

Abstract
Lessons Learned
Subcutaneous injection was an effective route of administration for envafolimab with a favorable pharmacokinetic profile in patients with previously treated advanced solid tumors.
Subcutaneous envafolimab was well tolerated and had durable antitumor activity at a wide range of doses and schedules.
Envafolimab has the potential to be a more convenient option than currently approved intravenous PD‐1/PD‐L1 inhibitors.
Background
Envafolimab is a novel fusion of a humanized single‐domain PD‐L1 antibody and human IgG1 Fc fragment formulated for subcutaneous injection. This study explored the safety and feasibility of subcutaneous administration of envafolimab as an alternative to intravenous administration of PD‐1/PD‐L1 inhibitors in the treatment of advanced, refractory solid tumors.
Methods
This was a first‐in‐human, open‐label phase I trial. In a dose‐escalation phase, patients received subcutaneous envafolimab 0.01–10 mg/kg once weekly following a modified 3+3 design. In a dose‐exploration phase, patients received subcutaneous envafolimab 300 mg once every 4 weeks.
Results
Twenty‐eight patients were enrolled (dose escalation n = 18, dose exploration n = 10, median age 66 years; 71% male; ECOG performance score = 0 [21%] or 1 [79%]). No dose‐limiting toxicities or injection‐site reactions were reported. Envafolimab demonstrated dose‐proportional increases in area under the time‐concentration curve and maximum plasma concentration. Median time to maximum plasma concentration was 4–7 days. In the dose‐exploration phase, terminal half‐life was 14 days after dose 1 in cycle 1 and 23 days at steady state. Three patients experienced a confirmed partial response.
Conclusion
Subcutaneous envafolimab had a favorable safety and pharmacokinetic profile, with promising preliminary antitumor activity in patients with advanced solid tumors.
Keywords: Envafolimab, Anti‐PD‐L1, Advanced solid tumors
Discussion
Envafolimab is a novel recombinant protein of a humanized single‐domain anti‐PD‐L1 antibody fused with a human IgG1 Fc fragment formulated for subcutaneous (SC) injection. This was a first‐in‐human phase I study to evaluate the safety and feasibility of SC administration of envafolimab as an alternative to intravenous administration of PD‐1/PD‐L1 inhibitors in the treatment of advanced, refractory solid tumors.
Twenty‐eight patients were included. The most common treatment‐emergent adverse events (reported in >3 patients) were fatigue (29%), nausea (18%), diarrhea (14%), and hypothyroidism (14%). No grade ≥4 study drug–related treatment‐emergent adverse events, dose‐limiting toxicities, or injection‐site reactions were reported. Antidrug antibodies were detected in 12 (43%) patients, although they were transient in most and did not appear to affect pharmacokinetic exposure to envafolimab.
Objective tumor responses were observed in three patients across several dose cohorts and at a dose as low as 0.3 mg/kg once weekly (QW; Fig. 1). These responses were durable (24.1+ to 59.9+ weeks), and two of the patients still had partial responses assessed at the time of data cutoff (November 25, 2019).
Figure 1.
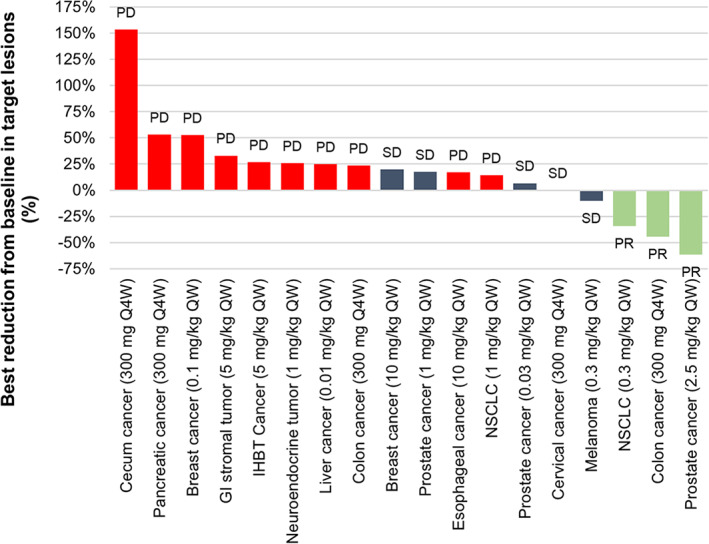
Waterfall plot of tumor reduction from baseline during the dose‐escalation and dose‐exploration phases (n = 18). Abbreviations: GI, gastrointestinal; IHBT, intrahepatic biliary tract; NSCLC, non‐small cell lung cancer; PD, progressive disease; PR, partial response; Q4W, once every 4 weeks; QW, once weekly; SD, stable disease.
Following a single SC administration in the dose‐escalation phase, the maximum plasma concentration (Cmax) and area under the curve (AUC) increased linearly over the dose range of 0.01 to 10 mg/kg (Fig. 2). At 0.3 mg/kg, two of three patients had a first‐dose Cmax that exceeded 0.5 μg/mL. Median time to reach Cmax was 4–7 days. Neither first‐dose Cmax nor AUC were significantly affected by injection site. During the dose‐exploration phase, in which all patients received envafolimab 300 mg SC once every 4 weeks, the mean Cmax after the first dose was 14 μg/mL, the AUC up to the last measured concentration (week 4) was 5,850 hours*μg/mL, and the median time to reach Cmax was 3 days. The first‐dose half‐life was estimated to be 14 days. At steady state, the mean effective half‐life was 23 days.
Figure 2.
