

Graphical abstract

Keyword: COVID-19, N-Arylhydroxamic Acids, Spike glycoprotein, +ssRNA, Molecular docking, Binding energy
Abstract
In the present investigation, molecular docking studies have been performed using AutoDock Vina to investigate the role of ligand-binding affinity at the hydrophobic pocket of COVID-19. The knowledge of the binding of protein receptors with ligand molecules is essential in drug discovery processes. Hydroxamic acids with reported biological activity, have been investigated for docking to an important target, SARS-CoV-2, in order to predict their therapeutic efficacy. The spike protein of the coronavirus is responsible for the attachment to host cells and a positive-sense single-strand RNA, (+)ssRNA, is a genetic material that can be translated into protein in the host cell. We modeled the structure of SARS-CoV-2 with the ligands, hydroxamic acids. They show binding capability with both, Spike protein and (+)ssRNA. The twain exhibit negative binding energies which signify that reactions are spontaneous, strong, and fast. The present research proposed hydroxamic acids as molecules which can be used for the development of anti-virals therapeutics against SARS-CoV-2.
1. Introduction
The present work is an approach to design new generation candidate drugs to inhibit SARS-CoV-2 through a molecular modeling approach. For structure-based drug design, molecular docking is a key tool in structural molecular biology and computer-assisted drug design to find the strongest candidate of the drug for various diseases. This technique involves the computational simulation of a ligand, binding to a receptor. To design a biologically active compound lots of money is required, (i) for their synthesis, (ii) to perform the physico-chemical/biological characterization of the huge number of molecules, and (iii) to separate the active product from the thousands of synthesized molecules followed by its testing and modification. Therefore, in-silico studies are important to save time and money. Molecular docking predicts the experimental binding modes and affinity of small molecules within the binding site of the particular receptor target which is generally DNA, RNA, or some other biopolymer [1]. In this technique, the 3D structure of a potential ligand (drug) is superimposed on the receptor target site to predict the structure of the intermolecular complexes thus formed. It also forecasts the strength of the binding and the binding affinity between ligand and receptor using scoring functions. If the binding specificity and strength of this protein can be mimicked by a small molecule, then RNA/DNA function can be artificially modulated, by binding this molecule.
COVID-19 is a positive sense, single-strand RNA, +ssRNA, associated with nucleoprotein within a capsid comprised of matrix protein and features the largest RNA genome known to date [2], [3]. SARS-CoV-2, SARS-CoV, and MERS-CoV are responsible for severe respiratory illness. The SARS-CoV-2, is of zoonotic origin and a member of the coronviridae genus, specifically of Betacoronavirus based on the nucleotide sequence unlike the MERS-CoV [4], [5], [6], [7], [8]. The spike protein of SARS-CoV-2 is responsible for the attachment at the host cell membrane, where it starts infecting the host [9], [10]. It is reported that the human receptor for COVID-19 is ACE-2/CD26 [11], [12], and the virus is SARS-CoV-2, a severe acute respiratory syndrome coronavirus. Treatment of COVID-19 is done by many therapeutics including some anti-virals but still, more effective therapeutics are needed especially for patients with co-morbidities [13], [14], [15]. Structures of SARS-CoV-2 and (+)ssRNA [16], [17], are portrayed in Fig. 1 .
Fig. 1.

(a) Structure of SARS-CoV-2 virus: Components of the structure of the SARS-CoV-2 virus labeled with spike proteins, M−proteins, HE, E, and RNA with Nucleocapsid (N) proteins. (b) Structure of trimer Spike glycoprotein of SARS-CoV-2 virus showing S2 Domain and S1 Domain (PDB ID-6VSB) (c) Structure of single-stranded ribonucleic acid showing the nucleobases and phosphate backbone (PDB ID-6NUD).
N-Arylhydroxamic acids, the excellent DNA and RNA binders [18], [19], [20], [21], [22], [23], [24], [25], of the general formula, R1-N-OH.R2-C O are neutral, polyfunctional molecules. The pharmacological property of these is associated with their characteristic feature, hydroxamic acid functional group, -N-OH, C O, which contains both Hydrogen-Bond Donor (HBD) and Hydrogen-Bond Acceptor (HBA) sites [19], [20].
In the present investigation, in-silico docking studies were performed, using AutoDock Vina, to investigate the role of N-arylhydroxamic acids as effective inhibitors of SARS-CoV-2 of COVID-19.
