

Abstract
The lack of efficient [18F]fluorination processes and target-specific organofluorine chemotypes remains the major challenge of fluorine-18 positron emission tomography (PET). We report here an ultrafast isotopic exchange method for the radiosynthesis of novel PET agent aryl [18F]fluorosulfate enabled by the emerging sulfur fluoride exchange (SuFEx) click chemistry. The method has been applied to the fully-automated 18F-radiolabeling of twenty-five structurally and functionally diverse aryl fluorosulfates with excellent radiochemical yield (83–100%, median 98%) and high molar activity (280 GBq µmol−1) at room temperature in 30 seconds. The purification of radiotracers requires no time-consuming HPLC, but rather a simple cartridge filtration. We further demonstrate the imaging application of a rationally designed poly(ADP-ribose) polymerase 1 (PARP1)-targeting aryl [18F]fluorosulfate by probing subcutaneous tumors in vivo.
Keywords: Click Chemistry, Sulfur Fluoride Exchange, Isotopic Exchange, Radiosynthesis, Aryl Fluorosulfate
Graphical Abstract
INTRODUCTION
The non-invasive molecular imaging technique – positron emission tomography (PET), especially that based on the radionuclide fluorine-18, is widely used for tracking biological processes in vivo1–6. PET imaging has found successful clinical applications in the diagnosis of malignant tumor or neurodegenerative diseases and the efficacy evaluation of therapeutic treatment. Ever-growing fluorination methodologies over the past decades with a focus on the formation of C–18F bonds sparked expansion of the 18F-based toolbox of radiotracers7–11.
Despite a few state-of-the-art methods12–19 reported to date, harsh reaction conditions and laborious purification that a common C–18F forming process requires have, nevertheless, significantly limited the substrate scope and clinical utility. [18F]Fluorodeoxyglucose (FDG), an agent developed half a century ago to map glucose metabolism20, remains the predominant shareholder of the clinically used PET radiopharmaceuticals. As noted by Fowler, radiochemists are working against time1 due to the short half-life of fluorine-18 (109.77 min). With these challenges21, an ideal 18F-radiosynthesis should be: 1) a rapid, mild, and clean 18F-incorporation in the final step of a synthetic sequence; 2) effective for a diverse spectrum of bioactive organofluorine compounds with reasonable in vivo stability; and 3) compatible with automation. These stringent criteria mirror those set for click chemistry22.
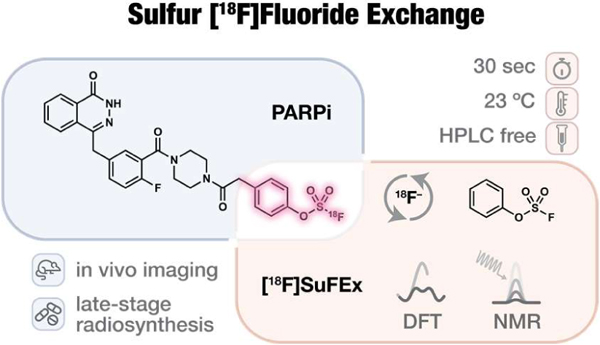
We envisioned23 that the newly developed sulfur(VI) fluoride exchange (SuFEx) reactions would naturally bridge click chemistry and 18F-radiosynthesis. As demonstrated in the first SuFEx manifesto24 and subsequent reports25–27, aryl fluorosulfate (ArOSO2F) is significantly more stable than aryl sulfonyl fluoride (krel = 0.035)25. This functional group may only become reactive in the presence of a proper catalyst in organic solvents or upon encountering a specific protein partner, if any, that possesses both a nucleophilic amino acid side chain and juxtaposed side chains, e.g. arginine to provide hydrogen bonding networks for the extraction and transport of the departing fluoride in the binding site. In most cases, nevertheless, aryl fluorosulfates simply remain intact in aqueous solutions near neutral conditions in the presence of nucleophilic amino acids, e.g. lysine, serine, threonine, and tyrosine (Figure 1b), and even in an entire proteome27. Its compatibility with most common medicinal chemistry transformations (Figure 1b) has also led to its use as a protecting group26. Importantly, our proof-of-principle pharmacokinetic evaluations have provided strong evidence of the inertness of several bioactive aryl fluorosulfates in vivo (Figure 1a)28–29.
Figure 1.
Unique properties of aryl fluorosulfates qualify them as potential radiopharmacophore. (a) In vivo validated non-covalent fluorosulfate inhibitors and strategies for their discovery or rational design. ER, estrogen receptor. hsEH, human soluble epoxide hydrolase. (S)-TPMBU, (S)-1-(1-(2-methylbutanoyl)piperidin-4-yl)-3-(4-(trifluoromethoxy)phenyl)urea. PARP, poly(ADP-ribose) polymerase. (b), Matrix showcasing fluorosulfate’s broad window of stability under the chemical stress of various conditions including the most used reactions in medicinal chemistry30. “Stable” denotes less than 10% decomposition after 2 h incubation under the indicated condition. “Conditional stable” denotes the stability has a case-by-case dependence on the substrate. “Unstable” denotes significant (> 10%) decomposition after 2 h incubation. TFA, trifluoroacetic acid. mCPBA, 3-chloroperoxybenzoic acid. (c) Empirical orbital analysis of the three strategies for nucleophilic 18F-incorporation and respective estimated17, 31 activation energies (Ea).
On the other hand, one can certainly imagine the possibility of an isotopic exchange event in which the 18F-enriched fluoride anion enables its own nucleophilic displacement of the other fluorine atom of aryl fluorosulfates32. This interchange process may actually be much less energy-demanding than a typical SuFEx reaction with amine or alcohol nucleophiles — the extraneous fluoride anion per se compensates the stringent requirements in the latter scenario for the stabilization or solvation of the departing fluoride from an SVI–F site. Significantly, compared to the traditional SN2-based C–F formation SuFEx with F− might engage the 3d-orbital of SVI, rendering a much lower kinetic barrier31, 33–37. Thus, we hypothesize that such a process may take place facilely at room temperature. Given the inertness of many aryl fluorosulfates under physiological conditions, such 18F-labeled aryl fluorosulfates may serve as ideal probes for PET imaging. Here we report the development of an ultrafast 18F-radiolabeling process of preparing aryl fluorosulfate-based probes and their application to PET imaging based on the above principles.
RESULTS
Computational evaluation of sulfur fluoride exchange between phenyl fluorosulfate and anionic fluoride.