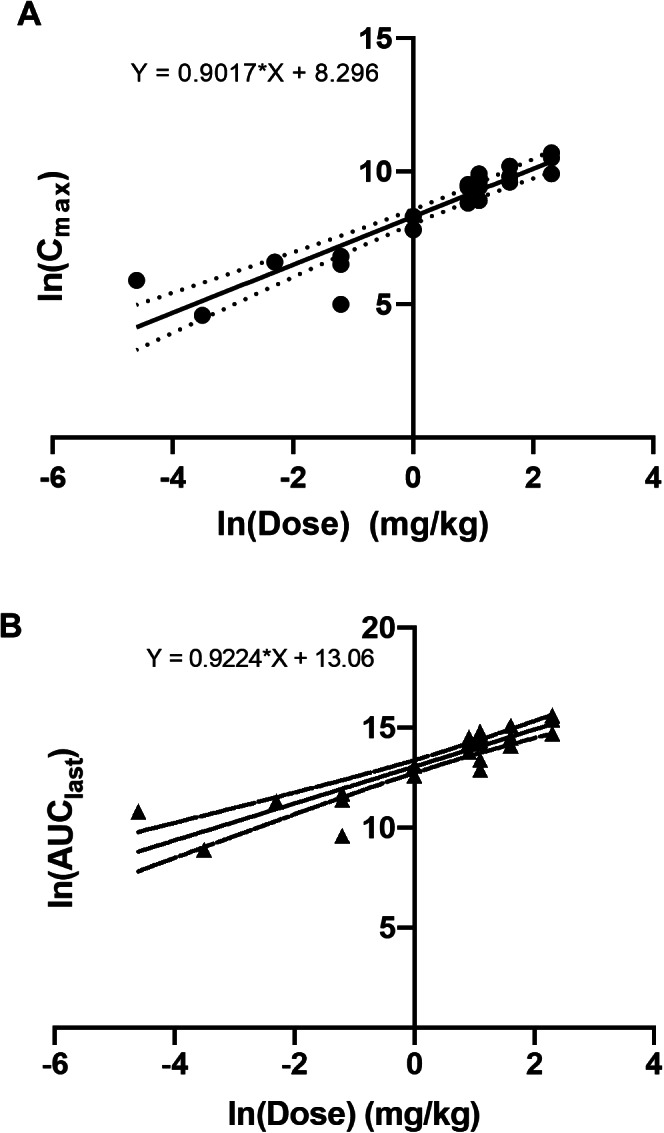
Relationship between natural log‐transformed dose and Cmax (A) and AUClast (B). Slopes were calculated by nonlinear regression analysis. Abbreviations: AUClast, area under the curve until the last measurement; Cmax, maximum plasma concentration.
The results show that SC injection of envafolimab was an effective route of administration, was well tolerated, and had durable antitumor activity at a wide range of doses and schedules in patients with previously treated advanced solid tumors (Table 1; Fig. 1).
Table 1.
Simulated pharmacokinetic data for envafolimab dosing regimens: predicted peak and trough concentrations
| Estimated blood concentration of envafolimab | ||||||
|---|---|---|---|---|---|---|
| Dosing regimen | Dose | Measurement | Week | Geometric mean, mg/L (95% CI) | 5th percentile, μg/mL | 10th percentile, μg/mL |
| 300 mg Q4W | 1 | Peak | 1 | 11.9 (11.8, 12.0) | 4.53 | 5.78 |
| 1 | Trough | 4 | 5.12 (5.07, 5.17) | 2.04 | 2.53 | |
| 8 | Peak | 29 | 20.4 (20.2, 20.6) | 7.34 | 9.29 | |
| 8 | Trough | 32 | 9.68 (9.55, 9.81) | 3.11 | 3.98 | |
| 300 mg Q3W | 1 | Peak | 1 | 11.9 (11.8, 12.0) | 4.53 | 5.78 |
| 1 | Trough | 3 | 6.79 (6.72, 6.86) | 2.80 | 3.47 | |
| 8 | Peak | 22 | 24.1 (23.8, 24.4) | 8.82 | 11.10 | |
| 8 | Trough | 24 | 14.2 (14.0, 14.4) | 4.86 | 6.16 | |
| 400 mg Q4W | 1 | Peak | 1 | 15.9 (15.7, 16.1) | 6.04 | 7.70 |
| 1 | Trough | 4 | 6.83 (6.76, 6.89) | 2.72 | 3.38 | |
| 8 | Peak | 29 | 27.2 (26.9, 27.5) | 9.93 | 12.40 | |
| 8 | Trough | 32 | 12.9 (12.7, 13.1) | 4.14 | 5.30 | |
| 150 mg QW | 1 | Peak | 1 | 5.97 (5.90, 6.03) | 2.28 | 2.90 |
| 1 | Trough | 2 | 5.82 (5.76, 5.87) | 2.39 | 2.97 | |
| 8 | Peak | 8 | 24.5 (24.2, 24.7) | 9.83 | 12.10 | |
| 8 | Trough | 9 | 21.9 (21.7, 22.2) | 8.86 | 10.90 | |
Abbreviations: CI, confidence interval; Q3W, once every 3 weeks; Q4W, once every 4 weeks; QW, weekly.