2. Materials & methods
2.1. Analysis of drug likeness of hydroxamic acids
The drug-likeness of hydroxamic acids was carried out by Lipinski filter (http://www.scfbio-iitd.res.in/software/drugdesign/lipinski.jsp), according to which an orally active drug should comply to a minimum of four of the five laid down criteria for drug-likeness. These are molecular mass, H-bond donor and H-bond acceptor sites, lipophilicity, clogP along with molecular refractive index [21]. This rule states that if a molecule has Molecular Weight ≤ 500, Lipophilicity ≤ 5, HBD sites≤ 5, and HBA sites ≤ 2 × 5, then a molecule can act as a drug in the biological system and worthy testing for biological assay. All the hydroxamic acids investigated here fulfill this rule.
2.1.1. Hydroxamic acids and H-Bonds
In life sciences, H-bonds are important as they are involved in the most specific molecular interactions of neutral molecules with the receptors and are the weak bonds, rapidly form and break [22], [23]. H-bond interactions play a crucial role in protein–ligand stability and are also an important parameter to determine which amino acid residues are necessary for docking interactions.
2.1.2. Hydroxamic acids and Lipophilicity, cLogP
Lipophilicity of compounds of therapeutic interest is an important parameter to understand the transport process across biological barriers [24]. It is a physicochemical parameter that depends on both, the molecule itself and intermolecular interactions. Collander [25], proposed that the rate of movement of organic molecules through the cellular material is α to lipophilicity. As per Lipinski [21], [26], lipophilicity should be less than “5. Substances with high LogP values are so hydrophobic that they partition very poorly into the aqueous components of biological systems. Such compounds remain with the lipid components and are generally poorly absorbed. These are generally bioaccumulated into fat tissue. Compounds with lower LogP do not bioaccumulate because of their low affinity for lipids.
Calculated lipophilicity, cLogP, were obtained following the Bio-Loom program of BioByte Company [27], [28], [29], and the values are presented in Table 1 .
Table 1.
Hydroxamic Acids: HBA, HBD Sites, and Calculated Lipophilicity.
| S. No. | Hydroxamic Acid | 2D Structure | 3D Structure | HBA | HBD | cLog P |
|---|---|---|---|---|---|---|
| 1 | N-Phenyl-benzo- |  |
 |
3 | 1 | 2.35 |
| 2 | N-Phenyl-2-chlorobenzo- |  |
 |
4 | 1 | 2.89 |
| 3 | N-Phenyl-4-chlorobenzo- |  |
 |
4 | 1 | 2.94 |
| 4 | N-Phenyl-2,4-dichlorobenzo- |  |
 |
5 | 1 | 3.54 |
| 5 | N-Phenyl-4-methylbenzo- |  |
 |
3 | 4 | 2.98 |
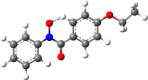
| 6 | N-Phenyl-4-nitrobenzo- | 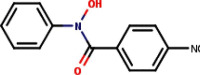 |
 |
6 | 1 | 1.68 |
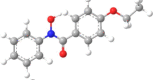
| 7 | N-Phenyl-4-ethoxybenzo- | 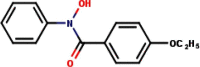 |
 |
4 | 6 | 2.85 |
| 8 | N-Phenyl-salicylic- | 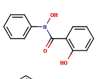 |
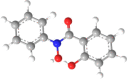 |
4 | 2 | 2.07 |
| 9 | N-Phenyl-naphtho- | 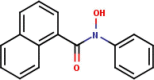 |
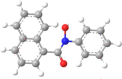 |
3 | 1 | 3.49 |
| 10 | N-Naphthyl-benzo- |  |
 |
3 | 1 | 3.31 |
| 11 | N-p-Tolylbenzo- |  |
 |
3 | 4 | 2.73 |
| 12 | N-p-Chloro-phenyl-benzo- |  |
 |
4 | 1 | 2.82 |
| 13 | N-p-Chlorophenyl-2,4-dichlorobenzo- |  |
 |
6 | 1 | 3.88 |
| 14 | N-p-Chlorophenyl-4-chlorobenzo- |  |
 |
5 | 1 | 3.54 |
| 15 | N-p-Chlorophenyl-4-bromobenzo- |  |
 |
5 | 1 | 3.46 |
Fifteen N-arylhdroxamic acids were chosen as ligands [30], [31], [32], [33], [34]. Their structure and relevant properties are focussed on in Table 1.
2.2. Molecular docking
The present work is an approach to screen new generation candidate drugs to inhibit SARS-CoV-2 through molecular docking method by applying the databases from Drug Bank, PubChem, Protein Data Bank, PDB, and software like AutoDock Vina, PyMOL 2.3.4, Chem3D Ultra 10.0, and Marvin Sketch version 16.12.5 were employed [35] for analysis.