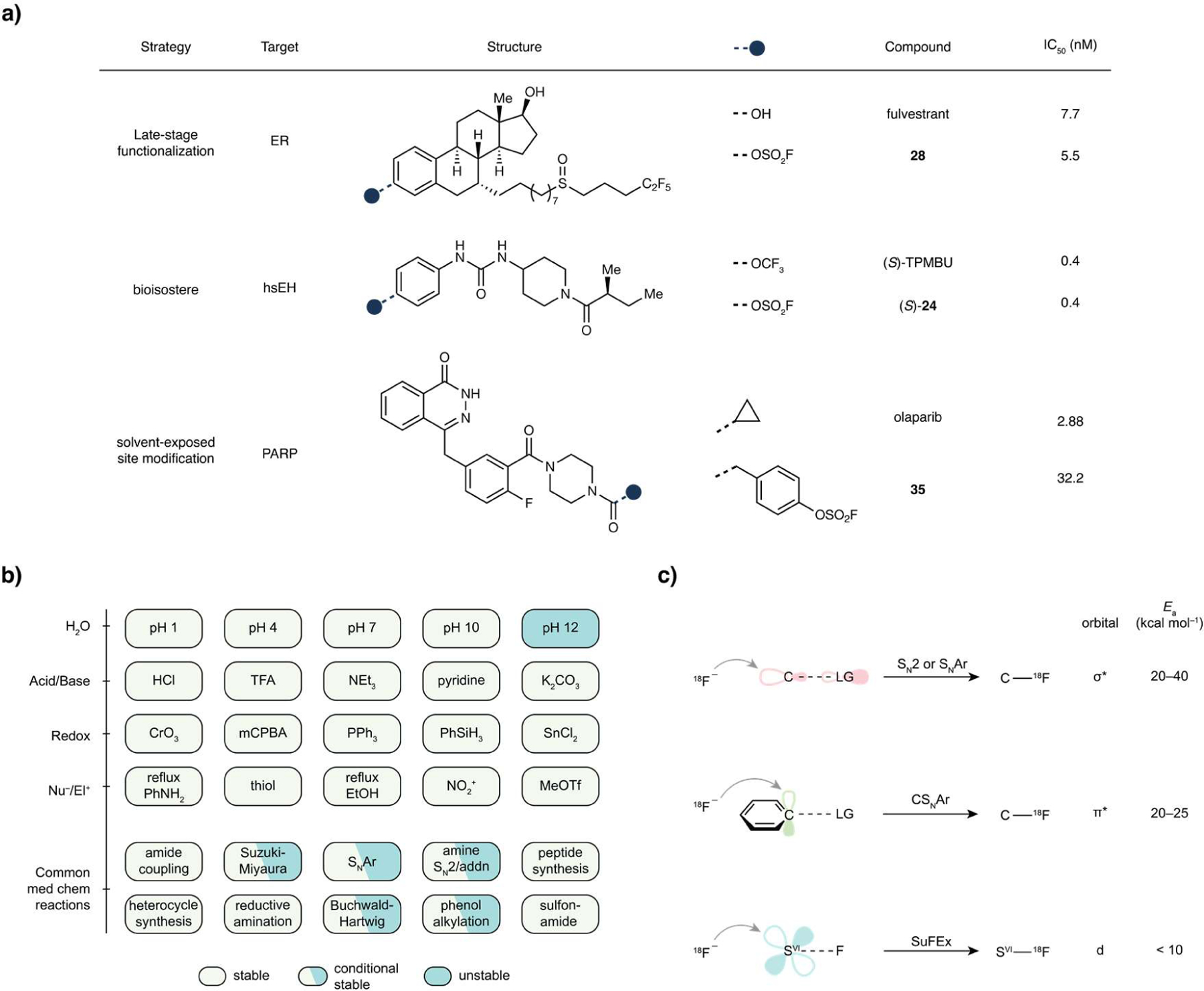
Computation by density functional theory (DFT) within the conductor like screening (COSMO) solvation model with the dielectric constant of acetonitrile (ε = 18.5) estimated a low barrier for the exchange between phenyl fluorosulfate (1) and anionic fluoride (Figure 2a, asterisk for clarity), ΔE‡ = 8.8 kcal mol−1 (ΔG‡ = 10.5 kcal mol−1). A pentacoordinated “ate”-complex intermediate formed in the reaction pathway with the two fluorine atoms occupying the two axial vertices (Figure 2b). By contrast, the undesired substitution with amines (e.g. MeNH2) or alcohols (e.g. MeOH) possesses a much higher calculated barrier (Supplementary Figure S70, ΔG‡ (MeNH2) = 23.2 kcal mol−1, ΔΔG‡ = 12.7 kcal mol−1), rendering at least 109-fold slower kinetics at room temperature.
Figure 2.
Investigation of sulfur fluoride exchange between phenyl fluorosulfate and a fluoride salt. (a) SuFEx of phenyl fluorosulfate (1). “F−” denotes any nucleophilic fluoride donor, exemplified by those shown below (i). (b), DFT calculated energy profile of a typical SuFEx process in acetonitrile and the optimized geometry of the highest transition state TS2. (c), Schematic illustration of TDST-NMR. (d), A representative plot of magnetization versus saturation time, and overlapped spectra (right) of the corresponding experiments with saturation time ranging from 0.01 s to 25 s. Conditions: phenyl fluorosulfate (1, 0.02 mol L−1), tetrabutylammonium bifluoride (2, 0.2 mol L−1), MeCN-d3, 298 K. kobs = 0.16 s−1 (R2 = 0.99). (e), Measurement of the second-order rate of the SuFEx reaction between 1 and 2, k298K = 0.43 L mol−1 s−1 (R2 = 0.99). (f) Erying plot of the SuFEx reaction between 1 and 2. The calculated activation enthalpy was determined, ΔH‡calc = 11.3 kcal mol−1 (R2 = 0.99). (g) Hammett plot of the SuFEx reactions between 2 and para-X-substituted phenyl fluorosulfates, ρ = 1.56 (R2 = 0.93). (h) Solvents effect. (i) Structure-activity relationship of various fluoride salts (2–10) on the SuFEx reaction of 1. †No exchange detected by NMR.
The inclusion of a counter ion, tetramethyl ammonium (TMA), to the fluoride exchange calculation led to a marginal change in activation energy (ΔΔE‡ = 0.31 kcal mol−1) and similar reaction profiles (Figure S69) compared to those of the “naked” fluoride calculation. The Gibbs energy of activation remained similarly low (ΔG‡ = 11.75 kcal mol−1), suggesting a rapid exchange at room temperature.
Investigation of sulfur fluoride exchange between an aryl fluorosulfate and a fluoride salt by NMR.
We used the 19F-NMR time-dependent saturation transfer (TDST) experiments38 to examine the interchange between an aryl fluorosulfate and a fluoride salt in solution. The TDST assay enables the differentiation of the “reactant” (left-hand side, Figure 2a) and the “product” (right-hand side) for kinetic rate measurements. In this bimolecular system, the fluoride salt signal was irradiated for a set of given saturation times (TS). If an intermolecular fluoride exchange process takes place, an apparent drop of the aryl fluorosulfate magnetization (ΔM) would be detected due to saturation transfer (Figure 2c).
Toward this end, SuFEx of phenyl fluorosulfate (PhOSO2F, 1, 0.02 mol L−1) and tetrabutylammonium bifluoride (n-Bu4N+FHF−, 2, 0.2 mol L−1, 10 equiv) was evaluated in acetonitrile-d3 (MeCN-d3). A set of saturation times (TSi, subscript i for ith measurement) was applied to the 19F-nuclei of 2 (–150 ppm relative to CFCl3) and the corresponding magnetizations (Mi) of 1 were recorded. Plotting Mi versus TSi, an exponential decay trend was indeed observed. The estimated pseudo-first order rate constant (kobs) was solved by Bloch equation with high coefficient of correlation (0.16 s−1, R2 = 0.99) (Figure 2d). By varying the concentration of 2, the second-order rate constant of the exchange between 1 and 2 at 298 K was determined, k298K = 0.43 L mol−1 s−1 (Figure 2e). Next, an estimation of the exchange barrier was obtained by performing TDST-NMR experiments at various temperatures ranging from 278 to 303 K (Figure 2f). From Eyring Equation, a low enthalpy of activation (11.3 kcal mol−1) was derived, suggesting the SuFEx process to be a facile reaction at room temperature. Furthermore, Hammett plot analysis was performed, which yielded a positive slope greater than unity (ρ = 1.56), indicating the emergence of negative charge during the reaction pathway (Figure 2g).
In parallel, we studied several factors that could influence the SuFEx process. Borosilicate glass, of which normal NMR tubes are made, showed no significant inhibition or acceleration when compared to a plastic reaction vessel (Figure S12). By contrast, solvent had significant impact on the rate of fluoride exchange. The use of a polar, aprotic solvent, such as N-Methyl-2-pyrrolidone (NMP), N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), or MeCN was found essential for achieving a fast SuFEx process (Figure 2h).
With tetrabutylammonium as the cation, we screened different fluoride anions (Figure 2i). We found that “basic” fluoride is more effective than its derivatives complexed by Brønsted or Lewis acids (2–5). Tetrabutylammonium fluoride (6•3H2O), although in its nucleophilicity-compromised trihydrate form, exhibited the highest exchange rate, which is approximately 50-fold faster than that of 2. Results from DFT calculation (Figure 2b) also suggested that the “naked” fluoride anion (6) is much more active than its bifluoride counterpart (2), ΔGcalc‡(6) = 10.5 kcal mol−1 (Cf. ΔGexp‡ (2) = 17.7 kcal mol−1, ΔΔG‡ = 7.2 kcal mol−1).
Subsequently, the structure-activity relationship of counter ions of bifluoride salts were investigated. Finely powdered potassium bifluoride alone did not effect the fluoride exchange in MeCN, albeit its complexes with 18-crown-6 (7) or [2.2.2]-cryptand (8) showed moderate exchange rates. In line with our earlier observation that tris(dimethylamino)sulfonium (TAS+) was a superior cation for SuFEx reactions39, the same salt (10) showed 10-fold increase in exchange rate compared with 2.
Automated radiosynthesis of aryl [18F]fluorosulfates.
