

Abstract
Esophageal cancer is a lethal malignancy with a high mortality rate, while the molecular mechanisms underlying esophageal cancer pathogenesis are still poorly understood. Here, we found that the N6-methyladenosine (m6A) methyltransferase-like 3 (METTL3) is significantly upregulated in esophageal squamous cell carcinoma (ESCC) and associated with poor patient prognosis. Depletion of METTL3 results in decreased ESCC growth and progression in vitro and in vivo. We further established ESCC initiation and progression models using Mettl3 conditional knockout mouse and revealed that METTL3-mediated m6A modification promotes ESCC initiation and progression in vivo. Moreover, using METTL3 overexpression ESCC cell model and Mettl3 conditional knockin mouse model, we demonstrated the critical function of METTL3 in promoting ESCC tumorigenesis in vitro and in vivo. Mechanistically, METTL3-catalyzed m6A modification promotes NOTCH1 expression and the activation of the Notch signaling pathway. Forced activation of Notch signaling pathway successfully rescues the growth, migration, and invasion capacities of METTL3-depleted ESCC cells. Our data uncovered important mechanistical insights underlying ESCC tumorigenesis and provided molecular basis for the development of novel strategies for ESCC diagnosis and treatment.
Keywords: METTL3, N6-methyladenosine, m6A, esophageal cancer, esophageal squamous cell carcinoma, ESCC, Notch signaling pathway
Graphical abstract
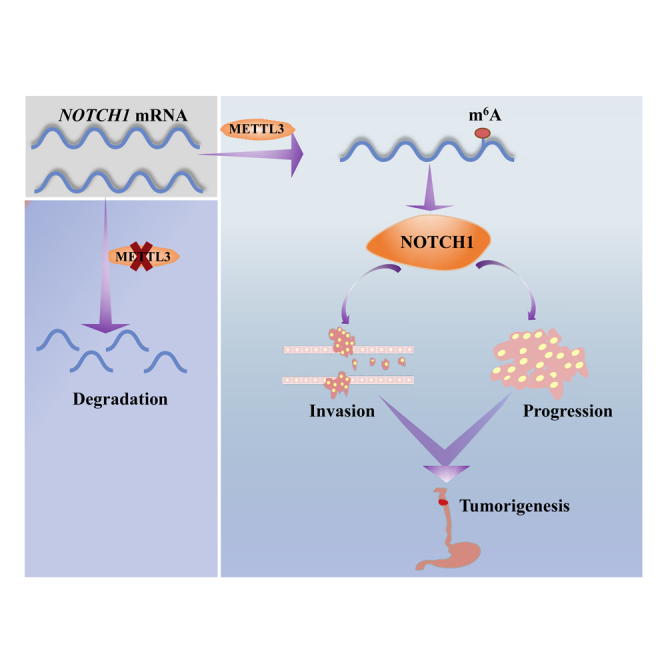
We provided strong evidence supporting that METTL3-mediated m6A mRNA modification is essential for esophageal squamous cell carcinoma (ESCC) initiation and progression in vitro and in vivo. Our data uncovered important mechanistical insights underlying ESCC tumorigenesis and provided molecular basis for development of novel strategies for ESCC diagnosis and treatment.
Introduction
Esophageal cancers, including esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC), are malignant tumors that seriously threaten human health.1,2 Esophageal cancer ranks as the seventh most common cancer worldwide and is a leading cause of cancer-related mortality.3,4 ESCC represents more than 90% of esophageal cancers and is often diagnosed at advanced stages.2,5 The high mortality rate of esophageal cancer is due to the lack of the effective diagnosis and therapeutic strategies.1,2,5 Therefore, better understanding of the molecular mechanisms underlying esophageal cancer pathogenesis is crucial for clinical diagnosis and treatment of esophageal cancer patients.
Abnormal genetic changes and/or epigenetic abnormalities often lead to ESCC pathogenesis.6, 7, 8 Mutations of key cancer genes, including P53, CCND1, and NOTCH receptors, are often associated with ESCC progression.8 In addition, mis-regulated DNA methylation, histone modifications, and non-coding RNAs also result in mis-regulated gene expression and ESCC development.7, 8, 9 In recent years, mRNA modifications were reported to play essential functions in regulation of posttranscriptional gene expression. N6-methyladenosine (m6A) is the most abundant internal modification on eukaryotic mRNAs.10,11 Methyltransferase-like 3 and 14 (METTL3 and METTL14) and their cofactors catalyze m6A modification on target mRNAs.10 In contrast, the m6A demethylases, including FTO and ALKBH5, can remove m6A modification from mRNAs.11,12 Different types of m6A readers recognize the m6A modification of a given target mRNA and regulate the stability, translation, splicing, or nuclear transport of the target mRNAs.13, 14, 15, 16 Given the important function of m6A modification in gene expression regulation, the mis-regulations of m6A modification often result in developmental diseases and cancers. Our previous studies have shown that the m6A methyltransferase METTL3 enhances the expression of various oncogenes and therefore promotes cancer progression.13, 14, 15, 16 Moreover, recent studies further uncovered the essential functions of m6A mRNA modification in regulation of different cancer types.17,18 METTL3 is upregulated in ESCC,19 however, the physiological functions and underlying molecular mechanisms of METTL3-mediated m6A modification in ESCC remain poorly understood.
In this study, we found that METTL3 expression is upregulated in human esophageal cancer samples and is associated with poor esophageal cancer progression. We further revealed the crucial functions of METTL3-mediated m6A modification in promoting ESCC initiation and progression in vitro and in vivo through activation of the NOTCH1 signaling pathway. Our work uncovered novel mechanisms underlying ESCC tumorigenesis and could provide new insights for the development of effective therapeutic strategies for ESCC treatment.
