

Abstract
Axonal Charcot-Marie-Tooth neuropathies (CMT type 2) are caused by inherited mutations in various genes functioning in different pathways. The types of genes and multiplicity of mutations reflect the clinical and genetic heterogeneity in CMT2 disease, which complicates its diagnosis and has inhibited the development of therapies. Here, we used CMT2 patient-derived pluripotent stem cells (iPSCs) to identify common hallmarks of axonal degeneration shared by different CMT2 subtypes. We compared the cellular phenotypes of neurons differentiated from CMT2 patient iPSCs with those from healthy controls and a CRISPR/Cas9-corrected isogenic line. Our results demonstrated neurite network alterations along with extracellular electrophysiological abnormalities in the differentiated motor neurons. Progressive deficits in mitochondrial and lysosomal trafficking, as well as in mitochondrial morphology, were observed in all CMT2 patient lines. Differentiation of the same CMT2 iPSC lines into peripheral sensory neurons only gave rise to cellular phenotypes in subtypes with sensory involvement, supporting the notion that some gene mutations predominantly affect motor neurons. We revealed a common mitochondrial dysfunction in CMT2-derived motor neurons, supported by alterations in the expression pattern and oxidative phosphorylation, which could be recapitulated in the sciatic nerve tissue of a symptomatic mouse model. Inhibition of a dual leucine zipper kinase could partially ameliorate the mitochondrial disease phenotypes in CMT2 subtypes. Altogether, our data reveal shared cellular phenotypes across different CMT2 subtypes and suggests that targeting such common pathomechanisms could allow the development of a uniform treatment for CMT2.
Keywords: Charcot-Marie-Tooth neuropathy, iPSC-derived motor and sensory neurons, phenotyping, mitochondrial dysfunction, dual leucine kinase inhibitor
See Müller and Horvath (doi:10.1093/brain/awab278) for a scientific commentary on this article.
Van Lent et al. use neurons differentiated from patient-derived iPSCs to identify common hallmarks of axonal degeneration, including impaired axonal transport and mitochondrial dysfunction, shared by different CMT2 subtypes. Targeting these pathomechanisms could provide new drug development opportunities for CMT2.
See Müller and Horvath (doi:10.1093/brain/awab278) for a scientific commentary on this article.
Introduction
Charcot-Marie-Tooth (CMT) disease is an inherited peripheral neuropathy associated with dominant or recessive mutations in a wide spectrum of genes, many of which fulfil ubiquitous and indispensable functions in almost every human cell. Yet, the impact of disease-causing mutations in these genes seems restricted to the PNS. Over 1000 mutations in more than 90 distinct CMT disease-associated genes have been reported to date.1,2 Intriguingly, the disease is not only genetically but also clinically heterogeneous. CMT is broadly classified into demyelinating (CMT1) and axonal (CMT2) forms, depending on whether primary deficits occur in the myelinating Schwann cell or the neuronal axon, respectively. CMT1A, the most common CMT subtype, accounts for 40–50% of all cases of CMT followed by CMT2, which accounts for ∼20%.3 Note also that a few intermediate CMT subtypes have been recognized because of the overlap between CMT1 and CMT2 clinical phenotypes occurring within the same family. Furthermore, some CMT2 clinical entities show phenotypic overlap with distal hereditary motor neuropathies and other related disorders, further complicating the classification of hereditary peripheral neuropathies.4
Over the past 10 years, significant advances in the biological understanding of the disease, acquired from cellular biology studies and transgenic rodent models, have led to the identification of several possible drug targets for CMT. However, despite some encouraging preclinical results, no clinically successful treatment has emerged. This suggests a need to extrapolate and validate key findings from existing cellular and murine models to other biologically and clinically relevant models that can recapitulate the human disease. Particularly for neurodegenerative diseases, neurons derived from patient-specific induced pluripotent stem cells (iPSCs) present a valuable resource for disease modelling and preclinical studies, which are otherwise only available upon post-mortem tissue sampling.4-6
Most of the efforts in the development of therapies are concentrated towards CMT1 owing to the larger disease burden, leaving an urgent unmet need for treatment in the more diverse group of CMT2 patients. The fragmentation of the CMT2 landscape, caused by the large number of different genes and the low number of patients per gene mutation, has prevented effective drug development. More than 50 causative genes have already been identified for CMT2 (the OMIM disease gene nomenclature begins with CMT2A and exceeds the alphabetical numbering, https://www.omim.org/), and yet nearly 60% of CMT2 patients are left without a genetic diagnosis.4,7 This makes CMT one of the most genetically diverse inherited disorders and poses unique challenges in identifying druggable targets, particularly for CMT2.
Therefore, in this study, we took advantage of iPSC technology to study multiple CMT2 subtypes, covering the most frequent CMT2 causal genes, and differentiated them into iPSC-derived motor and sensory neurons. Our data demonstrate that patient-specific motor and sensory neurons recapitulate the disease signatures of CMT2. We reveal a common trend towards a decrease in the neurite network density, along with alterations in excitability. We also report progressively affected axonal organelle trafficking as well as abnormalities in the mitochondrial expression pattern, morphology and oxidative phosphorylation. Identification of these common pathomechanism(s) may open attractive avenues for drug development, supported by the finding that inhibition of a dual leucine kinase (DLK) is able partially to restore mitochondrial dysfunction in two subtypes of CMT2.
Materials and methods
Generation and maintenance of iPSCs
Primary human fibroblast cultures were obtained from skin biopsies of five unrelated CMT2 patients and two healthy unrelated control subjects (Supplementary Table 1). Human iPSC lines (three clones/genotype) were generated and thoroughly characterized by the Stem Cell Institute, University of Leuven, Belgium as previously described.8 Most importantly, both patient and control lines were reprogrammed together using Sendai virus reprogramming. IPSCs were maintained on Matrigel® coated plates (734–1440, VWR Biotechnologies) in Essential 8TM Flex medium and supplement (A2858501, Thermo Fisher). RevitaCellTM supplement (A2644501, Thermo Fisher) was used additionally to revive iPSCs and enhance cell survival. The medium was refreshed every alternate day and upon reaching confluency, iPSCs were passaged using a brief incubation of 2 min with Versene (EDTA) (LO BE17-711E, Westburg) followed by scrapping the cells in the Essential 8TM Flex medium. The iPSC lines were maintained at 37°C in a humidified 5% carbon dioxide incubator. All the iPSC lines were tested negative for Mycoplasma. All experiments performed using patient material were approved by the Committee for Medical Ethics, University of Antwerp.
Differentiation of iPSCs into motor neurons
Differentiation of the iPSCs into motor neurons was based on a previously published protocol.9 Briefly, at the start of the differentiation, Essential 8TM Flex medium was replaced by neuronal medium, consisting of NeurobasalTM Medium (10888022, Thermo Fisher), DMEM/F12 medium (31330038, Thermo Fisher), N2 supplement (17502048, Thermo Fisher), B-27TM supplement (without vitamin A) (12587010, Thermo Fisher), GlutaMAXTM (35050061, Thermo Fisher), β-mercaptoethanol (11528926, Thermo Fisher) and ascorbic acid (A8960-5G, Sigma-Aldrich). RevitaCellTM was used to enhance cell survival. During the first 2 days, dual SMAD inhibitor SB431542 (1614/10, Bio-Techne), dual SMAD inhibitor LDN-193189 (QT106329, Selleck Chemicals) and WNT antagonist CHIR99021 (4423/10, Bio-Techne) were added to the neuronal medium. On the following days, half of the medium was replaced with media containing compounds necessary for motor neuron differentiation and maturation. Compounds used included: retinoic acid (R2625-50MG, Sigma Aldrich), smoothened agonist (SAG, 4366/1, Bio-Techne), brain derived neurotrophic factor (BDNF, 450-02, Peprotech), glial cell derived neurotrophic factor (GDNF, 450-10, Peprotech), γ-secretase inhibitor DAPT (2634/10, Bio-Techne), ciliary neurotrophic factor (CNTF, 450-13, Peprotech) and laminin (L2020-1MG, Sigma Aldrich). Motor neurons were plated on laminin-coated plates at a density of 105 cells/ml. Additionally, to enhance the purity of the motor neurons, a single treatment with 1 µM cytosine β-d-arabinofuranoside (Ara-C, C1768, Sigma Aldrich) was performed on Day 14.