2.2.1. Preparation of target
The leading part of targeted bio-macromolecules structure of SARS-CoV-2 Spike glycoprotein holding the heterotrimer chain A, B, and C, and +ssRNA, were downloaded from the PDB database from www.rcsb.org. The crystal structure of the targeted SARS-CoV-2 spike glycoprotein has the PDB ID of 6VSB [36] and +ssRNA has the PDB ID of 6NUD [37]. The targets spike glycoprotein and +ssRNA were optimized for the docking process. All the water molecules and heteroatom were removed and polar hydrogen atoms and charges were added to receptor molecules before initializing the molecular docking process. For the SARS-CoV-2, spike glycoprotein specifically, chain A is mainly kept into the grid as it is the receptor-binding domain. Docking was completed by taking all the rotatable bonds of the complex as rotatable and receptor as rigid. The grid spacing of 1.0 Å with a grid box size of 40 × 40 × 40 Å was used that incorporates the active site of SARS-CoV-2 spike glycoprotein and the nucleobases of (+)ssRNA. Default values were taken for other parameters needed for docking. The PDBQT file was generated for the target which was further used for the molecular docking process [38].
2.2.2. Preparation of ligand
3-D structures of the compounds 1 to 15, were prepared using Marvin Sketch version 16.12.5. The PDB structures were generated with the help of Chem3D Ultra 10.0. Using the MMT algorithm for optimization of the ligands, the lowest energy conformations for further docking were selected.
2.2.3. Molecular docking by AutoDock Vina
AutoDock Vina is an automated program applied to predict ligand and protein interaction. This software is developed and maintained by Scripps Research Institute [39]. It is open-source molecular modeling software mainly used for protein–ligand docking. It was optimized to use a model containing +ssRNA and different hydroxamic acids that include polar hydrogen atoms, but not the hydrogen atoms bonded to carbon atoms. The AutoDock tool (ADT) file with AutoDock Vina was used for molecular docking, which uses an empirical scoring function based on the free energy of binding [40], [41]. The docking analysis of ligands 1 to 15, which were selected as a ligand with SARS-CoV-2 spike glycoprotein and +ssRNA were carried out. The Lamarckian Genetic Algorithm (LGA) was used among the AutoDock suite. Employing the MGL Tool 1.5.6. and PyMOL 2.3.4.; the analysis of docking results and binding sites was performed, respectively. Various interactions like hydrogen bonding, hydrophobic interactions, π - π stacking, and halogen bonding along with the distance between the ligand and target were analyzed thoroughly.
2.2.4. 2-D interaction diagram for Protein-Ligand interactions
2-D interaction diagram of protein–ligand interaction was very worthy for the proper visualization of the actual residues, interacted to the ligand molecule. Here we used a protein–ligand interaction profiler-PLIP for the generation of a 2-D interaction diagram [42]. The docked structures were saved in the form of the PDB with the help of out and pdbqt files of the ligand and protein from PyMOL 2.3.4.
3. Results & discussion
In computational chemistry and molecular modeling, scoring functions are mathematical functions used to predict the binding affinity between two molecules after they have been docked. The smaller is the value of binding affinity, the greater is the affinity of the ligand to bind with the target. Binding free energy is the sum of all the intermolecular interactions that are present between the ligand and target. The docking score is the scoring function used to predict the binding affinity of both ligand and target once it is docked. Molecular docking is an important tool to know the protein–ligand interaction relationship. It plays a key role in stabilizing the complex and energetically favored the ligand in an open conformational environment of protein structures [43], [38].
In the present study, ligand-target complex interactions are derived from hydrophobic, van der Waals's contacts, polar interactions through H-bonds and electrostatic attraction. When the ligands were analyzed following In-Silico computational docking tools, they successfully docked against the inhibitor region of SARS-CoV-2 and generated negative values of binding energy suggesting the high affinity for binding pockets of COVID-19.
3.1. Molecular docking of N-Arylhydroxamic acids as ligand and SARS-CoV-2 spike glycoprotein as target
Docking predicted the experimental binding modes and affinity of small molecules with the binding sites of the particular receptor target. In the present study, spike glycoprotein is used as the target and hydroxamic acids as ligands. They interact, showing different poses at binding sites, and are positively docked into the receptor-binding domain specifically to the S1 domain of the spike glycoprotein. The docking results show that all the N-arylhydroxamic acids docked with a negative dock energy value and low acquired binding energy which support the spontaneous ligand-target interactions. The lowest binding energy was found for N-phenyl-naphtho- (ligand 9), and N-naphthyl-benzo- (ligand 10), hydroxamic acids which are −7.0 Kcal/mol to bind with spike protein. The result suggests strong and effective binding of these ligands, which stabilizes the target properly. Interactions involve are mainly supported by hydrophobic and hydrogen bondings like, -N—H—O-, -O—H—O-. Halogen bondings are accountable in chloro derivatives as -Cl-O- and π-π stacking, where rings and -C—C- bonds are employed. The molecular interaction study also indicates that more than one active site residue is involved for each ligand studied here. The distance between interacting residues and ligand ranges from 1.93 to 5.31 Å.
