

Abstract
Oxidative stress is a state of imbalance between oxidation and antioxidation. Excessive ROS levels are an important factor in tumor development. Damage stimulation and excessive activation of oncogenes cause elevated ROS production in cancer, accompanied by an increase in the antioxidant capacity to retain redox homeostasis in tumor cells at an increased level. Although moderate concentrations of ROS produced in cancer cells contribute to maintaining cell survival and cancer progression, massive ROS accumulation can exert toxicity, leading to cancer cell death. RNA modification is a posttranscriptional control mechanism that regulates gene expression and RNA metabolism, and m6A RNA methylation is the most common type of RNA modification in eukaryotes. m6A modifications can modulate cellular ROS levels through different mechanisms. It is worth noting that ROS signaling also plays a regulatory role in m6A modifications. In this review, we concluded the effects of m6A modification and oxidative stress on tumor biological functions. In particular, we discuss the interplay between oxidative stress and m6A modifications.
1. Introduction
Reactive oxygen species (ROS) are byproducts of the respiratory chain, which act as important signal transduction molecules in cells [1]. The production of ROS is regulated by a variety of intracellular and extracellular stimuli. These oxygen-based molecules contain unpaired electrons, and their instability can lead to the irreversible inactivation of intracellular targets such as proteins, nucleic acids, and lipids [2]. Under increased ROS production, cells protect themselves from ROS damage by producing enzymatic antioxidants (e.g., superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPX)) and nonenzymatic antioxidants (e.g., glutathione and thioredoxin) [3]. An imbalance in the relative abundance of ROS and antioxidants can lead to profound pathophysiological consequences. Oxidative stress is defined as a relative excess of ROS, which is closely associated with aging-related diseases, such as neurodegenerative disorders [4], cardiovascular diseases [5, 6], and normal senescence [7, 8], and with many other diseases, including cancer. Many oncogenes can affect ROS production in a direct or indirect manner; thus, cancer cells usually show elevated levels of ROS. To adapt to the relatively high levels of ROS and maintain survival and proliferative activity, the antioxidation capability of cancer cells is increased to neutralize the cytotoxicity caused by excessive ROS [9]. Significantly, oxidative stress impacts tumor development in a concentration-dependent manner. Elevated ROS levels generally participate in promoting cancer, but excessive ROS levels generate toxicity to cancer cells [10].
N6-methyladenosine (m6A) is the most common type of internal RNA modification in eukaryotes [11]. With the development of high-throughput sequencing, it has been found that m6A modifications are mainly enriched in 5′-untranslated regions (5′-UTRs), stop codons, and 3′-untranslated regions (3′-UTRs) and are located in specific RRACH (R=G/A, A=m6A, H=U/A/C) motifs in RNA [12, 13]. The location and distribution of m6A modifications at the transcriptome level are gradually being revealed. m6A marks are widely distributed in 6990 mRNAs and 250 noncoding RNAs that regulate maturation, transcription, translation, and metabolism and are involved in the modulation of various pathological and physiological activities. Changes in m6A levels have profound effects on numerous cellular processes, including autophagy [14], the DNA damage response [15], oxidative stress [16], and tumorigenesis [17]. m6A modifications and oxidative stress play complex and contradictory roles in tumorigenesis and development. Surprisingly, m6A modifications show widespread, close interrelationships with oxidative stress. m6A modifications affect oxidative stress-related genes' expression which have different effects on oxidative stress, thus affecting the generation and development of cancer [18–20]. On the other side, the expression and activity of m6A enzymes and m6A levels can be dynamically regulated by ROS [21, 22]. A systematic and in-depth understanding of m6A and oxidative stress in tumor formation and progression is thus of great significance for the diagnosis and treatment of cancer. Therefore, we describe the way in which oxidative stress and m6A modification influence biological functions in cancer and discuss the cross-talk between them in this review.
2. N6-Methyladenosine in Cancer
2.1. Writers
m6A regulatory factors can be classified into three categories: “writers”, “erasers” and “readers”. “Writers” are a set of proteins that participate in the formation of the m6A methyltransferase complex (MTC) and catalyze m6A modification by using S-adenosylmethionine as a methyl donor. The complex consists of methyltransferase-like 3 (METTL3), METTL14 and their cofactors WT1-associated protein (WTAP), Vir-like m6A methyltransferase associated (VIRMA), zinc finger CCCH-type containing 13 (ZC3H13), and RNA-binding motif protein 15/15B (RBM15/15B) [23–27]. METTL3 plays a major catalytic role in the MCT. Knockdown of METTL3 leads to almost complete loss of m6A modification activity [28]. METTL14 interacts with METTL14 to form a METTL3-METTL14 heterodimer through an extensive hydrogen-bonding network. It acts as an RNA-binding platform in m6A methylation and enhances the catalytic activity of METTL3 through allosteric activation. However, METTL14 itself cannot directly promote methyl transfer [29]. WTAP interacts with the METTL3-METTL14 heterodimer to stabilize MTC and promote the localization of the core complex to nuclear speckles. Knockdown targeting WTAP caused a significant reduction in the RNA-binding capability of METTL3, suggesting that WTAP plays a critical role in RNA modification by regulating the recruitment of MTC to RNA targets [25]. Other evidence has shown that RBM15/15B[27], VIRMA [30], and ZC3H13[31] may be components of the MTC and function as regulators to bind and recruit the complex to affect the stability and location of the MCT and thereby regulate the methylation modification process. METTL16 is a newly discovered m6A methyltransferase that can catalyze the m6A modification of U6 spliceosomal small nuclear RNA (snRNA) and participate in cotranscriptional and posttranscriptional splicing [32].
2.2. Erasers
The reversibility of the m6A modification relies on demethylases, also known as “erasers”. Fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5) are independent m6A demethylases that perform demethylation functions and require the involvement of the cofactor Fe(2+) and α-ketoglutarate (α-KG) or 2-oxoglutarate (2-OG). FTO partially colocalizes with nuclear speckles. The distribution of FTO in cells determines its effect on different RNA substrates [33]. FTO knockdown can increase the amount of m6A in mRNA, while FTO overexpression results in a decrease in m6A modifications [34]. ALKBH5 is another RNA demethylase that is expressed in most tissues, is mainly located in the nucleus, and can remove m6A residues from mRNA in vitro and in vivo. The deletion of the ALKBH5 gene leads to a remarkable reduction in mRNA levels in the cytoplasm, suggesting that its demethylation activity notably affects mRNA export as well as RNA metabolism [35].
2.3. Readers
“Readers” are a set of m6A-binding proteins that specifically recognize and mediate the biological functions of m6A modified RNA. Proteins with conserved m6A-binding domains, including YTHDC1-2 and YTHDF1-3, are the main m6A readers that belong to the YT521-B homology (YTH) domain family. YTHDC1 is the core member of the YTH domain family and can selectively recruit and modulate pre-mRNA splicing factors and regulate RNA alternative splicing after recognizing m6A in the nucleus [36]. YTHDC2 is an m6A-binding protein that contributes to enhancing the translation efficiency of target RNAs and simultaneously reduces their abundance [37]. YTHDF1 interacts with eukaryotic initiation factors (eIFs) and ribosomes to accelerate the translation of m6A-modified mRNAs [38]. YTHDF2 accelerates the degradation of m6A-modified transcripts by selectively binding with them and directing the complex to cellular RNA decay sites [39]. YTHDF3 not only works in conjunction with YTHDF1 to expedite mRNA translation but also participants in the process of YTHDF2-mediated mRNA decay [40, 41].
Heterogeneous nuclear ribonucleoproteins (HNRNPs), eIFs, and other special proteins also act as m6A readers. The HNRNP family is a series of nuclear RNA-binding proteins (including HNRNPC, HNRNPG, and HNRNPA2B1) that influence mRNA precursor processing [42]. In addition to participating in the maturation of mRNA, HNRNPA2B1 and METTL3 have also been proven to affect the beginning of microRNAs (miRNAs) biogenesis, primary miRNA processing, and alternative splicing, in the early stage of microRNA (miRNA) biogenesis by interacting with the microprocessors complex DiGeorge syndrome critical region 8 (DGCR8, [42–44]. eIF3 initiates translation in a cap-independent manner by directly binding to the m6A site of the mRNA 5′-UTR and recruiting the 43S complex [45]. Studies have revealed that insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) recognize the consensus m6A site GG (m6A) C and target mRNA transcripts to maintain their stability and thus increases the levels of its stored target mRNAs [46]. Furthermore, a novel m6A reader identified in recent research, proline-rich coiled-coil 2 A (Prrc2a), binds to a GG (m6A) CU motif of the target coding sequence and stabilizes its target mRNA in an m6A-dependent manner [47]. The FMRP Translational Regulator 1 (FMR1) and Leucine-Rich Pentatricopeptide Repeat-Containing (LRPPRC) proteins can also read m6A modifications and affect RNA behavior [48, 49]. Recent studies have reported that FMR1 directly interacts with YTHDF1 to inhibit the translation of target transcripts [50].
2.4. Role of m6A in Cancer
The dynamic reversibility of m6A affects gene expression and numerous cellular processes. A recent wave of studies has shown that m6A modification regulates RNA maturation, transcription, translation, and metabolism, which are involved in the modulation of various physiological activities. Therefore, imbalances in m6A levels lead to a variety of diseases, especially cancers. The abnormal modification and expression of m6A regulatory proteins can be detected in multiple tumor types and modulates the expression of tumor-related genes [51]. Changes in m6A levels may profoundly affect the processes of tumor growth, progression, and metastasis, including proliferation signaling [51], angiogenesis [52, 53], cell development and differentiation [54], cellular metabolic reprogramming [52, 55, 56], immune responses and evasion [57, 58], and inflammation [51, 59]. Due to the extensive and complex functions of m6A methylation, it plays dual roles in cancer: a high m6A level may lead to oncogenesis, but the deletion of m6A methylation modifications may lead to the progression of other tumors (Table 1).
Table 1.
Roles of m6A in cancer.
| m6A enzyme | Cancer type | Role | Change in target RNA | Function of m6A enzyme | References |
|---|---|---|---|---|---|
| METTL3 | Acute myeloid leukemia | Oncogene | MYC↑, BCL2↑, PTEN↑ | Inhibiting cell differentiation and apoptosis, increasing cell growth | [244] |
| Breast cancer | Oncogene | HBXIP↑ | Promoting cell proliferation, inhibiting apoptosis | [246, 277] | |
| Bladder cancer | Oncogene | miR-221/222↑, PTEN↓ | Promoting cell growth | [278] | |
| Oncogene | SETD7↓, KLF4↓ | Promoting cell proliferation and metastasis | [279] | ||
| Colorectal cancer | Oncogene | SOX2↓ | Promoting tumorigenesis and cell metastasis | [280] | |
| Tumor suppressor | p-p38↓, p-ERK↓ | Inhibiting cell proliferation, migration, and invasion | [281] | ||
| Glioblastoma | Oncogene | SRSFs↑ | Promoting tumor growth and progression | [282] | |
| Oncogene | ADAR1↑ | Promoting cancer progression | [283] | ||
| Lung cancer | Oncogene | JUNB↑ | Increasing TGF-β-induced EMT | [284] | |
| Oncogene | BRD4↑ | Promoting oncogenic transformation and tumor growth | [285] | ||
| Liver cancer | Oncogene | SOCS2↓ | Promoting cell proliferation, migration, and colony formation | [286] | |
| Oncogene | RDM1↓ | Increasing cell proliferation | [287] | ||
| METTL3, METTL14 | Endometrial cancer | Tumor suppressor | PHLPP2↑, mTORC2↓ | Inhibiting cell proliferation | [288] |
| Glioblastoma | Tumor suppressor | ADAM19↑, EPHA3↑, KLF4↑, CDKN2A↓, BRCA2↓, TP53I11↓ | Suppressing tumor genesis, growth, and self-renewal | [289] | |
| METTL14 | Acute myeloid leukemia | Oncogene | MYC↑, MYB↑ | Inhibiting cell differentiation, increasing cell proliferation | [290] |
| Breast cancer | Oncogene | DROSHA↑ | Enhancing breast cancer stem-like cell stemness maintenance | [291] | |
| Colorectal cancer | Tumor suppressor | SOX4↓ | Inhibiting EMT | [292] | |
| Tumor suppressor | lncRNA XIST↓ | Suppressing proliferation and metastasis | [293] | ||
| Pancreatic cancer | Oncogene | PERP↓ | Promoting tumor growth and metastasis | [294] | |
| WTAP | Liver cancer | Oncogene | ETS1↓ | Promoting cell proliferation and tumor growth | [295] |
| FTO | Acute myeloid leukemia | Oncogene | MYC↑, CEBPA↑ | Promoting cell proliferation and viability, inhibiting cell-cycle arrest and apoptosis | [296] |
| Oncogene | ASB2↓, RARA↓ | Enhancing cell transformation and leukemogenesis, inhibiting cell differentiation | [297] | ||
| Liver cancer | Oncogene | ALDOA↑ | Promoting cell growth under hypoxia | [298] | |
| Ovarian cancer | Tumor suppressor | PDE1C↓, PDE4B↓ | Inhibiting the tumor self-renewal, suppressing tumorigenesis | [299] | |
| ALKBH5 | Acute myeloid leukemia | Oncogene | TACC3↑ | Promoting tumor development and self-renewal | [300] |
| Lung cancer | Tumor suppressor | YAP↓ | Inhibiting tumor growth and metastasis | [301] | |
| Glioblastoma | Oncogene | FOXM1↑ | Promoting cell proliferation | [51] | |
| Pancreatic cancer | Tumor suppressor | PER1↑ | Reducing cell proliferation, migration, and invasion, suppressing tumor growth | [302] | |
| YTHDF1 | Lung cancer | Oncogene | CDK2↑, CDK4↑, cyclind1↑, Keap1↑ | Promoting cell proliferation and xenograft tumor formation | [215] |
| Liver cancer | Oncogene | EGFR↑ | Promoting cell viability and metastasis | [303] | |
| Ovarian cancer | Oncogene | EIF3C↑ | Facilitating tumor genesis and metastasis | [304] | |
| YTHDF2 | Glioblastoma | Oncogene | MYC↑, VEGFA↑ | Maintaining glioblastoma stem cell stemness | [241] |
| Liver cancer | Tumor suppressor | IL11↓, SERPINE2↓ | Reducing tumor inflammation and causing vascular abnormalities | [59] | |
| Prostate cancer | Oncogene | LHPP↓, NKX3-1↓ | Inducing tumor proliferation and migration | [305] | |
| YTHDF3 | Breast cancer | Oncogene | ST6GALNAC5↑, GJA1↑, EGFR↑ | Controlling the interaction of cancer and brain microenvironment, inducing brain metastasis | [306] |
| Colorectal cancer | Oncogene | LncRNA GAS5↓ | Promoting cancer progression | [307] | |
| IGF2BP1 | Colorectal cancer | Oncogene | c-Myc↑ | Promoting tumorigenesis | [308] |
| Ovarian, liver, and lung cancer | Oncogene | SRF↑, PDLIM7↑, FOXK1↑ | Promote cell growth and invasion | [309] | |
| IGF2BPs | Cervical and liver cancer | Oncogene | MYC↑ | Promoting tumorigenesis | [310] |
3. Overview of Oxidative Stress in Cancer
ROS are chemical species that form highly active radicals on the unpaired electrons of oxygen. They are generally considered to include reactive oxygen compounds such as superoxide (O2•−) and hydroxyl (HO•) free radicals and nonradical molecules such as hydrogen peroxide (H2O2), singlet oxygen (1O2), and ozone (O3) [60]. The most important source of ROS is the mitochondria. Approximately 2% of oxygen can receive single or double electrons from the middle portion of the electron transport chain (ETC) and be partially reduced to O2•−/H2O2 [61, 62]. NADPH oxidases (NOXs) are other sources of intracellular oxidants that are located on the cell membrane, nuclear membrane, or endoplasmic reticulum membrane. NOXs can transfer electrons from reduced NADPH to catalytic superoxide and other downstream ROS [63, 64]. In addition to NOXs and the ETC, ROS are produced by other enzymes in organelles such as the endoplasmic reticulum (ER) and peroxisomes, including xanthine oxidase (XO), endothelial nitric oxide synthase (eNOS), lipoxygenase (LOX), cyclooxygenase (COX), and cytochrome P450 reductase (POR) [65–67].
ROS are gradually coming to be considered important signal transduction molecules or regulators in biological systems, rather than only byproducts of metabolism [1]. Different ROS levels have different biological effects. Under physiological conditions, small amounts of ROS are produced in cells, which involve in cell proliferation and differentiation by activating stress-responsive survival signals, and their toxicity is easily offset by the antioxidant defense system. However, excessive ROS levels can exert toxicity, leading to cell dysfunction and even death. Consequently, the maintenance of normal physiological function depends on the balance between oxidants and antioxidants. The three-layer antioxidant defense system maintains the redox homeostasis of cells. Uric acid, glutathione (GSH), and vitamins C and E belong to small-molecule antioxidants which can scavenge ROS directly. The antioxidant enzymes that play roles in intermediate defense include SOD [68], CAT [69], GPX [70], thioredoxin (Trx) [71], and peroxiredoxin (Prx) [72], which catalyze the transformation of ROS into less cytotoxic products. Enzymes that repair or remove damaged biomolecules, such as 8-oxoguanine glycosylase (OGG1), apurinic/apyrimidinic endonuclease (APE1), and DNA polymerase, have generally been regarded as the last line of defense in the repair of oxidative damage [73–75].
