

Summary
Magnolia officinalis, a representative tall aromatic tree of the Magnoliaceae family, is a medicinal plant that is widely used in diverse industries from medicine to cosmetics. We report a chromosome-scale draft genome of M. officinalis, in which ∼99.66% of the sequences were anchored onto 19 chromosomes with the scaffold N50 of 76.62 Mb. We found that a high proportion of repetitive sequences was a common feature of three Magnoliaceae with known genomic data. Magnoliids were a sister clade to eudicots-monocots, which provided more support for understanding the phylogenetic position among angiosperms. An ancient duplication event occurred in the genome of M. officinalis and was shared with Lauraceae. Based on RNA-seq analysis, we identified several key enzyme-coding gene families associated with the biosynthesis of lignans in the genome. The construction of the M. officinalis genome sequence will serve as a reference for further studies of Magnolia, as well as other Magnoliaceae.
Subject areas: Biological sciences, Genetics, Evolutionary biology, Bioinformatics, Genomic analysis, Genomics
Graphical abstract
Highlights
-
•
We provide a chromosome-scale genome for Magnolia officinalis
-
•
We explore the phylogenetic position of Magnoliids
-
•
Analysis for lignan biosynthetic pathway is reported
-
•
We identify terpenes synthase in the floral fragrance composition of Magnolia
Biological sciences; Genetics; Evolutionary biology; Bioinformatics; Genomic analysis; Genomics
Introduction
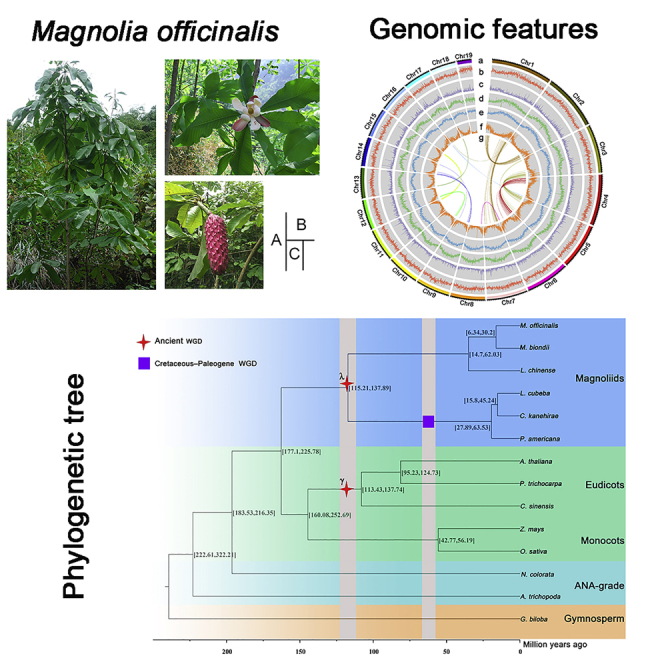
The Magnolia genus is very ancient in plant evolution and has a variety of medicinal, horticultural, and ornamental species. Magnolia officinalis Rehd. (also known as “Hou Po” in China) is one of the most significant plants in subgen Magnolia, and the fragrant and dazzling flowers are the attractive features of M. officinalis (Figure 1), which is mainly distributed in East and Southeast Asia (Cui et al., 2013; Lee et al., 2011). Originally recorded in the Shen Nong's Classic of Materia Medica, M. officinalis has been used historically in Chinese, Japanese, and Korean medicine as an alternative or complement to allopathic medicines (Cui et al., 2013; Poivre and Duez, 2017). Its barks and flowers can be used as traditional herbal medicines for treating gastrointestinal disorders, anxiety, allergic disease, and so on (Lee et al., 2011; Liu et al., 2007). Interestingly, the barks are also widely applied in cosmetics as an important ingredient (Li et al., 2007), and its flowers have been approved as a raw material in nutritional supplement. The main components of M. officinalis include lignans, alkaloids, volatile oils, and other constituents, of which magnolol, as well as honokiol are the two main active compounds (Guo et al., 2019; Luo et al., 2019). Given its multicomponent nature, numerous pharmacological activities of M. officinalis have been reported, such as anti-tumor, anti-inflammation, anti-virus, anti-microorganism, and so on (Yang et al., 2016). Despite M. officinalis is widely cultivated in China (Zhi-Lei and Yu-Shan, 2010) with great medicinal and economic values, its comprehensive utilization is still in a low situation, due to the deficiency of genetic resource, evolutionary history, secondary metabolites biosynthesis and other related molecular biological basis.
Figure 1.

Images of M. officinalis
Mature tree (A), flower (B), fruit (C).
In recent years, whole-genome sequencing has been widely performed in a number of magnoliid species, including Magnolia biondii, Liriodendron chinense, Cinnamomum kanehirae, Piper nigrum etc., which provides a certain support for the analyzing of the developmental relationship of angiosperms (Chaw et al., 2019; Chen et al., 2019; Dong et al., 2021; Hu et al., 2019). The position of magnoliids in plant evolution and taxonomy belongs to a relatively primitive class group, and there has long been considerable disagreement over the phylogenetic status of the phylogenetic position of the eudicots, monocots, and magnoliids (Chen et al., 2020a; Lv et al., 2020). For instance, the phylogenetic analysis of M. biondii, L. chinense, P. nigrum, Persea americana, and Phoebe bournei supported that the magnoliids were a sister clade of the eudicots and monocots (Chen et al., 2019, 2020a; Dong et al., 2021; Hu et al., 2019; Rendón-Anaya et al., 2019), whereas some other genomic studies on C. kanehirae, Litsea cubeba and Chimonanthus praecox, C. salicifolius recognized that magnoliids were a sister clade of eudicots (Chaw et al., 2019; Chen et al., 2020b; Lv et al., 2020; Shang et al., 2020). In addition, a phylogenetic summary including the ANA-grade angiosperms, eudicots, magnoliids, monocots, and a gymnosperm and timescale of 115 plant species (44 genomes and 71 transcriptomes) were summarized by Zhang et al. (Zhang et al., 2020), which suggested magnoliids were sister to the eudicots. Thus, more genomic data representing magnoliids are needed to clarify the conflicting phylogenetic positions.
Here, we completed the chromosome-scale genome assembly for M. officinalis (2n = 2x = 38) (Figure 2A) by the PacBio sequencing platform in Magnolia genus, and the high-quality of the genome will further enrich the research data of genetic evolution of M. officinalis. Additionally, phylogenetic analysis of the M. officinalis genome with other published magnoliids genomes provides new insights to address the phylogenetic position and genome evolution of magnoliids. In a word, this genome contributes to explore the biosynthesis of lignans and identify terpenes in the floral fragrance composition of Magnolia.
Figure 2.

Overview of the chromosomal features of M. officinalis
(A) M. officinalis genome features. Tracks from outside to inside are as follows: (a) The distribution of chromosome. (b) GC density. (c) LINE retrotransposons density. (d) LTR retrotransposons density. (e) DNA transposons density. (f) Gene density. (g) syntenic blocks.
(B) Hi-C interaction heatmap for M. officinalis genome showing interactions among 19 chromosomes. The deeper the color, the higher the frequency of interaction.
Results
Genome sequencing and assembly
The M. officinalis genome was sequenced using the PacBio, Illumina platforms and Hi-C technology. A total of 140.91 Gb (84-fold PacBio long reads coverage) of raw reads were obtained after low quality and short fragment filtering, with an average read length of 8.65 kb, an N50 read length of 13.78 kb, and a longest read of 128.49 kb (Table S1). The Hi-C library produced approximately 94.25 Gb clean reads (Table S2).
