

Abstract
Marinesco-Sjögren syndrome is a rare human disorder caused by biallelic mutations in SIL1 characterized by cataracts in infancy, myopathy and ataxia, symptoms which are also associated with a novel disorder caused by mutations in INPP5K. While these phenotypic similarities may suggest commonalties at a molecular level, an overlapping pathomechanism has not been established yet. In this study, we present six new INPP5K patients and expand the current mutational and phenotypical spectrum of the disease showing the clinical overlap between Marinesco-Sjögren syndrome and the INPP5K phenotype. We applied unbiased proteomic profiling on cells derived from Marinesco-Sjögren syndrome and INPP5K patients and identified alterations in d-3-PHGDH as a common molecular feature. d-3-PHGDH modulates the production of l-serine and mutations in this enzyme were previously associated with a neurological phenotype, which clinically overlaps with Marinesco-Sjögren syndrome and INPP5K disease. As l-serine administration represents a promising therapeutic strategy for d-3-PHGDH patients, we tested the effect of l-serine in generated sil1, phgdh and inpp5k a+b zebrafish models, which showed an improvement in their neuronal phenotype. Thus, our study defines a core phenotypical feature underpinning a key common molecular mechanism in three rare diseases and reveals a common and novel therapeutic target for these patients.
Keywords: SIL1, BiP, PHGDH, INPP5K, l-serine
Hathazi et al. use proteomics to measure the protein composition of cells derived from patients with two different but clinically overlapping rare diseases. They identify a common pathomechanism and a new therapeutic target, which shows promising results when tested in zebrafish models.
Introduction
Muscle diseases may be complicated with additional clinical hallmarks such as abnormalities of the eye, intellectual disability and neurodegeneration, leading to syndromic disorders. Marinesco-Sjögren syndrome (MSS) is an autosomal recessive disorder with infantile onset cataracts, cerebellar atrophy, microcephaly, intellectual disability (of varying degree) and progressive muscle wasting due to vacuolar myopathy.1-3 MSS is an autosomal recessive disorder with an infantile onset featuring cataracts, cerebellar atrophy, intellectual disability (of varying degrees), microcephaly and progressive muscle wasting due to vacuolar myopathy.1–3 MSS is caused by mutations in the SIL1 gene,4–6 which encodes a co-chaperone known as nucleotide exchange factor SIL1. This protein facilitates the efficient cycling of ADP and ATP for GRP78/BiP (encoded by HSPA5), one of the main chaperones of the endoplasmic reticulum (ER).7 Recently, recessive missense mutations in the INPP5K gene were linked to a new syndromic childhood onset neuromuscular disease.8,9 So far, the p.Ile50Thr mutation affecting the 5-phosphatase domain of INPP5K seems to represent the most frequent pathogenic amino acid substitution.8 INPP5K, a skeletal muscle and kidney enriched inositol phosphatase acts as a PI(4,5)P2 phosphatase and regulates the actin cytoskeleton, insulin signalling and cell migration.8,9 Patients with biallelic INPP5K variants present with bilateral cataracts, muscle weakness, and variable degree of intellectual disability.8,9 Similarities between MSS and the INPP5K-associated phenotype were observed not only at the clinical level but also on muscle pathology. Morphological examinations of muscle biopsies revealed the presence of vacuolated muscle fibres and at the ultrastructural level, electron-dense membranous structures surrounding degenerating myonuclei.2,8 Additionally, resemblance can be found at the molecular level: MSS is considered to be a disease of ER dysfunction, as loss of functional SIL14 impacts on regular BiP functions including protein folding and ER-stress modulation.10 Under basal conditions INPP5K localizes to the ER membrane where it also interacts with BiP (via its C-terminus). Once formed at the ER membrane, this functional BiP-INPP5K complex can be translocated to the plasma membrane in response to insulin stimulation.11 Functional studies in patient fibroblasts revealed that INPP5K missense mutants are still competent enough to bind to BiP and do not affect the cellular response to stress, as abundances of well-known unfolded protein response (UPR) markers seem to be mostly unchanged.8 However, the relevance of the functional INPP5K-BiP interplay has not been fully elucidated yet.
In this study, we describe six new patients with INPP5K mutations and hereby expand the mutational and clinical spectrum of the disease. Pathogenicity of two novel INPP5K missense mutations has been functionally confirmed. In addition, we systematically addressed the need to identify common molecular key players to further link MSS and the INPP5K-related phenotype as two rare diseases with considerable clinical overlap. For this purpose, using cells derived from patients with biallelic INPP5K and SIL1 variants, we performed proteomic profiling and identified that phosphoglycerate dehydrogenase (PHGDH) is significantly altered in abundance in the in vitro models of both diseases. Interestingly, autosomal recessive d-3-PHGDH or PHGDH mutations also result in a neurological phenotype clinically overlapping with MSS and INPP5K-related disease, and patients respond to l-serine treatment. Therefore, we addressed the effect of l-serine treatment preclinically in generated zebrafish models of these genes. Thus, our study builds a molecular bridge between three rare neurological diseases with overlapping clinical features, and more importantly, allows translation from preclinical models to develop treatments for these rare neurological diseases.
Materials and methods
Cell culture
Skin biopsies were taken from three genetically proven (c.149T>C; p.Ile50Thr) INPP5K patients8 as well as two genetically confirmed MSS patients [Patient MSS2: 645+1G→A, skipping exon 6, homozygous; and Patient MSS4: 947_948insT, L316fs (het); 1030–18G→A, M344fs (het4)] and five age-matched control subjects. Fibroblasts were grown in Dulbecco’s modified Eagle medium supplemented with 10% foetal bovine serum, 50 U/ml penicillin, 50 μg/ml streptomycin, 0.4% (v/v) amphotericin B (250 μg/ml), and 1 mm sodium pyruvate at 37°C in a 5% CO2 atmosphere. Fibroblasts were grown to 80% confluence prior to harvesting for proteomic profiling. Proteomic comparisons were carried out utilizing cells at similar passage numbers for each experiment. MSS patient-derived lymphoblastoid cells were generated and cultured as described before.12
Measurement of wild-type and mutant INPP5K activity and structural modelling of INPP5K
Wild-type and mutant (p.Asp310Gly, p.Val23Ala, p.Leu55Phe) INPP5K were expressed in BL21 pLysS cells and purified on GSA beads (Thermo Fisher Scientific) in assay buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM MgCl2, 1% TritonTM X-100). The protein activity was then measured as previously described by Wiessner et al.8
The structural model of INPP5K mutants was determined by threading INPP5K sequence on the closest available orthologous crystal structures, OCRL (catalytic domain, PDB: 4CMN) and NDP52 (SKITCH domain, PDB: 3VVW), using the Phyre2 server.13
Liquid chromatography coupled with mass spectrometry (LC-MS/MS) protein analysis
Further details are provided in the Supplementary material.
Zebrafish husbandry, morpholino knock-down and l-serine mediated phenotypic rescue
Zebrafish embryos and larvae were raised and staged according to standard procedures.14 A Chameleon digital camera (model CMLN-13S2M, Chameleon) mounted to a Leica stereomicroscope was used to capture video recordings of embryos and light microscopy images were taken with a Leica dissection stereomicroscope equipped with a Leica digital camera (model DFC 420C).
A translation-blocking morpholino oligonucleotide targeting phgdh was designed and manufactured by Gene Tools. Four previously published splice-blocking morpholinos were also used in our experiments. A sil1 exon 2 splice-blocking morpholino15 was purchased, as well as utilization of previously designed morpholinos directed against inpp5ka and inpp5kb.8 The Gene Tools standard control morpholino targeting a human β-haemoglobin gene was used as a negative control for the effects of morpholino injections. Morpholino sequence and injection concentrations are provided in Supplementary Table 1. Zygotes were injected following standard procedures.16 Efficient gene knockdown was verified by western blot for phgdh translation-blocking morpholino and RT-PCR for splice blocking morpholinos.8,15 Knockdowns were performed in zygotes of the Golden (slc24a5b1/+) (ZIRC) and transgenic (Isl1:GFP)17Danio rerio strains.
For attempted rescue of morpholino-induced phenotypes in zebrafish models, E3 embryo medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4, 0.1% methylene blue) was supplemented with l-serine (Fisher Scientific) to final concentrations of 50, 75 and 100 µM. At 24 h post-fertilization (hpf) movements within the chorion of uninjected control and injected zebrafish—both treated and untreated (l-serine)—were observed and recorded (10× fish per variable, recorded for 2 min). CNS neurons were imaged in treated and untreated transgenic (Isl1:GFP) models.
Free l-serine analysis in zebrafish morphants
We performed amino acid analysis in our zebrafish models (described above) to analyse the levels of free l-serine. Morphants were lysed using 1% SDS lysis buffer (100 µl of 50 mM Tris-HCl (pH 7.8) buffer containing 150 mM NaCl, 1% SDS, and Complete Mini using a manual glass grinder). After lysis ice cold ethanol (−20°C) was added to the samples at a ratio of 1:10. Samples were stored at −20°C overnight to allow the precipitation of macromolecules. Next, samples were centrifuged at 4°C and 18 000g for 30 min resulting in a precipitate that contains proteins and polypeptide chains, while free amino acids were left in the ethanol solution, which was evaporated using a SpeedVac.
Prior to analysis, the samples were further hydrolysed with 6 M HCl (Emprove® Expert, SAFC, Merck) at 110°C for 22 h (gas-phase hydrolysis). Following this, residues were derivatized according to Cohen and Michaud.18 Separation was done using reversed-phase high-performance liquid chromatography. A six-point calibration with amino acids standard solution (Amino Acid Standard H, Thermo Fisher) was used to determine the amount of each detectable amino acid. The instable amino acids tryptophan, methionine and cysteine were not taken into account.
Immunoblot analysis
Zebrafish and fibroblast protein extracts were separated by SDS-PAGE and transferred to Immobilon-FL PVDF membranes (Merck Millipore). Membranes were blocked for 1 h at room temperature with 5% milk in Tris-buffered saline-Tween and incubated with rabbit anti-PHGDH (GeneTex; dilution 1:1000) overnight at 4°C. Blots were developed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody (Invitrogen; dilution 1:5000) overnight at 4°C. Protein signals were detected with a SuperSignal™ West Femto Kit (Thermo Fisher Scientific) following the manufacturer’s protocol.
