

Abstract
Background
Microbial communities of wild animals are being increasingly investigated to provide information about the hosts’ biology and promote conservation. Loggerhead sea turtles (Caretta caretta) are a keystone species in marine ecosystems and are considered vulnerable in the IUCN Red List, which led to growing efforts in sea turtle conservation by rescue centers around the world. Understanding the microbial communities of sea turtles in the wild and how affected they are by captivity, is one of the stepping stones in improving the conservation efforts. Describing oral and cloacal microbiota of wild animals could shed light on the previously unknown aspects of sea turtle holobiont biology, ecology, and contribute to best practices for husbandry conditions.
Results
We describe the oral and cloacal microbiota of Mediterranean loggerhead sea turtles by 16S rRNA gene sequencing to compare the microbial communities of wild versus turtles in, or after, rehabilitation at the Adriatic Sea rescue centers and clinics. Our results show that the oral microbiota is more sensitive to environmental shifts than the cloacal microbiota, and that it does retain a portion of microbial taxa regardless of the shift from the wild and into rehabilitation. Additionally, Proteobacteria and Bacteroidetes dominated oral and cloacal microbiota, while Kiritimatiellaeota were abundant in cloacal samples. Unclassified reads were abundant in the aforementioned groups, which indicates high incidence of yet undiscovered bacteria of the marine reptile microbial communities.
Conclusions
We provide the first insights into the oral microbial communities of wild and rehabilitated loggerhead sea turtles, and establish a framework for quick and non-invasive sampling of oral and cloacal microbial communities, useful for the expansion of the sample collection in wild loggerhead sea turtles. Finally, our investigation of effects of captivity on the gut-associated microbial community provides a baseline for studying the impact of husbandry conditions on turtles’ health and survival upon their return to the wild.
Supplementary Information
The online version contains supplementary material available at 10.1186/s42523-021-00120-5.
Keywords: Microbiota, Bacterial diversity, Reptile, Rehabilitation, Adriatic Sea, Conservation
Background
Microbial communities associated with vertebrates can influence host’s evolution, development, immune system maturation, physiology, nutrient acquisition, health and disease [1, 2]. It is estimated that the host’s collection of bacteria could contain at least 100 times the genes as in the host’s genome, often adding to the metabolic functions’ repertoire, e.g. biochemical pathways in nutrient acquisition [3]. Moreover, we can consider the host and its microbial commensals as a distinct biological entity (holobiont and hologenome) susceptible to the processes of natural selection [4, 5].
Most studies of microbial communities have focused on the distal gut of humans or captive mammals [2, 6] but there are recent growing efforts in investigations of free-ranging wild animals. Wild animals are sensitive to environmental perturbations caused by climate change and anthropogenic habitat disruption, therefore investigating wild animal-associated microbial communities contributes to improving existing conservation efforts [7, 8]. Current research covering major vertebrate groups reveal evidence for co-phylogeny of mammals and their microbial communities, microbiome convergence in birds and bats, while microbial assemblages of non-mammalian hosts (e.g. reptiles) are mostly influenced through diet and the environment [9, 10]. Marine animals are permanently immersed in seawater environment, making the microbial dynamics different from those of land-dwelling animals [11]. As expected, marine mammals have been the focus of most vertebrate microbial community studies that undertook a wider sampling effort of body sites other than the distal gut or feces [12–16]. In comparison to other vertebrates, reptiles are still underrepresented in studies of their bacterial communities [6, 17], especially large marine reptiles, such as sea turtles. Sea turtles are large-bodied, long-lived marine top predators, considered as a keystone species, with critical roles in ecosystem processes such as bioturbation, bioaccumulation, energy flow, trophic status and mineral cycling [18]. Loss of foraging and nesting sites, increasing global temperatures, and bycatch are major threats for sea turtles’ survival. Currently, there are seven extant sea turtle species listed on the IUCN Red List of Threatened Species [19]: Kemp’s ridley (Lepidochelys kempii) and hawksbill sea turtles (Eretmochelys imbricata) are critically endangered; the green turtle (Chelonia mydas) is considered to be endangered; loggerhead (Caretta caretta), olive ridley (Lepidochelys olivacea) and leatherback (Dermochelys coriacea) sea turtles are listed as vulnerable, while data are deficient for the flatback sea turtle (Natator depressus). The efforts of sea turtle rescue and rehabilitation initiatives facilitate access for sea turtle-focused research [20] and, consequently, studies on microbial communities of sea turtles are increasing.
To date, microbial assemblages of the sea turtle gut have been described by sequencing the 16S rRNA genes of fecal or cloacal samples in wild, stranded [21, 22], and rehabilitated green sea turtles [23, 24], in juveniles undergoing an ontogenetic shift from pelagic to neritic habitats [25, 26], and mucosa-associated bacterial communities in stranded green turtles [27]. Additionally, there are reports on the gut microbiota of Kemp’s ridley turtles undergoing rehabilitation [28] and nesting flatback turtles [29, 30]. Loggerhead sea turtles’ fecal and gut microbial communities have been studied mostly in stranded animals or undergoing rehabilitation in the Mediterranean Sea [31–33] with recent reports on nesting females of the USA and Australian populations [30, 34]. Furthermore, Scheelings and colleagues have performed one of the most comprehensive studies on the distal gut microbial communities of all seven species of the sea turtles reporting phylogenetic aspects of sea turtle microbiome evolution [34].
The focus of this study is on the loggerhead sea turtles’ oral and distal gut microbiota in both recently caught and turtles undergoing rehabilitation at the Adriatic Sea turtle rescue centers. In addition to distal gut (cloacal) samples, we sampled the buccal (oral) cavity as there are no known reports on 16S rRNA profiling for oral microbial communities in sea turtles to the date of this study. Cultivation-based approaches have shown that oral bacterial communities of loggerhead sea turtles in the Mediterranean harbor antibiotic-resistant bacterial strains and common opportunistic pathogens [35, 36]. NGS amplicon sequencing of resistant bacterial isolates showed that injured Adriatic Sea loggerheads’ wounds contain bacteria with multiple antibiotic resistance genes [37]. Aforementioned reports emphasize the idea of sea turtles as sentinel species that can be studied as indicators of marine health and pollution [35]. To fill in the gap in understanding the loggerhead sea turtle microbiota, we provide data on loggerhead oral microbial communities as the oral cavity is the first line in transitioning from external to internal environments of the turtle. The aims of this study were to describe oral and cloacal microbial communities of loggerhead sea turtles and compare them between incidentally caught or stranded and captive animals undergoing rehabilitation. Additionally, we investigated the impact of short-term rehabilitation period on loggerhead microbiota, which could clarify the dynamics of the loggerhead sea turtles’ commensal microbes in relation to the turtles’ changing environment.
Methods
Target population
We sampled loggerhead sea turtles from the Adriatic Sea that were found floating, stranded on beaches or incidentally caught by fishing boats and then transported to the regional veterinary clinic or rescue center: The Sea Turtle Clinic (STC) of the Department of Veterinary Medicine of University of Bari “Aldo Moro” (Italy) and the Marine Turtle Rescue Center Aquarium Pula (Croatia). Samples collected immediately upon arrival to the treatment facility are considered “wild” as they were taken close to the time of turtle capture and marked as “before” samples in further analyses and text. All turtles were examined for injuries and relevant information were collected during sampling. Healthy individuals were released within 24 h, while others were kept under observation (“short-term rehabilitation”) or longer rehabilitation until recovered from injuries. List of sampled turtles is presented in Table 1 with an indication of release day.
Table 1.