Differentiation of iPSCs into sensory neurons
Differentiation of the iPSCs into sensory neurons was adapted from a previous study.10 Briefly, Essential 8TM Flex medium was replaced by knockout serum replacement medium (KSR) consisting of KnockOutTM DMEM (10829018, Thermo Fisher), KnockOutTM Serum Replacement (10828028, Thermo Fisher), GlutaMAXTM, non-essential amino acids (11140050, Thermo Fisher) and β-mercaptoethanol. RevitaCellTM was used to enhance cell survival. SB431542 and LDN-193189 were added to the KSR medium to promote anterior neuro-ectoderm specification. On Day 2, CHIR99021, DAPT and potent and selective vascular endothelial growth factor receptor and fibroblast growth factor receptor inhibitor SU5402 (SU, 3300, Tocris) were supplemented to the KSR medium. The medium was then progressively transitioned into neuronal medium consisting of neurobasal medium, N2 supplement, B-27TM supplement (without vitamin A), GlutaMAXTM and β-mercaptoethanol. Additional compounds used to promote neural crest cells and sensory neuron differentiation and maturation were: ascorbic acid, β-nerve growth factor (450-34, Peprotech), neurotrophin-3 (450-03, Peprotech), BDNF and GDNF. Sensory neurons were replated on Matrigel®-coated plates at a density of 2.5 × 105 cells/ml. Additionally, as in our motor neuron protocol, a single treatment with 1 µM Ara-C was performed on Day 14.
Microelectrode array analysis
Dissociated embryoid bodies were plated on microelectrode arrays at a final density of 50 000 cells/well. The microelectrode array plates (24W300/30G-288, Multichannel Systems) were composed of 24 wells, each containing 12 PEDOT-coated gold electrodes (300 µm electrode spacing and 30 µm electrode diameter). At Day 16 of the differentiation protocol, the medium was gradually shifted to BrainPhysTM Neuronal medium (05790, STEMCELL Technologies) to increase the proportion of synaptically active neurons. The spontaneous electrical activity of iPSC-derived motor neurons was recorded (300 s) at a sampling rate of 25 kHz during different development stages. Analysis of all microelectrode array activity and data processing was performed with custom-written MATLAB scripts (The Mathworks, MA, USA). Action potential and network-burst detection was carried out using the QSpike Tools package.11 In short, raw traces were filtered by a band-pass filter followed by peak detection of which the threshold is relative to the median of the recorded voltage. These threshold crossings were used in further analyses; each of these crossings represented a putative action potential detection on an electrode. If more than 0.02 of these occurrences were detected per second on a given microelectrode, the electrode was labelled active. When >30% of the active electrodes showed spiking activity in a 20 ms window, it was classified as a network burst—a network-wide synchronization epoch. The end of such bursts was determined when the network-wide activity was <80% of the maximum activity in the burst. To determine the burst start and end points accurately, a 1 ms spike time histogram was convolved with a Gaussian window. The start and end were then determined on the crossing of relative thresholds. Several other quality conditions applied; a burst was at least 100 ms long, bursts with an inter-burst interval shorter than 100 ms were taken together as one burst and at least 30% of the active electrodes participated in the burst.
Axonal transport and mitochondrial morphology analysis
Motor and sensory neurons were cultured in 8-well Ibidi plates (80826, Proxylab) and incubated with a final concentration of 250 nM MitoTracker™ Red CMH2Xros (M7513, Thermo Fisher) or 100 nM of LysoTrackerTM Red DND-99 (L7528, Thermo Fisher) in neuronal medium for 25 min at 37°C. Afterwards, the medium was removed, and fresh neuronal medium was added. Live-cell imaging at 540 nm was performed using a Carl Zeiss Axiovert 200 M microscope equipped with an incubation chamber (37°C, 5% CO2). Images were taken every 2 s for 3 min with an AxioCam MR Rev3 camera and 40× Plan-Neofluar (1.3 NA) objective, yielding 90 frames. Images were viewed using the AxioVision LE software (version 4.9) and analysed using the Fiji distribution of ImageJ.12,13 To analyse organellular transport, kymographs were generated by drawing segmented lines on the neurites and using the Fiji ‘Multi Kymograph’ function at line thickness 3. From the kymographs, organelle speeds were extracted by drawing straight lines on structures moving at constant speed (straight lines on the kymograph). The tangent of the angle of this line represented the speed (µm/s) after calibration (2 s/pixel in the vertical y-direction and 0.16125 µm/pixel in the horizontal x-direction). The percentage of moving organelles was calculated by dividing the moving organelles by the total number of organelles (including straight vertical lines in kymograph). At least five kymographs were made per recording. In the motor neurons, a mitochondrial speed >1.5 µm/s and lysosomal speed >2 µm/s were considered as outliers, whereas in the sensory neurons, a mitochondrial speed >1 µm/s and lysosomal speed >1.5 µm/s were considered as outliers. The first frame of the image sequence was also used to measure mitochondrial morphology by detecting individual mitochondria segments (as depicted previously14). Briefly, a segmentation procedure was executed using an ImageJ macro script that combined global (Median method) and local (Phansalkar method) automatic intensity thresholding and Boolean operations on the binary masks. The shape factors (aspect ratio and circularity) of mitochondrial segments were measured to describe the mitochondrial morphology.
Measuring mitochondrial oxygen consumption rates
Motor neurons were cultured on 8-well Seahorse XFp miniplates (103025-100, Agilent Technologies). Prior to the Cell Mito Stress Test assay (103010-100, Agilent Technologies), the neuronal medium was changed to Seahorse XF DMEM medium pH 7.4, including the Seahorse XF supplements (glucose, pyruvate and glutamine) (103680-100, Agilent Technologies). The Seahorse injection ports on the sensor cartridge were filled with 1 µM oligomycin (Port A), 2 µM carbonilcyanide p-triflouromethoxyphenylhydrazone (FCCP) (Port B) and 0.5 µM Rotenone/Antimycin A (Port C). All mitochondrial inhibitors were purchased from Agilent Technologies. During the run, the oxygen consumption rates were measured using the Seahorse XF HS Mini Analyzer (Agilent Technologies) and subsequently normalized to total protein levels and quantified following the manufacturer’s instructions.
Drug treatment
Motor neurons were treated with 1 µM of DLK inhibitor (GNE-8505, kindly produced and provided by Janssen Pharmaceutica, Beerse, Belgium) from Day 11 onwards and subsequently refreshed at every media change. An equivalent amount of dimethyl sulphoxide (DMSO) was used as a vehicle.
Statistical analysis
All experiments were performed at least in triplicate, collected from independent differentiations (except for the electron microscopy study and mtDNA copy number quantification). Statistical analysis was performed using GraphPad Prism (version 8.3.0). Normal distribution of the data was tested using the Shapiro-Wilk test. The comparison of two groups was either evaluated using the Student’s t-test (normal distribution) or the Mann-Whitney U-test. Data containing more than two groups were analysed with either a one-way ANOVA followed by Dunnett’s multiple comparisons test (normal distribution) or a Kruskal-Wallis non-parametric test with Dunn’s correction for multiple comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 were considered significant. Data values represent the mean ± standard error of the mean (SEM), except where otherwise mentioned.