Genome survey was conducted using Illumina data, M. officinalis genome size was estimated to be approximately 1.76 Gb, and the heterozygosity was as low as about 0.58% based on the K-mer analysis method (Figure S1). The PacBio and Hi-C data was used for chromosome-scale assembly, the final assembled genome size of M. officinalis was 1.68 Gb (99.95% sequence coverage), with a scaffold N50 of 76.62 Mb and a GC content of 40.65% (Table 1), and 1.67 Gb of assembled sequences were anchored onto 19 chromosomes, of which 1.53 Gb of sequences length were ordered and oriented, representing 91.21% (Table S5). To further check the genome quality, a genome-wide heatmap was generated by Hi-C assembly. The Hi-C assembly (Figures 2B and S4) showed that the 19 chromosomes of M. officinalis could be clearly distinguished, and the signal intensity of the diagonal interaction of each group was deeply higher than that of the non-diagonal position.
Table 1.
The major characteristics of M. officinalis genome
| M. officinalis | |
|---|---|
| Assembly | |
| Total length (bp) | 1,684,361,614 |
| Number of scaffolds | 2,892 |
| Scaffolds N50 (bp) | 76,619,249 |
| Number of contig | 11,562 |
| Contig N50 (bp) | 222,069 |
| GC content % | 40.65 |
| Complete BUSCOs % | 86.20 |
| Percent of CEGs % | 95.19 |
| Annotation | |
| Repeat sequences % | 81.44 |
| Number of predicted genes | 23,424 |
| Number of protein-coding genes | 23,050 |
| Number of non-coding RNAs | 1,096 |
| Average gene length (bp) | 10,206.68 |
| Average exon length (bp) | 242.75 |
| Average intron length (bp) | 1,673.73 |
To assess genome completeness, we combined both BUSCO and CEGMA approaches. BUSCO analysis determined that 86.2% (1,391/1,614) of complete BUSCO were found in the genome assembly (Table S3). Then, approximately 95.19% (436/458) conserved core genes of eukaryotes were detected in M. officinalis by CEGMA analysis (Table S4). This result confirmed that the M. officinalis genome assembly was nearly complete.
Genome annotation
Notably, the M. officinalis genome contains 1.37 Gb of repetitive elements, reaching an amazing ratio of 81.44% (Table S6), which was the highest proportion of sequenced repetitive sequences in magnoliids, compared with M. biondii (∼66.48% in a 2,252.5 Mb genome) (Dong et al., 2021), C. kanehirae (∼48% in a 730.7 Mb genome) (Chaw et al., 2019), L. chinense (∼62% in a 1742.4 Mb genome) (Chen et al., 2019), P. bournei (∼68.51% in a 989.19 Mb genome) (Chen et al., 2020a), and P. nigrum (∼54.85% in a 761.74 Mb genome) (Hu et al., 2019). Among these repetitive elements of M. officinalis, class I retrotransposons were the majority, with the LTR/Gypsy, LTR/Copia and LARDs classes accounting for 27.53%, 21.11% and 23.90% of the genome, respectively. The proportion of LTR/Gypsy was higher than that of LTR/Copia, as the results of genomic repeat annotation of magnoliids, such as M. biondii, P. nigrum, L. chinense, L. cubeba, and C. kanehirae. In addition, the LARDs elements of M. officinalis were also higher than that of the above species, and this item was the main elements responsible for the high repetition rate of M. officinalis compared with other magnoliid species. LARDs elements was originally identified in the barley and related genomes by Kalendar et al (Kalendar et al., 2004). Some researchers have suggested that LARDs were thought to be the remnants of deletion of autonomous LTR retrotransposons. LARDs (13% of the genome) in the pomegranate genome could affect the expression of genes for fruit coloration, e.g., flavonoid glucosyltransferase, MYB genes (Huang et al., 2021; Yuan et al., 2018). Therefore, we speculated that the abundant LARDs elements in the M. officinalis genome might also have a relationship with secondary metabolite accumulation, as in pomegranate. The large number of repetitive elements in the M. officinalis genome supported that the ongoing duplication of genetic material during plant evolution (Eichler and Sankoff, 2003).
For gene prediction, a total of 23,424 protein-coding genes and 1,096 non-coding RNA genes were predicted by ab initio-based and homology-based gene prediction methods. Non-coding RNAs included 72 microRNAs, 575 tRNAs, and 449 rRNAs (Table S7). In addition, we identified 23,050 annotated functional genes through blasting with NR, TrEMBL, KOG, GO, and KEGG databases (Table S8). Among these genes, 13,438 genes (57.37%) were annotated in GO terms (Figure S2A, Table S9). The most extensive GO term associated with biological process was metabolic process (7,709 genes). KEGG analysis revealed that M. officinalis genes were main involved in metabolic pathways (Figure S2B, Table S10), which were carbon metabolism (237 genes), biosynthesis of amino acids (217 genes), starch and sucrose metabolism (207 genes), and phenylpropanoid biosynthesis (190 genes). Further studies on KOG functional annotation revealed that most of the genes in M. officinalis were annotated for secondary metabolites, transport and catabolism, which were concentrated in cytochrome P450 (181 genes), and multicopper oxidases (56 genes) (Figure S2C, Table S11). Cytochrome P450s play an important role in the plant metabolic network (Mizutani and Ohta, 2010), e.g., CYP98 catalyzes a rate-limiting step in phenylpropanoid biosynthesis and C4H, a key enzyme in the general phenylpropanoid pathway, belongs to the CYP73 family (Alber et al., 2019; Li et al., 2020; Ma et al., 2015; Noel et al., 2005). We found the presence of CYP73A and CYP98A genes in the genome of M. officinalis (Table S10).
Gene family and phylogenomic analysis
Comparative genomic analysis indicated that total 21,966 of the 23,424 predicted protein-coding genes could be clustered into 14,082 gene families, of which 202 were unique gene families (Table S12). A total of 8,106 gene families were shared by magnoliids (M. officinalis, L. chinense, C. kanehirae, P. americana and L. cubeba). Among the gene families identified in M. officinalis, 784 gene families were shared with L. chinense, while much fewer gene families were shared compared with the Lauraceae family (Figure 3A).
Figure 3.

Comparative genomic analysis of M. officinalis and other species
(A) Venn diagram of the number of shared gene families within M. officinalis, L. chinense, L. cubeba, P. americana and C.kanehirae. The number below the species name is the total number of gene families. The number in the Venn diagram is the number of gene families.
(B) The analysis of gene copy number of species in each gene family.
To further explore the phylogenetic position of magnolias, a phylogenetic tree was constructed using 365 single-copy orthologs related to 14 species (Figure 4), the results showed that M. officinalis was most closely related to M. biondii. Based on the fossil calibration, we used MCMCtree to estimate the divergence time with 95% confidence intervals for the HPD and inferred that the divergence time between M. officinalis and M. biondii was about 6.3–30.2 mya (million years ago). To date, the phylogenetic position between the three groups, eudicots, monocots, and magnoliids, remain unclear. Based on the phylogenetic tree constructed by 365 single-copy genes, we found that the magnoliids were as sister groups to the eudicots and monocots. Together with the two genomes of Magnoliaceae (M. biondii and L. chinense) previously published, our phylogenomic analyses all indicated that magnoliids were independent of monocots and eudicots, which was consistent with the results observed in previous studies (Chen et al., 2019, 2020a; Dong et al., 2021; Hu et al., 2019; Rendón-Anaya et al., 2019).