The detailed interaction profile of the most potent ligand, N-phenyl-naphtho hydroxamic acid, ligand 9 is explored in Fig. 2 . The docked structures of other ligands in the binding pocket of spike protein residues, hydrogen-bond distance, interactions exhibited, and π-π stacking are presented in Fig. 3 . The binding energies are depicted in Table 2 and details of all ligands, including predicted key residues involved in the interactions, distance between interacting residues, and ligand atom are portrayed in Table S1.
Fig. 2.

Interaction of N-phenyl-naphthohydroxamic acid as ligand and SARS-CoV-2 Spike glycoprotein of COVID-19 (PDB-ID: 6VSB) as a target. (a) Structure of Spike protein with ligand showed in a zoomed view (b) Surface representation of the interacting ligand with SARS-CoV-2 Spike glycoprotein, (c) 2D interaction diagram showing the interaction of SARS-CoV-2 Spike glycoprotein with a ligand having all the interacting residues. Hydrogen bonds are depicted in blue color and hydrophobic interactions are shown in grey-colored dashes and π-π stacking is portrayed in green-colored dashes with white balls in aromatic rings.
Fig. 3.


Surface representation and 2D interaction diagram. Docked structures of Hydroxamic Acids with the target SARS-CoV-2 Spike glycoprotein of COVID-19 (PDB-ID: 6VSB).
Table 2.
Binding Energies of Hydroxamic Acids Employing AutoDock Vina Spike Protein (PDB ID-6VSB) and +ssRNA (PDB ID-6NUD) of COVID-19.
| S. No. | Hydroxamic Acid | Binding Energy (kcal/mol) |
|
|---|---|---|---|
| Spike Protein | +ssRNA | ||
| 1 | N-Phenyl-benzo- | −6.0 | −5.9 |
| 2 | N-Phenyl-2-chlorobenzo- | −6.0 | −5.7 |
| 3 | N-Phenyl-4-chlorobenzo- | −6.1 | −5.5 |
| 4 | N-Phenyl-2,4-dichloro-benzo- | −6.2 | −5.2 |
| 5 | N-Phenyl-4-methylbenzo- | −6.6 | −5.5 |
| 6 | N-Phenyl-4-nitrobenzo- | −6.5 | −6.1 |
| 7 | N-Phenyl-4-ethoxybenzo- | −6.0 | −5.2 |
| 8 | N-Phenyl-salicylic- | −6.1 | −6.0 |
| 9 | N-Phenyl-naphtho- | −7.0 | −6.1 |
| 10 | N-Naphthyl-benzo- | −7.0 | −6.0 |
| 11 | N-p-Tolylbenzo- | −6.0 | −5.2 |
| 12 | N-p-Chloro-phenyl-benzo- | −5.9 | −5.6 |
| 13 | N-p-Chloro-phenyl-2,4-dichloro-benzo- | −6.3 | −5.9 |
| 14 | N-p-Chlorophenyl-4-chlorobenzo- | −6.1 | −5.7 |
| 15 | N-p-Chlorophenyl-4-bromo-benzo- | −6.2 | −5.7 |
3.2. Molecular docking of N-Arylhydroxamic acids with the genome of COVID-19, +ssRNA
(+)ssRNA is the target of great importance for therapeutic interventions. Many diseases are caused due to the viral infection having the RNA as genetic material which is the core reason for the multiplication of the infected cells in the host body. In the present study, all the molecules interact with +ssRNA, which may eventually lead to inhibit the functionality of this particular genetic material.
The molecular docking results recommended that N-arylhydroxamic acids interact with the adenine (A14, A15, A16, or/and A17 as per demand) and uracil 14 (U14) bases of +ssRNA of COVID-19.
