

Abstract
Many aspects of cognition and behaviour are regulated by noradrenergic projections to the forebrain originating from the locus coeruleus, acting through alpha and beta adrenoreceptors. Loss of these projections is common in neurodegenerative diseases and contributes to their cognitive and behavioural deficits. We review the evidence for a noradrenergic modulation of cognition in its contribution to Alzheimer’s disease, Parkinson’s disease and other cognitive disorders. We discuss the advances in human imaging and computational methods that quantify the locus coeruleus and its function in humans, and highlight the potential for new noradrenergic treatment strategies.
Keywords: noradrenaline, locus coeruleus, neurodegeneration, cognition, dementia
Holland et al. review the role of the noradrenergic system in regulating cognition. They highlight the involvement of this system in many neurodegenerative diseases, including its contribution to clinical symptoms and its potential as a therapeutic target.
Introduction
Degeneration of the noradrenergic system is a pathological hallmark of many neurodegenerative diseases including Parkinson’s disease, Alzheimer’s disease, and Huntington disease.1 Indeed, pathology in the principal source of noradrenaline, the locus coeruleus (LC), can occur before the loss of other neurotransmitter systems commonly associated with such conditions or cerebral atrophy.2 Furthermore, the role of noradrenaline in diverse cognitive processes is well established, including vigilance, attention, and learning and memory.3-7 Yet, the degree to which noradrenergic systems contribute to the cognitive and behavioural changes resulting from neurological disease is often under-recognized8 despite early research reviews9 and more recent work to integrate the evidence into a coherent neurocognitive framework.10
In this review we bring together the evidence for a central role of noradrenaline in cognition and cognitive dysfunction in neurodegenerative diseases including Parkinson’s disease, Alzheimer’s disease, Huntington disease, frontotemporal lobar degeneration and multiple system atrophy. We draw on data from rodent and non-human primates, while acknowledging important species differences in the neurobiology of the LC. We review the evidence from recent advances in computational models and imaging of the LC in health and disease, and examine the potential for pharmacological and non-pharmacological treatments based on preclinical and clinical studies. Together, these point the way forward to new therapeutic strategies for selective and non-selective noradrenergic treatments.
The anatomy and pharmacology of noradrenaline and the locus coeruleus
The forebrain noradrenergic input is from a small bilateral collection of neurons called the locus coeruleus (LC), where the cells begin rostrally at the level of the inferior colliculus adjacent to the cerebral aqueduct and end caudally near the lateral wall of the fourth ventricle11; on axial brain slices, the anatomical landmarks are ∼1 mm under the fourth ventricle, ∼3 mm from the midline, and centred ∼14–21 mm above the ponto-medullary junction12 (Fig. 1). The LC is readily identified at post-mortem by its dark colour owing to the high neuromelanin content; the synthesis of the pigment, neuromelanin, is driven by excess levels of catecholamines in the cytosol and is thus crucially linked to the synthesis and metabolism of noradrenaline.13,14 Other conventional ways to identify noradrenaline producing neurons in the LC is by immunohistochemistry directed at tyrosine hydroxylase (TH), the enzyme that converts l-tyrosine to l-DOPA, or against dopamine beta-hydroxylase, which converts dopamine to noradrenaline.15 It is widely considered that the two markers are expressed by identical neuronal populations16 representing the noradrenergic neuronal population of LC which constitutes more than 95% of neurons in the LC in controls.17,18 However, a small proportion of large TH-positive neurons, which are more numerous in the rostral than caudal pons, lack pigmentation.15 Within the LC there is heterogeneity in both the population of the residing medium-sized neurons and neuronal numbers across the rostro-caudal gradient. The majority of medium-sized LC noradrenergic neurons are large multipolar cells (35–45 µm in diameter), with plump cell bodies and short dendrites; in the caudal LC and subcoeruleus, where the density of medium sized neurons is lower, the larger medium sized neurons are interspersed with smaller fusiform noradrenergic neurons (∼15 µm in diameter) with triangular cell bodies and two tufts of long dendrites.19 Along the LC, there is a spatially differentiated neuronal organization such that cells giving rise to hippocampal projections are located in more rostral segments while those that innervate the neocortex, cerebellum and spinal cord are located more caudally; subcortical projections of the LC are more scattered, with a proposed spatial bias towards the caudal portion.14,19 The observation of a rostrocaudal gradient has recently been confirmed in vivo and is important to consider when assessing for neuronal loss secondary to neurodegeneration where selective loss of rostral or caudal groups may be observed.20 We discuss this selective vulnerability later in the review.
Figure 1.

Neuroanatomical location and projections of the LC. (A) Schematic sagittal view of the brain, illustrating locus coeruleus anatomy, projections, and downstream cognitive dysfunction associated with disturbed LC projections. (B) Coronal and (C) axial views of the locus coeruleus obtained from magnetization transfer weighted sequences at 7 T MRI. Ant = anterior; Post = posterior. Image courtesy of Dr Rong Ye and Dr Claire O’Callaghan.
The LC is now increasingly studied in vivo, through MRI by utilizing the highly paramagnetic neuromelanin content,21–23 where the inferior colliculus and the recess of the fourth ventricle are used as key landmarks for its segmentation.20 Noradrenaline also arises from the subcoeruleus nucleus extending ventrolaterally from the caudal pole of the LC, innervating the brainstem and hypothalamus for neuroendocrine and autonomic regulation,11,24 but these projections are less relevant for higher cognitive functions.
Given its extensive projections to both cortical and subcortical areas25 (Fig.1A), the LC is surprisingly small. With variations in preparation and counting techniques, estimates vary in the range 20 000–98 000 neurons in humans,26–31 with the highest estimated neuronal numbers obtained by unbiased stereology.32 Within this collection of neurons lie subgroups that preferentially project to the primary motor cortex and the subregions of the prefrontal cortex33 leading to a non-uniform release of LC-mediated noradrenaline across the cortical mantle.34,35
Three factors contribute to the sophistication of noradrenergic transmission. First, the diversity of noradrenergic receptors (Fig. 2). Second, the distinction between tonic and phasic neurotransmission. Third, the non-linear relationship between innervation and performance (Fig. 3).
Figure 2.

Noradrenaline synthesis pathway, distribution of pre and postsynaptic adrenoreceptors, and available noradrenergic agonist and antagonists used in animal and human studies. Agonists are depicted by a plus symbol and dark green arrows, whilst antagonists are depicted by the letter ‘X’ and orange arrows. Drugs used in human studies and clinical trials are marked with an asterisk. Noradrenaline synthesis pathway: noradrenaline is synthesized from tyrosine, which is initially converted to l-DOPA through the action of tyrosine hydroxylase (TH); l-DOPA is further converted to dopamine by aromatic l-amino acid decarboxylase (AADC), before finally being converted to noradrenaline through the action of dopamine β-monooxygenase (DA-C; also known as dopamine β-hydroxylase). Noradrenaline is recycled through the norepinephrine transporter (NET) and degraded by monoamine oxidase (MOA), to the principal end product vanillylmandelic acid or a conjugated form of 3-methoxy-4-hydroxyphenylglycol (MHPG). Methylphenidate = mixed noradrenaline and dopamine reuptake inhibitor.
Figure 3.
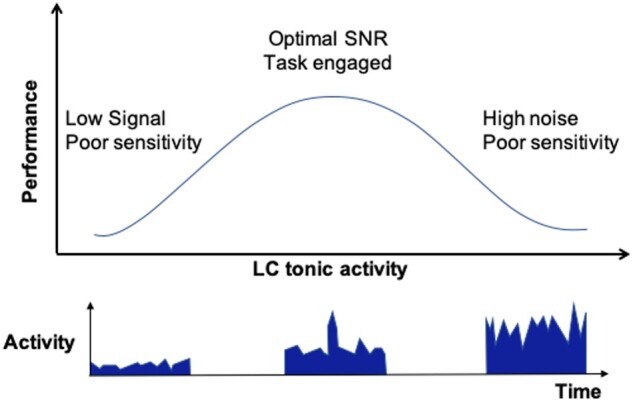
Schematic illustration of the non-linear function of performance versus locus coeruleus activity, analogous to the Yerkes-Dodson model of arousal and comparable to non-linear relationships in dopaminergic and serotonergic systems.
Noradrenaline exerts an excitatory action through the post-synaptic α1 and β adrenoceptors, and an inhibitory action through mainly presynaptic α2-adrenoreceptors.36 The distribution and affinity of adrenoreceptors is highly variable. For example, α2-adrenoreceptor are common in the prefrontal cortical areas, and noradrenaline has the highest affinity for these,37 and lower affinity for α1- and β-adrenoreceptors.38 As a consequence moderate levels of noradrenaline engage α2 receptors whilst higher levels (released during stress for example) engage the lower-affinity α1 and β receptors.39 This creates a non-linear relationship between noradrenergic transmission and performance, indicating that response to an excitatory input may be enhanced or suppressed depending on the receptor in action.40
The LC exhibits two broad firing patterns: tonic and phasic (Fig. 3).41 These have distinct properties and signal processing characteristics. For example, during direct physiological recordings in monkeys, in a visuo-motor task with reward and punishment, phasic responses followed salient stimuli but not distractors.41 Phasic responses were diminished or absent in poor performance trials suggesting a role as an attentional filter that selects for the occurrence of task-relevant stimuli. When not engaged in task performance the LC returns to a tonic firing rate. Within the same visuo-motor task, elevated tonic LC activity reduced the ability to discriminate stimuli from distractors; the monkeys were more distractible and made more errors. These observations are replicated in rats where stimulating LC tonic activity leads to increased decision noise and reduced task participation.42 The balance between tonic and phasic activity therefore, enables a gating signal function that regulates task engagement or disengagement according to salience and anticipated rewards or punishments, facilitating an adaptive behaviour. This is supported in human studies, where pupillometry has been used as a surrogate for LC activity43,44 such that a large baseline pupil diameter implies LC phasic activity and a smaller one implies tonic LC activity; for example, in an auditory discrimination task, phasic pupillary dilatation correlated with correct responses, whereas tonic pupillary dilatation correlated with periods of low reward value.45 In signal processing terms, dynamic LC activity regulates signal-to-noise ratio both at the level of the LC46 and at target neurons.47
Histological studies of the LC suggest that neuronal number and volume do not change significantly with age30,48; however, there is an increase in the LC neuromelanin content, which may reflect functional changes that contribute to variability in cognitive performance between healthy young and older adults.49,50 Until recently, studying the LC required invasive methods, limited mainly to preclinical models, or relied on indirect inferences based on the pupillometric response which is correlated with the activity of other neural networks besides the LC. However, advances in neuroimaging, by drawing on the paramagnetic features of the neuromelanin rich LC neurons, have aided the direct in vivo study of this structure and its functional connections in humans.
