

Abstract
The MYC gene regulates normal cell growth and is deregulated in many human cancers, contributing to tumor growth and progression. The Myc transcription factor activates RNA polymerases I, II, and III target genes that are considered housekeeping genes. These target genes are largely involved in ribosome biogenesis, fatty acid, protein and nucleotide synthesis, nutrient influx or metabolic waste efflux, glycolysis, and glutamine metabolism. Myc function as a driver of cell growth has been revealed through RNA sequencing, genome-wide chromatin immunoprecipitation, proteomics and importantly metabolomics, which is highlighted in this chapter.
Keywords: MYC, transcription, cancer metabolism, metabolomics, mass spectrometry
1. Introduction
The MYC gene encodes a basic-helix-loop-helix leucine zipper (bHLH-LZ) transcription factor which dimerizes with its partner bHLH-LZ Max to bind DNA and mediate transcriptional responses involved in cell growth, metabolism and proliferation (1, 2). The MYC gene emerges as a central regulator of cell growth and proliferation through its ability to increase metabolism, by inducing carbohydrate, lipid, nucleotide, and protein synthesis (1, 2). A key nexus that integrates many of these pathways is MYC-induced ribosome biogenesis, which requires ribosomal protein synthesis and ribosomal RNA synthesis from de novo production of ribose from glucose and nucleotide synthesis (Figure 1) (3, 4). These physiological and other roles of MYC, such as modulating inflammatory signaling, are hijacked when MYC is deregulated either downstream of oncogenic signaling or through MYC gene amplification, rendering MYC-driven tumorigenesis dependent on dysregulated, heightened anabolic metabolism (5).
Figure 1.
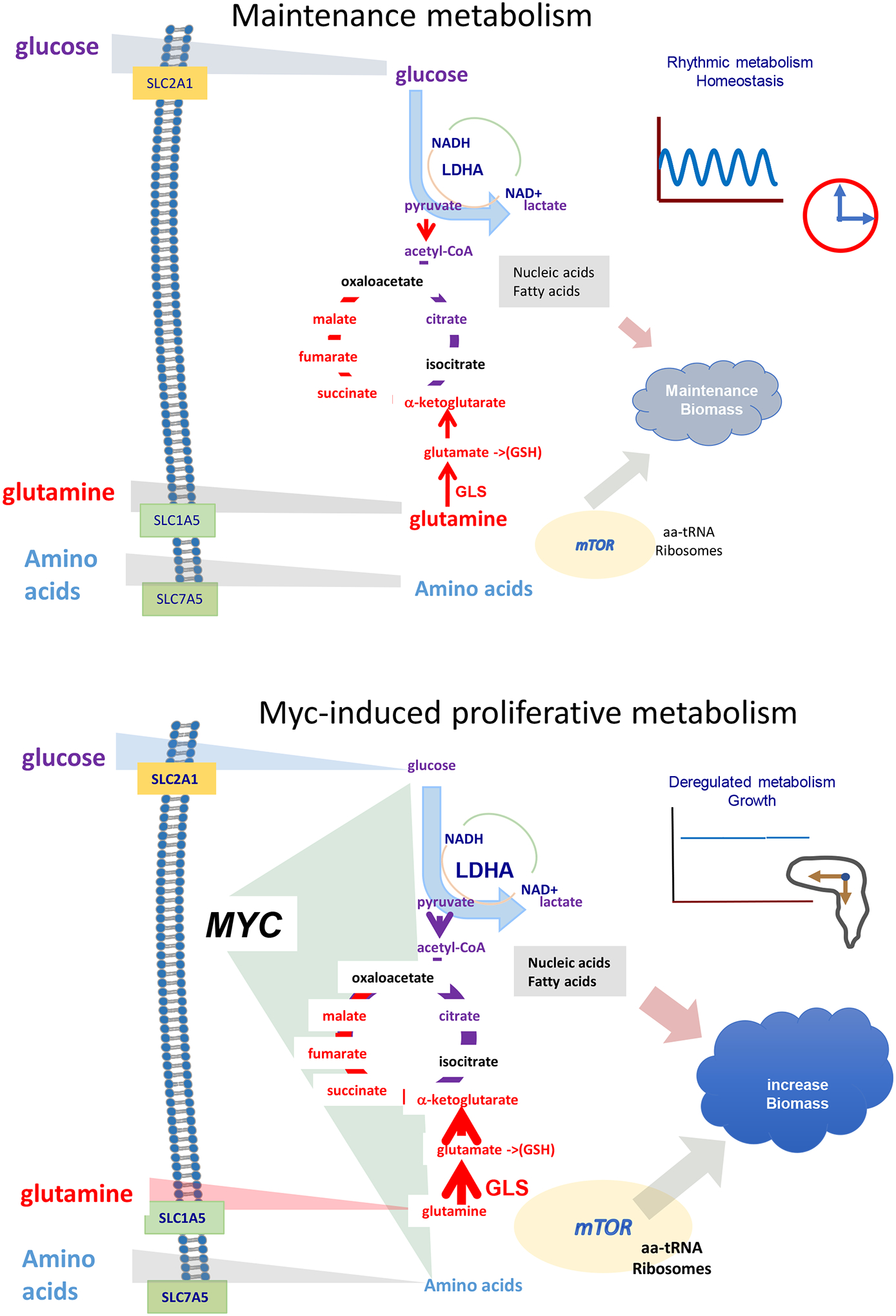
The diagram depicts maintenance metabolism versus Myc transcriptional amplification of proliferative metabolism through inducing metabolic genes and altering metabolism as gleaned from transcriptomic and metabolomic studies. While maintenance metabolism is largely influence by the circadian clock, which regulates hundreds of metabolic genes diurnally, MYC is able to disrupt the circadian clock to drive increased anabolic metabolism for cell growth and proliferation. The sizes of the arrows depict fluxes, the triangles from extracellular nutrients reflect the concentration gradients, and letter font sizes reflect expression or concentration levels. Notably, MYC induces many genes encoding nutrient transporters that allows for the import of building blocks as they are depleted intracellularly by enhanced incorporation into macromolecules and increased concentration gradient. Counter-intuitively, the intracellular levels of glucose, for example, is lowered by oncogenic MYC, which drives free intracellular glucose into biomass and thereby increasing the gradient and inward flux. Glucose, glutamine, and amino