The parent compound, ligand- 1, interacts with the A16 and U14 bases via hydrogen bonding, the hydrogen atom of ligand interacted with the oxygen atom of the base of RNA. A17 and U14 bonded via -C—H, and the interactions are hydrophobic. Binding of the A15 through a phosphate group and U14 through the oxygen of RNA to the oxygen atom of the ligand is found. The binding energy is 5.9 Kcal/mol, which indicates that the interaction is spontaneous. Ligands-2 and 3 possess the same interacting residues, however, the distance of interacting bases and ligand are different. All the interacting residues are the same for Ligands-4 and 12. The binding energy, observed is −5.6 Kcal/mol and −5.5 Kcal/mol, respectively. Ligand 5, forms a bond with A16 and A15. A17 forms -C—H bond and the interaction are hydrophobic. Ligand-6, shows the hydrogen bonding interaction with the bases A15, A16, A17, and U14, and the oxygen and hydrogen atoms participate in the binding. The smallest distance between the ligand and the base is 1.9 Aͦ and the largest distance is 2.9 Aͦ. The binding energy, evaluated is −6.1 Kcal/mol. In ligand-7, A16 and U14 interact through -O—H bond and A15 and U14 through --O—O- interaction. U14 and A17 amid -C—H, the hydrophobic interaction. The binding energy, observed is −5.2 Kcal/mol. Ligand-8 binds through oxygen and hydrogen atoms to A15. A16 bases interact via -O—H, -N—H, -C—H, and H—H bonds to the ligand. This ligand also shows oxygen–oxygen interactions by the U14 base. In ligand-9, interaction with the nucleobases through hydrogen bonding takes place. Here the residue involves is A16 via the -O—H bonding and the oxygen atom of the ligand is participating in the interaction. A15 and A17 are also attached to the ligand through -C—H bonding, involving hydrophobic and -O—O- interactions, respectively. U14 is also involved via -O—H and -C—H bonding. The binding energy, calculated is −6.1 Kcal/mol. Ligand-10 having the interacting residue A15 which forms hydrogen bonding via -O—H and oxygen atom of ligand. A16 also forms -O—H bond where PO2 of RNA and hydrogen of the ligand are involved. A17 and U14 via -C—H bonds are responsible for the hydrophobic interactions. Ligand-11 interacted through H-bonding with the residue A16, where the oxygen atom of ligand involves. A15 binds through its oxygen to the oxygen atom of the ligand. A17, U14, and A15 bind via -C—H bond through hydrophobic interactions. Ligand-13 shows the same residues as ligand-15, except for A17 which is absent in this case. In ligand-14, nucleobases A16 and U14 interact through -O—H bond and H-bonding. U14 interacts through the oxygen and phosphate groups. U14 and A17 amid -C—H, the hydrophobic interaction. Ligand-15 shows that A15 and A16 interact through -O—H and hydrogen bonds, A15 and U14 through oxygen and phosphate group through -O—O- bonding interaction. U14 and A17 amid C—H, the hydrophobic interaction.
Overall results demonstrate that all the N- arylhydroxamic acids interact with the +ssRNA base. The binding energies are tabulated in Table 2. The best molecular docked structure for ligand-9, N-phenyl-naphthohydroxamic acid is presented in Fig. 4 , and all other ligands with the target are demonstrated in Fig. 5 . The interacting bases with the ligand atom and the distance between them along with the type of interactions are summarized in Table S2.
Fig. 4.

Molecular Docked Structures +ssRNA with ligand docked. (a) Structure of +ssRNA with N-phenyl-naphthohydroxamic acid docked. (b) Surface representation and interaction of docked ligand as N-phenyl-naphthohydroxamic acid with target +ssRNA of COVID-19 (PDB-ID: 6NUD).
Fig. 5.

Molecular docked structures of Hydroxamic Acids showing binding sites with target +ssRNA of COVID-19 (PDB-ID: 6NUD).
In comparison to all the hydroxamic acids, both of the naphthyl derivatives possess the lowest binding energy of −6.1 Kcal/mol and the highest binding affinity towards a single-strand of (+)ssRNA. The above observations suggest that N-arylhydroxamic acids are wonderful ligands interacting with + ssRNA effectively and can be further used as therapeutic agents against the +ssRNA genome of COVID-19.
Hydroxamic acids are bioactive molecules [44], [45], [46], [47], and follow the “ Lipinski Rule of 5 ” [48], thus showing drug-likeness. Considering the values of lipophilicity calculated, the ligands show good transportability for oral and intestinal absorption and a good ability to cross the protein membrane. The accommodated H-bonds are also favorable. The hydroxamic acids studied are effective against SARS-CoV-2 spike glycoprotein, which is a viral component that helps in the internalization of the COVID-19 as well as they also interact with (+)ssRNA. All the ligands show negative docked energy onto the target spike protein and (+)ssRNA which exhibit the importance of these ligands. The outcome findings also indicate that the optimized hydrophobic interactions play an important role in stabilizing the ligand energetically at the interface of a protein structure. The interactions which are made or broken on binding are mainly hydrophobic effects and hydrogen bonds, in some cases π - π interactions and halogen bonds are active. The negative and lower values of binding energies demonstrate that the ligands bind spontaneously, strongly and fast, and appropriately with the virus during docking simulation. The lower the binding energy, the more stable is the complex. All the molecules exhibited well-established bonds with more than one amino acid and more than one active-site residue of each ligand. Binding poses and distance measurement of hydroxamic acid complexes with (+)ssRNA reveal that these ligands are in close proximity with the active site of coronavirus. Distance range from 1.4 to 3.0 Å. This confirms the tight binding between the small molecule and target. The novelty is the two-fold nature of the ligands These are potent against both, the spike protein and (+)ssRNA of coronavirus and thus, can be investigated further as the therapeutic agents against the SARS-CoV-2 of the COVID-19 target.