Information on sampled loggerhead sea turtles and their condition at the time of admission to the rescue centers in the Adriatic Sea
| Turtle ID | Origin | Sampling date (YYYY-MM-DD) | Days before sampling | Sampling site | CCL (cm) | Weight (kg) | Sex | Life stage | Reason for admission to rescue center | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Oral | Cloacal | Water | |||||||||
| ID010 | CRO, Korčula | 2018-12-11 | 3 | – | + | – | NA | 40 | F | adult | Old head injury; weight was measured prior to release 2019-11-04 |
| 2019-04-08 | 121 | – | + | + | 69.7 | 46.5 | |||||
| ID019 | IT, Barletta | 2019-01-09 | 0 | + | + | – | 50.7 | 31 | ND | juvenile | Trawling (32 m depth), treated for gas embolism |
| 2019-01-11 | 2R | + | + | – | |||||||
| ID022 | IT, Barletta-Trani | 2019-01-10 | 0R | + | + | – | 72 | 42 | M | adult | Trawling (22 m depth), released during the day |
| ID023 | IT, Trani | 2019-01-11 | 13 | + | + | – | 64 | 30.6 | ND | juvenile | Trawling (36 m depth) hospitalized at the WWF center in fresh water (7 days leeches removal treatment) |
| ID026 | IT, Molfetta | 2019-01-21 | 6R | + | + | + | 64.5 | 32.1 | M | adult | Found at the beach, not good clinical status |
| ID028 | IT, Barletta | 2019-01-17 | 0 | + | + | – | 74.5 | 46 | F | adult | Trawling (34 m depth), gas embolism |
| ID030 | IT, Barletta | 2019-01-17 | 0 | + | + | – | 63 | 29.5 | ND | juvenile | Trawling (34 m depth), gas embolism |
| 2019-01-21 | 4R | + | + | + | |||||||
| ID034 | IT, Bisceglie | 2019-01-22 | 0R | – | + | – | 72 | 45.6 | F | adult | Trawling (40 m depth), good clinical status |
| ID040 | IT, Barletta-Trani | 2019-01-28 | 4R | – | + | – | 66 | 35.2 | ND | juvenile | Trawling (40 m depth), good clinical status |
| ID041 | IT, Barletta-Trani | 2019-01-28 | 4R | + | + | – | 55.5 | 20 | ND | juvenile | Trawling (22 m depth), gas embolism |
| ID042 | IT, Barletta | 2019-01-29 | 0R | + | + | – | 67.5 | 36.4 | ND | juvenile | Trawling (40 m depth), good clinical status |
| ID044 | IT, Bisceglie | 2019-01-29 | 0R | + | + | – | 68 | 36.3 | ND | juvenile | Trawling (30 m depth), good clinical status |
Letter “R” in superscript next to the number of day before sampling indicates release into the wild on that day, if not present it means that the turtle was kept in the clinic or rescue center after the sampling
CCL curved carapace length, ND not determined
Sampling of 12 loggerhead turtles (Table 1) was performed by trained personnel during December 2018 and January 2019 in accordance with the 1975 Declaration of Helsinki, as revised in 2013, and the applicable national laws.
Loggerheads’ enclosure description
At the STC (Italy) the hospitalized turtles were kept in individual plastic tanks (approximately 1.5 m in diameter and 1 m in depth) with clean artificial saltwater (tap water with added NaCl at least up to 35 ppt salinity). The saltwater was changed every 2–3 days, with routine tank cleaning and disinfecting between saltwater changes. At the Marine Turtle Rescue Center Aquarium Pula (Croatia), the hospitalized turtle was kept in an individual plastic tank (2 m in diameter, 1.5 m in depth) with local seawater pumped and purified through the Aquarium’s filtration systems. The tank was occasionally cleaned by scrubbing the algal overgrowth and grime off the tank walls. All turtles in the study were fed with diverse foods ranging from frozen (herring, codfish, mullet) or fresh fish food (squid, pilchard, and mackerel).
Sample collection
The loggerheads’ cloacal and oral swab samples were collected either upon arrival of the turtle to the center (further regarded to as cloacal before, CB; oral before, OB) or within the rehabilitation period (after the turtle has spent time in the rescue center, further regarded to as cloacal rehabilitated, CR; oral rehabilitated, OR). When possible, we collected tank water during the rehabilitation period (further regarded to as tank water, W).
Oral swab samples were collected by gentle rotating of sterile dry cotton or synthetic swabs (Aptaca Nuova, Italy) on the tongue and palate mucosa, while cloacal samples were collected by inserting the swabs approximately 10 cm into the cloaca and rotating (Additional file 2: Figure S1). The swabs were collected in triplicate and stored individually in 97% ethanol at − 20 °C until DNA extraction. Samples of the tank water were collected prior to routine tank cleaning or during oral and cloacal sampling, in sterile containers and kept cool until arrival to the lab and filtering. Sampled tank water (250 ml) was vacuum filtered on a 45 mm in diameter, 0.2 μm pore-size sterile Whatman polycarbonate membrane filter (Sigma-Aldrich). Filters were carefully folded with sterile forceps and stored in 96% ethanol at − 20 °C until further processing. In total, 12 loggerhead turtles were sampled: three turtles were sampled twice (upon arrival and during rehabilitation), nine turtles were sampled once (five upon arrival, four during rehabilitation), and tank water samples were collected from three turtle enclosures (Table 1). Cloacal samples were collected from all turtles and sampling periods, while we could not obtain oral samples from three turtles (Table 1; ID010, ID034, and ID040).
DNA extraction and sequencing
Prior to DNA extraction the ethanol was removed from the tubes by pipetting (after centrifugation) and evaporation under laminar flow hood for 24 h. DNA from the filters and swabs was extracted with the DNeasy PowerSoil Kit (Qiagen) according to the manufacturer's instructions with several modifications: (1) after transferring the swabs and filters to the PowerBead Tube the samples were incubated for 15 min at 65 °C, (2) instead of beadbeating PowerBead Tubes were vortexed horizontally for 10 min at maximum speed, and (3) all downstream incubation times at 2–8 °C were increased to 15 min. DNA was extracted from each swab and filter individually, and DNA concentrations were measured by NanoDrop ND-1000 V3.8 spectrophotometer (ThermoFisher). For samples with low DNA yield, triplicate DNA isolates were pooled together and concentrated according to the troubleshoot section of the DNeasy Powersoil Kit instructions. Extracted DNA was sent for PCR, library preparation, and 250 × 2 paired-end Illumina MiSeq v2 setup sequencing of the V3-V4 region of 16S rDNA with primers 341F_ill (5′-CCTACGGGNGGCWGCAG-3′) and 805R_ill (5′-GACTACHVGGGTATCTAATCC-3′) [38] to Microsynth (Switzerland).
Sequence analysis
Demultiplexed sequences with removed adapters and linker sequences were obtained from the Microsynth sequencing facility and quality checked with FastQC [39]. Upon inspection, reverse sequences were shown to be of insufficient quality and length in some samples, therefore only forward reads were used in downstream analyses with QIIME 2 2020.2 [40]. Forward demultiplexed reads (Casava 1.8 single-end demultiplexed fastq format) were imported to QIIME 2 and summarized using q2-demux plugin followed by denoising with DADA2 q2-dada2 plugin [41]. Forward sequences were trimmed at 5’ end for 10 bp (primer removal) and truncated to 240 bp that produced a final sequence length of 230 bp. DADA2 dereplication produced amplicon sequence variants (ASVs) analogous to 100% operational taxonomic units (OTUs) [42]. ASVs were aligned with mafft [43] (via q2-alignment) and used to construct an unrooted phylogeny tree with fasttree2 [44] (via q2-phylogeny). Taxonomy was assigned to ASVs via q2-feature-classifier [45] classify-sklearn naïve Bayes taxonomy classifier against the SILVA ribosomal RNA sequence database (v. 132) [46]. Mitochondrial and chloroplast sequences were filtered out via q2-taxa prior to calculating alpha and beta diversity metrics via q2-diversity plugin.
Alpha diversity measurements, including Shannon’s diversity index, observed ASVs, and Faith’s phylogenetic diversity, were used for inspecting rarefaction curves to determine suitable sampling depth, and the differences between sampling sites were tested by Kruskal–Wallis H test. Beta diversity analyses were performed on rarefied dataset to 3200 sequences per sample to eliminate bias of different sampling depths [47, 48]. Comparisons of microbial communities were performed through Bray–Curtis, unweighted and weighted UniFrac [49, 50] Principal Coordinate Analyses (PCoA) via q2-diversity plugin. Due to intrinsic compositionality of microbial community datasets obtained by sequencing [51] we used an additional beta diversity analysis on non-rarified data through Robust Aitchison Principal Component Analysis (robust PCA; rPCA) via q2-deicode plugin [52]. Robust PCA is based on centered log-transformation and matrix completion, while retaining feature loadings that may discern between potential microbial niches. The analysis was performed after the exclusion of features with less than ten reads across samples. Log-ratios of rPCA feature loadings were inspected through q2-qurro plugin [53]. The permutational multivariate analysis of variance (PERMANOVA) was used to analyze beta diversity statistical differences via q2-diversity plugin, with the Benjamini–Hochberg false discovery rate (FDR) correction for multiple comparisons. Core features and genera (present in 80% or 85% of samples per sampling site) were determined via q2-feature-table plugin. All plots were visualized with ggplot2 [54] in RStudio (v. 1.3.959) and EMPeror [55].
