

Abstract
Oxaliplatin resistance is a challenge in the treatment of colorectal cancer (CRC) patients. Regulatory T cells (Tregs) are well known for their immunosuppressive roles, and targeting Tregs is an effective way to improve chemosensitivity. Exosome-delivered microRNA (miRNA) might be used as a potential biomarker for predicting chemosensitivity. However, the relationship between Tregs and exosomal miRNAs remains largely unknown. TaqMan low-density array was performed to screen the differentially expressed serum miRNAs from pooled serum of patients who had FOLFOX treatment. Differential expression was validated using qRT-PCR in individual samples. Exosomes were isolated by sequential differential centrifugation, and they were verified by transmission electron microscopy. The RNA and protein levels were determined by quantitative real-time PCR and western blotting. A mouse xenograft model was adopted to evaluate the correlation between exosome-derived miR-208b and Tregs in vivo. We demonstrated that circulating miR-208b is a non-invasive marker for predicting FOLFOX sensitivity in CRC. miR-208b in colon cancer was secreted by tumor cells in the pattern of exosomes, and oxaliplatin-resistant cells showed the most obvious phenomenon of miR-208b increase. Colon cancer cell-secreted miR-208b was sufficiently delivered into recipient T cells to promote Treg expansion by targeting programmed cell death factor 4 (PDCD4). Furthermore, in vivo studies indicated that Treg expansion mediated by cancer cell-secreted miR-208b resulted in tumor growth and oxaliplatin resistance. Our results demonstrate that tumor-secreted miR-208b promotes Treg expansion by targeting PDCD4, and it may be related to a decrease of oxaliplatin-based chemosensitivity in CRC. These findings highlight a potential role of exosomal miR-208b as a predictive biomarker for oxaliplatin-based therapy response, and they provide a novel target for immunotherapy.
Keywords: exosomes, miR-208b, oxaliplatin resistance, Tregs, PDCD4
Graphical abstract
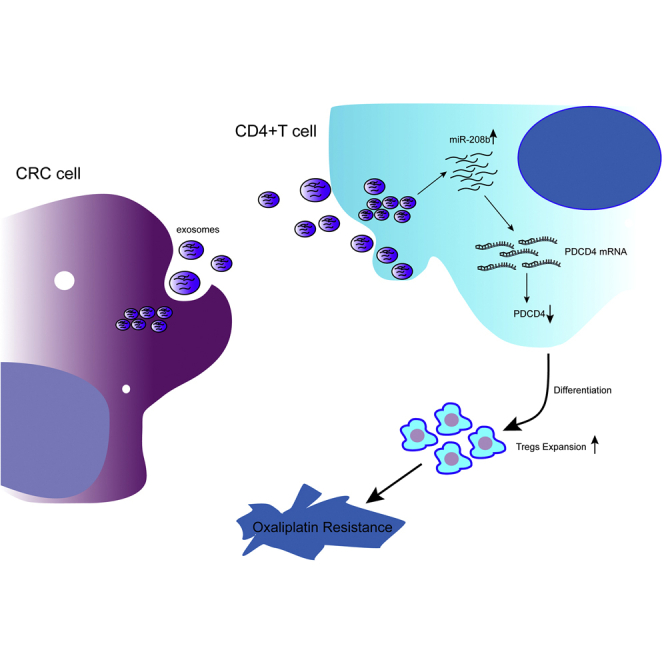
Ning et al. demonstrate that tumor-secreted miR-208b promotes Treg expansion by targeting PDCD4 and reduced oxaliplatin-based chemosensitivity in CRC. These findings highlight a potential role of exosomal miR-208b as a potential predictive biomarker for oxaliplatin-based therapy response, and they provide a novel target for immunotherapy.
Introduction
Colorectal cancer (CRC) is the third most common malignant disease and the second leading cause of cancer-related mortality worldwide.1 FOLFOX (oxaliplatin with l-leucovorin and 5-fluorouracil [5-FU]) is one of the mainstays used in a chemotherapeutic regimen following surgical resection or after recurrence and metastasis.2 Unfortunately, approximately 40%–50% of patients do not obtain an objective response at initial therapy or they acquire resistance during the course of therapy.3,4 Therefore, it is important to seek predictive biomarkers for effective therapy and achieve a deep understanding of the mechanisms for primary or acquired oxaliplatin resistance.
Oxaliplatin is a third-generation platinum compound that inhibits DNA synthesis and transcription, mainly through intrastrandal cross-links in DNA.4,5 Recent studies have shown that oxaliplatin can exert its anti-tumor effect by inducing potent immunogenic cell death, where the host immune system is activated to identify and eliminate tumor cells in response to the presentation of damage-associated molecular patterns (DAMPs) within cancer cells.6 Previous studies have shown that oxaliplatin treatment can increase the proportion of CD4+/CD8+ T lymphocytes and decrease the proportion of regulatory T cells (Tregs).7 However, some studies have reported that an increase of Tregs in peripheral blood was observed in patients receiving combined oxaliplatin and 5-FU treatment for CRC.8
Tregs are well known for their immunosuppressive roles, and multiple preclinical and clinical studies have suggested that Tregs may prevent the development of effective antitumor immunity in patients with established tumors, and possibly promote tumor progression.9 Therefore, targeting Tregs might be an effective way to improve the efficacy of oxaliplatin-based chemotherapy.
Exosomes are a type of small membrane vesicles with a diameter of 30–100 nm that are released from most cell types.10,11 Exosomes act as message transmitters in intercellular communication because they contain a variety of proteins, lipids, circular RNAs (circRNAs), and microRNAs (miRNAs).12 Our group has shown that exosomes play an important role in the establishment of the tumor microenvironment, and they can be used as nanocarriers for drugs or as biomarkers for diseases.13, 14, 15 Fast-growing tumor cells highly express certain miRNAs and release high levels of miRNA-containing exosomes.16 In addition, tumor cells can selectively package certain specific miRNAs into the exosomes, which are secreted under various pathophysiologic conditions.10,12 Additionally, a recent study revealed that tumor-derived miRNA can promote Treg expansion and induce tumor immune evasion.17 We postulate that CRC cells might actively regulate Tregs by delivering exosomal miRNAs and thus affect the anti-tumor activity of oxaliplatin.
The present study was designed to investigate the role of CRC cell-derived exosomes in the regulation of Tregs and to evaluate the predictive and therapeutic effects of exosomal miRNA on tumor growth and chemotherapy sensitization. We found that miR-208b was significantly elevated in the serum of FOLFOX-resistant CRC patients. In vitro data demonstrate that miR-208b was mainly packaged in the exosomes and was sufficiently delivered into recipient T cells, promoting Treg expansion by targeting programmed cell death factor 4 (PDCD4). An in vivo study demonstrated that tumor-derived miR-208b increases the ratio of Tregs, promotes tumor growth, and reduces chemotherapy efficacy. In conclusion, CRC-secreted miRNA-208b might be a promising biomarker for predicting chemosensitivity of an oxaliplatin-based regimen, and targeting miR-208b serves as a promising strategy to suppress immune evasion and improve chemosensitivity of tumor cells.