Using MRI enables both in vivo human quantification of LC size and neuromelanin content (Fig. 1B and C), and its functional connectivity, with good reliability.22,51 Better resolution and sensitivity of such sequences is being developed alongside post-mortem validation of the histological changes underlying each MRI contrast.21 Already, MRI has contributed to understanding the role of the LC in human cognition. For example, in a reversal learning task healthy adults (aged 65–84) were asked to make choices from single or double picture trials with reward and loss as feedback; this was then followed by MRI and a memory test of the pictures seen prior to the scanning session. Those adults with a higher LC signal intensity, performed better at the memory task especially for stimuli associated with negative feedback compared to younger adults (aged 20–31).49 A negative correlation has been proposed, between age and the connectivity between LC and ventral tegmental area, and a more complex non-linear relationship between age and the connectivity of LC to frontotemporal cortex.52 In the next section, we consider how these properties of the noradrenergic system are affected by neurodegenerative disorders.
Locus coeruleus neuronal loss in neurodegenerative diseases
LC neuronal loss is common and an early feature of neurodegenerative diseases. Post-mortem studies confirm that LC neuronal numbers are severely reduced in Alzheimer’s disease, Parkinson’s disease, and progressive supranuclear palsy (PSP) to a larger extent compared to neuronal loss in the nuclei commonly associated with these disorders.53Table 1 summarizes human studies reporting LC neuronal loss in the neurodegenerative diseases covered below.
Table 1.
Summary of human studies reporting evidence for LC pathology and noradrenergic deficiency in neurodegenerative diseases
| LC pathology/neuronal loss | Study type |
|---|---|
| Alzheimer’s disease | |
|
Neuronal loss in mild cognitive impairment54 |
Post-mortem stereology |
| Post-mortem stereology | |
|
Neurofibrillary tangle accumulation within the LC56 |
Post-mortem immunohistochemistry |
|
Reduced noradrenaline transporter PET radioligand uptake within the LC57 |
Post-mortem autoradiography |
|
LC neuronal loss correlates better with illness duration2,28 |
Post-mortem immunohistochemistry and stereology |
| Post-mortem stereology | |
|
LC signal intensity on MRI correlates with CSF Alzheimer’s disease biomarkers59 |
In vivo neuroimaging (MRI) and biochemistry |
| Parkinson’s disease | |
| Lewy body accumulation within the LC2,24,53 | Post-mortem immunohistochemistry |
|
Lewy body pathology within the LC preceding that within the SN60,61 |
Post-mortem immunohistochemistry and stereology |
|
Loss of LC neurons more severe at post-mortem than in the SN62 |
Post-mortem immunohistochemistry and stereology |
|
Lower LC signal intensity on MRI in Parkinson’s disease patients with cognitive impairment63 |
In vivo neuroimaging (MRI) |
|
Progressive LC signal loss with disease progression64 |
In vivo neuroimaging (PET) |
| Huntington’s disease | |
|
LC neuronal loss correlating with disease duration and severity of cognitive impairment65 |
Post-mortem immunohistochemistry and stereology |
| FTLD syndromes | |
| Progressive supranuclear palsy | |
| Post-mortem immunohistochemistry and stereology | |
|
LC neuronal loss negatively correlates with disease severity27 |
Post-mortem immunohistochemistry and stereology |
| Frontotemporal dementia | |
|
Tau accumulation within the LC68 |
Post-mortem immunohistochemistry |
|
Preserved LC neuronal density69 |
Post-mortem immunohistochemistry |
| CSF biochemistry; post-mortem high-performance liquid chromatography | |
| Multiple system atrophy | |
| Post-mortem immunocytochemistry |
FTLD = frontotemporal lobal degeneration; SN = substantia nigra.
Noradrenergic loss in Alzheimer’s disease and related disorders
Alzheimer’s disease is one of the commonest neurodegenerative disorders, characterized pathologically by abnormal tau accumulation in neurofibrillary tangles and the presence of extracellular plaques rich in amyloid-β deposition, and clinically by progressive memory loss and behavioural changes.74 Alzheimer’s disease has often been considered as a disorder of cholinergic dysfunction from degeneration of the nucleus basalis of Meynert (nbM)75; but there is significant LC neuronal loss which is more severe and better correlated with the duration of illness compared to the nucleus basalis2,28; and there may be selective loss of LC neurons projecting to the hippocampus, from the middle/rostral part of the LC.48,55 Alzheimer’s disease pathology is likely multifactorial and may lie in the interaction between noradrenaline (initiated from the LC) and acetylcholine (initiated from the nbM). This occurs at multiple levels:
(i)LC provided noradrenergic input to the nbM where dopamine beta-hydroxylase immunoreactive terminals make close contact with the choline acetyltransferase (ChAT) immunoreactive neurons.76,77
(ii)High densities of α2-adrenoreceptors are located in the basal forebrain of rat brain78 and α2-adrenoreceptor agonists inhibit the release of acetylcholine from the nbM.79 For example, infusion of the α2-adrenoreceptor antagonists dexefaroxan or idazoxan increase, whilst agonists at this receptor reduce the release of acetylcholine in the medial prefrontal cortex of conscious rats. The increase in acetylcholine response to dexefaroxan is diminished by noradrenergic depletion in response to DSP4 (a selective LC neurotoxin).80 Isolated saporin lesions of the cholinergic system, have been reported to cause little memory impairment despite a 90% loss in the cholinergic neurons in the basal nucleus and 90% of cortical ChAT81 while dual lesioning of the noradrenaline and cholinergic systems produce marked cognitive deficits.82
(iii)α1- and β-adrenoreceptors mediate excitatory effects of noradrenaline in basal forebrain cholinergic neurons in guinea pigs.83
Noradrenaline may also play an important neuro-inflammatory moderator role in the pathogenesis of Alzheimer’s disease via microglial activation. It negatively regulates the transcription of inflammatory genes in astrocytes and microglia, which both express adrenergic receptors.84 Lesions of the central noradrenergic systems, including LC pathways, using DSP4 in APP transgenic mice (TgAPP) that are subsequently injected with amyloid-β1–42, show an increased number of plaques in response to noradrenergic depletion, together with increased glial activation, reduced mRNA level for the amyloid-β degrading enzyme (metallopeptidase neprylisin, NEP) and reduced plaque degradation.85 Indeed, using molecular dynamics simulation, the addition of noradrenaline is shown to inhibit aggregation of amyloid-β1–42, and to promote disaggregation of amyloid-β protofibrils.86 Furthermore, in a multi-tracer PET imaging study in APP23 transgenic mice treated with DSP4, noradrenaline suppresses microglial transcription of pro-inflammatory genes, and cytokine and chemokine production, as well as regulating phagocytosis and microglia migration, leading to depletion of amyloid-β plaque burden.87,88
Pharmacological manipulation of the noradrenergic system in mouse models of Alzheimer’s disease provides some promise towards therapeutics. For example, treatment of the pro-inflammatory FAD mouse model [with combined mutations in the App gene and Psen1 (presenilin 1) gene] with droxipoda (a precursor for noradrenaline) and atomoxetine (reuptake inhibitor) improves learning in the water maze task. Similarly, treatment of this mouse model with vindeburnol (a derivative of the plant alkaloid vincamine), is associated with reduced inflammation in the LC, and increased noradrenaline levels from an increase in TH activity (the rate limiting step in noradrenaline synthesis); this experiment further shows these mutant mice have reduced anxiety-like behaviour associated with a reduction in amyloid burden in the hippocampus.89 However, the response to pharmacological modulation is highly dependent on baseline noradrenergic levels (Fig. 3). For example, stress-induced activation of β-adrenoreceptors in mouse models of Alzheimer’s disease causes further cognitive deficits with greater tau deposition and amyloid accumulation.90 The effects of pharmacological enhancement of noradrenaline, however, is yet to be fully evaluated.
These preclinical models are directly relevant to clinical studies. In humans, degeneration of LC neurons occurs in patients with mild cognitive impairment54 with accumulation of neurofibrillary tangles,56 and a linear progression of LC neuronal loss as the disease progresses into the advanced Braak stages.28,48,58 Additionally, there is reduced uptake of the noradrenaline transporter radioligand (S, S)-18F-FMeNER-D2, in the LC and thalamus of patients with Alzheimer’s disease compared to age-matched controls, which correlates with Alzheimer’s disease severity.57 More recently, Betts and colleagues59 have illustrated a negative association between the intensity of the LC signal on MRI and levels of amyloid-β in the CSF of patients with Alzheimer’s disease.
Alzheimer’s disease is prevalent in patients with Down syndrome (Trisomy 21), and indeed LC neuronal loss and subsequent noradrenergic deficit is seen in both human and mouse models of this condition.91 Patients with Down syndrome and Alzheimer’s disease and those with Down syndrome who subsequently develop Alzheimer’s disease, have lower plasma levels of noradrenaline breakdown products (MHPG; 3-methoxy-4-hydroxyphenylglycol), which significantly correlates with behavioural and psychological signs and symptoms of dementia.92,93 The transgenic mouse model of Down syndrome, Ts65n, exhibits an age-dependent reduction in noradrenaline concentration with poor contextual learning and memory which is partially reversible by droxipoda (noradrenaline prodrug). Treating these mice with formoterol (selective β2-adrenoreceptor agonist), leads to an improvement in contextual learning, reduced hyperactivity, and restored synaptic density.94 The use of such drugs in a clinical setting is limited by the abundance of β-adrenoreceptors in cardiac muscles and the associated cardiovascular abnormalities in patients with Down syndrome. More selective, direct manipulation of LC neurons is possible in mouse models using designer receptors exclusively activated by designer drugs (DREADDs). For example, stimulation of designer receptor hM3Dq by clozapine-N-oxide reduces hyperactivity and improves novel object recognition, comparable to the effect of droxipoda.95 Noradrenaline stimulation may therefore serve as a potential pathway in improving memory loss in selective individuals with Down syndrome and dementia.96 Although human literature is limited, in combination with preclinical evidence, it suggests a potential role for adjunctive noradrenergic therapy for selective patients with mild cognitive impairment and Alzheimer’s disease.
Noradrenergic loss in Parkinson’s disease
Parkinson’s disease is the second most common neurodegenerative disease, characterized pathologically by the presence of intraneuronal α-synuclein containing Lewy bodies97; clinically it presents with a characteristic movement disorder typically manifesting as bradykinesia, rigidity, tremor and postural instability98 and later cognitive decline.99 Lewy body pathology, the hallmark of Parkinson’s disease, occurs prominently in the substantia nigra pars compacta but it is also found in the LC and is associated with cell loss.2,24,53 Indeed, pathology in the LC is proposed to precede Lewy body formation in the substantia nigra pars compacta,60,61 suggesting that noradrenaline dysfunction may precede dopaminergic deficits in patients with Parkinson’s disease. Translational models of Parkinson’s disease also affect the LC. For example, in the A53T transgenic mouse model, expressing a mutant form of α-synuclein, reduction in noradrenaline but not dopamine is associated with synuclein pathology in the aged mice,100 suggesting that α-synuclein pathology is sufficient to cause noradrenergic deficiency. Such animal models also manifest cognitive dysfunction including learning and memory deficits that precede motor deficits,101 albeit confounded by anxiety-like behaviour. Pharmacological lesions induced by MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) in non-human primate models, cause both dopamine and noradrenergic deficits,102 in association with deficits in attention, attention set-shifting, working memory, cognitive flexibility, and problem solving.103
Human studies echo the above findings. The loss of neuronal density within the LC is greater in post-mortem cases of incidental Lewy body pathology compared to the neuronal loss in the substantia nigra,62 and dopamine beta-hydroxylase activity and the noradrenergic metabolite dihroxyphenylglycol (DHPG), are both reduced in the CSF.104 Similar findings are reported by neuroimaging studies. In patients with Parkinson’s disease with and without mild cognitive impairment, the LC signal on MRI is lower compared to age-matched controls, and is negatively associated with performance on the Trail Making Test B (a test of accuracy and cognitive flexibility).63 Using PET imaging in Parkinson’s disease patients exhibiting REM sleep disorder, Sommerauer and colleagues105 show a reduced uptake of 11C-MeNER (a PET reboxetine analogue with a high specificity for the noradrenaline transporter) in the LC compared with control. Markers of LC functional activity are also reduced in patients with Parkinson’s disease. In a longitudinal study of patients with early Parkinson’s disease, using 18F-DOPA PET imaging (a marker of amino acid decarboxylase activity; Fig. 2) Pavese et al.64 showed significant decline in the region of the LC over 3 years.