Data availability
The generated data are available from the corresponding author upon reasonable request. The Acapella script, written to quantify the images acquired with the Opera Phenix High Content Imaging system is available on GitHub (https://github.com/VerschuurenM/NeuronalConnectivity).
Results
Generation and characterization of CMT2 iPSCs
A particular challenge in using iPSCs to study a genetically diverse disease such as CMT stems not only from differences between genetic variants but also from differences introduced by the development of iPSCs in different laboratories. In this respect, and with the current technology, the same mutation generated at two different labs could produce very different experimental results. This has been prohibitive to the identification of shared molecular deficits between different CMT2 subtypes. To overcome this major challenge, we generated iPSC lines from five different CMT2 mutations along with two healthy controls (Fig. 1A, B and Supplementary Table 1) simultaneously and within the same research environment. In this study, we focused on the following gene mutations representing the respective common CMT2 subtypes: MFN2R94Q (CMT2A), NEFLP8R (CMT2E), HSPB8K141N (CMT2L), HSPB1G84R (CMT2F) and HSPB1P182L (CMT2F). These select genes and their respective mutations comprise the most frequent forms of axonal CMT identified to date.15–18 To account for potential variability, this list also contains two different mutations in the same gene (HSPB1G84R and HSPB1P182L) with different disease severity and affecting different protein domains of HSPB1.19 Reprogramming of fibroblasts was performed using a Sendai virus, consisting of four reprogramming factors (Oct4, Sox2, Klf4 and c-Myc). Pluripotency was confirmed by immunostaining for Nanog, Oct4, SSEA4, Sox2, Tra 1–60 and Tra 1–81 (Supplementary Fig. 1A). A quantitative analysis of self-renewal and tri-lineage differentiation potential (TaqMan® hPSC ScorecardTM assay) was performed (Supplementary Fig. 1B). Additionally, all iPSC lines expressed pluripotent stem cell markers demonstrated by RT-qPCR for Oct4, Sox2 and Nanog (Supplementary Fig. 1C). The absence of the relative viral expression of Sendai and c-Myc was confirmed by RT-qPCR (Supplementary Fig. 1D). Further characterization was performed by DNA fingerprinting, showing identical single nucleotide polymorphism profiles with the corresponding fibroblast sample (Supplementary Table 2). Finally, to account for potential variability, we generated an isogenic control line in which the MFN2R94Q mutation (MFN2ISO) was rescued using the CRISPR/Cas9 gene editing technology (Fig. 1B and Supplementary Fig. 8A). Together, this set of iPSC lines should provide the necessary tools to filter out natural variability and identify molecular changes truly stemming from the disease.
Figure 1.

Generation of CMT2 iPSCs and characterization of iPSC-derived motor neurons. (A) Graphic scheme of procedures for iPSC generation and the neuronal differentiation required to conduct this study. (B) Sequencing analysis confirming the mutations of the patient lines used in this study. (C) Protocol and small molecules used to obtain motor neurons. (D) Immunostaining at Day 25 with ChAT, Isl1, MAP2 and DAPI for patient-specific motor neurons. Scale bar = 100 µm. Ara-C = cytosine β-d-arabinofuranoside; BDNF = brain-derived neurotrophic factor; CHIR = CHIR99021; CNTF = ciliary neurotrophic factor; DAPT = a γ-secretase inhibitor; EB = embryoid body; GDNF = glial cell-derived neurotrophic factor; LDN = LDN-193189; MN = motor neuron; RA = retinoic acid; SAG = smoothened agonist; SB = SB431542.
Differentiation of iPSCs towards motor neurons
Differentiation of iPSCs into motor neurons was based on a previously published protocol.9 The different in vivo neuronal development stages were simulated by adding small molecules in vitro (Fig. 1C). This protocol, in combination with a single treatment with Ara-C on Day 14, ensured high-purity motor neuron cultures. Immunostaining revealed that all iPSC-derived motor neurons were positive for MAP2, Islt1 and ChAT (Fig. 1D). In addition, RT-qPCR demonstrated an increase in the relative expression of ChAT and HB9 over time from iPSC colonies (Day 0) to mature motor neurons (Supplementary Fig. 1E). No significant differences between mature patient lines and controls were observed. Moreover, the isogenic MFN2ISO line was also able to differentiate into motor neurons successfully. These patient and control iPSC-derived motor neuron lines were used in this study to seek common features that differentiated healthy from axonal CMT disease phenotypes.
To benchmark our lines, we first verified whether we would observe previously reported gene-specific phenotypes for certain CMT2 subtypes. For instance, mutations in the neurofilament light chain gene (NEFL), causing CMT2E, disturb the assembly of the intermediate filament network, by accumulating in the perikarya and neurites in particular.20 We were able to validate neurofilament light (NF-L) inclusions in the cell body as well as in the neurites of CMT2E-derived motor neurons by performing immunostaining for NF-L at Day 37 (Supplementary Fig. 2A). Blinded quantification revealed a significant increase in NF-L compared to a healthy controls, using NF-L signal thresholds (the total area of NF-L inclusions divided by a background of NF-L staining) (Supplementary Fig. 2B). In addition, filamentous accumulations were confirmed by ultrastructural analysis of the NEFLP8R motor neuron line (Supplementary Fig. 2C). A second example of a gene-specific phenotype is the formation of HSPB1-positive aggregates due to C-terminal HSPB1 mutations.21 We validated the presence of HSPB1 aggregates in our iPSC-derived motor neurons from the HSPB1P182L line, using immunostaining at Day 37 for HSPB1 (Supplementary Fig. 3A). While no aggregates were seen for the N-terminal HSPB1G84R mutation, a proportion of the HSPB1P182L cells contained large cytoplasmic aggregates (Supplementary Fig. 3A). Additionally, we confirmed a known reduction in α-tubulin acetylation levels for the HSPB1P182L patient line, first reported in a CMT2F mouse model (Supplementary Fig. 3B).22 Lastly, we recapitulated a decrease in BAG3 levels in iPSC-derived motor neurons for the HSPB8K141N mutation (Supplementary Fig. 4), a finding that has been observed and reported by us in the sciatic nerve of 12-month-old symptomatic Hspb8K141N/K141N knock-in mice and reflects alterations in autophagy.23 In conclusion, these CMT2-derived motor neurons recapitulated previously described gene-specific alterations. However, it remains unclear what unites these different CMT2 subtypes.
CMT2 motor neurons show neurite network deficits with altered extracellular electrophysiological properties
As alterations in the neurite network have been reported in iPSC-derived neuronal models as well as in animal models of peripheral neuropathies,24,25 we sought to explore whether a decrease in the neurite network would be a common phenotype in our CMT2 iPSC-derived motor neurons. We applied two methods to investigate this. First, we analysed phase-contrast images of neuronal cultures at Days 38–50 using pixel classification to segment and measure the number of neurites and cell bodies (Fig. 2A and B). Complementary to this, we studied the neuronal network at Days 25–30 by means of high-throughput automated fluorescence confocal microscopy (Fig. 2C). Of all iPSC-derived motor neuron lines, the two control lines on average had the densest neurite network in both types of analysis. With an equal number of cells after 25–30 days (Fig. 2D), a statistically significant decrease in neurite length was found for the MFN2R94Q and NEFLP8R lines (Fig. 2E). This suggested that CMT2 mutations negatively affect the neurite network of iPSC-derived motor neurons.
Figure 2.