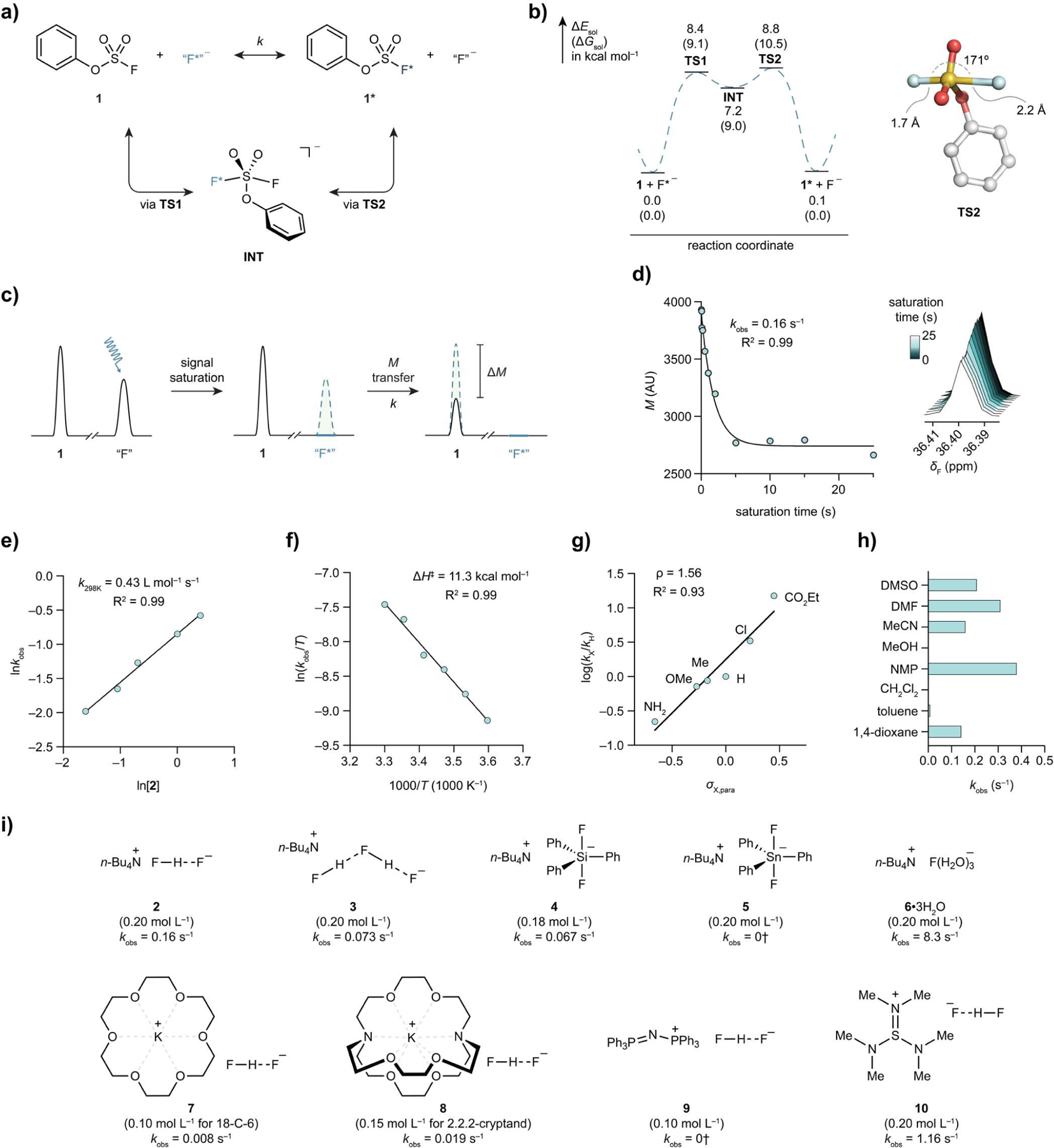
With kinetic parameters of [19F]SuFEx between phenyl fluorosulfate and fluoride salts determined, we embarked on the process development of the radioactive [18F]SuFEx. Compound 11, 3-Ethynylphenyl fluorosulfate, was chosen as the model substrate; potassium fluoride/[2.2.2]-cryptand (8) was selected as the [18F]fluoride source for its high elution efficiency and reproducibility. A few rounds of optimization based on the “cold” reaction results yielded a fully automated and highly reproducible method. In a typical experiment, 3-ethynylphenyl fluorosulfate (11, 0.1 mg, 0.5 µmol) in MeCN (0.5 mL) was added to azeotropically dried potassium [18F]fluoride (~3.7 GBq) in the presence of [2.2.2]-cryptand (8). The complete radiochemical conversion of [18F]F− was achieved in 30 s at room temperature, showing a single 18F-labeled 11 peak on the crude HPLC traces (Figure 3a,b). The radiochemical yield (RCY) based on HPLC40 was highly reproducible, 99.3 ± 0.6% (n = 4), followed by a less-than-one-minute C18-cartridge separation in which 3-ethynylphenyl [18F]fluorosulfate was isolated.
Figure 3.
[18F]SuFEx of aryl fluorosulfates. (a) [18F]SuFEx of 3-ethynylphenyl fluorosulfate (11). Conditions: Compound 11 (0.1 mg), K[18F]F (ca. 3.7 GBq), [2.2.2]-cryptand, K2CO3, MeCN (0.5 mL), 23 °C, 30 s. RCYHPLCs were determined by HPLC (n = 4) after the reaction crude been quenched by water (0.1 mL). (b) Representative HPLC chromatograms of a reaction crude of [18F]11, with 254 nm UV absorption (blue) and radioactivity (red) traces. (c) Substrate scope of the [18F]SuFEx reaction following the conditions described before in (a). Curie-scale synthesis of [18F]35. Conditions: Compound 35 (0.1 mg, 17.2 nmol), K[18F]F (~37 GBq or 111 GBq), [2.2.2]-cryptand, K2CO3, MeCN (10 mL), 23 °C, 30 s.
Using this protocol, we successfully synthesized twenty five aryl and heteroaryl [18F]fluorosulfates with excellent HPLC RCYs, 83–100% (median 98%, Figure 3c). Bifunctional [18F]fluorosulfate modules (14–20) covering a wide spectrum of (bio)orthogonally reactive groups could serve as positron-emitting tags for the in vitro or in vivo functionalization41 of biomacromolecules with native or preinstalled complementary handles such as alkyne (14), amine (15), thiol (16), hydroxyamine (17), trans-cyclooctene (18,19) and azide (20). In addition, [18F]SuFEx was applicable to fluorosulfate derivatives of both naturally occurring molecules (21, 23, 25, 26) and known phenolic medicines (22, 27, 29–3442–45). Importantly, the 18F-isotopologues of in vivo validated bioactive fluorosulfates (S)-24, 28 and 35 (vide infra) were synthesized in the final stage with excellent efficiency and fidelity.
While [18F]SuFEx was rarely interfered with substrates’ structures and functionalities, extensive reaction scope assessment revealed the inhibitory effects of acidic groups. Aryl fluorosulfates with carboxylic acid (pKaexp = 3.5) and NH-tetrazole gave lower RCYs, 50% and 28%, respectively (Figures S59, S60). Similarly, excess water was found to be a poison for the fluoride exchange. Only 14% RCY of [18F]11 was obtained when 50 equiv of water was added (Figure S28, Cf. 99% RCY with no water), which was in line with the NMR results (Figure S16).
Aryl [18F]fluorosulfate with high molar activity (Am) was prepared by leveraging the reaction stoichiometry. In two independent runs, “cold” fluorosulfate 35 (0.1 mg) was incubated with K[18F]F (ca. 37 GBq) at room temperature for 30 s rendered 93% and 85% HPLC RCY, respectively. The desired [18F]35 was purified by water quenching followed by C18-cartridge separation. Quantitative HPLC analysis of the final product with calibration curve revealed the molar activity 103 and 111 GBq µmol−1 (decay corrected to end-of-synthesis, E.O.S.), which is higher than the empirical threshold (74 GBq µmol−1)6 for a clinically useful radiotracer. Quantification also permits the measurement of the elution efficiency of a representative C18 cartridge separation to be 69–73% (n = 2). The isolated radiochemical yield (RCYisol) of [18F]35 was, depending on the gap between E.O.S. and purification, 21–42% (n = 2). Importantly, the same protocol afforded [18F]35 with even higher molar activity (280 GBq µmol−1) when 108 GBq of [18F]fluoride was used.
Stability examination of aryl fluorosulfate radiopharmacophores in aqueous and biological milieu.