Immunohistochemistry and immunofluorescence microscopy
Muscle biopsies
Routine histological examinations [including haematoxylin and eosin, nicotinamide adenine dinucleotide dehydrogenase tetrazolium reductase (NADH-TR), periodic acid-Schiff and succinate dehydrogenase (SDH) stainings] were carried out in addition to immunofluorescence-based studies of alpha-dystroglycan (Novus Biological 2238) and merosin (Abcam ab140482) on the muscle biopsy specimens derived from the index cases presented in our study. Moreover, muscle biopsy sections derived from three patients with homozygous c.149T>C p.Ile50Thr INPP5K mutations were analysed for abundance and distribution of INPP5K (SKIP C-19 Santa Cruz SC12073) and PHGDH (N-term antibody N1N2; GTX101948; GeneTex) proteins. In addition, also on the muscle biopsies of patients with the homozygous INPP5K p.Ile50Thr mutation, haematoxylin and eosin staining as well as fast and slow myosin fibre typing were carried out. Stainings were visualized using an Axio Imager Z1 fluorescent microscope (Zeiss). Tiled images were obtained using a 10× objective and stitched together using the image processing facility provided in the Zen software package. The stitched images were then analysed using the open-source image processing package Fiji.
Zebrafish
Whole-mount immunofluorescence of zebrafish embryos was performed as previously described.19,20 Briefly, injected embryos were dechorionated with Pronase E (Sigma Aldrich) and euthanized by anaesthetic overdose. Embryos were fixed in 4% paraformaldehyde/PBS overnight at 4°C, blocked at room temperature for 1 h in 5% horse serum/PBS containing 0.1% Tween-20 and then incubated overnight at 4°C with primary antibody mouse anti-synaptic vesicle protein 2 (Developmental Studies Hybridoma Bank; dilution 1:200). Incubation with secondary antibody Alexa Fluor® 488-conjugated goat anti-mouse IgG (Thermo Fisher; dilution 1:200) was performed for 1 h at room temperature. Acetylcholine receptor clusters were stained with Alexa Fluor® 594-conjugated α-bungarotoxin (Thermo Fisher; 1:1000). Alexa Fluor® 594-conjugated phalloidin (Thermo Fisher; 1:100) was used to visualize actin filaments. Z-stack images of zebrafish were obtained by scanning one-half of the trunk myotome. Digital images were captured with a Nikon A1R laser scanning confocal microscope (Nikon, UK).
For the live examination of Isl1-GFP zebrafish, florescence embryos were anaesthetized using 0.01% tricane and were mounted in 1.2% agar on a depression slide glass as previously described21 to avoid movement and drifts. Embryos were then visualized using a Zeiss florescence microscope with ApoTome attachment (Axio Imager Z1) using serial optical sections (z-stacks) of ∼4 mm intervals. The image was then reconstructed from the stacked microscope images using the Z project function (maximum intensity) in ImageJ.
Data availability
Data are available from the corresponding author upon reasonable request.
INPP5K fibroblasts: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD009272. To visualize the data please use the username: reviewer26477@ebi.ac.uk and password: RdNNHLlo.
MSS fibroblasts: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD009297. To visualize the data please use the username: reviewer67767@ebi.ac.uk and password: eOUI5Dcd.
The MSS lymphoblasts dataset has PXD003030 as an identifier and can be found online via ProteomeXchange.
Results
Expansion of the clinical and mutational spectrum in INPP5K-related disease
Patient 1 is a 25-year-old female (parents did not give consent for publication of photographs) born as the second child of non-consanguineous parents originating from a small town in southern Italy. The patient presented with cognitive delay since early infancy whereas motor milestones were regularly achieved. By the age of 10 years, she started to present with progressive proximal muscle weakness. The first neuromuscular examination was performed at the age of 22 years and showed microcephaly and short stature (below the 3rd centile), severe hyperlordosis, pseudohypertrophy of calves and hypotrophy of quadriceps. Notably, the patient presented with an unsteady and waddling gait. Muscle weakness also involved proximal muscles of the upper limbs and the patient was unable to raise her arms above her shoulders (serum creatine kinase was elevated up to about twice normal values). She also had a moderate intellectual disability. Brain MRI did not show cerebellar atrophy (Fig. 1). A biopsy of the quadriceps muscle showed dystrophic changes. Fibre diameter was variable with mildly increased perimysial connective tissue and centrally located myonuclei. Necrotic fibres were rarely detectable. Ophthalmological examination did not reveal the presence of cataracts.22
Figure 1.

Clinical and brain imaging findings in novel INPP5K patients. (A) Representative photographs of Patients 3 and 4 showing severe hyperlordosis. (B) Brain MRI of Patients 1–4 showing no brain abnormalities in Patients 1, 3 and 4, but a mild cerebellar atrophy in Patient 2. The size of the oculomotor muscles appears normal in Patients 3 and 4 and lenses have obviously been removed with vacuity of the eyes in Patient 3, whereas in Patient 4 the lenses have not been removed and are well visible within the eyeballs. 1 = lateral view; 2 = overhead view; 3 and 4 = ocular view.
Patient 2 is a 15-year-old male (parents did not give consent for publication of photographs) also originating from a small town in southern Italy. The patient is the child of non-consanguineous parents. At 3 years of age, he underwent surgery for bilateral cataracts. He presented with mild cognitive and motor delay by early infancy and acquired autonomous ambulation at the age of 2 years. The first neurological examination of the patient was performed at age 4 years and revealed an ataxic gait and mild weakness of proximal muscles with positive Gowers’ sign. Remarkably, brain MRI of Patient 2, performed at the age of 12 years, revealed mild cerebellar atrophy (Fig. 1). Serum creatine kinase was elevated up to about three times the normal values. A muscle biopsy was performed at the age of 5 years and its examination revealed myopathic changes characterized by myofibre size variability, and internalized myonuclei. In the progression of the disease, the patient developed steppage gait indicative of distal muscles weakness and at the age of 14 he underwent orthopaedic surgery for clubfoot correction.
Next generation sequencing revealed that Patients 1 and 2 harboured the previously described pathogenic homozygous nucleotide substitution c.67G>A (hg19: NM_016532.3), leading to the substitution of a non-polar aliphatic amino acid by a hydrophobic amino acid, p.Val23Met.9,22
Patients 3 and 4 are two sisters (Fig. 1) living on Reunion Island, born to a non-consanguineous couple of Creole descent. Both patients, aged 13 and 11, respectively, were investigated from early childhood for muscle weakness associated with learning difficulties. Bilateral cataracts were observed in only one sister and required surgery at the age of 4 years. Investigation of serum creatine kinase levels revealed an elevation up to about five times of normal values. Patients also presented with mild (Patient 3) and moderate (Patient 4) intellectual disability. Brain MRIs did not reveal pathological changes (Fig. 1). Examination of the muscle biopsy specimens revealed a dystrophic pattern strongly suggesting a congenital muscular dystrophy (see below). Various genes were subsequently screened including CAPN3 (a gene frequently involved muscular dystrophies in Reunion Island), DMPK (due to the association with cognitive deficits and early cataracts) and FKRP but did not show pathogenic sequence alterations. It is noteworthy that the mother developed early cataracts and underwent eye surgery at age 37.
The family was included within the ‘Myocapture project’, a French project based on exome sequencing of patients and relatives to identify new genes responsible for neuromuscular diseases. The analysis was performed using the bioinformatics pipeline Polyweb and led to the identification of a new variant c.68T>C; p.(Val23Ala) in INPP5K (hg19: NM_016532.3), affecting the same amino acid, but resulting in a different change than in Patients 1 and 2. Sanger sequencing confirmed the presence of the homozygous variant in the two affected females and its segregation within the family members (heterozygous variant in the father and mother). Family relatedness scores have been calculated and the results showed no consanguinity in the parents both originating from Reunion Island.
Patient 5 is now a 52-year-old female and was born after an uneventful pregnancy as the fourth child of non-consanguineous parents. Despite being able to walk independently at 14 months, she has had persistent walking difficulties throughout her childhood and has never being able to run. Myalgias and easy fatigability were also noticed in her infancy in addition to learning difficulties. She is also affected by high blood pressure and glaucoma. Her height is 145 cm (<5th centile) and her occipital frontal circumference is in the normal range for her height.
When first examined at age 42, Patient 5 could only walk unsupported for short distances with a waddling gait. She was unable to get up from a chair without external support. Furthermore, she could neither squat nor raise her arms above her head. Neurological examination detected bilateral Babinski sign and a tendency to fall backwards on Romberg’s manoeuvre. Mild bilateral upper limb dysmetria with eyes closed were also noticed in addition to reduced but present tendon reflexes. Muscle strength on the MRC scale showed weakness of the neck flexors = 3, neck extensors = 4, arm abductors = 4, hand finger extensors = 4, hip flexors = 2, hip extensors = 3, hip adductors and abductors = 3, knee flexors = 3, and ankle extensors = 4. The patient also reported intense muscle pain upon touch. No sensory deficits could be observed. Serum creatine kinase was elevated up to 10–12 times normal values. EMG/ENG (electromyography/electroneurography) revealed myopathic changes and a sensory axonal neuropathy. Moreover, somatosensory evoked potentials showed impairment of both peripheral and central somatosensory conduction. Brain MRI and spectroscopy as well as SPECT revealed no significant abnormalities (data not shown). IQ was scored at 60 (mild mental retardation). Microscopic investigation revealed myopathic changes (see below). All the glycolytic enzyme activities were in the normal range. Remarkably, ophthalmological examination showed no overt cataracts. Investigation of pulmonary function revealed moderate restrictive impairment (forced vital capacity 72%) while heart ultrasound was normal.22
In this patient, exome sequencing led to the identification of bi-allelic INPP5K mutations: compound heterozygosity was found for c.165G>T (hg19: NM_016532.3) resulting in a substitution of a non-polar hydrophobic by another non-polar hydrophobic amino acid (p.Leu55Phe) and c.753_756del (hg19: NM_016532.3) resulting in a frameshift (p.Arg251Serfs*24). In silico-based testing of pathogenicity of c.165G>T revealed a CADD score of 22.9 indicating that the variant is considered to be damaging. Mutations associated to the respective INPP5K-phenotypes are listed in Table 1.