Neurite network analysis of patient-specific motor neurons. (A) Example of neurite and cell body segmentation of phase contrast images using Weka-based pixel classification. (B) Quantification of normalized neurite length at Days 38–50 performed using phase-contrast images. Each dot represents a normalized microscopic field (n = 29–42; data pooled from two independent differentiations; mean ± SEM). (C) Example of a high-content image using MAP2 staining. (D and E) High-content imaging analysis at Days 25–30. Quantification of nuclear count and neurite length, respectively. Each dot represents the average value per well (n = 21–36; data pooled from five independent differentiations; mean ± SEM). (F) Representative raster plot of bursts recorded over 300 s. Each bar indicates a spike and each line an electrode. (G) Quantification of the burst rate (Days 31–32). Individual data-points represent the average value of one well (n = 9–16; data pooled from two independent differentiations; mean ± SEM). (B–H) Statistical significance was calculated using Kruskal-Wallis test with Dunn’s multiple comparison (*P < 0.05, **P < 0.01, ***P < 0.001, **** P < 0.0001). Scale bar = 100 µm.
To explore whether these small changes in the neurite network were coupled to alterations in neuronal activity, we performed microelectrode array experiments. The spontaneous electrical activity of iPSC-derived motor neurons was recorded at a sampling frequency of 25 kHz during different development stages. Representative traces that were collected over a 300 s recording period are shown and were characterized by isolated and periodic collective spiking events (Fig. 2F). Analysis of the traces indicated changes in the burst rate for all CMT2-derived motor neurons compared with the controls (Fig. 2G). The MFN2R94Q and NEFLP8R lines showed an increase in burst rate, suggesting hyper-connectivity. In contrast, the heat-shock protein mutants (HSPB8K141N, HSPB1G84R and HSPB1P182L) showed a trend towards a decrease in burst rate, suggesting a hypo-connectivity state. While gene-specific differences could explain the opposing trends between different CMT2 subtypes, changes either way could contribute to neuronal and axonal degeneration.9,20
Progressive axonal transport and morphological deficits in CMT2-derived motor neurons
Previous studies have reported axonal transport defects such as decreased mitochondrial speed in iPSC-derived neurons of CMT2A, CMT2E or CMTX6.20,26 However, it remains unknown whether these hallmarks of axonal degeneration are common to all CMT2 subtypes. To verify whether this was at least a shared feature in our five CMT2 lines, we performed live-cell imaging experiments to study mitochondrial and lysosomal trafficking in both control and patient iPSC-derived motor neurons over time using MitoTrackerTM and LysoTrackerTM dyes, respectively. We observed a significant decrease in mitochondrial speed in all patient-derived motor neurons at Days 38–50 (Fig. 3A), whereas at Days 22–28, only the MFN2R94Q motor neurons showed a decrease in mitochondrial speed (Supplementary Fig. 5A). The percentage of moving mitochondria was not significantly different; however, at Days 22–28 we observed a significant decrease in the MFN2R94Q patient line (Supplementary Fig. 5A). Similar to the mitochondrial speed, the lysosomal speed was significantly decreased in all patient motor neurons at Days 38–50 (Fig. 3B), whereas at Days 22–28, the MFN2R94Q, NEFLP8R and HSPB8K141N motor neurons showed a trend towards a decrease in lysosomal speed (Supplementary Fig. 5B). In addition, the percentage of moving lysosomes was significantly decreased at Days 38–50 (Supplementary Fig. 5B). These live-cell imaging experiments indicated that axonal transport was generally impaired and mitochondrial as well as lysosomal trafficking worsened over time.
Figure 3.

CMT2-derived motor neurons cause axonal transport deficits and altered mitochondrial morphology. (A) Mitochondrial transport analysis at Days 38–50 using kymographs (left). Quantification of mitochondrial speed (n = 182–295) and percentage of moving mitochondria (n = 8–11) was performed after MitoTrackerTM labelling and imaging for 3 min. (B) Lysosomal transport analysis at Days 38–50 using kymographs (left). Quantification of lysosomal speed (n = 161–433) and percentage of moving lysosomes (n = 6–10) was performed after LysoTrackerTM labelling and imaging for 3 min. (A and B) Examples of representative kymographs showing moving (yellow boxes and arrowheads) and stationary (red boxes and arrowheads) organelles. Dots represent either individual organelles (speed) or the average value of the kymographs per recording (% moving) (data pooled from two independent differentiations; mean ± SEM). (C) Representative fluorescence mask (MitoTrackerTM) used to study the mitochondrial shape. (D and E) Quantification of mitochondrial circularity and aspect ratio. Data-points represent the average values for the mitochondria present in the image (n = 8–10; data pooled from two independent differentiations; mean ± SEM). Statistical significance to evaluate speed was calculated using one-way ANOVA followed by Dunnett’s multiple comparisons test, while Kruskal-Wallis test with Dunn’s multiple comparison was used to perform statistics on the percentage of moving organelles and mitochondrial morphology (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Scale bar = 20 µm.
Given the intimate link between mitochondrial functionality and shape, we studied the effect of CMT2 mutations on mitochondrial shape (Fig. 3C). For MFN2 mutations, it is already known that fusion is impaired, causing mitochondrial fragmentation.27,28 However, for the other CMT2 mutants, this has not yet been systematically investigated. To assess whether alterations in mitochondrial morphology could form another common feature, we analysed the mitochondrial shape parameters in motor neuron cultures labelled with MitoTrackerTM dye. Our data revealed reduced mitochondrial elongation, with an increase in circularity (Fig. 3D) and a decrease in aspect ratio (Fig. 3E) in all patient-derived motor neurons between Days 38 and 50. Interestingly, for the MFN2R94Q and NEFLP8R mutant motor neurons, this was already observable at Days 22–28 (Supplementary Fig. 6A and B). Alterations in the mitochondrial morphology for the MFN2R94Q and NEFLP8R mutant motor neurons were further confirmed by transmission electron microscopy at Day 25 (Supplementary Fig. 6C and D). Altogether, our results demonstrated a progressively affected axonal transport and fragmented mitochondrial morphology in all the patient lines.
Motor neuron-specific subtypes of CMT2 do not affect iPSC-derived sensory neurons
Since the MFN2R94Q and NEFLP8R CMT2 patients displayed both motor and sensory symptoms (Supplementary Table 1), we investigated whether these mutations also affected the sensory neurons. Therefore, we differentiated iPSCs into peripheral sensory neurons using a previously published protocol10 (Fig. 4A), which produced a heterogeneous sensory neuronal culture.29 Immunostaining at Day 37 revealed that all iPSC-derived sensory neurons were positive for Beta-III-Tub, BRN3A and TRPV1 (Fig. 4B).
Figure 4.

Motor neuron-specific subtypes of CMT2 do not change axonal transport and mitochondrial morphology in iPSC-derived sensory neurons. (A) Protocol and small molecules used to obtain peripheral sensory neurons. (B) Immunostaining at Day 37 with Beta-III-Tub, BRN3A, TRPV1 and Hoechst 33342 for iPSC-derived sensory neurons. (C) Quantification of normalized neurite length at Days 38–50, performed using phase-contrast images. Each dot represents a normalized microscopic field (n = 26–36; data pooled from two independent differentiations; mean ± SEM). (D) Top: Mitochondrial transport analysis at Days 38–50 using kymographs. Quantification of mitochondrial speed (left) (n = 84–101) and percentage of moving mitochondria (right) (n = 8–10) was performed using MitoTrackerTM labelling. Bottom: Lysosomal transport analysis at Days 38–50 using kymographs. Quantification of lysosomal speed (left) (n = 129–258) and percentage of moving lysosomes (right) (n = 9–12) was performed using LysoTrackerTM labelling. Dots represent either individual mitochondria (speed) or the average value of the kymographs per recording (% moving) (data pooled from two independent differentiations; mean ± SEM). (E) Quantification of mitochondrial aspect ratio. Data-points represent the average values for the mitochondria present in the image (n = 11–19; data pooled from two independent differentiations; mean ± SEM). Statistical significance to evaluate speed was calculated using one-way ANOVA followed by Dunnett’s multiple comparisons test, while Kruskal-Wallis test with Dunn’s multiple comparison was used to perform statistics on the neurite network analysis, percentage of moving organelles and mitochondrial morphology (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Scale bar = 100 µm. AA = ascorbic acid; Ara-C = cytosine β-d-arabinofuranoside; β-NGF = β-nerve growth factor; BDNF = brain-derived neurotrophic factor; CHIR = CHIR99021; DAPT = a γ-secretase inhibitor; GDNF = glial cell-derived neurotrophic factor; KSR = knockout serum replacement medium; LDN = LDN-193189; MEF-CM = mouse embryonic fibroblast-conditioned medium; NCPC = neural crest stem cell; Neuronal = neuronal medium; NT-3 = neurotrophin-3; SB = SB431542; SU = SU5402; SN = sensory neuron.