Results
Circulating miR-208b is a non-invasive biomarker for predicting FOLFOX sensitivity in CRC
We designed a multiphase study to identify novel serum miRNAs as surrogate markers for predicting and monitoring the response to chemotherapy with FOLFOX (Figure 1). In the screening phase, compared with responders, serum from non-responders had 13 upregulated miRNAs and 15 downregulated miRNAs (fold change > 25). As a consequence, 28 miRNAs were identified and chosen for additional qRT-PCR validation (Table S1). The serum levels of miR-208b were normalized to U6 and the raw Ct value of U6 from TaqMan low-density array (TLDA) analysis (Table S3). In the testing phase, results showed that miR-208b was differentially expressed between these two groups (n = 12, Table S2). Subsequently, in the validating phase, miR-208b was selected as a candidate for further validation in a larger scale population (69 chemo-sensitive versus 47 chemo-insensitive patients). As expected, serum miR-208b was significantly decreased in responsive patients (p < 0.001, Figures 2A and 2B). Furthermore, serum carcinoembryonic antigen (CEA) levels were also detected at the same time (Figure 2B). ROC curve of the miRNA-208b and CEA levels in Figure 2B. The area under the curve (AUC) for serum miR-208b (95% confidence interval [CI], 0.771 [0.688, 0.855]) in comparison with the AUC for serum CEA (95% CI, 0.493 [0.385, 0.601]), and serum miR-208b has better behaves than serum CEA levels. We further evaluated the changing trend of miR-208b to chemotherapy. We found that the expression of miR-208b was significantly altered after chemotherapy, whose level was reduced in patients who were responsive to FOLFOX, but it was elevated in patients who were non-responsive to this regimen (Figure 2C). We also analyzed the progression-free survival (PFS) of these patients. There was a significantly longer PFS in the responder arm (p < 0.001), and patients with low levels of miR-208b before treatment were significantly associated with prolonged PFS (hazard ratio [HR] = 1.90; 95% CI, 1.24–2.90; p < 0.001) (Figure 2D). Taken together, these results demonstrated that serum miR-208b may serve as a non-invasive marker for predicting the chemosensitivity of FOLFOX in CRC.
Figure 1.

Flowchart depicting the experimental design
Figure 2.

Circulating miR-208b is a promising biomarker for predicting chemosensitivity of FOLFOX in CRC
(A) The level of serum miR-208b was significantly decreased in response patients compared with non-response patients (69 chemosensitivity versus 47 chemo-insensitivity). (B) Receiver operating characteristic (ROC) curve of the miRNA-208b and CEA levels. The area under the curve (AUC) for miRNA-208b was 0.771 (95% CI, 0.688–0.855), and the AUC for CEA was 0.493 (95% CI, 0.385, 0.601). (C) qRT-PCR analysis of the level of serum miR-208b. For response patients, it was significantly decreased after chemotherapy compared with before chemotherapy (n = 12). Additionally, the post-chemotherapy level of serum miR-208b was significantly increased in non-response patients compared with pre-chemotherapy (n = 12). (D) Kaplan-Meier analysis of progression-free survival (PFS) rate for response and non-response patients (p < 0.0001), and PFS with low and high serum miR-208b levels. ∗∗p < 0.01, ∗∗∗p < 0.001.
miR-208b is secreted by colon cancer cells in the pattern of exosomes
Many circulating miRNAs are correlated with the progression of CRC,18, 19, 20 and it is well known that cancer cells can release miRNAs through exosomes.10,21 Next, exosomes derived from NCM460, SW480, and SW480-OXA cells (a human oxaliplatin-resistant colon cancer cell line) were isolated from the sequential differentially centrifuged supernatants, and miR-208b expression was evaluated in these cells and cell-derived exosomes. As shown in Figures 3A–3C, cell-derived exosomes exhibited a typical round morphology, with a diameter of approximately 100 nm, and high expression of CD63, Alix, and Tsg101. Then, we detected the level of miR-208b in these cells and cell-derived exosomes using qRT-PCR. As expected, the SW480 cells showed an increase in the expression of miR-208b compared with the NCM460 cells, and the SW480-OXA cells showed the highest level of miR-208b (Figure 3D). In addition, miR-208b was enriched in CRC cell-derived exosomes, especially SW480-OXA (Figure 3E). These data suggested that miR-208b in colon cancer is secreted by tumor cells in the pattern of exosomes, and it may be related to oxaliplatin chemosensitivity.
Figure 3.

miR-208b is secreted by colon cancer cells in the pattern of exosomes
(A) Transmission electron microscopy showed exosomes purified from the supernatants of NCM460, SW480, and SW480-OXA cells. (B) Nanoparticle tracking analysis was used to determine the absolute exosome size distribution and concentration of exosomes (particles/mL). (C) Western blot analysis of the expression of exosomal markers (CD63, TSG101, and Alix) in cell lysates and exosomal protein purified from NCM460, SW480, and SW480-OXA cell supernatants. (D) Relative levels of miR-208b in NCM460, SW480, and SW480-OXA cells measured by qRT-PCR (n = 3). (E) qRT-PCR analysis of miR-208b levels in exosomes purified from NCM460, SW480, and SW480-OXA cell supernatants (n = 3). ∗∗p < 0.01.
SW480 cell-secreted exosomal miR-208b promotes Treg expansion
Previous studies have shown that oxaliplatin influences the tumor immune microenvironment.6,7 It has been reported that tumor-secreted miRNAs may induce immune evasion via promoting Treg expansion.17,22 Therefore, we subsequently evaluated the possible effect of CRC-secreted exosomal miR-208b on the differentiation of T cells. To remove miR-208b from exosomes, small interfering RNA (siRNA) of miR-208b was transfected into SW480 and SW480-OXA cells and exosomes were collected at 48 h post-transfection. Then, cell-derived exosomes containing different levels of miR-208b were incubated with primary CD4+ T cells in culture (Figure 4A), and the level of miR-208b in T cells was tested with qRT-PCR. As shown in Figure 4B, transfection of miR-208b inhibitors in SW480 and SW480-OXA cells resulted in decreased levels of miR-208b in exosomes. The levels of miR-208b in the recipient CD4+ T cells were markedly increased with cell-derived exosomes, especially in the SW480-OXA group, while no alterations were observed when treated with miR-208b-deficient exosomes (Figure 4C), suggesting that the elevation of miR-208 levels in CD4+ T cells was likely related to the exosome delivery of exogenous miR-208b. Next, the effect of cell-derived exosomes on recipient T cells was assessed. As is shown in Figure 4D, compared with the control group, SW480 and SW480-OXA cell-derived exosomes increased the percentage of CD4+CD25highFoxp3+ Tregs (p < 0.01), while miR-208b-deficient exosomes did not affect Treg expansion. Furthermore, the changes were apparent in the SW480-OXA group (Figure 4D). These results suggest that SW480 cell-secreted exosomal miR-208b promotes Treg expansion.
Figure 4.