Table 1.
Clinical features of individuals with bi-allelic INPP5K mutations
| Patient | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Sex | Female | Male | Female | Female | Female | Female |
| Age at molecular diagnosis | 25 years | 15 years | 11 years | 13 years | 51 years | 10 years |
| Origin | Italy | Italy | Reunion Island | Reunion Island | Italy | Italy |
| INPP5K variant | p.Val23Met | p.Val23Met | p.Val23Ala | p.Val23Ala | p.Leu55Phe and p.Arg251Serfs*24 | p.Val23Met |
| Initial presenting symptom | Cognitive delay (early infancy) | Mild cognitive and motor delay (early infancy) | Muscle weakness (early childhood) | Muscle weakness (early childhood) | Muscle weakness (early childhood) | Muscle weakness |
| Cataracts (at age) | Yes (?) | Yes (3 years) | Yes (4 years) | No | No | Yes (3 years) |
| Hypotonia | ? | ? | ? | ? | Not reported | Yes |
| Delayed motor milestones | No | Yes (mild) | ? | ? | No | Yes (mild) |
| Muscle weakness/atrophy | P > D (10 years), pseudohypertrophy of calves and hypotrophy of quadriceps (22 years) | P > D with positive Gowers’ sign | – | – | Proximal > distal | proximal muscle weakness with a positive Gowers |
| CK | ×2 | ×3 | ×5 | ×5 | × 10−12 | ? |
| Biopsy findings | Dystrophic changes, variable fibre diameter, mildly increased perimysial connective tissue, centrally located myonuclei | Myopathic changes characterized by myofibre size variability, and internalized myonuclei | Dystrophic pattern | Dystrophic pattern | Myopathy with glycogen accumulation | Dystrophic pattern |
| Intellectual disability | Moderate | ? | ? | ? | Mild | Mild |
| Brain anomaly | Not present | Mild cerebellar atrophy | Not present | Not present | Not present | Not present |
| Other findings | Microcephaly and short stature (below the 3rd centile), severe hyperlordosis | Clubfoot correction | – | – | Myalgias, short stature, sensory axonal neuropathy | Short stature, mild foot deformity and reduced mineral bone density |
CK = serum creatine kinase. P > D = proximal muscles more severely affected than distal muscles.
Patient 6 is a 10-year-old female who was recently examined and diagnosed. Her parents are not consanguineous, but both of their families come from the Puglia region in Southern Italy. She was reported to be hypotonic at birth, her motor milestones were slightly delayed with the ability to freely walk at 18 months of age. She was diagnosed with bilateral cataracts, which were operated on at age 3.5 years. In addition, short stature, a mild foot deformity and reduced mineral bone density were reported. She shows mild learning difficulties and receives special educational support. Neurological examination showed mild, proximal muscle weakness with a positive Gowers and difficulties to walk stairs. Brain MRI did not reveal any significant abnormalities.
Comparison of clinical findings obtained in Patients 1–6 is provided in Table 1 and shows that muscle vulnerability appears as a common clinical feature whereas cataracts is not always associated with causative variants in INPP5K. Moreover, additional clinical features such as mild cerebellar atrophy and sensory axonal neuropathy can be associated with the presence of INPP5K mutations expanding the currently known neurological spectrum of the disease. We hope that future description of INPP5K cases will aid will shed light to how often CNS pathologies occurs in these patients.
Testing of new INPP5K missense variants confirms pathogenicity and expands the pathomechanism of the disease
Given that INPP5K preferentially dephosphorylates PI(4,5)P2 at the D5 position23 and previously described mutants show reduced catalytic activity,8 we performed in vitro measurements to assess the catalytic activity of the full-length p.Val23Ala and of p.Leu55Phe mutant INPP5K. Results show an impaired release of phosphate from PI(4,5)P2 onto diC8 substrates for the p.Val23Ala mutant when compared with the full-length wild-type protein (Fig. 2A). The artificial mutant pAsp310Gly (catalytic dead mutation) was included as a control. This reduced enzymatic activity might be explained by the fact that the p.Val23Ala mutation is localized in the 5-phosphatase domain of the protein (Fig. 2B) and, as described for other mutants affecting the same region (including the previously reported p.Val23Met mutation identified in our Patients 1 and 2), this leads to a destabilization of the overall shape of active site.8 Studies of the catalytic activity of the p.Leu55Phe mutant form of INPP5K did not reveal a detrimental reduction in its activity against water soluble short-chain lipid substrate (Fig. 2A), although this amino acid is also localized in the vicinity of the 5-phosphatase domain (Fig. 2B). Extracting lipids, lipid immunostaining, and quantifying PI(4,5)P2 from patient material may help to determine, whether there is a difference in the total cellular levels of PI(4,5)P2 in the p.Leu55Phe-INPP5K mutant. However, there are currently no cellular models and the available muscle tissue is not sufficient to perform this analysis. Based on this finding we hypothesize that the amino acid substitution might trigger other pathophysiological mechanisms beyond its enzymatic activity, in association with the truncated p.Arg251Serfs*24 INPP5K present as compound heterozygous allele in Patient 5.
Figure 2.

Enzymatic activity of novel missense mutation in INPP5K. (A) Quantification of enzymatic activity of novel missense mutants of INPP5K. GST-INPP5K was bacterially expressed and purified and exposed to the water soluble substrate PI(4,5)P2 diC8. Phosphate released by hydrolysis of PI(4,5)P2 was measured by malachite assay, and the engineered catalytic null mutant INPP5KAsp310Gly was included as a negative control. GST-INPP5KVal23Met has significantly compromised enzymatic activity in this assay (n = 3 per condition, t-test), whereas GST-INPP5KLeu55Phe retains near wild-type levels of activity against the water soluble substrate (n = 4 wells per condition, t-test). (B) Predicted structure of the INPP5K catalytic domain, modelled using Phyre2.13 Val23 (yellow) is in the catalytic pocket of the enzyme (red), whereas Leu55 is an internal residue close to a predicted hydrophobic finger (hydrophobic residues coloured blue), which in the enzyme OCRL is thought to assist in associating the catalytic domain with highly curved membranes. n.s. = not significant.
Muscle biopsy findings in INPP5K patients are consistent with congenital muscular dystrophy
Haematoxylin and eosin, and periodic acid-Schiff reaction in a muscle biopsy specimen derived from Patient 3 revealed a dystrophic pattern with occasionally centralized myonuclei (Fig. 3A). The same dystrophic findings were observed in haematoxylin and eosin-stained muscle sections derived from Patient 5. Here, periodic acid-Schiff staining further revealed increased reactivity in the sarcoplasm of a proportion of muscle fibres consistent with glycogen accumulation (Fig. 3A). Several fibres with markedly increased SDH staining—suggestive for altered mitochondrial activity—could be observed in a proportion of muscle fibres (Fig. 3A). A number of lobulated fibres presented with focal increase of NADH-TR reactivity (Fig. 3A). For Patients 1–4 abundance and distribution of alpha-dystroglycan and laminin-2 was studied by immunofluorescence and minor alterations were observed as exemplified for Patients 1 and 2 (Fig. 3B).
Figure 3.
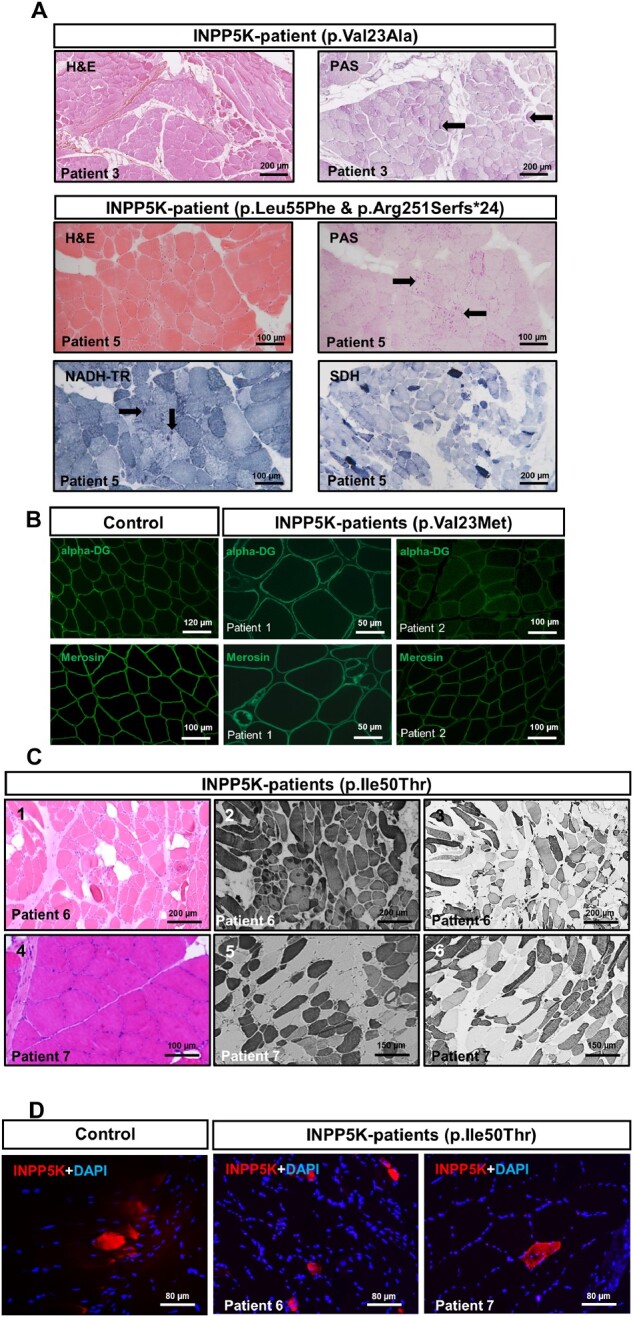
Muscle biopsy findings in INPP5K patients. (A) Representative histological findings in two (Patients 3 and 5) of the five novel INPP5K patients include variation in fibre size, rounding of fibres, increased endomysial collagen and some degree of fatty degeneration, as well as focal increase of glycogen as measured by periodic acid-Schiff (PAS) staining in some fibres (black arrows) and focal increase of mitochondria in a proportion of fibres as measured by NADH-TR reaction (black arrows). Moreover, high oxidative activity is indicated in a proportion of fibres by SDH reaction. (B) Immunofluorescence-based analyses of alpha-dystroglycan and merosin (shown for Patients 1 and 2) did not show changes that would accord with a vulnerability of both proteins upon the loss of functional INPP5K. (C) Findings in two INPP5K patients with p.Ile50Thr mutation.8 Haematoxylin and eosin stain (left) revealing variation in fibre size, rounding of fibres, increased endomysial collagen and some degree of fatty degeneration. Immunohistochemical analysis focusing on fast (middle) and slow (right) myosin revealed a predominance of fast fibres in Patient 6 compared with Patient 7. Atrophy of slow fibres some of which were grouped was variably seen in both patients together with co-expression of both myosin isoforms. (D) Immunofluorescence-based analysis of INPP5K expression/stability did not reveal changes in abundance or distribution between controls and Patients 6 and 7 (p.Ile50Thr mutation).