acids are shown as key nutrient sources driven into cells by mass action to provide the essential building blocks, generated through glycolysis (glucose conversion to pyruvate) and the tricarboxylic acid cycle. Translational amplification by mTOR, which is activated by amino acids, increases incorporation of building for biomass accumulation or cell growth and subsequent cell division. LDHA, lactate dehydrogenase A; GLS, glutaminase; GSH, glutathione; aa-tRNA, aminoacyl-tRNAs.
As a transcription factor, MYC was first found to induce genes involved and metabolism, such as lactate dehydrogenase and ornithine decarboxylase (1, 2). In the ensuing years after these initial discoveries, MYC was uncovered to broadly stimulate proliferative metabolism as compared to maintenance metabolism (5). In adult organisms, except for stem cell compartments, >90% of cells have differentiated into various tissues and no longer could proliferate. The terminally differentiated cells undergo maintenance metabolism that largely provides ATP for maintenance of membrane potentials and sufficient biosynthesis for replacement of damaged molecules and organelles. In maintaining the integrity of a non-proliferating cell, MYC does not appear to play an important role in ‘turbo-charging’ metabolic pathways to move large amounts nutrient-derived building blocks into the construction of new macromolecules and organelles for cell growth, DNA replication and cell division (5). By contrast, when MYC is stimulated in normal cells, such as T cells, or oncogenically activated in cancers, the open chromatin of metabolic genes permits MYC to bind these genes and amplify their expression (6, 7). The resulting production of enzymes across many intermediary pathways together with production of ribosomes allows for the growing cell to drive nutrient-derived building blocks into cell mass. A key difference between normal MYC versus oncogenic MYC is the ability of normal cells to return to maintenance metabolism after MYC is turned off and the cells no longer proliferate. By contrast, deregulated MYC induces deregulated metabolism and biosynthesis, rendering MYC-addicted cells dependent on continuous sources of nutrients (5).
Intriguingly, maintenance metabolism is largely affected by the circadian clock which regulates cellular metabolism in a 24-hour cycle (Figure 1), coupled with organismal fasting (sleep) and feeding (wake) cycles to optimize organismal metabolic fitness (8). During the sleep cycle, the cell intrinsic clock induces repair mechanisms to renew damaged molecules and organelles, while during the metabolic active wake phase, the clock allows for heightened metabolism to support the function of cells in various tissues and organs. Interestingly, oncogenic MYC (or MYCN) was found to profoundly disrupt the circadian clock and circadian metabolism as it induces proliferative metabolism (Figure 1) (9).