Results
A total of 744 531 high-quality reads were obtained for 15 cloacal, 11 oral, and three tank water samples (29 samples in total). The samples had a mean (± SE) 25 673 ± 3 265 sequences per sample that were clustered to 4476 ASVs (Additional file 1). Predominant phyla of cloacal samples consisted of Proteobacteria, Bacteroidetes, Kiritimatiellaeota, Firmicutes and Spirochaetes (> 90% of all cloacal sequences). Oral samples’ predominant phyla were Proteobacteria, Bacteroidetes and Planctomycetes (> 90% of all oral sequences), while tank water exhibited high prevalence of Proteobacteria, Bacteroidetes and Epsilonbacteraeota (> 90% of all tank water sequences). Taxa within phyla varied among individuals, sampling sites, and sampling periods (Additional file 1).
Alpha diversity metrics (Shannon’s diversity, observed features, Faith’s PD) were calculated for sampling sites; cloacal, oral, and tank water. No significant difference was observed (p > 0.05, Kruskal–Wallis H test) for different sampling sites in either of alpha diversity metrics tested (Additional file 2: Table S2). Tank water did exhibit higher variation than cloacal and oral samples, possibly due to sample size and differences in origin (artificial saltwater in Italy vs. filtered sea water in Croatia that showed greater diversity) (Fig. 1), but it was not significantly different from other sampling sites (Additional file 2: Table S2).
Fig. 1.
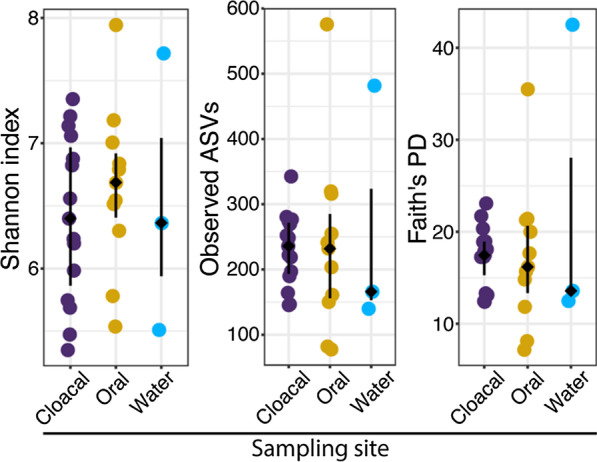
Alpha diversity (Shannon index, observed ASVs, Faith’s Phylogenetic Diversity) of loggerhead cloacal (purple), oral (yellow), and tank water (blue) samples. Filled diamond indicates sample median with lines extending to the upper and lower quartile of sample distribution
Bacterial communities of cloacal samples tended to cluster together, regardless of the sampling period, but oral sample communities showed some separation based on sampling before or during rehabilitation according to PCoA plots (Figs. 2a, 3) and rPCA biplot (Fig. 2b). Tank water samples did not show a visible pattern for Bray–Curtis PCoA or Robust Aitchison PCA (Fig. 2), but for UniFrac PCoA the samples tended to cluster near oral samples (Fig. 3). Feature loadings of Robust Aitchison PCA represent highly ranked individual ASVs, mostly uncultured Gammaproteobacteria, Rhodobacteraceae, and members of the Kiritimatiellae WCHB1-41 group (Fig. 2b).
Fig. 2.

Comparison of microbial diversity in loggerhead cloacal, oral and tank water samples a principal coordinate analysis (PCoA) plot of Bray–Curtis distances and b principal component analysis (PCA) biplot of robust Aitchison distances with loadings as individual highly ranked ASVs
Fig. 3.

Comparison of microbial diversity in loggerhead cloacal, oral and tank water samples. Principal component analysis (PCoA) plot of a Unweighted Unifrac and b Weighted Unifrac
Based on PERMANOVA (with 999 permutations) bacterial communities differed significantly (p < 0.05) between sampling sites and periods (cloacal before, CB; cloacal rehabilitated, CR; oral before, OB; oral rehabilitated, OR; tank water, W) for all used distance metrics tested (Bray–Curtis p = 0.001, pseudo-F = 2.37; Robust Aitchison p = 0.002, pseudo-F = 3.68; unweighted UniFrac p = 0.001, pseudo-F = 2.38; weighted UniFrac p = 0.001, psuedo-F = 3.59). Pairwise PERMANOVA testing for sampling site and period groups differed between metrics used with the most conservative result obtained from Robust Aitchison distance that detected a significant difference only between CR versus OB (p = 0.005, pseudo-F = 12.27) and CB versus OB (p = 0.005, pseudo-F = 10.40). Bray–Curtis distance, unweighted and weighted UniFrac distances pairwise test results showed a significant difference for CB versus OR and OR versus OB in addition to the same sampling site and period conditions that were observed with Robust Aitchison distance pairwise testing. No significant difference was detected between CB and CR samples. Out of all tested metrics, Bray–Curtis and unweighted UniFrac pairwise test results showed a significant difference (p < 0.05) among W versus CB, CR, and OB. We detected no significant difference between W and OR samples, which points to the effects of tank water on the oral microbiota of turtles in rehabilitation. Summary of pairwise PERMANOVA tests per distance metric is shown in Additional file 2: Table S3. Visual inspection of natural log ratios of up to 20% top and bottom feature loadings of the Robust Aitchison PCA biplot (Fig. 2b) shows clear segregation of oral before and oral rehabilitated samples (5%, 10%, and 20% top and bottom features on rPC1), and similar log-ratio values of all cloacal samples to oral rehabilitated samples (20% top and bottom features on rPC2) (Fig. 4).
Fig. 4.

Natural log-ratios (plotted by QURRO) of loggerhead samples’ top and bottom 5%, 10%, and 20% of feature loadings on rPC1 and rPC2 of Robust Aitchison PCA biplot feature loadings by sampling site and period: CB, cloacal before; CR, cloacal rehabilitated; OB, oral before; OR, oral rehabilitated; W, tank water
Bacterial communities were distributed across eleven dominant phyla present at > 1% relative abundance in at least one sampling site (Fig. 3). All sampling sites shared dominant phyla Proteobacteria, Bacteroidetes, and to a lesser extent Epsilonbacteraeota (Table 2). Firmicutes were shared between cloacal and oral samples, while tank water and oral samples shared Actinobacteria. Specific to cloacal samples were Kiritimatiellaeota, Spirochaetes and Lentisphaerae phyla, oral samples harbored Planctomycetes, and tank water Patescibacteria and Verrucomicrobia (Fig. 5).
Table 2.
Bacterial phyla of loggerhead sea turtle cloacal and oral samples, and tank water samples from the rescue centers present at > 1% relative abundance on average per sampling site
| Bacterial phyla | Cloacal (n = 15) | Oral (n = 11) | Tank water (n = 3) |
|---|---|---|---|
| Actinobacteria | 0.52 ± 0.18 | 1.37 ± 0.31 | 1.09 ± 0.87 |
| Bacteroidetes | 21.74 ± 2.16 | 33.88 ± 2.43 | 30.26 ± 4.33 |
| Epsilonbacteraeota | 2.48 ± 0.63 | 2.02 ± 0.57 | 4.15 ± 3.36 |
| Firmicutes | 6.74 ± 1.35 | 1.75 ± 0.37 | 0.22 ± 0.08 |
| Kiritimatiellaeota | 12.78 ± 4.04 | 0.46 ± 0.39 | 0.03 ± 0.03 |
| Lentisphaerae | 1.99 ± 0.72 | 0.14 ± 0.06 | 0.06 ± 0.03 |
| Patescibacteria | 0.17 ± 0.08 | 0.60 ± 0.24 | 1.83 ± 1.83 |
| Planctomycetes | 0.08 ± 0.04 | 1.65 ± 1.06 | 0.63 ± 0.61 |
| Proteobacteria | 48.60 ± 6.21 | 56.08 ± 3.19 | 58.62 ± 1.56 |
| Spirochaetes | 3.30 ± 1.47 | 0.44 ± 0.14 | 0.01 ± 0.01 |
| Verrucomicrobia | 0.03 ± 0.01 | 0.30 ± 0.14 | 1.14 ± 0.97 |
Values represent mean percentage ± SE, with mean values above 1% in bold
Fig. 5.