Trial Information
| Disease | Advanced cancer/solid tumor only |
| Stage of Disease/Treatment | Metastatic/advanced |
| Prior Therapy | No designated number of regimens |
| Type of Study | Phase I, dose escalation + dose exploration |
| Primary Endpoint | Safety, tolerability |
| Secondary Endpoints | Maximum tolerated dose, recommended phase II dose, pharmacodynamics, other |
| Additional Details of Endpoints or Study Design | The study included dose escalation and dose exploration. For the dose escalation, a modified 3+3 design was used, with dose‐limiting toxicity (DLT) evaluated up to 28 days after the first dose. Eight dose levels were evaluated. The first three dose levels were assessed in single‐patient cohorts. If a grade ≥2 drug‐related adverse event (AE) was observed during the DLT period, two additional patients were enrolled and administered the same dose. For the remaining dose levels, a standard 3+3 design was followed. In the dose exploration, patients received envafolimab 300 mg SC once every 4 weeks (Q4W). |
| Investigator's Analysis | Active and should be pursued further |
Drug Information: Dose Escalation
| Envafolimab | |
| Generic Name | Envafolimab |
| Company Name | KN035 |
| Drug Type | Antibody |
| Drug Class | Immune therapy |
| Dose | 0.01, 0.03, 0.1, 0.3, 1, 2.5, 5, and 10 mg/kg |
| Route | Subcutaneous |
| Schedule of Administration | In the dose escalation, envafolimab was administered on days 1, 8, 15, and 22 in each 28‐day cycle. |
Drug Information: Dose Exploration
| Envafolimab | |
| Generic Name | Envafolimab |
| Company Name | KN035 |
| Drug Type | Antibody |
| Drug Class | Immune therapy |
| Dose | 300 mg per flat dose |
| Route | Subcutaneous |
| Schedule of Administration | In the dose exploration, patients received envafolimab as a single fixed dose of 300 mg on day 1 in each 28‐day cycle. |
Patient Characteristics: Dose Escalation
| Number of Patients, Male | 13 |
| Number of Patients, Female | 5 |
| Stage |
III: n = 1 IV: n = 17 |
| Age | Median (range): 71 (53–79) years |
| Performance Status: ECOG |
0: n = 3 1: n = 15 |
| Cancer Types or Histologic Subtypes | Prostate cancer, 5 |
| Intrahepatic biliary tract cancer, 1 | |
| Non‐small cell lung cancer, 2 | |
| Breast cancer, 2 | |
| Cervical cancer, 1 | |
| Bladder cancer, 1 | |
| Esophageal cancer, 1 | |
| Head and neck cancer, 1 | |
| Liver cancer, 1 | |
| Melanoma, 1 | |
| Neuroendocrine tumor, 1 | |
| Gastrointestinal stromal tumor, 1 |
Patient Characteristics: Dose Exploration
| Number of Patients, Male | 7 |
| Number of Patients, Female | 3 |
| Stage | IV: n = 10 |
| Age | Median (range): 63 (35–77) years |
| Performance Status: ECOG |
0: n = 3 1: n = 7 |
| Cancer Types or Histologic Subtypes | Colorectal cancer, 5 |
| Intrahepatic biliary tract cancer, 2 | |
| Prostate cancer, 1 | |
| Cervical cancer, 1 | |
| Pancreatic cancer, 1 |
Patient Characteristics: Total
| Number of Patients, Male | 20 |
| Number of Patients, Female | 8 |
| Stage |
III: n = 1 IV: n = 27 |
| Age | Median (range): 66 (35–79) years |
| Performance Status: ECOG |
0: n = 6 1: n = 22 |
| Cancer Types or Histologic Subtypes | Prostate cancer, 6 |
| Colorectal cancer, 5 | |
| Intrahepatic biliary tract cancer, 3 | |
| Non‐small cell lung cancer, 2 | |
| Breast cancer, 2 | |
| Cervical cancer, 2 | |
| Bladder cancer, 1 | |
| Esophageal cancer, 1 | |
| Head and neck cancer, 1 | |
| Liver cancer, 1 | |
| Melanoma, 1 | |
| Neuroendocrine tumor, 1 | |
| Gastrointestinal stromal tumor, 1 | |
| Pancreatic cancer, 1 |
Secondary Assessment Method: Dose Escalation
| Title | Tumor response |
| Number of Patients Screened | 19 |
| Number of Patients Enrolled | 18 |
| Number of Patients Evaluable for Toxicity | 18 |
| Number of Patients Evaluated for Efficacy | 16 |
| Evaluation Method | RECIST 1.1 |
| Response Assessment CR | n = 0 (0%) |
| Response Assessment PR | n = 2 (11%) |
| Response Assessment SD | n = 5 (28%) |
| Response Assessment PD | n = 9 (50%) |
| Response Assessment OTHER | n = 2 (11%) |
| Median Duration of Treatment | 10.1 weeks |
| Outcome Notes | For Response Assessment, "OTHER" denotes not evaluable (efficacy could not be assessed in two patients because they had no postbaseline tumor assessment). |
Secondary Assessment Method: Dose Exploration
| Title | Tumor response |
| Number of Patients Screened | 19 |
| Number of Patients Enrolled | 10 |
| Number of Patients Evaluable for Toxicity | 10 |
| Number of Patients Evaluated for Efficacy | 8 |