To unravel the stability profile of aryl fluorosulfates in aqueous and biological milieu, we conducted a comprehensive examination of its propensity toward substitution and hydrolysis. Refluxing in the presence of aniline (bp 184 °C) for 3 h induced no change of phenyl fluorosulfate (1, bp 180 °C) (Figure S2). Incubating the same compound in aqueous HCl (1 mol L−1) and 50% trifluoroacetic acid in DCM, respectively, for 24 h resulted in neglectable degradation (Figure S6). While being inert under physiological conditions (pH 7.4, 37 °C) over a period of 1 week (Figure 4c), 1 was hydrolyzed slowly in a pH 10 buffer (approximately 10% hydrolysis after 24 h, Figure 4a) and rapidly by 0.025 mol L−1 aqueous sodium hydroxide (t1/2 = 20 min). We then incubated 1 in pH 7.4 buffer in the presence of a series of nucleophilic or reductive amino acids and found that only lysine and glutathione (GSH) caused approximately 10% of 1 hydrolysis after 48-h (Figure 4b). Further, intact protein MS indicated that Compound 1 (5 mM, 100 equiv) effected no covalent modification of bovine serum albumin (BSA, 50 µM) overnight at room temperature (Figure S10).
Figure 4.
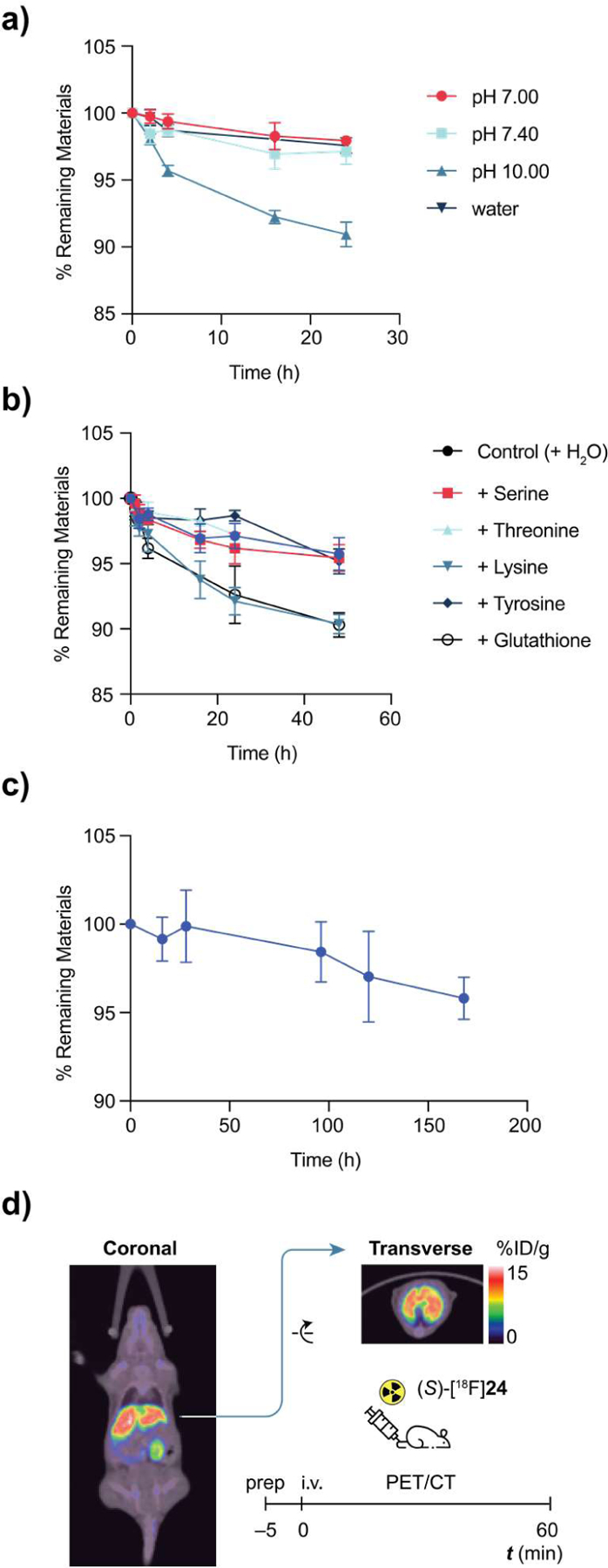
Stability characterization of aryl fluorosulfates in vitro and in vivo. (a) Stability of 1 (5 mmol L−1) in phosphate buffers, water and MeCN at room temperature over a course of 24 h. Percentage of remaining materials was quantified by HPLC (n = 3). (b) Stability of 1 (5 mmol L−1) in pH 7.4 buffer in the presence of nucleophilic amino acids and reduced glutathione (5 mmol L−1), respectively, at room temperature (n = 3). (c) Stability of 1 (5 mmol L−1) under physiological conditions, pH 7.4, 37 °C (n = 3). (d) PET images (60 min post-injection) of healthy mouse dosed by (S)-[18F]24 demonstrating its in vivo stability.
Subsequently, the 18F-labeled aryl fluorosulfate (S)-24 was used to assess the in vivo stability of the ArOSO2–F group. Upon injection into wild-type C57BL/6 mice via the intravenous (i.v.) route, [18F]-(S)-24 was found to be mainly enriched in liver where sEH is most abundantly expressed (Figure 4d). Importantly, we did not detect apparent 18F-associated signal in the bones — 18F bone deposition46 would indicate the release of [18F]F− from the aryl [18F]fluorosulfate via an undesired substitution or hydrolytic reaction. Similar in vivo stability was observed for the fulvestrant derivative [18F]28 (Figure S66). The absence of 18F-associated signal in the bone provides strong evidence of the in vivo stability of the S(VI)–F bonds of [18F]-(S)-24 and [18F]28, and suggests that aryl [18F]fluorosulfates, in general, may possess the required properties to be used for in vivo PET imaging.
Structure-based design and application of a PARP-targeting aryl [18F]fluorosulfate tracer
To apply aryl [18F]fluorosulfates to in vivo diagnostic PET imaging, we designed47 a [18F]fluorosulfate-functionalized analog of olaparib48–50 (35), an FDA-approved anti-cancer drug that targets tumor biomarker poly(ADP-ribose) polymerase 1 (PARP1), by extending the solvent-exposed site of olaparib (PDB 5DS3) with a pendent aryl fluorosulfate. Compound 35 maintained high binding affinity to PARP1 with a half maximal inhibitory concentration (IC50) of 32.2 nM (Figure 5a). When recombinant PARP1 was incubated with 35, no formation of covalent adduct was detected by tandem mass spectrometry (LC-MS/MS), suggesting that compound 35 functions as a non-covalent PARP1 inhibitor. Importantly, 35 exhibited similar stability profile as olaparib in the serum of healthy human donors (Figure 5b). Less than 25% of the small molecule was degraded after 3 h incubation, providing a useful time window for in vivo PET imaging.
Figure 5.