We first assessed whether gene-specific phenotypes were reproduced in iPSC-derived sensory neurons. We observed that NF-L accumulations were also present in patient iPSC-derived sensory neurons, as demonstrated by a significant increase of NF-L inclusions in the perikarya and neurites of the NEFLP8R patient line at Day 37 (Supplementary Fig. 2D-E). For HSPB1P182L, we only observed a decrease in α-tubulin acetylation in iPSC-motor neurons but not iPSC-sensory neurons at Day 37 (Supplementary Fig. 3C), which was in line with the predominant motor neuron phenotype in this type of CMT2 gene mutation. We also measured the neurite length in these iPSC-derived sensory neurons on phase contrast images of living unstained neurons, similarly as for the motor neurons. A significant decrease in neurite length occurred at Days 38–50 in the MFN2R94Q and NEFLP8R sensory neuron lines (Fig. 4C).
Next, we measured axonal transport using MitoTrackerTM and LysoTrackerTM to label the mitochondria and lysosomes, respectively. Kymograph analysis showed a significant reduction at Days 38–50 in the mitochondrial and lysosomal speeds for the MFN2R94Q and NEFLP8R sensory neuron lines (Fig. 4D). Furthermore, a reduced number of moving mitochondria and lysosomes was observed in these two patient lines compared with both the controls (Fig. 4D). We also found that the mitochondrial shape in the MFN2R94Q and NEFLP8R sensory neurons, similar to motor neurons, revealed a decrease in aspect ratio (Fig. 4E). Note that the speed distribution of mitochondrial and lysosomal trafficking was markedly different between the motor and sensory neurons: axonal transport speeds were significantly higher in the motor neurons compared with the sensory neurons of healthy controls (Supplementary Fig. 7A–D). This was in line with in vivo data in which axonal trafficking of signalling endosomes was significantly faster in motor versus sensory neurons of ChAT-eGFP mice.30 These differences in speed could potentially arise from differences in the axon thickness between motor and sensory neurons.
Combined, our results suggested that neurite length, axonal transport and mitochondrial shape were particularly disturbed in iPSC-derived sensory neurons from CMT2 patients with sensory symptoms, while the sensory neurons from CMT2 subtypes with a predominant motor-phenotype (HSPB1G84R, HSPB1P182L and HSPB8K141N) were indifferent from control iPSC-derived sensory neurons.
Correcting the MFN2R94Q mutation using gene editing restores disease phenotypes
In this study, we observed axonal degeneration features in differentiated motor and sensory neurons from the MFN2R94Q patient iPSC line. We applied CRISPR/Cas9 to edit the pathogenic MFN2R94Q patient iPSC line to generate a corrected MFN2ISO line (Fig. 1B and Supplementary Fig. 8A) and assess whether the correction rescues the characteristic disease phenotypes in motor and sensory neurons. This revealed that correction of the MFN2R94Q mutation rescued neurite length in iPSC-derived motor and sensory neurons (Supplementary Fig. 8B, C andG). Also the disturbances in extracellular electrophysiological properties were corrected, as demonstrated by a restoration of the burst rate (Supplementary Fig. 8D). The fragmented mitochondrial morphology and the altered mitochondrial and lysosomal trafficking at Days 38–50 were likewise corrected in both iPSC-derived motor and sensory neurons derived from the MFN2ISO line (Supplementary Fig. 8E, F, H and I). In summary, the isogenic MFN2ISO line restored the disease phenotype of the MFN2R94Q patient line, demonstrating that our results in iPSC-derived motor and sensory neurons are true hallmarks of CMT2A.
Altered mitochondrial expression profile in iPSC-derived motor neurons of CMT2
To globally delineate the transcriptomic profile between the two healthy control and five CMT2 patient iPSC-derived motor neurons, we performed bulk RNA-sequencing on Day 50 derived motor neurons. Principal component analysis (PCA) demonstrated that the axonal CMT samples clustered separately from the controls (Supplementary Fig. 9A). In the transcriptome of the five different CMT2 patient-derived motor neurons, 2846 genes were differentially expressed when compared with both controls (Fig. 5A). We employed RT-qPCR to validate some of the most common differentially expressed genes (NKX6-1, CNTN4 and KHDRBS2), which confirmed the differential expression of these transcripts between the CMT2 lines and controls (Fig. 5B). To discriminate between gene-specific versus shared differentially expressed transcripts, we performed a Venn diagram analysis (Fig. 5C). This allowed us to analyse the overlap between each of the CMT2 patient motor neuron lines and identify a core profile of 740 transcripts that were shared by all CMT2 gene mutations. Gene ontology (GO) analysis of these common differentially expressed genes identified enriched cellular component GO terms such as ‘respiratory chain’ and ‘axon’ (Supplementary Fig. 9BandC). Furthermore, in the top associated enriched pathways we found ‘axon guidance pathway’ and the ‘PI3K-Akt signalling pathway’ (Supplementary Fig. 9C). The latter pathway was also shown to be imbalanced in CMT rodent models.31
Figure 5.

Transcriptional profile of CMT2 iPSC-derived motor neurons at Day 50. (A) Volcano plot of log2 fold change versus mean comparing the control lines with the patients. Boxes are used to indicate genes that were validated using RT-qPCR. (B) RT-qPCR to confirm the relative expression of differentially expressed genes (DEGs) in iPSC-derived neurons at Day 50 (n = 3; data pooled from two independent differentiations; mean ± SEM). (C) Venn-diagram of the DEGs of CMT2 models compared with healthy donors. (D) Network analysis of the DEGs responsible for the top terms shown in Cellular Component terms. (E) PCA of all mitochondrial coded genes in healthy control and patient samples. Statistical significance of RT-qPCR results was calculated using one-way ANOVA followed by Dunnett’s multiple comparisons test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
One of the top hits detected was the ‘respiratory chain’, linking this core profile of differentially expressed genes to the mitochondria. We confirmed this with an additional PCA, showing that healthy controls and CMT2 samples clustered separately when analysing only the mitochondrial encoded genes (Fig. 5E). This was in line with our earlier results showing that mitochondrial transport and morphology are affected in all CMT2 patient iPSC lines. To construct a connectivity network of the differentially expressed genes underlying the top hits in the ‘respiratory chain’ term, we performed GeneMANIA gene network clustering (Fig. 5D) and this showed that many of the hits were interconnected at multiple levels, ranging from direct protein-protein interactions to proteins of the same pathway, further underscoring how different CMT2 genes could dysregulate the same mitochondrial processes.
Motor neurons generated from CMT2 patients have an altered bio-energetic profile
Our results to this point showed that mitochondrial transport and morphology were dysregulated and that this feature was common to all CMT2 motor neuron lines used in this study. Unsurprisingly, ‘PI3K-Akt signalling’ and ‘respiratory chain’ were therefore identified as associated GO terms in our transcriptomic analysis, indicating that mitochondrial dysfunction might be sensed by cytosolic stress integrators like Akt and AMPK. We therefore verified whether the Akt and AMPK phosphorylation levels were different in the CMT2 lines versus the controls. This confirmed a clear increase in phosphorylated Akt and AMPK in all CMT2 patient lines (Fig. 6A). To assess whether these in vitro findings would also extend to in vivo models, we isolated the sciatic nerve of 1-year-old symptomatic Hspb8K141N/K141N mice modelling CMT2L. Similar to our observations in the iPSC-motor neurons, we found an increase of phosphorylated Akt and AMPK in the sciatic nerve (Fig. 6B). Given that the sciatic nerve is not composed purely of motor neurons, as it also contains other cell types such as Schwann cells, fibroblasts and endothelial cells, the actual effect on motor neurons possibly remains underestimated.
