

Abstract
Gastric cancer (GC) is an aggressive malignancy that is the third leading cause of cancer mortality worldwide. Localized GC can be cured with surgery, but most patients present with more advanced non-operable disease. Until recently, treatment options for relapsed and refractory advanced GC have been limited to combination chemotherapy regimens, HER-2 directed therapy, and radiation, which lead to few durable responses. Over the past decade, there have been significant advances in our understanding of the molecular and immune pathogenesis of GC. The infectious agents Epstein-Barr virus and Helicobacter pylori perturb the gastric mucosa immune equilibrium, which creates a microenvironment that favors GC tumorigenesis and evasion of immune surveillance. Insights into immune mechanisms of GC have translated into novel therapeutics, including immune checkpoint inhibitors, which have become a treatment option for select patients with GC. Furthermore, chimeric antigen receptor T-cell therapies have emerged as a breakthrough treatment for many cancers, with recent studies showing this to be a potential therapy for GC. In this review, we summarize the current state of knowledge on immune mechanisms of GC and the status of emerging immunotherapies to treat this aggressive cancer, as well as outline current challenges and directions for future research.
Introduction
Gastric cancer (GC) is the fifth most common cancer worldwide, with more than 1 million new cases diagnosed in 2018, and GC is the third most common cause of cancer-related mortality [1]. GC exhibits a male predominance and there are striking regional variations in the incidence of this deadly cancer throughout the world, with Eastern and South-Eastern Asia having the world’s highest GC incidence of 32.1 cases per 100,000 men, followed by Central and Eastern Europe (17. 1 per 100,000 men), and South America (12.7 per 100,000 men) [1]. Within the United States, there is a markedly higher GC incidence among Alaska Native people as compared to the non-Hispanic White population (27.0 versus 7.6 per 100,000 men) [2,3].
GC has a notoriously poor prognosis. In the United States, the 5-year overall survival rate is among the lowest of all malignancies [4]. Definitive surgical resection is the only curative treatment for GC, and perioperative chemotherapy plays an important role in improving clinical outcomes [5]. While modern advances in the use of biologic therapies, neoadjuvant chemotherapy, and chemoradiation have modestly improved treatment outcomes, at least 50% of patients worldwide have unresectable, and thus incurable disease, with a 5-year overall survival (OS) of less than 20% [6]. A growing body of knowledge about the molecular pathogenesis of GC has emerged over the past decade, with a greater understanding of how immune mechanisms contribute to disease pathogenesis. More recently, therapies exploiting the host immune system have changed the landscape of medical oncology. Interruption of immune checkpoint interactions between molecules such as programmed cell death-1 (PD-1) and its ligand (PD-L1) have revolutionized the treatment of many solid tumors. Technologies such as chimeric antigen receptor (CAR) T-cell therapies have emerged as potential new breakthrough therapies for many cancers. This review will highlight recent advances in our understanding of immune mechanisms of GC pathogenesis, current immunotherapies, as well as emerging immune therapies for this devastating cancer, and outline conclusions and research challenges.
Immune Mechanisms of Gastric Cancer Pathogenesis
Cancer progression can be shaped by the interplay between tumor processes and the host immune response. In GC, the complexity of the tumor microenvironment is augmented by the presence of two infectious pathogens of gastric carcinogenesis, Helicobacter pylori (H. pylori) and Epstein-Barr virus (EBV). EBV-associated GC (EBVaGC) comprises approximately 10–20% of GC, with heterogeneity in prevalence throughout the world [7–10]. EBVaGC tumors often appear in the upper portion of the stomach and have a diffuse histology with lymphoid infiltration [11,12]. The Cancer Genome Atlas and others characterized the molecular features of EBVaGC as exhibiting over-expression of PD-L1/2, frequent alterations in the PIK3CA gene, amplification of the Janus kinase 2 gene, and a DNA methylation phenotype [2,8,12–18].
The precise mechanism of how EBV infects the gastric epithelium is unknown, however it is hypothesized that chronic inflammation such as in atrophic gastritis or co-infection with H. pylori serves as a lesion which enables “cell-in-cell” contact between latently infected B-lymphocytes and gastric epithelial cells (Figure 1) [19–21]. EBV infection alters immune response related genes in GC cells, such as major histocompatibility complex class II (MHC-II) and genes that regulate chemokine activity [22]. The presence of viral antigens and alterations to immune response genes allow EBVaGC to recruit reactive immune cells, leading to an inflamed tumor immune microenvironment. EBVaGCs elicit an interferon-gamma (IFN-γ) immune response/immune activated signatures, elevated tumor infiltrating immune stimulatory cells, and fewer CD204+ macrophages known to be associated with aggressive tumor behavior [13, 21–24]. Cytokine profiles from EBVaGC patients also exhibit increased T-cell activation via upregulation of IL-2, IL-12, IL-23, and IL-27 [7].
Figure 1. GC tumor microenvironment.
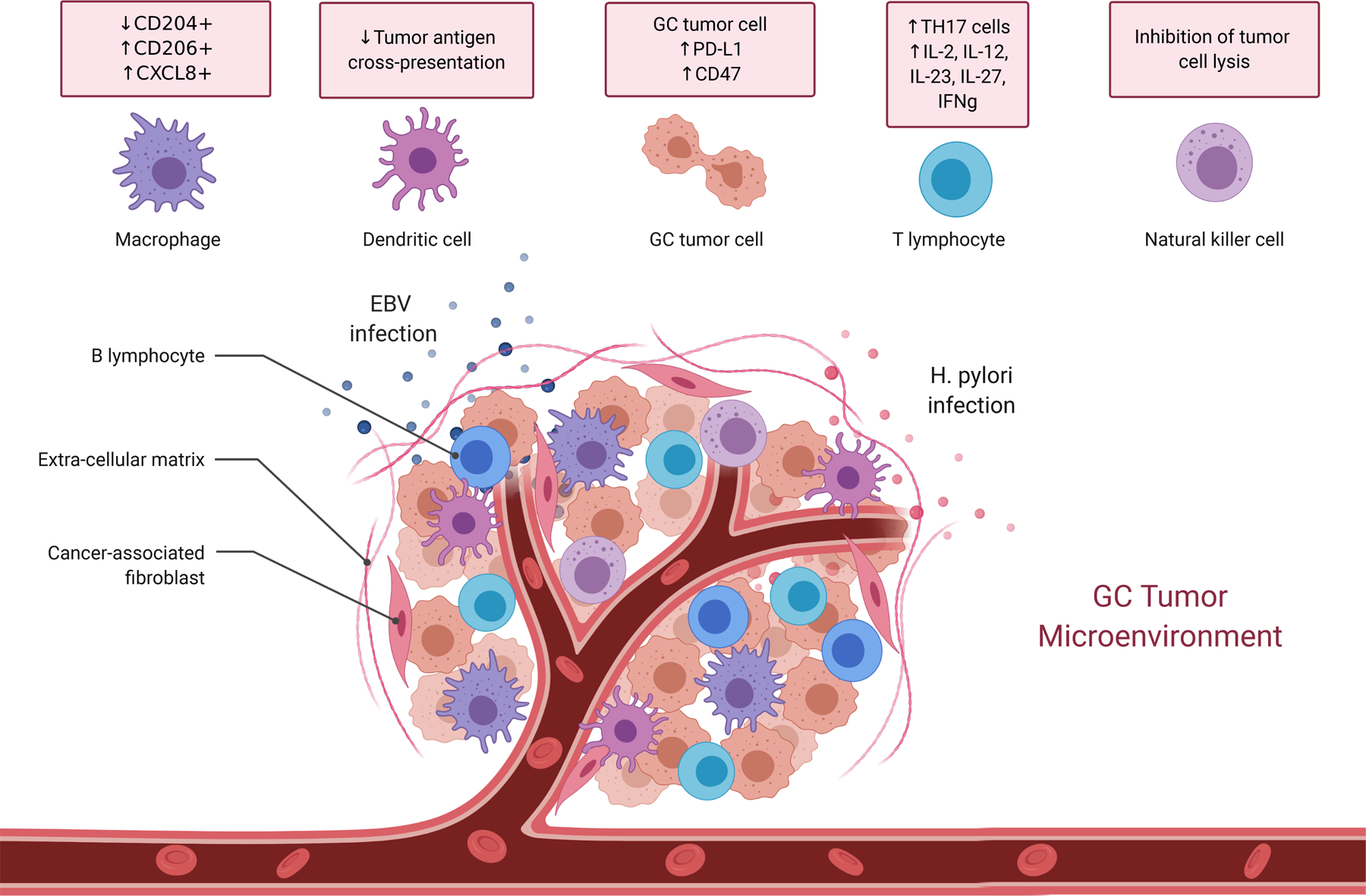
EBV and H. pylori perturb gastric mucosal immune equilibrium, favoring an innate immune phenotype characterized by macrophages exhibiting decreased CD204 expression and increased expression of CD206 and CXCL8, while eliciting tumor infiltrating lymphocytes associated with an IFN-γ response, Th17 cells, and activated T-cells that secrete IL-2, IL-12, IL-23, and IL-27. GC tumor cells escape immune surveillance through mechanisms such as increased expression of PD-L1 and CD47, decreased MHC class II antigen presentation, and inhibition of effector cell lysis.