Results
METTL3 is elevated in ESCCs and is associated with poor ESCC prognosis
To study the potential functions of METTL3-mediated m6A modification in esophageal cancer, we first determined the METTL3 expression in esophageal cancer patient samples using The Cancer Genome Atlas (TCGA) database (TCGA database: https://portal.gdc.cancer.gov/). Our analysis revealed that METTL3 mRNA expression is significantly upregulated in esophageal tumor tissues compared with those in normal tissues (Figure 1A). In addition, high METTL3 expression is associated with advanced tumor grades and cancer stages (Figures 1B and 1C) and high lymph node metastasis activity of esophageal cancer (Figure 1D). Moreover, patients with higher METTL3 expression have significantly poorer disease-free survival status (Figure 1E). To further verify the association between METTL3 and esophageal cancer progression, we performed immunohistochemistry (IHC) staining of METTL3 on tumor tissues from our cohort of ESCC patients. Consistently, we found a significantly higher expression of METTL3 in ESCC tumor tissues than those in peri-tumor tissues (Figures 1F–1H). Furthermore, data from our cohort also supported that high METTL3 expression is associated with poor prognosis of ESCC patients (Figures 1I and 1J). Overall, these data indicate the important functions of METTL3 in regulation of ESCC progression.
Figure 1.

METTL3 is elevated in ESCCs and is a negative prognostic factor for ESCC patients
(A) Quantification of METTL3 expression (transcripts per million [TPM]) in normal esophageal tissues and primary esophageal tumor. (B) METTL3 expression level in normal esophageal tissues and different grades of esophageal tumor tissues. (C) METTL3 expression level in normal esophageal tissues and different stages of esophageal tumor tissues. (D) METTL3 expression level in normal esophageal tissues and tumor tissues with different stages of lymph node metastasis. (E) Disease-free survival of ESCC patients with high or low METTL3 expression level. Kaplan-Meier was employed to analyze the survival fraction. (F and G) IHC staining of METTL3 in clinical samples and the quantification of METTL3 staining scores. (H) Proportion of high/low METTL3 expression cases in tumor tissues and the adjacent normal tissues. (I and J) Correlation between METTL3 expression and overall survival (I) or disease-free survival (J) of ESCC patients. Data are represented as means ± SDs. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (Student’s t test; Dunnett’s test; log rank test).
Targeting METTL3 expression impairs ESCC progression
To study the underlying molecular mechanisms of METTL3 in regulation of ESCC, we knocked down METTL3 expression in TE-9 and TE-10 ESCC cells using two distinct short hairpin RNAs (shRNA1 and shRNA2; hereafter referred to collectively as sh1 and sh2). Compared with the non-specific control (NC), both sh1 and sh2 significantly reduced METTL3 protein expression (Figure 2A). Knockdown of METTL3 inhibits proliferation (Figure 2B) and colony formation (Figures 2C and 2D) of ESCC cells, suggesting that METTL3 is essential for the ESCC growth. We further performed wound healing assay and migration assay and found that METTL3 depletion significantly reduces the migration of ESCC cells (Figures 2E–2H). Moreover, cell invasion assay also revealed that knockdown of METTL3 reduces the invasion ability (Figures 2I and 2J) of both TE-9 and TE-10 cells. Collectively, our in vitro results uncover the critical role of METTL3 in the regulation of ESCC progression.
Figure 2.

Targeting METTL3 expression impairs progression of ESCC in vitro
(A) Western blotting confirmation of METTL3 knockdown in TE-9 and TE-10 cells. (B) Growth of NC or shMETTL3 cells evaluated by CCK8. (C and D) Colony formation assay (C) and the quantification data (D) of TE-9 and TE-10 cells with or without METTL3 knockdown. (E and F) Wound healing assay (E) and the quantification data (F) of TE-9 and TE-10 cells with or without METTL3 knockdown. (G and H) Invasion assay (G) and the quantification data (H) of TE-9 and TE-10 cells with or without METTL3 knockdown. (I and J) Migration assay and the quantification data (J) of TE-9 and TE-10 cells with or without METTL3 knockdown. Data are represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (one-way ANOVA; Student’s t test).
METTL3-mediated m6A modification regulates Notch signaling pathway
We next performed m6A methylated RNA immunoprecipitation-sequencing (MeRIP-seq) to identify the downstream target and underlying molecular mechanisms of m6A in regulation of ESCC. Analysis of our m6A MeRIP-seq data revealed that m6A-modified sites are selectively located at the GGAC motif near the translation stop codon (Figures 3A and 3B). Gene Ontology (GO) analysis uncovered that the m6A-modified transcripts are significantly enriched in cancer gene pathways, including the Notch signaling pathway (Figure S1). Moreover, GO analysis of RNA-seq data revealed that the downregulated mRNAs in the METTL3 knockdown ESCC cells are also significantly enriched in the Notch signaling pathway (Figure S2), suggesting the potential function of METTL3 in regulation of Notch pathway in ESCC cells. Key Notch pathway component NOTCH1 receptor has strong and specific m6A modification located near its translation stop codon (Figure 3C). Moreover, gene set enrichment analysis (GSEA) uncovered that depletion of METTL3 in ESCC cells resulted in a significant change of Notch signaling pathway gene expression (Figure 3D; Tables S3 and S4). Notch signaling pathway is an important oncogenic pathway in cancers, therefore, we further studied the function of METTL3-mediated m6A mRNA modification in regulation of Notch pathway in ESCC. Expression analysis revealed that NOTCH1 expression is significantly upregulated in ESCC samples compared with the controls (Figure 3E). Moreover, the expressions of METTL3 and NOTCH1 are significantly correlated (Figure 3F), further confirming the potential role of METTL3-mediated m6A modification in regulation of the Notch signaling pathway in ESCC. We next knocked down METTL3 expression and found that depletion of METTL3 results in decrease of the m6A modification, mRNA, and protein expression levels of NOTCH1; moreover, the expression of Notch target P21 also decreases upon METTL3 depletion (Figures 3G–3I). Overall, our data uncover that METTL3 regulates the m6A modification and the expression of the NOTCH1 receptor in ESCC cells.