4. Conclusion
Docking is a molecular modeling technique that is used to predict how a protein interacts with the ligand. The present investigation shed a light on the potential interactions between hydroxamic acids and SARS-CoV-2 indicating the involvement of multiple interactions such as H-bonds, hydrophobic interactions, van der Waals forces, π stacking and halogen bonds, mostly depending on the binding sites of SARS-CoV-2 and the ligands. All ligands show better binding strength to SARS-CoV-2 and thus hold a promising potential to be explored for their anti-SARS-CoV-2 activity. These are worth further investigations in terms of both in-vitro and in-vivo studies. Based on docking scores it is concluded that all the hydroxamic acids display a better binding strength to SARS-CoV-2.
CRediT authorship contribution statement
Yamini Thakur: Conceptualization, Methodology, Writing - original draft. Rama Pande: Conceptualization, Data curation, Formal analysis, Supervision, Writing - review & editing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This research did not receive any financial support from funding agencies in the public, commercial, or not for - profit sectors.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.chemphys.2021.111354.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Berman H.M. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu C., Zhou Q., Li Y., Garner L.V., Watkins S.P., Carter L.J., Smoot J., Gregg A.C., Daniels A.D., Jervey S., Albaiu D. Research and development on therapeutic agents and vaccines for COVID-19 and related human Coronavirus diseases. ACS Cent. Sci. 2020;6(3):315–331. doi: 10.1021/acscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L., Ge J.i., Zheng L., Zhang Y., Wang H., Zhu Y., Zhu C., Hu T., Hua T., Zhang B., Yang X., Li J., Yang H., Liu Z., Xu W., Guddat L.W., Wang Q., Lou Z., Rao Z. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368(6492):779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan J.F.W., Lau S.K.P., To K.K.W., Cheng V.C.C., Woo P.C.Y., Yuen K.-Y. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS like disease. Clin. Microbiol. Rev. 2015;28(2):465–522. doi: 10.1128/CMR.00102-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elfiky A.A., Mahdy S.M., Elshemey W.M. Quantitative structure-activity relationship and molecular docking revealed a potency of anti-hepatitis C virus drugs against human corona viruses. J. Med. Virol. 2017;89(6):1040–1047. doi: 10.1002/jmv.24736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ibrahim I.M., Abdelmalek D.H., Elshahat M.E., Elfiky A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020;80(5):554–562. doi: 10.1016/j.jinf.2020.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lundstrom K., Seyran M., Pizzol D., Adadi P., El-Aziz T.M.A., Hassan S.S., Soares A., Kandimalla R., Tambuwala M.M., Aljabali A.A.A., Azad G.K., Choudhury P.P., Uversky V.N., Sherchan S.P., Uhal B.D., Rezaei N., Brufsky A.M. The importance of research on the origin of SARS-CoV-2. Viruses. 2020;12(1203):1–4. doi: 10.3390/v12111203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seyran M., Pizzol D., Adadi P., El‐Aziz T.M.A., Hassan S.S., Soares A., Kandimalla R., Lundstrom K., Tambuwala M., Aljabali A.A.A., Lal A., Azad G.K., Choudhury P.P., Uversky V.N., Sherchan S.P., Uhal B.D., Rezaei N., Brufsky A.M. Questions concerning the proximal origin of SARS-CoV-2. J. Med. Virol. 2021;93(3):1204–1206. doi: 10.1002/jmv.26478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pillaiyar T., Meenakshisundaram S., Manickam M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today. 2020;25(4):668–688. doi: 10.1016/j.drudis.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu T.Y., Frieman M., Wolfram J. Insights from nanomedicine into chloroquine efficacy against COVID-19. Nat. Nanotechnol. 