EBVaGC cells escape immune detection by developing adaptive immune resistance through multiple mechanisms. EBVaGC gene amplification of PD-L1, shown to occur during tumor progression, as well as upregulation of the PIK3CA/Akt pathway can both directly induce PD-L1 protein expression in cancer cells resulting in immune suppression (Figure 1) [7,12]. PD-L1 and the potent immune cell inhibitor indoleamine 2,3-dioxygenase (IDO) are both upregulated in response to IFN-γ secreting CD8+ T-cells, ultimately enabling the cancer cells to evade immune detection [25,26]. Furthermore, expression of CD47, a “do not eat me” signal, is upregulated in EBVaGC, and high expression correlates with a worse prognosis (Figure 1) [27].
The presence of H. pylori and EBV in the gastric mucosa increases the severity of the inflammatory response, thus increasing the overall risk of developing GC [28,29]. The pathogens cooperate in multiple mechanisms to promote growth of each other as well as enhance gastric inflammation and tissue damage [29–31]. In particular, H. pylori dependent stimulation of IFN-γ secretion, one of the key pro-inflammatory cytokines associated with disease severity [31], is implicated in EBV reactivation and intestinal GC [20]. Moreover, EBV-driven epigenetic modifications are enhanced in the presence of H. pylori, specifically the Cag A secretory antigen, resulting in increased cellular proliferation [32]. Furthermore, EBV and H. pylori associated gastric inflammation persistently activates Th17 T-cells which promote severe gastritis and GC [33–35]. Further understanding of the synergistic oncogenic effects of infectious agents in the gastric mucosa as well as the therapeutic significance of eradicating microorganisms for treatment of GC are needed.
Diffuse type GCs, including signet ring cell carcinomas, have been characterized as “cold tumors” which lack infiltrating immune cells [36–38]. Multiple mechanisms likely contribute to the “cold tumor” phenotype. Diffuse tumors are characterized as genomically stable with a lower mutational load and lack PD-L1 expression [7]. Diffuse tumors express lower levels of HLA-DR antigen [39], which may shape tumor antigen specific immune responses. Moreover, E-cadherin deficiency due to CDH1 mutations are implicated in the oncogenic initiation of diffuse and signet ring cell carcinomas [40] and the lack of CD8+ T-cell infiltrates [41]. Further investigation into mechanisms governing the “cold tumor” phenotype in diffuse GC may reveal targets which can be exploited in future immunotherapies.
Tumor immune surveillance can result in spontaneous cell mediated immune responses against cancer [42]. Analysis of the GC tumor microenvironment demonstrates that tumor infiltrating lymphocytes (TIL), including intratumoral T-cell and NK cells, correlate with improved survival [43,44]. However, the immune phenotype and the ability of immune cells to recognize and infiltrate tumors plays an important role in cancer detection and elimination. The transcription factor T-bet regulates mucosal homeostasis, promotes the Th1 phenotype, and prevents CD8+ T-cell exhaustion [45]. Patients with higher numbers of T-bet+ TIL exhibit longer survival [46]. The immune phenotype of macrophages also correlates with GC clinical outcomes, with CD11c+ cells being associated with improved survival [47], and CD206+ and CXCL8+ macrophages correlated with poor prognosis (Figure 1) [47,48]. Therefore, the TIL immune phenotype may be useful as a clinical prognostic factor and perturbing it may provide therapeutic utility.
Immune Directed Therapies for Gastric Cancer
Since the first approval for the immune checkpoint inhibitor ipilimumab for melanoma in 2011, immune therapies have revolutionized treatment for solid tumors, with a rapidly expanding number of indications for their use, and the United States Food and Drug Administration (FDA) approval of six additional agents [49]. Research in GC has lagged behind many solid tumors with respect to immune checkpoint inhibitor studies, but there is an increasing body of literature in this field (Table I). The initial GC clinical studies were in patients who progressed after chemotherapy. The PD-1 inhibitor nivolumab was compared to placebo in the randomized phase III trial ATTRACTION 02 conducted at 49 Asian clinical centers in 493 patients with non-operable advanced gastric and gastroesophageal junction (GEJ) cancers who had progressed after two or more lines of chemotherapy [50]. The ATTRACTION 02 trial reported 12-month overall survival rates of 26.2% (95% CI 20.7–32.0) with nivolumab and 10.9% (6.2–17.0) with placebo in an unselected patient population, demonstrating an encouraging response signal in this poor prognosis population [50]. A subset analysis of 226 Japanese patients enrolled in ATTRACTION 2 demonstrated a median OS that was longer with nivolumab versus placebo (5.4 months, 95% CI 4.6–7.4 versus 3.6 months, 95% CI 2.8–5.0) [51]. This trial led to the approval of nivolumab in Japan as a therapy for unresectable advanced or recurrent GC that has progressed on chemotherapy. A post-hoc analysis of variables associated with a favorable response to nivolumab on this trial demonstrated that the highest overall response rate (ORR) were observed in patients with an Eastern Cooperative Oncology Group (ECOG) performance status of 0; those whose tumors were deficient in DNA mismatch repair (dMMR); and those with PD-L1 positivity, PIK3CA mutations, a high tumor mutation burden, and Epstein-Barr virus positivity [52]. These variables warrant further investigation in prospective studies.
Table I.