Figure 3.

METTL3-mediated m6A modification regulates the Notch signaling pathway
(A) The sequence motif for m6A methylation identified in ESCC cells. (B) Metagene profiles of m6A distribution across the transcriptome in ESCC cells. (C) Representative m6A modification of NOTCH1 in ESCC cells. (D) GSEA analysis of transcriptome in NC cells or shMETTL3 cells with gene set from Notch signaling pathway. (E) Quantification of NOTCH1 expression (transcripts per million [TPM]) in normal esophageal tissues and primary esophageal tumor. (F) Correlation analysis of METTL3 and NOTCH1 expression in ESCC using TCGA database. (G) m6A-MeRIP-qPCR analysis showed the m6A level of NOTCH1 in ESCC cells with or without METTL3 knockdown. (H) Western blot of NOTCH1 and P21 protein level in METTL3-depleted ESCC cells. (I) qRT-PCR analysis of NOTCH1 and P21 mRNA level in METTL3-depleted ESCC cells. Data are represented as mean ± SD. ∗p < 0.05, ∗∗∗p < 0.001 (one-way ANOVA; Student’s t test; chi-square test).
Activation of the Notch pathway rescues ESCC progression in METTL3-depleted ESCC cells
To determine whether the Notch signaling pathway is a key downstream target of METTL3 in ESCC, we further performed rescue assay by forced activation of NOTCH1 in the METTL3-depleted cells. As shown in Figures 4A and 4B, overexpression of the active form of NOTCH1 (Intracellular domain of NOTCH1 [ICN1]) increased the expression of NOTCH1 in METTL3 knockdown cells, suggesting that we have successfully activated the Notch signaling pathway. Forced activation of the NOTCH1 signaling pathway rescued the growth (Figure 4C) and colony formation (Figures 4D and 4E) capacities of the METTL3 knockdown ESCC cells. In addition, wound healing (Figures 4F and 4G), migration, and invasion (Figures 4H and 4I) assays further revealed that NOTCH1 activation significantly promoted the migration and invasion of METTL3-depleted ESCC cells. These data support that the NOTCH1 signaling pathway is an important downstream target of METTL3 and is essential for its function in promoting ESCC progression.
Figure 4.

Activation of Notch pathway rescues ESCC progression in METTL3-depleted cells
(A) Western blot of ICN1 expression in METTL3-depleted ESCC cells. (B) qRT-PCR of ICN1 expression in METTL3-depleted ESCC cells. (C) CCK8 assay of METTL3-depleted ESCC cells with or without ICN1 overexpression. (D and E) Colony formation assay (D) and the quantification data (E) of METTL3-depleted ESCC cells with or without ICN1 overexpression. (F and G) Wound healing assay (F) and the quantification data (G) of METTL3-depleted ESCC cells with or without ICN1 overexpression. (H and I) Migration and invasion assay (H) and the quantification data (I) of METTL3-depleted ESCC cells with or without ICN1 overexpression. Data are represented as mean ± SD. ∗p < 0.05, ∗∗∗p < 0.001 (one-way ANOVA; Student’s t test).
Knockout (KO) of Mettl3 inhibits ESCC initiation in vivo
We next generated the Keratin14 promoter-driven Mettl3 conditional knockout (Keratin14-CreER; Mettl3fl/fl, cKO) mouse model to study the function of METTL3 in regulation of ESCC initiation and progression in vivo. To determine the role of Mettl3 in regulation of ESCC initiation, we first induced Mettl3 cKO by tamoxifen injection and then subjected the Mettl3 cKO and control mice to 16 weeks of 4-nitroquinoline N-oxide (4NQO) treatment to induced ESCC formation. On week 21, the mice were sacrificed for analysis of ESCC tumorigenesis in the Mettl3 cKO and control mice (Figures 5A and 5B). As shown in Figure 5C, Mettl3 cKO resulted in fewer and smaller lesions in the esophagus of mice compared with those in the control mice. Significantly decreased tumor burden as indicated by the reduced visible lesion area was observed on cKO mice (Figure 5D). Correspondingly, hematoxylin and eosin (H&E) staining revealed that Mettl3 cKO mice have a significantly decreased number of dysplasias and squamous cell carcinomas than those in the control mice (Figures 5E–5G). IHC staining further uncovered that the Ki67, NOTCH1, and HES1 expression levels are significantly decreased in the ESCC derived from the Mettl3 cKO mice (Figures 5H–5K), suggesting that Mettl3 cKO decreased the NOTCH1 signaling pathway activity and reduced the proliferation of ESCC in vivo. Interestingly, the expression of ESCC cancer stem cell marker SOX2 also decreased in the Mettl3 cKO mice (Figures S3A and S3B), suggesting that METTL3-mediated m6A modification could regulate the cancer stem cell function in vivo. Overall, these results support the critical function of METTL3 for the in vivo ESCC initiation.
Figure 5.