2020;15(4):247–249. doi: 10.1038/s41565-020-0674-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao Y., Li L., Feng Z., Wan S., Huang P., Sun X., Wen F., Huang X., Ning G., Wang W. Comparative genetic analysis of the novel corona virus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020;6:1–4. doi: 10.1038/s41421-020-0147-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalra R.S., Kandimalla R. Engaging the spikes: heparan sulfate facilitates SARS-CoV-2 spike protein binding to ACE2 and potentiates viral infection. Signal Transduct. Target. Ther. 2021;6:39. doi: 10.1038/s41392-021-00470-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vallamkondua J., Johnb A., Wanic W.Y., Ramadevia S.P., Jellad K.K., Reddye P.H., Kandimalla R. SARS-CoV-2 pathophysiology and assessment of coronaviruses in CNS diseases with a focus on therapeutic targets. BBA – Mol. Basis Dis. 2020;1866 doi: 10.1016/j.bbadis.2020.165889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hassan S.S., Attrish D., Ghosh S., Choudhury P.P., Uversky V.N., Aljabali A.A.A., Lundstrom K., Uhal B.D., Rezaei N., Seyran M., Pizzol D., Adadi P., Soares A., Abd El-Aziz T.M., Kandimalla R., Tambuwala M.M., Azad G.K., Sherchan S.P., Baetas-da-Cruz W., Lal A., Palù G., Takayama K., Serrano-Aroca Á., Barh D., Brufsky A.M. Notable sequence homology of the ORF10 protein introspects the architecture of SARS-CoV- 2. Int. J. Biol. Macro. 2021;181:801–809. doi: 10.1016/j.ijbiomac.2021.03.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.R. Kandimalla, A. John, C. Abburi, J. Vallamkondu, P. H. Reddy, Current status of multiple drug molecules, and vaccines: an update in SARS-CoV-2 therapeutics, Mol. Neurobiol. https://doi.org/10.1007/s12035-020-02022-0. [DOI] [PMC free article] [PubMed]
- 16.Lin S.-M., Lin S.-C., Hsu J.-N., Chang C.-k., Chien C.-M., Wang Y.-S., Wu H.-Y., Jeng U.-S., Kehn-Hall K., Hou M.-H. Structure-based stabilization of non-native protein−protein interactions of coronavirus nucleocapsid proteins in antiviral drug design. J. Med. Chem. 2020;63(6):3131–3141. doi: 10.1021/acs.jmedchem.9b01913. [DOI] [PubMed] [Google Scholar]
- 17.M. Seyran, K. Takayama, V.N. Uversky, K. Lundstrom, G. Palu, S. . Sherchan, D. Attrish, N. Rezaei, A.A.A. Aljabali, S. Ghosh, D. Pizzol, G. Chauhan, P. Adadi, T.M.A. El-Aziz1, A.G. Soares, R. Kandimalla, M. Tambuwala, S.S. Hassan, G.K. Azad, P.P.Choudhury, W. Baetas-da-Cruz, A. Serrano-Aroca, A.M. Brufsky, B.D. Uhal, The structural basis of accelerated host cell entry by SARS-CoV-2, FEBS J. 2020, 1-11.
- 18.Kumar A., Rajwade R.P., Pandey B.N., Pandey R., Mishra K.P. Mechanism of apoptotic death in ehrlich ascites tumor cells induced by derivatives of hydroxamic acids acid. Cell Tissue Res. 2007;7:943–948. [Google Scholar]
- 19.Neilands J.B. Hydroxamic acids in nature. Science. 1967;156(3781):1443–1447. doi: 10.1126/science.156.3781.1443. [DOI] [PubMed] [Google Scholar]
- 20.Vanjari H., Pande R. Hydroxamic acids: proton donor and acceptor strength for use in drug design. J. Pharmaceut. Biomed. 2003;33(4):783–788. doi: 10.1016/s0731-7085(03)00282-6. [DOI] [PubMed] [Google Scholar]
- 21.C.A. Lipinski. Lead and drug like compounds, the rule- of-five revolution, Drug Discov. Today Technol. 2004 337-341. [DOI] [PubMed]
- 22.Motiejunas D., Wade R.C. 4.09 - structural, energetic, and dynamic aspects of ligand-receptor interactions. Compreh. Med. Chem. II. 2007;4:193–213. [Google Scholar]
- 23.Tiwari V., Pande R. Pande molecular descriptors of N-arylhydroxamic acids: a tool in drug design. Chem. Biol. Drug. Des. 2006;68(4):225–228. doi: 10.1111/j.1747-0285.2006.00433.x. [DOI] [PubMed] [Google Scholar]
- 24.Waring M.J. Lipophilicity in drug discovery. Expert. Opin. Drug Discov. 2010;5(3):235–248. doi: 10.1517/17460441003605098. [DOI] [PubMed] [Google Scholar]
- 25.Collander R., Lindholm M., Haug C.M., Stene J., Sörensen N.A. The partition of organic compounds between higher alcohols and water. Acta Chem. Scand. 1951;5:774–780. [Google Scholar]
- 26.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 27.Leo A.J. Calculating log POa from structures. Chem. Rev. 1993;93:1281–1305. [Google Scholar]
- 28.clogP@biobyte.com.