Figure 6.
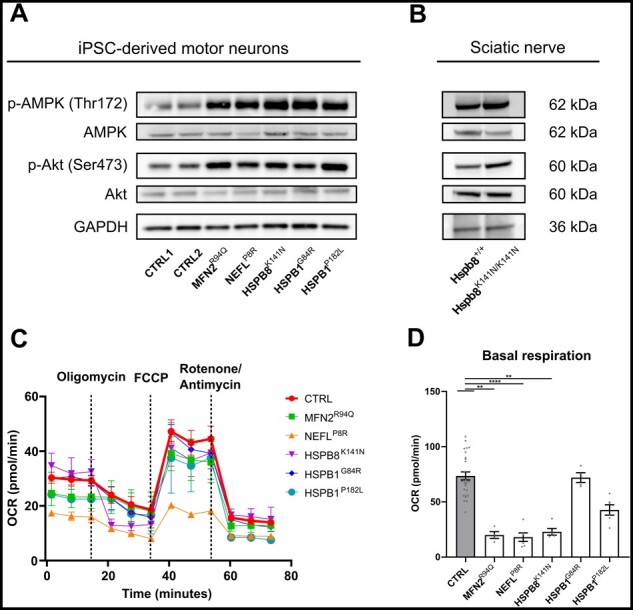
Mitochondrial function is impaired in CMT2 iPSC-derived motor neurons. (A) Expression levels of p-AMPK and p-Akt at Day 45 in iPSC-derived motor neurons determined using western blot. (B) Expression levels of p-AMPK and p-Akt in the sciatic nerve of 12-month-old Hspb8K141N/K141N mice. (C) Oxygen consumption rate (OCR) in iPSC-derived motor neurons at Days 24–25 using a Mito Stress Test [n = 4–25; data pooled from two independent differentiations; mean ± standard deviation (SD)]. (D) Quantification of basal respiration at Days 38–39. Individual data-points represent the average value per well (n = 4–27; data pooled from two independent differentiations; mean ± SEM). Statistical significance was calculated using one-way ANOVA followed by Dunnett’s multiple comparisons test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
As AMPK is a well-known sensor for cellular energy metabolism,32 we investigated whether these CMT2 mutations would exhibit deficits in oxidative phosphorylation. We studied mitochondrial respiration using a Seahorse analyser and assessed the mitochondrial function with the Cell Mito Stress Test assay (Fig. 6C). Measurements of the mitochondrial basal respiration at Days 38–39 revealed a decrease in the MFN2R94Q, NEFLP8R, HSPB8K141N and HSPB1P182L patient lines (Fig. 6D), which coincided with the altered mitochondrial shape (Supplementary Fig. 6A and B) and reduced mtDNA copy number (Supplementary Fig. 11) observed at this time point. At Days 24–25, a decrease in basal respiration and affected mitochondrial morphology could already be observed in the MFN2R94Q and NEFLP8R patient motor neuron lines (Supplementary Fig. 6A, B, D and Supplementary Fig. 10A). Finally, we also demonstrated that the isogenic MFN2ISO line was able to restore defective oxidative phosphorylation in the MFN2R94Q patient line, both in early and late time point experiments (Supplementary Fig. 10BandC).
DLK inhibition ameliorates mitochondrial dysfunction in motor neurons generated from CMT2A and CMT2E patients
As our data demonstrated that mitochondrial dysfunction was a common feature in our CMT2 lines and that stress pathways impinging on this are overactivated, we sought to explore whether modulating these stress pathways could ameliorate some of the disease-associated phenotypes. As MAPKs can directly and indirectly target mitochondria,33,34 we investigated whether we could target these stress-induced transcriptional changes (Fig. 7A) using a DLK (also known as MAP3K12) inhibitor. DLK acts upstream of the stress-responsive c-Jun N-terminal kinase (JNK) pathway (a member of the MAPK protein family) and is highly expressed in neuronal cells. In addition, mitochondrial dysfunction and DLK activation reinforce one another in inducing axonal damage.35 Furthermore, DLK and, by an extent, the JNK pathway are also intimately involved in axonal organelle transport.36 This DLK inhibitor is currently in a phase I clinical trial for amyotrophic lateral sclerosis (NCT02655614), as it has been shown not only to reduce the activation of JNK but also delay denervation at the neuromuscular junctions of SOD1G93A mice.37 Moreover, the loss of DLK is well tolerated.38 We focused on the MFN2R94Q and NEFLP8R patient motor neuron lines, since both display the most severely altered mitochondrial phenotype, and treated the motor neurons with 1 µM of DLK inhibitor from Day 11 onwards. To validate the effect of the treatment, total c-Jun levels were measured and the increase in these levels in the MFN2R94Q and NEFLP8R patient motor neurons lines decreased upon inhibition by DLK (Fig. 7B). To determine whether such a reduction in stress signalling would have a positive impact on mitochondrial functionality, we treated the motor neurons with the DLK inhibitor and assessed the mitochondrial morphology, mitochondrial respiration and mitochondrial transport at Days 38–50 (Fig. 7C–E). These experiments indicated that DLK inhibition led to an increase in the mitochondrial aspect ratio and basal respiration in both the MFN2R94Q and NEFLP8R lines. In addition, although not significant, it led to a slight increase in the mitochondrial transport speed and percentage of moving mitochondria. These results indicated that DLK inhibition was able partially to rescue mitochondrial dysfunction, demonstrating its broad therapeutic potential, and that targeting such shared pathways could pave the way for a universal CMT2 treatment.
Figure 7.

DLK inhibition restores mitochondrial dysfunction in motor neurons from CMT2A and CMT2E patients. (A) Venn diagram of the common DEGs of the MFN2R94Q and NEFLP8R mutations compared with healthy donors. Enriched pathways of the common overlapping differentially expressed genes (DEGs) are shown (bottom). (B) Total c-Jun levels at Day 40 in untreated (DMSO) and treated (DLK-inhibitor) iPSC-derived motor neurons determined using western blot. (C) Quantification of mitochondrial aspect ratio. Data-points represent the average values for the mitochondria present in the image (n = 19–21; data pooled from two independent differentiations; mean ± SEM). (D) Quantification of basal respiration at Days 38–39. Individual data-points represent the average value per well (n = 8–15; data pooled from two independent differentiations; mean ± SEM). (E) Quantification of mitochondrial speed (n = 107–187) and percentage of moving (n = 8–9) mitochondria was performed after MitoTrackerTM labelling and imaging for 3 min. Dots represent either individual mitochondria (speed) or the average value of the kymographs per recording (% moving) (data pooled from two independent differentiations; mean ± SEM). (C–E) Statistical significance of the rescue of mitochondrial morphology, basal respiration and percentage of moving mitochondria was calculated using the Mann-Whitney U-test, while the effect of the treatment on mitochondrial speed was evaluated using the unpaired t-test (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Discussion
Inherited disorders are often caused by a select group of genes. One of the best-known exceptions is the axonal form of CMT neuropathy, caused by a large number of mutations in more than 50 disease-associated genes. This genetic diversity, further complicated by clinical heterogeneity, has led to the association between axonal CMT and a vast number of pathways. One of the main reasons for the lag in the development of therapies for CMT2 is that the shared and gene-specific pathways remain largely unresolved. Our study has addressed this question and identified certain shared pathways.