Over the years, the role of MYC in metabolism has been enriched by many studies linking MYC to many different metabolic pathways. Many of these studies were enabled by metabolomics studies using mass spectrometry (10, 11). In this regard, to measure MYC-mediated metabolism, a number of factors should be considered for sample preparation, which is provided here.
Because metabolic pathways are highly dynamic and many metabolites have a fast turnover rate, rapid quenching and extraction of polar or non-polar compounds are critical steps for consistently measuring MYC-mediated metabolic changes at time of sample collection. The following provides protocols for extracting polar and non-polar metabolites so that they can be properly analyzed by mass spectrometry. With specific reference to MYC, experimental inducible systems are most desirable to determine the effect of MYC on metabolism in a controlled isogenic cell system.
2. Materials
2.1. Polar metabolite extraction
Methanol, LC-MS grade (Acros Organics).
Water, Milli-Q (Millipore).
Stable isotope-labeled (SIL) amino acid mix standard MSK-A2-1.2 (Cambridge Isotope Laboratories).
Dulbecco’s phosphate buffered saline (DPBS, Corning) or Hanks’ Balanced Salt Solution (HBSS, ThermoFisher Scientific) with Calcium and Magnesium.
1.5 ml polypropylene centrifuge tubes.
Disposable polyethylene cell lifters.
Positive displacement pipettes and tips.
Prepare extraction solution containing 80% methanol, 20% water, 0.2 μM SIL standard. Store at −20 °C until use. Transfer to dry ice on the day of extraction.
2.2. Lipid extraction
Chloroform, LC-MS grade (OmniSolv).
Methanol, LC-MS grade (Acros Organics).
Water, LC-MS grade (OmniSolv).
Dulbecco’s phosphate buffered saline (DPBS, Corning) or Hanks’ Balanced Salt Solution (HBSS, ThermoFisher Scientific) with Calcium and Magnesium.
EquiSPLASH Lipidomix internal standard (Avanti Polar Lipids).
Sodium chloride (Sigma).
Disposable polyethylene cell lifters (Fisherbrand).
Disposable glass tubes (10 ml or 15 ml) and caps with Teflon/PTFE septums (Corning).
Disposable glass pipets 10 ml (Fisherbrand).
Positive displacement pipettes and tips.
3. Methods
3.1. Extraction of polar metabolites for LC-MS/MS analysis
Effective, reproducible extraction of metabolites is critical to the success of any metabolomics experiment. The 80% methanol used in this protocol will extract a broad range of metabolites including amino acids, organic acids, phosphates, sugars, cofactors and nucleotides. Many small polar molecules involved in the central carbon metabolism are also efficiently extracted. Extracted metabolites are subsequently separated and analyzed using hydrophobic interaction liquid chromatography (HILIC) and liquid chromatography tandem mass spectrometry (LC-MS/MS). This protocol is suitable for quantifying steady state metabolite levels, or in a separate experiment, evaluation of metabolic fluxes using stable isotope tracers such as U-13C6-glucose or U-13C5-glutamine (see Note 1).
Prepare extraction solution containing 80% methanol, 20% water, 0.2 μM SIL standard. Store at −20 °C until use. Transfer to dry ice on the day of extraction.
Culture cells in appropriate dishes or flasks. A minimum of 3 biological replicates for extraction and a fourth parallel replicate to be used for cell counts are required per condition. Aim for 2 million cells or approximately 300 μg total protein per sample. In parallel, incubate several dishes or flasks with media but without cells to identify background compounds. (see Note 2).
For adherent cells, determine cell counts from a separate, parallel culture immediately before the end of the incubation period. (see Note 3 if working with suspension cells).
Place the culture dishes on a bed of ice and remove media completely by aspiration. Save an aliquot of the conditioned media if changes in metabolite levels in media are of interest (see Note 4).
Wash cells twice with ice-cold HBSS or DPBS while keeping the culture dishes on the bed of ice. After the final wash, completely remove all wash solution (see Note 5).
Add 700 μl of ice-cold extraction solution to each sample (see Note 6).
Scrape cells with a disposable cell lifter and transfer cell suspension to a pre-cooled 1.5 ml polypropylene centrifuge tube on dry ice.
Rinse the dish and cell lifter using 300 μl of ice-cold extraction solution. Transfer to the same 1.5 ml polypropylene tube on dry ice.
Vortex thoroughly for 30 sec. Place tube on dry ice for at least 30 min.