Similar to the pathogenesis in Alzheimer’s disease, LC impairment secondary to α-synuclein may also, in part, be immune mediated. T lymphocytes from patients with Parkinson’s disease recognize α-synuclein pathology, and neuromelanin containing organelles within the LC express human leukocyte antigens, leading to antigen presentation of endogenous or exogenous proteins to CD8 positive T lymphocytes; the latter in turn leads to cell death.106 The concept of an immune-mediated pathogenesis in Parkinson’s disease has led to the start of clinical trials targeting this pathway.107
Noradrenergic loss in Huntington’s disease
Huntington’s disease is a neurodegenerative disorder caused by unstable expansion of CAG repeats in the huntingtin gene resulting in pathology within both cortical and subcortical areas; clinically it manifests with motor, cognitive and psychiatric symptoms.108–111 Dysregulation in the monoamine oxidase A (MAOA) activity, which has a substrate selectivity for noradrenaline and serotonin, contributes to the common symptoms of depression and anxiety in this condition. In studying the YAC128 transgenic mouse model of Huntington’s disease, Garcia-Miralles et al.112 showed that treatment with clorgyline, restored noradrenaline levels and improved anxiety symptoms and behavioural manifestations of depression; targeting this pathway may therefore be a potential therapeutic option for addressing depression in this disease.
Data from human studies are limited. Post-mortem brains from patients with Huntington’s disease show lower LC neuronal counts associated with features of advanced disease, duration of illness, severity of dementia and impairment in activities of daily living.65 However, treatment of 20 patients with mild disease with atomoxetine did not improve performance on a battery of neuropsychological tests.113 This clinical study only included patients in the early stages of disease, and data from Parkinson’s disease literature suggests that premature noradrenergic treatment is likely to be ineffective.114
Noradrenergic loss in frontotemporal lobar degeneration syndromes
Several clinical disorders are associated with frontotemporal lobar degeneration, including PSP, corticobasal syndrome, motor neuron disease and frontotemporal dementia. They differ in the degree of evidence for a noradrenergic impairment.
PSP is a neurocognitive disorder, pathologically associated with accumulation of hyperphosphorylated 4-repeat tau, initially within the brainstem and basal ganglia before spreading to the cortex67; clinically it typically manifests with a movement disorder including axial rigidity, supranuclear gaze palsy and postural instability, with a dysexecutive, disinhibited and apathetic cognitive profile.115 Cognitive deficits in PSP are common and debilitating both for patients and carers. Fifty per cent of patients with PSP present with cognitive symptoms,116 and the majority will have a form of dementia during the course of their illness.117 The pathophysiology of impairment is multifactorial, but includes impairments of the noradrenergic system: post-mortem studies show PSP-related tau pathology in the LC, and LC neuronal loss,27,66,67 with α2-adrenoreceptor loss compared to age-matched controls.58 Moreover, the loss of pigmented LC neurons are negatively correlated with disease severity as measured by the PSP rating scale.27 Pharmacological manipulation in PSP has thus far focused on changing motor symptoms and not so much cognition118,119; however, given the prevalence of cognitive dysfunction, and beneficial effects of noradrenergic modulation in Parkinson’s disease,120,121 further research in this field is required.
Frontotemporal dementia is a clinical syndrome characterized by changes in personality, behaviour and language.122,123 It was formerly known clinically as Pick’s disease, although this term is now generally reserved for the particular pathological entity with 3-repeat tau aggregation (Pick’s disease).124 Other causes of frontotemporal dementia include TDP-43 inclusions or fused-in-sarcoma (FUS) pathology starting in but not limited to the frontal and temporal lobes and extending to include the rest of the brain with disease progression.125 Frontotemporal dementia lies phenotypically on a spectrum with motor neuron disease.126 The degree of change to the noradrenergic system in frontotemporal dementia and motor neuron disease remains unclear.127 Characteristic 3-repeat tau accumulation in Pick’s disease and TDP-43 inclusions have been reported in the LC of patients and the mSOD1 transgenic mouse model,68 although LC neuronal density may be preserved.69 Disruption in the noradrenaline breakdown (MHPG) product have been observed, albeit inconsistently, in the hippocampus, amygdala and frontal cortices of post-mortem of patients with behavioural variant frontotemporal dementia,71,128 and is reported to correlate with emotional lability, dementia severity and agitation.70,129 Pharmacological manipulation of the noradrenergic system may help with the common symptom of impulsivity in this patient group. In a within-subjects, double-blind placebo-controlled study of methylphenidate, a mixed dopamine/noradrenaline reuptake inhibitor, patients with frontotemporal dementia became less risk-taking in the Cambridge Gambling Task, and therefore less impulsive; of course, methylphenidate does not have a pure pharmacological target; however, the study provided some evidence for the role of noradrenergic modulation in this patient cohort.130
Noradrenergic loss in multiple system atrophy
Multiple system atrophy is a progressive neurodegenerative disease associated with oligodendroglial cytoplasmatic inclusions consisting of misfolded α-synuclein affecting the olivopontocerebellar and striatonigral systems.131,132 Clinically it presents with autonomic failure, parkinsonism and/or ataxia,133 with cognitive impairment in a minority of patients.134,135 The noradrenergic system in MSA is studied mainly in the context of autonomic dysfunction—a pathological hallmark of this disease.136,137 Loss of noradrenaline in both the LC and the caudal ventro-lateral medulla in patients with MSA has been reported,72 and post-mortem studies have shown severe loss of A5 noradrenergic neurons of the pontine tegmentum (projecting onto the medulla and spinal cord), comparable with that seen in the LC in these patients.73 Indeed in an open label study of patients with multiple system atrophy and postural hypotension, treatments with droxipoda significantly improved symptoms.138 In a recent interventional study in patients with autonomic failure secondary to multiple system atrophy, droxipoda was associated with irritability, confusion and memory impairment that improved on dose reduction or discontinuation; of note, however, none of the participants in this study had any background cognitive impairment on enrolment suggesting a baseline-dependent cognitive modulation in these patients.139
Towards new therapeutic approaches for cognition
Early comparative studies of the role of noradrenaline in cognition drew on two main paradigms: 6-hydroxydopamine (6-OHDA) lesions of the LC or the dorsal noradrenergic ascending bundle to the cerebral cortex and hippocampus; and pharmacological manipulations of adrenoreceptors using agonists (e.g. clonidine) or antagonists of presynaptic inhibitory autoreceptors (e.g. idazoxan). Later, these have been complemented by optogenetics,140 and other non-invasive measures. In humans, more recently the principal methods for the study of the noradrenergic system is through non-invasive functional MRI imaging with EEG monitoring,141 and 3 T and 7 T MRI imaging of the LC,21 or a narrower range of systemic pharmacological manipulations with noradrenergic reuptake inhibitors (e.g. atomoxetine, reboxetine). In this section, we will review the evidence for the role of the noradrenergic system in cognitive domains and review how noradrenergic modulation can benefit or deter performance in health and disease.
We draw the reader's attention to the potential limitations of evidence from non-human studies given the substantial changes the LC system and the target neocortex have been through across phylogeny. Both the LC neuronal number, and the size of one of its most important projection targets, the prefrontal cortex, have undergone considerable evolutionary change. The existence and location of the LC is consistent across non-human primates (chimpanzees, gorillas, gibbons, and macaque monkeys), although the number of TH immunoreactive neurons within the LC are increased in humans, out of proportion to the increase in size of the surrounding anatomy such as the medulla.142 Furthermore, the prefrontal expansion in humans is disproportionate to the increase in the size of other neocortical areas.143 The inhomogeneity of evolutionary development may underly some of the key behavioural differences between human and non-human primates. In addition, there are species differences in the expression of coexisting neuropeptides within the LC, such as galanin and its receptors, between humans and rodents.144 The significance of these neuropeptides for human cognition is not fully resolved. In reviewing the evidence for the role of the LC system in the following cognitive domains, one must remain aware of the caveats related to species differences even where homologies appear to exist.
Attention
The noradrenergic-LC pathway modulates alertness and attentiveness within a dynamic environment with evidence from cortical depletion of noradrenaline in rats,5 and lesions of the α2-adrenoreceptors in monkeys.41 Rowe et al.145 showed that systemic treatment with idazoxan (an α2-adrenoreceptor antagonist) impaired non-reversal task shifting but not task acquisition, indicating narrowed attention in lesioned animals. This result was complemented by a later study of effects of 6-OHDA lesions which selectively impaired extra-dimensional set-shifting.146 However, the effect of drugs depends on the noradrenergic state of the animal, which is critical for interpreting treatment potential in patients. For example, in saporin lesioned (noradrenaline depleted) rats, increasing synaptic noradrenaline by administering atomoxetine improved set-shifting but tended to impair this cognitive domain in non-lesioned rats.147 A similar baseline-dependent noradrenergic modulation of attention in rats is seen with a novel dopamine and noradrenergic modulator, SK609.148
The important balance between tonic and phasic stimulation influences the effect of drug interventions. For example, administering clonidine (an α2-adrenoreceptor agonist) to young healthy adults results in longer reaction times, indicating that inhibition of noradrenergic supply secondary to a global reduction in tonic noradrenergic input, especially in the frontal lobes where these receptors are abundant, may lead to reduced general alertness.149 This is echoed in the impairment seen in tasks requiring sustained attention (e.g. rapid visual information processing task) in response to clonidine.150 Howerver, this effect is reversed by increasing phasic alertness, and perceptual sensitivity through coupling accessory stimuli with task-related stimuli.149 In support of this phenomenon, using pupillometry as a surrogate marker of LC activity, Hoffing and Seitz151 show that in a task-irrelevant learning paradigm, healthy volunteers better memorize and recall scenes paired with task-relevant targets than distractors, and perform even better when scenes were paired with novel auditory stimuli. The noradrenaline-LC system is therefore involved in attending, or in computational terms improving the signal-to-noise ratio in the presence of novel or unexpected stimuli and enhances performance accordingly.