Gastric Cancer Immunotherapy Trials
| Immunotherapy Trial | Trial Design | Treatment | Patients | Median PFS months (95% CI) | Median OS months (95% CI) |
Ref |
|---|---|---|---|---|---|---|
| ATTRACTION-2 | Phase III Trial 3rd line |
nivolumab vs placebo |
330 163 |
1.61 (1.54–2.30) 1.45 (1.45–1.54) |
5.26 (4.60–6.37) 4.14 (3.42–4.86) |
50 |
| NCT01585987 | Phase II Trial 2nd line |
ipilumimab vs supportive care |
57 57 |
2.92 (1.61–5.16) 4.90 (3.45–6.54) |
12.7 (10.5–18.9) 12.1 (9.3-NA) |
53 |
| KEYNOTE-158 | Phase II Trial 2nd line, MSI-H |
pembrolizumab | 24 | 11.0 (2.1-NR) | NR (7.2-NR) | 54 |
| KEYNOTE-059 | Phase II Trial 3rd line |
pembrolizumab | 259 | 2.0 (2.0–2.1) | 5.6 (4.3–6.9) | 56 |
| KEYNOTE-061 | Phase III Trial 2nd line |
pembrolizumab paclitaxel |
196 199 |
1.5 (1.4–2.0) 4.1 (3.1–4.2) |
9.1 (6.2–10.7) 8.3 (7.6–9.0) |
58 |
| JAVELIN | Phase III Trial 3rd line |
avelumab chemotherapy |
185 186 |
1.4 (1.4–1.5) 2.7 (1.8–2.8) |
4.6 (3.6–5.7) 5.0 (4.5–6.3) |
59 |
| KEYNOTE-062 | Phase III Trial 1st line PD-L1 CPS 1+ |
pembrolizumab chemotherapy pembro/chemo |
256 250 257 |
2.0 (1.5–2.8) 6.4 (5.7–7.0) 6.9 (5.7–7.3) |
10.6 (7.7–13.8) 11.1 (9.2–12.8) 12.5 (10.8– 13.9) |
60 |
| Checkmate 032 | Phase I/II 3rd line |
nivolumab 3mg/kg nivolumab 1mg/kg + ipilumumab 3mg/kg nivolumab 3mg/kg + ipilumumab 1mg/kg |
59 49 52 |
1.4 (1.2–1.5) 1.4 (1.2–3.8) 1.6 (1.4–2.6) |
6.2 (3.4–12.4) 6.9 (3.7 to 11.5) 4.8 (3.0–8.4) |
61 |
| NCT02340975 | Phase IB/II 3rd line |
durvalumab tremelimumab durval/tremel |
24 12 52 |
1.6 (1.0–1.8) 1.7 (0.8–5.3) 1.8 (1.6–3.3) |
3.4 (1.7–4.4) 7.7 (2.1–13.7) 9.2 (5.4–12.6) |
62 |
| NCT02572687 | Phase Ib 2nd line |
durvalumab + ramucirumab | 29 | 2.6 (1.5–7.1) | 12.4 (5.5–16.9) | 63 |
| KEYNOTE-659 | IIb 1st line PDL-1+ |
pembrolizumab + SOX |
54 | 9.4 (6.6–NE) | NR | 64 |
The CTLA-4 inhibitor ipilimumab administered as monotherapy was investigated in a phase II study among 143 patients with non-operable advanced GC and GEJ cancer after having achieved an objective response to front line chemotherapy [53]. Patients were randomized to ipilimumab versus best supportive care. The study was stopped after the first planned analysis due to a lack of benefit of ipilimumab compared to supportive care, with both groups achieving an OS of approximately one year each [53].
The PD-1 inhibitor pembrolizumab was investigated in a “basket trial” of multiple tumor types that were histologically and cytologically confirmed to have microsatellite instability high (MSI-H) and to be deficient in dMMR, including 24 patients with advanced GC in the phase II trial KEYNOTE-158 [54]. The GC cohort exhibited an ORR of 45.8% (25.6 to 67.2%) a median progression free survival (PFS) of 11.0 months (2.1 to NR), and OS that was not reached (NR) (7.2 to NR) [54]. This trial prompted the FDA to approve pembrolizumab for patients with MSI-H/dMMR tumors regardless of histology. Pembrolizumab was studied in the open label multicenter phase Ib trial KEYNOTE-012 in patients with PD-L1-positive recurrent or metastatic GC or GEJ adenocarcinoma. In 36 evaluable patients there were 8 (22%, 95% CI 10–39) partial responses [55].
In the international phase II KEYNOTE-059 trial, 259 patients with non-operable advanced gastric and GEJ cancers were treated with pembrolizumab after three or more prior lines of chemotherapy [56]. The objective response (CR + PR) rate in those treated with pembrolizumab was 11.6% (95% CI, 8.0%−16.1%; 30 of 259 patients), with a complete response in 2.3% (95% CI, 0.9%−5.0%; 6 of 259 patients), and a median response duration of 8.4 months [56]. Outcomes were improved in patients whose tumors expressed PD-L1, with an ORR of 15.5% (95% CI, 10.1%−22.4%; 23 of 148 patients), while patients with PD-L1–negative tumors exhibited an ORR of 6.4% (95% CI, 2.6%−12.8%; 7 of 109 patients) [56]. The median response duration was 16.3 (1.6+ to 17.3+) months in PD-L1 positive patients and 6.9 (2.4 to 7.0+) months in patients with PD-L1−negative tumors [56]. This trial led to the FDA approval of pembrolizumab for patients who have received two previous lines of chemotherapy whose tumors express PD-L1. A second translational phase II study was conducted to better define molecular features that correlate with response to pembrolizumab [57]. Sixty one Korean patients were treated with pembrolizumab as second or third line treatment for metastatic GC with pre- and post-treatment biopsies performed, and an extensive molecular profiling of the tumors was conducted [57]. They observed high response rates in patients whose tumors were MSI-H (7 patients, 85.7% ORR) and EBV positive (6 patients, 100% ORR), and those whose tumors were PD-L1 positive (55 patients, 50% ORR) [57].
Pembrolizumab was compared to paclitaxel in 592 non-operable advanced GC and GEJ patients who progressed on front line fluoropyrimidine and platinum combinations studied in the randomized phase III KEYNOTE-061 trial [58]. 395 patients whose tumors expressed PD-L1 with a combined positive score (CPS) of 1% or higher were enrolled. Pembrolizumab did not significantly improve OS or PFS versus second-line paclitaxel therapy in patients with a PD-L1 CPS of 1 or higher, and in patients with a PD-L1 CPS of less than 1% pembrolizumab treatment resulted in a lower median OS of 4.8 (3.9–6.1) months compared to those treated with paclitaxel 8.2 (6.8–10.6) months [58]. The clinical responses in the subset of patients with tumors that are MSI-H/dMMR was not reported as a protocol defined endpoint.
The PD-L1 inhibitor avelumab was compared to physician’s choice of chemotherapy in unselected non-operable advanced GC and GEJ patients who had received two or more prior lines of chemotherapy in the randomized phase III trial JAVELIN Gastric 300 [59]. This trial did not meet the primary endpoints of improvements in OS or PFS compared with chemotherapy, although avelumab was better tolerated than chemotherapy [59].
More recently, front line pembrolizumab was investigated in the phase III KEYNOTE −062 trial of 763 patients with non-operable advanced gastric and GEJ cancers randomized 1:1:1 to chemotherapy, pembrolizumab, or a combination of the two [60]. The patient’s tumors had to be erythroblastic oncogene B2 (ERBB2/HER-2) negative and express PD-L1 with a CPS of 1 or greater. The investigators reported that pembrolizumab was non-inferior to chemotherapy for OS in patients with CPS of 1 or higher, and was better tolerated than chemotherapy with fewer adverse events [60]. They further reported that pembrolizumab prolonged OS versus chemotherapy in patients with a CPS of 10 or higher (median, 17.4 vs 10.8 months; HR, 0.69; 95% CI, 0.49–0.97), but this comparison was not statistically tested because it did not reach the protocol defined threshold for superiority [60]. Pembrolizumab plus chemotherapy was not superior to chemotherapy alone in terms of OS in patients with CPS of 1 or greater, or CPS of 10 or higher, or for PFS in patients with CPS of 1 or greater [60].