The production of ROS in malignant tumors increases and contributes to maintaining the cancer phenotype. Similar to normal cells, the intracellular sources of ROS produced by cancer cells contain mitochondrial ETC, NOX, ER, and LOX [10]. Damage stimulation and the excessive activation of oncogenes lead to elevated ROS production in cancer. Abnormal activation of Kirsten rat sarcoma viral oncogene homolog (KRAS) and the amplification of MYC protooncogene (MYC) in cancer enhance the catabolism of glutamine as the carbon source of the tricarboxylic acid cycle, increasing the ROS generation by ETC [76, 77]; mutant KRAS can also increasing mitochondrial ROS via decreasing the stabilization of electron transport and leading to the leakage of electrons [78]; B-cell lymphoma 2 (Bcl-2) is overexpressed in a variety of tumors which can affect the activity of ETC by interacting with cytochrome C oxidase [79]. Oncogenes also mediate ROS production by regulating NOXs expression. Signal transducer and activator of transcription 3 (STAT3) increases NOX4 expression [80]; mutant KRAS activates NOX1 and downregulates antioxidant enzymes, including SOD2, catalase, and Prxs [81, 82]; and Ras-related C3 botulinum toxin subunit 1 (Rac1) can stimulate the production of mitochondrial superoxide and participate in the assembly and activation of NOX1 and NOX3 [83–86]. Tumor suppressor p53 and forkhead box O (FOXO) family transcription factors can prevent oxidative stress by inducing the expression of antioxidant genes. The inactivation of these transcription factors in tumors may increase ROS production [9, 87]. In addition, transforming growth factor β (TGF-β) stimulates fibroblasts to induce NOX4 upregulation and elevate ROS production during tumor-related matrix remodeling [88].
One of the most important reasons for the enhanced antioxidant capacity in cancer is the regulation of the redox homeostasis of cancer cells by nuclear factor erythroid-2-related factor 2 (Nrf2) [89]. In the process of cancer development, Nrf2 expression can be increased by activating oncogenes (such as KRAS) or environmental signals (such as hypoxia) [90, 91], the loss of the negative regulator Kelch-like ECH-associated protein 1 (Keap1) directly activates Nrf2 [89], and elevated levels of ROS prevent the proteasome-mediated degradation of Nrf2. Nrf2 is a transcription factor whose increased expression and activity initiate the transcription of various antioxidant genes [92]. In addition, NADPH metabolism enzymes and NAD (P)H: quinone oxidoreductase 1 (NQO1), which inhibits the formation of free radicals, are regulated by Nrf2 [93, 94]. Therefore, Nrf2 is regarded as a stress reliever that maintains a high but balanced redox state in tumors and supports cancer cell survival. Another protein that restricts ROS is TP53-induced glycolysis regulatory phosphatase (TIGAR), a protein with bisphosphatase activity that is involved in activating the oxidized pentose phosphate pathway (PPP), thereby increasing the production of NADPH for antioxidant defense [95, 96].
4. Effect of Oxidative Stress on Cancer
Previous studies have evaluated the levels and production of ROS in cancer cells under different conditions, clarifying the relationship between ROS and tumor growth [97]. Excessive ROS play an antitumor role by inducing DNA damage, cell death, and aging, whereas elevated levels of oxidants and antioxidants support the proliferation and survival of cancer cells (Figure 1) [98, 99]. In different types of tumors, oxidative stress mediates anticarcinogenic effects or cancer-promoting effects through different mechanisms (Table 2).
Figure 1.
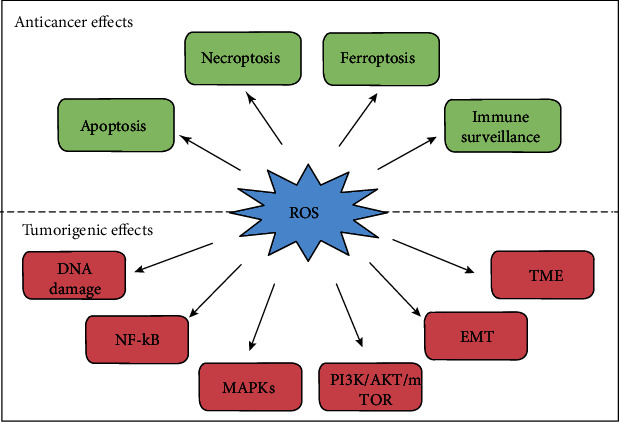
The dual effects of oxidative stress in cancer progression. ROS play an anticancer role by promoting apoptosis, necrosis, and ferroptosis of cancer cells and enhancing the immune surveillance ability of immune cells. Conversely, ROS promoting cancer progression by inducing DNA damage and genomic alterations, activating cell proliferation-related pathway (NF-B, MAPKs, and PI3K/AKT/mTOR), accelerating EMT, and altering the tumor microenvironment for cancer invasion and metastasis.
Table 2.
Effects of oxidative stress in cancers.
| Cancer type | Gene involved | Function of ROS | Description | References |
|---|---|---|---|---|
| Acute myeloid leukemia | TIGAR | Tumor suppressor | Knockdown of TIGAR promoting ROS-mediated apoptosis and antiproliferation | [311] |
| NOX | Tumor promoter | Activation of NOX increases extracellular ROS level promoting the proliferation of acute myeloid leukemia blasts | [312] | |
| Breast cancer | p53 | Tumor suppressor | p53 activation induced by ROS can promote necrosis and apoptosis of cancer cells | [313] |
| SOD2 | Tumor promoter | Upregulation of SOD2 induces elevated ROS to sustain AMPK-activated signal to promote aerobic glycolysis and malignant transformation | [314] | |
| ZEB1; GPX4 | Tumor promoter | ZEB1 inhibits transcription of GPX4, increases ROS accumulation and EMT, which promote breast cancer progression | [315] | |
| Colorectal cancer | HIF-2α | Tumor suppressor | Activated HIF-2α induces ROS production by an iron-dependent pathway which led colorectal cancer cell death | [316] |
| ANGPTL4; NOX4 | Tumor promoter | ANGPTL4/NOX4 axis maintains the metastatic ability of colorectal cancer cells via increasing ROS, MMP1, and MMP9 levels | [317] | |
| Glioblastoma | PRDX3 | Tumor suppressor | Prohibitin maintains the stability of PRDX3 to reduce the production of ROS, maintain glioblastoma stemness and promote the resistance of gliomas stem-like cells to radiotherapy | [318] |
| TRAP1; SIRT3 | Tumor suppressor | TRAP1 cooperate with SIRT3 to reduce ROS production and promotes stress adaptation of glioblastoma cancer stem cells | [319] | |
| Gastric cancer | NNT | Tumor suppressor | NNT deficiency can significantly reduce NADPH and significantly induce ROS production and apoptosis under stress. | [320] |
| GRIM-19; Nrf2 | Tumor promoter | GRIM-19 deficiency accelerates gastric cancer metastasis via abnormal oxidative stress and ROS-driven Nrf2 activation | [321] | |
| Hepatocellular carcinoma | UBQLN1; PGC1β | Tumor suppressor | Elevated expression of UBQLN1 induces PGC1β degradation to promote sorafenib resistance of hepatocellular carcinoma cells by reducing mitochondrial ROS production | [322] |
| PKCλ/ι; Nrf2 | Tumor promoter | Loss of PKCλ/ι induces ROS generation promoting hepatocellular carcinoma in a Nrf2-dependent manner | [323] | |
| Lung cancer | IL-15; mTOR | Tumor promoter | NK cells activate thioredoxin system through IL-15/mTOR axis to adapt to high ROS level in tumor microenvironment | [324] |
| AK4; HIF-1α | Tumor promoter | Upregulation of AK4 enhances expression of HIF-1α through increasing ROS production, and then EMT was induced in hypoxia condition | [325] | |
| AIM2; MAPK/ERK; MFN2 | Tumor promoter | Knockdown of AIM2 upregulates MFN2 and enhances the mitochondrial fusion, resulting in the reduction of mitochondrial ROS production, which in turn induces the inactivation of the MAPK/ERK pathway and hinders the progress of non-small cell lung cancer | [326] | |
| Nestin; Keap1; Nrf2; | Tumor suppressor | Nestin competed with Nrf2 for binding to Keap1, leading to Nrf2 escape and downstream antioxidant gene expression, which promotes the resistance of NSCLC to oxidative stress | [327] | |
| Melanoma | ANGPT2 | Tumor suppressor | Silence of Angpt2 expression significantly increases the level of intracellular ROS and activation of downstream MAPK pathway, thus resulting in the metastatic colonization of melanoma | [328] |
| Akt | Tumor promoter | Akt overexpression can induce the expression of NOX4, increase the level of ROS, increase the expression of VEGF, increase angiogenesis, and promote the aerobic glycolysis of melanoma cells | [329] | |
| Ovarian cancer | RAD51 | Tumor promoter | Loss of RAD51 accelerates mitochondrial ROS accumulation and DNA damage which can be weakened by treatment of antioxidant N-acetylcysteine | [330] |
| Pancreas cancer | UCP2; Akt; mTOR | Tumor suppressor | Inhibition of UCP2 plays an anticarcinogenic role in pancreatic adenocarcinoma cells via activating ROS/Akt/mTOR axis | [331] |
| Renal cell carcinoma | TAZ; EMP1; NOX4; | Tumor suppressor | Nuclear translocation of TAZ upregulates EMP1 expression, thereby increasing the mRNA level of NOX4 and inducing ferroptosis of renal cell carcinoma cells via elevated lipid ROS | [332] |
4.1. Tumorigenic Effect of Oxidative Stress
As we mentioned earlier, more elevated ROS levels showed in cancer cells than normal cells, and the antioxidant capacity is enhanced to counteract the toxicity of excessive ROS. An abnormal redox balance may play a carcinogenic role through different mechanisms.
4.1.1. Oxidative Stress Induces DNA Damage and Genomic Alterations
Carcinogenic stimulation, increased metabolic activity, and mitochondrial dysfunction lead to increased ROS levels in cancer cells [100]. As early as the 1990s, it was recognized that ROS-mediated DNA damage can induce gene mutation [101]. Excessive ROS can attack various components of DNA, resulting in DNA intrastrand adducts, oxidized bases, strand breaks, and DNA-protein crosslinks [102, 103]. Notably, mitochondrial DNA is more susceptible to damage than nuclear DNA because it is located closer to the site where ROS are produced. ROS-mediated mitochondrial DNA damage may lead to respiratory chain dysfunction, further amplifying oxidative stress and thereby destroying genome functions, inducing genome instability, and increasing the risk of mutations [100]. 8-Oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG) is an oxidative adduct produced by ROS-related DNA damage and is a common mutagenic structure in DNA that can be repaired mainly through OGG1-mediated base excision [104]. ROS affect DNA repair by inhibiting OGG1 activity [105]. In summary, ROS can alter the balance of DNA damage repair functions in cancer cells, causing ROS-mediated DNA damage to far exceed the repair ability, which leads to the accumulation of multiple mutations and, ultimately, carcinogenic mutations and tumorigenic transformation.
ROS can be involved in tumorigenesis by regulating oncogenes, tumor suppressor genes, and DNA repair genes [104, 106]. Elevated ROS promote the occurrence of cancer by inducing oxidation and base pair substitution mutations of these tumor-related genes [107]. The members of the rat sarcoma (RAS) viral oncogene family, which include HRAS, KRAS, and NRAS [108], are the most commonly mutated oncogenes in human cancers [109]. Activated KRAS has been proven to upregulate NOX and increase superoxide production and consequent malignant transformation [110, 111]. In addition, mitochondrial ROS are essential for KRASG12D-induced tumorigenesis [76]. Increased ROS conversely promote the expression of KRAS and Nrf2 under oxidative stress, and the ectopic expression of KRASG12D or KRASG12V stimulates Nrf2[112], supporting KRASG12D-driven tumor development [90]. The tumor suppressor gene tumor protein p53 (TP53) inhibits tumorigenesis by inducing cell growth arrest or apoptosis. ROS can induce G-to-T mutations in TP53 [113]. p53 protects the genome from ROS oxidation by enhancing DNA repair and upregulating the expression of antioxidant genes. Therefore, p53 loss-of-function mutations lead to further increases in intracellular ROS levels, cause excessive DNA oxidation, increase mutations and karyotype instability, and promote tumor development [114].
4.1.2. Oxidative Stress Promotes Tumor Cell Proliferation
Many classical pathways involved in the ROS-mediated proliferation of cancer cells. Nuclear factor-κB (NF-κB) (nuclear factor of kappa light polypeptide gene enhancer in B cells) plays a key role in multiple cellular processes including immune and inflammatory responses and cell proliferation and differentiation [115, 116]. The typical NF-κB pathway can be activated in response of oxidative stress [115]. ROS induce the phosphorylation of IκB kinase α (IκKα), which results in the ubiquitination and degradation of NF-κB inhibitor α (IκBα), thereby promoting translocation of NF-κB heterodimers to the nucleus and thus increasing the transcription of downstream target genes of the NF-κB pathway [117]. The abnormal activation of NF-κB promotes the growth, proliferation, and angiogenesis of a variety of cancers by upregulating the expression of antiapoptotic genes, cyclins, protooncogenes, matrix metalloproteinases (MMPs), and cell adhesion genes [118]. NF-κB is also conducive to conversion from oxidative phosphorylation to glycolysis in cancer cells [119] and promotes the survival and proliferation of cancer cells by regulating cellular components in the tumor microenvironment [120, 121]. In hepatocellular carcinoma cells, treatment with H2O2 and N-acetylcysteine (NAC, a kind of ROS scavenger) was shown to alter intracellular ROS levels, and the results showed that the activity of NF-κB increased after exposure to H2O2, while the opposite result was obtained after NAC treatment [122]. In internal stem cells (ISCs), Rac1-driven ROS and NF-κB signaling mediate progenitor cell proliferation and transformation [123].
ROS participate in PI3K/AKT/mTOR and MAPK/ERK signal-mediated activation of growth factors. Phosphatase and tensin homolog (PTEN), a lipid phosphatase that is sensitive to redox reactions, is one of the most frequently deleted and mutated antioncogenes in human cancers. PTEN can coordinate cell proliferation, growth, and survival by negatively regulating the PI3K/AKT/mTOR signaling pathway [124, 125]. H2O2 treatment results in the time- and concentration-dependent inactivation of purified PTEN in vitro [126]. Other studies have shown that H2O2 can oxidize cysteine residues of PTEN, resulting in its temporary inactivation, and can induce the activation of downstream protein kinase B (AKT) [127, 128]. Similarly, H2O2 catalyzes the reversible oxidation of protein tyrosine phosphatase 1B (PTP1B) [129]. Like PTEN, PTP1B is a negative regulator of phosphoinositide 3-kinase (PI3K) and AKT. ROS-mediated oxidative inactivation of PTEN and/or PTP1B can induce the overactivation of the PI3K/AKT/mTOR pathway, which is characteristic of malignant tumors [130, 131]. Endogenous antioxidants (e.g., Trxs and Prxs) regulate the intracellular redox state. An increase in Trx1 levels can cause Trx1 to bind to PTEN in a redox-dependent manner and inhibit its lipid phosphatase activity, leading to an increase in AKT activation in cells and thus promoting tumorigenesis [132]. However, PTEN can be reactive by thioredoxin-interacting protein- (TXNIP-) mediated pathways. [133]. Prxs regulate intracellular H2O2 levels by catalyzing hydrogen peroxide reduction [134]. Prx1 interacts with PTEN to protect and promote the antitumor function of PTEN under mild oxidative stress. However, Prx1 separates from PTEN and irreversibly loses its peroxidase activity under high concentrations of H2O2 (500 μM)[135].
MAPKs regulate proliferation, differentiation and apoptosis, and other cellular activities related to tumors. MAPKs mainly include four subgroups: c-Jun NH2 terminal kinase (JNK), extracellular signal-regulated kinase (ERK), big MAP kinase 1 (BMK1/ERK5), and p38 kinase (p38) [136]. Apoptosis signal-regulating kinase 1 (ASK1) is a kind of MAPK kinase kinase kinase (MAPKKK) that can activate MAPK cascades. The reduced form of the redox regulatory protein Trx binds to ASK1 to inhibit its activity, and this interaction can be reversed, thereby restoring ASK1 kinase activity, when ROS accumulation or a lack of antioxidants induces Trx oxidation [137]. ROS can also activate MAPKs directly by inhibiting MAPK phosphatases. The inhibition of JNK-inactivating phosphatases occurs through the reversible oxidation of cysteine residues to sulfonic acid by ROS, thereby maintaining the activation of JNK [138]. In addition, the oxidation of p53 cysteine residues has been shown to affect the ability of p53 to bind DNA, affecting downstream gene expression. Therefore, ROS may disrupt the cell cycle regulation function of p53, leading to uncontrolled cell proliferation [87]. In general, ROS can affect the abovementioned signal transduction pathways and stimulate cell proliferation.
4.1.3. Oxidative Stress Accelerates Tumor Invasion and Metastasis
The dissemination and colonization of primary tumor cells to invade distant organs are known as tumor metastasis. Epithelial-to-mesenchymal transition (EMT) is an early change that occurs during tumor metastasis. EMT is characterized by cytoskeletal reorganization, loss of epithelial morphology and markers (E-cadherin, desmoplakin, Muc-1, cytokeratin-18, γ-catenin, etc.), and increased expression of mesenchymal markers (N-cadherin, vimentin, fibronectin, α-smooth muscle actin (SMA), etc.) and MMPs [139–141]. ROS can participate in tumor metastasis by regulating EMT. TGF-β1 is considered to be an important inducer of EMT that activates NF-κB through ROS-dependent pathways, upregulates urokinase-type plasminogen activator (uPA) and MMP9, and thus promotes cell migration and invasion [142]. The deletion of TGF-β-activated kinase 1 (TAK1) enhances and accelerates EMT by negatively regulating RhoA through Rac-induced ROS [143]. Due to the uncontrolled division of cancer cells, there is an insufficient nutrition and oxygen supply in the tumor microenvironment; thus, cancer cells are in a state of hypoxia. Hypoxia-inducible factor (HIF) plays a key role in EMT in tumor cells. HIF targets the EMT promoter snail and promotes hypoxia-induced EMT in different tumor types [144–146]. Complex III of ECT is essential for stabilizing HIF-1α under hypoxic conditions by increasing ROS production [147]. ROS can also enhance the transcription of HIF-1α by phosphorylating ERK and PI3K/AKT during hypoxia [148, 149]. Inflammatory mediators also facilitate ROS-mediated alteration of HIF-1α transcription and translation. GSH depletion or exogenous ROS enhance the production of inflammatory mediators such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) [150], which induce HIF-1α transcription and continuous protein synthesis at room temperature [151]. Furthermore, mild oxidative stress increases the transcriptional activity of HIF-1α by regulating the stability and nuclear localization of Sentrin/SUMO-specific proteases (SENPs) [152]. Prolyl hydroxylase (PHD) family members play crucial roles in stabilizing HIF by acting as oxygen sensors. The prolyl residues of PHD are hydroxylated under aerobic conditions and bind to the tumor suppressor protein Von Hippel Lindau (pVHL), which is a component of a ubiquitin ligase complex. Therefore, proteasomal degradation of HIF-1α occurs in the presence of sufficient oxygen [153]. Exogenous H2O2 treatment can stabilize the HIF-1α protein by inhibiting the prolyl hydroxylation of PHD under normoxic conditions [147, 154], which can be reversed by vitamin C [155]. The loss of the antioxidant protein TIGAR is related to enhanced ROS production, which can promote the occurrence of EMT in pancreatic cancer cells by activating ERK. These phenotypes can be reversed by ROS limitation signaling [156].