Relative abundances (%) of bacterial phyla present (> 1% on average per sampling site) in loggerhead cloacal, oral and tank water samples. Turtle ID suffix indicates the sampling site and period (before or during/after rehabilitation) as follows: CB, cloacal before; CR, cloacal rehabilitated; OB, oral before; OR, oral rehabilitated; W, tank water
Further, bacterial taxa classified to genera (or the next available classification level) present at > 2% relative abundance in at least one sampling site and period conditions indicate taxa specificity to habitat and, on the other hand, the possibility of sharing bacterial taxa of the turtle endomicrobiota with the environment (e.g. Bizionia in oral rehabilitated and tank water bacterial communities) (Table 3). WCHB1-41 taxon (phylum Kiritimatiellaeota) was shown to be almost exclusive for cloacal samples (even though turtle ID010 has not had any sequences of that taxon detected), along with Treponema 2, Aeromonas, unclassified Aeromonadales, Desulfovibrio, unclassified Rikenellaceae, and Bacteroides genus. Oral samples often shared taxa with cloacal and tank water samples with noticeable differences in relative abundance of Pseudoalteromonas and unclassified Helieaceae that was not found at > 2% in cloacal samples or tank water. Interestingly, only tank water harbored Bermanella as it was not detected in cloacal nor oral samples (Table 3). Based on PERMANOVA results (Additional file 2: Table S3), wild oral samples (before) and oral microbiota during rehabilitation differ significantly, which is also reflected in relative abundances of microbial taxa abundance (Table 3). Wild oral samples harbored more Bacteroidales, Tenacibaculum, Rhodobacteraceae, Gammaproteobacteria and Halieaceae, in comparison to oral samples from turtles in rehabilitation, which showed greater abundance of Bizionia, Pseudoalteromonas, Shewanella, Pseudomonas, and Vibrio, similar to cloacal and tank water samples (Table 3).
Table 3.
Bacterial taxa of loggerhead sea turtle cloacal and oral, and tank water samples classified to the genus (or higher taxonomic level) present at > 2% average relative abundance in at least one sampling site and period category (before or wild and during rehabilitation)
| Bacterial taxa | Cloacal samples | Oral samples | Tank water | ||
|---|---|---|---|---|---|
| before (n = 9) | rehabilitated (n = 6) | before (n = 7) | rehabilitated (n = 4) | rehabilitated (n = 3) | |
| Phylum Bacteroidetes | |||||
| Bacteroidales; unclassified | 1.86 ± 0.60 | 2.45 ± 1.07 | 2.21 ± 0.28 | 0.19 ± 0.09 | 0.30 ± 0.24 |
| Bacteroides | 2.09 ± 0.61 | 1.73 ± 0.76 | 0.10 ± 0.10 | ND | 0.13 ± 0.13 |
| Marinifilum | 1.56 ± 0.55 | 4.23 ± 1.87 | 1.37 ± 0.54 | 0.74 ± 0.32 | 0.54 ± 0.37 |
| Rikenellaceae; unclassified | 3.03 ± 1.29 | 1.10 ± 0.73 | 0.14 ± 0.13 | ND | 0.03 ± 0.03 |
| Flavobacteriaceae; unclassified | 2.22 ± 0.88 | 1.94 ± 0.33 | 13.78 ± 2.93 | 11.54 ± 4.45 | 6.79 ± 1.70 |
| Bizionia | ND | 1.29 ± 0.77 | 0.03 ± 0.03 | 11.51 ± 4.27 | 6.25 ± 6.04 |
| Flavobacterium | 0.10 ± 0.05 | 0.84 ± 0.49 | 2.23 ± 2.12 | 2.16 ± 1.07 | 5.70 ± 5.26 |
| Tenacibaculum | 0.39 ± 0.13 | 0.50 ± 0.15 | 3.44 ± 1.96 | 0.81 ± 0.24 | 2.77 ± 2.18 |
| Phylum Kiritimatiellaeota | |||||
| WCHB1-41; unclassified | 15.45 ± 6.13 | 8.56 ± 4.26 | 0.69 ± 0.61 | 0.02 ± 0.02 | 0.03 ± 0.03 |
| Phylum Proteobacteria | |||||
| Rhodobacteraceae; unclassified | 1.69 ± 0.98 | 1.08 ± 0.24 | 8.18 ± 1.63 | 4.45 ± 1.32 | 3.47 ± 1.41 |
| Desulfovibrio | 2.76 ± 0.65 | 1.54 ± 0.59 | 0.37 ± 0.15 | 0.07 ± 0.07 | 0.08 ± 0.08 |
| Gammaproteobacteria; unclassified | 11.03 ± 5.02 | 22.69 ± 6.90 | 13.83 ± 2.50 | 3.33 ± 2.01 | 2.96 ± 2.10 |
| Aeromonadales; unclassified | 0.19 ± 0.19 | 4.74 ± 4.35 | ND | 0.01 ± 0.01 | ND |
| Aeromonas | 3.56 ± 3.46 | 0.01 ± 0.01 | 0.44 ± 0.44 | ND | 0.03 ± 0.03 |
| Colwellia | 0.21 ± 0.20 | 0.04 ± 0.03 | 0.32 ± 0.18 | 0.81 ± 0.40 | 3.29 ± 2.94 |
| Pseudoalteromonas | 2.16 ± 1.55 | 2.94 ± 1.12 | 1.70 ± 1.04 | 19.52 ± 7.69 | 3.13 ± 2.19 |
| Shewanella | 1.54 ± 0.55 | 7.15 ± 2.07 | 1.76 ± 1.74 | 3.86 ± 2.07 | 0.77 ± 0.68 |
| Halieaceae; unclassified | 0.07 ± 0.06 | 0.03 ± 0.02 | 2.16 ± 0.62 | 1.68 ± 0.92 | 0.02 ± 0.02 |
| Bermanella | ND | ND | ND | ND | 6.20 ± 6.20 |
| Pseudomonas | 0.68 ± 0.33 | 1.63 ± 0.81 | 2.95 ± 2.95 | 5.82 ± 1.99 | 14.10 ± 7.58 |
| Vibrio | 7.24 ± 3.09 | 3.07 ± 1.01 | 1.40 ± 0.50 | 8.43 ± 4.17 | 1.09 ± 0.44 |
| Phylum Spirochaetes | |||||
| Treponema 2 | 3.12 ± 2.29 | 2.22 ± 1.02 | 0.12 ± 0.11 | ND | ND |
Values represent mean percentage ± SE, with mean values above 2% in bold
ND not detected
Cloacal samples exhibited two core ASVs (present in more than 85% of samples (12/15)); Kiritimatiellae WCHB1-41 and Treponema 2. Oral samples did not show any core ASVs at 85% cutoff, but at 80% (8/11 samples) four putative core ASVs were detected, belonging to Gammaproteobacteria, Rhodobacteraceae, Pseudoalteromonas, and Halieaceae.
Core taxa collapsed to genus level (present in more than 85% of samples per sampling site) for cloacal samples consisted of uncultured WCHB-41, Desulfovibrio, Bacteroides, Shewanella, Treponema 2, Psychrobacter, uncultured Cardiobacteriaceae, uncultured Rikenellaceae, uncultured Clostridiales vadin BB60 group, and unassigned Lachnospiraceae. Oral samples putative core genera were Tenacibaculum, Flavobacterium, and unclassified Halieaceae. Genera present both in cloacal and oral samples are Vibrio, Marinifilum, Fusibacter and Arcobacter (see Additional file 3).
Discussion
The results of our research on the microbiota of loggerhead sea turtles show that oral and cloacal microbial communities differ, and that oral microbial assemblages are less stable than cloacal in regard to the turtles’ changing environment (wild versus veterinary clinic enclosures). We provide the first insights into oral bacterial communities of incidentally caught wild loggerhead sea turtles and deliver information on how the oral microbiome might respond to short-term rehabilitation in the recovery rescue centers. While most previous studies from the Mediterranean were based on gut microbiome from sick turtles found stranded or dead [31–33] this paper mostly encompasses loggerheads from the wild, incidentally caught during fishing activities. Thus, we consider microbial communities in samples taken prior to admission to the rescue center or clinic as a close representative of the wild microbiome, comparable to recent studies on wild, nesting, adult loggerhead females intestinal microbiome [30, 34]. Only two turtles in this study had to be hospitalized for longer periods due to head injuries (turtle code ID010) or leeches parasitization (turtle code ID023). On the other hand, oral microbiota of sea turtles has not yet been explored by 16S rRNA gene sequencing, while it has been investigated in the freshwater Krefft’s river turtle (Emydura macquarii krefftii) and pond slider turtle (Trachemys s. scripta) [56, 57].