In Parkinson’s disease, cognitive dysfunction in the form of dementia and attentional deficit are common.152,153 The Cambridgeshire Parkinson's Incidence from GP to Neurologist (CamPaIGN) study revealed that 46% of patients with Parkinson’s disease developed dementia over 10 years,153 and indeed manipulating noradrenaline levels with atomoxetine improves attention in this condition (see Table 3 in Kehagia et al.154). In Alzheimer’s disease prazosin (a postsynaptic α1-adrenoreceptor antagonist) has been studied in a randomized, double-blind, placebo-controlled study of 22 patients with agitation and aggression; results showed improvements on the Brief Psychiatric Rating Scale, Neuropsychiatric Inventory, and Clinical Global Impression of change. A current clinical trial, looking at the effect of formeterol (long acting β2-adrenoreceptor agonist) on cognition in mild to moderate Alzheimer’s disease is underway.155 In Huntington’s disease the evidence is sparse; a randomized controlled cross-over study targeting inattention with atomoxetine in 200 patients with Huntington’s disease, did not show a significant improvement (however, results may be confounded by concomitant use of other psychotropic drugs).113
Working memory
Working memory is the ability to transiently store and manipulate information to guide goal-directed behaviour. It depends on noradrenergic function in humans and animal studies.156–158 In rats stimulating postsynaptic α1 receptors (through inducing stress using tasks such as the learned helplessness task for example) impairs working memory—an effect that is seen in the fight/flight response, and is shown to be mediated through the activation of the PKC (phosphatidylinositol-protein kinase C) pathway. This effect is ameliorated through pretreatment with an α1-adrenoreceptor antagonist.39 In monkeys systemic and locally administered guanfacine,159 and clonidine160 improve spatial working memory of aged monkeys; similar results are seen in younger monkeys with experimental lesions of the prefrontal cortex or catecholamine depletion that are subsequently treated with clonidine or guanfacine.161,162 To echo this, Gamo and colleagues163 have further shown an improvement in a delayed response task in monkeys treated with the noradrenaline reuptake inhibitor atomoxetine.
Some of the early work linking the noradrenergic system to working memory stems from the disturbance to the noradrenergic pathways in patients with Korsakoff’s syndrome—an amnestic disorder resulting from thiamine deficiency found (mainly) in chronic alcohol use. Levels of the noradrenergic breakdown product MHPG are reduced in the CSF of patients with this condition and this reduction significantly correlates with short-term memory impairment in these individuals. Treatment with the alpha-2 receptor agonist clonidine improves mnemonic and attentional deficit in patients, but has the opposite effect in healthy volunteers—likely due to degeneration of presynaptic terminals and therefore a predominantly postsynaptic action in patients.164 The negative effect of noradrenergic modulation in young healthy adults is echoed by Coull and colleagues165 who illustrate an impairment in tests of visual working memory in healthy young volunteers in response to clonidine.
Working memory is impaired in both Parkinson’s disease and Alzheimer’s disease (reviewed in Zokaei and Husain166). In Parkinson’s disease, nearly 60% of patients have mild cognitive impairment with deficits in working memory at diagnosis, with 42% developing dementia, 6–8 years after diagnosis, even in the absence of Alzheimer’s disease pathology.167 Preliminary publications from two ongoing clinical trials, targeting mild cognitive impairment in Parkinson’s disease, have shown inconsistent results with droxipoda (NCT02066571), and atomoxetine (NCT01738191),168 although final published results are pending. In Alzheimer’s disease manipulating the noradrenergic system with monoamine oxidase (MAO) inhibitors has shown no overall clinical benefits in a Cochrane review and likewise no clear benefit from atomoxetine in a randomized placebo-controlled trial in Alzheimer’s disease.169 Given the complex nature of noradrenergic pathology in Alzheimer’s disease, trials investigating the more selective MAO inhibitor, rasagiline (NCT02359552), and also higher concentrations of atomoxetine in conjunction with other therapies are under way.
Impulsivity and response inhibition
Impulsivity is a complex construct, which includes risky behaviour in relation to reward, inappropriate and premature responses, and impairment of response inhibition. Noradrenaline modulates several of these components, but there is most evidence for its effect on response inhibition such as the stop-signal task that requires cancelation of an initiated action. Efficiency of response inhibition is often quantified by the stop-signal reaction time. It is dependent on the integrity and function of the inferior frontal gyrus (IFG) in humans and rats,170 and modulation by noradrenaline.
The complex pharmacology of the noradrenergic system, and its non-linear relationship to performance, determine whether agents that increase synaptic noradrenaline improve or impair response inhibition. In rats, increasing synaptic noradrenaline with atomoxetine or reboxetine typically improves response inhibition171,172 but this effect is more marked in impulsive animals173 in support of baseline-dependent effects.
In healthy adults, atomoxetine can enhance response inhibition using the stop-signal paradigm,174 in association with dose-dependent enhancement of activity in the right IFG.175 In contrast, an alternative form of response inhibition, using the NoGo paradigms, may be worsened by atomoxetine in health,176 noting that this type of inhibition is perhaps more closely associated with serotonergic transmission.177
Impulsivity may also be viewed as an urgency to make decisions. In a decision-making task, where healthy adults are asked to sample information before making a decision based on either fixed or decreasing (and negative) rewards, propranolol (a β-adrenoreceptor blocker) reduces information gathering compared to placebo. A Bayesian computation model suggests that the observed effect is likely due to increasing urgency to decide178; in other words noradrenergic blockade renders healthy subjects more impulsive.
This evidence is directly related to patients with Parkinson’s disease who are more impulsive than healthy controls even in the absence of severe impulse control disorders.179 Patients have longer stop-signal reaction times, less stop-related activation in the right IFG, and weaker functional connectivity between the right IFG and striatum compared with control subjects.114,180,181 In the Parkinson’s disease group, atomoxetine was found to enhance the stop-related right IFG activation, in proportion to disease severity, as well as restoring interactions between the supplementary motor cortex and the right IFG.120,121 These results are echoed in a double-blind randomized placebo-controlled trial of 25 patients with Parkinson’s disease, where atomoxetine improved stopping accuracy on the stop-signal task and reduced reflection impulsivity and risk taking.154
Evidence for noradrenergic modulation of response inhibition also comes from genetic polymorphisms of the noradrenergic transporter (NET), which modulates cortical noradrenaline levels.182 In 819 adolescents, this polymorphism influenced activity in the right IFG during response inhibition.183 Similarly, in a PET imaging study of 20 healthy individuals, using a NET radioligand, there appeared to be an association between higher scores of impulsivity and lower tracer uptake, albeit not statistically significant.184 However, genetic variance in metabolism of noradrenergic drugs like atomoxetine may affect its efficacy185 and how we interpret results of noradrenergic drug studies in cognition.
Cognitive flexibility
By regulating the signal-to-noise ratio during information processing, the noradrenergic system controls the maintenance versus shifting of task sets, which in turn affects cognitive flexibility and behavioural adaptation in a changing environment. During instrumental behaviours, stimulus-response associations may change in different ways: they may reverse within a stimulus set (reversal), or they may shift to new stimulus attributes that are orthogonal to the original associations; this shift may occur within the same feature space (intra-dimensional shift) or to a different stimulus dimension (extra-dimensional shift).157 The latter, extra-dimensional shift, is most strongly associated with the noradrenergic system,186 although the effect of noradrenergic modulation is once again dependent on the adrenoreceptors in action and their location.
Lesions of the noradrenergic bundle have variable effect on set-shifting. In rodents extra-dimensional set-shifting is selectively impaired through lesions of the dorsal-noradrenergic bundle,146 as is the case in rats exposed to the α2-adrenoreceptor antagonist, idazoxan.145 Moreover systemic infusion of atipamezole (another α2-adrenoreceptor antagonist) improved attentional set-shifting in another rodent experiment and its beneficial effects were blocked by the infusion of benoxathian, an α1-adrenoreceptor antagonist, into the medial prefrontal cortex; infusion of benoxathian alone did not have any effect.187 This is further echoed in an experiment by Snyder et al.188 who show that infusion of corticotropin-releasing factor into the LC improved extra-dimensional set-shifting whilst at higher doses impaired reversal learning. In support of a Yerkes-Dodson inverted U-shaped behaviour for the action of noradrenaline, atomoxetine has been shown to improve set-shifting in rats with noradrenergic lesions but impairs performance in intact rats.147 This result is supported further by Cain et al.189 who show that adolescent rats respond beneficially to a low dose of atomoxetine in a set-shifting task but not to high doses.
Similar mixed results are evident in human studies. In healthy human volunteers, administration of clonidine (α1/2-adrenoreceptor agonist) dramatically impairs extra-dimensional set-shifting in a task related to the Wisconsin Card Sort Test; an effect that was unexpectedly also seen with the α2-adrenoreceptor antagonist idazoxan, and potentiated with the addition of this to clonidine.190 This is in contrast to unequivocal effects on performance on a set-shifting task in young healthy male adults in response to the α2-adrenoreceptor agonist guanfacine.191 Data from blockade of the β-adrenoreceptor in improving cognitive flexibility is promising192; however, drawing conclusions from these results is difficult with the knowledge that such drugs as propranolol and atenolol reduce stress and anxiety levels and may therefore affect performance accordingly. The application to clinical groups with cognitive inflexibility from Alzheimer’s disease, PSP or Parkinson’s disease is warranted, but data are currently lacking.
Future directions and conclusions
We have presented evidence for noradrenergic loss in both animal models of neurodegenerative disease as well as in humans and the association with cognitive dysfunction seen in many debilitating neurodegenerative conditions. The noradrenaline reuptake inhibitor atomoxetine has proven well tolerated and promising in targeting impulsivity in patients with Parkinson’s disease; however, treatment studies for cognitive dysfunction in other neurodegenerative conditions are sparse. There are limitations to modulating the noradrenergic system in cognition and these include optimising the balance between too much and too little noradrenaline, the lack of specificity for tonic versus phasic activity, targeting the most symptomatic cognitive domain whilst not jeopardizing others, and personalizing treatment with genetic polymorphisms and baseline characteristics in mind.
We propose that pharmacotherapeutics that normalize noradrenaline levels, neurotransmission and signal-to-noise ratio provide useful strategies for enhancing cognitive function in the neurodegenerative conditions discussed. Noradrenergic effects on cardiovascular and respiratory functioning require careful selection and monitoring in trials, and a progress towards more centrally acting agents. Clinical trials must accommodate the marked individual differences in noradrenergic systems, including polymorphisms that affect noradrenaline synthesis or metabolism,193 which may also vary between populations in prevalence.194 Non-invasive biomarkers that can signal noradrenergic dysfunction could be used to identify and monitor those at risk. Advances in imaging the human LC, genetics, and relevant outcome measures, are leading towards new individualized noradrenergic treatment strategies for cognitive function in neurodegenerative disorders.
Funding
The authors are funded by the Wellcome Trust (220258), the National Institute for Health Research Cambridge Biomedical Research Centre (BRC-1215-20014) and the Association of British Neurologists (Patrick Berthoud Charitable Trust, RG99368).
Competing interests
The authors report no competing interests.