Knockout of Mettl3 inhibits occurrence of ESCC in vivo
(A) Experimental design for inducing mouse ESCC by 4NQO with or without epithelium-specific conditional Mettl3 KO. Keratin14-Cre was activated before the treatment of 4NQO. (B) Western blot with METTL3 antibody. (C) Representative image of esophageal lesions 21 weeks after 4NQO treatment. (D) Quantification of lesion areas in control group and cKO group. (E) Representative H&E staining of esophagus tissues in control group and cKO group. (F) Quantification of dysplasia numbers in control group and cKO group. (G) Quantification of ESCC numbers in control group and cKO group. (H) Representative images of METTL3 and Ki67 IHC staining of esophagus tissues in control group and cKO group. (I) Quantification of Ki67 staining scores in control group and cKO group. (J) Representative images of NOTCH1 and HES1 IHC staining in control group and cKO group. (K) Quantification of NOTCH1 and HES1 staining scores in control group and cKO group. Ctrl, control. Data are represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (Student’s t test).
Mettl3 is essential for ESCC progression in vivo
We further established an in vivo ESCC progression model to determine the role of METTL3 in ESCC progression using the Mettl3 cKO mice. Here we first treated the Keratin14-CreER; Mettl3fl/fl and control mice with 4NQO for 16 weeks to induce ESCC formation. After that, all the mice with the same condition of tumor bearing were injected with tamoxifen to induce Mettl3 cKO in the cKO mice, which allows us to study the function of METTL3 in regulation of the pre-existing ESCC in vivo (Figures 6A and 6B). At week 21, the mice were sacrificed for analysis of METTL3’s function in in vivo ESCC progression. Our data revealed that Mettl3 cKO in the tumor-bearing mice resulted in slower progression and milder tumor symptoms, as shown by the decreased visible lesion area in the cKO mice compared with those in the control mice (Figures 6C and 6D). In addition, Mettl3 cKO in mice with pre-existing ESCC significantly decreased the number of dysplasias and squamous cell carcinomas (Figures 6E–6G). Furthermore, KO of Mettl3 also resulted in the reduced Ki67 expression and NOTCH1 signaling pathway activity in the ESCC (Figures 6H–6K), suggesting that 3METTL3 is essential for the NOTCH1 activation and proliferation activity of the ESCC in vivo. Overall, these data support the essential function of METTL3 in promoting the progression of ESCC in vivo.
Figure 6.

Knockout of Mettl3 inhibits progression of ESCC in vivo
(A) Experimental design for inducing mouse ESCC by 4NQO with or without epithelium-specific conditional Mettl3 knockout. Keratin14-Cre was activated after the treatment of 4NQO for 16 weeks. (B) Western blot with METTL3 antibody. (C) Representative image of esophageal lesions 21 weeks after 4NQO treatment. (D) Quantification of lesion areas in esophageal tissues with or without Mettl3 cKO. (E) Representative H&E staining of esophagus in control group and cKO group. (F) Quantification of dysplasia numbers in control group and cKO group. (G) Quantification of ESCC numbers in control group and cKO group. (H) Representative images of METTL3 and Ki67 IHC staining of esophageal tissues in control group and cKO group. (I) Quantification of Ki67 staining scores of esophageal tissues in control group and cKO group. (J) Representative images of NOTCH1 and HES1 IHC staining of esophageal tissues in control group and cKO group. (K) Quantification of NOTCH1 and HES1 staining scores of esophageal tissues in control group and cKO group. Ctrl, control. Data are represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (Student’s t test).
Overexpression of METTL3 promotes ESCC tumorigenesis in vitro and in vivo in Mettl3 transgenic mice
Given that METTL3 expression is significantly upregulated in human ESCC samples, we determined whether overexpression of METTL3 could promote ESCC progression. Our data revealed that overexpression of METTL3 promotes the expression of NOTCH1 in ESCC cells (Figure 7A). Functionally, METTL3 overexpression promotes the proliferation of ESCC cells, as shown by the Cell Counting Kit-8 (CCK8) assays (Figures 7B and 7C). In addition, overexpression of METTL1 promotes the migration, invasion, and colony formation capacities of ESCC cells (Figures 7D–7O), further supporting the essential function of the METTL3-m6A-NOTCH1 axis in ESCC regulation of progression.
Figure 7.

Overexpression of METTL3 promotes ESCC progression in vitro
(A) Western blot with indicated antibodies of METTL3 overexpressed (OEM3) TE-9 and KYSE-30 cells. (B and C) Proliferation of OEM3 TE-9 cells (B) and KYSE-30 cells (C) by CCK8 assay. (D–G) Wound healing assay of migration ability OEM3 TE-9 cells (D and E) and KYSE-30 cells (F and G). (H–K) Transwell assay of OEM3 TE-9 cells (H and I) and KYSE-30 cells (J and K). (L–O) Colony formation assay of OEM3 TE-9 cells (L and M) and KYSE-30 cells (N and O). Data are mean ± SD. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001 against control group (Student’s t test).
We further generated the Mettl3 conditional knockin (cKI) mouse model to study the role of METTL33 in ESCC tumorigenesis in vivo. The Keratin14-cre mouse was crossed with Mettl3ki/WT mouse to generate the epithelium-specific overexpression of the METTL3 mouse model (Keratin14-Cre; Mettl3ki/WT, cKI). Then the Mettl3 cKI and control mice were treated with 4NQO for 16 weeks to induce ESCC tumorigenesis (Figures 8A and 8B). At week 20, the mice were sacrificed to study the role of METTL3 overexpression in ESCC tumorigenesis in vivo. Our data revealed that Mettl3 cKI mice developed massive tumor burden in esophagus, while only lighter tumor burden occurred in the esophagus of control (Ctrl) mice at week 20 (Figure 8C). The tumor burden as indicated by the visible lesion area in Mettl3 cKI mice was significantly increased compared with that in control mice (Figure 8D). H&E staining revealed that the cKI mice had a significantly increased number of dysplasias and squamous cell carcinomas compared with those in the control mice (Figures 8E–8G), which indicated that forced expression of METTL3 leads to increased tumorigenesis of ESCC. We then performed histological analysis upon the esophagus derived from the two groups of mice. IHC staining uncovered the significant upregulation of METTL3 and Ki67 levels in the cKI mice (Figures 8H and 8I), indicating that the tumors from the cKI mice are more aggressive. Moreover, the expressions of NOTCH1, HES1 and SOX2 are significantly upregulated in the Mettl3 cKI mice (Figures 8J, 8K, S3C, and S3D), confirming that METTL3 activates the NOTCH1 signaling pathway in ESCC in vivo. Overall, our data demonstrate the strong function of METTL3 in promoting ESCC tumorigenesis in vivo.