Taken together, these studies demonstrate that immune checkpoint blockade with pembrolizumab is emerging as an option for patients with non-operable advanced gastric and GEJ cancers with a prospective trial demonstrating non-inferiority to chemotherapy in the front line setting for patients whose tumors are ERBB2 negative and exhibit PD-L1 positivity with a CPS of 1 or higher. Furthermore, in the United States pembrolizumab is a treatment option for patients whose tumors exhibit MSI-H/dMMR who have received one prior line of chemotherapy, and for patients with PD-L1 positive tumors who have received two or more prior lines of chemotherapy. Other immune checkpoint inhibitors such as nivolumab and avelumab demonstrate objective responses in this poor risk population, and a favorable toxicity profile compared to chemotherapy. There are currently limited data on clinical biomarkers other than PD-L1 positivity that predict response to immune checkpoint inhibitors, although preliminary studies indicate that MSI-H/dMMR, PIK3CA mutations, a high TMB, and EBV positivity are promising indicators. Further work on identifying subsets of patients who benefit from immune blockade in prospective clinical trials is needed.
Emerging Immune Therapy Trials
With immune checkpoint inhibitor monotherapy now established as a treatment option for non-operable advanced GC and GEJ cancers, an emerging trend is to evaluate these compounds in combination with other agents. In the phase I/II Checkmate 032 trial, 160 patients with locally advanced non-operable and metastatic GC, GEJ cancer, and esophageal cancer were randomized to nivolumab 3 mg/kg (59 patients), nivolumab 1 mg/kg plus ipilimumab 3 mg/kg (49 patients), and nivolumab 3 mg/kg plus ipilimumab 1mg/kg (52 patients) [61]. Patients had received multiple lines of prior chemotherapy. At twelve months of follow up, there were objective responses in all patients groups with PFS rates of 8%, 17%, and 10% and OS rates of 39%, 35%, and 24% respectively. These encouraging results have led to larger phase III studies that are ongoing.
A more recent multicenter phase Ib/II study enrolled 113 non-operable advanced GC and GEJ cancer patients and randomized them to treatment with the PD-1 inhibitor durvalumab, the CTLA-4 inhibitor tremelimumab, or the combination of the two [62]. A tumor based IFN-γ gene signature was incorporated prospectively in this trial as well as on-treatment circulating tumor DNA levels. Response rates were low in all treatment arms with ORR ranging from 0%−8.3% [62]. Durvalumab was also studied in combination with ramucirumab in a multi-center single arm phase Ib study enrolling 29 patients with non-operable advanced GC and GEJ cancer [63]. The combination was well tolerated and was associated with a 21% ORR, and median PFS and OS of 2.6 and 12.4 months respectively [63]. Larger prospective studies are underway.
Recent studies have also explored the combination of immune therapies and chemotherapy in patients with non-operable advanced GC and GEJ cancers. Pembrolizumab was studied in combination with the oral fluoropyrimidine S-1 and oxaliplatin (SOX) administered as front line therapy to Japanese patients with non-operable advanced GC and GEJ cancers in the phase IIb KEYNOTE-659 study [64]. In an initial report published this year on the first cohort of 54 patients, after 10.1 months of follow up pembrolizumab/SOX resulted in an ORR of 72.2% (95% CI 58.4–83.5), with a PFS of 9.4 months and OS not reached [64]. The toxicity and safety profile of this combination was in line with the effects of these drugs given individually [64], indicating that further studies in larger numbers of patients are warranted.
Chimeric antigen receptor (CAR) T-cells have emerged as a powerful immune therapy for relapsed and refractory non-Hodgkin lymphomas and other hematologic malignancies, with many patients achieving durable long-term remissions [65,66]. CARs are constructed by introducing a single variable chain of an antibody (scFv) domain directed against a tumor-specific antigen as an extracellular molecule expressed in tandem with signal transduction domains of the T-cell receptor CD3 (Figure 2). Second and third generation CAR T-cell constructs also include genes encoding one or more co-stimulatory molecules such as CD28, CD138, or 4–1BB. The CAR construct is introduced into the patient’s autologous T-cells collected by apheresis and expanded in culture. This creates a targeted cellular therapy directed against the surface tumor specific antigen that kills upon binding without the requirement for self-antigen presentation. The CAR T-cells are then infused back into the patient where they elicit direct cytotoxicity, as well as proliferation of native T-cells and a cytokine response that leads to tumor destruction that can persist for months to years.
Figure 2.
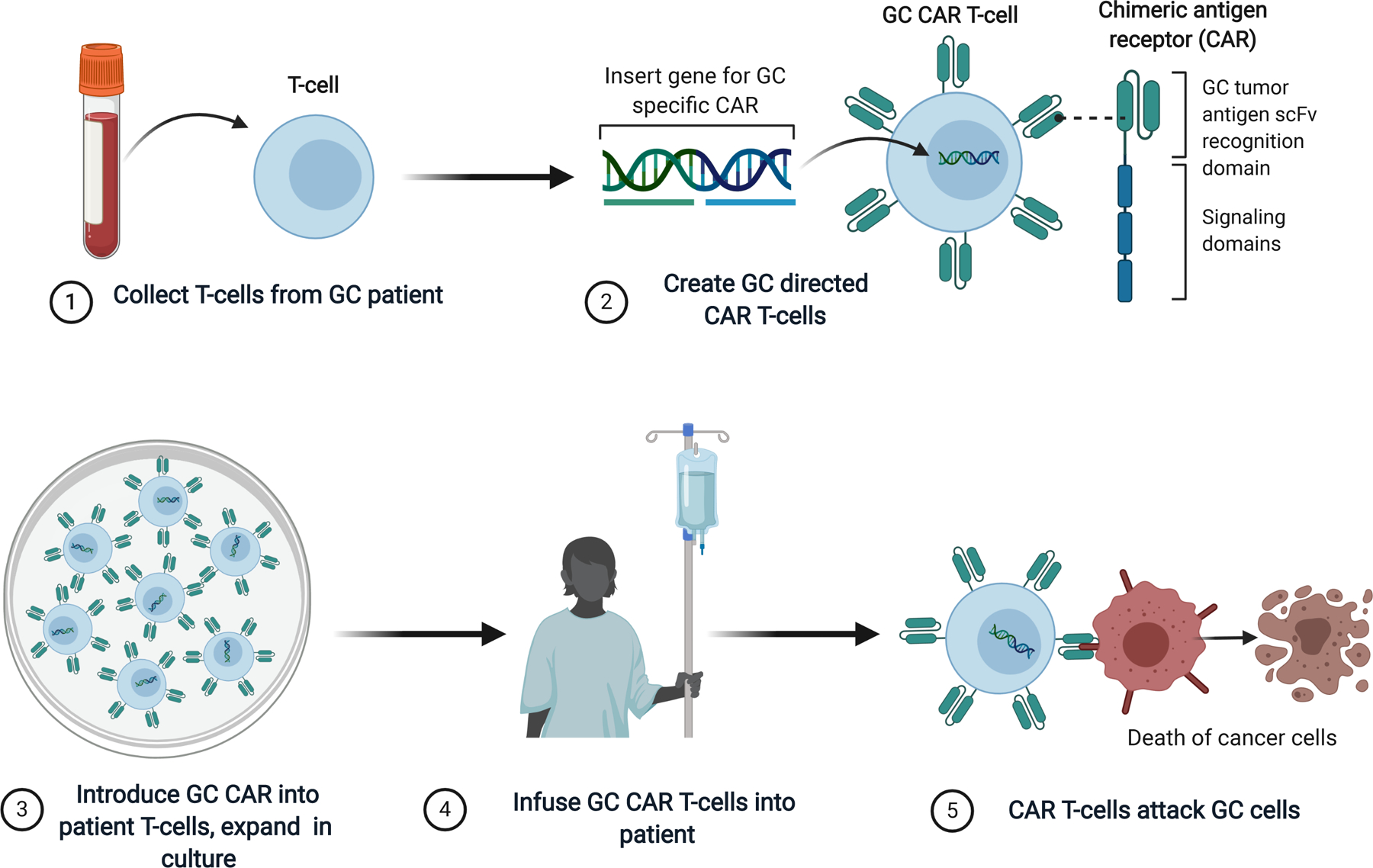
Schematic of GC CAR T-cell design and mechanism of action.