The development of the tumor microenvironment (TME) maintains an appropriate environment for tumor growth, invasion, and metastasis. Tumor-associated macrophages (TAMs) are critical regulators of tumorigenesis that drive aggressive cancer phenotypes [157]. Oxidative stress affects the differentiation of macrophages. A study indicated that ROS promote the differentiation of monocytes by activating ERK, while ROS inhibitors reduce the differentiation of M2-like tumor-promoting phenotypes [158]. ROS can also induce cancer cells to release the KRASG12D protein, which can be taken up by macrophages, leading to tumor-promoting transformation, thereby promoting the growth of pancreatic cancer [159]. Cancer-associated fibroblasts (CAFs), transformed from normal fibroblasts or fibroblast progenitor cells, can promote tumor proliferation, EMT, angiogenesis, tumor invasion, and immunosuppression by helping remodel the extracellular matrix (ECM) [160]. TGF-β is considered the main mediator of CAF activation [161]. The transcription factor JunD regulates antioxidant genes, and chronic oxidative stress caused by its deletion increases the migratory properties of stromal fibroblasts by inducing the accumulation of HIF-1α and CXC chemokine ligand 12 (CXCL12), thereby promoting tumor spreading [162]. The premetastatic niche determines whether circulating tumor cells can colonize and survive in distant sites, which is the final stage of successful tumor metastasis [163]. There is evidence that ROS play supporting roles in the tumor environment colonized by cancer cells. ROS inhibit the activity of cytotoxic CD8+ T cells, thereby promoting the survival of disseminated cancer cells at a secondary tumor site [164]. Lactic acid production lowers the pH value of the TME in colorectal cancer, leading to mitochondrial ROS accumulation and the apoptosis of liver NK cells, thereby promoting the occurrence of colorectal cancer liver metastasis [165].
4.2. Anticancer Effect of Oxidative Stress
4.2.1. Oxidative Stress Activates Apoptosis Pathways
Apoptosis is a kind of programmed cell death whose execution depends on apoptotic effector caspases [166]. The endogenous apoptotic pathway, also known as the mitochondrial apoptotic cascade, is regulated by mitochondrial Bcl-2 proteins. The Bcl-2 family increases the permeability of mitochondria, allowing the release of the proapoptotic factor cytochrome C, which then activates the caspase-9 signaling cascade and induces apoptosis [167, 168]. These are the key steps in the endogenous cell apoptosis process [169]. ROS stimulation facilitates cytochrome C release and induces the downstream apoptotic cascade by depleting the mitochondrial membrane potential, changing the permeability of the mitochondrial membrane, and oxidizing cardiolipin, leading to cytochrome C dissociation [170]. The overexpression of the antioxidant enzyme glutaredoxin 2 (GRX2) in HeLa cells can reduce cytochrome C dissociation and resist caspases [171]. ROS also increase mitochondrial outer membrane permeability by regulating the Bcl-2 family [172, 173]. In addition, ROS activate caspase-9 directly by increasing the interaction between oxidative-modified caspase-9 and apoptotic protease activating factor 1 (APAF-1), which in turn promotes caspase-9 activation and apoptosis [174].
The exogenous apoptotic pathway, also known as the death receptor pathway, is initiated by extracellular TNF superfamily members, which act as death ligands that bind to related cell surface death receptors and activate receptor clustering. The activated receptors recruit adaptor proteins, including Fas-associated via death domain (FADD) and TNFRSF1A associated via death domain (TRADD), and procaspase-8 forms a death-inducing signaling complex (DISC), which then induces apoptosis [175]. The cytosolic protein cellular FLICE-inhibitory protein (c-FLIP) is a main antiapoptotic regulator that inhibits DISC generation by inhibiting caspase 8 recruitment [176]. ROS activate the exogenous apoptotic signal by inducing the ubiquitination and degradation of c-FLIP. The pretreatment of prostate cancer cells with active oxygen scavengers can decrease the ROS-induced degradation of c-FLIP protein [177].
4.2.2. ROS Promote Tumor Cell Necroptosis
Necroptosis is a caspases-independent cell death form which mainly mediated by death receptors and their corresponding ligands [178, 179]. The death signal induces the activation of receptor-interacting proteins (RIPs) 1 and 3, which in turn phosphorylate mixed lineage kinase domain-like (MLKL), a specific protein driving cell necrosis. Phosphorylated MLKL (p-MLKL) is recruited into necrosomes and mediates the destruction of cell and organelle membrane integrity, resulting in the leakage of intracellular components and cell death [180–182]. An increasing number of studies have confirmed the role of necroptosis in mediating tumor death and limiting tumor metastasis [183–186]. ROS promote the autophosphorylation of RIP1 via the oxidative modification of RIP1 Cys-257, Cys-268, and Cys-586 residues and then recruit RIP3 to form necrosomes [187]. Mn (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP) removes mitochondrial superoxide, which can decrease ROS production and hinder RIP1 and RIP3 expression [188]. The removal of ROS by the antioxidant butyl hydroxyanisole (BHA) can significantly reduce the necroptosis of mouse fibrosarcoma cells [189]. In addition to the production of mitochondrial ROS, NOX1-induced oxidative stress has been shown to trigger necroptosis [190]. In summary, these results indicate that ROS promote tumor cell necroptosis by regulating the assembly of necrosomes and affecting the expression and activation of RIP1 as well as RIP3.
4.2.3. ROS Trigger Ferroptosis in Cancer
Ferroptosis is a type of cell death relying on iron and ROS. Ferroptosis can be distinguished from apoptosis, necrosis, and autophagy by morphology, biology, and genetics. Free redox-active iron in cells generates ROS through Fenton and/or increased lipoxygenase activity, causing the accumulation of peroxidated polyunsaturated fatty acids (PUFAs) and, thus, leading to cell death [191, 192]. The tumor suppressor gene p53 makes cells sensitive to ferroptosis by inhibiting the expression of solute carrier family 7 member 11 (SLC7A11). ROS treatment can downregulate the expression of SLC7A11 and maintain ferroptosis in p53 mutant inactivated cells, suggesting that ROS play a key role in ferroptosis [193]. Multiple studies have shown that ROS-triggered ferroptosis strongly inhibits various cancers, including colorectal cancer [194], nasopharyngeal cancer [195], melanoma [196], pancreatic tumors [197], and breast cancer [198].
4.2.4. Oxidative Stress Affects Immune Cells in the TME
The TME is a complex and dynamic environment. Earlier, we introduced the cancer promotion effect of ROS via the induction of malignant cell transformation and ECM remodeling in the TME. In contrast, T cells and natural killer (NK) cells participate in cancer immune surveillance [199, 200]. ROS act as signal mediators to activate T cells [201]. In mice with reduced mitochondrial ROS production, the antigen-specific expansion of T cells cannot be induced, indicating that mitochondrial ROS are key components in the activation of T cells [202]. Hydrogen peroxide influences lymphocyte activation by modulating negative regulatory phosphatases, and it plays an important role in initiating and amplifying signals at antigen receptors by acting as a second messenger [203]. Neutrophils and macrophages also exert tumor-killing effects through ROS [204], in which they release ROS, contributing to tumor killing [205, 206].
5. The Interplay between m6A Modification and Oxidative Stress
m6A methylation and oxidative stress are widely involved in the regulation of tumorigenesis and development. There is a complex relationship between the oxidative stress-related and m6A regulating signal pathways. m6A methylation of specific RNAs triggers or inhibits oxidative stress has been observed in cancers (Table 3). It is worth noting that ROS can act as an intracellular signal to affect the epigenetic modification of RNA (Table 4). On the one hand, m6A affects the survival and invasion of cancer cells by regulating oxidative stress; On the other hand, oxidative stress signals influence the overall and local levels of m6A, which may be an adaptive response of cancer cells to environmental changes and external stimuli. Some antioxidants exert regulatory effects on m6A modification have been confirmed to play an anticancer role in multiple cancer, while m6A-related signals may become a potential predictive or therapeutic marker in cancer for their regulatory role in redox homeostasis. Therefore, to discuss the crosslink between oxidative stress and m6A is helpful to understand the mechanism of tumorigenesis and provide the basis for the discovery of novel targets for tumor therapy.
Table 3.
m6A RNA methylation regulates oxidative stress in cancer.
| m6A enzyme | Change of m6A modification | ROS levels | Cancer type | Mechanism | Biofunction in cancer | References |
|---|---|---|---|---|---|---|
| METTL3; ALKBH5 | m6A level↑ | ↑ | Breast cancer | METTL3 enhances AK4 expression, increase the level of ROS in breast cancer cells and activate p38 Kinase | Promoting the resistance of breast cancer to Tamoxifen | [226] |
| METTL3 | Unknown | ↑ | Colorectal cancer | METTL3 facilitates the processing of miR-483, miR-676, and miR-877 which regulating the expression of mitochondrial related ETC genes | Promoting cancer growth and progression | [225] |
| METTL3; METTL14 | Unknown | ↓ | Colon carcinoma | METTL3/METTL14 catalyzes the m6A methylation of p21 and enhances p21 expression leading to elevated expression of Nrf2 | Inducing cell senescence | [208, 229] |
| FTO | Unknown | ↑ | Clear cell renal cell carcinoma | FTO increases stability and translation of PGC-1α mRNA thereby resulting in oxidative stress | Inducing ROS production and suppressing tumor growth | [227] |
| YTHDF1 | YTHDF1↓ | ↓ | Nonsmall cell lung cancer | Knockdown of YTHDF1 reduces the translation of keap, upregulates Nrf2 and its downstream antioxidant in response of cisplatin-induced ROS | Adapting to oxidative stress; Inducing cisplatin resistance in nonsmall cell lung cancer | [215] |
| METTL3; YTHDF2 | METTL3↑ SUMOylation of YTHDF2 |
↑ | Lung adenocarcinoma | YTHDF2 can be SUMOylated at K571 in hypoxia or oxidative stress condition | Promoting mRNA degradation and cancer progression | [21] |
| YTHDC2 | Unknown | ↑ | Lung adenocarcinoma | YTHDC2 regulates SLC7A11 mRNA decay, which leads to the inhibition system XC(-) function, thus impairing the antioxidant function | Inhibiting tumorigenesis | [233] |
Table 4.
ROS regulate m6A modification.
| Oxidative stress activators | Cell type | Change of m6A components | Biofunction | References |
|---|---|---|---|---|
| Low dose of NaAsO2 | Human keratinous HaCaT cells | METTL3↑; METTL14↑; WTAP↑; FTO↓ | Moderate level of ROS-facilitating cell survival via elevated m6A levels in HaCaT cells | [208] |
| High dose of NaAsO2 | Human keratinous HaCaT cells | METTL3↓; METTL14↓;WTAP↓;FTO↑ | High level of ROS inducing cell death by decreased m6A levels in HaCaT cells | [208] |
| Hypoxia | Breast cancer stem cells | ALKBH5↑ | Decreasing NANOG mRNA methylation, enhancing the expression of NANOG transcripts, and inducing breast cancer stem cell phenotype | [212] |
| H2O2 | Hematopoietic stem/progenitor cells | ALKBH5 m6A demethylase activity↓ | Participating in DNA damage repair and protecting genomic integrity of cells | [213] |
| Bmal1 deletion | Hepatic cells | METTL3↑; YTHDF2↑ | Increasing PPaRα m6A abundance, decreasing its expression, and promoting lipid accumulation | [214] |
| Hypoxia | NSCLC cells | YTHDF1↑ | Playing a role in hypoxia adaptation of NSCLC through Keap1-Nrf2-AKR1C1 axis | [215] |
| Hypoxia | Lung adenocarcinoma cells | YTHDF2 SUMOylation at the Lys571 site | Promoting degradation of transcriptome-wide mRNAs and cancer progression | [21] |
5.1. Oxidative Stress Regulates m6A RNA Methylation
In a study that used high-performance liquid chromatography tandem mass spectrometry to identify and quantify m6A modifications in highly purified yeast mRNA samples, researchers found that m6A-modified mRNA levels changed under oxidative stress [207]. Subsequent studies explored the effect of ROS on m6A modification levels in human keratinocytes. The treatment of human keratinous HaCaT cells with the environmental carcinogen arsenite can upregulate methyltransferase (METTL3/METTL14/WTAP) levels, increase the m6A modifications mediated by these enzymes, and inactivate the demethylase FTO, which protects cells from oxidative stress and promotes survival. NAC inhibits the increase in methyltransferase and m6A levels in human keratinocytes exposed to arsenite. In contrast, after high-dose arsenite treatment, increased oxidative stress induces the downregulation of m6A levels, showing an inhibitory effect on HaCaT cell viability (Figure 2(a)) [22, 208]. Oxidative damage induced by CdSO4 causes a significant reduction in m6A modifications in pancreatic β-cells. FTO and METTL3 mRNA levels also decrease in a concentration-dependent manner after CdSO4 treatment [209]. These results suggest that different degrees of oxidative stress may have different effects on the m6A modification.
Figure 2.
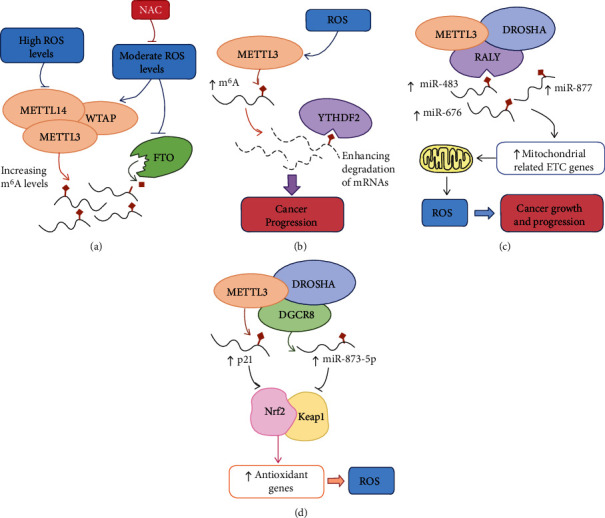
The interplay between m6A and oxidative stress. (a) ROS affect m6A modification in a dose-dependent manner. (b) ROS influence m6A-mediated RNA stability, which is involved in tumor progression. (c) m6A promotes tumor growth through mitochondrial function regulation and ROS production. (d) m6A regulates the antioxidant response through Nrf2/Keap1 signaling.
Hypoxia is the inducer of ROS production [210]. There are evidences show that ALKBH5 reducing the overall m6A level in response of hypoxia [211]. Expression of ALKBH5 can be promoted by HIF-1α and HIF-2α in a hypoxia condition. Elevated levels of ALKBH5 demethylate the transcripts of pluripotency-related gene Nanog homeobox (NANOG) thereby increasing its expression, and inducing breast cancer stem cell phenotype [212]. In addition, the activity of the demethylase ALKBH5 and the overall m6A methylation level is directly regulated by ROS. ROS can induce the posttranslational modification of ALKBH5 by activating the ERK/JNK signaling pathway and thus inhibit its activity, which helps to increase mRNA m6A levels and maintain the genomic integrity of cells [213].
Similarly, oxidative stress exerts different effects on m6A readers. The deletion of Bmal1 increases ROS production in hepatic cells, resulting in an increase in METTL3-mediated m6A mRNA methylation, particularly that of nuclear receptor peroxisome proliferator-activator α (PPaRα). YTHDF2 binds to PPaRα to mediate its mRNA stability to alter hepatic lipid metabolism [214]. YTHDF1 is considered to be a hypoxia-adaptive gene that is highly expressed in various cancers, including NSCLC. Under normoxic conditions, the overexpression of YTHDF1 increases the translation of m6A-modified transcripts and induces the proliferation of NSCLC cells. However, under conditions of hypoxia or chemotherapy-induced ROS accumulation, low expression of YTHDF1 in NSCLC can reduce the translation of keap, promote the upregulation of Nrf2 and its downstream antioxidant AKR1C1, and induce cisplatin resistance [215]. Hypoxia can induce the SUMOylation of YTHDF2 in vivo and in vitro at the Lys571 site, which is repressed by oxidative stress and SUMOylation inhibitors. The SUMOylation of YTHDF2 significantly increases its binding affinity for m6A-modified mRNAs, leading to the degradation of transcriptome-wide mRNAs and promoting cancer progression (Figure 2(b)) [21]. Stress granules (SGs) are dynamic structure in which translationally stalled mRNAs are deposited [216–218]. Oxidative stress induces the METTL3/METTL14/WTAP-mediated deposition of m6A on the 5′-UTR of SGs. The m6A reader protein YTHDF3 has been documented to triage m6A-modified mRNAs to SGs under oxidative stress in HEK293 and osteosarcoma U2OS cells [219].
Oxidative stress can alter the m6A level by affecting the expression and activity of m6A enzymes and, then, determinate cell fate and physiological functions. This regulation may be an adaptive response to harmful injury, suggesting the potential role of m6A in stress response. A variety of epigenetic mechanisms play regulatory roles in genes expression involved in stress stimulus response. Posttranslational modifications (PTMs) of histones are produced by nonenzymatic and enzymatic processes and produce a marked effect in controlling chromatin structure and gene expression. Histone H3 trimethylation at lys36 (H3K36me3) is a transcription elongating marker that can adjust m6A deposition at an overall level. There is evidence that the level of H3K36me3 is positively correlated with the m6A level. Further studies found that the core region of H3K36me3 can be recognized and bind by METTL14, which act as a gene regulation signal to promote the interaction of m6A methyltransferase complex and RNA polymerase II and then enhance on m6A deposition on nascent RNAs, suggesting that m6A RNA methylation is regulated by histone modification [220]. The results partially revealed the mechanism of m6A-specific deposition in the transcriptome. Free radicals as regulators can regulate histone PTMs in directly and indirectly manners and participate in the epigenetic landscape [221], implying that that oxidative stress may regulate m6A methylation by affecting the epigenetic regulations. Overall, the achievements above confirmed the crosslink between m6A and oxidative stress; however, its potential mechanism remains unclear and needs further research.