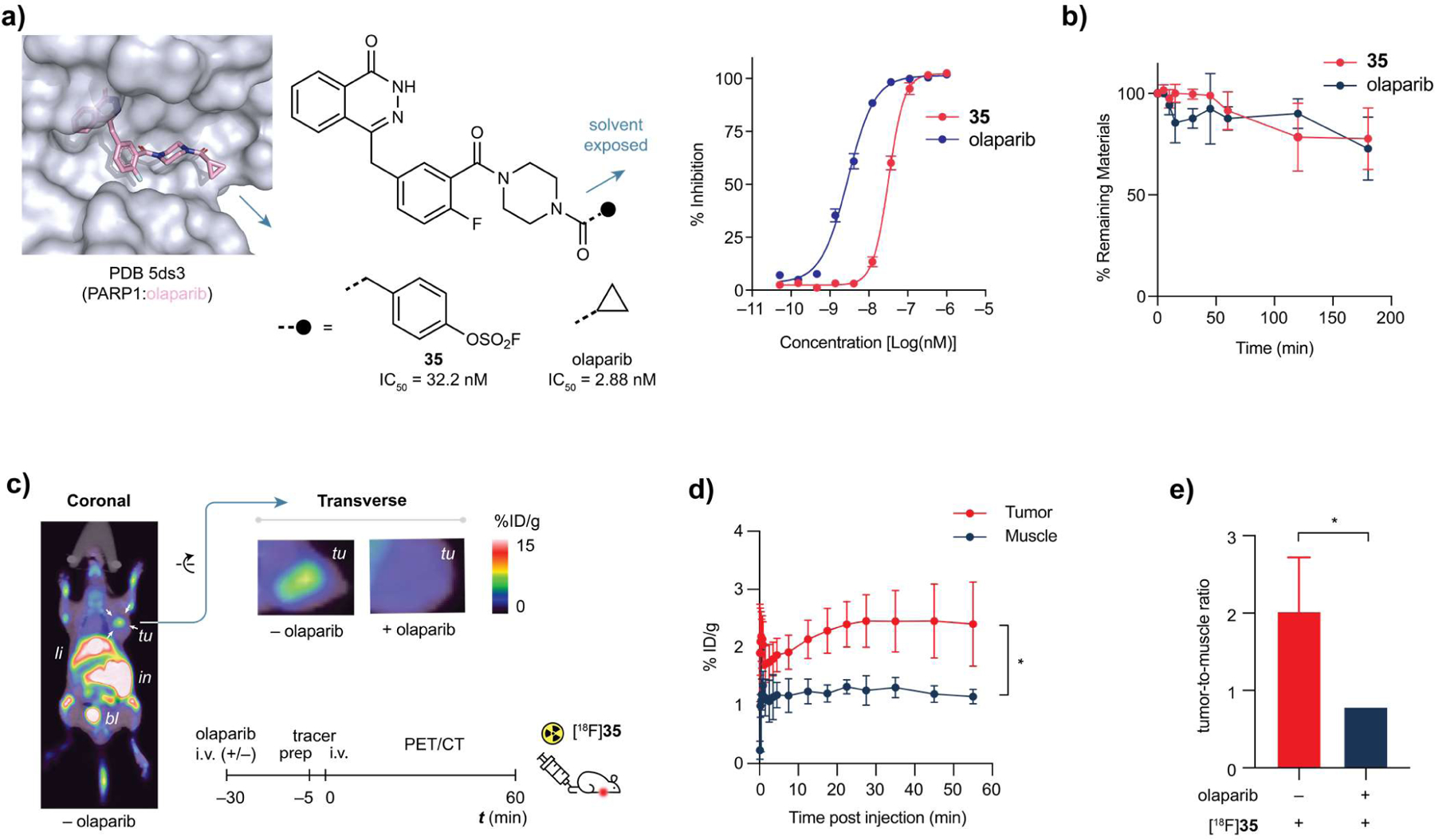
PET imaging in a tumor xenograft model using [18F]35. (a) Structure-based design of non-covalent PARP1 inhibitor 35 and its dose-response curve, olaparib as positive control. Error bars represent the mean plus standard deviation (SD, n = 3), IC50 = 32.2 nM. (b) Serum stability of 35 is comparable to that of olaparib. (c) Whole body coronal (left), and transverse (top-middle) PET/CT images of human MCF-7 bearing nude mice (transplanted under right shoulder, indicated by white arrows) acquired by performing a 55 min dynamic scan following [18F]35 administration. Transverse PET/CT image (top-right) of mice pre-treated by excess olaparib; No significant uptake of [18F]35 was observed at the tumor site. tu, tumor. li, liver. in, intestine. bl, bladder. (d) Time plot of percentage of injected dose per gram (%ID g−1) of tissue of interest. Error bars represent the mean plus standard deviation (SD, n = 3). P-values were calculated by unpaired Student’s t-test; *P < 0.05. (e) Comparison of pre-treatment with either vehicle (n = 3) or 50 mg kg−1 or olaparib (n = 3) on the %ID g−1 ratio of tumor versus muscle (healthy tissue). P-values were calculated by unpaired Student’s t-test; *P < 0.05.
A human breast cancer xenograft model was used to assess the applicability of the 18F-labeled 35 for in vivo PET imaging. [18F]35 was i.v. injected into nude mice of subcutaneous human breast cancer xenograft established using MCF-7, a human breast cancer cell line with upregulated PARP1 expression. The intensive accumulation of the 18F-labeled olaparib analog in tumors were clearly visualized with excellent target-to-background ratio after injection (Figure 5c,d). In a competition experiment, excess olaparib was dosed before administration of [18F]35, which blocked the accumulation of 35 in tumor, resulting in significant decrease in the ratio of % ID g−1 (injected dose per gram) of tumor-to-muscle from 2.02 ± 0.70 to 0.79 ± 0.04 (P < 0.05) (Figure 5c,e). These observations provide strong support for the specificity of this fluorosulfate-functionalized probe. Notably, in these experiments, we also detected significant 18F-associated signals in the bone marrow that has abundant PARP1 expression51–53.
DISCUSSION
Since its first introduction in 2014, aryl fluorosulfate-based SuFEx click chemistry has found a growing number of applications in chemical biology26–28, 47, 54–57. For example, the Kelly and Sharpless groups jointly discovered that biphenyl aryl fluorosulfate inactivates intracellular lipid binding protein(s) through chemoselective SuFEx reaction with a Tyr residue in the ligand-binding site27. Following this seminal work, highly selective fluorosulfate-based covalent small molecules were designed to target serine and lysine by the Jones56 and Pellecchia54 labs, respectively. Recently, by incorporating an O-fluorosulfonyl L-tyrosine, Wang and coworkers developed a proximity-enabled reactive PD-1 (programmed cell death protein-1) that covalently captures a proximal histidine of PD-L1 selectively, enabling irreversible binding of PD-1 to PD-L1 in vitro and in vivo58. Despite these pioneering studies that have demonstrated the potential of aryl fluorosulfates as covalent warheads for protein modifications, the reactivity-stability profile of S(VI)-F in a biological milieu remains elusive. Anyone who plans to use aryl fluorosulfates for biological applications should be cautious to presume their propensity to undergo expected covalent transformations under physiological conditions. As a matter of fact, aryl fluorosulfates are uniquely inert to disordered nucleophiles in bulk water due to its small net dipole. Works by others56–57, 59, and our own labs27, 60 have partially unraveled the stringent requirements for its activation. Only with the orientation of an aryl fluorosulfate and its reactive partner immobilized to just the right geometry of interaction, like a freeze-frame, by local electrostatic effects and an organized hydrogen bonding network, covalent capture would take place on the time scale of small molecule binding. Neither elongated contact nor perturbed nucleophile reactivity is dispensable. Accordingly, a protein, upon denaturing and losing its tertiary structure, immediately loses its reactivity with an aryl fluorosulfate-functionalized molecule that readily reacts with the same protein under native conditions27, 47.
On the other hand, the above covalent capture examples reflect only a tip of the reactivity landscape of aryl fluorosulfates. In a few extreme cases we have encountered, yet contrary to the biased common sense that acid halide is extremely labile, aryl fluorosulfates stay intact in refluxing aniline (184 °C), aqueous solutions (Figure 4) and even in the presence of an entire proteome27. The realization of the narrow reactivity-window has, thence, led to the scenario of taking advantage of the overlooked stability. As proof-of-principle, we have developed non-covalent inhibitors ((S)-24, 28) by bioisosteric replacement of trifluoromethoxy group28 and late-stage fluorosulfonylation of phenolic drugs29, 61–62, respectively, with improved potency and metabolic stability. Fluorosulfate is considered pharmacologically similar to trifluoromethoxy group in the hsEH case and superior to phenol precursor regarding oral bioavailability in the SERD case. That said, the current data set27, 47, 54, 57, 60 is still too small to make a priori inferences regarding the influence of fluorosulfate group on a molecule’s pharmacokinetic properties regarding its absorption, distribution, metabolism, and excretion (ADME).
Moreover, it is impossible at this moment to predict whether an aryl fluorosulfate-based hit molecule would function as a covalent inhibitor, and vice versa. Therefore, bioactive fluorosulfates should be studied case-by-case agnostically. Those characterized non-covalent inhibitors (e.g. (S)-24, 28, and 35) have the potential to serve as a tracer once radiolabeled, which not only could be used as a candidate for therapy but also for diagnosis and evaluation of their own treatment.
With that goal in mind, we discovered that fluoride anions circumvented the high kinetic barrier of a typical SuFEx reaction typically associated with amine or alcohol nucleophiles — the extraneous fluoride anion per se compensates the stringent requirements in the latter scenarios for the stabilization or solvation of the departing fluoride from an SVI–F site. In fact, anionic O-, or N-nucleophiles are rarely present in near-neutral aqueous environments, and their basicity is buffered by bulk water unless perturbed within the protein microenvironment27. This is presumably why aryl fluorosulfates undergo fast fluoride exchange in acetonitrile solution, while maintaining good stability in most biological milieu. Driven by this mechanistic rationale, we have successfully developed the ultrafast [18F]SuFEx process to incorporate 18F into aryl fluorosulfates as new radiotracers that, in the meantime, possess sufficient stability to be used for in vivo imaging with negligible drawbacks of hydrolysis and covalent bond formation.