Many initial attempts at CAR T-cell therapy for solid tumors were disappointing. However, persistent effort at developing more effective CAR T-cell constructs directed against solid tumors, including GC and GEJ cancers, has led to some encouraging clinical responses. A variety of GC tumor antigen targets are being investigated in CAR T-cell constructs [67]. Claudin18.2 (CLDN18.2), a gastric membrane protein expressed in 70% of GC tumors, was recently demonstrated to have potential utility as a target in a CAR T-cell construct. CLDN18.2-specific CAR T-cells were studied in a mouse GC xenograft model, which resulted in partial or complete tumor elimination, without any deleterious effect on the normal organs including the stomach [68]. An open label phase I first in human clinical trial (NCT03159819) utilizing this approach recently reported an ORR of 33% and a median PFS of 130 days (95% CI 38– 230) in 11 evaluable patients with advanced GC and pancreas cancer treated with CLDN 18.2-specific CAR T-cells [69]. Further clinical trials are ongoing.
The mucin transmembrane adhesion protein MUC1 has been extensively studied in epithelial cancers as a prognostic factor and a potential marker of tumor progression because of its role in stromal and endothelial cell adhesion and its effects on IL-11 secretion [70]. MUC1 is aberrantly glycosylated in tumors which creates a unique target for immune therapies [71] and it is over-expressed in GC [8]. A recent meta-analysis demonstrated that MUC1 expression in GC tumors is correlated with a higher rate of vascular and lymph node invasion and a lower 5 year OS [72,73]. A gene encoding an scFv directed against MUC1 has been incorporated into a CAR T-cell construct and shown in pre-clinical models to be effective in selective killing of tumor cells [74]. Clinical trials studying the use of MUC1 CAR T-cells in breast cancer patients are ongoing (NCT04020575), but GC clinical trials have not yet begun.
Epithelial cell adhesion molecule (EpCAM, CD326) is another transmembrane glycoprotein of interest in the study of CAR T-cell directed therapy for GC. EpCAM is highly expressed in epithelial carcinomas and is a potential tumor stem cell marker and target for precision cancer therapy [75]. Expression of EpCAM was recently demonstrated in a meta-analysis to be over-expressed in GC, and associated with larger tumor size, lymph node metastasis, and worse prognosis [76]. An initial trial exploring the use of EpCAM directed immune therapy utilized catumaxomab, a bispecific and trifunctional monoclonal antibody directed against EpCAM, the T-cell marker CD3, and the Fcγ receptors on innate immune cells [77]. Catumaxomab was administered to 31 patients with metastatic GC and peritoneal carcinomatosis- a group with a particularly poor prognosis- in a randomized phase II trial [77]. Patients were randomized to catumaxomab treatment followed by 5-florouracil, oxaliplatin, docetaxel (FLOT) chemotherapy versus FLOT alone [77]. Catumaxomab treatment was tolerable, but median PFS and OS were not significantly different between the two arms [77]. Anti-EpCAM CAR T-cells have been developed [78], and a single-arm multicenter Phase I/II trial treating patients with relapsed or refractory GC with CAR T-cells directed against EpCAM is ongoing (NCT02725125).
Another tumor antigen target being investigated in CAR T- cell therapies for GC is folate receptor 1 (FOLR1), also known as folate receptor alpha and folate binding protein, a glycosylphosphatidylinositol (GPI)-anchored membrane protein that is over-expressed in epithelial malignancies including ovarian, breast, renal, and lung cancers [79]. FOLR1 is over-expressed on 33% of GC and present at low levels on surfaces of epithelial cells in normal tissues [79]. FOLR1 CART-cells were recently studied in a xenograft mouse model and shown to recognize FOLR1- positive GC cells in a MHC-independent manner, induce secretion of cytokines, and induce tumor cell killing [80]. The clinical feasibility of this approach is being investigated in a cohort of patients with ovarian and primary peritoneal cancers (NCT03585764).
There are several other candidate tumor antigen directed scFv genes being investigated in CAR T-cell constructs in pre-clinical models and phase I trials (Table II), including the transmembrane receptor HER2/ERBB2 [81], the GPI-anchored protein mesothelin [82–84], carcinoembryonic antigen (CEA) [85], the transmembrane glycoprotein natural killer group 2D receptor [86,87], the surface glycoprotein CA 72–4 [88,89], and the natural killer cell activating receptor B7H6 [90]. While the field of CAR T-cell therapy for GC is still in its infancy, these preliminary results indicate the feasibility and potential clinical efficacy of this approach.
Table II.
Development of CAR T-cells directed against GC tumor antigens
| Tumor Antigen Target | Development Stage | CAR T- Cell Design | Clinical Trial | Reference |
|---|---|---|---|---|
| Claudin 18.2 | Phase I Trial | Anti- Claudin 18.2 scFv/CD28/CD3 |
NCT03159819
NCT03874897 |
68, 69 |
| MUC1 | Phase I Trial | Anti- MUC1 scFv/CD28/OX40/CD3ζ | NCT04020575 | 71, 74 |
| EpCAM | Phase I/II Trial | Anti-EpCAM scFv/ CD8α/ CD28/4–1BB/CD3ζ | NCT02725125 | 78 |
| FOLR1 | Phase I Trial | Anti-FOLR1 scFv/ CD28/CD3ζ | NCT03585764 | 80 |
| Mesothelin | Phase I Trial | Anti-mesothelin scFv/ CD3ζ/4–1BB | NCT01897415 NCT04503980 | 82, 83, 84 |
| CEA | Phase I Trial | Anti-CEA scFv/CD28/CD3ζ | NCT02349724 | 85 |
| CA 72–4 | Phase I Trial | Anti-CA 72–4 scFv/CD3ζ | — | 88,89 |
| NKG2D | Pre-Clinical | Anti-NKG2D scFv/CD3ζ | — | 86, 87 |
| ERBB2/HER2 | Pre-Clinical | Anti-HER2 scFv/CD137/CD3ζ | — | 81 |
| B7H6 | Pre-Clinical | Anti-B7H6 scFv/CD28/ CD3ζ | — | 90 |
|
|
||||
Conclusions
An expanding knowledge of the immune mechanisms of tumor pathogenesis has led to the development of promising therapies for many cancers. An attractive advantage of this treatment approach is the persistent anti-tumor effects of immune therapy compared to cytotoxic chemotherapy, with many patients achieving responses that last months to years [65]. Immune checkpoint inhibitors are effective in select GC patients, but challenges remain in identifying the most appropriate patients for this treatment. Recent studies have identified EBV positivity, MSI-H/dMMR, PIK3CA mutations, and a high TMB as promising predictive markers, but additional biomarkers are needed. The robust and complex network of inflammatory and immune cells within GC tumors raises the possibility that additional immunotherapies and predictive biomarkers may be identified in the future.
Another formidable challenge in the implementation of immune directed GC therapy is the ability of tumors to escape immune detection and attack through a variety of mechanisms, including decreasing tumor antigen expression, resistance to cytokine signaling, downregulation of major histocompatibility antigen proteins, and upregulation of multiple inhibitory checkpoint signals [91,92]. An emerging approach to overcome these barriers is to combine immune checkpoint inhibitors with CAR T-cell therapies in an effort to enhance sustained tumor cell killing [93]. Clinical trials combining immune checkpoint inhibitors with CAR T-cells to treat a variety of tumors are currently underway [93]. An improved understanding of immune mechanisms of GC pathogenesis may enable development of more effective therapies for this devastating malignancy.
Acknowledgements
Figures illustrations were created with BioRender.com. Research reported in this publication was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number 2P20GM103395. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the NIH.