| Evaluation Method | RECIST 1.1 |
| Response Assessment CR | n = 0 (0%) |
| Response Assessment PR | n = 1 (10%) |
| Response Assessment SD | n = 3 (30%) |
| Response Assessment PD | n = 4 (40%) |
| Response Assessment OTHER | n = 2 (20%) |
| Median Duration of Treatment | 8.4 weeks |
| Outcome Notes | For Response Assessment, "OTHER" denotes not evaluable (efficacy could not be assessed in two patients because they had no postbaseline tumor assessment). |
Secondary Assessment Method: Total
| Title | Tumor response |
| Number of Patients Screened | 38 |
| Number of Patients Enrolled | 28 |
| Number of Patients Evaluable for Toxicity | 28 |
| Number of Patients Evaluated for Efficacy | 24 |
| Evaluation Method | RECIST 1.1 |
| Response Assessment CR | n = 0 (0%) |
| Response Assessment PR | n = 3 (11%) |
| Response Assessment SD | n = 8 (29%) |
| Response Assessment PD | n = 13 (46%) |
| Response Assessment OTHER | n = 4 (14%) |
| Median PFS | 2.8 months, 95% CI: 1.8–7.6 |
| Median OS | 8.5 months, 95% CI: 3.1–17.4 |
| Median Response Duration | 24.9 weeks |
| Median Duration of Treatment | 8.6 weeks |
| Outcome Notes | For Response Assessment, "OTHER" denotes not evaluable (efficacy could not be assessed in four patients because they had no postbaseline tumor assessment). The three patients who achieved a PR comprised one patient with non‐small cell lung cancer who received 0.3 mg/kg envafolimab QW (response duration 24.9 weeks), one patient with microsatellite instability–high prostate cancer who received 2.5 mg/kg envafolimab QW (response duration 59.9+ weeks), and one patient with microsatellite stable, tumor mutation burden–high (16 mutations/Mb) colon cancer who received 300 mg Q4W (response duration 24.1+ weeks). At data cutoff (November 25, 2019), 24 of the 28 patients had discontinued treatment. The main reasons for treatment discontinuation were disease progression (n = 17) and unacceptable adverse events (n = 4). Duration of treatment ranged from 3.9 to 66.1+ weeks. |
Adverse Events: Dose Escalation
| All Dose Levels, All Cycles | |||||||
|---|---|---|---|---|---|---|---|
| Name | NC/NA | 1 | 2 | 3 | 4 | 5 | All grades |
| Fatigue | 61% | 22% | 17% | 0% | 0% | 0% | 39% |
| Nausea | 78% | 11% | 11% | 0% | 0% | 0% | 22% |
| Alanine aminotransferase increased | 83% | 6% | 0% | 11% | 0% | 0% | 17% |
| Aspartate aminotransferase increased | 83% | 6% | 0% | 11% | 0% | 0% | 17% |
| Diarrhea | 83% | 11% | 6% | 0% | 0% | 0% | 17% |
| Dry mouth | 83% | 17% | 0% | 0% | 0% | 0% | 17% |
| Abdominal pain | 89% | 0% | 0% | 11% | 0% | 0% | 11% |
| Blood alkaline phosphatase increased | 89% | 0% | 0% | 11% | 0% | 0% | 11% |
| Constipation | 89% | 6% | 6% | 0% | 0% | 0% | 11% |
| Decreased appetite | 89% | 6% | 6% | 0% | 0% | 0% | 11% |
| Hypokalemia | 89% | 11% | 0% | 0% | 0% | 0% | 11% |
| Hypomagnesemia | 89% | 11% | 0% | 0% | 0% | 0% | 11% |
| Hypophosphatemia | 89% | 6% | 0% | 6% | 0% | 0% | 11% |
| Hypothyroidism | 89% | 0% | 11% | 0% | 0% | 0% | 11% |
| Lymphopenia | 83% | 0% | 0% | 11% | 6% | 0% | 17% |
| Musculoskeletal chest pain | 89% | 11% | 0% | 0% | 0% | 0% | 11% |
| Musculoskeletal stiffness | 89% | 11% | 0% | 0% | 0% | 0% | 11% |
| Pain in extremity | 89% | 11% | 0% | 0% | 0% | 0% | 11% |
| Rash maculo‐papular | 89% | 11% | 0% | 0% | 0% | 0% | 11% |
| Salivary hypersecretion | 89% | 6% | 6% | 0% | 0% | 0% | 11% |
| Skin abrasion | 89% | 6% | 6% | 0% | 0% | 0% | 11% |
| Vomiting | 89% | 11% | 0% | 0% | 0% | 0% | 11% |
Adverse Events Legend
Adverse events occurring in ≥10% of patients are shown.
There were no DLTs.
Abbreviation: NC/NA, no change from baseline/no adverse event.
Serious Adverse Events: Dose Escalation
| Name | Grade | Attribution |
|---|---|---|
| Lung infection | 3 | Unrelated |
| Pneumonia | 3 | Unrelated |
| Pneumothorax | 3 | Unrelated |
| Pneumothorax | 3 | Unrelated |
| Compression fracture | 3 | Unrelated |
| Pneumonia | 3 | Unrelated |
| Gastroenteritis | 3 | Unrelated |
| Viral infection | 3 | Unrelated |
| Pancreatitis | 2 | Unrelated |
| Rectal hemorrhage | 3 | Unrelated |
| Deep vein thrombosis | 3 | Unrelated |
| Bile duct obstruction | 3 | Unrelated |
| Abdominal pain | 3 | Possible |
| Aspartate aminotransferase increased | 3 | Possible |
| Alanine aminotransferase increased | 3 | Possible |
Serious Adverse Events Legend
The two SAEs of pneumonia and two SAEs of pneumothorax all occurred in the same patient.
The SAEs of viral infection and pancreatitis occurred >30 days after the last dose of study drug.