Figure 4.

The Phylogenetic tree including 14 species was constructed based on 365 single-copy genes
The WGD shared between the Magnoliales and Laurales is indicated with a red star, and the purple box indicated that an extra recent WGD event was experienced in the Laurales.
Synthesis and regulation genes involved in lignans
Lignans occur in a variety of plants, including Magnolia L., Eleutherococcus L., Schisandra L., and Linum L. Significant biological activity has been proved for a few lignan compounds, such as secolariciresinol, lariciresinol, pinoresinol, matairesinol, magnolol, honokiol, and others (AdamskaSzewczyk and Zgórka, 2019). Magnolol and honokiol are natural biphenyl derivatives of M. officinalis that also pass through the lignans pathway (Shi et al., 2017). Considering the importance of magnolol and honokiol, we used high-performance liquid chromatography to quantify the two components (Figure 5) in M. officinalis five tissues (leaves, roots, stems, twigs, and fruits) and the RNA-seq of five tissues to analyze the genes within the lignan biosynthesis pathway.
Figure 5.

HPLC detection of magnolol and honokiol in M. officinalis five tissues (leaves, roots, stems, twigs, and fruits)
Based on the results of RNA-seq, we identified 65 genes lignans-related synthesis with at least one highly expressed member in five tissues (Figure 6B), such as PAL, C4H, 4CL, PLR, and so on, the expression of these genes could contribute to lignans biosynthesis. In the metabolic pathway of phenylalanine, a series of intermediates such as coumaric acid, caffeic acid, ferulic acid, and cinnamic acid are produced, which are then converted to lignans and other substances catalyzed by various enzymes. PAL, C4H, 4CL, and PLR are the most studied enzymes that play an important role in lignans biosynthesis (Chiang et al., 2019; Vanholme et al., 2019), and these related to lignans biosynthesis pathway genes were identified in our RNA-seq data set. PAL catalyzes the first step of phenylalanine reaction and converts phenylalanine to cinnamic acid by deamination. We detected high expression of PAL in five tissues of M. officinalis. C4H, belonging to cytochrome P450 family, was mainly expressed in roots and twigs. A variety of CYP450 enzyme families were also detected in the genome of M. officinalis (Table S11). These compounds specifically hydroxylate cinnamic acid to produce p-coumaric acid. The function of 4CL was to convert coumaric acid, caffeic acid and ferulic acid into CoA esters respectively, and it was also highly expressed in five tissues. PLR reduces pinoresinol to lariciresinol and then to secoisolariciresinol, which can be oxidized to matairesinol by SIDR (Figure 6A). The downstream lignan biosynthesis process initiated by matairesinol has not been fully clarified up to now. However, the step from phenylalanine to matairesinol is a common step in lignans biosynthesis pathway, and matairesinol is considered to be a central intermediate in the production of various lignans (Xia et al., 2001). In conclusion, our work extends previous studies of related candidate genes involved in the biosynthesis of neolignans (magnolol and honokiol) in M. officinalis, and further studies are needed to elucidate their potential molecular regulatory mechanisms.
Figure 6.

Expression profiling of lignan biosynthesis pathway genes in five tissues of M. officinalis by RNA-Seq
(A) The general biosynthetic pathway of lignans. The solid arrows are reactions catalyzed by known enzymes, whereas the dashed arrows are the predicted lignan biosynthetic steps in M. officinalis.
(B) Expression values of lignan biosynthesis pathway genes. R, roots; F, fruits; S, stems; L, leaves; T, twigs; C4H, cinnamate 4-hydroxylase; COMT, caffeic acid 3-O-methyltransferase; CCOAMT, caffeoyl-CoA O-methyltransferase; HCT, shikimate O-hydroxycinnamoyltransferase; SIRD, secoisolariciresinol dehydrogenase; CAD, cinnamyl-alcohol dehydrogenase; CCR, cinnamoyl-CoA reductase; 4CL, 4-coumarate-CoA ligase; PLR, pinoresinol-lariciresinol reductase; PAL, phenylalanine ammonia-lyase.
Whole-genome duplication analysis and synteny analysis
Whole-genome duplication events are thought to be an important driver of evolution (Song et al., 2020). We investigated the whole genome duplication (WGD) events of M. officinalis vs. L. chinense, C. kanehirae, and L. cubeba. The distribution of Ks values for M. officinalis paralogs showed only one peak with Ks = ∼0.73, indicating an ancient WGD event. In addition, the Ks distributions of L. chinense, C. kanehirae and L. cubeba paralogs showed peaks at Ks2, which were about 0.68, 0.76 and 0.8, respectively (Figure 7A). WGD analyses indicated the Magnoliaceae and Lauraceae shared this ancient whole-genome duplication event, and Lauraceae have experienced an extra additional WGD event (named as Cretaceous–Paleogene WGD (Zhang et al., 2020)) since then (Ks1 = ∼0.5). First ancient duplication was called lambda (λ) events, and during the early diversification of magnoliids at about 120 mya (Zhang et al., 2020). Thus, these indicated that all four magnoliid species experienced a λ multiplication event about 120 million years ago. This point was consistent with previously sequenced genomes of M. biondii, L. chinense, C. kanehirae, and L. cubeba (Chaw et al., 2019; Chen et al., 2019, 2020b; Dong et al., 2021). While the Ks values of one-to-one orthologs analysis of M. officinalis with L. chinense, C. kanehirae and L. cubeba were 0.13, 0.75, and 0.76, respectively. The result confirmed that M. officinalis underwent a WGD event just following the divergence of the common ancestor of Magnoliaceae and Lauraceae, which differs from a recent genome evolution study on the same genus M. biondii (Dong et al., 2021). We inferred the Lauraceae family, represented by C. kanehirae and L. cubeba, diverged from the Magnoliaceae (M. officinalis and L. chinense) during the Cretaceous period about 116–120 million years ago.
Figure 7.

Evolutionary and Synteny genomic analyses of M. officinalis
(A) The red dashed boxes represent comparisons within a species and the blue dashed boxes represent comparisons between species. Peaks of intraspecies Ks distribution indicate ancient WGD events, and peaks of interspecies Ks distribution indicate speciation events.
(B) Synteny patterns show that M. officinalis can be tracked to up to two regions in C. kanehirae and to up to one region in L. chinense. Gray edges in the background highlight major syntenic blocks spanning the genomes (highlighted by one syntenic set shown in color).
We performed a synteny analysis of M. officinalis genome with L. chinense and C. kanehirae. Based on the syntenic blocks, we found that 23,122 homologous genes exist in M. officinalis and L. chinense, accounting for 39.39% of the total number of genes. Among the syntenic blocks of M. officinalis and C. kanehirae, including 15,417 genes that accounted for 30.86% of the total number of genes. Meanwhile, we conducted a comparative genomic analysis of M. officinalis genome with L. chinense and C. kanehirae, which inferred 1:1and 1:2 syntenic depth ratios in the M. officinalis-L. chinense and M. officinalis-C. kanehirae comparisons (Figure 7B), respectively, and this result was consistent with the fact that L. chinense experienced an ancient WGD event and two ancient WGD events of C. kanehirae.