Figure 8.

Knockin of Mettl3 promotes in vivo ESCC tumorigenesis
(A) Experimental design for inducing mouse ESCC by 4NQO with or without epithelium-specific Mettl3 knockin. (B) Western blot with METTL3 antibody. (C) Representative image of esophageal lesions 20 weeks after 4NQO treatment. (D) Quantification of lesion areas in control group and cKI group. (E) Representative H&E staining of esophagus tissues in control group and cKI group. (F and G) Quantification of dysplasia numbers (F) and ESCC numbers (G) in esophagus with or without Mettl3 cKI. (H) Representative images of 3METTL3 and Ki67 IHC staining in esophageal tissues with or without Mettl3 cKI. (I) Quantification ofMETTL3 and Ki67 staining scores in control group and cKI group. (J) Representative images of NOTCH1 and HES1 IHC staining in control group and cKI group. (K) Quantification of NOTCH1 and HES1 staining scores in control group and cKI group. Ctrl, control. Data are represented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 (Student’s t test).
Discussion
Here we uncovered that the m6A methyltransferase METTL3 is significantly upregulated in ESCC and associated with poor ESCC prognosis. Our data revealed that depletion of METTL3 decreases human esophageal carcinoma cells’ proliferation, invasion, and migration capacities, while overexpression of METTL3 promotes ESCC progression in vitro. Most importantly, our in vivo ESCC models using epithelium-specific Mettl3 knockout and knockin mice further supported the strong physiological functions of METTL3 in ESCC occurrence and progression in vitro. Mechanistically, we found that the NOTCH1 signal pathway is a crucial m6A target that facilitates METTL3’s function in ESCC progression. Overall, our work provided strong in vitro and in vivo evidence supporting the important function of m6A in the regulation of ESCC tumorigenesis and progression.
Studies in the past few years have revealed that m6A modification plays a key role in different bioprocesses, including RNA processing, tissue development, DNA damage or heat shock responses, circadian clock control, spermatogenesis, maternal-to-zygotic transition, angiogenesis, as well as self-renewal and differentiation of embryonic stem cells.20, 21, 22 More recently, extensive efforts have been made to study the biological impacts of dysregulated m6A modification and the related mechanisms in various cancers. Emerging evidence supported that m6A modification on mRNAs can promote the development and progression of cancers, such as bladder cancer, lung cancer, and breast cancer.16,23, 24, 25, 26 Here we revealed the important oncogenic function of METTL3 in regulation of ESCC in vitro and in vivo. Most importantly, we established an in vivo ESCC tumor model using Mettl3 cKO and cKI mouse models to provide direct and strong evidence supporting the critical function of METTL3 ESCC initiation and progression.
Mechanistically, we uncovered that the NOTCH1 signaling pathway is a critical m6A target that mediates METTL3’s function in promoting ESCC progression. The Notch signaling pathway is a highly conserved pathway that regulates many aspects of cancer biology.27 NOTCH1 is significantly upregulated in ESCC, and mis-regulation of the Notch pathway promotes ESCC initiation and progression; therefore, targeting the Notch pathway is a promising therapeutic strategy for the treatment of ESCC.28, 29, 30 METTL3-mediated m6A modification could regulate downstream mRNA’s splicing, transport, stability, and translation. Our data revealed that NOTCH1 mRNA has strong and specific m6A modification, and knockdown of METTL3 reduces the mRNA levels of NOTCH1, suggesting that METTL3-mediated m6A modification regulates the stability of NOTCH1 mRNA. Furthermore, we found that forced activation of the NOTCH1 pathway can rescue the growth, migration, and invasion capacities of METTL3 knockdown cells, suggesting that METTL3-mediated m6A modification promotes ESCC progression by regulation of the Notch signaling pathway.
Overall, we demonstrate strong in vitro and in vivo evidence supporting the critical function of METTL3-mediated m6A modification in promoting ESCC initiation and progression. Our data uncovered novel molecular mechanisms regulating the ESCC pathogenesis and provided new insights for the development of effective therapeutic strategies for ESCC treatment.
Materials and methods
Animals
All animal care and experiments protocols were approved by the Institutional Ethics Committee for Clinical Research and Animal Trials of the First Affiliated Hospital, Sun Yat-sen University. The study was in compliance with all relevant ethical regulations regarding Reporting of In Vivo Experiments (ARRIVE) guidelines. Mice were euthanized when they met with the institutional euthanasia criteria for tumor size and overall health condition. Experiments were carried out using mixed male and female animals.
Clinical samples
Paraffin-embedded specimens of tumor and peri-tumor tissues from patients with esophageal carcinoma in this study were obtained from Sun Yat-sen University Cancer Center (Guangzhou, China). Ethical approval for research on human subjects was obtained from the Institutional Review Board of Sun Yat-sen University Cancer Center, and written consent was obtained from all patients prior to analysis.
Cell culture
All the cells were cultured in DMEM (GIBCO, USA) with 10% fetal bovine serum (FBS; GIBCO, USA), 1% streptomycin (GIBCO, USA), and 1% penicillin (GIBCO, USA). Cells were cultured in a water-saturated atmosphere under 5% CO2 at 37°C.