SW480 cell-secreted exosomal miR-208b promotes Treg expansion
(A) Co-culture model for the addition of exosomes purified from SW480 cells to CD4+ T cells. (B) qRT-PCR analysis of miR-208b levels in exosomes purified from SW480, SW480-208b-del, SW480-OXA, and SW480-OXA-208b-del cells. (C) Confocal microscopy image of the internalization of fluorescently labelled exosomes in CD4+ T cells. (D) qRT-PCR analysis of miR-208b levels in CD4+ T cells co-cultured with different exosomes (n = 3). (E) Flow cytometry analysis of the percentage of CD4+CD25highFoxp3+ Tregs in CD4+ T cells co-cultured with different exosomes (n = 3). ∗∗p < 0.01.
miR-208b directly targets and inhibits PDCD4 expression in CD4+ T cells
To further explore the molecular mechanisms responsible for the function of miR-208b, we searched for the downstream target genes of miR-208 by using miRNA target prediction software (TargetScan, PicTar, and RNAhybrid), and PDCD4 was chosen for further experimental confirmation. According to the bioinformatics prediction, miR-208b can directly target the 3′ UTR of PDCD4 mRNA (Figure 5A). Then, we efficiently overexpressed or knocked down the miR-208b level in 293T cells with miR-208b mimics or inhibitors, respectively (Figure 5B). The luciferase assay showed that the relative luciferase activity was clearly inhibited when miR-208b mimics were co-transfected with the luciferase reporters, while the relative luciferase activity was noticeably increased when miR-208b inhibitors were used (Figure 5C). In SW480-derived exosomes (especially in SW480-OXA-derived exosomes) there was also a sharp decrease in luciferase activity compared to the control group, but this inhibition was lost when miR-208b was removed from the exosomes (miR-208b del) (Figure 5D). In addition, there was no enhancement or inhibition of luciferase activity following transfection with miR-208b in the mutant group, which did not contain the predicted binding sites. Next, we detected protein level by western blot. As shown in Figure 5E, the expression of PDCD4 in T cells was downregulated when treated with SW480 exosomes, and this inhibition was more obvious in the SW480-OXA group; however, this phenomenon disappeared when miR-208b was deleted from the exosomes. Additionally, PDCD4 protein dramatically decreased upon the addition of miR-208b mimics, whereas the inhibition of miR-208b relatively enhanced the expression of PDCD4. Furthermore, there were no obvious changes in PDCD4 expression at the mRNA level (Figure 5F). We then evaluated the effect of miR-208b on the expansion of CD4+ T cells. It was shown that transfection of miR-208b mimics noticeably enhanced Treg expansion, whereas suppressing miR-208b levels inhibited Treg expansion (Figures 5G and 5H). These results indicate that miR-208b directly targets PDCD4, leading to the differentiation of Tregs.
Figure 5.

miR-208b promotes Treg expansion by directly targeting and inhibiting PDCD4 expression
(A) Predicted binding region between PDCD4 and miR-208b. (B) Relative levels of miR-208b in 293T cells transfected with miR-208b mimics and inhibitors (n = 3). (C) Relative levels of luciferase in 293T cells co-transfected with firefly luciferase reporter plasmid containing either wild-type (WT) or mutant PDCD4 3′ UTR and miR-208b mimics/inhibitors (n = 3). (D) Cell-derived exosomal miR-208b directly binds to the 3′ UTR of PDCD4 mRNA (n = 3). (E) Western blot analysis of PDCD4 expression in CD4+ T cells transfected with miR-208b mimics and inhibitors (n = 3). (F) Relative levels of PDCD4 mRNA in cell-derived exosomes (n = 3). (G) Flow cytometry analysis of the CD4+CD25highFoxp3+ Tregs transfected with miR-208b mimics and inhibitors (n = 3). (H) Quantitative analysis of the percentage of CD4+CD25highFoxp3+ Tregs in cells transfected with miR-208b mimics and inhibitors, respectively (n = 3). ∗∗p < 0.01, ∗∗∗p < 0.001.
Validation of the role of PDCD4 in regulating Treg expansion
We next investigated the effects of PDCD4 on Treg expansion in CD4+ T cells. As shown in Figures 6A–6C, the expression levels of both PDCD4 protein and mRNA were markedly upregulated in the PDCD4 overexpression (OE.PDCD4) group and were sharply downregulated by siRNA of PDCD4 (si.PDCD4) as compared with the control. When CD4+ T cells were co-transfected with miR-208b inhibitors and PDCD4 siRNA simultaneously, the expression of PDCD4 protein was partly rescued (Figure 6A). CD4+ T cells in the OE.PDCD4 group exhibited a lower rate of Treg expansion, but they exhibited a notably higher rate of Treg expansion in the si.PDCD4 group (Figures 6D and 6E). Thus, it was confirmed that PDCD4 is a key gene that negatively regulates Treg expansion.
Figure 6.

PDCD4 is closely associated with Treg expansion
(A) Western blot analysis of PDCD4 expression in cells transfected with PDCD4 overexpression (OE.PDCD4), siRNA of PDCD4 (si.PDCD4), and si.PDCD4+miR-208b inhibitors (si.PDCD4 inhibitors.208b) (n = 3). (B) Quantitative analysis of (A) (n = 3). (C) Relative levels of PDCD4 mRNA in cells transfected with OE.PDCD4, si.PDCD4, and si.PDCD4 inhibitors.208b (n = 3). (D) Flow cytometry analysis percentage of CD4+CD25highFoxp3+ Tregs in OE.PDCD4, si.PDCD4, and si.PDCD4+ inhibitors.208b groups (n = 3). ∗∗p < 0.01(E) Histogram analysis of (D) (n=3).
Tumor-derived exosomal miR-208b affects chemotherapy and tumor growth in vivo
We next determined the effects of tumor-secreted miR-208b on Tregs and tumor growth in vivo. CT26 cells were transfected with lentivirus to generate miR-208b-overexpressing (miR-208b.OE) and miR-208b-knockdown (miR-208b.KD) cell lines. A diagram of the animal experimental design is shown in Figure 7A. Experiments were performed using six groups (10 mice each), and three groups of them received oxaliplatin. BALB/c mice were implanted with CT26 cells to create a tumor-implanted model. As shown in Figures 7B–7D, the miR-208b.OE group presented with a high rate of tumor growth. However, the growth of tumors in the miR-208.KD group was shown to be slower than that in the control group. The groups (OXA group, miR-208b.OE OXA group, and miR-208b.KD OXA group) received oxaliplatin. The miR-208b.OE OXA group presented with a high rate of tumor growth and miR-208b. The KD OXA group presented with a low rate of tumor growth compared with the OXA group. This suggests that the miR-208b level may be important for reducing chemosensitivity upon oxaliplatin treatment. However, in vitro and vivo studies showed that miR-208b has little effect on the growth of CT26 cells (Figures S1 and S2). Additionally, we investigated the tumor-infiltrated Tregs in BALB/c mice by immunohistochemistry analysis, as shown in Figure S3. Compared with the group of miR-208b.KD and the negative control (NC), the miR-208b.OE group had more obvious tumor-infiltrated Tregs. These results indicating that miR-208b promotes tumor growth in vivo by inducing increased numbers of Tregs. The peripheral CD4+ T cells isolated from these mice showed a significant downregulation of PDCD4 (Figures 7E and 7F) in the miR-208b.OE group, and the result was the opposite in the miR-208.KD group. However, no difference was observed in the PDCD4 mRNA levels (Figure 7G). Then, exosomes were isolated from the serum and the miR-208b level was detected (Figure 7H). As expected, miR-208b was markedly increased in the miR-208b.OE group, while the level of miR-208b was decreased in the miR-208b.KD group (Figure 7I). These results demonstrated that tumor-secreted miR-208b is sufficiently delivered into CD4+ T cells and inhibits PDCD4 expression in vivo. Moreover, the phenomenon was more apparent in the groups treated with oxaliplatin (miR-208b.OE+OXA and miR-208.KD+OXA).