Abbreviations
- AN
Alaska Native
- CI
confidence interval
- CLDN18.2
Claudin 18.2
- BART
Bam-HI-A rightward transcripts
- BARF1
BAM-HI A rightward frame 1
- CEA
carcinoembryonic antigen
- CPS
combined positive score
- CR
complete response
- dMMR
deficient in mismatch repair
- EBV
Epstein-Barr virus
- EBVaGC
EBV-associated gastric cancer
- EpCAM
epithelial cell adhesion molecule
- FLOT
5-florouracil, oxaliplatin, docetaxel
- FDA
Food and Drug Administration
- ERBB2
erythroblastic oncogene B2
- FOLR1
folate receptor 1
- GC
gastric cancer
- GEJ
gastroesophageal junction
- GPI
glycosylphosphatidylinositol
- H. pylori
Helicobacter pylori
- IDO
indoleamine 2,3-dioxygenase
- IFNγ
interferon gamma
- MHCII
major histocompatibility complex class II
- MSI-H
microsatellite instability high
- NR
not reached
- ORR
overall response rate
- OS
overall survival
- ORR
overall response rate
- PD-1
programmed death-1
- PD-L1
programmed death ligand-1
- PR
partial response rate
- PFS
progression free survival
- TMB
tumor mutation burden
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global Cancer Statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin, in press. The online GLOBOCAN 2018 database is accessible at http://gco.iarc.fr/, as part of IARC’s Global Cancer Observatory. [DOI] [PubMed] [Google Scholar]
- 2.Melkonian SC, Pete D, Jim MA, Haverkamp D, Wiggins CL, Bruce MG, et al. Gastric Cancer Among American Indian and Alaska Native Populations in the United States, 2005–2016. Am J Gastroenterol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinson HA, Shelby NJ, Alberts SR, Olnes MJ. Gastric cancer in Alaska Native people: A cancer health disparity. World J Gastroenterol. 2018; 24(25): 2722–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thrift AP, and El-Serag HB. Burden of Gastric Cancer. Clin Gastroentero. and Hepatol. 2020; 18: 545–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cunningham D, Allum WH, Stenning SP, Thompson JN, Van De Velde CJH, Nicholson M et al. Perioperative Chemotherapy versus Surgery Alone for Resectable Gastroesophageal Cancer. N Engl J Med. 2006; 355: 11–20. [DOI] [PubMed] [Google Scholar]
- 6.Wagner AD, Syn NL, Moehler M, Grothe W, Yong WP, Tai BC, et al. Chemotherapy for advanced gastric cancer. Cochrane Database Syst Rev. 2017August29;8(8):CD004064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research Network. 2014. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature. 2014; 513(7517):202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martinson HA, Mallari D, Richter C, Wu TT, Tiesinga J, Alberts SR et al. Molecular Classification of Gastric Cancer among Alaska Native People. Cancers. 2020; 12:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Owen GI, Pinto MP, Retamal IN, Fernádez FM, Cisternas B, Mondaca S et al. Chilean Gastric Cancer Task Force: a study protocol to obtain a clinical and molecular classification of a cohort of gastric cancer patients. Medicine. 2018; 97(16):e0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cordova-Delgado M, Pinto MP, Retamal IN, Muñoz-Medel M, Bravo ML, Fernández MF, et al. High Proportion of Potential Candidates for Immunotherapy in a Chilean Cohort of Gastric Cancer Patients: Results of the FORCE1 Study. Cancers (Basel). 2019; 11(9):1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy G, Pfeiffer R, Camargo MC, Rabkin CS. Meta-analysis shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology. 2009; 137(3):824–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saito R, Abe H, Kunita A, Yamashita H, Seto Y, Fukayama M. Overexpression and gene amplification of PD-L1 in cancer cells and PD-L1+ immune cells in Epstein-Barr virus-associated gastric cancer: the prognostic implications. Mod Pathol. 2017March;30(3):427–439. [DOI] [PubMed] [Google Scholar]
- 13.Derks S, Liao X, Chiaravalli AM, Xu X, Camargo MC, Solcia E, et al. Abundant PD-L1 expression in Epstein-Barr Virus-infected gastric cancers. Oncotarget. 2016;7(22):32925–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sukawa Y, Yamamoto H, Nosho K, Kunimoto H, Suzuki H, Adachi Y, et al. Alterations in the human epidermal growth factor receptor 2-phosphatidylinositol 3-kinase-v-Akt pathway in gastric cancer. World J Gastroenterol. 2012;18(45):6577–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang WL, Huang KH, Lan YT, Lin CH, Chang SC, Chen MH, et al. Mutations in PI3K/AKT pathway genes and amplifications of PIK3CA are associated with patterns of recurrence in gastric cancers. Oncotarget. 2016; 7(5):6201–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang MS, Uozaki H, Chong JM, Ushiku T, Sakuma K, Ishikawa S, et al. CpG island methylation status in gastric carcinoma with and without infection of Epstein-Barr virus. Clin Cancer Res. 2006; 12(10):2995–3002. [DOI] [PubMed] [Google Scholar]
- 17.Kang GH, Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, et al. Epstein-barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am J Pathol. 2002; 160(3):787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chong JM, Sakuma K, Sudo M, Ushiku T, Uozaki H, Shibahara J, et al. Global and non-random CpG-island methylation in gastric carcinoma associated with Epstein-Barr virus. Cancer Sci. 2003; 94(1):76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yue W, Zhu M, Zuo L, Xin S, Zhang J, Liu L, et al. Early Pattern of Epstein-Barr Virus Infection in Gastric Epithelial Cells by “Cell-in-cell”. Virol Sin. 2019; 34(3):253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cárdenas-Mondragón MG, Torres J, Sánchez-Zauco N, Gómez-Delgado A, Camorlinga-Ponce M, Maldonado-Bernal C, et al. Elevated Levels of Interferon-γ Are Associated with High Levels of Epstein-Barr Virus Reactivation in Patients with the Intestinal Type of Gastric Cancer. J Immunol Res. 2017; 2017:7069242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yau TO, Tang CM, Yu J. Epigenetic dysregulation in Epstein-Barr virus-associated gastric carcinoma: disease and treatments. World J Gastroenterol. 2014; 20(21):6448–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SY, Park C, Kim HJ, Park J, Hwang J, et al. Deregulation of immune response genes in patients with Epstein-Barr virus-associated gastric cancer and outcomes. Gastroenterology. 2015;148(1):137–147.e9. [DOI] [PubMed] [Google Scholar]
- 23.Kang BW, Seo AN, Yoon S, Bae HI, Jeon SW, Kwon OK, et al. Prognostic value of tumor-infiltrating lymphocytes in Epstein-Barr virus-associated gastric cancer. Ann Oncol. 2016; 27(3):494–501. [DOI] [PubMed] [Google Scholar]
- 24.Chiaravalli AM, Feltri M, Bertolini V, Bagnoli E, Furlan D, Cerutti R, et al. Intratumour T cells, their activation status and survival in gastric carcinomas characterised for microsatellite instability and Epstein-Barr virus infection. Virchows Arch. 2006; 448(3):344–53. [DOI] [PubMed] [Google Scholar]
- 25.Cho J, Kang MS, Kim KM. Epstein-Barr Virus-Associated Gastric Carcinoma and Specific Features of the Accompanying Immune Response. J Gastric Cancer. 2016; 16(1):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014; 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abe H, Saito R, Ichimura T, Iwasaki A, Yamazawa S, Shinozaki-Ushiku A, et al. CD47 expression in Epstein-Barr virus-associated gastric carcinoma: coexistence with tumor immunity lowering the ratio of CD8+/Foxp3+ T cells. Virchows Arch. 2018; 472(4):643–651. [DOI] [PubMed] [Google Scholar]