5.2. m6A Modifications Affect Oxidative Stress
m6A, as the most common form of RNA epigenetic transcription regulation, participates in a variety of biological processes, involving oxidative stress, by affecting RNA alternative splicing, stability, translation, and subcellular localization. m6A RNA modification can control redox homeostasis by regulating the production of ROS, altering antioxidant genes expression, or affecting oxidative stress-related signal pathways.
5.2.1. ROS Production
Mitochondria is the main source of ROS in cells [222]. m6A can affect the redox balance of cells by directly modifying the pathway of ROS production. The microprocessor complex, composed of Type III RNase DROSHA and RNA-binding protein DGCR8, involves in the processing of primary microRNAs (pri-miRNAs). m6A writer METTL3 has been proven to increase the m6A level of pri-miRNAs and promote their recognition and processing by DGCR8, participating in the first step of miRNA biogenesis [223, 224]. The RALY heterogeneous nuclear ribonucleoprotein (RALY, also known as hnRNPCL2) is a novel RNA-binding protein that is an important regulatory component of the DROSHA complex. It regulates the expression of mitochondrial-related ETC genes by promoting the posttranscriptional modification of specific miRNA subsets and then reprograms the mitochondrial metabolism in cancer cells. METTL3-dependent m6A modification is necessary for RALY-mediated miRNA maturation. METTL3 enhances m6A methylation in the terminal loop of pri-mir483, pri-mir877, and pri-mir676, so as to increase their interaction with the RALY complex and promote miRNA processing. The deficiency of METTL3 resulted in a significant decrease in the levels of these three pri-miRNAs and significantly affected the growth and progress of colorectal cancer. Further studies showed that METTL3 depletion induces the expression of mitochondrion-related ETC genes ATP synthase membrane subunit e (ATP5) I, ATP5G1, ATP5G3, and cytochrome c1 (CYC1) at the protein and RNA levels led to the promotion of mitochondrial respiration and accumulation of ROS. The overexpression of miR-877 can partially reverse the effect caused by METTL3 gene knockdown, indicating that METTL3-mediated m6A methylation of pri-miRNAs influences the mitochondrial metabolism and cell fate of colorectal cancer cells (Figure 2(c)). The overexpression of miR-877 can partially reverse the effect caused by METTL3 gene knockdown, indicating that METTL3-mediated m6A methylation of pri-miRNAs influence the mitochondrial metabolism and cell fate of colorectal cancer cells (Figure 2(c)) [225]. Tamoxifen has been widely used in the treatment of patients with estrogen receptor-positive breast cancer, but the effect of drug resistance has affected the patient's benefit. Adenylate kinase 4 (AK4) is a key enzyme located in the mitochondrial matrix and involved in the regulation of cell energy metabolism. In Tamoxifen-resistant breast cancer cells, the methylation level of m6A-specific motifs in AK4 increased significantly. Methyltransferase METTL3 can enhance AK4 expression, increase the level of ROS in breast cancer cells and activate p38 Kinase, and promote the resistance of breast cancer to Tamoxifen, and the role of demethylation ALKBH5 is opposite to that of METTL3 [226]. FTO is also involved in the oxidative stress response in clear cell renal cell carcinoma by acting as an m6A easer. FTO induces oxidative stress by reducing m6A levels of peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α) mRNA, a major regulator of mitochondrial metabolism, which leads to increased stability and translation of PGC-1α mRNA. The elevated expression of PGC-1α restores mitochondrial activity, induces ROS production, and suppresses tumor growth [227]. In the hepatic ischemia-reperfusion injury model, FTO plays a protective role by demethylating dynamic-related protein 1 (Drp1) mRNA and inhibits Drp1-mediated oxidative stress and mitochondrial fragmentation in the liver [228].
5.2.2. Expression of Antioxidant Genes
m6A RNA methylation can affect the expression of antioxidant-related genes, thus regulating oxidative stress. As mentioned above, METTL3 interacts with the RNA-binding protein DGCR8 and may participate in regulating DGCR8 recognition and binding primary miRNAs, thereby influencing the maturation of miRNAs [223]. Experiments showed that the maturation of miR-873-5p modulated in a METTL3-dependent manner could activate the Nrf2 antioxidant pathway and inhibit its negative regulator Keap1 to resist colistin-induced oxidative stress and apoptosis [20]. In human HeLa cells and colon carcinoma HCT116 cells, METTL3/METTL14 catalyzes m6A methylation in the 3′-UTR of p21 and enhances p21 expression, leading to elevated expression of Nrf2 and inducing cell senescence under ROS stress (Figure 2(d))[229]. Other studies have shown that the endocrine-disrupting chemical di-(2-ethylhexyl) phthalate (DEHP) can increase the overall level of m6A methylation and increase the m6A modification level of Nrf2 transcripts by altering the expression of FTO and YTHDC2, thereby inhibiting the Nrf2-mediated antioxidant pathway and inducing oxidative stress [230].
In a study of human hepatoma HepG2 cells treated with different concentrations of fumonisinB1 (FB1, a common carcinogen), it was investigated whether there is cross-talk between FB1-induced oxidative stress and m6A. The results showed that FB1 induced intracellular ROS accumulation, and the increase in the m6A level was accompanied by increases in both m6A writers, including METLL3 and METLL14, and readers, including YTHDF1, YTHDF2, YTHDF3, and YTHDC2, and decreases in FTO and ALKBH5. FB1-induced m6A RNA methylation results in a decrease in Keap1 expression and an increase in Nrf2 expression [18]. System XC(-), a cystine/glutamate transporter, promotes cystine import and participates in GSH synthesis in response to oxidative stress [231]. The activity of System XC(-) can be regulated by SLC7A11 [232]. A study has found that the antitumor mechanism of YTHDC2 is related to the impairment of antioxidant function caused by inhibition of system XC(-) function in lung adenocarcinoma. The mechanism depends on m6A-mediated SLC7A11 mRNA decay, which leads to the inhibition of the downstream antioxidant process by reducing cystine uptake [233]. MALAT1 is a long noncoding RNA that has been shown to be involved in oxidative stress. Antagonism of MALAT1 can lead to transcriptional activation of Keap1 and reduction of Nrf1/2, and they mediated antioxidant gene expression and ROS accumulation [234]. A study showed that MALAT1 level was positively correlated with m6A modification [209], suggesting that m6A modification may also be involved in the regulation of oxidative stress by a MALAT1-mediated pathway.
5.2.3. Upstream of ROS Generation in Cancer
Oncogenes and tumor suppressor gene pathways such as KRAS, MYC, Bcl-2, and p53 account for the abnormal oxidative stress state of cancer. The abnormal activation of KRAS occurs in pancreatic ductal adenocarcinoma, nonsmall cell lung cancer NSCLC, colorectal cancer, and other tumors which can promote ROS generation and the metabolic reprogramming of cancer cells by regulating mitochondria function [78, 235]. A bioinformatics analysis, including 1017 NSCLC patients with copy number variation (CNV) data, has shown that the high expression of FTO is positively correlated with the activation of the KRAS signal transduction pathway [236]. Overexpression of m6A reader YTHDF2 inhibits the activation of MEK and ERK which are downstream of Ras and impairs hepatocellular carcinoma progression [237]. Moreover, B-Raf protooncogene (BRAF) and MEK inhibitors can remodel mRNA translation in melanoma which is related to m6A level, suggesting that m6A affects tumor growth and drug resistance by participating in Ras signal regulation [238].
Expression of endogenous oncogenic alleles of MYC can increase Nrf2 transcription and its mediated antioxidant program to detoxify intracellular ROS [90]. Another study has shown that MYC oncoprotein increases ROS produced by mitochondrial ETC [77]. These results suggest that MYC plays a complex role in oxidative stress through different mechanisms. m6A can directly modify MYC mRNA. A research exhibits that MYC act as a direct target of m6A in haematopoietic stem cells by RNA sequencing [239]. The increase of m6A modifications of MYC mRNA facilitates the binding of YTHDF1, then promotes MYC expression and subsequent metabolic reprogramming and proliferation in lung adenocarcinoma [240]. YTHDF2 can stabilize MYC transcripts to mediate the viability of glioblastoma stem cells in an m6A-dependent manner [241]. Similarly, IGF2BP1 recognizes m6A modification of MYC mRNA to increase the stability and expression MYC, thereby promoting tumorigenesis [242]. In addition to directly affecting the transcription and translation of MYC, m6A can also regulate MYC expression by lncRNA. Lung Cancer Associated Transcript 3 (LCAT3) is a newly identified oncogenic lncRNA, which is upregulated in lung adenocarcinoma through METTL3 mediated m6A methylation. LCAT3 can activate the transcription of MYC and promote the survival and progression of lung adenocarcinoma [243].
Bcl-2 is one of the most important oncogenes in the field of apoptosis which can maintain mild a prooxidant state by enhancing mitochondrial respiration to sustain the survival of cancer cells [79]. Single-nucleotide-resolution mapping combining ribosome profiling shows that the translation of Bcl-2 mRNAs is governed by m6A methylation in acute myeloid leukemia cells [244]. Other studies have also demonstrated that Bcl-2 expression is regulated by m6A-dependent pathways in NSCLC [245] and breast cancer [246].
p53 is an important tumor suppressor gene participating in cell cycle arrest, apoptosis, and senescence. As a crucial transcription factor, wild-type p53 plays a coordinating role in oxidative stress. p53 showed antioxidant capacity in response to low levels of oxidative stress to clear ROS and maintain cell survival; in the face of high levels of oxidative stress, p53 can promote ROS production and further induce cell death [247, 248]. p53R273H is the missense mutation form of p53. It has been observed that there is m6A methylation at the 273 mutated codons (G to A) of p53 pre-mRNA which can increase the splicing of pre-mRNA and, then, promote the expression of p53R273H protein. Subsequent studies confirmed this process is mediated by METTL3. Upregulation of METTL3 can promote the expression of p53R273H and mediate the drug resistance of colon cancer cells [249]. A study uses 1 μM arsenite to induce an overall increase in m6A RNA modifications showed that decreased activity, phosphorylation, acetylation, and nuclear expression levels of p53 in human keratinocyte HaCaT cells with elevated m6A levels. METTL3 knockout confirmed that m6A modification influences the expression and activity of p53 by downregulating positive regulatory factors and upregulating negative regulatory factors of p53 [250]. p21 is a cyclin-dependent kinases inhibitor downstream of p53. 3′UTR of p21 occurs the combined modification of NOP2/Sun RNA methyltransferase 2- (NSUN2-) mediated 5-methylcytosine (m5C) and METTL3/METTL4-mediated m6A methylation. They synergistically enhance the expression of p21 at the translation level in response of oxidative stress-induced cellular senescence [229].
m6A modification alters the redox state by regulating ROS production, governing antioxidant gene expression, and affecting oncogene signals that induce oxidative stress. In general, these findings suggest that the mutual regulation does occur between oxidative stress regulators and m6A modifications (Tables 4 and 3). However, the interplay of m6A modification and oxidative stress is complex and dynamic, and its regulatory mechanism needs to be systematically and deeply studied.
5.3. Role of Oxidative Stress and m6A Modification in Cancer Therapy
Conventional anticancer therapies, such as radiotherapy and chemotherapy, are closely related to ROS generation. Cytotoxic drugs and ionizing radiation can cause DNA damage and apoptosis mediated by ROS. Therefore, the regulation of oxidative stress plays an important role in cancer therapy. As mentioned earlier, elevated ROS levels induce tumor growth and development, so antioxidant supplementation may be a potential therapeutic strategy for ROS-induced cancer. Vitamin C is one of the most common antioxidants. In multiple myeloma [251], pancreatic cancer [252], and ovarian cancer [253], vitamin C can function either in cooperation with chemotherapy or alone to play an anticancer role. The role of vitamin C in the anticancer immune response has been studied. In preclinical models, a high dose of vitamin C can regulate immune cell infiltration in the tumor microenvironment, enhance the cytotoxicity of adoptively transferred T cells, and cooperate with immune checkpoint blockers to treat a variety of cancers [254]. Vitamin E is also considered to be an effective antioxidant that plays a role in reducing the risk of hepatocellular carcinoma [255], prostate cancer [256], and breast cancer [257]. Nrf2 is a major regulator of the tumor antioxidant response [33]. Many natural and synthetic compounds, including curcumin, resveratrol, sulforaphane, and RTA 405, are considered to be Nrf2 activators and participate in the chemoprevention of cancer [258].
It is worth noting that some studies have shown that antioxidant treatment may increase the risk of certain cancers and promote their progression. For example, the antioxidants vitamin E and N-acetylcysteine can improve oxidative damage in the lung tissue of mice but may induce the development and progression of lung cancer [259, 260]. A large-sample, multicenter, prospective cohort study that explored the role of vitamin E in prostate cancer prevention found that the long-term use of supplemental vitamin E may increase the risk of prostate cancer in healthy men [261]. These results suggest that some types of cancer may survive under the protective effects of antioxidants and that prooxidants may be effective in such cases. Common chemotherapy drugs, including paclitaxel, platinum complex, adriamycin, and antimetabolites, can cause oxidative stress. Arsenic trioxide can induce the apoptosis of many kinds of cancer cells by increasing ROS levels [262]. The cytotoxicity of 5-fluorouracil is related to the production of ROS. Cancer cells are resistant to 5-fluorouracil through antioxidant mechanisms [263]. ROS also mediates oxidative damage and cell death induced by ionizing radiation [264]. In addition, the anticancer effects of many drugs directly targeting ROS metabolic pathways have been confirmed. These drugs fall into two categories [265]. The first is drugs that target glutathione; for example, NOV-002 is a kind of oxidized glutathione that can inhibit DNA repair and play a direct anticancer role. In addition, it can be used as a substrate to participate in s-glutathionation and affect a variety of signaling pathways. Clinical data show that the combination of NOV-002 and cisplatin can increase the survival of patients with NSCLC and increase the tolerance of patients to chemotherapy [266]. The second kind of drug targets thioredoxin, including auranofin, PX-12, and dimesna. PX-12 is an irreversible inhibitor of thioredoxin-1 that can inhibit cell growth, increase ROS levels, and induce apoptosis in acute lymphoblastic leukemia [267], gastric cancer, and hepatocellular carcinoma [268]. At present, some redox regulatory drugs have entered the clinical trial stage and are expected to become candidates for cancer treatment [269].
Some agents targeting oxidative stress may play a role in regulating m6A modifications. Sulforaphane (SFN) is not only an oxidative stress regulator but also an epigenetic modulator. On the one hand, SFN plays an antioxidation role by activating Nrf2; on the other hand, it can mediate the depletion of GSH and increase the production of ROS [270]. Studies have found that the effect of SFN is concentration dependent. Using low-dose SFN to treat breast cancer cell lines can reduce the overall m6A level, and induce oxidative stress, G2/M cell cycle arrest, and apoptosis which benefit to cancer inhibition [271]. Epigallocatechin gallate (EGCG) is a kind of green tea polyphenols which play a role in antioxidation [272]. EGCG treated adipocytes shown a decrease of FTO expression and increase of total m6A-methylated RNA levels resulting in inhibition of adipocyte differentiation [273]. Both resveratrol and curcumin are natural antioxidants which have been considered as effective agents for cancer chemoprevention [274]. The combination of them can lead to the downregulation of the level of m6A [275]. Another study had shown that resveratrol can reducing the ROS accumulation induced by mycotoxin Aflatoxin B1 thereby decreasing m6A-modified RNA levels [276]. These results exhibit the association between oxidative stress and m6A modification. The extensive and profound effect of ROS on the level of m6A implies that m6A modification may play a role in the effects of many anticancer drugs related to oxidative stress. On the other side, using oxidative modulators to affect m6A modification of specific transcripts may be a potential cancer treatment. m6A enzymes and levels may become biomarkers of anticancer therapy. The discovery and development of new drugs targeting oxidative stress is a very promising research direction.
6. Conclusions and Future Perspectives
One feature that distinguishes cancer cells from normal cells is their aberrant oxidation and antioxidant system. The study of the mechanisms of oxidative stress in cancer may give rise to a new field of redox medicine in which oxidants and antioxidants may become effective treatment strategies for cancer. However, due to the extensive and complex effects of ROS in cancer, their roles as signaling molecules cannot be simply defined. Increasing evidence demonstrates that oxidative stress has the ability to regulate m6A RNA methylation, which may be correlated with the degree of ROS accumulation and can affect tumor progression. Similarly, m6A methylation has proven to affect the biological functions of cancer cells, including growth, progression, senescence, and apoptosis, by affecting ROS levels. In this review, we summarize the dual functions and effects of oxidative stress in cancer cells and the general mechanisms underlying its effects. The changes in m6A methylation levels in response of oxidative stress and the regulation of intracellular ROS levels via m6A modification are emphasized. These findings not only provide new clues for elucidating the mechanisms of different cell responses to oxidative stress but also give rise to new prospects for targeting m6A modification pathways to regulate oxidative stress.
