] [PubMed] [Google Scholar]
- 6.Kaczorowska M, Kuczynski D, Jurkiewicz E, Scheper G C, van der Knaap M S, Jozwiak S. Acute fright induces onset of symptoms in vanishing white matter disease-case report. Eur J Paediatr Neurol. 2006;10(04):192–193. doi: 10.1016/j.ejpn.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Leegwater P A, Vermeulen G, Könst A A. Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat Genet. 2001;29(04):383–388. doi: 10.1038/ng764. [DOI] [PubMed] [Google Scholar]
- 8.van der Knaap M S, Leegwater P A, Könst A A. Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann Neurol. 2002;51(02):264–270. doi: 10.1002/ana.10112. [DOI] [PubMed] [Google Scholar]
- 9.van der Knaap M S, Fogli A, Boespflug-Tanguy O.2003. Feb 20 [Updated 2019 Apr 4].Seattle, WA: University of Washington; 1993–2020. [Google Scholar]
- 10.Labauge P. Magnetic resonance findings in leucodystrophies and MS. Int MS J. 2009;16(02):47–56. [PubMed] [Google Scholar]
- 11.Serafini G, Pompili M, Innamorati M. White matter hyperintensities and self-reported depression in a sample of patients with chronic headache. J Headache Pain. 2012;13(08):661–667. doi: 10.1007/s10194-012-0493-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiffmann R, van der Knaap M S. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72(08):750–759. doi: 10.1212/01.wnl.0000343049.00540.c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hyun S E, Choi B S, Jang J H, Jeon I, Jang D H, Ryu J S. Correlation between vanishing white matter disease and novel heterozygous EIF2B3 variants using next-generation sequencing: a case report. Ann Rehabil Med. 2019;43(02):234–238. doi: 10.5535/arm.2019.43.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gulati S, Jain P, Chakrabarty B, Kumar A, Gupta N, Kabra M. The spectrum of leukodystrophies in children: experience at a tertiary care centre from North India. Ann Indian Acad Neurol. 2016;19(03):332–338. doi: 10.4103/0972-2327.179975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravishankar S, Sinha S, Taly A B, Panicker J. Vanishing white matter disease: Phenotypic, MR imaging and H spectroscopic observation. Ann Indian Acad Neurol. 2006;9:172–174. [Google Scholar]
- 16.Gowda V K, Srinivasan V M, Bhat M, Benakappa A. Case of childhood ataxia with central nervous system hypomyelination with a novel mutation in EIF2B3 gene. J Pediatr Neurosci. 2017;12(02):196–198. doi: 10.4103/jpn.JPN_183_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee H N, Koh S H, Lee K Y, Ki C S, Lee Y J. Late-onset vanishing white matter disease with compound heterozygous EIF2B5 gene mutations. Eur J Neurol. 2009;16(03):e42–e43. doi: 10.1111/j.1468-1331.2008.02395.x. [DOI] [PubMed] [Google Scholar]
- 18.Maletkovic J, Schiffmann R, Gorospe J R. Genetic and clinical heterogeneity in eIF2B-related disorder. J Child Neurol. 2008;23(02):205–215. doi: 10.1177/0883073807308705. [DOI] [PubMed] [Google Scholar]
- 19.Güngör G, Güngör O, Çakmaklı S. Vanishing white matter disease with different faces. Childs Nerv Syst. 2020;36(02):353–361. doi: 10.1007/s00381-019-04334-6. [DOI] [PubMed] [Google Scholar]
- 20.Turón-Viñas E, Pineda M, Cusí V. Vanishing white matter disease in a spanish population. J Cent Nerv Syst Dis. 2014;6:59–68. doi: 10.4137/JCNSD.S13540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamilton E MC, van der Lei H DW, Vermeulen G. natural history of vanishing white matter. Ann Neurol. 2018;84(02):274–288. doi: 10.1002/ana.25287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fontenelle L M, Scheper G C, Brandão L, van der Knaap M S.Atypical presentation of vanishing white matter disease Arq Neuropsiquiatr 200866(3A)549–551. [DOI] [PubMed] [Google Scholar]
- 23.Barros S R, Parreira S CR, Miranda A FB, Pereira A MB, Campos N MP. New insights in vanishing white matter disease: isolated bilateral optic neuropathy in adult onset disease. J Neuroophthalmol. 2018;38(01):42–46. doi: 10.1097/WNO.0000000000000565. [DOI] [PubMed] [Google Scholar]
- 24.van der Knaap M S, Pronk J C, Scheper G C. Vanishing white matter disease. Lancet Neurol. 2006;5(05):413–423. doi: 10.1016/S1474-4422(06)70440-9. [DOI] [PubMed] [Google Scholar]
- 25.Zhou L, Zhang H, Chen N. Similarities and differences between infantile and early childhood onset vanishing white matter disease. J Neurol. 2018;265(06):1410–1418. doi: 10.1007/s00415-018-8851-6. [DOI] [PubMed] [Google Scholar]
- 26.van der Lei H D, Steenweg M E, Barkhof F. Characteristics of early MRI in children and adolescents with vanishing white matter. Neuropediatrics. 2012;43(01):22–26. doi: 10.1055/s-0032-1307456. [DOI] [PubMed] [Google Scholar]
- 27.Singh R R, Livingston J, Lim M, Berry I R, Siddiqui A. An unusual neuroimaging finding and response to immunotherapy in a child with genetically confirmed vanishing white matter disease. Eur J Paediatr Neurol. 2017;21(02):410–413. doi: 10.1016/j.ejpn.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 28.Wilson C J, Pronk J C, Van der Knaap M S.Vanishing white matter disease in a child presenting with ataxia J Paediatr Child Health 200541(1-2):65–67. [DOI] [PubMed] [Google Scholar]