Adverse Events: Dose Exploration
| All Cycles | |||||||
|---|---|---|---|---|---|---|---|
| Name | NC/NA | 1 | 2 | 3 | 4 | 5 | All Grades |
| Dehydration | 70% | 10% | 10% | 10% | 0% | 0% | 30% |
| Enterocolitis infectious | 80% | 0% | 20% | 0% | 0% | 0% | 20% |
| Hypothyroidism | 80% | 0% | 20% | 0% | 0% | 0% | 20% |
| Allergic rhinitis | 90% | 10% | 0% | 0% | 0% | 0% | 10% |
| Back pain | 90% | 0% | 0% | 10% | 0% | 0% | 10% |
| Blepharitis | 90% | 0% | 10% | 0% | 0% | 0% | 10% |
| Cough | 90% | 10% | 0% | 0% | 0% | 0% | 10% |
| Dermatitis acneiform | 90% | 10% | 0% | 0% | 0% | 0% | 10% |
| Diarrhea | 90% | 0% | 0% | 10% | 0% | 0% | 10% |
| Diverticulitis | 90% | 0% | 0% | 10% | 0% | 0% | 10% |
| Dyspnea | 90% | 0% | 0% | 0% | 0% | 10% | 10% |
| Fatigue | 90% | 10% | 0% | 0% | 0% | 0% | 10% |
| Fecaloma | 90% | 0% | 0% | 10% | 0% | 0% | 10% |
| Hypokalemia | 90% | 0% | 10% | 0% | 0% | 0% | 10% |
| Hypomagnesemia | 90% | 0% | 10% | 0% | 0% | 0% | 10% |
| Nausea | 90% | 0% | 10% | 0% | 0% | 0% | 10% |
| Pneumonitis | 90% | 10% | 0% | 0% | 0% | 0% | 10% |
| Rectal tenesmus | 90% | 0% | 10% | 0% | 0% | 0% | 10% |
| Sepsis | 90% | 0% | 0% | 0% | 10% | 0% | 10% |
| Urinary tract infection | 90% | 0% | 10% | 0% | 0% | 0% | 10% |
| Vomiting | 90% | 0% | 10% | 0% | 0% | 0% | 10% |
| Wound | 90% | 0% | 10% | 0% | 0% | 0% | 10% |
Adverse Events Legend
Adverse events occurring in ≥10% of patients are shown.
Abbreviation: NC/NA, no change from baseline/no adverse event.
Serious Adverse Events: Dose Exploration
| Name | Grade | Attribution |
|---|---|---|
| Sepsis | 4 | Unlikely |
| Sepsis | 3 | Unlikely |
| Sepsis | 4 | Unlikely |
| Diverticulitis | 3 | Unrelated |
| Fecaloma | 3 | Unrelated |
| Back pain | 3 | Unrelated |
| Dehydration | 3 | Unrelated |
| Urinary tract infection | 2 | Unrelated |
| Dyspnea | 5 | Unrelated |
Serious Adverse Events Legend
The three SAEs of sepsis all occurred in the same patient.
Adverse Events: Total
| All Dose Levels, All Cycles | |||||||
|---|---|---|---|---|---|---|---|
| Name | NC/NA | 1 | 2 | 3 | 4 | 5 | All grades |
| Fatigue | 71% | 18% | 11% | 0% | 0% | 0% | 29% |
| Nausea | 82% | 7% | 11% | 0% | 0% | 0% | 18% |
| Diarrhea | 86% | 7% | 4% | 4% | 0% | 0% | 14% |
| Hypothyroidism | 86% | 0% | 14% | 0% | 0% | 0% | 14% |
| Alanine aminotransferase increased | 89% | 4% | 0% | 7% | 0% | 0% | 11% |
| Aspartate aminotransferase increased | 89% | 4% | 0% | 7% | 0% | 0% | 11% |
| Dehydration | 89% | 4% | 4% | 4% | 0% | 0% | 11% |
| Dry mouth | 89% | 11% | 0% | 0% | 0% | 0% | 11% |
| Hypokalemia | 89% | 7% | 4% | 0% | 0% | 0% | 11% |
| Hypomagnesemia | 89% | 7% | 4% | 0% | 0% | 0% | 11% |
| Lymphopenia | 89% | 0% | 0% | 7% | 4% | 0% | 11% |
| Vomiting | 89% | 7% | 4% | 0% | 0% | 0% | 11% |
Adverse Events Legend
Adverse events occurring in ≥10% of patients are shown.
Abbreviation: NC/NA, no change from baseline/no adverse event.
Serious Adverse Events: Total
| Name | Grade | Attribution |
|---|---|---|
| Lung infection | 3 | Unrelated |
| Pneumonia | 3 | Unrelated |
| Pneumothorax | 3 | Unrelated |
| Pneumothorax | 3 | Unrelated |
| Compression fracture | 3 | Unrelated |
| Pneumonia | 3 | Unrelated |
| Gastroenteritis | 3 | Unrelated |
| Viral infection | 3 | Unrelated |
| Pancreatitis | 2 | Unrelated |
| Rectal hemorrhage | 3 | Unrelated |
| Deep vein thrombosis | 3 | Unrelated |
| Bile duct obstruction | 3 | Unrelated |
| Abdominal pain | 3 | Possible |
| Aspartate aminotransferase increased | 3 | Possible |
| Alanine aminotransferase increased | 3 | Possible |
| Sepsis | 4 | Unlikely |
| Sepsis | 3 | Unlikely |
| Sepsis | 4 | Unlikely |
| Diverticulitis | 3 | Unrelated |
| Fecaloma | 3 | Unrelated |
| Back pain | 3 | Unrelated |
| Dehydration | 3 | Unrelated |
| Urinary tract infection | 2 | Unrelated |
| Dyspnea | 5 | Unrelated |
Serious Adverse Events Legend
The two SAEs of pneumonia and two SAEs of pneumothorax all occurred in the same patient.
The SAEs of viral infection and pancreatitis occurred >30 days after the last dose of study drug.
The three SAEs of sepsis all occurred in the same patient.