Abbreviations
- ADAM19:
ADAM metallopeptidase domain 19
- ADAR1:
Adenosine deaminase RNA specific
- AIM2:
Absent in melanoma 2
- ALDOA:
AldolaseA
- AMPK:
AMP-activated protein kinase
- ANGPT2:
Angiopoietin 2
- ANGPTL4:
Angiopoietin-like 4
- ASB2:
Ankyrin repeat and SOCS box containing 2
- BRCA2:
BRCA2 DNA repair associated
- BRD4:
Bromodomain-containing protein 4
- CDK:
Cyclin-dependent kinase
- CDKN2A:
Cyclin-dependent kinase inhibitor 2A
- CEBPA:
CCAAT enhancer-binding protein alpha
- DROSHA:
Drosha ribonuclease III
- EGFR:
Epidermal growth factor receptor
- EIF3C:
Eukaryotic translation initiation factor 3 subunit C
- EPHA3:
EPH receptor A3
- ETS1:
ETS protooncogene 1
- FOXM1:
Forkhead box M1
- GAS5:
Growth arrest-specific 5
- GJA1:
Gap junction protein alpha 1
- GRIM-19:
Gene associated with retinoic and interferon-induced mortality 19 protein
- HBXIP:
Hepatitis B X-interacting protein
- IL11:
Interleukin 11
- JUNB:
JunB protooncogene
- KLF4:
Kruppel like factor 4
- LHPP:
Phospholysine phosphohistidine inorganic pyrophosphate phosphatase
- LncRNA:
Long noncoding RNA
- MALAT1:
Metastasis-associated lung adenocarcinoma transcript 1
- MFN2:
Mitofusin 2
- mTOR:
Mechanistic target of rapamycin kinase
- MYB:
MYB protooncogene
- MYC:
MYC protooncogene
- NKX3-1:
NK3 homeobox 1
- NNT:
Nicotinamide nucleotide transhydrogenase
- PDE:
Phosphodiesterase
- PER1:
Period circadian regulator 1
- PERP:
p53 apoptosis effector related to PMP22
- PGC1β:
Peroxisome proliferator-activated receptor γ coactivator 1β
- PHLPP2:
PH domain and leucine-rich repeat protein phosphatase 2
- PKCλ/ι:
Protein kinase C λ/ι
- PRDX3:
Peroxiredoxin3
- RAD51:
RAD51 recombinase
- RARA:
Retinoic acid receptor alpha
- RDM1:
RAD52 motif 1
- SERPINE2:
Serpin family E member 2
- SETD7:
SET domain containing 7
- SIRT3:
Sirtuin3
- SOCS2:
Suppressor of cytokine signaling 2
- SOX2:
SRY (sex determining region Y)-box 2
- SRSFs:
serine- and arginine-rich splicing factors
- ST6GALNAC5:
ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 5
- TACC3:
Transforming acidic coiled-coil containing protein 3
- TP53I11:
Tumor protein p53 inducible protein 11
- TRAP1:
Tumor necrosis factor-receptor-associated protein 1
- UBQLN1:
Ubiquilin 1
- UCP2:
Mitochondrial uncoupling protein 2
- UTRs:
Untranslated regions
- VEGF:
Vascular endothelial growth factor
- VEGFA:
Vascular endothelial growth factor A
- VHL:
Von Hippel-Lindau
- XIST:
X inactive-specific transcript
- YAP:
Yes1 associated transcriptional regulator
- ZEB1:
Zinc finger E-box binding homeobox 1.
Data Availability
No data were used to support this study
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- 1.Janssen-Heininger Y. M. W., Mossman B. T., Heintz N. H., et al. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radical Biology and Medicine. 2008;45(1):1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balaban R. S., Nemoto S., Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharyya A., Chattopadhyay R., Mitra S., Crowe S. E. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiological Reviews. 2014;94(2):329–354. doi: 10.1152/physrev.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dionisio P. A., Amaral J. D., Rodrigues C. M. P. Oxidative stress and regulated cell death in Parkinson's disease. Ageing Research Reviews. 2021;67, article 101263 doi: 10.1016/j.arr.2021.101263. [DOI] [PubMed] [Google Scholar]
- 5.Forstermann U., Xia N., Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circulation Research. 2017;120(4):713–735. doi: 10.1161/CIRCRESAHA.116.309326. [DOI] [PubMed] [Google Scholar]
- 6.Drummond G. R., Selemidis S., Griendling K. K., Sobey C. G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nature Reviews Drug Discovery. 2011;10(6):453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vatner S. F., Zhang J., Oydanich M., Berkman T., Naftalovich R., Vatner D. E. Healthful aging mediated by inhibition of oxidative stress. Ageing Research Reviews. 2020;64, article 101194 doi: 10.1016/j.arr.2020.101194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo J., Mills K., le Cessie S., Noordam R., van Heemst D. Ageing, age-related diseases and oxidative stress: what to do next? Ageing Research Reviews. 2020;57, article 100982 doi: 10.1016/j.arr.2019.100982. [DOI] [PubMed] [Google Scholar]
- 9.Gorrini C., Harris I. S., Mak T. W. Modulation of oxidative stress as an anticancer strategy. Nature Reviews Drug Discovery. 2013;12(12):931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 10.Hayes J. D., Dinkova-Kostova A. T., Tew K. D. Oxidative stress in cancer. Cancer Cell. 2020;38(2):167–197. doi: 10.1016/j.ccell.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desrosiers R., Friderici K., Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proceedings of the National Academy of Sciences. 1974;71(10):3971–3975. doi: 10.1073/pnas.71.10.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer K. . D., Saletore Y., Zumbo P., Elemento O., Mason C. . E., Jaffrey S. . R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell. 2012;149(7):1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Y., Dominissini D., Rechavi G., He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nature Reviews Genetics. 2014;15(5):293–306. doi: 10.1038/nrg3724. [DOI] [PubMed] [Google Scholar]
- 14.Jin S., Zhang X., Miao Y., et al. m6A RNA modification controls autophagy through upregulating ULK1 protein abundance. Cell Research. 2018;28(9):955–957. doi: 10.1038/s41422-018-0069-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiang Y., Laurent B., Hsu C.-H., et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543(7646):573–576. doi: 10.1038/nature21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan Q., Zhu H., Liu H., Wang M., Chu H., Zhang Z. METTL3 regulates PM2.5-induced cell injury by targeting OSGIN1 in human airway epithelial cells. Journal of Hazardous Materials. 2021;415, article 125573 doi: 10.1016/j.jhazmat.2021.125573. [DOI] [PubMed] [Google Scholar]
- 17.Lin X., Chai G., Wu Y., et al. RNA m6A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nature Communications. 2019;10(1):p. 2065. doi: 10.1038/s41467-019-09865-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Arumugam T., Ghazi T., Chuturgoon A. A. Fumonisin B1 alters global m6A RNA methylation and epigenetically regulates Keap1-Nrf2 signaling in human hepatoma (HepG2) cells. Archives of Toxicology. 2021;95(4):1367–1378. doi: 10.1007/s00204-021-02986-5. [DOI] [PubMed] [Google Scholar]
- 19.Fu Y., Zhuang X. m6A-binding YTHDF proteins promote stress granule formation. Nature Chemical Biology. 2020;16(9):955–963. doi: 10.1038/s41589-020-0524-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J., Ishfaq M., Xu L., Xia C., Chen C., Li J. METTL3/m(6)A/miRNA-873-5p attenuated oxidative stress and apoptosis in colistin-induced kidney injury by modulating Keap1/Nrf2 pathway. Frontiers in Pharmacology. 2019;10 doi: 10.3389/fphar.2019.00517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou G., Zhao X., Li L., et al. SUMOylation of YTHDF2 promotes mRNA degradation and cancer progression by increasing its binding affinity with m6A-modified mRNAs. Nucleic Acids Research. 2021;49(5):2859–2877. doi: 10.1093/nar/gkab065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao T., Li X., Sun D., Zhang Z. Oxidative stress: One potential factor for arsenite-induced increase of N6-methyladenosine in human keratinocytes. Environmental Toxicology and Pharmacology. 2019;69:95–103. doi: 10.1016/j.etap.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Bokar J. A., Shambaugh M. E., Polayes D. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3(11):1233–1247. [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J., Yue Y., Han D., et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N 6-adenosine methylation. Nature Chemical Biology. 2014;10(2):93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ping X.-L., Sun B.-F., Wang L., et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Research. 2014;24(2):177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwartz S., Mumbach M. . R., Jovanovic M., et al. Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5′ Sites. Cell Reports. 2014;8(1):284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patil D. P., Chen C.-K., Pickering B. F., et al. m6A RNA methylation promotes XIST -mediated transcriptional repression. Nature. 2016;537(7620):369–373. doi: 10.1038/nature19342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geula S., Moshitch-Moshkovitz S., Dominissini D., et al. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347(6225):1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- 29.Wang X., Feng J., Xue Y., et al. Structural basis of N 6-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534(7608):575–578. doi: 10.1038/nature18298. [DOI] [PubMed] [Google Scholar]
- 30.Yue Y., Liu J., Cui X., et al. VIRMA mediates preferential m6A mRNA methylation in 3′ UTR and near stop codon and associates with alternative polyadenylation. Cell Discovery. 2018;4(1) doi: 10.1038/s41421-018-0019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knuckles P., Lence T., Haussmann I. U., et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m6A machinery component Wtap/Fl(2)d. Genes & Development. 2018;32(5-6):415–429. doi: 10.1101/gad.309146.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pendleton K. E., Chen B., Liu K., et al. The U6 snRNA m6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell. 2017;169(5):824–835.e14. doi: 10.1016/j.cell.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei J., Liu F., Lu Z., et al. Differential m6A, m6Am, and m1A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Molecular Cell. 2018;71(6):973–985.e5. doi: 10.1016/j.molcel.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jia G., Fu Y., Zhao X., et al. Correction: Corrigendum: N 6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nature Chemical Biology. 2012;8(12) doi: 10.1038/nchembio1212-1008a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng G., Dahl J. . A., Niu Y., et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Molecular Cell. 2013;49(1):18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiao W., Adhikari S., Dahal U., et al. Nuclear m6A Reader YTHDC1 Regulates mRNA Splicing. Molecular Cell. 2016;61(4):507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 37.Hsu P. J., Zhu Y., Ma H., et al. Ythdc2 is an N 6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Research. 2017;27(9):1115–1127. doi: 10.1038/cr.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X., Zhao B. . S., Roundtree I. . A., et al. N 6 -methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161(6):1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X., Lu Z., Gomez A., et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi H., Wang X., Lu Z., et al. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Research. 2017;27(3):315–328. doi: 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li A., Chen Y.-S., Ping X.-L., et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Research. 2017;27(3):444–447. doi: 10.1038/cr.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu N., Dai Q., Zheng G., He C., Parisien M., Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alarcón C. R., Goodarzi H., Lee H., Liu X., Tavazoie S., Tavazoie S. F. HNRNPA2B1 Is a Mediator of m6A-Dependent Nuclear RNA Processing Events. Cell. 2015;162(6):1299–1308. doi: 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou K. I., Shi H., Lyu R., et al. Regulation of Co-transcriptional Pre-mRNA Splicing by m6A through the Low-Complexity Protein hnRNPG. Molecular Cell. 2019;76(1):70–81.e9. doi: 10.1016/j.molcel.2019.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer K. D., Patil D. P., Zhou J., et al. 5′ UTR m6A Promotes Cap-Independent Translation. Cell. 2015;163(4):999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang H., Weng H., Sun W., et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nature Cell Biology. 2018;20(3):285–295. doi: 10.1038/s41556-018-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu R., Li A., Sun B., et al. A novel m6A reader Prrc2a controls oligodendroglial specification and myelination. Cell Research. 2019;29(1):23–41. doi: 10.1038/s41422-018-0113-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arguello A. E., DeLiberto A. N., Kleiner R. E. RNA chemical proteomics reveals the N6-Methyladenosine (m6A)-Regulated protein-RNA interactome. Journal of the American Chemical Society. 2017;139(48):17249–17252. doi: 10.1021/jacs.7b09213. [DOI] [PubMed] [Google Scholar]
- 49.Edupuganti R. R., Geiger S., Lindeboom R. G. H., et al. N 6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nature Structural & Molecular Biology. 2017;24(10):870–878. doi: 10.1038/nsmb.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Worpenberg L., Paolantoni C., Longhi S., et al. Ythdf is a N6-methyladenosine reader that modulates Fmr1 target mRNA selection and restricts axonal growth in Drosophila. The EMBO Journal. 2021;40(4, article e104975) doi: 10.15252/embj.2020104975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang S., Zhao B. S., Zhou A., et al. m6A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell. 2017;31(4):591–606.e6. doi: 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Q., Chen C., Ding Q., et al. METTL3-mediated m6A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69(7):1193–1205. doi: 10.1136/gutjnl-2019-319639. [DOI] [PubMed] [Google Scholar]
- 53.Wang H., Deng Q., Lv Z., et al. N6-methyladenosine induced miR-143-3p promotes the brain metastasis of lung cancer via regulation of VASH1. Molecular Cancer. 2019;18(1):p. 181. doi: 10.1186/s12943-019-1108-x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Vu L. P., Cheng Y. M., Kharas M. G. The biology of m6A RNA methylation in normal and malignant hematopoiesis. Cancer Discovery. 2019;9(1):25–33. doi: 10.1158/2159-8290.CD-18-0959. [DOI] [PubMed] [Google Scholar]
- 55.Liu Y., You Y., Lu Z., et al. N6-methyladenosine RNA modification-mediated cellular metabolism rewiring inhibits viral replication. Science. 2019;365(6458):1171–1176. doi: 10.1126/science.aax4468. [DOI] [PubMed] [Google Scholar]
- 56.Shen C., Xuan B., Yan T., et al. m6A-dependent glycolysis enhances colorectal cancer progression. Molecular Cancer. 2020;19(1):p. 72. doi: 10.1186/s12943-020-01190-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Su R., Dong L., Li Y., et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 2020;38(1):79–96.e11. doi: 10.1016/j.ccell.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang L., Hui H., Agrawal K., et al. m6A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. The EMBO Journal. 2020;39(20, article e104514) doi: 10.15252/embj.2020104514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hou J., Zhang H., Liu J., et al. YTHDF2 reduction fuels inflammation and vascular abnormalization in hepatocellular carcinoma. Molecular Cancer. 2019;18(1):p. 163. doi: 10.1186/s12943-019-1082-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bayir H. Reactive oxygen species. Critical Care Medicine. 2005;33:S498–S501. doi: 10.1097/01.CCM.0000186787.64500.12. [DOI] [PubMed] [Google Scholar]
- 61.Bleier L., Wittig I., Heide H., Steger M., Brandt U., Dröse S. Generator-specific targets of mitochondrial reactive oxygen species. Free Radical Biology and Medicine. 2015;78:1–10. doi: 10.1016/j.freeradbiomed.2014.10.511. [DOI] [PubMed] [Google Scholar]
- 62.Brand M. D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radical Biology and Medicine. 2016;100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 63.Lambeth J. D., Neish A. S. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annual Review of Pathology: Mechanisms of Disease. 2014;9(1):119–145. doi: 10.1146/annurev-pathol-012513-104651. [DOI] [PubMed] [Google Scholar]
- 64.Jiang F., Zhang Y., Dusting G. J. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacological Reviews. 2011;63(1):218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 65.Li H. G., Horke S., Forstermann U. Oxidative stress in vascular disease and its pharmacological prevention. Trends in Pharmacological Sciences. 2013;34(6):313–319. doi: 10.1016/j.tips.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 66.Leung T. M., Nieto N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. Journal of Hepatology. 2013;58(2):395–398. doi: 10.1016/j.jhep.2012.08.018. [DOI] [PubMed] [Google Scholar]
- 67.Kuang F., Liu J., Tang D., Kang R. Oxidative damage and antioxidant defense in ferroptosis. Frontiers in Cell and Development Biology. 2020;8 doi: 10.3389/fcell.2020.586578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Buettner G. R. Superoxide dismutase in redox biology: the roles of superoxide and hydrogen peroxide. Anti-Cancer Agents in Medicinal Chemistry. 2011;11(4):341–346. doi: 10.2174/187152011795677544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kirkman H. N., Gaetani G. F. Mammalian catalase: a venerable enzyme with new mysteries. Trends in Biochemical Sciences. 2007;32(1):44–50. doi: 10.1016/j.tibs.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 70.Brigelius-Flohe R., Maiorino M. Glutathione peroxidases. Biochimica et Biophysica Acta. 2013;1830(5):3289–3303. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 71.Fujino G., Noguchi T., Takeda K., Ichijo H. Thioredoxin and protein kinases in redox signaling. Seminars in Cancer Biology. 2006;16(6):427–435. doi: 10.1016/j.semcancer.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 72.Detienne G., De Haes W., Mergan L., Edwards S. L., Temmerman L., Van Bael S. Beyond ROS clearance: peroxiredoxins in stress signaling and aging. Ageing Research Reviews. 2018;44:33–48. doi: 10.1016/j.arr.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 73.Lei X. G., Zhu J.-H., Cheng W.-H., et al. Paradoxical roles of antioxidant enzymes: basic mechanisms and health implications. Physiological Reviews. 2016;96(1):307–364. doi: 10.1152/physrev.00010.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Snezhkina A. V., Kudryavtseva A. V., Kardymon O. L., et al. ROS generation and antioxidant defense systems in normal and malignant cells. Oxidative Medicine and Cellular Longevity. 2019;2019:17. doi: 10.1155/2019/6175804.6175804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jang S., Kumar N., Beckwitt E. C., et al. Damage sensor role of UV-DDB during base excision repair. Nature Structural & Molecular Biology. 2019;26(8):695–703. doi: 10.1038/s41594-019-0261-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Weinberg F., Hamanaka R., Wheaton W. W., et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proceedings of the National Academy of Sciences. 2010;107(19):8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wise D. R., DeBerardinis R. J., Mancuso A., et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences. 2008;105(48):18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hu Y., Lu W., Chen G., et al. K-rasG12V transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Research. 2012;22(2):399–412. doi: 10.1038/cr.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Z. X., Pervaiz S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death and Differentiation. 2010;17(3):408–420. doi: 10.1038/cdd.2009.132. [DOI] [PubMed] [Google Scholar]