To investigate the pathomechanism of INPP5K mutations, muscle biopsy specimens derived from patients carrying the most common homozygous mutation, p.Ile50Thr8 were investigated on the histological and immunological level. Results of haematoxylin and eosin staining accord with a muscular dystrophy and fast/slow myosin staining did not indicate fibre-type grouping (Fig. 3C). As missense mutations can influence structural and functional properties of proteins and consequently their stability, we explored the abundance/stability of mutant INPP5K protein by immunofluorescence studies in p.Ile50Thr-mutant INPP5K8 and control muscle biopsies. We observed no reduced stability of p.Ile50Thr-mutant INPP5K (Fig. 3D) according to our previous findings obtained in in vitro models over-expressing the wild-type and mutant forms of INPP5K (8).
PHGDH is a common molecular player in Marinesco-Sjögren syndrome and INPP5K-related disease
Given that clinical features of our five INPP5K patients overlap with MSS features (Table 2), we aimed to identify biochemical key players linking MSS and the INPP5K phenotype. We therefore performed proteomic profiling utilizing lymphoblastoid and fibroblast cells derived from MSS patients caused by SIL1 mutations,4 as well as fibroblasts derived from INPP5K patients.8 The overall findings of our profiling using MSS lymphoblastoid cells have already been described.12 Proteomic analysis of MSS patient-derived fibroblasts allowed the quantification of 2996 proteins, of which 139 showed altered abundances and have been quantified based on at least two unique peptides. The proteomic response of INPP5K p.Ile50Thr mutant fibroblasts revealed a statistically significant (PANOVA ≥ 0.05) altered abundance of 44 proteins (22 are increased and 22 are decreased) of a total of 3018 identified proteins. The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE24 partner repository with the dataset identifiers PXD003030 (SIL1 lymphoblasts), PDX009297 (SIL1 fibroblasts), PXD009272 (INPP5K fibroblasts). After individual proteomic profiling, obtained data were filtered for proteins vulnerable in all three experiments. This approach allowed us to identify d-3-PHGDH as a protein decreased in MSS patient derived cells but increased in p.Ile50ThrINPP5K mutated fibroblasts (Fig. 4A). Immunoblot analysis of PHGDH levels in p.Ile50ThrINPP5K and control fibroblasts confirmed the increase in the patient derived cells (Fig. 4A, right column). Quantitative analysis of PHGDH in cells is consistent with a statistically significant increase in Ile50Thr-INPP5K fibroblasts while in MSS fibroblasts, a decrease was observed (Supplementary Fig. 1). The focus on PHGDH was additionally prompted by the fact that recessive PHGDH mutations have already directly been linked to a neuropaediatric phenotype with features common to MSS and INPP5K (Table 2). To verify the proteomic results obtained in the patient-derived cells (p.Ile50Thr-INPP5K), we investigated the abundances and distribution of PHGDH in muscle biopsy specimens derived from patients harbouring the p.Ile50Thr, p.Val23Met and p.Val23Ala-INPP5K variants.8 Results of the immunofluorescence studies revealed an increase in PHGDH intensity in a proportion of muscle fibres in two of the three p.Ile50Thr-INPP5K patients analysed and in both of the p.Val23Met-INPP5K patients (Fig. 4B and C). Overall quantification of fluorescence intensity confirmed a statistically significant (t-test < 0.05) PHGDH increase in the INPP5K patient-derived biopsies compared to the two investigated control biopsies (Fig. 4C). Unfortunately, biopsy material or fibroblasts from patients carrying the p.Leu55Phe and Arf251Serfs*24 mutations were not available.
Table 2.
Clinical comparison of MSS (SIL1 patients), and INPP5K mutation- and PHGDH mutation-associated phenotypes
| Symptoms | SIL1 patients | INPP5K patients | PHGDH patients |
|---|---|---|---|
| Vulnerability of skeletal muscle | Progressive vacuolar myopathy | Congenital muscular dystrophy | Not described |
| Cataracts | Congenital or infantile | Congenital | Congenital |
| Intellectual disability | Present (varying degree) | Present (mild) | Present |
| Ataxia | Cerebellar ataxia | First case of cerebellar atrophy is described in this study | Cerebellar ataxia |
| Neuropathy | One described case with motor neuropathy and axonal vulnerability and two cases with axonal degeneration on the ultra-morphological level | First case of neuropathy is described in this study | Axonal |
| Microcephaly | Present | Not described | Present |
Figure 4.

Workflow depicting our protein studies on fibroblasts derived from MSS and INPP5K patients. (A) For patients with MSS, immortalized lymphoblastoid cells and primary fibroblasts were used for iTRAQ-labelling and subsequent peptide fractionation followed by LC-MS/MS analyses (left and middle column). For primary fibroblast cells derived from INPP5K patients (all with p.Ile50Thr mutation), label-free proteomic profiling was carried out (right column). In all three experiments, as shown by the precursor and reporter ion intensities of the respective experiments (exemplified by the unique peptides of PHGDH, i.e. GTIQVITQGTSLK in label-free and QADVNLVNAK in labelling approaches, respectively), PHGDH was altered in abundance: in SIL1 patient-derived cells, a statistically significant decrease could be detected whereas in INPP5K patient-derived fibroblasts, a statistically significant increase could be identified. This increase in INPP5K fibroblasts was also confirmed by immunoblotting. The volcano plots highlight our overall proteomic findings: each red and green dot represents a protein altered with a PANOVA ≥ 0.05. (B) Representative PHGDH staining in a muscle biopsy specimen of a control subject (top) and of an INPP5K patient harbouring the p.Ile50Thr and p.Val23Met mutation (bottom) showing an increase of PHGDH in a proportion of diseased muscle fibres. Scale bars = 200 µm. (C and D) Box plots depicting the fluorescence data-points measured in two controls and three p.Ile50Thr patients, respectively, and two (p.Val23Met and p.Val23Ala) patients. The central band represents the median, the lower and upper hinges correspond to the first and third quartiles (25 and 75%) while the whiskers extend to the highest and lowest points within the data (1.5× the interquartile range, IQR). Results were observed in one (n = 1) independent experiments (because of the limited amount of sample). Statistical analysis unpaired Student’s t‐test was employed where *P ≤ 0.05 was considered statistically significant. (E) Immunohistochemistry analysis of PHGDH on paraffin sections from paraformaldehyde-fixed quadriceps muscle specimens of 26-week-old wild-type and three woozy mouse muscle. Scale bars = 22 µm. (F) Optical density (OD) determination of F where graph show the results of our three independent experiments resulted from the analysis of three controls and three woozy mice. Data represent mean ± standard error of the mean (SEM), statistical analysis unpaired Student's t‐test was employed where *P ≤ 0.05 was considered statistically significant. Similar results were observed in at least three (n = 3) independent experiments.
Immunohistochemistry analysis of PHGDH in skeletal muscle derived from 26 weeks woozy mice (Fig. 4E and F), the mouse model for MSS, shows a significant decrease (t-test ≤ 0.05) of PHGDH compared with the controls, thus supporting our data obtained from proteomics and immunofluorescence analysis of MSS fibroblasts.
The dysregulation of other proteins identified in our analysis (Fig. 1A) localized to the ER or other subcellular compartments such as the mitochondria might reflect the concomitant activation of further pro-survival or detrimental mechanisms. We might have to consider that these mechanisms can be tissue specific.
Zebrafish morphants sil1, inpp5k and phgdh show neurological phenotypes with overlapping pathology
To compare the phenotypical consequences of Sil1, Inpp5k and Phgdh depletion in vivo, respective zebrafish models (morphants) were generated. The choice of zebrafish as animal models was prompted by the recent descriptions of inpp5k and sil1 morphants as suitable in vivo systems for the human phenotypes.8,9,15 To the best of our knowledge, no zebrafish model for PHGDH has been previously described, therefore, we introduce here the first fish model for PHGDH deficiency (Fig. 5A and B). A comparison of live embryo images of non-injected, control and phgdh morpholino-injected zebrafish at 48 hpf revealed a spectrum of moderate to severe alterations of the tail at the light microscope level (Fig. 5A). At 48 hpf, the decreased expression of phgdh in injected zebrafish was confirmed by RT-PCR (data not shown) and given that PHGDH represents a common molecular denominator for MSS and INPP5K-CMD, as described above, Phgdh protein levels were examined in the three different fish models by immunoblotting. As expected, no detectable levels of Phgdh protein were found in the phgdh morphants (Fig. 5C and D), whereas a slight increase of Phgdh in the inpp5k morphants and a decrease in the sil1 morphants was detected. Additionally, the Phgdh expression pattern is also supported by the amounts of free l-serine as phgdh and sil1 morphants present with lower amounts compared to control embryos while inpp5k a+b zebrafish show an increase consistent with higher levels of Phgdh (Fig. 5E). Our analysis was performed on whole embryos at 48 hpf. These results are in accordance with the proteomic findings obtained from the patient-derived cells (Fig. 4A).