Glossary
- IFG
inferior frontal gyrus
- LC
locus coeruleus
- PSP
progressive supranuclear palsy
References
- 1.Marien MR, Colpaert FC, Rosenquist AC.. Noradrenergic mechanisms in neurodegenerative diseases: A theory. Brain Res Brain Res Rev. 2004;45(1):38–78. [DOI] [PubMed] [Google Scholar]
- 2.Del Tredici K, Braak H.. Dysfunction of the locus coeruleus-norepinephrine system and related circuitry in Parkinson's disease-related dementia. J Neurol Neurosurg Psychiatry. 2013;84(7):774–783. [DOI] [PubMed] [Google Scholar]
- 3.Aston-Jones G, Chiang C, Alexinsky T.. Discharge of noradrenergic locus coeruleus neurons in behaving rats and monkeys suggests a role in vigilance. Prog Brain Res. 1991;88:501–520. [DOI] [PubMed] [Google Scholar]
- 4.Berridge CW, Arnsten AF, Foote SL.. Noradrenergic modulation of cognitive function: Clinical implications of anatomical, electrophysiological and behavioural studies in animal models. Psychol Med. 1993;23(3):557–564. [DOI] [PubMed] [Google Scholar]
- 5.Cole BJ, Robbins TW.. Forebrain norepinephrine: Role in controlled information processing in the rat. Neuropsychopharmacology. 1992;7(2):129–142. [PubMed] [Google Scholar]
- 6.Devauges V, Sara SJ.. Activation of the noradrenergic system facilitates an attentional shift in the rat. Behav Brain Res. 1990;39(1):19–28. [DOI] [PubMed] [Google Scholar]
- 7.Harley CW.A role for norepinephrine in arousal, emotion and learning?: Limbic modulation by norepinephrine and the Kety hypothesis. Prog Neuropsychopharmacol Biol Psychiatry. 1987;11(4):419–458. [DOI] [PubMed] [Google Scholar]
- 8.Sara SJ.The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci. 2009;10(3):211–223. [DOI] [PubMed] [Google Scholar]
- 9.Amaral DG, Sinnamon HM.. The locus coeruleus: Neurobiology of a central noradrenergic nucleus. Prog Neurobiol. 1977;9(3):147–196. [DOI] [PubMed] [Google Scholar]
- 10.Aston-Jones G, Cohen JD.. An integrative theory of locus coeruleus-norepinephrine function: Adaptive gain and optimal performance. Annu Rev Neurosci. 2005;28:403–450. [DOI] [PubMed] [Google Scholar]
- 11.German DC, Walker BS, Manaye K, Smith WK, Woodward DJ, North AJ.. The human locus coeruleus: Computer reconstruction of cellular distribution. J Neurosci. 1988;8(5):1776–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandes P, Regala J, Correia F, Gonçalves-Ferreira AJ.. The human locus coeruleus 3-D stereotactic anatomy. Surg Radiol Anatomy. 2012;34(10):879–885. [DOI] [PubMed] [Google Scholar]
- 13.Benarroch EE.Locus coeruleus. Cell Tissue Res. 2018;373(1):221–232. [DOI] [PubMed] [Google Scholar]
- 14.Berridge CW, Waterhouse BD.. The locus coeruleus-noradrenergic system: Modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. 2003;42(1):33–84. [DOI] [PubMed] [Google Scholar]
- 15.Iversen LL, Rossor MN, Reynolds GP, et al. Loss of pigmented dopamine-beta-hydroxylase positive cells from locus coeruleus in senile dementia of Alzheimer's type. Neurosci Lett. 1983;39(1):95–100. [DOI] [PubMed] [Google Scholar]
- 16.Chan-Palay V, Asan E.. Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson's disease with and without dementia and depression. J Comp Neurol. 1989;287(3):373–392. [DOI] [PubMed] [Google Scholar]
- 17.Baker KG, Törk I, Hornung JP, Halasz P.. The human locus coeruleus complex: An immunohistochemical and three dimensional reconstruction study. Exp Brain Res. 1989;77(2):257–270. [DOI] [PubMed] [Google Scholar]
- 18.Oh J, Eser RA, Ehrenberg AJ, et al. Profound degeneration of wake-promoting neurons in Alzheimer's disease. Alzheimers Dement. 2019;15(10):1253–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarz LA, Luo L.. Organization of the locus coeruleus-norepinephrine system. Curr Biol. 2015;25(21):R1051–R1056. [DOI] [PubMed] [Google Scholar]
- 20.Ye R, Rua C, O'Callaghan C, et al. An in vivo probabilistic atlas of the human locus coeruleus at ultra-high field. Neuroimage. 2021;225:117487- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Betts MJ, Kirilina E, Otaduy MCG, et al. Locus coeruleus imaging as a biomarker for noradrenergic dysfunction in neurodegenerative diseases. Brain. 2019;142(9):2558–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Priovoulos N, Jacobs HIL, Ivanov D, Uludağ K, Verhey FRJ, Poser BA.. High-resolution in vivo imaging of human locus coeruleus by magnetization transfer MRI at 3T and 7T. Neuroimage. 2018;168:427–436. [DOI] [PubMed] [Google Scholar]
- 23.Sulzer D, Cassidy C, Horga G, et al. Neuromelanin detection by magnetic resonance imaging (MRI) and its promise as a biomarker for Parkinson's disease. NPJ Parkinsons Dis. 2018;4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halliday GM, Li YW, Blumbergs PC, et al. Neuropathology of immunohistochemically identified brainstem neurons in Parkinson's disease. Ann Neurol. 1990;27(4):373–385. [DOI] [PubMed] [Google Scholar]
- 25.Swanson LW, Hartman BK.. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J Comp Neurol. 1975;163(4):467–505. [DOI] [PubMed] [Google Scholar]
- 26.Arendt T, Brückner MK, Morawski M, Jäger C, Gertz HJ.. Early neurone loss in Alzheimer's disease: Cortical or subcortical? Acta Neuropathol Commun. 2015;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaalund SS, Passamonti L, Allinson KSJ, et al. Locus coeruleus pathology in progressive supranuclear palsy, and its relation to disease severity. Acta Neuropathol Commun. 2020;8(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kelly SC, He B, Perez SE, Ginsberg SD, Mufson EJ, Counts SE.. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer's disease. Acta Neuropathol Commun. 2017;5(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marner L, Søborg C, Pakkenberg B.. Increased volume of the pigmented neurons in the locus coeruleus of schizophrenic subjects: A stereological study. J Psychiatr Res. 2005;39(4):337–345. [DOI] [PubMed] [Google Scholar]
- 30.Mouton PR, Pakkenberg B, Gundersen HJ, Price DL.. Absolute number and size of pigmented locus coeruleus neurons in young and aged individuals. J Chem Neuroanat. 1994;7(3):185–190. [DOI] [PubMed] [Google Scholar]
- 31.Ohm TG, Busch C, Bohl J.. Unbiased estimation of neuronal numbers in the human nucleus coeruleus during aging. Neurobiol Aging. 1997;18(4):393–399. [DOI] [PubMed] [Google Scholar]
- 32.Theofilas P, Polichiso L, Wang X, et al. ; Brazilian Aging Brain Study Group. A novel approach for integrative studies on neurodegenerative diseases in human brains. J Neurosci Methods. 2014;226:171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandler DJ, Gao WJ, Waterhouse BD.. Heterogeneous organization of the locus coeruleus projections to prefrontal and motor cortices. Proc Natl Acad Sci U S A. 2014;111(18):6816–6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agster KL, Mejias-Aponte CA, Clark BD, Waterhouse BD.. Evidence for a regional specificity in the density and distribution of noradrenergic varicosities in rat cortex. J Comp Neurol. 2013;521(10):2195–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aston-Jones G, Waterhouse B.. Locus coeruleus: Rom global projection system to adaptive regulation of behavior. Brain Res. 2016;1645:75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogawski MA, Aghajanian GK.. Activation of lateral geniculate neurons by locus coeruleus or dorsal noradrenergic bundle stimulation: Selective blockade by the alpha 1-adrenoceptor antagonist prazosin. Brain Res. 1982;250(1):31–39. [DOI] [PubMed] [Google Scholar]
- 37.Aoki C, Go CG, Venkatesan C, Kurose H.. Perikaryal and synaptic localization of alpha 2A-adrenergic receptor-like immunoreactivity. Brain Res. 1994;650(2):181–204. [DOI] [PubMed] [Google Scholar]
- 38.Avery RA, Franowicz JS, Studholme C, van Dyck CH, Arnsten AF.. The alpha-2A-adrenoceptor agonist, guanfacine, increases regional cerebral blood flow in dorsolateral prefrontal cortex of monkeys performing a spatial working memory task. Neuropsychopharmacology. 2000;23(3):240–249. [DOI] [PubMed] [Google Scholar]
- 39.Birnbaum SG, Yuan PX, Wang M, et al. Protein kinase C overactivity impairs prefrontal cortical regulation of working memory. Science. 2004;306(5697):882–884. [DOI] [PubMed] [Google Scholar]
- 40.Devilbiss DM, Waterhouse BD.. Norepinephrine exhibits two distinct profiles of action on sensory cortical neuron responses to excitatory synaptic stimuli. Synapse. 2000;37(4):273–282. [DOI] [PubMed] [Google Scholar]
- 41.Rajkowski J, Kubiak P, Aston-Jones G.. Locus coeruleus activity in monkey: Phasic and tonic changes are associated with altered vigilance. Brain Res Bull. 1994;35(5-6):607–616. [DOI] [PubMed] [Google Scholar]
- 42.Kane GA, Vazey EM, Wilson RC, et al. Increased locus coeruleus tonic activity causes disengagement from a patch-foraging task. Cogn Affect Behav Neurosci. 2017;17(6):1073–1083. [DOI] [PubMed] [Google Scholar]
- 43.Gilzenrat MS, Nieuwenhuis S, Jepma M, Cohen JD.. Pupil diameter tracks changes in control state predicted by the adaptive gain theory of locus coeruleus function. Cogn Affect Behav Neurosci. 2010;10(2):252–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shine JM, Bissett PG, Bell PT, et al. The dynamics of functional brain networks: Integrated network states during cognitive task performance. Neuron. 2016;92(2):544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beatty J.Phasic not tonic pupillary responses vary with auditory vigilance performance. Psychophysiology. 1982;19(2):167–172. [DOI] [PubMed] [Google Scholar]
- 46.Foote SL, Morrison JH.. Extrathalamic modulation of cortical function. Annu Rev Neurosci. 1987;10:67–95. [DOI] [PubMed] [Google Scholar]
- 47.Mather M, Clewett D, Sakaki M, Harley CW.. Norepinephrine ignites local hotspots of neuronal excitation: How arousal amplifies selectivity in perception and memory. Behav Brain Sci. 2016;39:e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Theofilas P, Ehrenberg AJ, Dunlop S, et al. Locus coeruleus volume and cell population changes during Alzheimer's disease progression: A stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimers Dementia. 2017;13(3):236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hammerer D, Callaghan MF, Hopkins A, et al. Locus coeruleus integrity in old age is selectively related to memories linked with salient negative events. Proc Natl Acad Sci U S A. 2018;115(9):2228–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu KY, Kievit RA, Tsvetanov KA, et al. ; Cam-CAN. Noradrenergic-dependent functions are associated with age-related locus coeruleus signal intensity differences. Nat Commun. 2020;11(1):1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tona KD, Keuken MC, de Rover M, et al. In vivo visualization of the locus coeruleus in humans: Quantifying the test-retest reliability. Brain Struct Funct. 2017;222(9):4203–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacobs HIL, Muller-Ehrenberg L, Priovoulos N, Roebroeck A.. Curvilinear locus coeruleus functional connectivity trajectories over the adult lifespan: A 7T MRI study. Neurobiol Aging. 2018;69:167–176. [DOI] [PubMed] [Google Scholar]