Terpenes synthase gene families
M. officinalis, as an ornamental tree, has a unique floral fragrance and large, elegant flowers. Some other species of the Magnoliaceae family, such as M. biondii (Dong et al., 2021), M. champaca (Dhandapani et al., 2017) and Michelia alba (Sanimah et al., 2008), showed the fragrance to be intense. Previous studies on the floral volatile organic compounds of M. officinalis at different flowering stages revealed that terpenes were the main constituents, with the highest content of monoterpenes and sesquiterpenes such as camphene (C10H16), D-limonene (C10H16), myrcene (C10H16), and caryophyllene (C15H24) (Wang et al., 2011). Terpenes are the largest class of floral components, of which the terpene synthase (TPS) genes are the key genes that determine the spatiotemporal release of volatile compounds (Gao et al., 2018). The involvement of TPS in the production of floral fragrance has been demonstrated in various plants (Muhlemann et al., 2014). A total of 40 putative TPS proteins in the M. officinalis genome were annotated according to the conserved domains (PF01397 and PF03936) using HMMER (Potter et al., 2018). We constructed a phylogenetic tree by aligning the TPS proteins among M. officinalis, M. biondii, L. chinense, and A. thaliana to confirm the classification of TPS family proteins in M. officinalis (Figure 8B). According to the protein phylogenetic tree, the 40 TPS proteins were divided into five groups, which contain TPS-a, TPS-b, TPS-c, TPS-e, and TPS-f. Most MoTPS cluster in the TPS-a and TPS-b group, which contain all known sesquiterpene synthases and monoterpene synthases from angiosperm (Aubourg et al., 2002). Compared with the multiple closely related MoTPS in the TPS-a and TPS-b groups, only a few members were found in each of the three subfamilies TPS-c, TPS-e, and TPS-f. However, we did not observe the presence of the TPS-g subfamily in the assembled genome of M. officinalis. It was found that terpenes produced by TPS-g were more volatile than those produced by other TPS subfamilies due to the acyclic monoterpenes synthesized by TPS-g (Dudareva et al., 2003; Martin et al., 2004). M. officinalis produces fewer acyclic monoterpenes among the volatile organic compounds, and its floral fragrance is faint. Therefore, we speculate that the TPS-g subfamily may have been lost during the evolution of this species. This difference might be involved in the biosynthesis of floral volatile organic compounds. Additionally, we observed that TPS genes were mainly distributed on 10 chromosomes of M. officinalis, with many clusters of TPS genes on chromosomes 1, 2, 15, and 16 (Figure 8A), which may be the result of tandem duplication of TPS genes.
Figure 8.

Phylogenetic tree and localization of TPS gene families in M. officinalis
(A) Phylogenetic tree of TPS proteins among M. officinalis, M. biondii, L. chinense and A. thaliana.
(B) Chromosome localization of M. officinalis TPS genes.
Discussion
Despite the size and complexity of plant genomes, with the development of genome sequencing and bioinformatics technology, as well as the reduction of sequencing cost and the improvement of analysis methods, the genome sequencing research of M. officinalis have been greatly promoted.
We combined the long-read sequences from PacBio with highly accurate reads from Hi-C technology to construct a high-quality chromosome-scale genome assembly of M. officinalis. The high heterozygosity and high duplication content were the features of the M. officinalis genome. Combining the published genomic information of Magnoliaceae and Lauraceae species, we found that the genome size of all Magnoliaceae species were relatively large, e.g., M. officinalis (1.68 Gb), M. biondii (2.25 Gb), L. chinense (1.74 Gb), compared with other species in the Lauraceae family (0.73–1.32Gb) (Chaw et al., 2019; Chen et al., 2019, 2020b; Rendón-Anaya et al., 2019). This phenomenon was particularly related to transposable elements (TEs) in repetitive sequences, and a strong correlation between TEs content and genome size has been found in the genomes of angiosperms, ranging from 20–30% in the Arabidopsis thaliana genome (115M) (Arabidopsis Genome Initiative, 2000), about 70–80% in oat genus repeats (Liu et al., 2019), and more than 85% in the maize genome (2G) (Schnable et al., 2009). The proportion of TEs in each of the three Magnoliaceae species exceeds more than 60% of their genome size. Repeat sequences proliferation provides a broad source of variation for genome evolution; however, our current research fails to explain whether TEs bursts increased the genetic diversity of species thus they could adapt quickly to the environment (Kidwell and Lisch, 1997; Niu et al., 2019). WGD events are particularly common in angiosperms, and the large size and complex structure of the genomes of many species are associated with WGD events, also known as polyploidy event. We showed evidence for two ancient WGD events found in magnoliids, one shared with Laurales and Magnoliales, and another additional WGD in Lauraceae. We also noted evidence of synteny between the genomes of M. officinalis and other magnoliids, e.g., we found syntenic blocks of 1:1 and 1:2 of M. officinalis vs L. chinense and M. officinalis vs C. kanehirae. Thus, compared with Lauraceae (C. kanehirae) that was known to experience two polyploidy events, the Magnoliaceae (M. officinalis and L. chinense) only underwent one polyploidy event shared with all angiosperms.
Magnoliids are an early branch of the angiosperm lineage. However, the phylogenetic relationships of magnoliids with eudicots and monocots have not been finally resolved, and there have been proposed three main phylogenetic topologies, e.g., (1) sister to the eudicots, (2) sister to the monocots, and (3) sister to the group of eudicots-monocots. Why these researches provide three different relationships? Many factors may contribute to these topological differences, e.g., small sample sizes (Soltis and Soltis, 2019), inadequate sampling of taxa and genes (Jim et al., 2005), absence key lineages of mesangiosperms and so on. There is still no available genomic data of Canellales in magnoliids, more plant lineages genomic data are essential to comprehend the developmental position of these long-isolated angiosperms. In our comparative genomics studies, magnoliids were considered to be a sister group of eudicots-monocots by the 365 single-copy orthologs of 14 species, which contained six Magnoliids, three Eudicots, two Monocots, two ANA-grade angiosperms, and one Gymnosperms. This phylogenetic topology was in agreement with M. biondii and L. chinense of the same family, respectively (Chen et al., 2019; Dong et al., 2021), and also with the results of several other studies (Chen et al., 2020a; Hu et al., 2019; Rendón-Anaya et al., 2019).
Moreover, our work also focused on exploring the biological processes associated with the biosynthesis of magnolol and honokiol. Through the multi-omics analysis, we detected some key enzymes in phenylalanine biosynthesis that play a significant role in the process from the deamination of phenylalanine to the formation of matairesinol. Unfortunately, we have not yet clarified how matairesinol, the important intermediate in the downstream pathway of lignans, produces magnolol and honokiol, which means more studies are required to explore the specific mechanism. The floral fragrance of M. officinalis is mainly derived from its terpenes. We identified a total of 40 putative genes of the terpene biosynthetic pathway, which helped to determine the TPS gene family classification. Compared with M. biondii, the TPS-g subfamilies were not present in the genome of M. officinalis. Because the TPS-g group synthesizes acyclic terpenoids with strong volatility, we speculate that the TPS-g group may have been lost during the evolutionary process, so the flower fragrance of M. officinalis is very light.
Taken together, our works provided a chromosome-scale reference genome about M. officinalis, which was conducive to understand the evolutionary history of magnoliids and the underlying genes involved in the lignans biosynthesis.