Our data show that there are common alterations in mitochondrial morphology, mitochondrial gene expression patterns, extracellular electrophysiology, oxidative phosphorylation and mtDNA copy number in five different axonal CMT iPSC lines representing a diverse set of autosomal dominant CMT2 genes. Of note, an altered mitochondrial metabolic defect was recently reported in iPSC-derived motor neurons from CMTX6 patients.26 These results indicate that mitochondrial energy deficits could form a common hallmark of axonal CMT neuropathies. Given the length of these neurons, which can be up to 1 m in adults, this may be further exacerbated by axonal transport defects. While high levels of ATP are required to support axonal survival and nerve conduction at, for instance, the nodes of Ranvier, deficits in both transport and energy production could form a double hit that prevents the axon from sustaining this high energy demand and ultimately leads to axonal dysfunction. It is therefore not surprising that there have been observations of axonal transport defects or mitochondrial dysfunction in many models of CMT disease (as reviewed previously by Beijer et al.39). However, whether these factors truly contribute to the pathogenesis of the disease or whether the impairment of these functions is simply an end point of a degenerating axon has remained elusive.
Our results show that, besides gene-specific phenotypes, deficits in mitochondria and axonal transport are common across different CMT2 subtypes and these are not simply downstream end points of axonal degeneration. This is supported by the fact that these defects are progressive and coincide with general electrophysiological and neurite network deficits. These results therefore also imply that therapies oriented towards restoring axonal transport or mitochondrial activity may hold the potential to rescue axonal degeneration in multiple CMT2 subtypes. However, it remains unclear which of these parameters is essential to rescuing axonal degeneration or whether both transport and mitochondrial activity must be rescued to prevent axonal degeneration.
The complex interplay between axonal transport and mitochondrial activity is currently incompletely understood. Studies using the mito-KillerRed molecule, which leads to the release of reactive oxygen species upon photoactivation, have shown that local activation in a subdomain of the neurite leads to mitochondrial stalling.40 Presumably, the nerve attempts to prevent the spreading of oxidative damage by quarantining the mitochondria at the site of insult. Similarly, mitophagy of dysfunctional mitochondria along the neurite also occurs in the axon itself.41 It thus appears that local mitochondrial deficits can lead directly to mitochondrial stalling. How this is controlled at the molecular level is less clear, but it could involve a switch from dynamic kinesin and dynein transporters to static syntaphilin anchoring proteins.42 Moreover, inter-organelle contacts may further determine the fate of dysfunctional mitochondria. Specifically, mitochondria-associated membranes are contact sites between mitochondria and the endoplasmic reticulum and are widely implicated in neurodegeneration.43 In addition, for CMT2 it has been shown that defects in mitochondria-associated membranes contribute to mutant MFN2 pathology.44
An important sensor of such local oxidative damage is AMPK.45 Our data show an increased activation of AMPK in the mutant CMT lines suggests a primary mitochondrial deficit. Moreover, we observed that both axonal transport and mitochondrial activity deficits occur at the same time, which further suggests that they are interconnected, and restoring one could lead to amelioration of the other. This notion is supported by the fact that DLK inhibition ameliorated both mitochondrial transport, morphology and functionality in MFN2R94Q and NEFLP8R patient motor neuron lines. Additionally, in three CMT2 mouse models it has been shown that HDAC6 inhibitors can restore tubulin acetylation and consequently improve axonal transport and the total number of mitochondria.22,46 However, for HDAC6 inhibitors, their primary mode-of-action is less clear. They facilitate axonal transport through the control of tubulin acetylation levels, but HDAC6 also tightly controls the acetylation of Miro1 and thus mitochondrial transport.47 Moreover, the loss of Miro1 is sufficient to induce axonal degeneration in hippocampal neurons.48 Thus, not just mitochondrial dysfunction can cause axonal degeneration, mitochondrial transport is equally important to maintaining axonal integrity by removing damaged organelles and replenishing the axon with healthy mitochondria. Thus, in future it will be important to unravel the molecular details of potential interconnectedness between axonal transport and mitochondrial dysfunction in CMT2. Furthermore, other common pathways may exist, such as endoplasmic reticulum-phagy, which appears to be a common pathway for a group of genes linked to sensory neuropathies.49,50
In summary, we have identified common disease mechanisms in neurons differentiated from CMT2 patient-derived iPSC lines and this opens new, additional opportunities to develop drugs for inherited neuropathies, which will potentially be beneficial to a larger group of CMT patients.
Supplementary Material
Acknowledgements
The authors thank the patients and control individuals who volunteered to provide skin biopsies to generate iPSC lines for research purposes with informed consent. We also appreciate the longstanding collaboration with the clinicians who documented these patients in previous phenotype-genotype correlation studies, those being professors Angelika Hahn, Michaela Auer-Grumbach, Pavel Seeman, Peter De Jonghe and Jonathan Baets. We have appreciated the help of Dr Manisha Juneja in the initial training of the first author and initiation of some motor neuron differentiation experiments. We thank Marlies Verschuuren for assistance with the image data analysis. We also thank professor Jean-Pierre Timmermans, Dr Isabel Pintelon and Ms Karen Sterck from the Antwerp Centre for Advanced Microscopy (ACAM) for assistance with spinning disk and electron microscopy. Furthermore, we would also like to thank Mike Wijnants for critical discussions during the MEA data analysis.
Funding
This work was supported in part by the University of Antwerp (DOC-PRO4 PhD fellowship to J.V.L., TOP-BOF research grant N° 38694 to V.T., BOF research grant N° 41739 and VLAIO grant HBC.2016.0534 to W.D.V.), the Fund for Scientific Research (FWO-Flanders research grant N° G041416N to V.T., grant N° G017618N to W.D.V, and FWO postdoc fellowship to EA), the “Association Belge contre les Maladies Neuromusculaires” (ABMM grants to E.A. and V.T.), the Medical Foundation Queen Elisabeth (GSKE grant to V.T.), the American Muscular Dystrophy Association (MDA research grant N° 577497 to V.T.), and the EU projects NEUROMICS (FP7 under grant agreement number N° 2012–305121) and Solve-RD (Horizon 2020 under grant agreement N° 779257). The UltraView ERS confocal spinning disk microscope and Tecnai G2 Spirit Bio Twin Electron Microscope was supported by FWO-HERCULES large infrastructure grants N° 23714 and N° 25340. The Seahorse XF HS Mini Analyzer was supported by the University of Antwerp Basic Research Infrastructure grant N° 41438. V.T. and W.D.V. are members of the µNeuro Center of Excellence at the University of Antwerp.