Figure 7.

miR-208b facilitates tumor growth by promoting Treg expansion in vivo
CT26 cells were transfected with lentivirus to generate miR-208b-overexpressing (miR-208b.OE) and miR-208b-knockdown (miR-208b.KD) cell lines. Then, 1 × 107 cells were subcutaneously injected into the groins of BALB/c mice to create a tumor-implanted model. (A) Flow diagram for creating the implanted tumor model. (B) Analysis of tumor diameters in each group (n = 10). (C) The comparison of the tumors taken from different groups (n = 10). (D) Analysis of the weight of tumors taken from different groups (n = 10). (E) Western blotting analysis of PDCD4 in each group (n = 10). (F) Quantitative analysis of (E) (n = 10). (G) Relative levels of PDCD4 mRNA in different groups (n = 10). (H) Scanning electron microscopy of exosomes isolated from mouse serum. (I) Relative levels of miR-208b in serum exosomes isolated from different groups (n = 10). ∗p < 0.05, ∗∗p < 0.01.
We next determined the effects of secreted miR-208b on Tregs and survival in vivo. As expected (Figures 8A and 8B), the percentage of CD4+CD25+Foxp3+ Tregs in the total CD4+ T cell population isolated from the peripheral blood and spleen was significantly higher in the miR-208b.OE and miR-208b.OE+OXA groups, whereas a significant decrease in the percentage of Tregs in the CD4+ T cells was shown in the miR-208b-KD and miR-208.KD+OXA groups. Consistent with the clinical data, the survival rate of the low miR-208b expression group was consistently higher than that of the group with high expression (Figure 8C). Taken together, these results indicate that tumor-secreted miR-208b increases the percentage of Tregs and promotes tumor growth in vivo. Also, oxaliplatin can regulate the number of Tregs through miR-208b in vivo, which is related to chemosensitivity of oxaliplatin ingredients.
Figure 8.

Cancer cell-secreted miR-208b increases the number of Tregs in vivo
(A) Flow cytometry analysis of the CD4+CD25highFoxp3+ Tregs in six groups (n = 10). (B) Quantitation percentage of the CD4+CD25highFoxp3+ Tregs in different group (n = 10). (C) Survival curve analysis of survival rate in six groups (n = 10). ∗p < 0.05, ∗∗p < 0.01.
Discussion
Primary and acquired resistance of oxaliplatin remains a major challenge to the long-term management of advanced CRC patients. There is a need to identify markers to predict drug efficiency and spare patients of the side effects of ineffective treatment. It is well established that oxaliplatin can induce systemic immune responses and the immune-microenvironment, which are related with the efficacy. However, few reports have shown the relationship between Tregs and chemotherapy. In the present study, we report that tumor-secreted miR-208b plays a fundamental role in tumorigenesis by suppressing the immune response in host cells, which in turn accelerates tumor growth. This result reveals a new mechanism of oxaliplatin resistance and provides a novel biomarker for predicting chemotherapy oxaliplatin sensitivity in CRC.
miR-208b may serve as a predictor and modifier of oxaliplatin-based chemotherapy in metastatic CRC. Aberrant function and expression profiles of miRNAs have been reported in many types of cancers, including CRC. However, the importance of miRNAs involved in drug resistance has only been noted during the past few years.23,24 Most miRNAs have been demonstrated to alter the cellular response to chemotherapy via modulation or by negatively regulating targeted mRNAs that are involved in survival or by an apoptotic pathway.25 In the present study, we performed studies analyzing distinctive expression of circulating miRNA profiles in association with the response to first-line FOLFOX treatment in CRC patients. There was a higher expression level of circulating miRNAs in the non-responders than that of responders before treatment, indicating that the association of these increased miRNA expression with drug resistance and poor clinical outcomes. In addition, by comparing the paired blood samples before and after treatment, we further found that these circulating miRNAs were consistently altered in different populations responsive to chemotherapy. Thus, using the profile of miRNA in each combination therapy may assist oncologists in selecting the most appropriate therapy for each patient, as well as monitoring the therapy response during treatment. A previous similar study demonstrated changes in miRNA expression following exposure to 5-FU/oxaliplatin in colon cancer cells.3 Previous studies have evaluated the correlation of miRNA expression with chemotherapy response in patients treated with first-line 5-FU/oxaliplatin-based treatment.3,26 Both studies identified a profile of circulating miRNA expression in patients responding and not responding to 5-FU/oxaliplatin-based treatment as a predictor for the response to chemotherapy, but with inconsistent results. The considerable inconsistency in these two studies may be attributed to the lack of a validated method for sample collection, RNA isolation, and consistent normalization. Appropriate normalization using stably expressed reference genes is important and critical for the accurate quantification of RNA levels with qRT-PCR. Our laboratory has clearly demonstrated that a combination of let-7d, let-7g, and let-7i can be a suitable reference for the normalization of serum miRNAs and is statistically superior to the commonly used reference genes.27 In this study, we used let-7d/g/i as an endogenous control for normalization of serum miRNA levels.
Tumor-secreted miR-208b induces Tregs in CRC. Tregs are well known for their immunosuppressive roles in maintaining self-tolerance and in controlling inflammatory responses. The presence of elevated frequencies of Tregs in tumors correlates with disease progression and poor survival in patients with cancer.28 Elevated frequencies of Tregs have been reported in many types of tumors, including breast,29 lung,30 and ovarian carcinoma,31 and their high frequencies correlate with poor prognosis. However, in CRC, the prognostic role of Tregs remains controversial. In the present study, we showed Tregs in colon cancer could induce tumor immune evasion, and this efficacy was correlated with the expression of miR-208b derived from tumor cells. Multiple factors, such as interleukin (IL)-2,32, 33, 34 CCR2, and transforming growth factor (TGF)-β,32,33 can be responsible for the Treg expansion in the tumors. Herein, we present evidence suggesting that tumor-secreted miR-208b could expand the CD4+CD25highFoxp3+ Treg population in CRC. Our results showed that the miR-208b level was increased with tumor growth. The current results still do not support that CD4+ T cell conversion to Tregs is only dependent on miR-208b. Although the proportion of CD25+Foxp3+ cells with treatments compared to controls was only slightly increased (Figures 4 and 5), it also shows statistical significance. It would be better that other functional data be provided to show whether and how the Treg expansion contributes to tumor growth and oxaliplatin resistance. There are still many other regulation pathways through which CD4+ T cells are converted to Tregs, and what we have discovered is only one of them. In addition, the expression level of miR-208b could influence the sensitivity of oxaliplatin-based chemotherapy. Some studies have reported that oxaliplatin treatment caused a reduction in the proportion of Tregs compared to the vehicle-treated cohort, while other studies have found an increase of Tregs in blood samples from patients receiving combined oxaliplatin and 5-FU treatment for CRC.