Limitations of the study
We sequenced the genome of M. officinalis, which identified serval genes related to the biosynthesis of lignan. However, we were unable to explain the synthetic pathways of the active components of M. officinalis. More metabolomics studies are needed to explain how magnolol and honokiol are produced.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Cheng Peng (peng_cutcm@126.com).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
This study does not include experiments or subjects.
Method details
Plant materials
The materials of M. officinalis were collected from the Medicinal Botanical Garden of Chengdu University of Traditional Chinese Medicine, Chengdu, China (103°48′16″E, 030°41′29″ N). Genomic DNA was extracted from fresh leaves of M. officinalis using a modified CTAB protocol (Raimundo et al., 2018), and leaves, stems, roots, twigs and fruits from M. officinalis were used for RNA-seq with three biological repetitions. Total RNA was extracted using Biomarker Plant Total RNA Isolation Kit (China) according to the manufacturer's instructions. All materials were stored at −80°C in the State Bank of Chinese Drug Germplasm Resources.
Genome library construction, sequencing and genome size estimation
Genomic DNA used for library construction following the PacBio SMRT library construction protocol. Preparation of the SMRTbell template involved DNA fragmentation, DNA concentration, damage repair, end repair, adapter ligation and template purification. Then the genome was sequenced on the PacBio Sequel platform (Pacific Biosciences, Menlo Park, CA, USA), and the paired-end 150 (PE150) sequencing was performed on Illumina Hiseq X Ten platform (Illumina, San Diego, CA, USA). Hi-C library was constructed by the standard procedures, and sequenced on Illumina Hiseq X Ten platform.
We conducted the M. officinalis's genomic survey to estimate the genome size, heterozygosity level and duplication content based on K-mer (k = 21) distribution curve by Illumina Hiseq X Ten platform.
RNA sequencing
The cDNA library was constructed using an NEB Next Ultra RNA Library Prep Kit (USA). Libraries were sequenced using the Illumina NovaSeq 6000 platform with PE150 read layout (Biomarker Technologies Corporation, Beijing, China). For the transcriptome-based method, the mixed samples (leaves, stems, roots, twigs and fruits) generated the RNA-seq reads were assembled using Trinity v2.1.1 (Grabherr et al., 2011).
De novo genome assembly and Hi-C assembly
De novo assembly was carried out using Canu v1.8 (Koren et al., 2017), and combined with WTDBG (Ruan and Li, 2020)accomplished correction assembly. The genome assemble results by two methods was optimized by using the merging idea of Quickmerge (Chakraborty et al., 2016). Finally, using Pilon (Walker et al., 2014) software to corrected the draft assembly combined the Illumina reads. In order to anchor the scaffold onto the chromosome (Zhang et al., 2019), we used the Illumina HiSeq X Ten platform to construct the Hi-C library. The clean Hi-C data were aligned to initial genome assembly segments using BWA v0.78 (Li and Durbin, 2009). To evaluate the Hi-C library quality, the HiC-Pro v2.7.1 (Servant et al., 2015) was used to identify the valid interaction pairs based on unique mapped read pairs. Then, these corrected genome scaffolds were be clustered, ordered and oriented onto a chromosomal genome by LACHESIS (Burton et al., 2013). The genome completeness was assessed by CEGMA v2.5 (Parra et al., 2007) and BUSCO v 5.0 (Simão et al., 2015) to evaluated.
Repeat annotation
Annotation of repetitive sequences in the M. officinalis genome using a combination of ab initio and homology-based approaches. LTR_Finder v1.05 (Xu and Wang, 2007), RepeatScout v1.05 (Price et al., 2005) and PILER v2.4 (Edgar and Myers, 2005) were used to identify the ab initio-based repeat. The homology-based repeat was classified by PASTEClassifier v1.0 (Wicker et al., 2007) and RepeatMasker v4.06 (Tarailo-Graovac and Chen, 2009) to search on the repeat sequence database Repbase (Jurka et al., 2005) (https://www.girinst.org/repbase/).
Gene prediction
The M. officinalis protein-coding genes were detected by combination two strategies. Based on ab initio prediction strategy using Genscan v3.1 (Burge and Karlin, 1997), Augustus v2.4 (Stanke and Waack, 2003), GlimmerHMM v3.0.4 (Majoros et al., 2004), GeneID v1.4 (Alioto et al., 2018), and SNAP v2006-07-28 (Johnson et al., 2008). The four homologous species (Arabidopsis thaliana, Oryza sativa, Helianthus annuus, Nelumbo nucifera) protein sequences were obtained from NCBI database, and mapped using TblastN, then GeMoMa v1.3.1 (Keilwagen et al., 2016) was employed for the homology-based prediction. At last, EVM v1.1.1 (Haas et al., 2008) was used to integrate the results of the above strategies.
Non-coding RNA annotation and functional annotation
According to the structure characteristics of different non-coding RNAs, rRNA and microRNA were predicted by Infernal v1.1.2 (Nawrocki and Eddy, 2013) through queried Rfam (Griffiths-Jones et al., 2005) and miRbase (Griffiths-Jones et al., 2006) database, and tRNA genes were identified using tRNAscan-SE v1.3.1 (Lowe and Eddy, 1997). Functional annotation was conducted based on aligning functional databases with BLASTP (E-value<1e-5) (Camacho et al., 2009), including NR (Marchler-Bauer et al., 2011), KOG (Tatusov et al., 2001), KEGG (Kanehisa and Goto, 2000), TrEMBL (Boeckmann et al., 2003) and other functional databases. Functional annotation of GO (Harris et al., 2004) was employed with Blast2GO (Conesa et al., 2005). Furthermore, KEGG pathway enrichment analysis (Kanehisa et al., 2016) were used to identify the M. officinalis' predicted genes.
Comparative genomics and phylogenetic tree
To explore gene evolution patterns and specific genes of M. officinalis, we selected the other thirteen sequenced genomes for multispecies alignments, including five Magnoliids (C. kanehirae, L. chinense, M. biondii, L. cubeba, P. Americana), three Eudicots (Arabidopsis thaliana, Populus trichocarpa, Camellia sinensis), two Monocots (Oryza sativa, Zea mays), two ANA-grade angiosperms (Amborella trichopoda, Nymphaea colorate) and one Gymnosperms (Ginkgo biloba) (Figure S3). The orthologous gene families was conducted in OrthoFinder v2.3.8 (Emms and Kelly, 2019).
The single-copy protein sequences were aligned by MAFFT v7.394 (Katoh and Standley, 2013). Then PAL2NAL (version 14) (Suyama et al., 2006) converted the protein sequence into the coding sequence, and IQ-TREE v2.0.7 (Nguyen et al., 2015) constructed the phylogenetic trees with JTT + F + I + G4 model. The bootstrap support values were calculated on 1000 replicates. MCMCTREE, a program of PAML (Puttick, 2019), was applied to calculate divergence time of above species with parameters (burnin = 5,000,000, nsample = 5,000,000 and sampfreq = 30). Two calibration points from the TimeTree database (http://www.timetree.org/) (Kumar et al., 2017) were selected as normal priors to constrain the age of the nodes, such as published divergence times for A. trichopoda-L. chinense (∼173–199 Mya), O. sativa-Z. mays (∼42–52 Mya).