- 29.Marciniec K., Bafeltowska J., Maślankiewicz M.J., Buszman E., Boryczka S. Determination of the lipophilicity of quinolinesulfonamides by reversed-phase HPLC and theoretical calculations. J. Liq. Chromatogr. Relat. Technol. 2016;39(15):702–709. [Google Scholar]
- 30.Tandon S.G., Bhattacharyya S.C. Preparation and properties of Some N-Aryl hydroxamic acids. J. Chem. Eng. Data. 1962;7(4):553–555. [Google Scholar]
- 31.Roshania R., Agrawal Y.K. Synthesis of N-arylhydroxamic acids. J. Chem. Eng. Data. 1978;23(3):259–260. [Google Scholar]
- 32.Choudhary T.R., Tandon S.G. Preparation and properties of N-Methyl- and N-(4-Chlorophenyl)-substituted hydroxamic acids. J. Chem. Eng. Data. 1985;30(2):237–239. [Google Scholar]
- 33.Gupta V.K., Tandon S.G. Studies on hydroxamic acids: preparation and properties of N-1-naphthylhydroxamic acids. J. Chem. Eng. Data. 1972;17:248–249. [Google Scholar]
- 34.Pande K.R., Tandon S.G. Preparation and properties of N-arylhydroxamic acids. J. Chem. Eng. Data. 1979;24(1):72–74. [Google Scholar]
- 35.Pagadala N.S., Syed K., Tuszynski J. Software for molecular docking: a review. Biophys. Rev. 2017;9(2):91–102. doi: 10.1007/s12551-016-0247-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.-L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo M., Zhang K., Zhu Y., Pintilie G.D., Guan X., Li S., Schmid M.F., Ma Z., Chiu W., Huang Z. Coupling of +ssRNA cleavage with DNase activity in Type III-A CRISPR-Csm revealed by Cryo-EM and biochemistry. Cell Res. 2019;29(4):305–312. doi: 10.1038/s41422-019-0151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D.W. Ritchie, V. Venkatraman. Ultra-fast FFT protein docking on graphics processors, Bioinformatics 26 2010 2398–2405. [DOI] [PubMed]
- 39.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huey R., Morris G.M., Olson A.J., Goodsell D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007;28(6):1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh P., Banerjee P. How paramagnetic and diamagnetic LMOCs detect picric acid from surface water and the intracellular environment: a combined experimental and DFT-D3 study. Phys. Chem. Chem. Phys. 2016;18:22805–22815. doi: 10.1039/c6cp01620k. [DOI] [PubMed] [Google Scholar]
- 42.Salentin S., Schreiber S., Haupt V.J., Adasme M.F., Schroeder M. PLIP: fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015;43:W443–W447. doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itoh Y., Nakashima Y., Tsukamoto S., Kurohara T., Suzuki M., Sakae Y., Oda M., Okamoto Y., Suzuki T. N+-C-H···O Hydrogen bonds in protein-ligand complexes. Sci. Rep. 2019;9:1–12. doi: 10.1038/s41598-018-36987-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajwade R.P., Pande R. Evaluation of lipophilicity of N-Arylhydroxamic acids by reversed phase-high performance liquid chromatographic method and self-organizing molecular field analysis. Anal. Chim. Acta. 2008;630:205–210. doi: 10.1016/j.aca.2008.09.062. [DOI] [PubMed] [Google Scholar]
- 45.Singh P., Khare D., Pande R. Springers; Chemical Paper: 2014. Evaluation of Antioxidant Activity and DNA Cleavage Protection Effect of Naphthylhydroxamic Acid Derivatives Through Conventional and Fluorescence Microscopic Methods; pp. i–vii. [Google Scholar]
- 46.Singh P., Pande R. Determination of volumetric, steric and excess properties of naphthylhydroxamic acid derivatives in ethanol at 298.15 and 313.15 K. J. Mol. Liq. 2017;248:91–99. [Google Scholar]
- 47.Shi Y.K., Li Z.H., Han X.Q., Yi J.H., Wang Z.H., Hou J.L., Feng C.R., Fang Q.H., Wang H.H., Zhang P.F., Wang F.S., Shen J., Wang P. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances taxol-induced cell death in breast cancer. Cancer Chemother. Pharmacol. 2010;66:1131–1140. doi: 10.1007/s00280-010-1455-1. [DOI] [PubMed] [Google Scholar]
- 48.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.