- 80.Igelmann S., Neubauer H. A., Ferbeyre G. STAT3 and STAT5 activation in solid cancers. Cancers (Basel) 2019;11(10, article 1428) doi: 10.3390/cancers11101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Park Y. H., Kim S. U., Lee B. K., et al. Prx I suppresses K-ras-driven lung tumorigenesis by opposing redox-sensitive ERK/cyclin D1 pathway. Antioxidants & Redox Signaling. 2013;19(5):482–496. doi: 10.1089/ars.2011.4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adachi Y., Shibai Y., Mitsushita J., Shang W. H., Hirose K., Kamata T. Oncogenic Ras upregulates NADPH oxidase 1 gene expression through MEK-ERK- dependent phosphorylation of GATA-6. Oncogene. 2008;27(36):4921–4932. doi: 10.1038/onc.2008.133. [DOI] [PubMed] [Google Scholar]
- 83.Cheng G., Diebold B. A., Hughes Y., Lambeth J. D. Nox1-dependent Reactive Oxygen Generation Is Regulated by Rac1. Journal of Biological Chemistry. 2006;281(26):17718–17726. doi: 10.1074/jbc.M512751200. [DOI] [PubMed] [Google Scholar]
- 84.Ueyama T., Geiszt M., Leto T. L. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Molecular and Cellular Biology. 2006;26(6):2160–2174. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kheradmand F., Werner E., Tremble P., Symons M., Werb Z. Role of Rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science. 1998;280(5365):898–902. doi: 10.1126/science.280.5365.898. [DOI] [PubMed] [Google Scholar]
- 86.Werner E., Werb Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. The Journal of Cell Biology. 2002;158(2):357–368. doi: 10.1083/jcb.200111028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vurusaner B., Poli G., Basaga H. Tumor suppressor genes and ROS: complex networks of interactions. Free Radical Biology & Medicine. 2012;52(1):7–18. doi: 10.1016/j.freeradbiomed.2011.09.035. [DOI] [PubMed] [Google Scholar]
- 88.Sampson N., Koziel R., Zenzmaier C., et al. ROS signaling by NOX4 drives fibroblast-to-myofibroblast differentiation in the diseased prostatic stroma. Molecular Endocrinology. 2011;25(3):503–515. doi: 10.1210/me.2010-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rojo de la Vega M., Chapman E., Zhang D. D. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34(1):21–43. doi: 10.1016/j.ccell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.DeNicola G. M., Karreth F. A., Humpton T. J., et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mitsuishi Y., Taguchi K., Kawatani Y., et al. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22(1):66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 92.Zimta A. A., Cenariu D., Irimie A., et al. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers (Basel) 2019;11(11):p. 1755. doi: 10.3390/cancers11111755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hayes J. D., McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends in Biochemical Sciences. 2009;34(4):176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 94.Nioi P., Hayes J. D. Contribution of NAD(P)H:quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutation Research. 2004;555(1-2):149–171. doi: 10.1016/j.mrfmmm.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 95.Bensaad K., Tsuruta A., Selak M. A., et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126(1):107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 96.Cheung E. C., Athineos D., Lee P., et al. TIGAR is required for efficient intestinal regeneration and tumorigenesis. Developmental Cell. 2013;25(5):463–477. doi: 10.1016/j.devcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aggarwal V., Tuli H. S., Varol A., et al. Role of reactive oxygen species in cancer progression: molecular mechanisms and recent advancements. Biomolecules. 2019;9(11):p. 735. doi: 10.3390/biom9110735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Prasad S., Gupta S. C., Tyagi A. K. Reactive oxygen species (ROS) and cancer: role of antioxidative nutraceuticals. Cancer Letters. 2017;387:95–105. doi: 10.1016/j.canlet.2016.03.042. [DOI] [PubMed] [Google Scholar]
- 99.Raza M. H., Siraj S., Arshad A., et al. ROS-modulated therapeutic approaches in cancer treatment. Journal of Cancer Research and Clinical Oncology. 2017;143(9):1789–1809. doi: 10.1007/s00432-017-2464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pelicano H., Carney D., Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resistance Updates. 2004;7(2):97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 101.Dizdaroglu M. Chemical determination of free radical-induced damage to DNA. Free Radical Biology & Medicine. 1991;10(3-4):225–242. doi: 10.1016/0891-5849(91)90080-M. [DOI] [PubMed] [Google Scholar]
- 102.Barnes R. P., Fouquerel E., Opresko P. L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mechanisms of Ageing and Development. 2019;177:37–45. doi: 10.1016/j.mad.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Srinivas U. S., Tan B. W. Q., Vellayappan B. A., Jeyasekharan A. D. ROS and the DNA damage response in cancer. Redox Biology. 2019;25, article 101084 doi: 10.1016/j.redox.2018.101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Poetsch A. R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Computational and Structural Biotechnology Journal. 2020;18:207–219. doi: 10.1016/j.csbj.2019.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bravard A., Vacher M., Gouget B., et al. Redox regulation of human OGG1 activity in response to cellular oxidative stress. Molecular and Cellular Biology. 2006;26(20):7430–7436. doi: 10.1128/MCB.00624-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sadikovic B., al-Romaih K., Squire J. A., Zielenska M. Cause and consequences of genetic and epigenetic alterations in human cancer. Current Genomics. 2008;9(6):394–408. doi: 10.2174/138920208785699580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liao Z. H., Chua D., Tan N. S. Reactive oxygen species: a volatile driver of field cancerization and metastasis. Molecular Cancer. 2019;18(1):p. 65. doi: 10.1186/s12943-019-0961-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lowy D. R., Willumsen B. M. Function and regulation of ras. Annual Review of Biochemistry. 1993;62(1):851–891. doi: 10.1146/annurev.bi.62.070193.004223. [DOI] [PubMed] [Google Scholar]
- 109.Prior I. A., Lewis P. D., Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Research. 2012;72(10):2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Irani K., Xia Y., Zweier J. L., et al. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science. 1997;275(5306):1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 111.Park M. T., Kim M. J., Suh Y., et al. Novel signaling axis for ROS generation during K-Ras-induced cellular transformation. Cell Death and Differentiation. 2014;21(8):1185–1197. doi: 10.1038/cdd.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ferino A., Rapozzi V., Xodo L. E. The ROS- KRAS -Nrf2 axis in the control of the redox homeostasis and the intersection with survival-apoptosis pathways: Implications for photodynamic therapy. Journal of Photochemistry and Photobiology B: Biology. 2020;202, article 111672 doi: 10.1016/j.jphotobiol.2019.111672. [DOI] [PubMed] [Google Scholar]
- 113.Tudek B., Winczura A., Janik J., Siomek A., Foksinski M., Oliński R. Involvement of oxidatively damaged DNA and repair in cancer development and aging. American Journal of Translational Research. 2010;2(3):254–284. [PMC free article] [PubMed] [Google Scholar]
- 114.Sablina A. A., Budanov A. V., Ilyinskaya G. V., Agapova L. S., Kravchenko J. E., Chumakov P. M. The antioxidant function of the p53 tumor suppressor. Nature Medicine. 2005;11(12):1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hayden M. S., Ghosh S. Shared Principles in NF-κB Signaling. Cell. 2008;132(3):344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 116.Vallabhapurapu S., Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annual Review of Immunology. 2009;27(1):693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 117.Morgan M. J., Liu Z. G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Research. 2011;21(1):103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Perkins N. D. The diverse and complex roles of NF-κB subunits in cancer. Nature Reviews. Cancer. 2012;12(2):121–132. doi: 10.1038/nrc3204. [DOI] [PubMed] [Google Scholar]
- 119.Kawauchi K., Araki K., Tobiume K., Tanaka N. p53 regulates glucose metabolism through an IKK-NF-κB pathway and inhibits cell transformation. Nature Cell Biology. 2008;10(5):611–618. doi: 10.1038/ncb1724. [DOI] [PubMed] [Google Scholar]
- 120.Taniguchi K., Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nature Reviews. Immunology. 2018;18(5):309–324. doi: 10.1038/nri.2017.142. [DOI] [PubMed] [Google Scholar]
- 121.Quail D. F., Joyce J. A. Microenvironmental regulation of tumor progression and metastasis. Nature Medicine. 2013;19(11):1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huang Q. C., Zhan L., Cao H. Y., et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy. 2016;12(6):999–1014. doi: 10.1080/15548627.2016.1166318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Myant K. B., Cammareri P., McGhee E. J., et al. ROS Production and NF-κB Activation Triggered by RAC1 Facilitate WNT-Driven Intestinal Stem Cell Proliferation and Colorectal Cancer Initiation. Cell Stem Cell. 2013;12(6):761–773. doi: 10.1016/j.stem.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Luo J., Manning B. D., Cantley L. C. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4(4):257–262. doi: 10.1016/S1535-6108(03)00248-4. [DOI] [PubMed] [Google Scholar]
- 125.Stambolic V., Suzuki A., de la Pompa J. L., et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95(1):29–39. doi: 10.1016/S0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 126.Lee S. R., Yang K. S., Kwon J., Lee C., Jeong W., Rhee S. G. Reversible Inactivation of the Tumor Suppressor PTEN by H2O2. The Journal of Biological Chemistry. 2002;277(23):20336–20342. doi: 10.1074/jbc.M111899200. [DOI] [PubMed] [Google Scholar]
- 127.Kwon J., Lee S. R., Yang K. S., et al. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(47):16419–16424. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang Y., Park J., Han S. J., et al. Redox regulation of tumor suppressor PTEN in cell signaling. Redox Biology. 2020;34, article 101553 doi: 10.1016/j.redox.2020.101553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Salmeen A., Andersen J. N., Myers M. P., et al. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl- amide intermediate. Nature. 2003;423(6941):769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 130.Lee Y. R., Chen M., Pandolfi P. P. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nature Reviews. Molecular Cell Biology. 2018;19(9):547–562. doi: 10.1038/s41580-018-0015-0. [DOI] [PubMed] [Google Scholar]
- 131.Julien S. G., Dubé N., Read M., et al. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nature Genetics. 2007;39(3):338–346. doi: 10.1038/ng1963. [DOI] [PubMed] [Google Scholar]
- 132.Meuillet E. J., Mahadevan D., Berggren M., Coon A., Powis G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN's lipid phosphatase activity and membrane binding: a mechanism for the functional loss of PTEN's tumor suppressor activity. Archives of Biochemistry and Biophysics. 2004;429(2):123–133. doi: 10.1016/j.abb.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 133.Lee S., Kim S. M., Lee R. T. Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxidants & Redox Signaling. 2013;18(10):1165–1207. doi: 10.1089/ars.2011.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rhee S. G. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312(5782):1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 135.Cao J., Schulte J., Knight A., et al. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. The EMBO Journal. 2009;28(10):1505–1517. doi: 10.1038/emboj.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Peluso I., Yarla N. S., Ambra R., Pastore G., Perry G. MAPK signalling pathway in cancers: olive products as cancer preventive and therapeutic agents. Seminars in Cancer Biology. 2019;56:185–195. doi: 10.1016/j.semcancer.2017.09.002. [DOI] [PubMed] [Google Scholar]
- 137.Saitoh M., Nishitoh H., Fujii M., et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. The EMBO Journal. 1998;17(9):2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kamata H., Honda S., Maeda S., Chang L., Hirata H., Karin M. Reactive Oxygen Species Promote TNFα-Induced Death and Sustained JNK Activation by Inhibiting MAP Kinase Phosphatases. Cell. 2005;120(5):649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 139.Yilmaz M., Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Reviews. 2009;28(1-2):15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- 140.Wang Z., Li Y., Sarkar F. H. Signaling mechanism(s) of reactive oxygen species in epithelial-mesenchymal transition reminiscent of cancer stem cells in tumor progression. Current Stem Cell Research & Therapy. 2010;5(1):74–80. doi: 10.2174/157488810790442813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mandal M., Rajput M., Anura A., Pathak T., Chatterjee J. Regulation of epithelial mesenchymal transition under compliant polydimethylsiloxane substrate. Biophysical Journal. 2019;116(3):p. 549a. doi: 10.1016/j.bpj.2018.11.2954. [DOI] [Google Scholar]
- 142.Tobar N., Villar V., Santibanez J. F. ROS-NFkappaB mediates TGF-beta1-induced expression of urokinase-type plasminogen activator, matrix metalloproteinase-9 and cell invasion. Molecular and Cellular Biochemistry. 2010;340(1-2):195–202. doi: 10.1007/s11010-010-0418-5. [DOI] [PubMed] [Google Scholar]
- 143.Lam C. R., Tan C., Teo Z., et al. Loss of TAK1 increases cell traction force in a ROS-dependent manner to drive epithelial-mesenchymal transition of cancer cells. Cell Death & Disease. 2013;4(10, article e848) doi: 10.1038/cddis.2013.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Luo D. C., Wang J. X., Li J., Post M. Mouse snail is a target gene for HIF. Molecular Cancer Research. 2011;9(2):234–245. doi: 10.1158/1541-7786.MCR-10-0214. [DOI] [PubMed] [Google Scholar]
- 145.Bao B., Azmi A. S., Ali S., et al. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2012;1826(2):272–296. doi: 10.1016/j.bbcan.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yang S. W., Zhang Z. G., Hao Y. X., et al. HIF-1α induces the epithelial-mesenchymal transition in gastric cancer stem cells through the Snail pathway. Oncotarget. 2017;8(6):9535–9545. doi: 10.18632/oncotarget.14484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Guzy R. D., Hoyos B., Robin E., et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metabolism. 2005;1(6):401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 148.Koshikawa N., Hayashi J., Nakagawara A., Takenaga K. Reactive Oxygen Species-generating Mitochondrial DNA Mutation Up-regulates Hypoxia-inducible Factor-1α Gene Transcription via Phosphatidylinositol 3-Kinase-Akt/Protein Kinase C/Histone Deacetylase Pathway. The Journal of Biological Chemistry. 2009;284(48):33185–33194. doi: 10.1074/jbc.M109.054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.du J., Xu R., Hu Z., et al. PI3K and ERK-induced Rac1 activation mediates hypoxia-induced HIF-1α expression in MCF-7 breast cancer cells. PLoS One. 2011;6(9, article e25213) doi: 10.1371/journal.pone.0025213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Haddad J. J. Redox regulation of pro-inflammatory cytokines and IκB-α/NF-κB nuclear translocation and activation. Biochemical and Biophysical Research Communications. 2002;296(4):847–856. doi: 10.1016/S0006-291X(02)00947-6. [DOI] [PubMed] [Google Scholar]
- 151.Westra J., Brouwer E., Bos R., et al. Regulation of cytokine-induced HIF-1 Expression in rheumatoid synovial fibroblasts. Annals of the New York Academy of Sciences. 2007;1108(1):340–348. doi: 10.1196/annals.1422.035. [DOI] [PubMed] [Google Scholar]
- 152.Huang C., Han Y., Wang Y. M., et al. SENP3 is responsible for HIF-1 transactivation under mild oxidative stress via p300 de-SUMOylation. EMBO Journal. 2009;28(18):2748–2762. doi: 10.1038/emboj.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kaelin W. G., Jr., Ratcliffe P. J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Molecular Cell. 2008;30(4):393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 154.Brunelle J. K., Bell E. L., Quesada N. M., et al. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metabolism. 2005;1(6):409–414. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 155.Gerald D., Berra E., Frapart Y. M., et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118(6):781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 156.Cheung E. C., DeNicola G. M., Nixon C., et al. Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell. 2020;37(2):168–182.e4. doi: 10.1016/j.ccell.2019.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Condeelis J., Pollard J. W. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 158.Zhang Y., Choksi S., Chen K., Pobezinskaya Y., Linnoila I., Liu Z. G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Research. 2013;23(7):898–914. doi: 10.1038/cr.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dai E., Han L., Liu J., et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16(11):2069–2083. doi: 10.1080/15548627.2020.1714209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nieto M. A., Huang R. Y., Jackson R. A., Thiery J. P. Emt: 2016. Cell. 2016;166(1):21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 161.Sampson N., Brunner E., Weber A., et al. Inhibition of Nox4-dependent ROS signaling attenuates prostate fibroblast activation and abrogates stromal-mediated protumorigenic interactions. International Journal of Cancer. 2018;143(2):383–395. doi: 10.1002/ijc.31316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Toullec A., Gerald D., Despouy G., et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Molecular Medicine. 2010;2(6):211–230. doi: 10.1002/emmm.201000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Joyce J. A., Pollard J. W. Microenvironmental regulation of metastasis. Nature Reviews. Cancer. 2009;9(4):239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sceneay J., Parker B. S., Smyth M. J., Möller A. Hypoxia-driven immunosuppression contributes to the pre-metastatic niche. Oncoimmunology. 2013;2(1, article e22355) doi: 10.4161/onci.22355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Harmon C., Robinson M. W., Hand F., et al. Lactate-mediated acidification of tumor microenvironment induces apoptosis of liver-resident NK cells in colorectal liver metastasis. Cancer Immunology Research. 2019;7(2):335–346. doi: 10.1158/2326-6066.CIR-18-0481. [DOI] [PubMed] [Google Scholar]
- 166.Hengartner M. O. The biochemistry of apoptosis. Nature. 2000;407(6805):770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 167.Burke P. J. Mitochondria, bioenergetics and apoptosis in cancer. Trends in Cancer. 2017;3(12):857–870. doi: 10.1016/j.trecan.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Dorstyn L., Akey C. W., Kumar S. New insights into apoptosome structure and function. Cell Death and Differentiation. 2018;25(7):1194–1208. doi: 10.1038/s41418-017-0025-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kagan V. E., Tyurin V. A., Jiang J. F., et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nature Chemical Biology. 2005;1(4):223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 170.Orrenius S., Gogvadze A., Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annual Review of Pharmacology. 2007;47(1):143–183. doi: 10.1146/annurev.pharmtox.47.120505.105122. [DOI] [PubMed] [Google Scholar]