Figure 5.

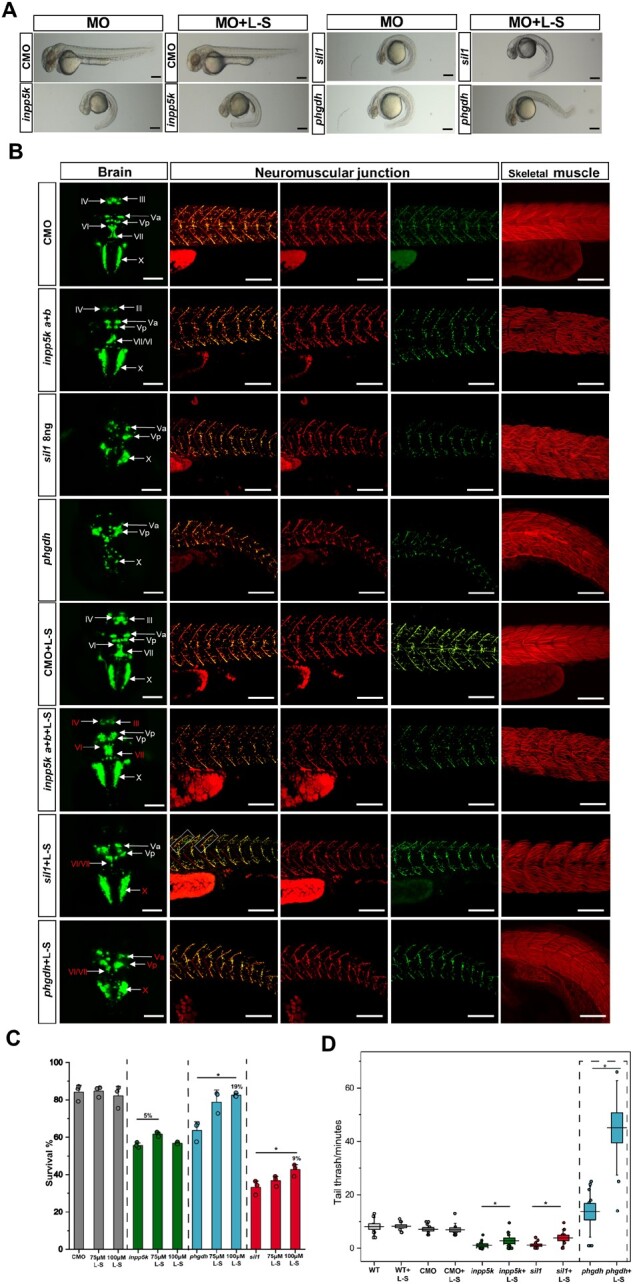
Phenotyping of phgdh and sil1 morphants. (A) Morpholino-based depletion of phgdh and sil1 in 48 hpf zebrafish resulted in abnormal curvature of the tails of both models compared to wild-type (WT) and control morpholino (CMO) injected fish. (B) Immunofluorescence-based studies of tissue morphologies revealed disintegration of muscle fibres visualized by phalloidin staining (top row), abnormal patterning of the presynaptic neuromuscular junctions (NMJ) visualized by reduced SV2-immunoreactivity (rows 2–4) and abnormal development of the brain affecting the cranial nerves (bottom row) in both fish models. Scale bars = 50 µm. Regarding brain abnormalities, in phgdh morphants, nerves Va, Vp, VI, VII, X, and in sil1 morphants, nerves III, IV, VI, VII and X, are vulnerable to the loss of the respective protein expression. Scale bars = 100 µm. Similar results were observed in at least four (n = 4) independent experiments. (C) Western blot studies of Phgdh protein level in the three fish models revealed a slight increase in whole protein extracts of inpp5k morphants but a considerable decrease in the sil1 morphants as well as in the phgdh morphants. (D) Decrease of the Phgdh protein in the phgdh morphants reflects the successful morpholino-based depletion of the expression of the corresponding gene as also depicted quantitatively. Data represent mean ± SEM, statistical analysis unpaired Student’s t‐test was employed where *P ≤ 0.05 was considered statistically significant. Similar results were observed in at least three (n = 3) independent results. (E) Free l-serine analysis in whole embryos at 48 hpf by liquid chromatography. Results accord with the Phgdh expression in zebrafish. Data represent mean ± SEM, statistical analysis unpaired Student’s t‐test was employed where *P ≤ 0.05 was considered statistically significant. Similar results were observed in at least two (n = 2) independent experiments because of the large number of embryos needed for this analysis. (F and G) Depletion of phgdh and sil2 is also associated with reduced survival of the fish at 48 hpf. The central band represents the median, the lower and upper hinges correspond to the first and third quartiles (25 and 75%) while the whiskers extend to the highest and lowest points within the data (1.5× IQR). Statistical analysis unpaired Student’s t‐test was employed where *P ≤ 0.05 was considered statistically significant. Similar results were observed in at least four (n = 4) independent experiments.
Injection of the phgdh morpholino led to an 8% increase in lethality of embryos compared to those injected with control morpholino (Fig. 5F). Phalloidin-staining (labelling of actin cytoskeleton) was carried out to investigate skeletal muscle pathology and showed disruption of the regular chevron shape of somites, wavy fibres and, in some cases, absent notochord in the phgdh morpholino fish compared to control morpholino-injected zebrafish (Fig. 5B). Moreover, neuromuscular junction development is perturbed in the phgdh morpholino-injected embryos as seen by the reduced immunoreactivity of synaptic vesicle glycoprotein 2 (SV2) suggesting a significant reduction of neuromuscular synapses in the dorsal region of the myotome segment, whereas the pre-patterning of the acetylcholine receptor (AChR) clusters seems to be mostly unaffected (Fig. 5B). Live imaging of the phgdh morpholino-injected transgenic Isl1:GFP zebrafish showed a strong effect of Phgdh depletion on the development of the hindbrain visualized by a reduction of GFP-fluorescence intensity of the nerves Va, Vp, VI, VII and X (Fig. 5B). Brain malformations have also been observed in the Phgdh-deficient mouse model,25 thus confirming the suitability of this newly generated phgdh fish model.
The zebrafish model for MSS was recently introduced by Kawahara and Hayashi.15 The exact splice blocking morpholino targeting the splice acceptor site of exon 2 was employed for the generation of the sil1 depleted zebrafish described in this study. A comparison of live embryo images of non-injected, control morpholino and sil1 morpholino injected zebrafish at 48 hpf revealed a spectrum of mild to severe alterations of the tail already on the light microscopic level (Fig. 5A). Embryos were injected with 6 ng of the morpholino, which reduced the expression of sil1 compared to controls (data not shown). Injection of the sil1 morpholino led to a 11% increase in lethality of embryos compared to those injected with control morpholino (Fig. 5G). Labelling of actin cytoskeleton by phalloidin in control morpholino and sil1 fish at 48 hpf displayed almost no abnormality of muscle fibre integrity. Although Kawahara and Hayashi15 stated that at this morpholino concentration they are able to observe a disturbed muscle pattern and an affection of muscle fibre integrity, this finding could not be confirmed upon careful inspection in our experiment, which revealed only a very mild muscle phenotype (Fig. 5B). Perturbed neuromuscular junction morphology in sil1 morpholino-injected fish was recently described by our group26 and could be recapitulated in this experiment: neuromuscular junctions of sil1 zebrafish show disruption of synapse formation along the myosepta, particularly at the presynaptic part of the neuromuscular junction (Fig. 5B). This finding is in agreement with affected neuromuscular junctions in both MSS patients and woozy mice (mouse model of MSS26). Live imaging of the cranial motor neurons using the transgenic Isl1:GFP fish showed an altered brain morphology with nerves III (oculomotor), IV (trochlear), VI (abduces) and VII (facial) being most vulnerable to the altered sil1 expression (Fig. 6B).
Figure 6.
l-serine treatment studies in phgdh, sil1 and inpp5k morphants. (A) The study of the effect of l-serine (L-S) treatment (100 µM) on the phenotype of fish did not reveal changes on the curvature of the tail in the inpp5k and sil1 morphants (MO), whereas a slight beneficial effect could be observed in the phgdh morphants; 30 fish were analysed per condition. Scale bars = 500 µm. (B) Results of immunofluorescence-based studies of l-serine treatment (100 µM) in tissues vulnerable against the depletion of inpp5k, sil1 and phgdh expression. A slight amelioration of muscle fibre integrity can only be identified in the phgdh morphants but not in the two other models. A minimum of 10 fish were analysed per condition. Scale bars = 50 µm. In contrast, the presynaptic phenotype was ameliorated in the sil1 as well as in the phgdh morphants, as exemplified by an increase of SV2-immunoreactivity (inpp5k morphants did not show a neuromuscular junction phenotype per se). A minimum of 10 fish were analysed per condition. Scale bars = 50 µm. In addition, GFP fluorescence intensity suggests that the cranial nerves III, IV, VI and VII improved in inpp5k morphants. The phenotype in sil1 morphants was ameliorated in the nerves VI, VII as well as X, and in phgdh morphants for nerves Va, Vp, VI, VII and X after treatment based on increased GFP fluorescence intensity. A minimum of 10 fish were analysed per condition. Scale bars = 100 µm. (C) Survival rates of zebrafish disease models treated with 75 µM and 100 µM of l-serine, respectively. Whereas l-serine treatment has no impact on survival of wild-type fish, an effect on morphant survival can be observed for the three different disease models Data represent mean ± SEM, statistical analysis unpaired Student’s t‐test was employed where *P ≤ 0.05 was considered statistically significant. (D) l-serine treatment did not show an effect on the number of tail thrashes per minute in the wild-type (WT) and control morpholino (CMO) fish but a statistically significant increase in sil1, inpp5k and phgdh morphants, n = 12 fish from three different experiments were investigated per condition. The central band represents the median, the lower and upper hinges correspond to the first and third quartiles (25 and 75%) while the whiskers extend to the highest and lowest points within the data (1.5× IQR). Statistical analysis unpaired Student’s t‐test was employed where *P ≤ 0.05 was considered statistically significant. Similar results were observed in at least three (n = 3) independent experiments.