In our study, alpha diversity metrics did not show significant differences between oral and cloacal body sites or sampling periods (before and during rehabilitation). This could be explained by the size of our target population (relatively small), with juveniles and adults of similar nutritional status, which is insufficient for discovering potential characteristics that could be associated with microbial diversity of samples on this level. In oral microbiomes, ID023 turtle sample is an outlier with much higher microbial diversity, which may be linked with its rehabilitation in the WWF care facility where it was undergoing freshwater treatment for leeches removal prior to admission to the rescue center where it was sampled. Tank water samples from the Aquarium Pula local circulating seawater showed much higher diversity with frequent marine microbial taxa, in comparison to water from the STC in Bari that harbored non-circulating artificial saltwater. Further, aquarium seawater tank exhibited a similar trait to seawater samples in a study by Biagi et al. [33], having a higher diversity of low abundance phyla. Aquarium tank water also had higher abundances of phyla Planctomycetes and Patescibacteria which were observed mostly in oral samples before hospitalization. Therefore, the aquarium recirculating tank water could present a more “natural” marine habitat rather than the tanks with artificial seawater.
Beta diversity metrics consistently showed separation of cloacal and oral microbiomes but with different significance detection between sampling period depending on the metric tested by PERMANOVA. Beta diversity measures used in most sea turtle microbiome studies are still Bray–Curtis dissimilarity, unweighted, and weighted Unifrac even though they do not account for the compositionality of microbiome datasets obtained by sequencing [51]. Due to data compositionality, we decided on presenting already widely accepted beta diversity metrics PCoA along with the robust Aitchison distance PCA,argued to be a better choice for compositional data [52, 58]. Our combined results indicate strong differences between wild cloacal microbiota versus both oral sample periods. Moreover, no significant differences were found among tank water and oral rehabilitated microbiota, which emphasizes the impact of the environment on oral microbiota of loggerhead sea turtles.
Reptile oral microbiomes were considered to be influenced by the prey fecal microbiome but Zancolli et al. [57] observed distinct species-specific patterns in snakes and freshwater turtles that undermine the assumption that reptiles’ oral cavity is a passive reservoir of microbes. As sea turtles are mostly submerged and in close contact with the water medium (sea), we hypothesized that oral microbiome would resemble the environment. As expected, oral samples clustered based on sampling period with samples before rehabilitation clustered closer to the aquarium free-circulating tank water while oral rehabilitated clustered closer to tank water of enclosures with non-filtered artificial seawater (Bray–Curtis and unweighted Unifrac PCoA). No significant differences were observed between oral and tank water samples, but specific bacterial taxa not found in tank water suggest that the oral microbiome consists, at least partially, of endogenous and transitional microbes from the environment. Proteobacteria and Bacteroidetes, abundant in oral microbiota in our study, were also dominantly present in the oral microbiome of Krefft’s river turtle, which was markedly different from the external turtle microbiome and the environment [56]. Comparisons beyond phylum level show that Krefft’s river turtle and pond slider turtle share Burkholderiaceae and Weeksellaceae families not detected in our study [56, 57] while Flavobacteriaceae are shared between Kreffts and loggerheads.
In our study, we detected high abundances of ASVs which could not be classified to genera but only to higher taxonomic ranks: Bacteroidales, Flavobacteriaceae, Rhodobacteraceae, and Gammaproteobacteria. Highly abundant oral ASVs often overlapped in classification with highly abundant taxa in water tanks, but the actual taxonomic diversity between those groups remains to be determined as overly unclassified reads could implicate a high incidence of yet undiscovered bacteria, or insufficient sequence length required for successful taxonomical identification. Interestingly, the genus Pseudoalteromonas was more abundant in oral microbiomes of rehabilitated turtles, while unclassified Halieaceae were more abundant in oral microbiomes before rehabilitation than in any other sample type. Halieaceae are often found in coastal, neritic environment, deep-sea waters, and in demersal animals (e.g. sponges) [59, 60], hence, they could be easily transported from the marine environment and into the oral cavity. Pseudoalteromonas spp. are marine bacteria known for production of antimicrobial substances with many of the species found in association with marine eukaryotes [61] which has been proposed as beneficial to its marine hosts [62]. It is possible that the low abundance taxa in wild oral microbiota are enriched by the veterinary clinic’s enclosure environment conditions; temperature and nutrient availability are relatively stable in comparison to the turtle’s natural habitat. Other taxa that had higher abundances were also notably present in cloacal (Vibrio spp., Shewanella spp.) or tank water samples (Pseudomonas spp., Bizionia spp.), which could be transient and non-specific for oral microbiome. At this point, little data are available to compare aquatic turtles’ oral microbiomes beyond the superficial taxonomic levels, and according to our results habitat has a significant effect on the sea turtle oral microbiota. Additional sampling across many different groups of turtles and their habitats would be needed to decouple the effects of the habitat from the intrinsic and possibly representative oral microbes. Even though effects of oral microbial communities on the host have been described in humans and other mammals, it is unknown what roles reptile microbiome may have, especially in marine species [15, 63].
Cloacal microbiome samples did not show any significant clustering of different sample traits in our study design, which is consistent with previous reports [31, 32], but there have been reports on effects of the CCL on cloacal microbiota clustering [33]. As sea turtles often exhibit ontogenetic habitat shift and transit from pelagic to neritic prey, the change in the microbiota regarding to the size and age of the individual could be explained by changed preferences in habitat and food. In juvenile green turtles, there is a significant variation in cloacal microbiomes between pelagic and neritic habitats and transition to herbivorous lifestyle [25]. Additionally, green turtles in rescue centers exhibit a microbiome shift depending on the type of food they receive during rehabilitation, where the fecal microbiome constitutes of bacterial communities prepped for higher protein content as the recovering turtles are fed mostly seafood, but the community shifts to communities known for metabolization of plant polysaccharide upon introduction of plant food near the end of the recovery [24]. The developmental shift from pelagic to neritic habitats of loggerheads in the central Mediterranean Sea is more relaxed, where juveniles have a short epipelagic stage but later choose the habitat opportunistically, according to food availability and oceanographic features [64]. Consequently, shallow north-central Adriatic Sea enables early recruitment to the neritic habitats where rich and diverse benthic pray is available even to small juveniles (< 30 cm) [64]. Presented microbiome of Adriatic Loggerheads seems to confine with satellite tracking and tagging studies that suggest long-term residence of both adults and juveniles in the shallow neritic Adriatic, with seasonal migrations along the Italian coast to the south during winter [65]. Hence, the differences observed in fecal, cloacal and intestinal microbial communities between loggerheads sampled in the central Mediterranean [31, 32], Australia, or USA [30, 34], may be partially explained by highly opportunistic feeding nature and food availability for sampled turtle populations.
The most comprehensive loggerhead microbiota studies from geographically and genetically distinct healthy nesting females [30, 34], usually linked to neritic feeding grounds, reported that microbial communities of the sea turtle gut are dominated by Proteobacteria, followed by Spirochaetes, Bacteroidetes and Actinobacteria. This coincides with our results on wild and early rehabilitation microbial profiles of cloacal samples. On the other hand, microbial communities dominated by higher proportions of Fusobacteria, Firmicutes, and Bacteroidetes with low abundance of Proteobacteria may be considered atypical and describe fecal microbiota of rehabilitated or stranded turtles, connected with the turtle health status [31–33].
The only study on loggerhead microbiome from the Adriatic Sea [33] on fecal microbial communities of stranded or turtles captured in trawling nets showed high abundance of Firmicutes and Fusobacteria, while Bacteroidetes and Proteobacteria were not pronounced. A significant portion of microbial taxa they reported belonged to Cetobacterium and Clostridium genera, which were not observed in our study. Since these Adriatic loggerheads shared a similar ecological niche and foraging habitats, described non-Proteobacteria dominated microbiome [33] probably arises from their health status, changes in immunity, rehabilitation treatments, recorded period of starvation, and sampling feces rather than the intestine or cloaca. In our study, we detected two putative core cloacal ASVs belonging to WCHB1-41 and Treponema 2; while uncultured Clostridiales and Lachnospiraceae were detected as putative core taxa and were not overly abundant. Within phylum Bacteroidetes, major components were Bacteroides, which have been observed in loggerhead fecal microbiomes [32, 33] and mammalian microbiome [66], Marinifilum spp. (commonly found in seawater), unclassified Rikenellaceae (specialized for the digestive tract of different animals) [67] and unclassified Flavobacteriaceae found in a wide variety of habitats. Interestingly, a major proportion of reads in cloacal samples belonged to the novel Kiritimatiellaeota phylum (formerly in Verrucomicrobia) and were identified as uncultured eubacterium WCHB1-41 [68]. Uncultured WCHB1-41 have been found in equine vaginal and distal gut microbiome, and rumen of cattle [69–71]. Verrucomicrobia have been found in loggerhead cloacal and fecal samples [33, 34] and it is possible that at least a portion of Verrucomicrobia reads would be classified as Kiritimatiellaeota if SILVA v.132 was used to assign the taxonomy, as in this study and study by Arizza et al. [32].