- 171.Enoksson M., Fernandes A. P., Prast S., Lillig C. H., Holmgren A., Orrenius S. Overexpression of glutaredoxin 2 attenuates apoptosis by preventing cytochrome c release. Biochemical and Biophysical Research Communications. 2005;327(3):774–779. doi: 10.1016/j.bbrc.2004.12.067. [DOI] [PubMed] [Google Scholar]
- 172.Kalkavan H., Green D. R. MOMP, cell suicide as a BCL-2 family business. Cell Death and Differentiation. 2018;25(1):46–55. doi: 10.1038/cdd.2017.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Pan J. S., Hong M. Z., Ren J. L. Reactive oxygen species: a double-edged sword in oncogenesis. World Journal of Gastroenterology. 2009;15(14):1702–1707. doi: 10.3748/wjg.15.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zuo Y., Xiang B. G., Yang J., et al. Oxidative modification of caspase-9 facilitates its activation via disulfide-mediated interaction with Apaf-1. Cell Research. 2009;19(4):449–457. doi: 10.1038/cr.2009.19. [DOI] [PubMed] [Google Scholar]
- 175.Kiraz Y., Adan A., Kartal Yandim M., Baran Y. Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biology. 2016;37(7):8471–8486. doi: 10.1007/s13277-016-5035-9. [DOI] [PubMed] [Google Scholar]
- 176.Safa A. R. c-FLIP, a master anti-apoptotic regulator. Experimental Oncology. 2012;34(3):176–184. [PMC free article] [PubMed] [Google Scholar]
- 177.Wilkie-Grantham R. P., Matsuzawa S., Reed J. C. Novel Phosphorylation and Ubiquitination Sites Regulate Reactive Oxygen Species-dependent Degradation of Anti-apoptotic c-FLIP Protein. The Journal of Biological Chemistry. 2013;288(18):12777–12790. doi: 10.1074/jbc.M112.431320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhou W., Yuan J. Y. Necroptosis in health and diseases. Seminars in Cell & Developmental Biology. 2014;35:14–23. doi: 10.1016/j.semcdb.2014.07.013. [DOI] [PubMed] [Google Scholar]
- 179.Feltham R., Silke J. The small molecule that packs a punch: ubiquitin-mediated regulation of RIPK1/FADD/caspase-8 complexes. Cell Death and Differentiation. 2017;24(7):1196–1204. doi: 10.1038/cdd.2017.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Choi M. E., Price D. R., Ryter S. W., Choi A. M. K. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight. 2019;4(15) doi: 10.1172/jci.insight.128834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Cai Z. Y., Jitkaew S., Zhao J., et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nature Cell Biology. 2014;16(1):55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sun L. M., Wang H. Y., Wang Z. G., et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1-2):213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 183.Su Z. Y., Yang Z. Z., Xu Y. Q., Chen Y., Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Molecular Cancer. 2015;14(1):p. 48. doi: 10.1186/s12943-015-0321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fu Z. Z., Deng B. Y., Liao Y. X., et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer. 2013;13(1) doi: 10.1186/1471-2407-13-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ogretmen B. Sphingolipid metabolism in cancer signalling and therapy. Nature Reviews Cancer. 2018;18(1):33–50. doi: 10.1038/nrc.2017.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hsu S. K., Chang W. T., Lin I. L., et al. The role of necroptosis in ROS-mediated cancer therapies and its promising applications. Cancers. 2020;12(8):p. 2185. doi: 10.3390/cancers12082185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhang Y., Su S. S., Zhao S., et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nature Communications. 2017;8 doi: 10.1038/ncomms14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lu B., Gong X., Wang Z. Q., et al. Shikonin induces glioma cell necroptosis in vitro by ROS overproduction and promoting RIP1/RIP3 necrosome formation. Acta Pharmacologica Sinica. 2017;38(11):1543–1553. doi: 10.1038/aps.2017.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Vanlangenakker N., vanden Berghe T., Bogaert P., et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death and Differentiation. 2011;18(4):656–665. doi: 10.1038/cdd.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kim Y. S., Morgan M. J., Choksi S., Liu Z. G. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Molecular Cell. 2007;26(5):675–687. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 191.Yang J., Carroll K. S., Liebler D. C. The Expanding Landscape of the Thiol Redox Proteome. Molecular & Cellular Proteomics. 2016;15(1):1–11. doi: 10.1074/mcp.O115.056051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Stockwell B. R., Friedmann Angeli J. P., Bayir H., et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Jiang L., Kon N., Li T., et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sui X. B., Zhang R. N., Liu S. P., et al. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Frontiers in Pharmacology. 2018;9 doi: 10.3389/fphar.2018.01371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Li Y., Chen F., Chen J., et al. Disulfiram/copper induces antitumor activity against both nasopharyngeal cancer cells and cancer-associated fibroblasts through ROS/MAPK and ferroptosis pathways. Cancers. 2020;12(1):p. 138. doi: 10.3390/cancers12010138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Luo M. Y., Wu L. F., Zhang K. X., et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death and Differentiation. 2018;25(8):1457–1472. doi: 10.1038/s41418-017-0053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Badgley M. A., Kremer D. M., Maurer H. C., et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368(6486):85–89. doi: 10.1126/science.aaw9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhang J., Yang J., Zuo T. T., et al. Heparanase-driven sequential released nanoparticles for ferroptosis and tumor microenvironment modulations synergism in breast cancer therapy. Biomaterials. 2021;266, article 120429 doi: 10.1016/j.biomaterials.2020.120429. [DOI] [PubMed] [Google Scholar]
- 199.Cózar B., Greppi M., Carpentier S., Narni-Mancinelli E., Chiossone L., Vivier E. Tumor-infiltrating natural killer cells. Cancer Discovery. 2021;11(1):34–44. doi: 10.1158/2159-8290.CD-20-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Lo Presti E., Dieli F., Fourniè J. J., Meraviglia S. Deciphering human γδ T cell response in cancer: lessons from tumor-infiltrating γδ T cells. Immunological Reviews. 2020;298(1):153–164. doi: 10.1111/imr.12904. [DOI] [PubMed] [Google Scholar]
- 201.Franchina D. G., Dostert C., Brenner D. Reactive oxygen species: involvement in T cell signaling and metabolism. Trends in Immunology. 2018;39(6):489–502. doi: 10.1016/j.it.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 202.Lin W., Shen P., Song Y., Huang Y., Tu S. Reactive oxygen species in autoimmune cells: function, differentiation, and metabolism. Frontiers in Immunology. 2021;12, article 635021 doi: 10.3389/fimmu.2021.635021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nature Immunology. 2002;3(12):1129–1134. doi: 10.1038/ni1202-1129. [DOI] [PubMed] [Google Scholar]
- 204.Forman H. J., Torres M. Reactive oxygen species and cell signaling: respiratory burst in macrophage signaling. American Journal of Respiratory and Critical Care Medicine. 2002;166(12, Part 2):S4–S8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- 205.Peng H., Chen B., Huang W., et al. Reprogramming tumor-associated macrophages to reverse EGFRT790MResistance by dual-targeting codelivery of gefitinib/vorinostat. Nano Letters. 2017;17(12):7684–7690. doi: 10.1021/acs.nanolett.7b03756. [DOI] [PubMed] [Google Scholar]
- 206.Mensurado S., Rei M., Lança T., et al. Tumor-associated neutrophils suppress pro-tumoral IL-17+ γδ T cells through induction of oxidative stress. PLoS Biology. 2018;16(5, article e2004990) doi: 10.1371/journal.pbio.2004990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Tardu M., Jones J. D., Kennedy R. T., Lin Q., Koutmou K. S. Identification and quantification of modified nucleosides inSaccharomyces cerevisiaemRNAs. ACS Chemical Biology. 2019;14(7):1403–1409. doi: 10.1021/acschembio.9b00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Chen H. Y., Zhao T. H., Sun D. L., Wu M., Zhang Z. Changes of RNA N6-methyladenosine in the hormesis effect induced by arsenite on human keratinocyte cells. Toxicology in Vitro. 2019;56:84–92. doi: 10.1016/j.tiv.2019.01.010. [DOI] [PubMed] [Google Scholar]
- 209.Qu T., Mou Y., Dai J., et al. Changes and relationship of N6-methyladenosine modification and long non-coding RNAs in oxidative damage induced by cadmium in pancreatic β-cells. Toxicology Letters. 2021;343:56–66. doi: 10.1016/j.toxlet.2021.02.014. [DOI] [PubMed] [Google Scholar]
- 210.Fuhrmann D. C., Brüne B. Mitochondrial composition and function under the control of hypoxia. Redox Biology. 2017;12:208–215. doi: 10.1016/j.redox.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wang Y. J., Yang B., Lai Q., et al. Reprogramming of m6A epitranscriptome is crucial for shaping of transcriptome and proteome in response to hypoxia. RNA Biology. 2021;18(1):131–143. doi: 10.1080/15476286.2020.1804697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhang C., Samanta D., Lu H., et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proceedings of the National Academy of Sciences of the United States of America. 2016;113(14):E2047–E2056. doi: 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Yu F., Wei J., Cui X., et al. Post-translational modification of RNA m6A demethylase ALKBH5 regulates ROS-induced DNA damage response. Nucleic Acids Research. 2021;49(10):5779–5797. doi: 10.1093/nar/gkab415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhong X., Yu J., Frazier K., et al. Circadian Clock Regulation of Hepatic Lipid Metabolism by Modulation of m6A mRNA Methylation. Cell Reports. 2018;25(7):1816–1828.e4. doi: 10.1016/j.celrep.2018.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Shi Y., Fan S., Wu M., et al. YTHDF1 links hypoxia adaptation and non-small cell lung cancer progression. Nature Communications. 2019;10(1, article 4892) doi: 10.1038/s41467-019-12801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Buchan J. R., Parker R. Eukaryotic stress granules: the ins and outs of translation. Molecular Cell. 2009;36(6):932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kedersha N., Ivanov P., Anderson P. Stress granules and cell signaling: more than just a passing phase? Trends in Biochemical Sciences. 2013;38(10):494–506. doi: 10.1016/j.tibs.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Jain S., Wheeler J. R., Walters R. W., Agrawal A., Barsic A., Parker R. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell. 2016;164(3):487–498. doi: 10.1016/j.cell.2015.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Anders M., Chelysheva I., Goebel I., et al. Dynamic m6A methylation facilitates mRNA triaging to stress granules. Life Science Alliance. 2018;1(4):p. e201800113. doi: 10.26508/lsa.201800113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Huang H., Weng H., Zhou K., et al. Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature. 2019;567(7748):414–419. doi: 10.1038/s41586-019-1016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.García-Giménez J. L., Garcés C., Romá-Mateo C., Pallardó F. V. Oxidative stress-mediated alterations in histone post-translational modifications. Free Radical Biology & Medicine. 2021;170:6–18. doi: 10.1016/j.freeradbiomed.2021.02.027. [DOI] [PubMed] [Google Scholar]
- 222.Hernansanz-Agustín P., Enríquez J. A. Generation of reactive oxygen species by mitochondria. Antioxidants (Basel) 2021;10(3) doi: 10.3390/antiox10030415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Alarcón C. R., Lee H., Goodarzi H., Halberg N., Tavazoie S. F. N 6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519(7544):482–485. doi: 10.1038/nature14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Knuckles P., Carl S. H., Musheev M., Niehrs C., Wenger A., Bühler M. RNA fate determination through cotranscriptional adenosine methylation and microprocessor binding. Nature Structural & Molecular Biology. 2017;24(7):561–569. doi: 10.1038/nsmb.3419. [DOI] [PubMed] [Google Scholar]
- 225.Sun L., Wan A., Zhou Z., et al. RNA-binding protein RALY reprogrammes mitochondrial metabolism via mediating miRNA processing in colorectal cancer. Gut. 2021;70(9):1698–1712. doi: 10.1136/gutjnl-2020-320652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Liu X., Gonzalez G., Dai X., et al. Adenylate Kinase 4 Modulates the Resistance of Breast Cancer Cells to Tamoxifen through an m6A-Based Epitranscriptomic Mechanism. Molecular Therapy. 2020;28(12):2593–2604. doi: 10.1016/j.ymthe.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zhuang C., Zhuang C., Luo X., et al. N6-methyladenosine demethylase FTO suppresses clear cell renal cell carcinoma through a novel FTO-PGC-1α signalling axis. Journal of Cellular and Molecular Medicine. 2019;23(3):2163–2173. doi: 10.1111/jcmm.14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.du Y. D., Guo W. Y., Han C. H., et al. N6-methyladenosine demethylase FTO impairs hepatic ischemia-reperfusion injury via inhibiting Drp1-mediated mitochondrial fragmentation. Cell Death & Disease. 2021;12(5):p. 442. doi: 10.1038/s41419-021-03622-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Li Q., Li X., Tang H., et al. NSUN2-mediated m5C methylation and METTL3/METTL14-mediated m6A methylation cooperatively enhance p21 translation. Journal of Cellular Biochemistry. 2017;118(9):2587–2598. doi: 10.1002/jcb.25957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zhao T. X., Wang J. K., Shen L. J., et al. Increased m6A RNA modification is related to the inhibition of the Nrf2-mediated antioxidant response in di-(2-ethylhexyl) phthalate-induced prepubertal testicular injury. Environmental Pollution. 2020;259, article 113911 doi: 10.1016/j.envpol.2020.113911. [DOI] [PubMed] [Google Scholar]
- 231.Habib E., Linher-Melville K., Lin H. X., Singh G. Expression of xCT and activity of system xc− are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biology. 2015;5:33–42. doi: 10.1016/j.redox.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Ottestad-Hansen S., Hu Q. X., Follin-Arbelet V. V., et al. The cystine-glutamate exchanger (xCT, Slc7a11) is expressed in significant concentrations in a subpopulation of astrocytes in the mouse brain. Glia. 2018;66(5):951–970. doi: 10.1002/glia.23294. [DOI] [PubMed] [Google Scholar]
- 233.Ma L., Chen T., Zhang X., et al. The m6A reader YTHDC2 inhibits lung adenocarcinoma tumorigenesis by suppressing SLC7A11-dependent antioxidant function. Redox Biology. 2021;38, article 101801 doi: 10.1016/j.redox.2020.101801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Amodio N., Stamato M. A., Juli G., et al. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia. 2018;32(9):1948–1957. doi: 10.1038/s41375-018-0067-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kerk S. A., Papagiannakopoulos T., Shah Y. M., Lyssiotis C. A. Metabolic networks in mutant KRAS-driven tumours: tissue specificities and the microenvironment. Nature Reviews. Cancer. 2021;21(8):510–525. doi: 10.1038/s41568-021-00375-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Shi H., Zhao J., Han L., et al. Retrospective study of gene signatures and prognostic value of m6A regulatory factor in non-small cell lung cancer using TCGA database and the verification of FTO. Aging (Albany NY) 2020;12(17):17022–17037. doi: 10.18632/aging.103622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Zhong L., Liao D., Zhang M., et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Letters. 2019;442:252–261. doi: 10.1016/j.canlet.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 238.Shen S., Faouzi S., Bastide A., et al. An epitranscriptomic mechanism underlies selective mRNA translation remodelling in melanoma persister cells. Nature Communications. 2019;10(1, article 5713) doi: 10.1038/s41467-019-13360-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Lee H., Bao S., Qian Y., et al. Stage-specific requirement for Mettl3 -dependent m6A mRNA methylation during haematopoietic stem cell differentiation. Nature Cell Biology. 2019;21(6):700–709. doi: 10.1038/s41556-019-0318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Yang X., Shao F., Guo D., et al. WNT/β-catenin-suppressed FTO expression increases m6A of c-Myc mRNA to promote tumor cell glycolysis and tumorigenesis. Cell Death & Disease. 2021;12(5):p. 462. doi: 10.1038/s41419-021-03739-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Dixit D., Prager B. C., Gimple R. C., et al. The RNA m6A reader YTHDF2 maintains oncogene expression and is a targetable dependency in glioblastoma stem cells. Cancer Discovery. 2021;11(2):480–499. doi: 10.1158/2159-8290.CD-20-0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zhu S., Wang J. Z., Chen D., et al. An oncopeptide regulates m6A recognition by the m6A reader IGF2BP1 and tumorigenesis. Nature Communications. 2020;11(1, article 1685) doi: 10.1038/s41467-020-15403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Qian X., Yang J., Qiu Q., et al. LCAT3, a novel m6A-regulated long non-coding RNA, plays an oncogenic role in lung cancer via binding with FUBP1 to activate c-MYC. Journal of Hematology & Oncology. 2021;14(1):p. 112. doi: 10.1186/s13045-021-01123-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Vu L. P., Pickering B. F., Cheng Y., et al. The N 6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nature Medicine. 2017;23(11):1369–1376. doi: 10.1038/nm.4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Zhang Y., Liu S., Zhao T., Dang C. METTL3-mediated m6A modification of Bcl-2 mRNA promotes non-small cell lung cancer progression. Oncology Reports. 2021;46(2) doi: 10.3892/or.2021.8114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Wang H., Xu B., Shi J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene. 2020;722, article 144076 doi: 10.1016/j.gene.2019.144076. [DOI] [PubMed] [Google Scholar]
- 247.Ladelfa M. F., Toledo M. F., Laiseca J. E., Monte M. Interaction of p53 with tumor suppressive and oncogenic signaling pathways to control cellular reactive oxygen species production. Antioxidants & Redox Signaling. 2011;15(6):1749–1761. doi: 10.1089/ars.2010.3652. [DOI] [PubMed] [Google Scholar]
- 248.Liu D., Xu Y. p53, oxidative stress, and aging. Antioxidants & Redox Signaling. 2011;15(6):1669–1678. doi: 10.1089/ars.2010.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Uddin M. B., Roy K. R., Hosain S. B., et al. An N6 -methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochemical Pharmacology. 2019;160:134–145. doi: 10.1016/j.bcp.2018.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Zhao T., Sun D., Zhao M., Lai Y., Liu Y., Zhang Z. N6-methyladenosine mediates arsenite-induced human keratinocyte transformation by suppressing p53 activation. Environmental Pollution. 2020;259, article 113908 doi: 10.1016/j.envpol.2019.113908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Xia J., Xu H., Zhang X., et al. Multiple myeloma tumor cells are selectively killed by pharmacologically-dosed ascorbic acid. eBioMedicine. 2017;18:41–49. doi: 10.1016/j.ebiom.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Espey M. G., Chen P., Chalmers B., et al. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Radical Biology & Medicine. 2011;50(11):1610–1619. doi: 10.1016/j.freeradbiomed.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ma Y., Chapman J., Levine M., Polireddy K., Drisko J., Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Science Translational Medicine. 2014;6(222, article 222ra218) doi: 10.1126/scitranslmed.3007154. [DOI] [PubMed] [Google Scholar]