Morpholino-based depletion of inpp5ka and inpp5kb expression recapitulated the already described pathology in living embryos, including altered length and curvature of tails, differences in eye-to-head ratios,8,9 muscle fibre defects and loss of the chevron shape of somites and deformation of myosepta (Fig. 6B). The live imaging of inpp5k a+b morphant brains using Isl1:GFP transgenic zebrafish displayed mild abnormalities of the cranial nerves III, IV, VI and VII, exemplified by reduced fluorescence intensity (Fig. 6B). As described previously, depletion of inpp5ka+inpp5kb does not alter neuromuscular junction integrity in zebrafish (Fig. 6B).8
l-serine treatment ameliorates the neuropathological phenotype in sil1, inpp5k and phgdh zebrafish morphants
Numerous publications have underlined the importance of l-serine (produced by PHGDH) in the development of the CNS27–29 and supplementation of l-serine was found to have a beneficial effect on neurodegeneration in CNS disorders.30,31 Thus, rescue experiments were performed, in which l-serine was supplemented in the growth media for the respective fish models, and the phgdh morphants included as proof-of-principle controls. Doing so, first the optimal dosage of l-serine (ranging from 75 to 200 µM) was determined using the non-injected and control morpholino-injected fish since previous reports of d-serine highlighted that a concentration of 1000 ppm has a detrimental effect on muscle and neuromuscular junction integrity.32 Studies of skeletal muscle phenotype and survival rates (assessed at 48 hpf) revealed no differences between the 75 and 100 µM l-serine (Fig. 6C). Given that sil1 morpholino (6 ng) injected embryos did not present a considerable muscle pathology, a possible improvement of myopathology upon l-serine treatment would have been difficult to monitor. Thus, injection of 8 ng of the sil1 morpholino was carried out, resulting in a more pronounced muscle pathology and impact on the overall survival of the MSS fish (Fig. 5C).
Treatment of the three different fish models (sil1 fish with 6 ng and 8 ng morpholino injection, and 20 ng phgdh morpholino) with 100 µM l-serine revealed that, in sil1 and phgdh morphants, the mean survival ratio increased by 19% and 18%, respectively, when compared to the mock-treated group (Fig. 6C). In contrast, the survival rates remain mostly unchanged in the inpp5k treated and untreated morphants (Fig. 6C).
To verify the potential beneficial effects of l-serine treatment, a comparative analysis of the morphology of treated and untreated fish was carried out: on the light microscopic level, inpp5k, sil1 and phgdh morpholino-injected fish treated with l-serine for 48 h presented no differences in comparison with the respective morpholino-injected fish models without treatment (Fig. 6A). However, further immunofluorescence-based morphological studies of vulnerable tissues revealed an amelioration of the slight decrease of fluorescence intensity of nerves VI and VII in inpp5k morphants upon l-serine treatment (Fig. 6B). Also, in the sil1 morphants amelioration of decreased fluorescence intensity for cranial nerves the VI, VII and X (corresponding to abduces, facial and vagal nerves) could be detected upon l-serine treatment (Fig. 6B). In the phgdh morphants, upon l-serine treatment, a marked improvement of cranial nerves VI, VII and X in addition to Va and Vp, as exemplified by an increase of the respective fluorescence intensities, was observed (Fig. 6B). Recovery of neuromuscular integrity upon l-serine treatment was also assessed in the zebrafish models: immunofluorescence studies revealed a slight improvement of motor axon growth and synaptogenesis/axon extension along myosepta in treated sil1 and phgdh morpholino-injected embryos (Fig. 6B) as shown by the Pearson’s coefficient, which expresses an increase in co-localization of SV2 with AChR (bungarotoxin) (Supplementary Fig. 2B). As expected, inpp5k morphants present no changes in neuromuscular junction morphology (Fig. 6B and Supplementary Fig. 2B) upon l-serine treatment. Investigation of the effect of l-serine based intervention of the muscular phenotype in all three fish models revealed no effect in the inpp5k morphants but did reveal a mild amelioration in the sil1 morphant with a more positive effect on the myopathology in the phgdh morphants (Fig. 6B).
We performed quantification of chorion movements33 to monitor if l-serine treatment could have a beneficial effect. Zebrafish embryos were recorded for 2 min and the normal tail movements (tail thrashes) were counted at 24 hpf. Non-injected zebrafish moved on average 4.7 times per minute and injection of the control morpholino reduced this to 4.1. No movements were recorded in the inpp5ka+b morphants; however, the supplementation of l-serine increased the movements to 2.4 per minute. The sil1 morphants (8 ng of injected morpholino) treated with l-serine presented with 4.1 movements per minute, a statistically significant increase (t-test < 0.05) compared with the mock treated sil1 morphants, which showed 50% less movements (Fig. 6D). The phgdh morphant embryos per se presented with an irregular increase of tail movements catalogued as alternating side tail thrash (18.6 movements per minute; Fig. 6D) as a result of the severe neuronal phenotype. Despite the severe phenotype observed in this model, l-serine treatment resulted in a statistically significant increase of tail trashes (41.2 movements per minute; Fig. 6D).
Prompted by our zebrafish results we have additionally assessed the cellular survival in three INPP5K8 and two MSS4 fibroblasts (Supplementary Fig. 3) lines, which showed up to 20% increase in cellular viability in l-serine MSS-treated cells compared to non-treated cells, while in INPP5K-treated fibroblasts, the viability increased by 5% in line with the embryo viability we have obtained in our fish models (Fig. 6C).
Discussion
Genetic, clinical and muscle biopsy findings
Different pathogenic variants with a predominance of missense mutations in INPP5K have been recently associated with a syndromic form of congenital muscular dystrophy.8,9,22 Applying whole exome sequencing in five patients, we identified two homozygous mutations affecting amino acid position 23 of INPP5K while the p.Val23Met missense mutation has already been described,9,22 the p.Val23Ala mutation tested to be pathogenic by our further functional studies (Fig. 2A and B) has not yet been reported. Additionally, a missense mutation affecting amino acid 55 (p.Leu55Phe) and a frameshift mutation affecting amino acid 251 (p.Arg251Serfs*24)—both also not reported so far in the literature—were identified. Hence, our clinical findings expand the current mutational spectrum of the disease which, based on the current literature, is defined by the presence of congenital/early onset cataracts, mild intellectual disability and congenital myopathy. We now expand the INPP5K clinical phenotype that overlaps with MSS, as degeneration of the cerebellum represents a consistent finding in MSS patients as well as in woozy mice.6,34,35 In the same context, absence of cataracts, as observed in one of our patients, has also been already described as an unusual finding in MSS.6 With the presence of myalgias and an axonal neuropathy, our index patient 5 also contributes to the current spectrum of the INPP5K phenotype. It is important to note that motor neuropathy has also been described in one MSS patient36 and axonal vulnerability was recently shown in MSS, sil1 morphant zebrafish and woozy mice.26
A comprehensive study in patients with INPP5K variants reported on biochemical vulnerability of components of the dystrophin-associated glycoprotein complex (DGC), defining the phenotype as a recessive form of congenital muscular dystrophy overlapping with MSS and dystroglycanopathy.9 The histological findings in our patients cannot confirm the above-described feature, rather they support our previous findings that proteins of the DGC are not necessarily affected by INPP5K pathophysiology.8
Consequently, the combined clinical findings in our patient cohort suggest that both rare neuropaediatric diseases represent a clinical continuum, which might be caused by common pathophysiological cascades and mechanisms preventing the manifestation of clinical hallmarks in a minority of the patients.
Pathophysiological cascades and common treatment concept
A molecular link between MSS and the INPP5K phenotype has already been suggested based on previous studies highlighting that both proteins bind to BiP and are involved in BiP-related processes controlling ER organization and function.37 While SIL1 loss results in perturbed protein processing associated with the massive build-up of protein aggregates,38,39 these pathophysiological findings could not be identified in INPP5K patient-derived fibroblasts.8 A possible explanation for this difference includes the localization and function of these proteins within the ER and distinct roles that result from the interaction of INPP5K, SIL1 with BiP respectively. While INPP5K is localized within the ER tubules (including distal portions) and is suggested to fine tune ER organization,37 SIL1, is localized within the ER lumen and acts as a nucleotide exchange factor.40 In skeletal muscle the interaction of INPP5K with BiP is necessary not only for its ER localization but has also been shown to regulate insulin signalling,37,41 while the BiP-SIL1 interaction is necessary for protein production and folding.42 Additionally, the presence of a residual INPP5K activity in patient muscle and fibroblasts can still modulate BiP related processes thus acting as a compensatory mechanism.