When discussing the representative microbiome of the turtle gut, it is important to discern between the fecal microbiome that is often affected by food composition [24] and is a better descriptor of gut lumen microbiome, versus the microbial communities attached to the mucosa and in direct contact with the host, which might or might not be influenced by the shifts in habitat, environment and food type availability [72]. In our study, we used swabs for both oral and cloacal samples rather than feces, as collecting swab samples is less time-consuming in comparison to collecting fecal samples, relatively non-invasive to the turtle and may be performed during fieldwork or within rescue centers. Our results show that cloacal swabs might be sufficient to describe microbial communities as a proxy to feces and intestinal samples, which would allow for wider and less invasive sampling of loggerheads. Sampling wild microbial communities of loggerheads (among other sea turtles and reptiles in general) is necessary to gain basic insights into reptile microbiomes. A recent study in bacterial communities of wild animals via de-novo metagenome assembly showed that wild microbiomes are a resource for novel bacterial species and biological functions [17]. Furthermore, when identifying unknown bacterial genomes of Reptilia microbiota consisted predominantly of novel microbial members and are under sampled in most meta-microbiome studies [6, 9, 17]. Higher abundances of unclassified members of Proteobacteria, Bacteroidetes and other phyla might then prove to be reservoirs of novel bacterial species with interesting features.
Microbial community studies should inform conservation efforts and rehabilitation facilities in ways to improve treatments, housing conditions, and preparation for the release of rehabilitated turtles. In this study, we show that the oral microbiota is potentially less stable and more prone to the acquisition of external microbial taxa in comparison to the relatively stable cloacal microbiota. Implications and effects of long-term rehabilitation of turtles in tanks with non-circulating artificial seawater on the turtles are still unknown and should be investigated further. Due to the sensitivity of oral microbiota to external conditions it should be noted that local circulating seawater should be preferred in rescue centers whenever possible, to preserve and enrich bacterial communities.
Conclusions
Our work provided the first insights into oral and cloacal microbiota of incidentally caught and mostly healthy loggerhead sea turtles before admission to the rescue center or clinic and after rehabilitation. Other studies focused on hospitalized, dead, and stranded Mediterranean loggerheads [31–33] while our research provided mostly healthy, wild microbiota information as in recent studies on nesting female loggerheads [30, 34]. We showed that cloacal microbiota remains relatively stable during short-term hospitalization, which has been shown in previous studies. Even though loggerhead oral microbial communities do not completely resemble the microbiota of the turtle's environment, they are dynamic and change swiftly as they accommodate taxa from a new environment. Furthermore, cloacal and oral swabs are sufficient for description of microbial communities of loggerheads and allow quick and non-invasive sampling. As reptile microbial communities are still less investigated, wild sea turtle microbiota characterization provides essential information for the expansion of our knowledge on sea turtle biology and guidelines on how to improve on the conservation efforts for these vulnerable, and highly important keystone species in marine ecosystems.
Supplementary Information
Additional file 1: Table S1. Sequencing results (raw counts) and taxonomy assignments per ASV for all samples in this study.
Additional file 2: Figure S1. Oral (A) and cloacal (B) sampling of loggerhead sea turtle at the Sea Turtle Clinic of the Department of Veterinary Medicine of University of Bari (Italy). Courtesy of Adriana Trotta. Table S2. Alpha diversity measures (Shannon’s diversity, observed ASVs. Faith’s Phylogenetic Diversity) for cloacal, oral and tank water sampling sites with Kruskal–Wallis H test results. Values are represented as mean ± SD, with significance level α < 0.05. Table S3. A comparison of differences in microbial communities of different sampling sites and periods by pairwise PERMANOVA for Bray–Curtis, Robust Aitchison, unweighted and weighted UniFrac distance metrics. SampSling sites and periods are marked as follows: CB, cloacal before; CR, cloacal rehabilitated; OB, oral before; OR, oral rehabilitated; W, tank water. P-values shown have been FDR corrected. Significance levels are indicated by an asterisk: p ≤ 0.05*, p ≤ 0.01** with all significant values bolded.
Additional file 3: Table S4. Core taxa of cloacal and oral microbiota at ASV level (85% and 80% cutoff, respectably) and collapsed to the genus level (85% cutoff).
Acknowledgements
For the Croatian sample collection, we are thankful to Milena Mičić, Karin Gobić Medica and the rest of the staff from the Marine Turtle Rescue Center (Aquarium Pula).
Abbreviations
- ASV
Amplicon sequence variants
- CB
Cloacal before
- CCL
Curved carapace length
- CR
Cloacal rehabilitated
- DNA
Deoxyribonucleic acid
- IUCN
International Union for Conservation of Nature
- OB
Oral before
- OR
Oral rehabilitated
- PCoA
Principal coordinate analysis
- rPCA
Robust Principal component analysis
- rRNA
Ribosomal ribonucleic acid
- STC
Sea Turtle Clinic
- USA
United States of America
- W
Tank water
- WWF
World Wildlife Fund
Authors' contributions
AT, RG, AB, MC, and SB concepted and designed the study; AT and SB collected the samples; AT and KF carried out the laboratory work; KF conducted the bioinformatics, statistical analyses, and data visualization and interpretation; KF wrote the first draft of the manuscript; All authors revised the paper and approved the final version of the manuscript.
Funding
This work has been fully supported by the Croatian Science Foundation under the project number UIP-2017-05-5635. The work of doctoral student K. Filek has been fully supported by the “Young Researchers’ Career Development Project – Training of Doctoral Students” of the Croatian Science Foundation funded by the European Union from the European Social Fund.
Availability of data and materials
The amplicon sequencing data obtained in this study are available in the European Nucleotide Archive under accession number PRJEB46638. Additional information about data analysis and extended metadata are available in Mendeley Data repository (10.17632/45skyv5hzy.1).
Declarations
Ethical approval and Consent to participate
Sampling was performed in accordance with the 1975 Declaration of Helsinki, as revised in 2013 and the applicable national laws. The sampling at the Sea Turtle Clinic (Bari, Italy) was conducted with the permission of the Department of Veterinary Medicine Animal Ethic Committee (Authorization # 4/19), while sampling in Croatia was done in accordance with the authorization of the Marine Turtle Rescue Center by the Ministry of Environment and Energy of the Republic of Croatia.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Klara Filek, Email: klara.filek@biol.pmf.hr.
Adriana Trotta, Email: adriana.trotta@uniba.it.
Romana Gračan, Email: romana.gracan@biol.pmf.hr.
Antonio Di Bello, Email: antonio.dibello@uniba.it.
Marialaura Corrente, Email: marialaura.corrente@uniba.it.
Sunčica Bosak, Email: suncica.bosak@biol.pmf.hr.