- 28.Cárdenas-Mondragón MG, Torres J, Flores-Luna L, Camorlinga-Ponce M, Carreón-Talavera R, Gomez-Delgado A, et al. Case–control study of Epstein–Barr virus and Helicobacter pylori serology in Latin American patients with gastric disease. Br J Cancer. 2015;112(12):1866–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cárdenas-Mondragón MG, Carreón-Talavera R, Camorlinga-Ponce M, Gomez-Delgado A, Torres J, Fuentes-Pananá EM. Epstein Barr virus and Helicobacter pylori co-infection are positively associated with severe gastritis in pediatric patients. PLoS One. 2013April24;8(4):e62850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minoura-Etoh J, Gotoh K, Sato R, Ogata M, Kaku N, Fujioka T, et al. Helicobacter pylori-associated oxidant monochloramine induces reactivation of Epstein-Barr virus (EBV) in gastric epithelial cells latently infected with EBV. J Med Microbiol. 2006; 55(Pt 7):905–911. [DOI] [PubMed] [Google Scholar]
- 31.Allison CC, Ferrand J, McLeod L, Hassan M, Kaparakis-Liaskos M, Grubman A, Bhathal PS, et al. Nucleotide oligomerization domain 1 enhances IFN-γ signaling in gastric epithelial cells during Helicobacter pylori infection and exacerbates disease severity. J Immunol. 2013; 190(7):3706–15. [DOI] [PubMed] [Google Scholar]
- 32.De Re V, Caggiari L, De Zorzi M, Fanotto V, Miolo G, Puglisi F, et al. Epstein-Barr virus BART microRNAs in EBV- associated Hodgkin lymphoma and gastric cancer. Infect Agent Cancer. 2020June; 15:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eaton KA, Mefford M, Thevenot T. The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J Immunol. 2001; 166(12):7456–61. [DOI] [PubMed] [Google Scholar]
- 34.Zhuang Y, Shi Y, Liu XF, Zhang JY, Liu T, Xet Fan et al. Helicobacter pylori-infected macrophages induce Th17 cell differentiation. Immunobiology. 2011; 216(1–2):200–7. [DOI] [PubMed] [Google Scholar]
- 35.Su Z, Sun Y, Zhu H, Liu Y, Lin X, Shen H, et al. Th17 cell expansion in gastric cancer may contribute to cancer development and metastasis. Immunol Res. 2014; 58(1):118–24. [DOI] [PubMed] [Google Scholar]
- 36.Pernot S, Terme M, Radosevic-Robin N, Castan F, Badoual C, Marcheteau E, et al. Infiltrating and peripheral immune cell analysis in advanced gastric cancer according to the Lauren classification and its prognostic significance. Gastric Cancer. 2020; 23(1):73–81. [DOI] [PubMed] [Google Scholar]
- 37.Li R, Zhang H, Cao Y, Liu X, Chen Y, Qi Y, et al. Lauren classification identifies distinct prognostic value and functional status of intratumoral CD8+ T cells in gastric cancer. Cancer Immunol Immunother. 2020; 69(7):1327–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim TS, da Silva E, Coit DG, Tang LH. Intratumoral Immune Response to Gastric Cancer Varies by Molecular and Histologic Subtype. Am J Surg Pathol. 2019; 43(6):851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teh M, Lee YS. HLA-DR antigen expression in intestinal-type and diffuse-type gastric carcinoma. Cancer. 1992; 69(5):1104–7. [DOI] [PubMed] [Google Scholar]
- 40.Humar B, Blair V, Charlton A, More H, Martin I, Guilford P. E-cadherin deficiency initiates gastric signet-ring cell carcinoma in mice and man. Cancer Res. 2009; 69(5):2050–6. [DOI] [PubMed] [Google Scholar]
- 41.Hofmann M, Pircher H. E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc Natl Acad Sci U S A. 2011; 108(40):16741–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finn OJ. Cancer immunology. N Engl J Med. 2008; 358(25):2704–15. [DOI] [PubMed] [Google Scholar]
- 43.Zheng X, Song X, Shao Y, Xu B, Chen L, Zhou Q, et al. Prognostic role of tumor-infiltrating lymphocytes in gastric cancer: a meta-analysis. Oncotarget. 2017; 8(34):57386–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li F, Sun Y, Huang J, Xu W, Liu J, Yuan Z. CD4/CD8 + T cells, DC subsets, Foxp3, and IDO expression are predictive indictors of gastric cancer prognosis. Cancer Med. 2019; 8(17):7330–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lazarevic V, Glimcher L & Lord G T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol. 13, 777–789 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L, Zheng X, Shen Y, Zhu Y, Li Q, Chen J, et al. Higher numbers of T-bet(+) intratumoral lymphoid cells correlate with better survival in gastric cancer. Cancer Immunol Immunother CII. 2013; 62(3):553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, Wang X, Shen Z, Xu J, Qin J, Sun Y. Infiltration of diametrically polarized macrophages predicts overall survival of patients with gastric cancer after surgical resection. Gastric Cancer. 2015; 18(4):740–50. [DOI] [PubMed] [Google Scholar]
- 48.Lin C, He H, Liu H, Li R, Chen Y, Qi Y, et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut. 2019; 68(10):1764–73. [DOI] [PubMed] [Google Scholar]
- 49.Vaddepally RK, Kharel P, Pandey R, Garje R, Chandra AB. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers (Basel). 2020; 20;12(3):738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K, et al. Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538–12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017; 390(10111):2461–2471. [DOI] [PubMed] [Google Scholar]
- 51.Kato K, Satoh T, Muro K, Yoshikawa T, Tamura T, Hamamoto Y, et al. A subanalysis of Japanese patients in a randomized, double-blind, placebo-controlled, phase 3 trial of nivolumab for patients with advanced gastric or gastro-esophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538–12, ATTRACTION-2). Gastric Cancer. 2019; 22(2):344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mishima S, Kawazoe A, Nakamura Y, Sasaki A, Kotani D, Kuboki Y, et al. Clinicopathological and molecular features of responders to nivolumab for patients with advanced gastric cancer. J Immunother Cancer. 2019; 7(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bang YJ, Cho JY, Kim YH, Kim JW, Di Bartolomeo M, Ajani JA, et al. Efficacy of Sequential Ipilimumab Monotherapy versus Best Supportive Care for Unresectable Locally Advanced/Metastatic Gastric or Gastroesophageal Junction Cancer. Clin Cancer Res. 2017; 23(19):5671–5678. [DOI] [PubMed] [Google Scholar]
- 54.Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J Clin Oncol. 2020; 38(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S, et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol. 2016; 17(6):717–726. [DOI] [PubMed] [Google Scholar]
- 56.Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients With Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018; 4(5):e180013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim ST, Cristescu R, Bass AJ, Kim K-M, Odegaard JI, Kim K et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med. 2018; 24(9):1449–1458. [DOI] [PubMed] [Google Scholar]
- 58.Shitara K, Özgüroğlu M, Bang YJ, Di Bartolomeo M, Mandalà M, Ryu MH, et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): a randomised, open-label, controlled, phase 3 trial. Lancet. 2018; 392(10142):123–133. [DOI] [PubMed] [Google Scholar]