To obtain further insights into the molecular connection of MSS and INPP5K phenotypes, unbiased proteomic studies using patient-derived cells were carried out and data intersection allowed the definition of PHGDH as a protein with decreased abundance in MSS patient-derived fibroblasts but increased abundance in INPP5K patient-derived fibroblasts (Fig. 4). The decrease of PHGDH has recently been described by us in lymphoblastoid cells derived from MSS patients as well as in SIL1-vulnerable tissue derived from the woozy mouse model.12 In addition, the same study confirmed the increase of PHGDH upon the presence of ER stress,12 supporting the idea that PHGDH expression is modulated by ER stress.43
PHGDH is a 3-phosphoglycerate dehydrogenase that catalyses the transformation of 3-phosphoglycerate to 3-hydroxyphosphopyruvate, which is the first step in the de novo biosynthesis of l-serine from carbohydrates.44 Patients suffering from recessive mutations in the PHGDH gene have either decreased levels of the corresponding protein or show a reduction of the catalytic activity of this enzyme, resulting in decreased serine levels that in turn, lead to phenotypical consequences, such as intellectual disability, microcephaly and progressive neuropathy, as well as occasionally encephalopathy with spasticity or seizures.45,46 Recurrent muscle contractions called infantile spasms are typical in this disorder, a primary muscle pathology has not been linked to PHGDH deficiency. Most affected individuals have an infantile onset, which is the most severe form of the disease spectrum.46–48 Recessive PHGDH mutations have been linked to the manifestation of another (neurological) syndromic phenotype characterized by microcephaly, cataracts, intellectual disability, mild cerebellar ataxia and axonal sensorimotor polyneuropathy.49 Although of varying degrees of severity, these phenotypical findings show an overlap with the clinical presentation of the INPP5K phenotype and MSS (Table 2). Consequently, our biochemical findings combined with the clinical manifestation of these diseases (also see above) lend further support to the hypothesis of a clinical continuum of rare neuropaediatric diseases based on common pathophysiological cascades. Although primary involvement of skeletal muscle has not been explicitly described in PHDGH patients, results of our zebrafish experiments reveal vulnerability of skeletal muscle upon reduction of PHGDH (Figs 5 and 6) expression in vivo. This observation is in agreement with the fact that PHGDH is crucial for muscle cell growth and has been recently found to be increased in skeletal muscle of patients with reversible myopathy.50,51 In our zebrafish models, PHGDH protein levels decreased in the sil1 morpholino-injected zebrafish (and in phgdh morphants as a proof-of-principle) whereas the INPP5K depletion correlated with a slight increase in PHGDH (Fig. 5C and D). Free l-serine levels quantified in our zebrafish models are also in accordance with the expression of PHGDH in these morphants (Fig. 5C–E). While the differences observed are relatively small, they may have biologically relevant effect. Free amino acids represent ∼3% of the total amino acid pools and are potent activators of different signalling pathways at very low concentrations.52
Concordantly, our immunofluorescence studies, using INPP5K patient-derived muscle biopsies, also revealed an increased abundance of PHGDH in a proportion of fibres (Fig. 4B–D). l-serine mediates neuroprotection also through the modulation of the ER stress response.53 Given that this cellular defence mechanism is perturbed in MSS pathophysiology (not in INPP5K) and that l-serine treatment has already been successfully applied to ameliorate the PHGDH phenotype in humans,49 we preclinically tested l-serine treatment as a novel therapeutic strategy for INPP5K- and SIL1-related diseases in the respective zebrafish models. Overall l-serine supplementation in zebrafish models demonstrated a hormetic effect of this amino acid’s concentration, which is a biphasic dose response characterized by a beneficial effect at controlled doses (75 and 100 µm) and an inhibitory or toxic effect at higher doses, leading to embryonic lethality (over 200 µm). The small improvements seen in inpp5k a+b morphants might correlate with the observed increase of PHGDH in INPP5K patients (and whole protein extracts from inpp5k a+b morphants) suggesting that an increase of l-serine is already at a therapeutic level in these patients and acts as a cellular stress defence mechanism thus further elevation of this amino acid has no beneficial effect in terms of saturated compensation. Moreover, we observed that l-serine treatment of MSS fibroblasts has a beneficial effect with an up to 20% increase in cellular survival, while INPP5K fibroblasts showed a minor increase only (5%) (Supplementary Fig. 3) This may support our hypothesis that further supplementation with l-serine will in INPP5K disease models will not lead to major improvements as the levels of this amino acid are already at ‘therapeutic’ levels through an endogenous compensatory mechanism.
Regarding the myopathic phenotypes, l-serine treatment had no effect in the inpp5k morphants, whereas in sil1 and phgdh morphants, a mild amelioration of muscle fibre disintegration could be detected (Fig. 6B). Given this, one might assume that in SIL1 myopathy, PHGDH reduction is a stronger general contributor to the overall muscle pathophysiology than in INPP5K myopathy, where, in contrast, the levels of this protein are elevated.
Cranial nerves in all sil1 and phgdh fish models greatly benefit from the increased concentration of l-serine while the inpp5k a+b morphants show minor changes (Fig. 6B). Numerous publications have underlined the importance of l-serine in CNS development, as supplementation of l-serine was found to have a beneficial effect on neurodegeneration in CNS disorders.30,54l-serine was shown to be an astroglia-derived trophic factor for Purkinje neurons,47 a population highly affected by the loss of SIL1.35,47 Here we show that the same cellular population might also be affected in INPP5K patients as mild cerebellar atrophy was shown in one patient. We hypothesize based on our data that an elevation of PHGDH in INPP5K patients contribute to the morphological rescue of the brain and that lower levels of this enzyme might contribute to a neurological phenotype as seen in Patient 2 (Table 1).
Combined results of our preclinical interventional study suggest that MSS and INPP5K patients might benefit from l-serine intake whereby a greater effect on the nervous system phenotype than on the muscle phenotype can be predicted. l-serine has been used successfully without any major side effects in patients with amyotrophic lateral sclerosis which have received approximatively twice a day 15 g of l-serine.29 Recent studies have demonstrated that l-serine administration might be also beneficial for patients with Alzheimer’s disease.30 It is important to note here that the FDA states l-serine is generally regarded as safe as long as it consists of no more than 8.4% of total protein in the diet.29
Conclusions
Within this study, the description of six new patients suffering from INPP5K mutations expanded both the mutational and phenotypical spectrum of the underlying disease. The latter aspect is of particular importance for clinical evaluation and suggestion of candidate genes. Results of our proteomic profiling experiments allowed the definition of a first ‘molecular puzzle’ of three rare neuromuscular diseases with striking phenotypical (syndromic) overlap (Table 2) via altered abundance of PHGDH. A therapeutic potential of this new acquired knowledge has been directly implicated by treatment of our zebrafish models phenocopying the human diseases with l-serine and thus suggests that this approach might be beneficial for MSS and INPP5K patients, respectively.
Supplementary Material
Acknowledgements
We thank Mrs Nancy Meyer for her technical expert assistance. Prof. Dr Jan Senderek (Friedrich-Baur Institute Munich, Germany) kindly provided the cells derived from MSS patients.
Funding
This work was supported by a grant from the Deutsche Forschungsgemeinschaft (to R.P.Z.; ZA 639/1–1), by the Deutsche Gesellschaft für Muskelkranke (to A.R.). Moreover, financial support by the French Muscular Dystrophy Association (AFM-Téléthon; grant 21466 to A.R., grant 22429 to L.S. and postdoctoral fellowship to E.O.), the Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfalen, the Der Regierende Bürgermeister von Berlin, Senatskanzlei Wissenschaft und Forschung is gratefully acknowledged. The Institut National de la Santé et de la Recherche Médicale, the Sorbonne Université- Faculté de Médecine, the Association Française contre les Myopathies and France Génomique (Myocapture Project), National Research Agency, Investment for the Future (Grant No. ANR-10-INBS-09) also supported this work. H.L. receives support from the Canadian Institutes of Health Research (Foundation Grant FDN-167281), the Canadian Institutes of Health Research and Muscular Dystrophy Canada (Network Catalyst Grant for NMD4C), the Canada Foundation for Innovation (CFI-JELF 38412), and the Canada Research Chairs program (Canada Research Chair in Neuromuscular Genomics and Health, 950–232279). R.H. was supported by the European Research Council [309548], the Wellcome Investigator Award [109915/Z/15/Z]. the Medical Research Council (UK) [MR/N025431/1]; the Wellcome Trust Pathfinder Scheme [201064/Z/16/Z], the Newton Fund [UK/Turkey, MR/N027302/1], the Lily Foundation and the Evelyn Trust.