Centrifuge at maximum speed in a microfuge (>16,000 × g) for 15 min at 4°C to pellet the cell debris.
Carefully transfer supernatant to LC vials while taking care to not disturb the pellet. Air dry the pellet and determine the protein concentration using a commercial colorimetric protein assay (see Note 7).
Analyze extracted metabolites in supernatant using a suitable LC-MS/MS system with chromatographic separation on a HILIC column (see Note 8).
3.2. Extraction of non-polar lipids for LC-MS/MS analysis
This protocol describes the extraction of non-polar lipids from cells using liquid-liquid extraction based on the modified Folch method (12, 13). Extraction with chloroform: methanol: 0.88% NaCl results in formation of a biphasic solution with non-polar lipids partitioned into the lower phase and a protein pellet suspended between the phases. Extracted lipids are analyzed by high resolution reversed phase LC-MS/MS. More than 1000 lipid species spanning more than 25 classes are routinely identified and quantified.
Prepare 0.88% (w/v) NaCl in water. Filter (0.2 μm) and store at 4 °C.
Prepare synthetic lower phase by mixing chloroform: methanol: 0.88% NaCl at 2:1:1 ratio. Using a glass pipet, transfer lower phase to a glass bottle with Teflon/PTFE-lined cap. Store synthetic lower phase, chloroform and methanol at −20 °C (see Note 9).
Culture cells in appropriate dishes or flasks. A minimum of 3 biological replicates are required per condition for extraction plus a fourth replicate for cell counting. Aim for at least 4 million cells or approximately 600 μg total protein per sample. In parallel, incubate several dishes or flasks with media without cells for extraction and identification of background compounds (see Note 2).
For adherent cells, determine cell counts from a separate, parallel culture immediately before the end of the incubation period (see Note 3 if working with suspension cells).
Place the culture dishes on a bed of ice and remove media by aspiration. Save an aliquot of the conditioned media if changes in non-polar metabolite levels in media are of interest (see Note 4).
Wash cells twice with ice-cold HBSS or DPBS while keeping the culture dishes on the bed of ice. After the final wash, completely remove all wash solution (see Note 5).
Add 0.75 ml of ice-cold methanol per 4 million cells to the dish. Scrape cells with a disposable cell lifter and transfer cell suspension to a 10 ml glass tube.
Rinse dish and cell lifter with 0.75 ml of ice-cold methanol and transfer to the same 10 ml glass tube.
Add 5 μl EquiSPLASH to each sample (see Note 10).
Add 1.5 ml of ice-cold 0.88% NaCl and 3 ml of ice-cold chloroform.
Vortex vigorously for 30 sec.
Sonicate in ice-cooled water bath for 5 min. Vortex vigorously for another 30 sec.
Centrifuge at 250 × g, at 4 °C, for 15 min.
Transfer lower phase containing non-polar lipids into a new glass tube using 9” glass Pasteur pipets.
Re-extract upper phase with 3 ml of synthetic lower phase by repeating steps 11–13.
Combine this lower phase with the previously collected lower phase (step 14) and dry under nitrogen.
Solubilize dried lipids in 100 μl 1:9 chloroform: methanol (see Note 11.)
Centrifuge for 2 min and transfer samples to glass LC vials with Teflon/PTFE-lined caps for reversed phase LC-MS/MS analysis (see Note 12).
Air dry protein pellet and determine the protein concentration (see Note 7).
4. Notes
For stable isotope tracing experiments, replace media with the same media where a natural metabolite of interest has been replaced with its stable isotope version (such as U-13C6-glucose or U-13C5-glutamine). Culture cells until steady state labeling is reached; typically, 24 hr is sufficient. Use the appropriate isotopic tracer for the pathway of interest (13).
For smaller sized cells (e.g. T-cells), aim for the number of cells per replicate that is equivalent to ~300 μg protein for polar metabolites and ~600 μg protein for non-polar metabolites. If cells are limiting, proportionally scale down the volume of extraction solution to achieve the targeted concentration of extracted metabolites.
For suspension cells, count cells in each sample. Pellet cells by centrifugation at 180 × g for 5 min at 4 °C. Wash cells twice with cold DPBS and transfer to 1.5 ml polypropylene tube. To extract polar metabolites, add 1 ml of ice-cold extraction solution to cell pellet. Continue with steps 9 to 12 (section 3.1). For non-polar metabolites, add 1.5 ml of methanol to cell pellet, vortex and transfer to a 10 ml glass tube. Continue with steps 9 to 19 (section 3.2).