Whole-genome duplication and Synteny analysis
We used Wgd (version 1.1.1) (Zwaenepoel and Van de Peer, 2019) to conduct the synonymous substitutions rate (Ks) distribution analysis, which calculated the distribution of the transversion rate on Ks with the methods of cross-and monophyletic-species analyses. The obvious peaks in Ks distribution curve represented species separations or WGD events. Using the formula T = Ks/2r (r = 3.21 × 10−9) to calculated the WGDs events and orthology divergence. We used BLASTP (E-value < 1e−5) to perform homolog searches with M. officinalis and other genomes by DIAMOND (Buchfink et al., 2015). Then, MCScanX (Wang et al., 2012) was used to analyze chromosome collinearity, detected the number of the syntenic blocks in the genome.
Quantification and statistical analysis
The statistical analyses were performed in BLAST, with the E-value < 1e−5.
Additional resources
This study does not include additional resources.
Acknowledgments
This work was supported by the Genomics Innovation platform of Distinctive Traditional Chinese Medicine Resources in Southwest characteristics (No. 2020ZYD058), Talent Project in Chengdu University of Traditional Chinese Medicine (QNXZ2018017, QNXZ2019001), Research on key technologies and demonstration application of innovative products of Magnolia officinalis antibacterial disinfectant solution and efficient antibacterial paper towel (NO. 2021MS236), and the first batch of key projects for the construction of TCM disciplines in Sichuan Province: medicinal botany, Sichuan TCM Letter [2020] No. 84.
Author contributions
C.P. and J.H.G. designed the project. H.J.Z., L.J.Z., and F.X.H. collected plant materials. Y.P.Y., J.P., B.R., and J.R.C. conducted experiments. Y.P.Y., X.M.Y., L.W., and X.D.S. analyzed data and conducted bioinformatic analysis; Y.P.Y., F.P., X.F.X., and J.H.G. wrote and modified the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: September 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102997.
Contributor Information
Cheng Peng, Email: peng_cutcm@126.com.
Jihai Gao, Email: gaojihai@cdutcm.edu.cn.
Supplemental information
Data and code availability
The accession number for the genome assembly and raw reads reported in this paper is GenBank: PRJNA752832 and PRJNA752923, respectively. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- AdamskaSzewczyk A., Zgórka G. Plant polyphenols in cosmetics–a review. EJMT. 2019;3:24. [Google Scholar]
- Alber A.V., Renault H., Basilio-Lopes A., Bassard J.-E., Liu Z., Ullmann P., Lesot A., Bihel F., Schmitt M., Werck-Reichhart D., Ehlting J. Evolution of coumaroyl conjugate 3-hydroxylases in land plants: lignin biosynthesis and defense. Plant J. 2019;99:924–936. doi: 10.1111/tpj.14373. [DOI] [PubMed] [Google Scholar]
- Alioto T., Blanco E., Parra G., Guigó R. Using geneid to identify genes. Curr. Protoc. Bioinformatics. 2018;64:e56. doi: 10.1002/cpbi.56. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Aubourg S., Lecharny A., Bohlmann J. Genomic analysis of the terpenoid synthase ( AtTPS) gene family of Arabidopsis thaliana. Mol. Genet. Genomics. 2002;267:730–745. doi: 10.1007/s00438-002-0709-y. [DOI] [PubMed] [Google Scholar]
- Boeckmann B., Bairoch A., Apweiler R., Blatter M.-C., Estreicher A., Gasteiger E., Martin M.J., Michoud K., O'Donovan C., Phan I. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B., Xie C., Huson D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- Burton J.N., Adey A., Patwardhan R.P., Qiu R., Kitzman J.O., Shendure J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013;31:1119–1125. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty M., Baldwin-Brown J.G., Long A.D., Emerson J.J. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 2016;44:e147. doi: 10.1093/nar/gkw654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaw S.-M., Liu Y.-C., Wu Y.-W., Wang H.-Y., Lin C.-Y.I., Wu C.-S., Ke H.-M., Chang L.-Y., Hsu C.-Y., Yang H.-T. Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution. Nat. Plants. 2019;5:63–73. doi: 10.1038/s41477-018-0337-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Hao Z., Guang X., Zhao C., Wang P., Xue L., Zhu Q., Yang L., Sheng Y., Zhou Y. Liriodendron genome sheds light on angiosperm phylogeny and species-pair differentiation. Nat. Plants. 2019;5:18–25. doi: 10.1038/s41477-018-0323-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.-P., Sun W.-H., Xiong Y.-F., Jiang Y.-T., Liu X.-D., Liao X.-Y., Zhang D.-Y., Jiang S.-Z., Li Y., Liu B. The genome sheds light on the evolution of magnoliids. Hortic. Res. 2020;7:146. doi: 10.1038/s41438-020-00368-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.-C., Li Z., Zhao Y.-X., Gao M., Wang J.-Y., Liu K.-W., Wang X., Wu L.-W., Jiao Y.-L., Xu Z.-L. The Litsea genome and the evolution of the laurel family. Nat. Commun. 2020;11:1675. doi: 10.1038/s41467-020-15493-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang N.T., Ma L.T., Lee Y.R., Tsao N.W., Yang C.K., Wang S.Y., Chu F.H. The gene expression and enzymatic activity of pinoresinol-lariciresinol reductase during wood formation in Taiwania cryptomerioides Hayata. Holzforschung Int. J. Biol. Chem. Phys. Technol. Wood. 2019;73:197–208. [Google Scholar]
- Conesa A., Götz S., García-Gómez J.M., Terol J., Talón M., Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Cui W., Wang Y., Chen Q., Sun W., Cai L., Tan Y., Kim K.-S., Kim K.H., Kim Y.H. Magnolia extract (BL153) ameliorates kidney damage in a high fat diet-induced obesity mouse model. Oxid. Med. Cell Longev. 2013;2013:367040. doi: 10.1155/2013/367040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhandapani S., Jin J., Sridhar V., Sarojam R., Chua N.-H., Jang I.-C. Integrated metabolome and transcriptome analysis of Magnolia champaca identifies biosynthetic pathways for floral volatile organic compounds. BMC Genomics. 2017;18:463. doi: 10.1186/s12864-017-3846-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S., Liu M., Liu Y., Chen F., Yang T., Chen L., Zhang X., Guo X., Fang D., Li L. The genome of Magnolia biondii Pamp. provides insights into the evolution of Magnoliales and biosynthesis of terpenoids. Hortic. Res. 2021;8:38. doi: 10.1038/s41438-021-00471-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudareva N., Martin D., Kish C.M., Kolosova N., Gorenstein N., Fäldt J., Miller B., Bohlmann J. (E)-beta-ocimene and myrcene synthase genes of floral scent biosynthesis in snapdragon: function and expression of three terpene synthase genes of a new terpene synthase subfamily. Plant Cell. 2003;15:1227–1241. doi: 10.1105/tpc.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C., Myers E.W. PILER: identification and classification of genomic repeats. Bioinformatics. 2005;21:i152–i158. doi: 10.1093/bioinformatics/bti1003. [DOI] [PubMed] [Google Scholar]
- Eichler E.E., Sankoff D. Structural dynamics of eukaryotic chromosome evolution. Science. 2003;301:793–797. doi: 10.1126/science.1086132. [DOI] [PubMed] [Google Scholar]