The most unique feature of [18F]SuFEx is its efficiency and fidelity without sacrificing the probe’s stability. The traditional radiosynthesis of an 18F-tagged N-hydroxysuccinimide ester (NHS) requires sequential SNAr, saponification, and esterification, and a 20-min HPLC purification63. By contrast, [18F]SuFEx fulfills 18F installation and purification at room temperature within 2 min ([18F]15). To our knowledge, [18F]SuFEx represents the most efficient 18F-incorporation process to date.
The mild exchange condition and chemical orthogonality of SuFEx also rendered a much broader substrate scope than the previously known radiolabeling protocols (Figure 3). Thus, the radiolabeling could be accomplished in the final step of the 18F-labeled compound synthesis with significantly less activity going futile. The chemical inertness of aryl fluorosulfates also offers much flexibility in planning the “cold” precursor preparation such that the fluorosulfate handle may be installed in either early (e.g. 35 and fluorosulfonylated peptides26) or late stages (e.g. 33, 34) in a synthetic scheme. In either scenario, [18F]SuFEx could accomplish the [18F]fluorination in the final step without tedious protecting group manipulations.
Like earlier isotopic exchange-based radiosynthesis31, 33, 35–37, 64–65, [19F]fluoride in the substrate limits the theoretical maximum molar activity66. Noteworthy, molar activity represents the major caveat of this process, especially when the “cold” substrate is in large excess. Good molar activity was achieved in this case at the cost of high [18F]fluoride activity (> 37 GBq) that is uncommon in conventional nucleophilic [18F]fluorination processes. Even though, the percentage of [18F]fluorosulfate/[19F]fluorosulfate was less than 1% which may cause low signal-to-noise ratio when targeting less abundant proteins. In all three Curie-scale syntheses, the obtained molar activities (decay corrected to E.O.S.) were roughly half of their theoretical values, which may indicate the isotopic exchange is relatively less sensitive to system-induced [19F]fluoride than nucleophilic [19F]fluorination. The latter often requires microvolume reactor66 or flow-based system67 to minimize “cold” contamination while the influence of highly dilute condition to the current [18F]SuFEx synthesis is small.
Lastly, we showcased the first PET imaging application of an aryl fluorosulfate-based probe targeting the aberrantly overexpressed PARP1 in a murine model of human tumor xenograft. The on-target engagement of radiolabeled compound [18F]35 was apparent in the olaparib blocking experiments (Figure 5). Although we have demonstrated the non-covalent binding of 35 to PARP1 and its stability in serum and to albumin, at this moment, off-target engagement with other protein(s) via an either covalent or non-covalent way could not be 100% excluded. We are not certain yet whether compound 35 inherited olaparib’s high selectivity for PARP168, but the interrogation of its protein target is current under way.
To conclude, we have developed an ultrafast process to radiosynthesize unprecedented aryl [18F]fluorosulfate and showcased the first PET imaging application of an S–18F-based probe. Aryl [18F]fluorosulfates complement the current C–18F toolkit by unlocking an uncharted radiochemical space. We envisage that it is possible to design additional aryl [18F]fluorosulfate-containing radiotracers to probe proteins beyond the ones described here, such as radiotracers for targeting the central nervous system.
Supplementary Material
ACKNOWLEDGMENT
We thank Huayi Isotopes Co. (Changshu, Jiangsu Province, China) for access of cyclotron and PET-CT, Dr. Dee-Hua Huang (Scripps Research NMR Facility) for assistance on TDST-NMR experiments, Prof. Jin-Quan Yu (Scripps Research) for access of glovebox and GC-EI-MS, Dr. Yongxuan Su (UC San Diego) for assistance on APCI-MS, and Yunfei Cheng (Scripps Research) for NMR characterizations. We are grateful to Prof. Jiajia Dong (Shanghai Institute of Organic Chemistry) and Dr. John R. Cappiello (Scripps Research) for helpful discussions.
Funding Sources
This work is supported by the U.S. National Institutes of Health (R01GM117145 to K.B.S., R35GM139643 to P.W., and R01GM100934 to L.N.), the U.S. National Science Foundation (CHE-1610987 to K.B.S.), the National Natural Science Foundation of China (21977070 to H.Xu), ShanghaiTech University startup fund (G.Y.), Ellen Browning Scripps Foundation (graduate fellowship to Q.Z.).
Footnotes
ASSOCIATED CONTENT
Supporting Information
Experimental procedures for chemical and radiosynthesis, 1H-NMR, 13C-NMR, 19F-NMR, 19F-TDST-NMR spectra, analytical HPLC radioactivity and UV chromatograms, PET-CT images, immunohistochemistry, and DFT calculations are available in the Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
Supplementary text, figures, tables and equations (PDF)
The authors declare no competing financial interest.
REFERENCES
- 1.Fowler JS; Wolf AP, Working against time: Rapid radiotracer synthesis and imaging the human brain. Acc. Chem. Res 1997, 30 (4), 181–188. [Google Scholar]
- 2.Phelps ME, Positron emission tomography provides molecular imaging of biological processes. Proc. Natl. Acad. Sci. USA 2000, 97 (16), 9226–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gambhir SS, Molecular imaging of cancer with positron emission tomography. Nat. Rev. Cancer 2002, 2 (9), 683–693. [DOI] [PubMed] [Google Scholar]
- 4.Tsien RY, Imagining imaging’s future. Nat. Rev. Mol. Cell Bio 2003, SS16–SS21. [PubMed]
- 5.Willmann JK; van Bruggen N; Dinkelborg LM; Gambhir SS, Molecular imaging in drug development. Nat. Rev. Drug Discov 2008, 7 (7), 591–607. [DOI] [PubMed] [Google Scholar]
- 6.Ametamey SM; Honer M; Schubiger PA, Molecular imaging with PET. Chem. Rev 2008, 108 (5), 1501–1516. [DOI] [PubMed] [Google Scholar]
- 7.Cai LS; Lu SY; Pike VW, Chemistry with [18F]fluoride ion. Eur. J. Org. Chem 2008, (17), 2853–2873. [Google Scholar]
- 8.Tredwell M; Gouverneur V, 18F Labeling of Arenes. Angew. Chem. Int. Ed 2012, 51 (46), 11426–11437. [DOI] [PubMed] [Google Scholar]
- 9.Brooks AF; Topczewski JJ; Ichiishi N; Sanford MS; Scott PJH, Late-stage [18F]fluorination: new solutions to old problems. Chem. Sci 2014, 5 (12), 4545–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang SH; Vasdev N, C(sp3)-18F Bond Formation by Transition-Metal-Based [18F]Fluorination. Angew. Chem. Int. Ed 2014, 53 (43), 11416–11418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Preshlock S; Tredwell M; Gouverneur V, 18F-Labeling of arenes and heteroarenes for applications in positron emission tomography. Chem. Rev 2016, 116 (2), 719–766. [DOI] [PubMed] [Google Scholar]
- 12.Lee E; Kamlet AS; Powers DC; Neumann CN; Boursalian GB; Furuya T; Choi DC; Hooker JM; Ritter T, A fluoride-derived electrophilic late-stage fluorination reagent for PET imaging. Science 2011, 334 (6056), 639–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huiban M; Tredwell M; Mizuta S; Wan ZH; Zhang XM; Collier TL; Gouverneur V; Passchier J, A broadly applicable [18F]trifluoromethylation of aryl and heteroaryl iodides for PET imaging. Nat. Chem 2013, 5 (11), 941–944. [DOI] [PubMed] [Google Scholar]
- 14.Graham TJA; Lambert RF; Ploessl K; Kung HF; Doyle AG, Enantioselective radiosynthesis of positron emission tomography (PET) tracers containing [18F]fluorohydrins. J. Am. Chem. Soc 2014, 136 (14), 5291–5294. [DOI] [PubMed] [Google Scholar]
- 15.Rotstein BH; Stephenson NA; Vasdev N; Liang SH, Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat. Commun 2014, 5, 4365. [DOI] [PubMed] [Google Scholar]
- 16.Sergeev ME; Morgia F; Lazari M; Wang C; van Dam RM, Titania-catalyzed radiofluorination of tosylated precursors in highly aqueous medium. J. Am. Chem. Soc 2015, 137 (17), 5686–5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neumann CN; Hooker JM; Ritter T, Concerted nucleophilic aromatic substitution with 19F- and 18F-. Nature 2016, 538 (7624), 369–373. [DOI] [PubMed] [Google Scholar]
- 18.Levin MD; Chen TQ; Neubig ME; Hong CM; Theulier CA; Kobylianskii IJ; Janabi M; O’Neil JP; Toste FD, A catalytic fluoride-rebound mechanism for C(sp3)-CF3 bond formation. Science 2017, 356 (6344), 1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen W; Huang Z; Tay NES; Giglio B; Wang M; Wang H; Wu Z; Nicewicz DA; Li Z, Direct arene C-H fluorination with 18F- via organic photoredox catalysis. Science 2019, 364 (6446), 1170–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ido T; Wan CN; Casella V; Fowler JS; Wolf AP; Reivich M; Kuhl DE, Labeled 2-deoxy-D-glucose analogs. 18F-labeled 2-deoxy-2-fluoro-D-glucose, 2-deoxy-2-fluoro-D-mannose and 14C-2-deoxy-2-fluoro-D-glucose. J. Labelled Compd. Rad 1978, 14 (2), 175–183. [Google Scholar]
- 21.Campbell MG; Mercier J; Genicot C; Gouverneur V; Hooker JM; Ritter T, Bridging the gaps in F-18 PET tracer development. Nat. Chem 2017, 9 (1), 1–3. [DOI] [PubMed] [Google Scholar]
- 22.Kolb HC; Finn MG; Sharpless KB, Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed 2001, 40 (11), 2004–2021. [DOI] [PubMed] [Google Scholar]
- 23.Dong J; Sharpless KB; Kelly JW; Chen W Sulfur (VI) fluoride compounds and methods for the preparation thereof 10,117,840, 2018.