Tumor-derived miR-208b induces Treg expansion by targeting PDCD4. PDCD4 has recently been demonstrated to be a new tumor suppressor gene involved in programmed cell death.35,36 Its expression has been reported, for example, in human IL-12-induced natural killer (NK) and T cells,37 normal lung tissues from humans,38 senescent human fibroblasts,39 and growth-suppressed human neuroendocrine pancreatic carcinoma cells.40 Previous studies have shown that normal mucosa demonstrated strong nuclear Pdcd4, which was reduced significantly in adenomas (p < 0 0.01) and was almost lost in tumors (p < 0.01).41 PDCD4 is an independent prognostic factor in CRC and is regulated by a miRNA.42 Recently, some studies showed that PDCD4 is related to inflammation and can influence the differentiation of partial T lymphocyte subsets.43, 44, 45 However, when T cells were transfected with miR-208b, an clear change of PDCD4 was observed. It is likely that PDCD4 in tumors is not regulated by one miRNA and that it is a target of miR-208b in T cells. It is suggested that different regulation mechanisms may existed between T cells and tumors. In addition, different tumor types may have different targets. For instance, Yin et al.17 reported that tumor-derived miR-214 efficiently downregulated phosphatase and tensin homolog (PTEN) and promoted Treg expansion in lung cancer.
In conclusion, our study provides evidence that tumor-derived miR-208b promotes Treg expansion by directly targeting PDCD4 and influences the oxaliplatin-based chemosensitivity in CRC. These findings highlight a potential role of miR-208b as a predictive biomarker for oxaliplatin-based chemosensitivity and provide insight into understanding the association between miRNAs and the chemotherapy response. Current results still do not support that the conversion of CD4+ T cells to Tregs is only dependent on miR-208b, and there are still many other regulation pathways waiting to be exploited. Through directly targeting immune cells, tumor-secreted exosomal miRNAs manipulate the immune response and promote Treg expansion.
Materials and methods
Patients
From 2009 to 2013, 116 CRC patients in the first-line FOLFOX regimen (oxaliplatin, leucovorin, fluoropyrimidine) therapy from Tianjin Medical University Cancer Institute and Hospital were recruited. Eligible patients were previously untreated with systemic treatment for CRC. Previous adjuvant chemotherapy was permitted if it had been completed at least 6 months before inclusion. All patients showed a performance status (Eastern Cooperative Oncology Group) score of 0 or 1. Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 guidelines were used to assess tumor response. We considered patients showing complete response (CR) and partial response (PR) as responders (chemosensitivity), and stable disease (SD) and progressive disease (PD) as non-responders (chemo-insensitivity). PFS was defined as the time from the first date of chemotherapy to the date of confirmed disease progression. Written informed consent was obtained from all patients prior to the study. The study was approved by the Institutional Review Board of Tianjin Medical University Cancer Institute and Hospital.
TLDA analysis of serum miRNA
Venous blood samples were collected from 12 responders and 12 non-responders before they received first-line therapy. After centrifugation, equal volumes of serum from these responders or non-responders were pooled separately, and total RNA was extracted by using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). For the TLDA analysis, reverse transcription was performed using the TaqMan miRNA reverse transcription kit and Megaplex RT primers as described previously.46 Reverse transcription was performed using a thermal cycler (UNO-Thermoblock, Biometra, Göttingen, Germany). miRNA profiling of human miRNAs was then performed using TLDA analysis (TaqMan array human miRNA A+B cards set v3.0) according to the manufacturer’s instructions.
RNA isolation and qRT-PCR
Total RNA was isolated from serum, cells, or exosomes using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Quantification of miRNAs and mRNAs was performed using methods as described previously.47 miRNA determination was performed using TaqMan miRNA probes (Applied Biosystems, Foster City, CA, USA). Reverse transcription and qPCR were performed on an Applied Biosystems AB7500 real-time PCR system. U6 snRNA was used as an internal control for miRNAs, and mRNA levels were normalized to GAPDH. Primer sequences for PDCD4 and GAPDH were as follows: PDCD4 sense, 5′-TTGCC GCAAAGTGTGTAACG-3′ and PDCD4 antisense, 5′-GTCAC CCCTAAATGCCACCG-3′; GAPDH sense, 5′-AGAAGGCT GGGGCTCATTTG-3′ and GAPDH antisense, 5′-AGGGGC CATCCACAGTCTTC-3′.
Serum CEA detection
The serum values of CEA were measured using a commercial kit (CEA fluoroimmunometric assay, Beckman Coulter, Fullerton, CA, USA) on the Beckman Coulter UniCel DxI 800 access immunoassay system.
Cell lines and cell culture
The human colon cancer cell line SW480 and human oxaliplatin-resistant colon cancer cell line SW480-OXA were purchased from the Shanghai Institute of Cell Biology of the Chinese Academy of Sciences (Shanghai, China), and the normal human colon epithelial cell line NCM460 was obtained from INCELL. The mouse colon cancer cell line CT26 was purchased from the Shanghai Institute of Cell Biology of the Chinese Academy of Sciences (Shanghai, China). CT26, SW480, and SW480-OXA cells were cultured in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (Gibco) with routine addition of ampicillin and streptomycin. NCM460 cells were cultured in DMEM/high-glucose medium (HyClone, GE Healthcare Life Sciences) supplemented in the same manner as for RPMI 1640.
Cell transfection
miRNA overexpression was achieved by transfecting cells with miRNA mimics (RiboBio, Guangzhou, China), whereas knockdown was achieved by transfecting the cells with miRNA inhibitors (RiboBio), and Lipofectamine 2000 (Invitrogen) was used for cell transfection. PDCD4 overexpression lentiviral particles were used for PDCD4 upregulation, and an empty lentiviral vector was used as a negative control. A siRNA targeting PDCD4 (sc-39554, Santa Cruz Biotechnology) was used for PDCD4 downregulation, and a scrambled siRNA was used as a negative control.
Isolation of exosomes from serum and medium
Exosomes in serum, as well as in cell medium, were isolated by gradient centrifugation as described previously.14 Briefly, the serum and cell medium were initial centrifuged at 3,000 × g to remove cells and other debris, and then the supernatants were centrifuged at 10,000 × g to remove shedding vesicles and other vesicles that were larger than exosomes. Finally, the supernatants were centrifuged at 110,000 × g for 70 min in 4°C. Exosomes were harvested, resuspended in PBS, and stored at −80°C.