- Emms D.M., Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:1–14. doi: 10.1186/s13059-019-1832-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F., Liu B., Li M., Gao X., Fang Q., Liu C., Ding H., Wang L., Gao X. Identification and characterization of terpene synthase genes accounting for volatile terpene emissions in flowers of Freesia x hybrida. J. Exp. Bot. 2018;69:4249–4265. doi: 10.1093/jxb/ery224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., Moxon S., Marshall M., Khanna A., Eddy S.R., Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–D124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S., Grocock R.J., van Dongen S., Bateman A., Enright A.J. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–D144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo K., Tong C., Fu Q., Xu J., Shi S., Xiao Y. Identification of minor lignans, alkaloids, and phenylpropanoid glycosides in Magnolia officinalis by HPLC‒DAD‒QTOF-MS/MS. J. Pharm. Biomed. Anal. 2019;170:153–160. doi: 10.1016/j.jpba.2019.03.044. [DOI] [PubMed] [Google Scholar]
- Haas B.J., Salzberg S.L., Zhu W., Pertea M., Allen J.E., Orvis J., White O., Buell C.R., Wortman J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008;9:R7. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M.A., Clark J., Ireland A., Lomax J., Ashburner M., Foulger R., Eilbeck K., Lewis S., Marshall B., Mungall C. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L., Xu Z., Wang M., Fan R., Yuan D., Wu B., Wu H., Qin X., Yan L., Tan L. The chromosome-scale reference genome of black pepper provides insight into piperine biosynthesis. Nat. Commun. 2019;10:4702. doi: 10.1038/s41467-019-12607-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Liang J., Tan Q., Ou L., Li X., Zhong C., Huang H., Møller I.M., Wu X., Song S. Insights into triterpene synthesis and unsaturated fatty-acid accumulation provided by chromosomal-level genome analysis of Akebia trifoliata subsp. australis. Hortic. Res. 2021;8:33. doi: 10.1038/s41438-020-00458-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jim L.M., Raubeson L.A., Cui L., Kuehl J.V., Fourcade M.H., Chumley T.W., Boore J.L., Jansen R.K., Depamphilis C.W. Identifying the basal angiosperm node in chloroplast genome phylogenies: sampling one's way out of the Felsenstein zone. Mol. Biol. Evol. 2005:1948–1963. doi: 10.1093/molbev/msi191. [DOI] [PubMed] [Google Scholar]
- Johnson A.D., Handsaker R.E., Pulit S.L., Nizzari M.M., O'Donnell C.J., de Bakker P.I.W. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J., Kapitonov V.V., Pavlicek A., Klonowski P., Kohany O., Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- Kalendar R., Vicient C.M., Peleg O., Anamthawat-Jonsson K., Bolshoy A., Schulman A.H. Large retrotransposon derivatives: abundant, conserved but nonautonomous retroelements of barley and related genomes. Genetics. 2004;166:1437–1450. doi: 10.1534/genetics.166.3.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:D457–D462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilwagen J., Wenk M., Erickson J.L., Schattat M.H., Grau J., Hartung F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016;44:e89. doi: 10.1093/nar/gkw092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell M.G., Lisch D. Transposable elements as sources of variation in animals andplants. Proc. Natl. Acad. Sci. U S A. 1997;94:7704–7711. doi: 10.1073/pnas.94.15.7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M. Canu: scalable and accurate long-read assembly via adaptive -mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Suleski M., Hedges S.B. TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017;34:1812–1819. doi: 10.1093/molbev/msx116. [DOI] [PubMed] [Google Scholar]
- Lee Y.-J., Lee Y.M., Lee C.-K., Jung J.K., Han S.B., Hong J.T. Therapeutic applications of compounds in the Magnolia family. Pharmacol. Ther. 2011;130:157–176. doi: 10.1016/j.pharmthera.2011.01.010. [DOI] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N., Song Y., Zhang W., Wang W., Chen J., Wong A.W., Roberts A. Evaluation of the in vitro and in vivo genotoxicity of magnolia bark extract. Regul. Toxicol. Pharmacol. 2007;49:154–159. doi: 10.1016/j.yrtph.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Li G., Liu X., Zhang Y., Muhammad A., Cai Y. Cloning and functional characterization of two cinnamate 4-hydroxylase genes from Pyrus bretschneideri. Plant Physiol. Biochem. 2020;156:135–145. doi: 10.1016/j.plaphy.2020.07.035. [DOI] [PubMed] [Google Scholar]
- Liu Z., Zhang X., Cui W., Zhang X., Li N., Chen J., Wong A.W., Roberts A. Evaluation of short-term and subchronic toxicity of magnolia bark extract in rats. Regul. Toxicol. Pharmacol. 2007;49:160–171. doi: 10.1016/j.yrtph.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Liu Q., Li X., Zhou X., Li M., Heslop-Harrison J.S. The repetitive DNA landscape in Avena (Poaceae): chromosome and genome evolution defined by major repeat classes in whole-genome sequence reads. BMC Plant Biol. 2019;19:226. doi: 10.1186/s12870-019-1769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H., Wu H., Yu X., Zhang X., Lu Y., Fan J., Tang L., Wang Z. A review of the phytochemistry and pharmacological activities of Magnoliae officinalis cortex. J. Ethnopharmacol. 2019;236:412–442. doi: 10.1016/j.jep.2019.02.041. [DOI] [PubMed] [Google Scholar]
- Lv Q., Qiu J., Liu J., Li Z., Zhang W., Wang Q., Fang J., Pan J., Chen Z., Cheng W. The Chimonanthus salicifolius genome provides insight into magnoliid evolution and flavonoid biosynthesis. Plant J. 2020;103:1910–1923. doi: 10.1111/tpj.14874. [DOI] [PubMed] [Google Scholar]
- Ma X.-H., Ma Y., Tang J.-F., He Y.-L., Liu Y.-C., Ma X.-J., Shen Y., Cui G.-H., Lin H.-X., Rong Q.-X. The biosynthetic pathways of tanshinones and phenolic acids in Salvia miltiorrhiza. Molecules. 2015;20:16235–16254. doi: 10.3390/molecules200916235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros W.H., Pertea M., Salzberg S.L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A., Lu S., Anderson J.B., Chitsaz F., Derbyshire M.K., DeWeese-Scott C., Fong J.H., Geer L.Y., Geer R.C., Gonzales N.R. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–D229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin D.M., Fäldt J., Bohlmann J. Functional characterization of nine Norway Spruce TPS genes and evolution of gymnosperm terpene synthases of the TPS-d subfamily. Plant Physiol. 2004;135:1908–1927. doi: 10.1104/pp.104.042028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani M., Ohta D. Diversification of P450 genes during land plant evolution. Annu. Rev. Plant Biol. 2010;61:291–315. doi: 10.1146/annurev-arplant-042809-112305. [DOI] [PubMed] [Google Scholar]
- Muhlemann J.K., Klempien A., Dudareva N. Floral volatiles: from biosynthesis to function. Plant Cell Environ. 2014;37:1936–1949. doi: 10.1111/pce.12314. [DOI] [PubMed] [Google Scholar]