IHC
Paraffin sections with a thickness of 5 μm were baked for 1 h at 65°C. Prior to immunostaining, paraffin sections were deparaffinized and rehydrated in an alcohol series, followed by antigen retrieval with microwave (MW) heating in Citrate Antigen Retrieval Solution. Sections were blocked with 5% normal goat serum and incubated with the primary antibodies against METTL3 or Ki67 at 4°C overnight. Then immunoreactions were detected with an UltraSensitive S-P Ultrasensitive Kit (horseradish peroxidase [HRP], anti-rabbit) and visualized with 3, 3' -Diaminobenzidine (DAB) substrate. Images were acquired with a microscope (ZEISS, Germany). The protein expression level was evaluated by a semiquantitative immunoreactivity scoring system. The staining intensity was scored as 0 (absent), 1 (weak), 2 (moderate), and 3 (strong), and the proportion of positive staining cells was scored as 0, 1, 2, and 3 for absent, <10%, 10%–50%, and >50%, respectively. The product of intensity score and proportion score resulted in an immunoreactivity scoring system index and was used to evaluate the immunoreactivity of METTL3.
Lentiviral transduction
Lentiviral vectors expressing pLKO.1 shRNAs targeting GFP and METTL3 were obtained from Open Biosystems, Inc. The lentiviral vectors were co-transfected with packaging vector pCMVΔR8.9 and enveloped vector pCMV-VSVG into 293T cells using Lipofectamine 2000 reagent (Invitrogen, USA) for lentivirus production. Viruses were collected to infect TE-9 and TE-10 cells with 8 μg/mL Polybrene (Solarbio, China). The stable infected cells were selected with puromycin (Solarbio, China) (3 μg/mL) for 48 h. The sequences of shRNAs used in this study were listed in Table S1.
Plasmid construction
METTL3 expression plasmid was generated by cloning the full-length open reading frame (ORF) of human METTL3 gene (GenBank: NM_019852.5) into pFLAG-CMV2 vector as previously described.15 Overexpression of METTL3 was performed using Lipofectamine 2000 reagent (Invitrogen, USA) following the manufacturer’s instructions.
RNA isolation and qRT-PCR
RNA isolation and qRT-PCR were performed as previously described.31 In brief, total RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s instructions. Reverse transcription was performed with PrimeScript RT reagent Kit (Takara, Japan) using 2 μg total RNA in a 20-μL reaction system. After reverse transcription, the cDNA samples were diluted by 1:20 for the analysis of gene expression using qRT-PCR. qRT-PCR was carried out by TB Green Premix Ex Taq II (Takara, Japan) in StepOnePlus real-time PCR system (Thermo Scientific, USA). Each sample was replicated three times, and relative mRNA expression was calculated using β-ACTIN as an internal control. Primers used in this study were listed in Table S1.
m6A MeRIP-seq and MeRIP qRT-PCR
The m6A MeRIP-seq and MeRIP qRT-PCR assays were performed as previously described.15 In brief, total RNA was fragmented using ZnCl2 and incubated with anti-m6A polyclonal antibody (202003; SynapticSystems) at 4°C for 2 h and then treated with protein A/G magnetic beads (88802; Thermo Fisher Scientific) at 4°C for an additional 2 h. The bound RNA was purified and then used for sequencing library construction and qRT-PCR. The library was sequenced with the Illumina Next-Seq 500 sequencer and analyzed as previously described.15 Methylated sites on RNAs (peaks) were identified with Model-based Analysis of ChIP-Seq (MACS) software.
Western blotting analysis
SDS loading buffer was added to samples and boiled for 5 min at 95°C. Samples were loaded on 10% SDS-PAGE gels and performed with electrophoresis followed by transfer to polyvinylidene fluoride membrane subjected to immunoblotting with different antibodies. After incubation with HRP-conjugated secondary antibodies (CST) at room temperature for 2 h, immunoreactions were visualized using ECL Plus (Thermo Scientific, USA). Antibodies used in this study were listed in Table S2.
Cell proliferation assay
Fifteen hundred cells were seeded into the well of a 96-well culture plate and cultured in 100 μL of culture medium in a humidified, 5% CO2 atmosphere. CCK8 Reagent (Dojindo, Japan) was used to detect cell proliferation following the manufacturer’s instructions. The plate was incubated at 37°C for 1.5 h in a humidified, 5% CO2 atmosphere. Finally, the absorbance was recorded at 450 nm using an Automatic microplate reader (Bio-Tek, USA).
Clone formation assay
Five hundred cells were seeded in six-well plates with 2 mL fresh medium and grown for 2 weeks. Visible colonies were stained with 0.1% crystal violet, and colony numbers were calculated.
Wound healing assay
One million cells were seeded into the well of six-well culture plates in 3 mL culture medium with FBS and culture until confluent. Use a pipette tip to make a straight scratch in each well, and replace the medium by DMEM (GIBCO, USA) without FBS. Count and record the width of the scratch at 0, 12, and 24 h under a microscope (Leica, Germany).
Cell migration assay
Migration assays were performed using 24-well Transwell inserts with an 8-μm pore size (Corning, USA). One hundred thousand cells in 200 μL of culture medium without FBS were plated in the upper chambers, and 600 μL of culture medium containing 20% FBS was placed in each bottom chamber. After incubation for 24 h at 37°C, the invaded cells were fixed with 100% methanol for 20 min and stained with 0.1% crystal violet solution for 15 min at room temperature. The invaded cells were counted and recorded under a microscope (Leica, Germany).
Cell invasion assay
Transwell Matrigel invasion assays were performed using 24-well Transwell inserts with an 8-μm pore size (Corning, USA). A 24-well permeable support plate was coated with 200 μg/mL Corning Matrigel matrix (Corning, USA) followed by incubation at 37°C for 5 h to solidify the matrigel matrix. The following steps beginning with seeding cells were performed as cell migration assay.