References
- 1.McFall-Ngai M, Hadfield MG, Bosch TC, Carey HV, Domazet-Lošo T, Douglas AE, et al. Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci. 2013;110:3229–3236. doi: 10.1073/pnas.1218525110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol. 2008;6:776–788. doi: 10.1038/nrmicro1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilbert J, Blaser MJ, Caporaso GJ, Jansson J, Lynch SV, Knight R. Current understanding of the human microbiome. Nat Med. 2018;24:392–400. doi: 10.1038/nm.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg E, Zilber-Rosenberg I. The hologenome concept of evolution after 10 years. Microbiome. 2018;6:78. doi: 10.1186/s40168-018-0457-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moran NA, Sloan DB. The hologenome concept: Helpful or hollow? PLoS Biol. 2015;13:e1002311. doi: 10.1371/journal.pbio.1002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colston TJ, Jackson CR. Microbiome evolution along divergent branches of the vertebrate tree of life: what is known and unknown. Mol Ecol. 2016;25:3776–3800. doi: 10.1111/mec.13730. [DOI] [PubMed] [Google Scholar]
- 7.Trevelline BK, Fontaine SS, Hartup BK, Kohl KD. Conservation biology needs a microbial renaissance: a call for the consideration of host-associated microbiota in wildlife management practices. Proc R Soc B Biol Sci. 2019;286:20182448. doi: 10.1098/rspb.2018.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.West AG, Waite DW, Deines P, Bourne DG, Digby A, McKenzie VJ, et al. The microbiome in threatened species conservation. Biol Conserv. 2019;229:85–98. doi: 10.1016/j.biocon.2018.11.016. [DOI] [Google Scholar]
- 9.Youngblut ND, Reischer GH, Walters W, Schuster N, Walzer C, Stalder G, et al. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat Commun. 2019;10:2200. doi: 10.1038/s41467-019-10191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song SJ, Sanders JG, Delsuc F, Metcalf J, Amato K, Taylor MW, et al. Comparative Analyses of Vertebrate Gut Microbiomes Reveal Convergence between Birds and Bats. MBio. 2020;11:e02901–e2919. doi: 10.1128/mBio.02901-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apprill A. Marine animal microbiomes: toward understanding host-microbiome interactions in a changing ocean. Front Mar Sci. 2017;4:222. doi: 10.3389/fmars.2017.00222. [DOI] [Google Scholar]
- 12.Miller CA, Holm HC, Horstmann L, George JC, Fredricks HF, Van Mooy BAS, et al. Coordinated transformation of the gut microbiome and lipidome of bowhead whales provides novel insights into digestion. ISME J. 2020;14:688–701. doi: 10.1038/s41396-019-0549-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Apprill A, Miller CA, Moore MJ, Durban JW, Fearnbach H, Barrett-Lennard LG. Extensive core microbiome in drone-captured whale blow supports a framework for health monitoring. mSystems. 2017;2:e00119–e1117. doi: 10.1128/mSystems.00119-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soares-Castro P, Araújo-Rodrigues H, Godoy-Vitorino F, Ferreira M, Covelo P, López A, et al. Microbiota fingerprints within the oral cavity of cetaceans as indicators for population biomonitoring. Sci Rep. 2019;9:1–15. doi: 10.1038/s41598-019-50139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudek NK, Sun CL, Burstein D, Kantor RS, Aliaga Goltsman DS, Bik EM, et al. Novel microbial diversity and functional potential in the marine mammal oral microbiome. Curr Biol. 2017;27:3752–3762.e6. doi: 10.1016/j.cub.2017.10.040. [DOI] [PubMed] [Google Scholar]
- 16.Hooper R, Brealey JC, van der Valk T, Alberdi A, Durban JW, Fearnbach H, et al. Host-derived population genomics data provides insights into bacterial and diatom composition of the killer whale skin. Mol Ecol. 2019;28:484–502. doi: 10.1111/mec.14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levin D, Raab N, Pinto Y, Rothschild D, Zanir G, Godneva A, et al. Diversity and functional landscapes in the microbiota of animals in the wild. Science. 2021;372:eabb552. doi: 10.1126/science.abb5352. [DOI] [PubMed] [Google Scholar]
- 18.Lovich JE, Ennen JR, Agha M, Gibbons JW. Where have all the turtles gone, and why does it matter? Bioscience. 2018;68:771–781. doi: 10.1093/biosci/biy095. [DOI] [Google Scholar]
- 19.IUCN 2020. IUCN Red List of Threatened Species. https://www.iucnredlist.org. Accessed 24 Feb 2021.
- 20.Ullmann J, Stachowitsch M. A critical review of the Mediterranean sea turtle rescue network: a web looking for a weaver. Nat Conserv. 2015;10:45–59. doi: 10.3897/natureconservation.10.4890. [DOI] [Google Scholar]
- 21.Ahasan MS, Waltzek TB, Huerlimann R, Ariel E. Fecal bacterial communities of wild-captured and stranded green turtles (Chelonia mydas) on the Great Barrier Reef. FEMS Microbiol Ecol. 2017;93:1–11. doi: 10.1093/femsec/fix139. [DOI] [PubMed] [Google Scholar]
- 22.McDermid KJ, Kittle RP, Veillet A, Plouviez S, Muehlstein L, Balazs GH. Identification of gastrointestinal microbiota in Hawaiian green turtles (Chelonia mydas) Evol Bioinforma. 2020;16:1176934320914603. doi: 10.1177/1176934320914603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahasan MS, Waltzek TB, Huerlimann R, Ariel E. Comparative analysis of gut bacterial communities of green turtles (Chelonia mydas) pre-hospitalization and post-rehabilitation by high-throughput sequencing of bacterial 16S rRNA gene. Microbiol Res. 2018;207:91–99. doi: 10.1016/j.micres.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 24.Bloodgood JCG, Hernandez SM, Isaiah A, Suchodolski JS, Hoopes LA, Thompson PM, et al. The effect of diet on the gastrointestinal microbiome of juvenile rehabilitating green turtles (Chelonia mydas) PLoS ONE. 2020;15:e022060. doi: 10.1371/journal.pone.0227060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price JT, Paladino FV, Lamont MM, Witherington BE, Bates ST, Soule T. Characterization of the juvenile green turtle (Chelonia mydas) microbiome throughout an ontogenetic shift from pelagic to neritic habitats. PLoS ONE. 2017;12:1–13. doi: 10.1371/journal.pone.0177642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campos P, Guivernau M, Prenafeta-Boldú FX, Cardona L. Fast acquisition of a polysaccharide fermenting gut microbiome by juvenile green turtles Chelonia mydas after settlement in coastal habitats. Microbiome. 2018;6:69. doi: 10.1186/s40168-018-0454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahasan MS, Waltzek TB, Owens L, Ariel E. Characterisation and comparison of the mucosa-associated bacterial communities across the gastrointestinal tract of stranded green turtles Chelonia mydas. AIMS Microbiol. 2020;6:361–378. doi: 10.3934/microbiol.2020022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samuelson MM, Pulis EE, Ray C, Arias CR, Samuelson DR, Mattson EE, et al. Analysis of the fecal microbiome in Kemp’s ridley sea turtles Lepidochelys kempii undergoing rehabilitation. Endanger Species Res. 2020;43:121–131. doi: 10.3354/esr01043. [DOI] [Google Scholar]
- 29.Scheelings TF, Moore RJ, Van TTH, Klaassen M, Reina RD. No correlation between microbiota composition and blood parameters in nesting flatback turtles (Natator depressus) Sci Rep. 2020;10:8333. doi: 10.1038/s41598-020-65321-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheelings TF, Moore RJ, Van TTH, Klaassen M, Reina RD. The gut bacterial microbiota of sea turtles differs between geographically distinct populations. Endanger Species Res. 2020;42:95–108. doi: 10.3354/esr01042. [DOI] [Google Scholar]
- 31.Abdelrhman KFA, Bacci G, Mancusi C, Mengoni A, Serena F, Ugolini A. A first insight into the gut microbiota of the sea turtle Caretta caretta. Front Microbiol. 2016;7:1–5. doi: 10.3389/fmicb.2016.01060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arizza V, Vecchioni L, Caracappa S, Sciurba G, Berlinghieri F, Gentile A, et al. New insights into the gut microbiome in loggerhead sea turtles Caretta caretta stranded on the Mediterranean coast. PLoS ONE. 2019;14:e0220329. doi: 10.1371/journal.pone.0220329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biagi E, D’Amico F, Soverini M, Angelini V, Barone M, Turroni S, et al. Faecal bacterial communities from Mediterranean loggerhead sea turtles (Caretta caretta) Environ Microbiol Rep. 2019;11:361–371. doi: 10.1111/1758-2229.12683. [DOI] [PubMed] [Google Scholar]
- 34.Scheelings TF, Moore RJ, Van TTH, Klaassen M, Reina RD. Microbial symbiosis and coevolution of an entire clade of ancient vertebrates: the gut microbiota of sea turtles and its relationship to their phylogenetic history. Anim Microbiome. 2020;2:17. doi: 10.1186/s42523-020-00034-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pace A, Dipineto L, Fioretti A, Hochscheid S. Loggerhead sea turtles as sentinels in the western Mediterranean: antibiotic resistance and environment-related modifications of Gram-negative bacteria. Mar Pollut Bull. 2019;149:110575. doi: 10.1016/j.marpolbul.2019.110575. [DOI] [PubMed] [Google Scholar]
- 36.Alduina R, Gambino D, Presentato A, Gentile A, Sucato A, Savoca D, et al. Is Caretta Caretta a carrier of antibiotic resistance in the Mediterranean Sea? Antibiotics. 2020;9:116. doi: 10.3390/antibiotics9030116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trotta A, Cirilli M, Marinaro M, Bosak S, Diakoudi G, Ciccarelli S, et al. Detection of multi-drug resistance and AmpC β-lactamase/extended-spectrum β-lactamase genes in bacterial isolates of loggerhead sea turtles (Caretta caretta) from the Mediterranean Sea. Mar Pollut Bull. 2021;164:112015. doi: 10.1016/j.marpolbul.2021.112015. [DOI] [PubMed] [Google Scholar]
- 38.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andrews S. FastQC: a quality control tool for high throughput sequencing data. 2010. https://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed 27 Mar 2020.