Pharmacokinetics/Pharmacodynamics: Dose Escalation
| Dose level | Dose of drug: envafolimab | No. enrolled | Cmax (μg/mL) mean (CV) | Tmax (hours) median (min–max) | AUC0–last (hours*μg/mL) mean (CV) |
|---|---|---|---|---|---|
| 1 | 0.01 mg/kg | 1 | 0.370 | 167 | 48.0 |
| 2 | 0.03 mg/kg | 1 | 0.095 | 96.0 | 7.60 |
| 3 | 0.1 mg/kg | 1 | 0.702 | 168 | 81.5 |
| 4 | 0.3 mg/kg | 3 | 0.588 (68%) | 97.1 (95.8–144) | 75.9 (73%) |
| 5 | 1 mg/kg | 3 | 2.87 (30%) | 97.4 (50.7–168) | 367 (32%) |
| 6 | 2.5 mg/kg | 3 | 10.8 (35%) | 96.3 (48.8–168) | 1,494 (33%) |
| 7 | 5 mg/kg | 3 | 19.4 (29%) | 121 (97.0–167) | 2,330 (54%) |
| 8 | 10 mg/kg | 3 | 32.9 (37%) | 95.9 (95.9–167) | 4,483 (43%) |
Pharmacokinetics/Pharmacodynamics: Dose Exploration
| Dose level | Dose of drug: envafolimab | No. enrolled | Cmax (μg/mL) mean (CV) | Tmax (hours) median (min–max) | AUC0–last (hours*μg/mL) mean (CV) | t½ (hours) mean (CV) | CI F (mL/hour) mean (CV) | V/F (L) mean (CV) |
|---|---|---|---|---|---|---|---|---|
| 1 (cycle 1, day 1) | 300 mg | 10 | 14.0 (32%) | 71.9 (47‐3‐167) | 5,850 (39%) | 362 (36%) | 36.5 (25%) | 18.7 (44%) |
| 1 (cycle 5, day 1) | 300 mg | 3 | 23.1 (6.3%) | 70.6 (46.9‐93.5) | 10,533 (4.8%) | 546 (8.9%) | 28.5 (4.6%) | 16.7 (20%) |
Assessment, Analysis, and Discussion
| Completion | Study completed |
| Investigator's Assessment | Active and should be pursued further |
Currently approved anti‐PD‐1/PD‐L1 antibodies are administered intravenously (IV). The potential benefits of subcutaneous (SC) administration facilitated the discovery and development of envafolimab, a recombinant protein of a humanized single‐domain anti‐PD‐L1 antibody fused with a human IgG1 Fc fragment [1]. In a first‐in‐human phase I study, the safety, tolerability, pharmacokinetics, and antitumor activity of SC envafolimab (200 mg/mL) were evaluated in 28 adults with advanced, refractory solid tumors.
In a dose‐escalation phase, no dose‐limiting toxicities were reported, the maximum tolerated dose was not reached, and the maximum dose administered was 10 mg/kg once weekly (QW). As of the data cutoff date, there were no infusion‐related or injection‐site reactions in any treated patient. Treatment‐emergent adverse events (TEAEs) reported in >3 patients included fatigue (n = 8), nausea (n = 5), diarrhea (n = 4), and hypothyroidism (n = 4). Grade ≥3 TEAEs occurred in 10 of 18 patients in the dose‐escalation phase and 4 of 10 in the dose‐exploration phase. Fourteen patients overall had TEAEs considered to be drug related, most of which were grade ≤2. The most common of these were expected and previously reported for other PD‐1/PD‐L1 antibodies in patients with solid tumors [2, 3, 4]. Three patients reported grade 3 study drug–related TEAEs, including lymphocytopenia (n = 1, 0.1 mg/kg), abdominal pain (n = 1, 10 mg/kg), and increased alanine aminotransferase, aspartate aminotransferase, and blood alkaline phosphatase (n = 2, 10.0 mg/kg). No grade ≥4 study drug–related TEAEs were reported in either the dose‐escalation or dose‐exploration phase. A single patient who received 10 mg/kg envafolimab in the dose‐escalation phase had three serious TEAEs considered related to the study drug (grade 3 events of abdominal pain, increased alanine aminotransferase, and increased aspartate aminotransferase). These were treated with corticosteroids and resolved with sequelae. Five patients (18%) experienced TEAEs leading to treatment discontinuation. In one patient, these events (increased alanine aminotransferase and aspartate aminotransferase) were considered to be drug related.
Three patients had confirmed partial responses according to RECIST version 1.1 (Fig. 1), of whom two had an ongoing response at data cutoff. Eight patients who received envafolimab had a best overall response of stable disease. The disease control rate was 39.3% (95% confidence interval [CI], 21.5–59.4), and the objective response rate was 10.7% (95% CI, 2.3–28.2), in line with the efficacy of other anti‐PD‐1/PD‐L1 antibodies in previously treated patients with advanced solid tumors [3, 5, 6]. Best reductions in tumor size from baseline are shown in Figure 1. The median progression‐free survival was 2.8 months (95% confidence interval, 1.8–7.6) and median overall survival was 8.5 months (95% confidence interval, 3.1–17.4). A recently completed phase II trial in patients with microsatellite instability–high tumors (NCT03667170) provided confirmatory evidence of the efficacy of SC envafolimab, with an objective response rate of 42.7% at a dose of 150 mg QW [7].
Following a single SC administration in the dose‐escalation phase, envafolimab could be detected in the serum of each patient for at least one time point at all dose levels. The maximum plasma concentration (Cmax) and area under the curve (AUC) increased linearly over the dose range of 0.01–10 mg/kg (Fig. 2). At 0.3 mg/kg, two of three patients had a first‐dose Cmax that exceeded 0.5 μg/mL. The median time to reach Cmax was 4–7 days. Neither first‐dose Cmax nor AUC were significantly affected by injection site (Fig. 3). During the dose‐exploration phase, in which all patients received envafolimab 300 mg SC once every 4 weeks (Q4W), the mean Cmax after the first dose was 14 μg/mL, and the median time to reach Cmax was 3 days. The first‐dose half‐life was estimated to be 14 days, and at steady state (first day of cycle 5), the mean effective half‐life was 23 days. Pharmacokinetics simulations estimated that most patients would attain steady state after five cycles and that >90% of those receiving envafolimab 300 mg once every 3 weeks (Q3W) and 400 mg Q4W would maintain trough concentration above 5 μg/mL (Table 1), which is at least 10‐fold higher than the minimum pharmacologically active concentration (0.5 μg/mL [1]).