Transmission electron microscopy (TEM) assay
The exosomes were placed in 2.5% glutaraldehyde and fixed overnight at pH 7.2 at 4°C. After rinsing in PBS three times (10 min each) and post-fixing in 1% osmium tetroxide for 60 min at room temperature, the samples were embedded in 10% gelatin, fixed in glutaraldehyde at 4°C, and cut into several blocks (less than 1 mm). Then, the samples were dehydrated in increasing concentrations of alcohol (30%, 50%, 70%, 90%, 95%, and 100% × 3, 10 min, each step). Specimens were then infiltrated with increasing concentrations of Quetol-812 epoxy resin mixed with propylene oxide (25%, 50%, 75%, and 100%, at least 3 h for each step). Specimens were embedded in fresh pure Quetol-812 epoxy resin. After polymerization at 35°C for 12 h, 45°C for 12 h, and 60°C for 24 h, a Leica UC6 ultramicrotome was used to cut ultrathin sections (100 nm). Then, the specimens were stained with uranyl acetate for 10 min and lead citrate for 5 min before they were observed in an FEI Tecnai T20 transmission electron microscope.
Nanoparticle tracking analysis (NTA)
The sizes of exosomes were measured using the NanoSight NS300 system (NanoSight Technology, Malvern, UK) as described previously.23,24 Briefly, after resuspension in PBS (5 μg/mL), samples were further diluted 100- to 500-fold to achieve 20–100 objects per frame and manually injected into the sample chamber at room temperature. Particles were then configured with a high-sensitivity sCMOS camera and a 488-nm laser. Each sample was monitored in triplicate at a camera setting of 13 with a detection threshold setting of 7 and an acquisition time of 30 s. At least 200 completed tracks were analyzed per video. Finally, the data were analyzed using NTA analytical software (version 2.3)
Isolation and culture of CD4+ T lymphocytes
Freshly isolated mice spleens and peripheral blood were placed in cold PBS and homogenized immediately. The lymphocyte subsets were purified using a CD4+ T cell isolation kit (Miltenyi Biotec) according to the manufacturer’s instructions. The CD4+ T lymphocyte were cultured in RPMI 1640 medium supplemented with 1 mM HEPES, 1 μg/mL anti-mouse CD3e (BD Biosciences), 1 μg/mL anti-mouse CD28 (BD Biosciences), and 12% FBS in a 5% CO2 water-saturated atmosphere.
Flow cytometry
A mouse Treg staining kit (eBioscience) was used to analyze the CD4+CD25highFoxp3+ Tregs. The cells were analyzed with a BD FACSCalibur flow cytometer using CellQuest Pro software (BD Biosciences).
Western blotting
The PDCD4 protein expression was assessed by western blotting and its level was normalized to GAPDH. In addition, CD63, TSG101, and Alix were used as markers and internal controls for exosomes. Total proteins were extracted from cells or exosomes in radioimmunoprecipitation assay (RIPA) buffer with freshly added protease inhibitor. Then, total lysates were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). After blocking with 2% bovine serum albumin (BSA) for 1 h, the membranes were incubated overnight at 4°C with anti-PDCD4 (1:2,000, Abcam), anti-CD63 (1:1,000, Santa Cruz Biotechnology), anti-TSG101 (1:1,000, Santa Cruz Biotechnology), anti-Alix (1:1,000, Santa Cruz Biotechnology), and anti-GAPDH (1:3,000, Abcam) antibodies, respectively. Then, the membranes were incubated with the corresponding secondary antibodies for 2 h and visualized with an enhanced chemiluminescence system kit (Millipore, Bedford, MA, USA) according to the manufacturer’s protocol.
miRNA target prediction and luciferase reporter assay
miRNA target prediction was analyzed with the bioinformatics tools of TargetScan (http://www.targetscan.org/), miRanda (http://www.microrna.org/), and PicTar (https://pictar.mdc-berlin.de/). Then, the intersection of prediction results was used for verification. The expression sequence of miR-208b was obtained from microRNA.org (http://www.microrna.org/). The reporter plasmid p-MIR-PDCD4 containing the predicted miR-208b targeting regions was designed by GenScript (Nanjing, China), and β-galactosidase expression vector (Ambion) was used as a transfection control. For the luciferase reporter assays, luciferase reporter plasmid (2 mg), β-galactosidase expression plasmid (2 mg), and equal amounts (200 pmol) of the miR-208b mimic, inhibitor, or scrambled negative control RNA were transfected into the prepared CRC cells using Lipofectamine 2000. Similarly, the prepared cells were also cotransfected with luciferase reporter plasmid, β-galactosidase expression plasmid (2 mg), and equal amounts of exosomes derived from SW480 or SW480-OXA. Twenty-four hours later, cells were analyzed using a Dual-Luciferase assay kit (Promega) according to the manufacturer’s instructions.
Animal experiments
The lentiviruses containing miR-208b overexpressing sequences or short hairpin RNA (shRNA) (GeneChem, Shanghai, China) were used to increase or decrease the expression of miR-208b in the CT26 cells according to the protocol. Tumor-implanted mice were divide into six groups (10 mice per group), with three of them having received oxaliplatin. Then, these cells were injected subcutaneously into each BALB/c mouse (1 × 107 cells per mouse). Mice were sacrificed 28 days after injection to remove the xenografted tumors, and the volumes and weights of the tumors were recorded. Before mice were sacrificed, peripheral blood was collected. In a separate survival experiment, the survival time of mice (10 mice per group) was determined to evaluate the life-prolonging effect. All experimental procedures were approved by the Institutional Animal Care and Research Advisory Committee of Tianjin Medical University Cancer Institute and Hospital.
Immunohistochemical (IHC) analysis
IHC straining was performed in formalin-fixed, paraffin-embedded tissues using standard methods. In brief, sections were heat treated for antigen retrieval in 10 mmol/L citrate solution and then incubated with 5% BSA to block unspecific binding sites. For IHC analysis, sections were incubated with Foxp3+ primary antibody (1:500, Cell Signaling Technology) at 4°C overnight. A negative control was performed by replacing the primary antibody with PBS. The sections were then incubated with a secondary antibody labeled with horseradish peroxidase (1:100, Dingguo Changsheng Biotechnology) at room temperature for 60 min. Finally, the signal was developed for visualization with 3,3′-diaminobenzidine (DAB) tetrahydrochloride, and all of the slides were counterstained with hematoxylin.
PKH26 staining
PKH26 red fluorescent cell linker kits (Sigma) were used for lipid bilayer labeling. Exosomes were first resuspended in 100 μL of diluent C. A dye solution (4 × 10−6 M) was prepared by adding 0.4 μL of PKH26 ethanolic dye solution to 100 μL of diluent C. The 100 μL of exosome suspension was then mixed with 100 μL of dye solution by pipetting. After incubating the cell/dye suspension for 3–5 min with periodic mixing, the staining was stopped by adding 200 μL of serum and incubating for 1 min. The stained exosomes were finally washed twice with 1× PBS and resuspended in a fresh sterile conical polypropylene tube. Eventually, moderate amounts of stained exosomes were added into cell medium to be coincubated for 4 h before imaging.