- 53.Zarow C, Lyness SA, Mortimer JA, Chui HC.. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60(3):337–341. [DOI] [PubMed] [Google Scholar]
- 54.Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM.. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer's disease. Neurobiol Aging. 2007;28(3):327–335. [DOI] [PubMed] [Google Scholar]
- 55.German DC, Manaye KF, White CL III, et al. Disease-specific patterns of locus coeruleus cell loss. Ann Neurol. 1992;32(5):667–676. [DOI] [PubMed] [Google Scholar]
- 56.Theofilas P, Ehrenberg AJ, Nguy A, et al. Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer's disease Braak stages: A quantitative study in humans. Neurobiol Aging. 2018;61:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gulyas B, Brockschnieder D, Nag S, et al. The norepinephrine transporter (NET) radioligand (S,S)-[18F]FMeNER-D2 shows significant decreases in NET density in the human brain in Alzheimer's disease: A post-mortem autoradiographic study. Neurochem Int. 2010;56(6-7):789–798. [DOI] [PubMed] [Google Scholar]
- 58.Eser RA, Ehrenberg AJ, Petersen C, et al. Selective vulnerability of brainstem nuclei in distinct tauopathies: A postmortem study. J Neuropathol Exp Neurol. 2018;77(2):149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Betts MJ, Cardenas-Blanco A, Kanowski M, et al. Locus coeruleus MRI contrast is reduced in Alzheimer's disease dementia and correlates with CSF Aβ levels. Alzheimers Dement (Amst). 2019;11:281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E.. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24(2):197–211. [DOI] [PubMed] [Google Scholar]
- 61.van Dijk KD, Berendse HW, Drukarch B, et al. The proteome of the locus ceruleus in Parkinson's disease: Relevance to pathogenesis. Brain Pathol. 2012;22(4):485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dickson DW, Fujishiro H, DelleDonne A, et al. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson's disease. Acta Neuropathol. 2008;115(4):437–444. [DOI] [PubMed] [Google Scholar]
- 63.Li Y, Wang C, Wang J, et al. Mild cognitive impairment in de novo Parkinson's disease: A neuromelanin MRI study in locus coeruleus. Mov Disord. 2019;34(6):884–892. [DOI] [PubMed] [Google Scholar]
- 64.Pavese N, Rivero-Bosch M, Lewis SJ, Whone AL, Brooks DJ.. Progression of monoaminergic dysfunction in Parkinson's disease: A longitudinal 18F-dopa PET study. Neuroimage. 2011;56(3):1463–1468. [DOI] [PubMed] [Google Scholar]
- 65.Zweig RM, Ross CA, Hedreen JC, et al. Locus coeruleus involvement in Huntington's disease. Arch Neurol. 1992;49(2):152–156. [DOI] [PubMed] [Google Scholar]
- 66.Passamonti L, Lansdall C, Rowe J.. The neuroanatomical and neurochemical basis of apathy and impulsivity in frontotemporal lobar degeneration. Curr Opin Behav Sci. 2018;22:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kovacs GG, Lukic MJ, Irwin DJ, et al. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathologica. 2020;140(2):99–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iwanaga K, Wakabayashi K, Honma Y, Takahashi H.. Amyotrophic lateral sclerosis: Occurrence of Bunina bodies in the locus ceruleus pigmented neurons. Clin Neuropathol. 1997;16(1):23–26. [PubMed] [Google Scholar]
- 69.Yang Y, Schmitt HP.. Frontotemporal dementia: Evidence for impairment of ascending serotoninergic but not noradrenergic innervation. Immunocytochemical and quantitative study using a graph method. Acta Neuropathol. 2001;101(3):256–270. [DOI] [PubMed] [Google Scholar]
- 70.Engelborghs S, De VK, Van de Casteele T, et al. Diagnostic performance of a CSF-biomarker panel in autopsy-confirmed dementia. Neurobiol Aging. 2008;29(8):1143–1159. [DOI] [PubMed] [Google Scholar]
- 71.Vermeiren Y, Janssens J, Aerts T, et al. Brain serotonergic and noradrenergic deficiencies in behavioral variant frontotemporal dementia compared to early-onset Alzheimer's disease. J Alzheimers Dis. 2016;53(3):1079–1096. [DOI] [PubMed] [Google Scholar]
- 72.Benarroch EE, Schmeichel AM, Parisi JE.. Depletion of mesopontine cholinergic and sparing of raphe neurons in multiple system atrophy. Neurology. 2002;59(6):944–946. [DOI] [PubMed] [Google Scholar]
- 73.Benarroch EE, Schmeichel AM, Low PA, Sandroni P, Parisi JE.. Loss of A5 noradrenergic neurons in multiple system atrophy. Acta Neuropathol. 2008;115(6):629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Montine TJ, Phelps CH, Beach TG, et al. ; Alzheimer’s Association. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012;123(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Coyle JT, Price DL, DeLong MR.. Alzheimer's disease: A disorder of cortical cholinergic innervation. Science. 1983;219(4589):1184–1190. [DOI] [PubMed] [Google Scholar]
- 76.Mesulam MM.Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer's disease. J Comparat Neurol. 2013;521(18):4124–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zaborszky L, Cullinan WE, Luine VN.. Catecholaminergic-cholinergic interaction in the basal forebrain. Prog Brain Res. 1993;98:31–49. [DOI] [PubMed] [Google Scholar]
- 78.Hume SP, Lammertsma AA, Opacka-Juffry J, et al. Quantification of in vivo binding of [3H]RX 821002 in rat brain: Evaluation as a radioligand for central alpha 2-adrenoceptors. Int J Rad Appl Instrum B. 1992;19(8):841–849. [DOI] [PubMed] [Google Scholar]
- 79.Siniscalchi A, Badini I, Bianchi C, Beani L.. Monoamines modulate the electrically-evoked efflux of 3H-choline from slices of guinea pig nucleus basalis magnocellularis. Naunyn Schmiedebergs Arch Pharmacol. 1994;350(1):10–14. [DOI] [PubMed] [Google Scholar]
- 80.Tellez S, Colpaert F, Marien M.. Alpha2-adrenoceptor modulation of cortical acetylcholine release in vivo. Neuroscience. 1999;89(4):1041–1050. [DOI] [PubMed] [Google Scholar]
- 81.Rossner S, Ueberham U, Schliebs R, Perez-Polo JR, Bigl V.. The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling. Prog Neurobiol. 1998;56(5):541–569. [DOI] [PubMed] [Google Scholar]
- 82.Haroutunian V, Kanof PD, Tsuboyama G, Davis KL.. Restoration of cholinomimetic activity by clonidine in cholinergic plus noradrenergic lesioned rats. Brain Res. 1990;507(2):261–266. [DOI] [PubMed] [Google Scholar]
- 83.Fort P, Khateb A, Pegna A, Muhlethaler M, Jones BE.. Noradrenergic modulation of cholinergic nucleus basalis neurons demonstrated by in vitro pharmacological and immunohistochemical evidence in the guinea-pig brain. Eur J Neurosci. 1995;7(7):1502–1511. [DOI] [PubMed] [Google Scholar]
- 84.Feinstein DL, Heneka MT, Gavrilyuk V, Dello RC, Weinberg G, Galea E.. Noradrenergic regulation of inflammatory gene expression in brain. Neurochem Int. 2002;41(5):357–365. [DOI] [PubMed] [Google Scholar]
- 85.Kalinin S, Gavrilyuk V, Polak PE, et al. Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer's disease. Neurobiol Aging. 2007;28(8):1206–1214. [DOI] [PubMed] [Google Scholar]
- 86.Zou Y, Qian Z, Chen Y, Qian H, Wei G, Zhang Q.. Norepinephrine inhibits Alzheimer’s amyloid-β peptide aggregation and destabilizes amyloid-β protofibrils: A molecular dynamics simulation study. ACS Chem Neurosci. 2019;10(3):1585–1594. [DOI] [PubMed] [Google Scholar]
- 87.Heneka MT, Nadrigny F, Regen T, et al. Locus ceruleus controls Alzheimer's disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci U S A. 2010;107(13):6058–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Winkeler A, Waerzeggers Y, Klose A, et al. Imaging noradrenergic influence on amyloid pathology in mouse models of Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2008;35(Suppl 1):S107–S113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Braun D, Feinstein DL.. The locus coeruleus neuroprotective drug vindeburnol normalizes behavior in the 5xFAD transgenic mouse model of Alzheimer's disease. Brain Res. 2019;1702:29–37. [DOI] [PubMed] [Google Scholar]
- 90.Yu JT, Wang ND, Ma T, Jiang H, Guan J, Tan L.. Roles of beta-adrenergic receptors in Alzheimer's disease: Implications for novel therapeutics. Brain Res Bull. 2011;84(2):111–117. [DOI] [PubMed] [Google Scholar]
- 91.Mann DM, Yates PO, Marcyniuk B, Ravindra CR.. Pathological evidence for neurotransmitter deficits in Down's syndrome of middle age. J Ment Defic Res. 2008;29 (2):125–135. [DOI] [PubMed] [Google Scholar]
- 92.Dekker AD, Coppus AM, Vermeiren Y, et al. Serum MHPG strongly predicts conversion to Alzheimer's disease in behaviorally characterized subjects with Down syndrome. J Alzheimers Dis. 2015;43(3):871–891. [DOI] [PubMed] [Google Scholar]
- 93.Dekker AD, Strydom A, Coppus AM, et al. Behavioural and psychological symptoms of dementia in Down syndrome: Early indicators of clinical Alzheimer's disease? Cortex. 2015;73:36–61. [DOI] [PubMed] [Google Scholar]
- 94.Dang V, Medina B, Das D, et al. Formoterol, a long-acting beta2 adrenergic agonist, improves cognitive function and promotes dendritic complexity in a mouse model of Down syndrome. Biol Psychiatry. 2014;75(3):179–188. [DOI] [PubMed] [Google Scholar]
- 95.Fortress AM, Hamlett ED, Vazey EM, et al. Designer receptors enhance memory in a mouse model of Down syndrome. J Neurosci. 2015;35(4):1343–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Whittle N, Sartori SB, Dierssen M, Lubec G, Singewald N.. Fetal Down syndrome brains exhibit aberrant levels of neurotransmitters critical for normal brain development. Pediatrics. 2007;120(6):e1465–e71. [DOI] [PubMed] [Google Scholar]
- 97.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M.. Alpha-synuclein in Lewy bodies. Nature. 1997;388(6645):839–840. [DOI] [PubMed] [Google Scholar]
- 98.Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord. 2015;30(12):1591–1601. [DOI] [PubMed] [Google Scholar]