- 40.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Callahan BJ, McMurdie PJ, Holmes SP. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017;11:2639–2643. doi: 10.1038/ismej.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katoh K. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Price MN, Dehal PS, Arkin AP. FastTree 2 - Approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6:90. doi: 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011;5:169–172. doi: 10.1038/ismej.2010.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiss S, Xu ZZ, Peddada S, Amir A, Bittinger K, Gonzalez A, et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome. 2017;5:27. doi: 10.1186/s40168-017-0237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lozupone C, Knight R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lozupone CA, Hamady M, Kelley ST, Knight R. Quantitative and qualitative β diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol. 2007;73:1576–1585. doi: 10.1128/AEM.01996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome datasets are compositional: and this is not optional. Front Microbiol. 2017;8:1–6. doi: 10.3389/fmicb.2017.02224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martino C, Morton JT, Marotz CA, Thompson LR, Tripathi A, Knight R, et al. A novel sparse compositional technique reveals microbial perturbations. mSystems. 2019;4:1–13. doi: 10.1128/mSystems.00016-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fedarko MW, Martino C, Morton JT, González A, Rahman G, Marotz CA, et al. Visualizing ’omic feature rankings and log-ratios using Qurro. NAR Genom Bioinform. 2020;2:lqa023. doi: 10.1093/nargab/lqaa023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wickham, H. ggplot2: elegant graphics for data analysis. Springer, New York; 2016. https://ggplot2.tidyverse.org. Accessed 21 Jul 2020.
- 55.Vázquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. GigaScience. 2013;2:16. doi: 10.1186/2047-217X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McKnight DT, Zenger KR, Alford RA, Huerlimann R. Microbiome diversity and composition varies across body areas in a freshwater turtle. Microbiology. 2020;166:440–452. doi: 10.1099/mic.0.000904. [DOI] [PubMed] [Google Scholar]
- 57.Zancolli G, Mahsberg D, Sickel W, Keller A. Reptiles as reservoirs of bacterial infections: Real threat or methodological bias? Microb Ecol. 2015;70:579–584. doi: 10.1007/s00248-015-0618-3. [DOI] [PubMed] [Google Scholar]
- 58.Gloor GB, Reid G. Compositional analysis: a valid approach to analyze microbiome high-throughput sequencing data. Can J Microbiol. 2016;62:692–703. doi: 10.1139/cjm-2015-0821. [DOI] [PubMed] [Google Scholar]
- 59.Suzuki T, Yazawa T, Morishita N, Maruyama A, Fuse H. Genetic and physiological characteristics of a novel marine propylene-assimilating Halieaceae bacterium isolated from seawater and the diversity of its alkene and epoxide metabolism genes. Microb Environ. 2019;34:33–42. doi: 10.1264/jsme2.ME18053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Podell S, Blanton JM, Oliver A, Schorn MA, Agarwal V, Biggs JS, et al. A genomic view of trophic and metabolic diversity in clade-specific Lamellodysidea sponge microbiomes. Microbiome. 2020;8:97. doi: 10.1186/s40168-020-00877-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holmström C, Kjelleberg S. Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol Ecol. 1999;30:285–293. doi: 10.1016/S0168-6496(99)00063-X. [DOI] [PubMed] [Google Scholar]
- 62.Offret C, Desriac F, Le Chevalier P, Mounier J, Jégou C, Fleury Y. Spotlight on antimicrobial metabolites from the marine bacteria Pseudoalteromonas: chemodiversity and ecological significance. Mar Drugs. 2016;14:129. doi: 10.3390/md14070129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. 2018;16:745–759. doi: 10.1038/s41579-018-0089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Casale P, Abbate G, Freggi D, Conte N, Oliverio M, Argano R. Foraging ecology of loggerhead sea turtles Caretta caretta in the central Mediterranean Sea: evidence for a relaxed life history model. Mar Ecol Prog Ser. 2008;372:265–276. doi: 10.3354/meps07702. [DOI] [Google Scholar]
- 65.Luschi P, Casale P. Movement patterns of marine turtles in the Mediterranean Sea: a review. Ital J Zool. 2014;81:478–495. doi: 10.1080/11250003.2014.963714. [DOI] [Google Scholar]
- 66.Wexler AG, Goodman AL. An insider’s perspective: Bacteroides as a window into the microbiome. Nat Microbiol. 2017;2:1–11. doi: 10.1038/nmicrobiol.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Graf J. The Family Rikenellaceae. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, editors. The prokaryotes: other major lineages of bacteria and the Archaea. Berlin: Springer; 2014. pp. 857–859. [Google Scholar]
- 68.Spring S, Bunk B, Spröer C, Schumann P, Rohde M, Tindall BJ, et al. Characterization of the first cultured representative of Verrucomicrobia subdivision 5 indicates the proposal of a novel phylum. ISME J. 2016;10:2801–2816. doi: 10.1038/ismej.2016.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barba M, Martínez-Boví R, Quereda JJ, Mocé ML, Plaza-Dávila M, Jiménez-Trigos E, et al. Vaginal microbiota is stable throughout the estrous cycle in arabian mares. Animals. 2020;10:2020. doi: 10.3390/ani10112020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edwards JE, Shetty SA, van den Berg P, Burden F, van Doorn DA, Pellikaan WF, et al. Multi-kingdom characterization of the core equine fecal microbiota based on multiple equine (sub)species. Anim Microbiome. 2020;2:1–16. doi: 10.1186/s42523-020-0023-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ozutsumi Y, Tajima K, Takenaka A, Itabashi H. The effect of protozoa on the composition of rumen bacteria in cattle using 16S rRNA gene clone libraries. Biosci Biotechnol Biochem. 2005;69:499–506. doi: 10.1271/bbb.69.499. [DOI] [PubMed] [Google Scholar]
- 72.Ingala MR, Simmons NB, Wultsch C, Krampis K, Speer KA, Perkins SL. Comparing microbiome sampling methods in a wild mammal: fecal and intestinal samples record different signals of host ecology, evolution. Front Microbiol. 2018;9:803. doi: 10.3389/fmicb.2018.00803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Sequencing results (raw counts) and taxonomy assignments per ASV for all samples in this study.
Additional file 2: Figure S1. Oral (A) and cloacal (B) sampling of loggerhead sea turtle at the Sea Turtle Clinic of the Department of Veterinary Medicine of University of Bari (Italy). Courtesy of Adriana Trotta. Table S2. Alpha diversity measures (Shannon’s diversity, observed ASVs. Faith’s Phylogenetic Diversity) for cloacal, oral and tank water sampling sites with Kruskal–Wallis H test results. Values are represented as mean ± SD, with significance level α < 0.05. Table S3. A comparison of differences in microbial communities of different sampling sites and periods by pairwise PERMANOVA for Bray–Curtis, Robust Aitchison, unweighted and weighted UniFrac distance metrics. SampSling sites and periods are marked as follows: CB, cloacal before; CR, cloacal rehabilitated; OB, oral before; OR, oral rehabilitated; W, tank water. P-values shown have been FDR corrected. Significance levels are indicated by an asterisk: p ≤ 0.05*, p ≤ 0.01** with all significant values bolded.
Additional file 3: Table S4. Core taxa of cloacal and oral microbiota at ASV level (85% and 80% cutoff, respectably) and collapsed to the genus level (85% cutoff).
Data Availability Statement
The amplicon sequencing data obtained in this study are available in the European Nucleotide Archive under accession number PRJEB46638. Additional information about data analysis and extended metadata are available in Mendeley Data repository (10.17632/45skyv5hzy.1).