Statistical analysis
All experiments were performed in triplicate, and the results are presented as the mean value ± standard deviation. SPSS 20.0 was used to analyze the data. Difference between the groups were performed using an unpaired Student’s t test and analysis of variance (ANOVA). Survival curves were conducted according to the Kaplan-Meier method. The statistical significance of p value was defined as p < 0.05 (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (nos. 82072664, 81702437, 81772629, 81802363, 81702431, 81772843, and 81974374) and by the Demonstrative Research Platform of Clinical Evaluation Technology for New Anticancer Drugs (no. 2018ZX09201015). This work was also supported by the Tianjin Science Foundation (nos. 18JCQNJC81900, 18JCYBJC92000, 18JCYBJC25400, 16PTSYJC00170, and 18JCYBJC92900) and by the Science & Technology Development Fund of the Tianjin Education Commission for Higher Education (nos. 2018KJ046 and 2017KJ227). The funders had no role in the study design, the data collection and analysis, the interpretation of the data, the writing of the report, and the decision to submit this article for publication.
Author contributions
T.N., J.L., and Y.H. performed most of the experiments, analyzed the data, and wrote the manuscript, H.Z. and T.D. reviewed and edited the manuscript. T.D., Q.F., and K.Z. performed some experiments. X.W., H.L., M.B., and R.L. contributed to experimental procedures and clinical sample collection. Y.B. and G.Y. designed the experiments and edited the manuscript. Y.B. is the guarantor of this work, had full access to all data reported in the study, and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.04.028.
Contributor Information
Guoguang Ying, Email: yingguoguang163@163.com.
Yi Ba, Email: bayi@tjmuch.com.
Supplemental information
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2018. CA Cancer J. Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Hong Y.S., Kim S.Y., Lee J.S., Nam B.H., Kim K.P., Kim J.E., Park Y.S., Park J.O., Baek J.Y., Kim T.Y. Oxaliplatin-based adjuvant chemotherapy for rectal cancer after preoperative chemoradiotherapy (ADORE): Long-term results of a randomized controlled trial. J. Clin. Oncol. 2019;37:3111–3123. doi: 10.1200/JCO.19.00016. [DOI] [PubMed] [Google Scholar]
- 3.Zhou J., Zhou Y., Yin B., Hao W., Zhao L., Ju W., Bai C. 5-Fluorouracil and oxaliplatin modify the expression profiles of microRNAs in human colon cancer cells in vitro. Oncol. Rep. 2010;23:121–128. [PubMed] [Google Scholar]
- 4.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 5.Jardim D.L., Rodrigues C.A., Novis Y.A.S., Rocha V.G., Hoff P.M. Oxaliplatin-related thrombocytopenia. Ann. Oncol. 2012;23:1937–1942. doi: 10.1093/annonc/mds074. [DOI] [PubMed] [Google Scholar]
- 6.Zhao X., Yang K., Zhao R., Ji T., Wang X., Yang X., Zhang Y., Cheng K., Liu S., Hao J. Inducing enhanced immunogenic cell death with nanocarrier-based drug delivery systems for pancreatic cancer therapy. Biomaterials. 2016;102:187–197. doi: 10.1016/j.biomaterials.2016.06.032. [DOI] [PubMed] [Google Scholar]
- 7.Stojanovska V., Prakash M., McQuade R., Fraser S., Apostolopoulos V., Sakkal S., Nurgali K. Oxaliplatin treatment alters systemic immune responses. BioMed Res. Int. 2019;2019:4650695. doi: 10.1155/2019/4650695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vizio B., Novarino A., Giacobino A., Cristiano C., Prati A., Ciuffreda L., Montrucchio G., Bellone G. Potential plasticity of T regulatory cells in pancreatic carcinoma in relation to disease progression and outcome. Exp. Ther. Med. 2012;4:70–78. doi: 10.3892/etm.2012.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakaguchi S., Yamaguchi T., Nomura T., Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 10.Théry C., Zitvogel L., Amigorena S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 11.Shao H., Im H., Castro C.M., Breakefield X., Weissleder R., Lee H. New technologies for analysis of extracellular vesicles. Chem. Rev. 2018;118:1917–1950. doi: 10.1021/acs.chemrev.7b00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z., Chen J.Q., Liu J.L., Tian L. Exosomes in tumor microenvironment: Novel transporters and biomarkers. J. Transl. Med. 2016;14:297. doi: 10.1186/s12967-016-1056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ohno S., Takanashi M., Sudo K., Ueda S., Ishikawa A., Matsuyama N., Fujita K., Mizutani T., Ohgi T., Ochiya T. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013;21:185–191. doi: 10.1038/mt.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bai M., Li J., Yang H., Zhang H., Zhou Z., Deng T., Zhu K., Ning T., Fan Q., Ying G., Ba Y. miR-135b delivered by gastric tumor exosomes inhibits FOXO1 expression in endothelial cells and promotes angiogenesis. Mol. Ther. 2019;27:1772–1783. doi: 10.1016/j.ymthe.2019.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Zhang H., Deng T., Liu R., Bai M., Zhou L., Wang X., Li S., Wang X., Yang H., Li J. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat. Commun. 2017;8:15016. doi: 10.1038/ncomms15016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Al-Nedawi K., Meehan B., Rak J. Microvesicles: Messengers and mediators of tumor progression. Cell Cycle. 2009;8:2014–2018. doi: 10.4161/cc.8.13.8988. [DOI] [PubMed] [Google Scholar]
- 17.Yin Y., Cai X., Chen X., Liang H., Zhang Y., Li J., Wang Z., Chen X., Zhang W., Yokoyama S. Tumor-secreted miR-214 induces regulatory T cells: A major link between immune evasion and tumor growth. Cell Res. 2014;24:1164–1180. doi: 10.1038/cr.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hur K., Toiyama Y., Okugawa Y., Ide S., Imaoka H., Boland C.R., Goel A. Circulating microRNA-203 predicts prognosis and metastasis in human colorectal cancer. Gut. 2017;66:654–665. doi: 10.1136/gutjnl-2014-308737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toiyama Y., Hur K., Tanaka K., Inoue Y., Kusunoki M., Boland C.R., Goel A. Serum miR-200c is a novel prognostic and metastasis-predictive biomarker in patients with colorectal cancer. Ann. Surg. 2014;259:735–743. doi: 10.1097/SLA.0b013e3182a6909d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imaoka H., Toiyama Y., Fujikawa H., Hiro J., Saigusa S., Tanaka K., Inoue Y., Mohri Y., Mori T., Kato T. Circulating microRNA-1290 as a novel diagnostic and prognostic biomarker in human colorectal cancer. Ann. Oncol. 2016;27:1879–1886. doi: 10.1093/annonc/mdw279. [DOI] [PubMed] [Google Scholar]
- 21.Wang X., Luo G., Zhang K., Cao J., Huang C., Jiang T., Liu B., Su L., Qiu Z. Hypoxic tumor-derived exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kγ to promote pancreatic cancer metastasis. Cancer Res. 2018;78:4586–4598. doi: 10.1158/0008-5472.CAN-17-3841. [DOI] [PubMed] [Google Scholar]