- 24.Dong J; Krasnova L; Finn MG; Sharpless KB, Sulfur(VI) Fluoride Exchange (SuFEx): Another good reaction for click chemistry. Angew. Chem. Int. Ed 2014, 53 (36), 9430–9448. [DOI] [PubMed] [Google Scholar]
- 25.Zheng Q; Woehl JL; Kitamura S; Santos-Martins D; Smedley CJ; Li G; Forli S; Moses JE; Wolan DW; Sharpless KB, SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc. Natl. Acad. Sci. USA 2019, 116 (38), 18808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen W; Dong J; Li S; Liu Y; Wang Y; Yoon L; Wu P; Sharpless KB; Kelly JW, Synthesis of sulfotyrosine-containing peptides by incorporating fluorosulfated tyrosine using an Fmoc-based solid-phase strategy. Angew. Chem. Int. Ed 2016, 55 (5), 1835–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen W; Dong J; Plate L; Mortenson DE; Brighty GJ; Li S; Liu Y; Galmozzi A; Lee PS; Hulce JJ; Cravatt BF; Saez E; Powers ET; Wilson IA; Sharpless KB; Kelly JW, Arylfluorosulfates inactivate intracellular lipid binding protein(s) through chemoselective SuFEx reaction with a binding site Tyr residue. J. Am. Chem. Soc 2016, 138 (23), 7353–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong J; Krasnova L; Sharpless KB Fluorosulfonyl sEH Inhibitors. WO2015188060A1, 2015.
- 29.Liu ZL; Li J; Li SH; Li GC; Sharpless KB; Wu P, SuFEx click chemistry enabled late-stage drug functionalization. J. Am. Chem. Soc 2018, 140 (8), 2919–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown DG; Boström J, Analysis of past and present synthetic methodologies on medicinal chemistry: Where have all the new reactions gone? J. Med. Chem 2016, 59 (10), 4443–4458. [DOI] [PubMed] [Google Scholar]
- 31.Bernard-Gauthier V; Wangler C; Schirrmacher E; Kostikov A; Jurkschat K; Wangler B; Schirrmacher R, 18F-labeled silicon-based fluoride acceptors: potential opportunities for novel positron emitting radiopharmaceuticals. BioMed Res. Int 2014, 2014, 454503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alauddin MM; Conti PS; Mathew T; Fissekis JD; Prakash GKS; Watanabe KA, Stereospecific fluorination of 1,3,5-tri-O-benzoyl-alpha-D-ribofuranose-2-sulfonate esters: preparation of a versatile intermediate for synthesis of 2’-[18F]-fluoro-arabinonucleosides. J. Fluorine Chem 2000, 106 (1), 87–91. [Google Scholar]
- 33.Bernard-Gauthier V; Bailey JJ; Liu ZB; Wangler B; Wangler C; Jurkschat K; Perrin DM; Schirrmacher R, From unorthodox to established: The current status of 18F-trifluoroborate- and 18F-SiFA-based radiopharmaceuticals in PET nuclear imaging. Bioconjugate Chem 2016, 27 (2), 267–279. [DOI] [PubMed] [Google Scholar]
- 34.Pascali G; Matesic L; Zhang B; King AT; Robinson AJ; Ung AT; Fraser BH, Sulfur - fluorine bond in PET radiochemistry. EJNMMI Radiopharm. Chem 2017, 2 (1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ting R; Adam MJ; Ruth TJ; Perrin DM, Arylfluoroborates and alkylfluorosilicates as potential PET imaging agents: High-yielding aqueous biomolecular F-18-labeling. J. Am. Chem. Soc 2005, 127 (38), 13094–13095. [DOI] [PubMed] [Google Scholar]
- 36.Schirrmacher R; Bradtmoller G; Schirrmacher E; Thews O; Tillmanns J; Siessmeier T; Buchholz HG; Bartenstein P; Waengler B; Niemeyer CM; Jurkschat K, F-18-labeling of peptides by means of an organosilicon-based fluoride acceptor. Angew. Chem. Int. Ed 2006, 45 (36), 6047–6050. [DOI] [PubMed] [Google Scholar]
- 37.Liu ZB; Pourghiasian M; Radtke MA; Lau J; Pan JH; Dias GM; Yapp D; Lin KS; Benard F; Perrin DM, An organotrifluoroborate for broadly applicable one-step 18F-Labeling. Angew. Chem. Int. Ed 2014, 53 (44), 11876–11880. [DOI] [PubMed] [Google Scholar]
- 38.Taitelbaum H; Weiss GH; Spencer RGS, Optimization of magnetization-transfer experiments for kinetic rate measurements. NMR Biomed 1994, 7 (6), 287–292. [DOI] [PubMed] [Google Scholar]
- 39.Gao B; Zhang LD; Zheng QH; Zhou F; Klivansky LM; Lu JM; Liu Y; Dong JJ; Wu P; Sharpless KB, Bifluoride-catalysed sulfur(VI) fluoride exchange reaction for the synthesis of polysulfates and polysulfonates. Nat. Chem 2017, 9 (11), 1083–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coenen HH; Gee AD; Adam M; Antoni G; Cutler CS; Fujibayashi Y; Jeong JM; Mach RH; Mindt TL; Pike VW; Windhorst AD, Consensus nomenclature rules for radiopharmaceutical chemistry - Setting the record straight. Nucl. Med. Biol 2017, 55, V–Xi. [DOI] [PubMed] [Google Scholar]
- 41.Meyer JP; Adumeau P; Lewis JS; Zeglis BM, Click chemistry and radiochemistry: The first 10 years. Bioconjugate Chem 2016, 27 (12), 2791–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong DF; Rosenberg PB; Zhou Y; Kumar A; Raymont V; Ravert HT; Dannals RF; Nandi A; Brasić JR; Ye W; Hilton J; Lyketsos C; Kung HF; Joshi AD; Skovronsky DM; Pontecorvo MJ, In vivo imaging of amyloid deposition in Alzheimer disease using the radioligand 18F-AV-45 (florbetapir [corrected] F 18). J. Nucl. Med 2010, 51 (6), 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang W; Oya S; Kung M-P; Hou C; Maier DL; Kung HF, F-18 Polyethyleneglycol stilbenes as PET imaging agents targeting Aβ aggregates in the brain. Nucl. Med. Biol 2005, 32 (8), 799–809. [DOI] [PubMed] [Google Scholar]
- 44.Klunk WE; Engler H; Nordberg A; Wang Y; Blomqvist G; Holt DP; Bergström M; Savitcheva I; Huang G-F; Estrada S; Ausén B; Debnath ML; Barletta J; Price JC; Sandell J; Lopresti BJ; Wall A; Koivisto P; Antoni G; Mathis CA; Långström B, Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol 2004, 55 (3), 306–319. [DOI] [PubMed] [Google Scholar]
- 45.Rami-Mark C; Zhang M-R; Mitterhauser M; Lanzenberger R; Hacker M; Wadsak W, [18F]FMeNER-D2: Reliable fully-automated synthesis for visualization of the norepinephrine transporter. Nucl. Med. Biol 2013, 40 (8), 1049–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grant FD; Fahey FH; Packard AB; Davis RT; Alavi A; Treves ST, Skeletal PET with 18F-fluoride: applying new technology to an old tracer. J. Nucl. Med 2008, 49 (1), 68–78. [DOI] [PubMed] [Google Scholar]