GO and pathway analysis
GO analysis was performed as previously described.31 In brief, GO of TE-decreased genes was conducted using the ToppFun module of the ToppGene Suite (https://toppgene.cchmc.org/enrichment.jsp). The ontology terms with Benjamini-Hochberg adjusted p value (false discovery rate [FDR]) < 0.05 were considered as significantly enriched. Pathway analysis was conducted using the GSEA toolkit by pidpathway.
Generation of epithelium-specific conditional Mettl3 cKO/cKI mice
K14-Cre mice or K14-CreER mice were crossed with Mettl3ki/ki or Mettl3fl/fl mice to obtain K14-Cre; Mettl3ki/WT mice and K14-CreER; Mettl3fl/WT as heterozygous epithelial-specific Mettl3 cKI mice or heterozygous epithelial-specific Mettl3 cKO mice. The mice were further crossed to generate K14-CreER; Mettl3fl/fl mice, which were ready for experiment. The genotypes of transgenic mice were identified by PCR amplification of target DNA and agarose gel electrophoresis. Genomic DNA was extracted from mouse tails by Mouse Tail Direct PCR Kit (Foregene, China). Primers for floxed Mettl3 cKO/cKI allele genotyping were listed in Table S1.
ESCC tumorigenesis mouse models
For induction of ESCC, 4-week-old mice were treated with drinking water containing 50 μg/mL 4NQO (Sigma-Aldrich, USA) for 16 weeks and then given normal drinking water for another 4–5 weeks. Cre was activated by the intraperitoneal injection of tamoxifen (Sigma-Aldrich, USA) at a dose of 9 mg per 40 g body weight every other day for a total of three injections. For tumor measurement, mice were sacrificed, and the esophagus was dissected immediately. The surface areas of tumors were measured as described previously.14 Intact esophagus was fixed in 4% paraformaldehyde for 48 h, embedded in paraffin, and sectioned into 5-μm sections. The dysplasia and ESCC were determined and counted in the H&E-stained sections according to the following criteria: epithelial dysplasia confined to esophageal epidermis without obvious invasion, and the basement membrane was relatively intact (dysplasia); distinct invasion, unclearness, or loss of basement membrane; drop; and diffuse or extensive infiltration into the muscle layer (ESCC).
Statistical analysis
Error bars denote standard deviations (SDs). Two groups of independent samples were compared by two-sample t test. The results of the cell proliferation and cell migration experiments were analyzed using repeated-measures ANOVA. Protein expression data for esophageal carcinoma clinical tissue sections were analyzed using the chi-square test. Pearson’s correlation was used for correlation analysis. For a test with a size of α = 0.05, p < 0.05 was considered statistically significant (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Data and code availability
Additional information is available at Molecular Therapy-Nucleic Acids’s website (https://www.sciencedirect.com/journal/molecular-therapy-nucleic-acids). The accession number of raw sequencing data deposited to NCBI Gene Expression Omnibus (GEO) is GEO: GSE179267 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179267).
Acknowledgments
This work was supported by grants from National Natural Science Foundation of China (81772999, 81974435, 81872409, and 81922052) and Natural Science Foundation of Guangdong Province (2019B151502011 and 2018A030313610).
Author contributions
D.C., Y.-Z.J., and S.L. conceived the study, designed the experiments, and supervised the project. H.H., C.Y., S.Z., M.C., S.G., Y.Z., J.M., Y.L., L.W., S.Z., and Z.W. performed the experiment and analyzed the data. H.H., D.C., and S.L. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.07.007.
Contributor Information
Demeng Chen, Email: chendm29@mail.sysu.edu.cn.
Yi-Zhou Jiang, Email: jiangyz@szu.edu.cn.
Shuibin Lin, Email: linshb6@mail.sysu.edu.cn.
Supplemental information
References
- 1.Reichenbach Z.W., Murray M.G., Saxena R., Farkas D., Karassik E.G., Klochkova A., Patel K., Tice C., Hall T.M., Gang J., et al. Clinical and translational advances in esophageal squamous cell carcinoma. Adv. Cancer Res. 2019;144:95–135. doi: 10.1016/bs.acr.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y.M., Hong P., Xu W.W., He Q.Y., Li B. Advances in targeted therapy for esophageal cancer. Signal Transduct. Target. Ther. 2020;5:229. doi: 10.1038/s41392-020-00323-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 4.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 5.Gaur P., Kim M.P., Dunkin B.J. Esophageal cancer: Recent advances in screening, targeted therapy, and management. J. Carcinog. 2014;13:11. doi: 10.4103/1477-3163.143720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng L., Cheng S., Lin Y., Cui Q., Luo Y., Chu J., Shao M., Fan W., Chen Y., Lin A., et al. CCGD-ESCC: A Comprehensive Database for Genetic Variants Associated with Esophageal Squamous Cell Carcinoma in Chinese Population. Genomics Proteomics Bioinformatics. 2018;16:262–268. doi: 10.1016/j.gpb.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Talukdar F.R., di Pietro M., Secrier M., Moehler M., Goepfert K., Lima S.S.C., Pinto L.F.R., Hendricks D., Parker M.I., Herceg Z. Molecular landscape of esophageal cancer: implications for early detection and personalized therapy. Ann. N Y Acad. Sci. 2018;1434:342–359. doi: 10.1111/nyas.13876. [DOI] [PubMed] [Google Scholar]
- 8.Gao Y.B., Chen Z.L., Li J.G., Hu X.D., Shi X.J., Sun Z.M., Zhang F., Zhao Z.R., Li Z.T., Liu Z.Y., et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014;46:1097–1102. doi: 10.1038/ng.3076. [DOI] [PubMed] [Google Scholar]