- 22.Gallimore A., Godkin A. Regulatory T cells and tumour immunity—Observations in mice and men. Immunology. 2008;123:157–163. doi: 10.1111/j.1365-2567.2007.02748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Azmi A.S., Bao B., Sarkar F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013;32:623–642. doi: 10.1007/s10555-013-9441-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riquelme I., Letelier P., Riffo-Campos A.L., Brebi P., Roa J.C. Emerging role of miRNAs in the drug resistance of gastric cancer. Int. J. Mol. Sci. 2016;17:424. doi: 10.3390/ijms17030424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng T., Wang J., Chen X., Liu L. Role of microRNA in anticancer drug resistance. Int. J. Cancer. 2010;126:2–10. doi: 10.1002/ijc.24782. [DOI] [PubMed] [Google Scholar]
- 26.Liang X., Shi H., Yang L., Qiu C., Lin S., Qi Y., Li J., Zhao A., Liu J. Inhibition of polypyrimidine tract-binding protein 3 induces apoptosis and cell cycle arrest, and enhances the cytotoxicity of 5-fluorouracil in gastric cancer cells. Br. J. Cancer. 2017;116:903–911. doi: 10.1038/bjc.2017.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen X., Liang H., Guan D., Wang C., Hu X., Cui L., Chen S., Zhang C., Zhang J., Zen K., Zhang C.Y. A combination of Let-7d, Let-7g and Let-7i serves as a stable reference for normalization of serum microRNAs. PLoS ONE. 2013;8:e79652. doi: 10.1371/journal.pone.0079652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohos A., Sebestyén T., Liszkay G., Plótár V., Horváth S., Gaudi I., Ladányi A. Immune cell profile of sentinel lymph nodes in patients with malignant melanoma—FOXP3+ cell density in cases with positive sentinel node status is associated with unfavorable clinical outcome. J. Transl. Med. 2013;11:43. doi: 10.1186/1479-5876-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bates G.J., Fox S.B., Han C., Leek R.D., Garcia J.F., Harris A.L., Banham A.H. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. 2006;24:5373–5380. doi: 10.1200/JCO.2006.05.9584. [DOI] [PubMed] [Google Scholar]
- 30.Petersen R.P., Campa M.J., Sperlazza J., Conlon D., Joshi M.B., Harpole D.H., Jr., Patz E.F., Jr. Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer. 2006;107:2866–2872. doi: 10.1002/cncr.22282. [DOI] [PubMed] [Google Scholar]
- 31.Curiel T.J., Coukos G., Zou L., Alvarez X., Cheng P., Mottram P., Evdemon-Hogan M., Conejo-Garcia J.R., Zhang L., Burow M. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 32.Chen W., Jin W., Hardegen N., Lei K.J., Li L., Marinos N., McGrady G., Wahl S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noack M., Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014;13:668–677. doi: 10.1016/j.autrev.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 34.Spolski R., Li P., Leonard W.J. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat. Rev. Immunol. 2018;18:648–659. doi: 10.1038/s41577-018-0046-y. [DOI] [PubMed] [Google Scholar]
- 35.Yin H., Xiong G., Guo S., Xu C., Xu R., Guo P., Shu D. Delivery of anti-miRNA for triple-negative breast cancer therapy using RNA nanoparticles targeting stem cell marker CD133. Mol. Ther. 2019;27:1252–1261. doi: 10.1016/j.ymthe.2019.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan T., Jia P., Chen N., Fang Y., Liang Y., Guo M., Ding X. Delayed remote ischemic preconditioning confers renoprotection against septic acute kidney injury via exosomal miR-21. Theranostics. 2019;9:405–423. doi: 10.7150/thno.29832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Azzoni L., Zatsepina O., Abebe B., Bennett I.M., Kanakaraj P., Perussia B. Differential transcriptional regulation of CD161 and a novel gene, 197/15a, by IL-2, IL-15, and IL-12 in NK and T cells. J. Immunol. 1998;161:3493–3500. [PubMed] [Google Scholar]
- 38.Chen Y., Knösel T., Kristiansen G., Pietas A., Garber M.E., Matsuhashi S., Ozaki I., Petersen I. Loss of PDCD4 expression in human lung cancer correlates with tumour progression and prognosis. J. Pathol. 2003;200:640–646. doi: 10.1002/path.1378. [DOI] [PubMed] [Google Scholar]
- 39.Kang M.J., Ahn H.S., Lee J.Y., Matsuhashi S., Park W.Y. Up-regulation of PDCD4 in senescent human diploid fibroblasts. Biochem. Biophys. Res. Commun. 2002;293:617–621. doi: 10.1016/S0006-291X(02)00264-4. [DOI] [PubMed] [Google Scholar]
- 40.Stålberg P., Lopez-Egido J.R., Wang S., Gobl A., Oberg K., Skogseid B. Differentially expressed cDNAs in PLCβ3-induced tumor suppression in a human endocrine pancreatic tumor cell line: activation of the human mismatch repair protein 3 gene. Biochem. Biophys. Res. Commun. 2001;281:227–231. doi: 10.1006/bbrc.2001.4329. [DOI] [PubMed] [Google Scholar]
- 41.Fassan M., Pizzi M., Giacomelli L., Mescoli C., Ludwig K., Pucciarelli S., Rugge M. PDCD4 nuclear loss inversely correlates with miR-21 levels in colon carcinogenesis. Virchows Arch. 2011;458:413–419. doi: 10.1007/s00428-011-1046-5. [DOI] [PubMed] [Google Scholar]
- 42.Allgayer H. Pdcd4, a colon cancer prognostic that is regulated by a microRNA. Crit. Rev. Oncol. Hematol. 2010;73:185–191. doi: 10.1016/j.critrevonc.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Yang H.S., Matthews C.P., Clair T., Wang Q., Baker A.R., Li C.C., Tan T.H., Colburn N.H. Tumorigenesis suppressor Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase kinase 1 expression to suppress colon carcinoma cell invasion. Mol. Cell. Biol. 2006;26:1297–1306. doi: 10.1128/MCB.26.4.1297-1306.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Q., Sun Z., Yang H.S. Downregulation of tumor suppressor Pdcd4 promotes invasion and activates both β-catenin/Tcf and AP-1-dependent transcription in colon carcinoma cells. Oncogene. 2008;27:1527–1535. doi: 10.1038/sj.onc.1210793. [DOI] [PubMed] [Google Scholar]
- 45.Zhang S., Li J., Jiang Y., Xu Y., Qin C. Programmed cell death 4 (PDCD4) suppresses metastastic potential of human hepatocellular carcinoma cells. J. Exp. Clin. Cancer Res. 2009;28:71. doi: 10.1186/1756-9966-28-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen C., Tan R., Wong L., Fekete R., Halsey J. Quantitation of microRNAs by real-time RT-qPCR. Methods Mol. Biol. 2011;687:113–134. doi: 10.1007/978-1-60761-944-4_8. [DOI] [PubMed] [Google Scholar]
- 47.Zhang H., Bai M., Deng T., Liu R., Wang X., Qu Y., Duan J., Zhang L., Ning T., Ge S. Cell-derived microvesicles mediate the delivery of miR-29a/c to suppress angiogenesis in gastric carcinoma. Cancer Lett. 2016;375:331–339. doi: 10.1016/j.canlet.2016.03.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.