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Glossary
- ER
endoplasmic reticulum
- MSS
Marinesco-Sjögren syndrome
References
- 1.Marinesco G, Draganesco SDV.. Nouvelle maladie familiale caractérisée par une cataracte congénitale et un arrêt du développement somato-neuro-psychique. Encephale. 1931;26:97–109. [Google Scholar]
- 2.Sewry CA, Voit T, Dubowitz V.. Myopathy with unique ultrastructural feature in Marinesco-Sjogren syndrome. Ann Neurol. 1988;24(4):576–580. [DOI] [PubMed] [Google Scholar]
- 3.Sjogren T.Hereditary congenital spinocerebellar ataxia accompanied by congenital cataract and oligophrenia; a genetic and clinical investigation. Conf Neurol. 1950;10(5):293–308. [PubMed] [Google Scholar]
- 4.Senderek J, Krieger M, Stendel C, et al. Mutations in SIL1 cause Marinesco-Sjogren syndrome, a cerebellar ataxia with cataract and myopathy. Nat Genet. 2005;37(12):1312–1314. [DOI] [PubMed] [Google Scholar]
- 5.Anttonen AK, Mahjneh I, Hamalainen RH, et al. The gene disrupted in Marinesco-Sjogren syndrome encodes SIL1, an HSPA5 cochaperone. Nat Genet. 2005;37(12):1309–1311. [DOI] [PubMed] [Google Scholar]
- 6.Krieger M, Roos A, Stendel C, et al. SIL1 mutations and clinical spectrum in patients with Marinesco-Sjogren syndrome. Brain. 2013;136(Pt 12):3634–3644. [DOI] [PubMed] [Google Scholar]
- 7.Munro S, Pelham HR.. An Hsp70-like protein in the ER: Identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell. 1986;46(2):291–300. [DOI] [PubMed] [Google Scholar]
- 8.Wiessner M, Roos A, Munn CJ, et al. Mutations in INPP5K, encoding a phosphoinositide 5-phosphatase, cause congenital muscular dystrophy with cataracts and mild cognitive impairment. Am J Hum Genet. 2017;100(3):523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Osborn DP, Pond HL, Mazaheri N, et al. Mutations in INPP5K cause a form of congenital muscular dystrophy overlapping Marinesco-Sjogren syndrome and dystroglycanopathy. Am J Hum Genet. 2017;100(3):537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hendershot LM.The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71(5):289–297. [PubMed] [Google Scholar]
- 11.Ijuin T, Hatano N, Takenawa T.. Glucose-regulated protein 78 (GRP78) binds directly to PIP3 phosphatase SKIP and determines its localization. Genes Cells. 2016;21(5):457–465. [DOI] [PubMed] [Google Scholar]
- 12.Kollipara L, Buchkremer S, Coraspe JAG, et al. In-depth phenotyping of lymphoblastoid cells suggests selective cellular vulnerability in Marinesco-Sjogren syndrome. Oncotarget. 2017;8(40):68493–68516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ.. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF.. Stages of embryonic development of the zebrafish. Dev Dyn. 1995;203(3):253–310. [DOI] [PubMed] [Google Scholar]
- 15.Kawahara G, Hayashi YK.. Characterization of zebrafish models of Marinesco-Sjogren syndrome. PLoS One. 2016;11(10):e0165563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosen JN, Sweeney MF, Mably JD.. Microinjection of zebrafish embryos to analyze gene function. J Vis Exp. 2009;(25):1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Higashijima S, Hotta Y, Okamoto H.. Visualization of cranial motor neurons in live transgenic zebrafish expressing green fluorescent protein under the control of the islet-1 promoter/enhancer. J Neurosci. 2000;20(1):206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cohen SA, Michaud DP.. Synthesis of a fluorescent derivatizing reagent, 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate, and its application for the analysis of hydrolysate amino acids via high-performance liquid chromatography. Anal Biochem. 1993;211(2):279–287. [DOI] [PubMed] [Google Scholar]
- 19.Müller JS, Jepson CD, Laval SH, Bushby K, Straub V, Lochmüller H.. Dok-7 promotes slow muscle integrity as well as neuromuscular junction formation in a zebrafish model of congenital myasthenic syndromes. Hum Mol Genet. 2010;19(9):1726–1740. [DOI] [PubMed] [Google Scholar]
- 20.Senderek J, Müller JS, Dusl M, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet. 2011;88(2):162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Westerfield M, The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio), 3rd Edition, 1995, Eugene, OR, University of Oregon Press. [Google Scholar]
- 22.D'Amico A, Fattori F, Nicita F, et al. A recurrent pathogenic variant of INPP5K underlies autosomal recessive congenital muscular dystrophy with cataracts and intellectual disability: Evidence for a founder effect in Southern Italy. Front Genet. 2020;11:565868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmid AC, Wise HM, Mitchell CA, Nussbaum R, Woscholski R.. Type II phosphoinositide 5-phosphatases have unique sensitivities towards fatty acid composition and head group phosphorylation. FEBS Lett. 2004;576(1-2):9–13. [DOI] [PubMed] [Google Scholar]
- 24.Deutsch EW, Csordas A, Sun Z, et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017;45(D1):D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang JH, Wada A, Yoshida K, et al. Brain-specific Phgdh deletion reveals a pivotal role for L-serine biosynthesis in controlling the level of D-serine, an N-methyl-D-aspartate receptor co-agonist, in adult brain. J Biol Chem. 2010;285(53):41380–41390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Phan V, Cox D, Cipriani S, et al. SIL1 deficiency causes degenerative changes of peripheral nerves and neuromuscular junctions in fish, mice and human. Neurobiol Dis. 2019;124:218–229. [DOI] [PubMed] [Google Scholar]
- 27.Yoshida K, Furuya S, Osuka S, et al. Targeted disruption of the mouse 3-phosphoglycerate dehydrogenase gene causes severe neurodevelopmental defects and results in embryonic lethality. J Biol Chem. 2004;279(5):3573–3577. [DOI] [PubMed] [Google Scholar]
- 28.de Koning TJ, Snell K, Duran M, Berger R, Poll-The BT, Surtees R.. L-serine in disease and development. Biochem J. 2003;371(Pt 3):653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabatabaie L, Klomp LW, Berger R, de Koning TJ.. L-serine synthesis in the central nervous system: A review on serine deficiency disorders. Mol Genet Metab. 2010;99(3):256–262. [DOI] [PubMed] [Google Scholar]
- 30.Levine TD, Miller RG, Bradley WG, et al. Phase I clinical trial of safety of L-serine for ALS patients. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(1-2):107–111. [DOI] [PubMed] [Google Scholar]
- 31.Le Douce J, Maugard M, Veran J, et al. Impairment of glycolysis-derived l-serine production in astrocytes contributes to cognitive deficits in Alzheimer's disease. Cell Metab. 2020;31(3):503–517.e8. [DOI] [PubMed] [Google Scholar]
- 32.Chen XG, Wang YH, Wen CC, Chen YH.. Overdose of D-serine induces movement disorder and neuromuscular changes of zebrafish larvae. J Toxicol Pathol. 2014;27(1):19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menelaou E, Husbands EE, Pollet RG, Coutts CA, Ali DW, Svoboda KR.. Embryonic motor activity and implications for regulating motoneuron axonal pathfinding in zebrafish. Eur J Neurosci. 2008;28(6):1080–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao L, Longo-Guess C, Harris BS, Lee JW, Ackerman SL.. Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nat Genet. 2005;37(9):974–979. [DOI] [PubMed] [Google Scholar]
- 35.Buchkremer S, Gonzalez Coraspe JA, Weis J, Roos A.. Sil1-mutant mice elucidate chaperone function in neurological disorders. J Neuromuscul Dis. 2016;3(2):169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byrne S, Dlamini N, Lumsden D, et al. SIL1-related Marinesco-Sjoegren syndrome (MSS) with associated motor neuronopathy and bradykinetic movement disorder. Neuromuscul Disord. 2015;25(7):585–588. [DOI] [PubMed] [Google Scholar]
- 37.Dong R, Zhu T, Benedetti L, et al. The inositol 5-phosphatase INPP5K participates in the fine control of ER organization. J Cell Biol. 2018;217(10):3577–3592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roos A, Buchkremer S, Kollipara L, et al. Myopathy in Marinesco-Sjogren syndrome links endoplasmic reticulum chaperone dysfunction to nuclear envelope pathology. Acta Neuropathol. 2014;127(5):761–777. [DOI] [PubMed] [Google Scholar]
- 39.Roos A, Kollipara L, Buchkremer S, et al. Cellular signature of SIL1 depletion: disease pathogenesis due to alterations in protein composition beyond the ER machinery. Mol Neurobiol. 2016;53(8):5527–5541. [DOI] [PubMed] [Google Scholar]
- 40.Dudek J, Benedix J, Cappel S, et al. Functions and pathologies of BiP and its interaction partners. Cell Mol Life Sci. 2009;66(9):1556–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ijuin T, Hatano N, Hosooka T, Takenawa T.. Regulation of insulin signaling in skeletal muscle by PIP3 phosphatase, SKIP, and endoplasmic reticulum molecular chaperone glucose-regulated protein 78. Biochim Biophys Acta. 2015;1853(12):3192–3201. [DOI] [PubMed] [Google Scholar]
- 42.Ichhaporia VP, Kim J, Kavdia K, et al. SIL1, the endoplasmic-reticulum-localized BiP co-chaperone, plays a crucial role in maintaining skeletal muscle proteostasis and physiology. Dis Model Mech. 2018;11(5):dmm033043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dombroski BA, Nayak RR, Ewens KG, Ankener W, Cheung VG, Spielman RS.. Gene expression and genetic variation in response to endoplasmic reticulum stress in human cells. Am J Hum Genet. 2010;86(5):719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pacold ME, Brimacombe KR, Chan SH, et al. Corrigendum: A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12(8):656. [DOI] [PubMed] [Google Scholar]
- 45.Tabatabaie L, Klomp LW, Rubio-Gozalbo ME, et al. Expanding the clinical spectrum of 3-phosphoglycerate dehydrogenase deficiency. J Inherit Metab Dis. 2011;34(1):181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van der Crabben SN, Verhoeven-Duif NM, Brilstra EH, et al. An update on serine deficiency disorders. J Inherit Metab Dis. 2013;36(4):613–619. [DOI] [PubMed] [Google Scholar]
- 47.Furuya S, Tabata T, Mitoma J, et al. L-serine and glycine serve as major astroglia-derived trophic factors for cerebellar Purkinje neurons. Proc Natl Acad Sci U S A. 2000;97(21):11528–11533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pind S, Slominski E, Mauthe J, et al. V490M, a common mutation in 3-phosphoglycerate dehydrogenase deficiency, causes enzyme deficiency by decreasing the yield of mature enzyme. J Biol Chem. 2002;277(9):7136–7143. [DOI] [PubMed] [Google Scholar]
- 49.Brassier A, Valayannopoulos V, Bahi-Buisson N, et al. Two new cases of serine deficiency disorders treated with l-serine. Eur J Paediatr Neurol. 2016;20(1):53–60. [DOI] [PubMed] [Google Scholar]
- 50.Brown DM, Williams H, Ryan KJ, et al. Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) and serine biosynthetic pathway genes are co-ordinately increased during anabolic agent-induced skeletal muscle growth. Sci Rep. 2016;6(1):28693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hathazi D, Griffin H, Jennings MJ, et al. Metabolic shift underlies recovery in reversible infantile respiratory chain deficiency. EMBO J. 2020;39(23):e105364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Falco F, Barra M, Cammarata M, et al. Amino acid composition in eyes from zebrafish (Danio rerio) and sardine (Sardina pilchardus) at the larval stage. Springerplus. 2016;5:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dunlop RA, Powell JT, Metcalf JS, Guillemin GJ, Cox PA.. L-serine-mediated neuroprotection includes the upregulation of the ER stress chaperone protein disulfide isomerase (PDI). Neurotox Res. 2018;33(1):113–122. [DOI] [PubMed] [Google Scholar]
- 54.Garofalo K, Penno A, Schmidt BP, et al. Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. J Clin Invest. 2011;121(12):4735–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding author upon reasonable request.
INPP5K fibroblasts: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD009272. To visualize the data please use the username: reviewer26477@ebi.ac.uk and password: RdNNHLlo.
MSS fibroblasts: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD009297. To visualize the data please use the username: reviewer67767@ebi.ac.uk and password: eOUI5Dcd.
The MSS lymphoblasts dataset has PXD003030 as an identifier and can be found online via ProteomeXchange.