Figure 3.
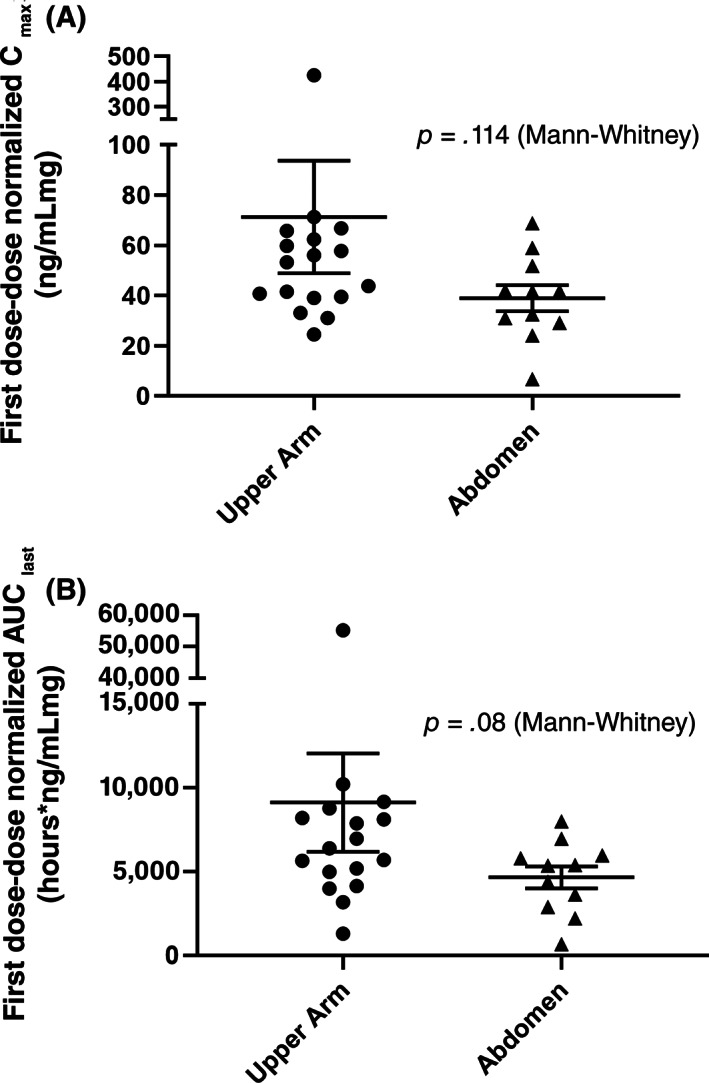
Effect of subcutaneous injection site on first‐dose dose‐normalized Cmax (A) Or dose‐normalized AUClast (B). Abbreviations: AUClast, area under the curve until the last measurement; Cmax, maximum plasma concentration.
Antidrug antibodies (ADAs) were detected in 12 of the 28 patients, of whom 2 had pre‐existing ADAs. The frequency of de novo ADA production (36%) is in the range reported for IV administered nivolumab and atezolizumab but higher than for pembrolizumab and cemiplimab [8]. For the 10 patients who developed ADAs following treatment, the median time to first detection was 4.1 weeks and the median duration of positivity was 4.1 weeks (Table 2). Nine of these patients had additional ADA data, of whom three had only negative tests and two had a negative test at the last assessment. Dose‐normalized steady‐state trough concentrations did not significantly differ between patients without and with ADAs, irrespective of when they were detected (Fig. 4).
Table 2.
Antidrug antibodies
| Measure | Overall ADA analysis population (n = 28) |
|---|---|
| Baseline test result, n (%) | |
| Positive | 2 (7) |
| Negative | 26 (93) |
| Test result during treatment, n (%) | |
| Positive | 12 (43) |
| Positive but negative at baseline | 10 (36) |
| Time to positivity in participants negative at baseline, weeks | |
| Mean (SD) | 3.6 (1.0) |
| Median | 4.1 |
| Min–max | 2.0–4.4 |
| Duration of positivity in participants negative at baseline, weeks | |
| Median | 4.1 |
| Min, max | 0.14+, 31.14+ |
| Participants who were negative at baseline, had a positive postbaseline test, and had ≥1 subsequent test result, n (%) | |
| Data available | 9 (32) |
| All subsequent test results were negative | 3 (11) |
| ≥1 subsequent test result was positive | 6 (21) |
| Last test was positive | 4 (14) |
| Last test was negative | 2 (7) |
Abbreviation: ADA, antidrug antibodies.
Figure 4.
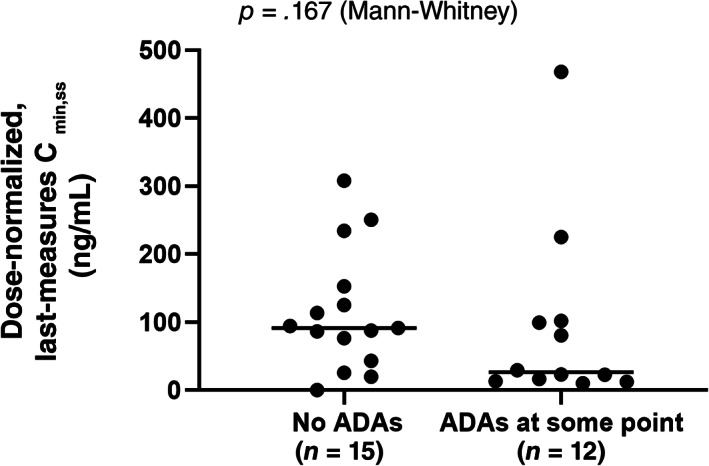
Dose‐normalized steady‐state serum concentrations of envafolimab in patients without and with antidrug antibodies. Abbreviations: ADA, antidrug antibody; Cmin,ss, minimum serum concentration at steady state.