- 59.Bang YJ, Ruiz EY, Van Cutsem E, Lee KW, Wyrwicz L, Schenker M, et al. Phase III, randomised trial of avelumab versus physician’s choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: primary analysis of JAVELIN Gastric 300. Ann Oncol. 2018; 29(10):2052–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shitara K, Van Cutsem E, Bang YJ, Fuchs C, Wyrwicz L, Lee KW, et al. Efficacy and Safety of Pembrolizumab or Pembrolizumab Plus Chemotherapy vs Chemotherapy Alone for Patients With First-line, Advanced Gastric Cancer: The KEYNOTE-062 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020; 6(10):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Janjigian YY, Bendell J, Calvo E, Kim JW, Ascierto PA, Sharma P, et al. CheckMate-032 Study: Efficacy and Safety of Nivolumab and Nivolumab Plus Ipilimumab in Patients With Metastatic Esophagogastric Cancer. J Clin Oncol. 2018;36(28):2836–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kelly RJ, Lee J, Bang Y-J, Almhanna K, Blum-Murphy M, Catenacci DVT, et al. Safety and Efficacy of Durvalumab and Tremelimumab Alone or in Combination in Patients with Advanced Gastric and Gastroesophageal Junction Adenocarcinoma. Clin Cancer Res. 2020; (26) (4): 846–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bang YJ, Golan T, Dahan L, Fu S, Moreno V, Park K, et al. Ramucirumab and durvalumab for previously treated, advanced non-small-cell lung cancer, gastric/gastro-oesophageal junction adenocarcinoma, or hepatocellular carcinoma: An open-label, phase Ia/b study (JVDJ). Eur J Cancer. 2020; 137:272–284. [DOI] [PubMed] [Google Scholar]
- 64.Kawazoe A, Yamaguchi K, Yasui H, Negoro Y, Azuma M, Amagai K, et al. Safety and efficacy of pembrolizumab in combination with S-1 plus oxaliplatin as a first-line treatment in patients with advanced gastric/gastroesophageal junction cancer: Cohort 1 data from the KEYNOTE-659 phase IIb study. Eur J Cancer. 2020;129:97–106. [DOI] [PubMed] [Google Scholar]
- 65.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. 2018; Science, 359: 1361–1365. [DOI] [PubMed] [Google Scholar]
- 66.Jain T, Bar M, Kansagra AJ, Chong EA, Hashmi SK, Neelapu SS, et al. Use of Chimeric Antigen Receptor T Cell Therapy in Clinical Practice for Relapsed/Refractory Aggressive B Cell Non-Hodgkin Lymphoma: An Expert Panel Opinion from the American Society for Transplantation and Cellular Therapy. Biol Blood Marrow Transplant. 2019;25(12):2305–2321. [DOI] [PubMed] [Google Scholar]
- 67.Bębnowska D, Grywalska E, Niedźwiedzka-Rystwej P, Sosnowska-Pasiarska B, Smok-Kalwat J Pasiarski M et al. CAR-T Cell Therapy—An Overview of Targets in Gastric Cancer. J Clin Med 2020; 9: 1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang H, Shi Z, Wang P, Wang C, Yang L, Du G et al. Claudin18.2-Specific Chimeric Antigen Receptor Engineered T Cells for the Treatment of Gastric Cancer. J Natl Cancer Inst. 2019: 111:409–418. [DOI] [PubMed] [Google Scholar]
- 69.Zhan X, Wang B, Li Z, Li J, Wang H, Chen H, et al. Phase I trial of Claudin 18.2-specific chimeric antigen receptor T cells for advanced gastric and pancreatic adenocarcinoma. J Clin Oncol 2019; 37:15_suppl, 2509–2509. [Google Scholar]
- 70.Wang X, Che X, Liu C, Fan Y, Bai M, Hou K, et al. Cancer-associated fibroblasts-stimulated interleukin-11 promotes metastasis of gastric cancer cells mediated by upregulation of MUC1. Exp Cell Res. 2018; 368: 184–193. [DOI] [PubMed] [Google Scholar]
- 71.Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008; 180: 4901–9. [DOI] [PubMed] [Google Scholar]
- 72.Xu F, Liu F, Zhao H, An G, Feng G. Prognostic Significance of Mucin Antigen MUC1 in Various Human Epithelial Cancers: A Meta-Analysis. Medicine (Baltimore). 2015; 94 (50):e2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang XT, Kong FB, Mai W, Li L, Pang LM. MUC1 Immunohistochemical Expression as a Prognostic Factor in Gastric Cancer: Meta-Analysis. Dis Markers. 2016; 2016:9421571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Maher J, Wilkie S. CAR mechanics: Driving T cells into the MUC of cancer. Cancer Res. 2009; 69:4559–4562. [DOI] [PubMed] [Google Scholar]
- 75.Fong D, Seeber A, Terracciano L, Kasal A, Mazzoleni G, Lehne F, et al. Expression of EpCAMMF and EpCAMMT variants in human carcinomas. J Clin Pathol. 2014; 67:408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dai M, Yuan F, Fu C, Shen G, Hu S, Shen G. Relationship between epithelial cell adhesion molecule (EpCAM) overexpression and gastric cancer patients: A systematic review and meta-analysis. PLoS ONE 2017; 12, e0175357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Knödler M, Körfer J, Kunzmann V, Trojan J, Daum S, Schenk M, et al. Randomised phase II trial to investigate catumaxomab (anti-EpCAM × anti-CD3) for treatment of peritoneal carcinomatosis in patients with gastric cancer. Br J Cancer 2018; 119: 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang B-L, Li D, Gong Y-L, Huang Y, Qin D-Y, Jiang L, et al. Preclinical Evaluation of Chimeric Antigen Receptor-Modified T Cells Specific to Epithelial Cell Adhesion Molecule for Treating Colorectal Cancer. Human Gene Therapy. 2019; 4:402–412. [DOI] [PubMed] [Google Scholar]
- 79.Cheung A, Bax HJ, Josephs DH, Ilieva KM, Pellizzari G, Opzoomer J, et al. Targeting folate receptor alpha for cancer treatment. Oncotarget. 2016; 7: 52553–52574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim M, Pyo S, Kang CH, Lee CO, Lee HK, Choi SU et al. Folate receptor 1 (FOLR1) targeted chimeric antigen receptor (CAR) T cells for the treatment of gastric cancer. PLoS ONE 2018; 13(6): e0198347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Song Y, Tong C, Wang Y, Gao Y, Dai H, Guo Y, et al. Effective and persistent antitumor activity of HER2-directed CAR-T cells against gastric cancer cells in vitro and xenotransplanted tumors in vivo. Protein Cell. 2018; 9: 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morello A, Sadelain M, Adusumilli PS. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016; 6: 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur. J. Cancer 2008; 44:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014; 6:261ra151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Holzinger A, Abken H. CAR T cells targeting solid tumors: Carcinoembryonic antigen (CEA) proves to be a safe target. Cancer Immunol Immunother. 2017; 66: 1505–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tao K, He M, Tao F, Xu G, Ye M, Zheng Y, et al. Development of NKG2D-based chimeric antigen receptor-T cells for gastric cancer treatment. Cancer Chemother Pharmacol. 2018; 82: 815–827. [DOI] [PubMed] [Google Scholar]
- 87.Demoulin B, Cook WJ, Murad J, Graber DJ, Sentman ML, Lonez C et al. Exploiting natural killer group 2D receptors for CAR T-cell therapy. Future Oncol. 2017; 13: 1593–1605. [DOI] [PubMed] [Google Scholar]
- 88.Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017; 5: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murad JP, Kozlowska AK, Lee HJ, Ramamurthy M, Chang WC, Yazaki P, et al. Effective Targeting of TAG72+ Peritoneal Ovarian Tumors via Regional Delivery of CAR-Engineered T Cells. Front Immunol. 2018; 9:2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wu MR, Zhang T, DeMars LR, Sentman CL. B7H6-specific chimeric antigen receptors lead to tumor elimination and host anti-tumor immunity. Gene Ther. 2015; 22: 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 2002; 3: 991–998. [DOI] [PubMed] [Google Scholar]
- 92.Sharma P, Hu-Lieskovan S, Wargo JS, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017; 168: 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell. 2019; 36(5):471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]