- 99.Williams-Gray CH, Evans JR, Goris A, et al. The distinct cognitive syndromes of Parkinson's disease: 5 year follow-up of the CamPaIGN cohort. Brain. 2009;132(Pt 11):2958–2969. [DOI] [PubMed] [Google Scholar]
- 100.Sotiriou E, Vassilatis DK, Vila M, Stefanis L.. Selective noradrenergic vulnerability in alpha-synuclein transgenic mice. Neurobiol Aging. 2010;31(12):2103–2114. [DOI] [PubMed] [Google Scholar]
- 101.Xu Y, Yan J, Zhou P, et al. Neurotransmitter receptors and cognitive dysfunction in Alzheimer's disease and Parkinson's disease. Prog Neurobiol. 2012;97(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schneider JS, Kovelowski CJ.. Chronic exposure to low doses of MPTP. I. Cognitive deficits in motor asymptomatic monkeys. Brain Res. 1990;519(1-2):122–128. [DOI] [PubMed] [Google Scholar]
- 103.Decamp E, Schneider JS.. Attention and executive function deficits in chronic low-dose MPTP-treated non-human primates. Eur J Neurosci. 2004;20(5):1371–1378. [DOI] [PubMed] [Google Scholar]
- 104.Goldstein DS, Holmes C, Sharabi Y.. Cerebrospinal fluid biomarkers of central catecholamine deficiency in Parkinson's disease and other synucleinopathies. Brain. 2012;135(6):1900–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sommerauer M, Fedorova TD, Hansen AK, et al. Evaluation of the noradrenergic system in Parkinson's disease: An 11C-MeNER PET and neuromelanin MRI study. Brain. 2018;141(2):496–504. [DOI] [PubMed] [Google Scholar]
- 106.Sulzer D, Alcalay RN, Garretti F, et al. T cells from patients with Parkinson's disease recognize alpha-synuclein peptides. Nature. 2017;546(7660):656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Greenland JC, Cutting E, Kadyan S, et al. Azathioprine immunosuppression and disease modification in Parkinson’s disease (AZA-PD): a randomised double-blind placebo-controlled phase II trial protocol. BMJ Open. 2020;10:e040527. [DOI] [PMC free article] [PubMed]
- 108.Lawrence AD, Hodges JR, Rosser AE, et al. Evidence for specific cognitive deficits in preclinical Huntington's disease. Brain. 1998;121 (7):1329–1341. [DOI] [PubMed] [Google Scholar]
- 109.MacDonald ME, Ambrose CM, Duyao MP, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72(6):971–983. [DOI] [PubMed] [Google Scholar]
- 110.Paulsen JS, Zhao H, Stout JC, et al. ; Huntington Study Group. Clinical markers of early disease in persons near onset of Huntington's disease. Neurology. 2001;57(4):658–662. [DOI] [PubMed] [Google Scholar]
- 111.Rüb U, Seidel K, Heinsen H, Vonsattel JP, den Dunnen WF, Korf HW.. Huntington's disease (HD): The neuropathology of a multisystem neurodegenerative disorder of the human brain. Brain Pathol. 2016;26(6):726–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Garcia-Miralles M, Ooi J, Ferrari BC, et al. Treatment with the MAO-A inhibitor clorgyline elevates monoamine neurotransmitter levels and improves affective phenotypes in a mouse model of Huntington disease. Exp Neurol. 2016;278:4–10. [DOI] [PubMed] [Google Scholar]
- 113.Beglinger LJ, Adams WH, Paulson H, et al. Randomized controlled trial of atomoxetine for cognitive dysfunction in early Huntington disease. J Clin Psychopharmacol. 2009;29(5):484–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ye Z, Rae CL, Nombela C, et al. Predicting beneficial effects of atomoxetine and citalopram on response inhibition in Parkinson's disease with clinical and neuroimaging measures. Hum Brain Mapp. 2016;37(3):1026–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Höglinger GU, Respondek G, Stamelou M, et al. ; Movement Disorder Society-endorsed PSP Study Group. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord. 2017;32(6):853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Respondek G, Stamelou M, Kurz C, et al. ; Movement Disorder Society-endorsed PSP Study Group. The phenotypic spectrum of progressive supranuclear palsy: A retrospective multicenter study of 100 definite cases. Mov Disord. 2014;29(14):1758–1766. [DOI] [PubMed] [Google Scholar]
- 117.Burrell JR, Hodges JR, Rowe JB.. Cognition in corticobasal syndrome and progressive supranuclear palsy: A review. Mov Disord. 2014;29(5):684–693. [DOI] [PubMed] [Google Scholar]
- 118.Ghika J, Tennis M, Hoffman E, Schoenfeld D, Growdon J.. Idazoxan treatment in progressive supranuclear palsy. Neurology. 1991;41(7):986–991. [DOI] [PubMed] [Google Scholar]
- 119.Matsuo H, Takashima H, Kishikawa M, et al. Pure akinesia: An atypical manifestation of progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 1991;54(5):397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Borchert RJ, Rittman T, Passamonti L, et al. Atomoxetine enhances connectivity of prefrontal networks in Parkinson's disease. Neuropsychopharmacol. 2016;41(8):2171–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rae CL, Nombela C, Rodríguez PV, et al. Atomoxetine restores the response inhibition network in Parkinson's disease. Brain. 2016;139(Pt 8):2235–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(9):2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Irwin DJ, Brettschneider J, McMillan CT, et al. Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol. 2016;79(2):272–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rohrer JD, Lashley T, Schott JM, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain. 2011;134(Pt 9):2565–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Murphy J, Factor-Litvak P, Goetz R, et al. ; ALS COSMOS. Cognitive-behavioral screening reveals prevalent impairment in a large multicenter ALS cohort. Neurology. 2016;86(9):813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Murley AG, Rowe JB.. Neurotransmitter deficits from frontotemporal lobar degeneration. Brain. 2018;141(5):1263–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sjogren M, Minthon L, Passant U, Blennow K, Wallin A.. Decreased monoamine metabolites in frontotemporal dementia and Alzheimer's disease. Neurobiol Aging. 1998;19(5):379–384. [DOI] [PubMed] [Google Scholar]
- 129.Engelborghs S, Vloeberghs E, Le BN, et al. The dopaminergic neurotransmitter system is associated with aggression and agitation in frontotemporal dementia. Neurochem Int. 2008;52(6):1052–1060. [DOI] [PubMed] [Google Scholar]
- 130.Rahman S, Robbins TW, Hodges JR, et al. Methylphenidate ('Ritalin') can ameliorate abnormal risk-taking behavior in the frontal variant of frontotemporal dementia. Neuropsychopharmacol. 2006;31(3):651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Adams RD, Vanbogaert L, Vandereecken H. STRIATO-NIGRAL DEGENERATION. J Neuropathol Exp Neurol. 1964;23:584–608. [PubMed]
- 132.Fanciulli A, Wenning GK.. Multiple-system atrophy. N Engl J Med. 2015;372(3):249–263. [DOI] [PubMed] [Google Scholar]
- 133.Stankovic I, Quinn N, Vignatelli L, et al.; Movement Disorder Society Multiple System Atrophy Study Group. A critique of the second consensus criteria for multiple system atrophy. Mov Disord. 2019;34(7):975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brown RG, Lacomblez L, Landwehrmeyer BG, et al.; NNIPPS Study Group. Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy. Brain. 2010;133(Pt 8):2382–2393. [DOI] [PubMed]
- 135.Koga S, Parks A, Uitti RJ, et al. Profile of cognitive impairment and underlying pathology in multiple system atrophy. Mov Disord. 2017;32(3):405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kaufmann H.L-dihydroxyphenylserine (Droxidopa): A new therapy for neurogenic orthostatic hypotension: The US experience. Clin Auton Res. 2008;18(Suppl 1):19–24. [DOI] [PubMed] [Google Scholar]
- 137.Mathias CJ.L-dihydroxyphenylserine (Droxidopa) in the treatment of orthostatic hypotension: The European experience. Clin Auton Res. 2008;18(Suppl 1):25–29. [DOI] [PubMed] [Google Scholar]
- 138.Kaufmann H, Freeman R, Biaggioni I, et al. ; On behalf of the NOH301 Investigators. Droxidopa for neurogenic orthostatic hypotension: A randomized, placebo-controlled, phase 3 trial. Neurology. 2014;83(4):328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McDonell KE, Shibao CA, Biaggioni I, Hartman A, Robertson D, Claassen DO.. Cognitive and behavioral changes in patients treated with droxidopa for neurogenic orthostatic hypotension: A retrospective review. Cogn Behav Neurol. 2019;32(3):179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Glennon E, Carcea I, Martins ARO, et al. Locus coeruleus activation accelerates perceptual learning. Brain Res. 2019;1709:39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nieuwenhuis S, Aston-Jones G, Cohen JD. Decision making, the P3, and the locus coeruleus-norepinephrine system. Psychol Bull. 2005;131(4):510–532. [DOI] [PubMed] [Google Scholar]
- 142.Sharma Y, Xu T, Graf WM, et al. Comparative anatomy of the locus coeruleus in humans and nonhuman primates. J Compar Neurol. 2010;518(7):963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Smaers JB, Gómez-Robles A, Parks AN, Sherwood CC. Exceptional evolutionary expansion of prefrontal cortex in great apes and humans. Curr Biol. 2017;27(5):714–720. [DOI] [PubMed] [Google Scholar]
- 144.Le Maître E, Barde SS, Palkovits M, Diaz-Heijtz R, Hökfelt TGM. Distinct features of neurotransmitter systems in the human brain with focus on the galanin system in locus coeruleus and dorsal raphe. Proc Natl Acad Sci U S A. 2013;110(6):E536-E545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rowe JB, Saunders JR, Durantou F, Robbins TW. Systemic idazoxan impairs performance in a non-reversal shift test: Implications for the role of the central noradrenergic systems in selective attention. J Psychopharmacol. 1996;10(3):188–194. [DOI] [PubMed] [Google Scholar]
- 146.Tait DS, Brown VJ, Farovik A, Theobald DE, Dalley JW, Robbins TW. Lesions of the dorsal noradrenergic bundle impair attentional set-shifting in the rat. Eur J Neurosci. 2007;25(12):3719–3724. [DOI] [PubMed] [Google Scholar]
- 147.Newman LA, Darling J, McGaughy J. Atomoxetine reverses attentional deficits produced by noradrenergic deafferentation of medial prefrontal cortex. Psychopharmacology (Berl). 2008;200(1):39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Marshall CA, Brodnik ZD, Mortensen OV, et al. Selective activation of Dopamine D3 receptors and norepinephrine transporter blockade enhances sustained attention. Neuropharmacology. 2019;148:178–188. [DOI] [PMC free article] [PubMed]