- 9.Feng Q., Zhang H., Yao D., Chen W.D., Wang Y.D. Emerging Role of Non-Coding RNAs in Esophageal Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019;21:E258. doi: 10.3390/ijms21010258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X., et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y.G., He C. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng G., Dahl J.A., Niu Y., Fedorcsak P., Huang C.M., Li C.J., Vågbø C.B., Shi Y., Wang W.L., Song S.H., et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang F., Jin H., Que B., Chao Y., Zhang H., Ying X., Zhou Z., Yuan Z., Su J., Wu B., et al. Dynamic m6A mRNA methylation reveals the role of METTL3-m6A-CDCP1 signaling axis in chemical carcinogenesis. Oncogene. 2019;38:4755–4772. doi: 10.1038/s41388-019-0755-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choe J., Lin S., Zhang W., Liu Q., Wang L., Ramirez-Moya J., Du P., Kim W., Tang S., Sliz P., et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561:556–560. doi: 10.1038/s41586-018-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin S., Choe J., Du P., Triboulet R., Gregory R.I. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell. 2016;62:335–345. doi: 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu L., Wu Y., Li Q., Liang J., He Q., Zhao L., Chen J., Cheng M., Huang Z., Ren H., Chen J., et al. METTL3 Promotes Tumorigenesis and Metastasis through BMI1 m6A Methylation in Oral Squamous Cell Carcinoma. Mol. Ther. 2020;28:2177–2190. doi: 10.1016/j.ymthe.2020.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J., Harada B.T., He C. Regulation of Gene Expression by N6-methyladenosine in Cancer. Trends Cell Biol. 2019;29:487–499. doi: 10.1016/j.tcb.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang H., Weng H., Chen J. m6A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell. 2020;37:270–288. doi: 10.1016/j.ccell.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia T.L., Yan S.M., Yuan L., Zeng M.S. Upregulation of METTL3 Expression Predicts Poor Prognosis in Patients with Esophageal Squamous Cell Carcinoma. Cancer Manag. Res. 2020;12:5729–5737. doi: 10.2147/CMAR.S245019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao B.S., Roundtree I.A., He C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao M.D., Jiang Q., Ma Y., Liu C., Zhu C.Y., Sun Y.N., Shan K., Ge H.M., Zhang Q.Y., Zhang H.Y., Yao J., et al. Role of METTL3-Dependent N6-Methyladenosine mRNA Modification in the Promotion of Angiogenesis. Mol. Ther. 2020;28:2191–2202. doi: 10.1016/j.ymthe.2020.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang X., Li M., Yu T., Liu G., Wang J. Reversible N6-methyladenosine of RNA: The regulatory mechanisms on gene expression and implications in physiology and pathology. Genes Dis. 2020;7:585–597. doi: 10.1016/j.gendis.2020.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jin H., Ying X., Que B., Wang X., Chao Y., Zhang H., Yuan Z., Qi D., Lin S., Min W., et al. N6-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMedicine. 2019;47:195–207. doi: 10.1016/j.ebiom.2019.07.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin D., Guo J., Wu Y., Du J., Yang L., Wang X., Di W., Hu B., An J., Kong L., et al. m6A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J. Hematol. Oncol. 2019;12:135. doi: 10.1186/s13045-019-0830-6. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Shi Y., Zheng C., Jin Y., Bao B., Wang D., Hou K., Feng J., Tang S., Qu X., Liu Y., et al. Reduced Expression of METTL3 Promotes Metastasis of Triple-Negative Breast Cancer by m6A Methylation-Mediated COL3A1 Up-Regulation. Front. Oncol. 2020;10:1126. doi: 10.3389/fonc.2020.01126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu X., Gonzalez G., Dai X., Miao W., Yuan J., Huang M., Bade D., Li L., Sun Y., Wang Y. Adenylate Kinase 4 Modulates the Resistance of Breast Cancer Cells to Tamoxifen through an m6A-Based Epitranscriptomic Mechanism. Mol. Ther. 2020;28:2593–2604. doi: 10.1016/j.ymthe.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Majumder S., Crabtree J.S., Golde T.E., Minter L.M., Osborne B.A., Miele L. Targeting Notch in oncology: the path forward. Nat. Rev. Drug Discov. 2021;20:125–144. doi: 10.1038/s41573-020-00091-3. [DOI] [PubMed] [Google Scholar]
- 28.Li Y., Li Y., Chen X. NOTCH and Esophageal Squamous Cell Carcinoma. Adv. Exp. Med. Biol. 2021;1287:59–68. doi: 10.1007/978-3-030-55031-8_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Natsuizaka M., Whelan K.A., Kagawa S., Tanaka K., Giroux V., Chandramouleeswaran P.M., Long A., Sahu V., Darling D.S., Que J., et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat. Commun. 2017;8:1758. doi: 10.1038/s41467-017-01500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lubin D.J., Mick R., Shroff S.G., Stashek K., Furth E.E. The notch pathway is activated in neoplastic progression in esophageal squamous cell carcinoma. Hum. Pathol. 2018;72:66–70. doi: 10.1016/j.humpath.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Lin S., Liu Q., Lelyveld V.S., Choe J., Szostak J.W., Gregory R.I. Mettl1/Wdr4-Mediated m7G tRNA Methylome Is Required for Normal mRNA Translation and Embryonic Stem Cell Self-Renewal and Differentiation. Mol. Cell. 2018;71:244–255.e5. doi: 10.1016/j.molcel.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Additional information is available at Molecular Therapy-Nucleic Acids’s website (https://www.sciencedirect.com/journal/molecular-therapy-nucleic-acids). The accession number of raw sequencing data deposited to NCBI Gene Expression Omnibus (GEO) is GEO: GSE179267 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179267).