These results provide data to select the dosing of SC envafolimab. When administered QW, SC envafolimab was safe up to the maximum administered dose of 10 mg/kg and was active at doses as low as 0.3 mg/kg. SC administration at a dose of 300 mg Q4W was also feasible and resulted in a similar tumor response and safety as SC administration of doses ≥0.3 mg/kg administered QW. As with other PD‐1 and anti‐PD‐L1 antibodies [9], increasing the dose of envafolimab was not associated with an improvement in objective response or increased toxicity. These results support use of a fixed‐dose schedule administered Q3W or Q4W for the future clinical development of SC envafolimab. In ongoing studies, 300 mg Q3W and 400 mg Q4W is being investigated.
This phase I study showed that SC injection of envafolimab at 200 mg/mL was an effective route of administration, was well tolerated, and had durable antitumor activity at a wide range of doses and schedules in patients with previously treated advanced solid tumors. A recent phase I trial of the humanized PD‐1 monoclonal antibody PF‐068‐1591 showed that it was well tolerated and had antitumor activity when administered SC, although three separate 2‐mL injections of 50 mg/mL were required to deliver the full dose [10]. Like envafolimab, SC administration of PF‐068‐1591 resulted in prolonged absorption (median time to Cmax ~8 days). It also resulted in a lower Cmax and correspondingly fewer grade ≥3 TEAEs than IV administration. Therefore, the slower absorption, lower Cmax, and prolonged half‐life of envafolimab may offer advantages over IV administration.
In conclusion, envafolimab is the first‐in‐class PD‐1/PD‐L1 antibody that can be administered at a therapeutic dose in a single SC injection of under 2 mL. As such, envafolimab has the potential to be a more convenient option than currently approved IV PD‐1/PD‐L1 inhibitors.
Disclosures
Kyriakos P. Papadopoulos: Basilia (C/A), Abbvie, Amgen, ADC Therapeutics, Anheart, 3D Medicines, Basilia, Bayer, Calithera Biosciences, Daiichi Sankyo, Eli Lilly and Company, EMD Serono, F‐star, Incyte, Jounce Therapeutics, Linnaeus, Mabspace Biosciences, Merck, Mirati Therapeutics, MedImmune, Mersana, Peleton Therapeutics, Regeneron, Syros Pharmaceuticals, Pfizer, Treadwell Therapeutics, Tempest Therapeutics (RF); Siying Xu: 3D Medicines (E, OI); Haolan Lu: 3D Medicines (E, OI); Ni Lu: 3D Medicines (E, OI); Yue He: 3D Medicine (E, OI); Ting Xu: Alphamab Co., Ltd., (E, OI, IP); John Gong: 3D Medicines (E, OI, IP); David Liu: 3D Medicines (E, OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Figures and Tables
Acknowledgments
We thank Xiaojia Xu, Sunny Wang, Kevin Chen, and Karen Xie for the data management, project management, and statistical programming support in the data analysis of this article. We also thank the patients and their families, investigators, research nurses, study coordinators, and operations staff who contributed to this study. They received no compensation for their contributions. Medical writing was provided by Katie Crosslin, Ph.D., Stephen Gilliver, Ph.D., and Phillip Leventhal, Ph.D. (Evidera), and was funded by 3D Medicines.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
Footnotes
- ClinicalTrials.govIdentifier: NCT02827968
- Sponsor: 3D Medicines Co., Ltd
- Principal Investigator: Wael Harb
- IRB Approved: Yes
References
- 1.Zhang F, Wei H, Wang X et al. Structural basis of a novel pd‐l1 nanobody for immune checkpoint blockade. Cell Discov 2017;3:17004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Hodi FS, Brahmer JR et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012;366:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patnaik A, Kang SP, Rasco D et al. Phase I study of pembrolizumab (MK‐3475; anti‐PD‐1 monoclonal antibody) in patients with advanced solid tumors. Clin Cancer Res 2015;21:4286–4293. [DOI] [PubMed] [Google Scholar]
- 4.Shi Y, Su H, Song Y et al. Safety and activity of sintilimab in patients with relapsed or refractory classical Hodgkin lymphoma (ORIENT‐1): A multicentre, single‐arm, phase 2 trial. Lancet Haematol 2019;6:e12–e19. [DOI] [PubMed] [Google Scholar]
- 5.Brahmer JR, Drake CG, Wollner I et al. Phase I study of single‐agent anti‐programmed death‐1 (MDX‐1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol 2010;28:3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heery CR, O'Sullivan‐Coyne G, Madan RA et al. Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): A phase 1a, multicohort, dose‐escalation trial. Lancet Oncol 2017;18:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen L, Li J, Deng Y, et al. 70MO ‐ efficacy and safety of envafolimab (KN035) in advanced tumours with mismatch‐repair deficiency. Ann Oncol 2020;31:S1270–S1272. [Google Scholar]
- 8.Davda J, Declerck P, Hu‐Lieskovan S et al. Immunogenicity of immunomodulatory, antibody‐based, oncology therapeutics. J Immunother Cancer 2019;7:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renner A, Burotto M, Rojas C. Immune checkpoint inhibitor dosing: Can we go lower without compromising clinical efficacy? J Glob Oncol 2019;5:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson ML, Braiteh F, Grilley‐Olson JE et al. Assessment of subcutaneous vs intravenous administration of anti‐PD‐1 antibody PF‐06801591 in patients with advanced solid tumors: A phase 1 dose‐escalation trial. JAMA Oncol 2019;5:999–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]