Metabolites from conditioned media can be extracted and analyzed in parallel with cell extracts. For polar metabolites, add 950 μl of ice-cold extraction solution to 50 μl of media in a 1.5 ml polypropylene tube. Continue with steps 9 to 12 (section 3.1). For non-polar metabolites, transfer 500 μl of media to a 15 ml glass tube and add 2.5 ml of methanol. Continue with steps 9 to 19 (section 3.2) but use 2 ml of 0.88% NaCl, 5 ml of chloroform, and 5 ml of synthetic lower phase.
Use HBSS for poorly adhering cells. It is critical to perform this step as quickly as possible and on ice with ice-cold solutions to minimize perturbation of metabolites. Retention of excess HBSS or DPBS after centrifugation could potentially interfere with LC-MS analysis.
80% methanol will lyse cells and stop cellular metabolism. The relative broad range of metabolites effectively extracted makes this protocol suitable for untargeted metabolomic analysis. For targeted analysis of specific metabolites, alternative extraction solutions could be more appropriate. SIL internal standards in the extraction solution are used to assess extraction reproducibility and LC-MS/MS system performance.
Do not over-dry protein pellet and ensure the pellet is completely solubilized in appropriate buffer (e.g. 1% SDS, 150 mM NaCl, 1 mM EDTA, 50 mM Tris-Cl, pH 7.5). Protein concentration determined using a colorimetric protein assay, such as the BCA assay, can be used for normalizing metabolite and lipid levels across samples.
Our LC-MS/MS system consists of a high mass resolution Q Exactive HF-X mass spectrometer and Vanquish Horizon UHPLC (ThermoFisher Scientific). Polar metabolites are separated by HILIC chromatography on a ZIC-pHILIC 2.1-mm × 150 mm column (EMD Millipore). However, recent batches of this column were observed to contain high levels of 212.1 m/z contaminant and displayed poor separation of phosphate-containing metabolites. An alternative HILIC column with good performance is the Amide XBridge column (Waters).
Chloroform is toxic and incompatible with plastics. All transfers involving chloroform should be conducted in a chemical fume hood. Solutions and samples containing chloroform should only be handled using glass pipets and stored in glass tubes with Teflon/PTFE-lined caps. All organic solvents should be added using positive displacement pipettes.
EquiSPLASH contains a mixture of 13 deuterated lipid internal standards from different lipid classes that are used to assess extraction reproducibility, LC-MS/MS system performance, and normalization of lipid quantitation across samples.
To effectively solubilize dried lipids, first add 10 μl of chloroform to samples, vortex, then add 90 μl of methanol and vortex again.
We typically separate lipids by reversed phase chromatography on an Accucore C30 2.1-mm × 150 mm column (ThermoFisher Scientific).
References
- 1.Dang CV (2012) MYC on the path to cancer, Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eilers M, and Eisenman RN (2008) Myc’s broad reach, Genes & development 22, 2755–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnston LA et al. (1999) Drosophila myc regulates cellular growth during development, Cell 98, 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Riggelen J, Yetil A, and Felsher DW (2010) MYC as a regulator of ribosome biogenesis and protein synthesis, Nature reviews. Cancer 10, 301–309. [DOI] [PubMed] [Google Scholar]
- 5.Dang CV (2016) A Time for MYC: Metabolism and Therapy. Cold Spring Harb Symp Quant Biol 81, 79–83. [DOI] [PubMed] [Google Scholar]
- 6.Wang R, et al. (2011) The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kress TR, Sabò A, Amati B (2015) MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer. 15, 593–607. [DOI] [PubMed] [Google Scholar]
- 8.Bass J, Takahashi JS (2010) Circadian integration of metabolism and energetics. Science. 330, 1349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Altman BJ, et al. (2015) MYC Disrupts the Circadian Clock and Metabolism in Cancer Cells. Cell Metab 22, 1009–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le A, et al. (2012) Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells, Cell Metab 15, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy TA, Dang CV, Young JD (2013) Isotopically nonstationary 13C flux analysis of Myc-induced metabolic reprogramming in B-cells. Metab Eng. 15, 206–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Folch J, Lees M,. Sloane Stanley GH. (1957) A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 226, 497–509. [PubMed] [Google Scholar]
- 13.Schug ZT, et al. (2015) Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang C, Chen L, Rabinowitz JD (2018) Metabolomics and Isotope Tracing. Cell 173, 822–837. [DOI] [PMC free article] [PubMed] [Google Scholar]