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Glossary
- CMT
Charcot-Marie-Tooth
- DLK
dual leucine kinase
- iPSC
induced pluripotent stem cell
References
- 1.Baets J, De Jonghe P, Timmerman V.. Recent advances in Charcot-Marie-Tooth disease. Curr Opin Neurol. 2014;27(5):532–540. [DOI] [PubMed] [Google Scholar]
- 2.Pisciotta C, Shy ME.. Neuropathy. Handbook Clin Neurol. 2018;148:653–665. [DOI] [PubMed] [Google Scholar]
- 3.Pareyson D.Axonal Charcot-Marie-Tooth disease: The fog is only slowly lifting. Neurology. 2007;68(20):1649–1650. [DOI] [PubMed] [Google Scholar]
- 4.Pipis M, Rossor AM, Laura M, Reilly MM.. Next-generation sequencing in Charcot–Marie–Tooth disease: Opportunities and challenges. Nat Rev Neurol. 2019;15(11):644–656. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi K, Yamanaka S.. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. [DOI] [PubMed] [Google Scholar]
- 6.Juneja M, Burns J, Saporta MA, Timmerman V.. Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J Neurol Neurosurg Psychiatry. 2019;90(1):58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossor AM, Polke JM, Houlden H, Reilly MM.. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol. 2013;9(10):562–571. [DOI] [PubMed] [Google Scholar]
- 8.Juneja M, Azmi A, Baets J, et al. PFN2 and GAMT as common molecular determinants of axonal Charcot-Marie-Tooth disease. J Neurol Neurosurg Psychiatry. 2018;89(8):870–878. [DOI] [PubMed] [Google Scholar]
- 9.Guo W, Naujock M, Fumagalli L, et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat Commun. 2017;8(1):861- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chambers SM, Qi Y, Mica Y, et al. Combined small-molecule inhibition accelerates developmental timing and converts human pluripotent stem cells into nociceptors. Nat Biotechnol. 2012;30(7):715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahmud M, Pulizzi R, Vasilak E, Giugliano M.. QSpike tools: A generic framework for parallel batch preprocessing of extracellular neuronal signals recorded by substrate microelectrode arrays. Front Neuroinform. 2014;8(26). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schneider CA, Rasband WS, Eliceiri KW.. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: An open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Estrada-Cuzcano A, Martin S, Chamova T, et al. Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain. 2017;140(2):287–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jordanova A, De Jonghe P, Boerkoel CF, et al. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease. Brain. 2003;126(Pt 3):590–597. [DOI] [PubMed] [Google Scholar]
- 16.Züchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36(5):449–451. [DOI] [PubMed] [Google Scholar]
- 17.Irobi J, Van Impe K, Seeman P, et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 2004;36(6):597–601. [DOI] [PubMed] [Google Scholar]
- 18.Evgrafov OV, Mersiyanova I, Irobi J, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36(6):602–606. [DOI] [PubMed] [Google Scholar]
- 19.Adriaenssens E, Geuens T, Baets J, Echaniz-Laguna A, Timmerman V.. Novel insights in the disease biology of mutant small heat shock proteins in neuromuscular diseases. Brain. 2017;140(10):2541–2549. [DOI] [PubMed] [Google Scholar]
- 20.Saporta MA, Dang V, Volfson D, et al. Axonal Charcot-Marie-Tooth disease patient-derived motor neurons demonstrate disease-specific phenotypes including abnormal electrophysiological properties. Exp Neurol. 2015;263:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alderson TR, Adriaenssens E, Asselbergh B, et al. A weakened interface in the P182L variant of HSP27 associated with severe Charcot-Marie-Tooth neuropathy causes aberrant binding to interacting proteins. EMBO J. 2021;e103811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.d'Ydewalle C, Krishnan J, Chiheb DM, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med. 2011;17(8):968–974. [DOI] [PubMed] [Google Scholar]
- 23.Bouhy D, Juneja M, Katona I, et al. A knock-in/knock-out mouse model of HSPB8-associated distal hereditary motor neuropathy and myopathy reveals toxic gain-of-function of mutant Hspb8. Acta Neuropathol. 2018;135(1):131–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ponomareva OY, Eliceiri KW, Halloran MC.. Charcot-Marie-Tooth 2b associated Rab7 mutations cause axon growth and guidance defects during vertebrate sensory neuron development. Neural Dev. 2016;11:2- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujimori K, Ishikawa M, Otomo A, et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat Med. 2018;24(10):1579–1589. [DOI] [PubMed] [Google Scholar]
- 26.Perez-Siles G, Cutrupi A, Ellis M, et al. Energy metabolism and mitochondrial defects in X-linked Charcot-Marie-Tooth (CMTX6) iPSC-derived motor neurons with the p.R158H PDK3 mutation. Sci Rep. 2020;10(1):9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Filadi R, Pendin D, Pizzo P.. Mitofusin 2: From functions to disease. Cell Death Dis. 2018;9(3):330- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Y, Carmona S, Muhammad AKMG, et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J Clin Invest. 2019;129(4):1756–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartzentruber J, Foskolou S, Kilpinen H, et al. ; HIPSCI Consortium. Molecular and functional variation in iPSC-derived sensory neurons. Nat Genet. 2018;50(1):54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sleigh JN, Tosolini AP, Gordon D, et al. Mice carrying ALS mutant TDP-43, but not mutant FUS, display in vivo defects in axonal transport of signaling endosomes. Cell Rep. 2020;30(11):3655–3662.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fledrich R, Stassart RM, Klink A, et al. Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nat Med. 2014;20(9):1055–1061. [DOI] [PubMed] [Google Scholar]
- 32.Hinchy EC, Gruszczyk AV, Willows R, et al. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J Biol Chem. 2018;293(44):17208–17217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ballard-Croft C, Kristo G, Yoshimura Y, et al. Acute adenosine preconditioning is mediated by p38 MAPK activation in discrete subcellular compartments. Am J Physiol - Hear Circ Physiol. 2005;288(3):H1359-H1366. [DOI] [PubMed] [Google Scholar]
- 34.Wall JA, Wei J, Ly M, et al. Alterations in oxidative phosphorylation complex proteins in the hearts of transgenic mice that overexpress the p38 MAP kinase activator, MAP kinase kinase 6. Am J Physiol - Hear Circ Physiol. 2006;291(5):H2462–2472. [DOI] [PubMed] [Google Scholar]
- 35.Summers DW, Frey E, Walker LJ, Milbrandt J, DiAntonio A.. DLK activation synergizes with mitochondrial dysfunction to downregulate axon survival factors and promote SARM1-dependent axon degeneration. Mol Neurobiol. 2020;57(2):1146–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horiuchi D, Collins CA, Bhat P, et al. Control of a kinesin-cargo linkage mechanism by JNK pathway kinases. Curr Biol. 2007;17(15):1313–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Pichon CE, Meilandt WJ, Dominguez S, et al. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci Transl Med. 2017;9(403):eaag0394. [DOI] [PubMed] [Google Scholar]
- 38.Pozniak CD, Ghosh AS, Gogineni A, et al. Dual leucine zipper kinase is required for excitotoxicity-induced neuronal degeneration. J Exp Med. 2013;210(12):2553–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beijer D, Sisto A, Van Lent J, Baets J, Timmerman V.. Defects in axonal transport in inherited neuropathies. J Neuromuscul Dis. 2019;6(4):401–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grimm A, Cummins N, Götz J.. Local oxidative damage in the soma and dendrites quarantines neuronal mitochondria at the site of insult. iScience. 2018;6:114–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL.. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014;206(5):655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sheng ZH.The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends Cell Biol. 2017;27(6):403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krols M, van Isterdael G, Asselbergh B, et al. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 2016;131(4):505–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernard-Marissal N, van Hameren G, Juneja M, et al. Altered interplay between endoplasmic reticulum and mitochondria in Charcot-Marie-Tooth type 2A neuropathy. Proc Natl Acad Sci U S A. 2019;116(6):2328–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rabinovitch RC, Samborska B, Faubert B, et al. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 2017;21(1):1–9. [DOI] [PubMed] [Google Scholar]
- 46.Benoy V, Van Helleputte L, Prior R, et al. HDAC6 is a therapeutic target in mutant GARS-induced Charcot-Marie-Tooth disease. Brain. 2018;141(3):673–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kalinski AL, Kar AN, Craver J, et al. Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J Cell Biol. 2019;218(6):1871–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.López-Doménech G, Higgs NF, Vaccaro V, et al. Loss of dendritic complexity precedes neurodegeneration in a mouse model with disrupted mitochondrial distribution in mature dendrites. Cell Rep. 2016;17(2):317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilkinson S.Emerging principles of selective ER autophagy. J Mol Biol. 2020;432(1):185–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hübner CA, Dikic I.. ER-phagy and human diseases. Cell Death Differ. 2020;27(3):833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The generated data are available from the corresponding author upon reasonable request. The Acapella script, written to quantify the images acquired with the Opera Phenix High Content Imaging system is available on GitHub (https://github.com/VerschuurenM/NeuronalConnectivity).