- 254.Magrì A., Germano G., Lorenzato A., et al. High-dose vitamin C enhances cancer immunotherapy. Science Translational Medicine. 2020;12(532):p. eaay8707. doi: 10.1126/scitranslmed.aay8707. [DOI] [PubMed] [Google Scholar]
- 255.Singh S., Singh P. P., Roberts L. R., Sanchez W. Chemopreventive strategies in hepatocellular carcinoma. Nature Reviews. Gastroenterology & Hepatology. 2014;11(1):45–54. doi: 10.1038/nrgastro.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Thompson I. M., Jr., Cabang A. B., Wargovich M. J. Future directions in the prevention of prostate cancer. Nature Reviews. Clinical Oncology. 2014;11(1):49–60. doi: 10.1038/nrclinonc.2013.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Dandawate P. R., Subramaniam D., Jensen R. A., Anant S. Targeting cancer stem cells and signaling pathways by phytochemicals: novel approach for breast cancer therapy. Seminars in Cancer Biology. 2016;40-41:192–208. doi: 10.1016/j.semcancer.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Milkovic L., Zarkovic N., Saso L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biology. 2017;12:727–732. doi: 10.1016/j.redox.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Breau M., Houssaini A., Lipskaia L., et al. The antioxidant N-acetylcysteine protects from lung emphysema but induces lung adenocarcinoma in mice. JCI Insight. 2019;4(19) doi: 10.1172/jci.insight.127647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Sayin V. I., Ibrahim M. X., Larsson E., Nilsson J. A., Lindahl P., Bergo M. O. Antioxidants accelerate lung cancer progression in mice. Science Translational Medicine. 2014;6(221, article 221ra215) doi: 10.1126/scitranslmed.3007653. [DOI] [PubMed] [Google Scholar]
- 261.Klein E. A., Thompson I. M., Jr., Tangen C. M., et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT) JAMA. 2011;306(14):1549–1556. doi: 10.1001/jama.2011.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Miller WH Jr, Schipper H. M., Lee J. S., Singer J., Waxman S. Mechanisms of action of arsenic trioxide. Cancer Research. 2002;62(14):3893–3903. [PubMed] [Google Scholar]
- 263.Hwang I. T., Chung Y. M., Kim J. J., et al. Drug resistance to 5-FU linked to reactive oxygen species modulator 1. Biochemical and Biophysical Research Communications. 2007;359(2):304–310. doi: 10.1016/j.bbrc.2007.05.088. [DOI] [PubMed] [Google Scholar]
- 264.Havaki S., Kotsinas A., Chronopoulos E., Kletsas D., Georgakilas A., Gorgoulis V. G. The role of oxidative DNA damage in radiation induced bystander effect. Cancer Letters. 2015;356(1):43–51. doi: 10.1016/j.canlet.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 265.Montero A. J., Jassem J. Cellular redox pathways as a therapeutic target in the treatment of cancer. Drugs. 2011;71(11):1385–1396. doi: 10.2165/11592590-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 266.Townsend D. M., He L., Hutchens S., Garrett T. E., Pazoles C. J., Tew K. D. NOV-002, a glutathione disulfide mimetic, as a modulator of cellular redox balance. Cancer Research. 2008;68(8):2870–2877. doi: 10.1158/0008-5472.CAN-07-5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Ehrenfeld V., Fulda S. Thioredoxin inhibitor PX-12 induces mitochondria-mediated apoptosis in acute lymphoblastic leukemia cells. Biological Chemistry. 2020;401(2):273–283. doi: 10.1515/hsz-2019-0160. [DOI] [PubMed] [Google Scholar]
- 268.Li G. Z., Liang H. F., Liao B., et al. PX-12 inhibits the growth of hepatocelluar carcinoma by inducing S-phase arrest, ROS-dependent apoptosis and enhances 5-FU cytotoxicity. American Journal of Translational Research. 2015;7(9):1528–1540. [PMC free article] [PubMed] [Google Scholar]
- 269.Ramanathan R. K., Kirkpatrick D. L., Belani C. P., et al. A Phase I pharmacokinetic and pharmacodynamic study of PX-12, a novel inhibitor of thioredoxin-1, in patients with advanced solid tumors. Clinical Cancer Research. 2007;13(7):2109–2114. doi: 10.1158/1078-0432.CCR-06-2250. [DOI] [PubMed] [Google Scholar]
- 270.Mangla B., Javed S., Sultan M. H., et al. Sulforaphane: a review of its therapeutic potentials, advances in its nanodelivery, recent patents, and clinical trials. Phytotherapy Research. 2021 doi: 10.1002/ptr.7176. [DOI] [PubMed] [Google Scholar]
- 271.Lewinska A., Adamczyk-Grochala J., Deregowska A., Wnuk M. Sulforaphane-induced cell cycle arrest and senescence are accompanied by DNA hypomethylation and changes in microRNA profile in breast cancer cells. Theranostics. 2017;7(14):3461–3477. doi: 10.7150/thno.20657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kao Y. H., Hiipakka R. A., Liao S. Modulation of endocrine systems and food intake by green tea epigallocatechin gallate. Endocrinology. 2000;141(3):980–987. doi: 10.1210/endo.141.3.7368. [DOI] [PubMed] [Google Scholar]
- 273.Wu R., Yao Y., Jiang Q., et al. Epigallocatechin gallate targets FTO and inhibits adipogenesis in an mRNA m6A-YTHDF2-dependent manner. International Journal of Obesity. 2018;42(7):1378–1388. doi: 10.1038/s41366-018-0082-5. [DOI] [PubMed] [Google Scholar]
- 274.Patra S., Pradhan B., Nayak R., et al. Chemotherapeutic efficacy of curcumin and resveratrol against cancer: chemoprevention, chemoprotection, drug synergism and clinical pharmacokinetics. Seminars in Cancer Biology. 2021;73:310–320. doi: 10.1016/j.semcancer.2020.10.010. [DOI] [PubMed] [Google Scholar]
- 275.Gan Z., Wei W., Wu J., et al. Resveratrol and curcumin improve intestinal mucosal integrity and decrease m6A RNA methylation in the intestine of weaning piglets. ACS Omega. 2019;4(17):17438–17446. doi: 10.1021/acsomega.9b02236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Wu J., Gan Z., Zhuo R., Zhang L., Wang T., Zhong X. Resveratrol attenuates aflatoxin B(1)-induced ROS formation and increase of m(6)A RNA methylation. Animals (Basel) 2020;10(4) doi: 10.3390/ani10040677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Cai X., Wang X., Cao C., et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Letters. 2018;415:11–19. doi: 10.1016/j.canlet.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 278.Han J., Wang J. Z., Yang X., et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Molecular Cancer. 2019;18(1):p. 110. doi: 10.1186/s12943-019-1036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Xie H., Li J., Ying Y., et al. METTL3/YTHDF2 m6A axis promotes tumorigenesis by degrading SETD7 and KLF4 mRNAs in bladder cancer. Journal of Cellular and Molecular Medicine. 2020;24(7):4092–4104. doi: 10.1111/jcmm.15063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Li T., Hu P. S., Zuo Z., et al. METTL3 facilitates tumor progression via an m6A-IGF2BP2-dependent mechanism in colorectal carcinoma. Molecular Cancer. 2019;18(1):p. 112. doi: 10.1186/s12943-019-1038-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Deng R., Cheng Y. K., Ye S. B., et al. m6A methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. Oncotargets and Therapy. 2019;12:4391–4402. doi: 10.2147/OTT.S201052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Li F. X., Yi Y., Miao Y. Y., et al. N6-Methyladenosine modulates nonsense-mediated mRNA decay in human glioblastoma. Cancer Research. 2019;79(22):5785–5798. doi: 10.1158/0008-5472.CAN-18-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Tassinari V., Cesarini V., Tomaselli S., et al. ADAR1 is a new target of METTL3 and plays a pro-oncogenic role in glioblastoma by an editing-independent mechanism. Genome Biology. 2021;22(1):p. 51. doi: 10.1186/s13059-021-02271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Wanna-udom S., Terashima M., Lyu H., et al. The m6A methyltransferase METTL3 contributes to Transforming Growth Factor- beta-induced epithelial-mesenchymal transition of lung cancer cells through the regulation of JUNB. Biochemical and Biophysical Research Communications. 2020;524(1):150–155. doi: 10.1016/j.bbrc.2020.01.042. [DOI] [PubMed] [Google Scholar]
- 285.Choe J., Lin S. B., Zhang W. C., et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561(7724):556–560. doi: 10.1038/s41586-018-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Chen M. N., Wei L., Law C. T., et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–2270. doi: 10.1002/hep.29683. [DOI] [PubMed] [Google Scholar]
- 287.Chen S. L., Liu L. L., Wang C. H., et al. Loss of RDM1 enhances hepatocellular carcinoma progression via p53 and Ras/Raf/ERK pathways. Molecular Oncology. 2020;14(2):373–386. doi: 10.1002/1878-0261.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Liu J., Eckert M. A., Harada B. T., et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nature Cell Biology. 2018;20(9):1074–1083. doi: 10.1038/s41556-018-0174-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Cui Q., Shi H. L., Ye P., et al. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Reports. 2017;18(11):2622–2634. doi: 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Weng H. Y., Huang H. L., Wu H. Z., et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m6A Modification. Cell Stem Cell. 2018;22(2):191–205.e9. doi: 10.1016/j.stem.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Peng F., Xu J., Cui B., et al. Oncogenic AURKA-enhanced N 6-methyladenosine modification increases DROSHA mRNA stability to transactivate STC1 in breast cancer stem-like cells. Cell Research. 2021;31(3):345–361. doi: 10.1038/s41422-020-00397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Chen X., Xu M., Xu X., et al. METTL14-mediated N6-methyladenosine modification of SOX4 mRNA inhibits tumor metastasis in colorectal cancer. Molecular Cancer. 2020;19(1):p. 106. doi: 10.1186/s12943-020-01220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Yang X., Zhang S., He C., et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Molecular Cancer. 2020;19(1):p. 46. doi: 10.1186/s12943-020-1146-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Wang M., Liu J., Zhao Y., et al. Upregulation of METTL14 mediates the elevation of PERP mRNA N6 adenosine methylation promoting the growth and metastasis of pancreatic cancer. Molecular Cancer. 2020;19(1):p. 130. doi: 10.1186/s12943-020-01249-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Chen Y. H., Peng C. H., Chen J. R., et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Molecular Cancer. 2019;18(1):p. 127. doi: 10.1186/s12943-019-1053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Su R., Dong L., Li C., et al. R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m6A/MYC/CEBPA Signaling. Cell. 2018;172(1-2):90–105.e23. doi: 10.1016/j.cell.2017.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Li Z. J., Weng H. Y., Su R., et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N 6-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31(1):127–141. doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Niu Y., Lin Z., Wan A., et al. Loss-of-function genetic screening identifies ALDOA as an essential driver for liver cancer cell growth under hypoxia. Hepatology. 2021 doi: 10.1002/hep.31846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Huang H., Wang Y. N., Kandpal M., et al. FTO-DependentN6-Methyladenosine modifications inhibit ovarian cancer stem cell self-renewal by blocking cAMP signaling. Cancer Research. 2020;80(16):3200–3214. doi: 10.1158/0008-5472.CAN-19-4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Shen C., Sheng Y., Zhu A. C., et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell. 2020;27(1):64–80.e9. doi: 10.1016/j.stem.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Jin D., Guo J. W., Wu Y., et al. m6A demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2-mediated YAP activity in NSCLC. Molecular Cancer. 2020;19(1) doi: 10.1186/s12943-020-01161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Guo X. Y., Li K., Jiang W. L., et al. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A-YTHDF2-dependent manner. Molecular Cancer. 2020;19(1):p. 91. doi: 10.1186/s12943-020-01158-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Su T., Huang M., Liao J., et al. Insufficient radiofrequency ablation promotes hepatocellular carcinoma metastasis through m6A mRNA methylation dependent mechanism. Hepatology. 2021 doi: 10.1002/hep.31766. [DOI] [PubMed] [Google Scholar]
- 304.Liu T., Wei Q., Jin J., et al. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Research. 2020;48(7):3816–3831. doi: 10.1093/nar/gkaa048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Li J., Xie H., Ying Y., et al. YTHDF2 mediates the mRNA degradation of the tumor suppressors to induce AKT phosphorylation in N6-methyladenosine-dependent way in prostate cancer. Molecular Cancer. 2020;19(1):p. 152. doi: 10.1186/s12943-020-01267-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Chang G. Q., Shi L., Ye Y. Q., et al. YTHDF3 Induces the Translation of m6A-Enriched Gene Transcripts to Promote Breast Cancer Brain Metastasis. Cancer Cell. 2020;38(6):857–871.e7. doi: 10.1016/j.ccell.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Ni W., Yao S., Zhou Y. X., et al. Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m6A reader YTHDF3. Molecular Cancer. 2019;18(1):p. 143. doi: 10.1186/s12943-019-1079-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Zhu S., Wang J.-Z., De Chen Y.-T. H., et al. An oncopeptide regulates m(6)A recognition by the m(6)A reader IGF2BP1 and tumorigenesis. Nature Communications. 2020;11(1) doi: 10.1038/s41467-020-15403-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Müller S., Glaß M., Singh A. K., et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Research. 2019;47(1):375–390. doi: 10.1093/nar/gky1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Huang H. L., Weng H. Y., Sun W. J., et al. Recognition of RNA N 6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nature Cell Biology. 2018;20(3):285–295. doi: 10.1038/s41556-018-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Qian S., Li J., Hong M., et al. TIGAR cooperated with glycolysis to inhibit the apoptosis of leukemia cells and associated with poor prognosis in patients with cytogenetically normal acute myeloid leukemia. Journal of Hematology & Oncology. 2016;9(1):p. 128. doi: 10.1186/s13045-016-0360-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Hole P. S., Zabkiewicz J., Munje C., et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood. 2013;122(19):3322–3330. doi: 10.1182/blood-2013-04-491944. [DOI] [PubMed] [Google Scholar]
- 313.Montero J., Dutta C., van Bodegom D., Weinstock D., Letai A. p53 regulates a non-apoptotic death induced by ROS. Cell Death and Differentiation. 2013;20(11):1465–1474. doi: 10.1038/cdd.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Hart P. C., Mao M., de Abreu A. L., et al. MnSOD upregulation sustains the Warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nature Communications. 2015;6(1, article 6053) doi: 10.1038/ncomms7053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Han X., Duan X., Liu Z., et al. ZEB1 directly inhibits GPX4 transcription contributing to ROS accumulation in breast cancer cells. Breast Cancer Research and Treatment. 2021;188(2):329–342. doi: 10.1007/s10549-021-06301-9. [DOI] [PubMed] [Google Scholar]
- 316.Singhal R., Mitta S. R., Das N. K., et al. HIF-2α activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. Journal of Clinical Investigation. 2021;131(12) doi: 10.1172/JCI143691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Shen C. J., Chang K. Y., Lin B. W., et al. Oleic acid-induced NOX4 is dependent on ANGPTL4 expression to promote human colorectal cancer metastasis. Theranostics. 2020;10(16):7083–7099. doi: 10.7150/thno.44744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Huang H., Zhang S., Li Y., et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nature Communications. 2021;12(1, article 3720) doi: 10.1038/s41467-021-24108-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Park H. K., Hong J. H., Oh Y. T., et al. Interplay between TRAP1 and sirtuin-3 modulates mitochondrial respiration and oxidative stress to maintain stemness of glioma stem cells. Cancer Research. 2019;79(7):1369–1382. doi: 10.1158/0008-5472.CAN-18-2558. [DOI] [PubMed] [Google Scholar]
- 320.Li S., Zhuang Z., Wu T., et al. Nicotinamide nucleotide transhydrogenase-mediated redox homeostasis promotes tumor growth and metastasis in gastric cancer. Redox Biology. 2018;18:246–255. doi: 10.1016/j.redox.2018.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Wang X., Ye T., Xue B., et al. Mitochondrial GRIM-19 deficiency facilitates gastric cancer metastasis through oncogenic ROS-NRF2-HO-1 axis via a NRF2-HO-1 loop. Gastric Cancer. 2021;24(1):117–132. doi: 10.1007/s10120-020-01111-2. [DOI] [PubMed] [Google Scholar]
- 322.Xu J., Ji L., Ruan Y., et al. UBQLN1 mediates sorafenib resistance through regulating mitochondrial biogenesis and ROS homeostasis by targeting PGC1β in hepatocellular carcinoma. Signal Transduction and Targeted Therapy. 2021;6(1):p. 190. doi: 10.1038/s41392-021-00594-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Kudo Y., Sugimoto M., Arias E., et al. PKCλ/ι loss induces autophagy, oxidative phosphorylation, and NRF2 to promote liver cancer progression. Cancer Cell. 2020;38(2):247–262.e211. doi: 10.1016/j.ccell.2020.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Yang Y., Neo S. Y., Chen Z., et al. Thioredoxin activity confers resistance against oxidative stress in tumor-infiltrating NK cells. The Journal of Clinical Investigation. 2020;130(10):5508–5522. doi: 10.1172/JCI137585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Jan Y. H., Lai T. C., Yang C. J., Lin Y. F., Huang M. S., Hsiao M. Adenylate kinase 4 modulates oxidative stress and stabilizes HIF-1α to drive lung adenocarcinoma metastasis. Journal of Hematology & Oncology. 2019;12(1):p. 12. doi: 10.1186/s13045-019-0698-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Qi M., Dai D., Liu J., et al. AIM2 promotes the development of non-small cell lung cancer by modulating mitochondrial dynamics. Oncogene. 2020;39(13):2707–2723. doi: 10.1038/s41388-020-1176-9. [DOI] [PubMed] [Google Scholar]
- 327.Wang J., Lu Q., Cai J., et al. Nestin regulates cellular redox homeostasis in lung cancer through the Keap1-Nrf2 feedback loop. Nature Communications. 2019;10(1):p. 5043. doi: 10.1038/s41467-019-12925-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Abdul Pari A. A., Singhal M., Hübers C., et al. Tumor cell-derived angiopoietin-2 promotes metastasis in melanoma. Cancer Research. 2020;80(12):2586–2598. doi: 10.1158/0008-5472.CAN-19-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Govindarajan B., Sligh J. E., Vincent B. J., et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. The Journal of Clinical Investigation. 2007;117(3):719–729. doi: 10.1172/JCI30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Xu L., Wu T., Lu S., et al. Mitochondrial superoxide contributes to oxidative stress exacerbated by DNA damage response in RAD51-depleted ovarian cancer cells. Redox Biology. 2020;36, article 101604 doi: 10.1016/j.redox.2020.101604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Dando I., Pacchiana R., Pozza E. D., et al. UCP2 inhibition induces ROS/Akt/mTOR axis: Role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Free Radical Biology & Medicine. 2017;113:176–189. doi: 10.1016/j.freeradbiomed.2017.09.022. [DOI] [PubMed] [Google Scholar]
- 332.Yang W.-H., Ding C.-K. C., Sun T., et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Reports. 2019;28(10):2501–2508.e2504. doi: 10.1016/j.celrep.2019.07.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data were used to support this study