- 149.Brown SB, Tona KD, van Noorden MS, Giltay EJ, van der Wee NJ, Nieuwenhuis S.. Noradrenergic and cholinergic effects on speed and sensitivity measures of phasic alerting. Behav Neurosci. 2015;129(1):42–49. [DOI] [PubMed] [Google Scholar]
- 150.Coull JT, Middleton HC, Robbins TW, Sahakian BJ.. Clonidine and diazepam have differential effects on tests of attention and learning. Psychopharmacology (Berl). 1995;120(3):322–332. [DOI] [PubMed] [Google Scholar]
- 151.Hoffing RC, Seitz AR.. Pupillometry as a glimpse into the neurochemical basis of human memory encoding. J Cogn Neurosci. 2015;27(4):765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nombela C, Rowe JB, Winder-Rhodes SE, et al.; ICICLE-PD study group. Genetic impact on cognition and brain function in newly diagnosed Parkinson's disease: ICICLE-PD study. Brain. 2014;137(Pt 10):2743–2758. [DOI] [PMC free article] [PubMed]
- 153.Williams-Gray CH, Mason SL, Evans JR, et al. The CamPaIGN study of Parkinson's disease: 10-year outlook in an incident population-based cohort. J Neurol Neurosurg Psychiatry. 2013;84(11):1258–1264. [DOI] [PubMed] [Google Scholar]
- 154.Kehagia AA, Housden CR, Regenthal R, et al. Targeting impulsivity in Parkinson's disease using atomoxetine. Brain. 2014;137(Pt 7):1986–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Phillips C, Fahimi A, Das D, Mojabi FS, Ponnusamy R, Salehi A.. Noradrenergic system in Down syndrome and Alzheimer's disease a target for therapy. Curr Alzheimer Res. 2016;13(1):68–83. [DOI] [PubMed] [Google Scholar]
- 156.Arnsten AF.Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10(6):410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Chamberlain SR, Robbins TW.. Noradrenergic modulation of cognition: Therapeutic implications. J Psychopharmacol. 2013;27(8):694–718. [DOI] [PubMed] [Google Scholar]
- 158.Robbins TW, Arnsten AF.. The neuropsychopharmacology of fronto-executive function: Monoaminergic modulation. Annu Rev Neurosci. 2009;32:267–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mao ZM, Arnsten AF, Li BM. Local infusion of an alpha-1 adrenergic agonist into the prefrontal cortex impairs spatial working memory performance in monkeys. Biol Psychiatry. 1999;46(9):1259–1265. [DOI] [PubMed] [Google Scholar]
- 160.Arnsten AF, Goldman-Rakic PS.. Analysis of alpha-2 adrenergic agonist effects on the delayed nonmatch-to-sample performance of aged rhesus monkeys. Neurobiol Aging. 1990;11(6):583–590. [DOI] [PubMed] [Google Scholar]
- 161.Arnsten AF, Goldman-Rakic PS.. Alpha 2-adrenergic mechanisms in prefrontal cortex associated with cognitive decline in aged nonhuman primates. Science. 1985;230(4731):1273–1276. [DOI] [PubMed] [Google Scholar]
- 162.Pu X, Ma Y, Cai J.. A study on the effect of lesions of area 7 of the parietal cortex on the short-term visual spatial memory of rhesus monkeys (Macaca mulatta). Brain Res. 1993;600(2):187–192. [DOI] [PubMed] [Google Scholar]
- 163.Gamo NJ, Wang M, Arnsten AF.. Methylphenidate and atomoxetine enhance prefrontal function through alpha2-adrenergic and dopamine D1 receptors. J Am Acad Child Adolesc Psychiatry. 2010;49(10):1011–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mair RG, McEntee WJ.. Cognitive enhancement in Korsakoff's psychosis by clonidine: A comparison with L-dopa and ephedrine. Psychopharmacology (Berl). 1986;88(3):374–380. [DOI] [PubMed] [Google Scholar]
- 165.Coull JT, Middleton HC, Robbins TW, Sahakian BJ.. Contrasting effects of clonidine and diazepam on tests of working memory and planning. Psychopharmacology (Berl). 1995;120(3):311–321. [DOI] [PubMed] [Google Scholar]
- 166.Zokaei N, Husain M.. Working memory in Alzheimer's disease and Parkinson's disease. Curr Top Behav Neurosci. 2019;41:325–344. [DOI] [PubMed] [Google Scholar]
- 167.Hurtig HI, Trojanowski JQ, Galvin J, et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson's disease. Neurology. 2000;54(10):1916–1921. [DOI] [PubMed] [Google Scholar]
- 168.Turner TH, Renfroe JB, Elm J, Duppstadt-Delambo A, Hinson VK.. Robustness of reliable change indices to variability in Parkinson's disease with mild cognitive impairment. Appl Neuropsychol Adult. 2016;23(6):399–402. [DOI] [PubMed] [Google Scholar]
- 169.Mohs RC, Shiovitz TM, Tariot PN, Porsteinsson AP, Baker KD, Feldman PD.. Atomoxetine augmentation of cholinesterase inhibitor therapy in patients with Alzheimer disease: 6-month, randomized, double-blind, placebo-controlled, parallel-trial study. Am J Geriatr Psychiatry. 2009;17(9):752–759. [DOI] [PubMed] [Google Scholar]
- 170.Eagle DM, Baunez C, Hutcheson DM, Lehmann O, Shah AP, Robbins TW.. Stop-signal reaction-time task performance: Role of prefrontal cortex and subthalamic nucleus. Cereb Cortex. 2008;18(1):178–188. [DOI] [PubMed] [Google Scholar]
- 171.Eagle DM, Bari A, Robbins TW.. The neuropsychopharmacology of action inhibition: Cross-species translation of the stop-signal and go/no-go tasks. Psychopharmacology (Berl). 2008;199(3):439–456. [DOI] [PubMed] [Google Scholar]
- 172.Liu YP, Huang TS, Tung CS, Lin CC.. Effects of atomoxetine on attention and impulsivity in the five-choice serial reaction time task in rats with lesions of dorsal noradrenergic ascending bundle. Prog Neuropsychopharmacol Biol Psychiatry. 2015;56:81–90. [DOI] [PubMed] [Google Scholar]
- 173.Kumar U, Medel-Matus JS, Redwine HM, et al. Effects of selective serotonin and norepinephrine reuptake inhibitors on depressive- and impulsive-like behaviors and on monoamine transmission in experimental temporal lobe epilepsy. Epilepsia. 2016;57(3):506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chamberlain SR, Muller U, Robbins TW, Sahakian BJ.. Neuropharmacological modulation of cognition. Curr Opin Neurol. 2006;19(6):607–612. [DOI] [PubMed] [Google Scholar]
- 175.Chamberlain SR, Hampshire A, Muller U, et al. Atomoxetine modulates right inferior frontal activation during inhibitory control: A pharmacological functional magnetic resonance imaging study. Biol Psychiatry. 2009;65(7):550–555. [DOI] [PubMed] [Google Scholar]
- 176.Graf H, Abler B, Freudenmann R, et al. Neural correlates of error monitoring modulated by atomoxetine in healthy volunteers. Biol Psychiatry. 2011;69(9):890–897. [DOI] [PubMed] [Google Scholar]
- 177.Macoveanu J, Hornboll B, Elliott R, et al. Serotonin 2A receptors, citalopram and tryptophan-depletion: A multimodal imaging study of their interactions during response inhibition. Neuropsychopharmacol. 2013;38(6):996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Hauser TU, Moutoussis M, Purg N, Dayan P, Dolan RJ.. Beta-blocker propranolol modulates decision urgency during sequential information gathering. J Neurosci. 2018;38(32):7170–7178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nombela C, Rittman T, Robbins TW, Rowe JB.. Multiple modes of impulsivity in Parkinson's disease. PLoS One. 2014;9(1):e85747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ye Z, Altena E, Nombela C, et al. Selective serotonin reuptake inhibition modulates response inhibition in Parkinson's disease. Brain. 2014;137(Pt 4):1145–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ye Z, Altena E, Nombela C, et al. Improving response inhibition in Parkinson's disease with atomoxetine. Biol Psychiatry. 2015;77(8):740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jonsson EG, Nothen MM, Gustavsson JP, et al. Polymorphisms in the dopamine, serotonin, and norepinephrine transporter genes and their relationships to monoamine metabolite concentrations in CSF of healthy volunteers. Psychiatry Res. 1998;79(1):1–9. [DOI] [PubMed] [Google Scholar]
- 183.Whelan R, Conrod PJ, Poline JB, et al. ; IMAGEN Consortium. Adolescent impulsivity phenotypes characterized by distinct brain networks. Nat Neurosci. 2012;15(6):920–925. [DOI] [PubMed] [Google Scholar]
- 184.Hesse S, Muller U, Rullmann M, et al. The association between in vivo central noradrenaline transporter availability and trait impulsivity. Psychiatry Res Neuroimaging. 2017;267:9–14. [DOI] [PubMed] [Google Scholar]
- 185.Choi CI, Bae JW, Lee YJ, Lee HI, Jang CG, Lee SY.. Effects of CYP2C19 genetic polymorphisms on atomoxetine pharmacokinetics. J Clin Psychopharmacol. 2014;34(1):139–142. [DOI] [PubMed] [Google Scholar]
- 186.Kehagia AA, Barker RA, Robbins TW.. Revisiting the effects of Parkinson's disease and frontal lobe lesions on task switching: The role of rule reconfiguration. J Neuropsychol. 2014;8(1):53–74. [DOI] [PubMed] [Google Scholar]
- 187.Lapiz MD, Morilak DA.. Noradrenergic modulation of cognitive function in rat medial prefrontal cortex as measured by attentional set shifting capability. Neuroscience. 2006;137(3):1039–1049. [DOI] [PubMed] [Google Scholar]
- 188.Snyder K, Wang WW, Han R, McFadden K, Valentino RJ.. Corticotropin-releasing factor in the norepinephrine nucleus, locus coeruleus, facilitates behavioral flexibility. Neuropsychopharmacol. 2012;37(2):520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cain RE, Wasserman MC, Waterhouse BD, McGaughy JA.. Atomoxetine facilitates attentional set shifting in adolescent rats. Dev Cogn Neurosci. 2011;1(4):552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Middleton HC, Sharma A, Agouzoul D, Sahakian BJ, Robbins TW. Idazoxan potentiates rather than antagonizes some of the cognitive effects of clonidine. Psychopharmacology (Berl). 1999;145(4):401–411. [DOI] [PubMed] [Google Scholar]
- 191.Muller U, Clark L, Lam ML, et al. Lack of effects of guanfacine on executive and memory functions in healthy male volunteers. Psychopharmacology (Berl). 2005;182(2):205–213. [DOI] [PubMed] [Google Scholar]
- 192.Alexander JK, Hillier A, Smith RM, Tivarus ME, Beversdorf DQ.. Beta-adrenergic modulation of cognitive flexibility during stress. J Cogn Neurosci. 2007;19(3):468–478. [DOI] [PubMed] [Google Scholar]
- 193.Combarros O, Warden DR, Hammond N, et al. The dopamine beta-hydroxylase -1021C/T polymorphism is associated with the risk of Alzheimer's disease in the Epistasis Project. BMC Med Genet. 2010;11(1):162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lee YH, Song GG. COMT Val158Met and PPARgamma Pro12Ala polymorphisms and susceptibility to Alzheimer's disease: A meta-analysis. Neurol Sci. 2014;35(5):643–651. [DOI] [PubMed] [Google Scholar]