- 47.Jones LH; Kelly JW, Structure-based design and analysis of SuFEx chemical probes. RSC Med. Chem 2020, 11 (1), 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dawicki-McKenna JM; Langelier MF; DeNizio JE; Riccio AA; Cao CD; Karch KR; McCauley M; Steffen JD; Black BE; Pascal JM, PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol. Cell 2015, 60 (5), 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carney B; Carlucci G; Salinas B; Di Gialleonardo V; Kossatz S; Vansteene A; Longo VA; Bolaender A; Chiosis G; Keshari KR; Weber WA; Reiner T, Non-invasive PET imaging of PARP1 expression in glioblastoma models. Mol. Imaging Biol 2016, 18 (3), 386–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carney B; Kossatz S; Lok BH; Schneeberger V; Gangangari KK; Pillarsetty NVK; Weber WA; Rudin CM; Poirier JT; Reiner T, Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nat. Commun 2018, 9 (1), 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uhlen M; Fagerberg L; Hallstrom BM; Lindskog C; Oksvold P; Mardinoglu A; Sivertsson A; Kampf C; Sjostedt E; Asplund A; Olsson I; Edlund K; Lundberg E; Navani S; Szigyarto CA; Odeberg J; Djureinovic D; Takanen JO; Hober S; Alm T; Edqvist PH; Berling H; Tegel H; Mulder J; Rockberg J; Nilsson P; Schwenk JM; Hamsten M; von Feilitzen K; Forsberg M; Persson L; Johansson F; Zwahlen M; von Heijne G; Nielsen J; Ponten F, Proteomics. Tissue-based map of the human proteome. Science 2015, 347 (6220), 1260419. [DOI] [PubMed] [Google Scholar]
- 52.Human Protein Atlas PARP1. https://www.proteinatlas.org/ENSG00000143799-PARP1/tissue (accessed September 16).
- 53.Schöder H; França PDDS; Nakajima R; Burnazi E; Roberts S; Brand C; Grkovski M; Mauguen A; Dunphy MP; Ghossein RA; Lyashchenko SK; Lewis JS; Donoghue JA; Ganly I; Patel SG; Lee NY; Reiner T, Safety and feasibility of PARP1/2 imaging with 18F-PARPi in patients with head and neck cancer. Clinical Cancer Research 2020, 26 (13), 3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baggio C; Udompholkul P; Gambini L; Salem AF; Jossart J; Perry JJP; Pellecchia M, Aryl-fluorosulfate-based lysine covalent pan-inhibitors of apoptosis protein (IAP) antagonists with cellular efficacy. J. Med. Chem 2019, 62 (20), 9188–9200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baranczak A; Liu Y; Connelly S; Du WGH; Greiner ER; Genereux JC; Wiseman RL; Eisele YS; Bradbury NC; Dong JJ; Noodleman L; Sharpless KB; Wilson IA; Encalada SE; Kelly JW, A fluorogenic aryl fluorosulfate for intraorganellar transthyretin imaging in living cells and in caenorhabditis elegans. J. Am. Chem. Soc 2015, 137 (23), 7404–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fadeyi OO; Hoth LR; Choi C; Feng X; Gopalsamy A; Hett EC; Kyne RE; Robinson RP; Jones LH, Covalent enzyme inhibition through fluorosulfate modification of a noncatalytic serine residue. ACS Chem. Biol 2017, 12 (8), 2015–2020. [DOI] [PubMed] [Google Scholar]
- 57.Jones LH, Emerging utility of fluorosulfate chemical probes. ACS Med. Chem. Lett 2018, 9 (7), 584–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Q; Chen Q; Klauser PC; Li M; Zheng F; Wang N; Li X; Zhang Q; Fu X; Wang Q; Xu Y; Wang L, Developing covalent protein drugs via proximity-enabled reactive therapeutics. Cell 2020, 182 (1), 85–97. [DOI] [PubMed] [Google Scholar]
- 59.Wang N; Yang B; Fu C; Zhu H; Zheng F; Kobayashi T; Liu J; Li S; Ma C; Wang PG; Wang Q; Wang L, Genetically encoding fluorosulfate-l-tyrosine to react with lysine, histidine, and tyrosine via SuFEx in proteins in vivo. J. Am. Chem. Soc 2018, 140 (15), 4995–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mortenson DE; Brighty GJ; Plate L; Bare G; Chen W; Li S; Wang H; Cravatt BF; Forli S; Powers ET; Sharpless KB; Wilson IA; Kelly JW, “Inverse drug discovery” strategy to identify proteins that are targeted by latent electrophiles as exemplified by aryl fluorosulfates. J. Am. Chem. Soc 2018, 140 (1), 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou H; Mukherjee P; Liu R; Evrard E; Wang D; Humphrey JM; Butler TW; Hoth LR; Sperry JB; Sakata SK; Helal CJ; Am Ende CW, Introduction of a crystalline, shelf-stable reagent for the synthesis of sulfur(VI) fluorides. Org. Lett 2018, 20 (3), 812–815. [DOI] [PubMed] [Google Scholar]
- 62.Guo TJ; Meng GY; Zhan XJ; Yang Q; Ma TC; Xu L; Sharpless KB; Dong JJ, A new portal to SuFEx click chemistry: A stable fluorosulfuryl imidazolium salt emerging as an “F-SO2+” donor of unprecedented reactivity, selectivity, and scope. Angew. Chem. Int. Ed 2018, 57 (10), 2605–2610. [DOI] [PubMed] [Google Scholar]
- 63.Shao X, Synthesis of N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB). In Radiopharmaceuticals for Positron Emission Tomography, 1 ed.; Scott PJH; Hockley BG, Eds. Wiley: Hoboken, N.J., 2012; Vol. 1, pp 81–85. [Google Scholar]
- 64.Ting R; Harwig C; auf dem Keller U; McCormick S; Austin P; Overall CM; Adam MJ; Ruth TJ; Perrin DM, Toward [18F]-labeled aryltrifluoroborate radiotracers: In vivo positron emission tomography imaging of stable aryltrifluoroborate clearance in mice. J. Am. Chem. Soc 2008, 130 (36), 12045–12055. [DOI] [PubMed] [Google Scholar]
- 65.Liu Z; Lin K-S; Bénard F; Pourghiasian M; Kiesewetter DO; Perrin DM; Chen X, One-step 18F labeling of biomolecules using organotrifluoroborates. Nature Protocols 2015, 10 (9), 1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sergeev M; Lazari M; Morgia F; Collins J; Javed MR; Sergeeva O; Jones J; Phelps ME; Lee JT; Keng PY; van Dam RM, Performing radiosynthesis in microvolumes to maximize molar activity of tracers for positron emission tomography. Communications Chemistry 2018, 1 (1), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee C-C; Sui G; Elizarov A; Shu CJ; Shin Y-S; Dooley AN; Huang J; Daridon A; Wyatt P; Stout D; Kolb HC; Witte ON; Satyamurthy N; Heath JR; Phelps ME; Quake SR; Tseng H-R, Multistep synthesis of a radiolabeled imaging probe using integrated microfluidics. Science 2005, 310 (5755), 1793. [DOI] [PubMed] [Google Scholar]
- 68.Knezevic CE; Wright G; Rix LLR; Kim W; Kuenzi BM; Luo Y; Watters JM; Koomen JM; Haura EB; Monteiro AN; Radu C; Lawrence HR; Rix U, Proteome-wide profiling of clinical PARP inhibitors reveals compound-specific secondary targets. Cell Chemical Biology 2016, 23 (12), 1490–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.