- Nawrocki E.P., Eddy S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.-T., Schmidt H.A., von Haeseler A., Minh B.Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu X.-M., Xu Y.-C., Li Z.-W., Bian Y.-T., Hou X.-H., Chen J.-F., Zou Y.-P., Jiang J., Wu Q., Ge S. Transposable elements drive rapid phenotypic variation in. Proc. Natl. Acad. Sci. U S A. 2019;116:6908–6913. doi: 10.1073/pnas.1811498116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel J.P., Austin M.B., Bomati E.K. Structure-function relationships in plant phenylpropanoid biosynthesis. Curr. Opin. Plant Biol. 2005;8:249–253. doi: 10.1016/j.pbi.2005.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G., Bradnam K., Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- Poivre M., Duez P. Biological activity and toxicity of the Chinese herb Magnolia officinalis Rehder & E. Wilson (Houpo) and its constituents. J. Zhejiang Univ. Sci. B. 2017;18:194–214. doi: 10.1631/jzus.B1600299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter S.C., Luciani A., Eddy S.R., Park Y., Lopez R., Finn R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018;46:W200–W204. doi: 10.1093/nar/gky448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A.L., Jones N.C., Pevzner P.A. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:i351–i358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- Puttick M.N. MCMCtreeR: functions to prepare MCMCtree analyses and visualize posterior ages on trees. Bioinformatics. 2019;35:5321–5322. doi: 10.1093/bioinformatics/btz554. [DOI] [PubMed] [Google Scholar]
- Raimundo J., Reis C.M.G., Ribeiro M.M. Rapid, simple and potentially universal method for DNA extraction from Opuntia spp. fresh cladode tissues suitable for PCR amplification. Mol. Biol. Rep. 2018;45:1405–1412. doi: 10.1007/s11033-018-4303-8. [DOI] [PubMed] [Google Scholar]
- Rendón-Anaya M., Ibarra-Laclette E., Méndez-Bravo A., Lan T., Zheng C., Carretero-Paulet L., Perez-Torres C.A., Chacón-López A., Hernandez-Guzmán G., Chang T.-H. The avocado genome informs deep angiosperm phylogeny, highlights introgressive hybridization, and reveals pathogen-influenced gene space adaptation. Proc. Natl. Acad. Sci. U S A. 2019;116:17081–17089. doi: 10.1073/pnas.1822129116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan J., Li H. Fast and accurate long-read assembly with wtdbg2. Nat. Methods. 2020;17:1–4. doi: 10.1038/s41592-019-0669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanimah S., Suri R., Azizun R.N., Hazniza A., Radzali M., Rusli I., Hassan M. Volatile compounds of essential oil from different stages of Michelia alba (cempaka putih) flower development. J. Trop. Agric. Food Sci. 2008:109–119. [Google Scholar]
- Schnable P.S., Ware D., Fulton R.S., Stein J.C., Wei F., Pasternak S., Liang C., Zhang J., Fulton L., Graves T.A. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. doi: 10.1126/science.1178534. [DOI] [PubMed] [Google Scholar]
- Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.-J., Vert J.-P., Heard E., Dekker J., Barillot E. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J., Tian J., Cheng H., Yan Q., Li L., Jamal A., Xu Z., Xiang L., Saski C.A., Jin S. The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol. 2020;21:200. doi: 10.1186/s13059-020-02088-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X., Yang L., Gao J., Sheng Y., Li X., Gu Y., Zhuang G., Chen F. Deep sequencing of Magnoliae officinalis reveals upstream genes related to the lignan biosynthetic pathway. J. For. Res. 2017;28:671–681. [Google Scholar]
- Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Soltis D.E., Soltis P.S. Nuclear genomes of two magnoliids. Nat. Plants. 2019;5:6–7. doi: 10.1038/s41477-018-0344-1. [DOI] [PubMed] [Google Scholar]
- Song X.-M., Wang J.-P., Sun P.-C., Ma X., Yang Q.-H., Hu J.-J., Sun S.-R., Li Y.-X., Yu J.-G., Feng S.-Y. Preferential gene retention increases the robustness of cold regulation in and other plants after polyploidization. Hortic. Res. 2020;7:20. doi: 10.1038/s41438-020-0253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M., Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19:ii215–ii225. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- Suyama M., Torrents D., Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarailo-Graovac M., Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics. 2009;Chapter 4 doi: 10.1002/0471250953.bi0410s25. Unit 4.10. [DOI] [PubMed] [Google Scholar]
- Tatusov R.L., Natale D.A., Garkavtsev I.V., Tatusova T.A., Shankavaram U.T., Rao B.S., Kiryutin B., Galperin M.Y., Fedorova N.D., Koonin E.V. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanholme R., De Meester B., Ralph J., Boerjan W. Lignin biosynthesis and its integration into metabolism. Curr. Opin. Biotechnol. 2019;56:230–239. doi: 10.1016/j.copbio.2019.02.018. [DOI] [PubMed] [Google Scholar]
- Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C.A., Zeng Q., Wortman J., Young S.K., Earl A.M. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Yang Z., Yang X., He Z. Analysis and comparison of aroma constituents from pistil-stamen and petal of Magnolia officinalis at different flowering stages. J. Plant Resour. Environ. 2011;20:42–48. [Google Scholar]
- Wang Y., Tang H., Debarry J.D., Tan X., Li J., Wang X., Lee T.-h., Jin H., Marler B., Guo H. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T., Sabot F., Hua-Van A., Bennetzen J.L., Capy P., Chalhoub B., Flavell A., Leroy P., Morgante M., Panaud O. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007;8:973–982. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Xia Z.Q., Costa M.A., Pelissier H.C., Davin L.B., Lewis N.G. Secoisolariciresinol dehydrogenase purification, cloning, and functional expression. Implications for human health protection. J. Biol. Chem. 2001;276:12614–12623. doi: 10.1074/jbc.M008622200. [DOI] [PubMed] [Google Scholar]
- Xu Z., Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–W268. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C., Zhi X., Xu H. Advances on semisynthesis, total synthesis, and structure-activity relationships of honokiol and magnolol derivatives. Mini Rev. Med. Chem. 2016;16:404–426. doi: 10.2174/1389557516666151120115558. [DOI] [PubMed] [Google Scholar]
- Yuan Z., Fang Y., Zhang T., Fei Z., Han F., Liu C., Liu M., Xiao W., Zhang W., Wu S. The pomegranate (Punica granatum L.) genome provides insights into fruit quality and ovule developmental biology. Plant Biotechnol. J. 2018;16:1363–1374. doi: 10.1111/pbi.12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.-C., Guo L., Guo H.-Y., Zhu K.-C., Li S.-Q., Zhang Y., Zhang N., Liu B.-S., Jiang S.-G., Li J.-T. Chromosome-level genome assembly of golden pompano (Trachinotus ovatus) in the family Carangidae. Sci. Data. 2019;6:216. doi: 10.1038/s41597-019-0238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Wu S., Chang X., Wang X., Zhao Y., Xia Y., Trigiano R.N., Jiao Y., Chen F. The ancient wave of polyploidization events in flowering plants and their facilitated adaptation to environmental stress. Plant Cell Environ. 2020;43:2847–2856. doi: 10.1111/pce.13898. [DOI] [PubMed] [Google Scholar]
- Zhi-Lei Z., Yu-Shan Z. Development and utilization Status,Problems and strategies of medicinal plant Magnolia officinalis. J. Fujian For. ence Technol. 2010;37:103–109. https://kns.cnki.net/kcms/detail/detail.aspx?FileName=FJLK201001027&DbName=CJFQ2010 [Google Scholar]
- Zwaenepoel A., Van de Peer Y. wgd-simple command line tools for the analysis of ancient whole-genome duplications. Bioinformatics. 2019;35:2153–2155. doi: 10.1093/bioinformatics/bty915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the genome assembly and raw reads reported in this paper is GenBank: PRJNA752832 and PRJNA752923, respectively. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.