

Abstract
The efficiencies offered by C4 photosynthesis have motivated efforts to understand its biochemical, genetic, and developmental basis. Reactions underlying C4 traits in most C4 plants are partitioned between two cell types, bundle sheath (BS), and mesophyll (M) cells. RNA-seq has been used to catalog differential gene expression in BS and M cells in maize (Zea mays) and several other C4 species. However, the contribution of translational control to maintaining the distinct proteomes of BS and M cells has not been addressed. In this study, we used ribosome profiling and RNA-seq to describe translatomes, translational efficiencies, and microRNA abundance in BS- and M-enriched fractions of maize seedling leaves. A conservative interpretation of our data revealed 182 genes exhibiting cell type-dependent differences in translational efficiency, 31 of which encode proteins with core roles in C4 photosynthesis. Our results suggest that non-AUG start codons are used preferentially in upstream open reading frames of BS cells, revealed mRNA sequence motifs that correlate with cell type-dependent translation, and identified potential translational regulators that are differentially expressed. In addition, our data expand the set of genes known to be differentially expressed in BS and M cells, including genes encoding transcription factors and microRNAs. These data add to the resources for understanding the evolutionary and developmental basis of C4 photosynthesis and for its engineering into C3 crops.
Ribo-seq and RNA-seq analysis of maize bundle sheath and mesophyll cells provides evidence that translational control and microRNA expression contribute to the distinct proteomes in the two cell types.
Introduction
Plant species are classified as C3 or C4 according to their mechanism of photosynthetic carbon fixation. C4 photosynthesis offers advantages under hot, dry conditions (Hatch, 1987), and it evolved many times from a C3 progenitor (Sage et al., 2011; Schlüter and Weber, 2020). Many C4 plants are characterized by a specialized leaf anatomy, denoted Kranz anatomy, that partitions enzymes of the C4 pathway between two cell types: mesophyll (M) and bundle sheath (BS). M cells surround BS cells, which in turn surround vascular bundles. Dissolved atmospheric CO2, , is first fixed by phosphoenolpyruvate (PEP) carboxylase (PEPC) in M cells to produce four-carbon acids. These diffuse to BS cells, where they are decarboxylated, providing CO2 for fixation by ribulose bisphosphate carboxylase/oxygenase (Rubisco) according to the C3 scheme. This organization reduces Rubisco’s wasteful oxygenation reaction by increasing local CO2 concentration and decreasing local O2 concentration.
Research into the genetic and developmental mechanisms underlying C4 traits has been propelled by interest in introducing C4 traits into C3 crops (Sedelnikova et al., 2018; Ermakova et al., 2020). A thorough understanding of patterns of gene expression during C4 differentiation is essential to achieve this goal. Numerous studies have approached this problem through transcriptome analysis (Wang et al., 2016; Schlüter and Weber, 2020), including studies that profiled transcriptomes during development of C4 leaves (Li et al., 2010; Pick et al., 2011; Liu et al., 2013; Wang et al., 2013b, 2014; Ding et al., 2015; Mattiello et al., 2015; Denton et al., 2017), or compared transcriptomes between BS and M cells (Li et al., 2010; Chang et al., 2012; Aubry et al., 2014; John et al., 2014; Tausta et al., 2014; Denton et al., 2017), among different C4 lineages (Aubry et al., 2014; Chen et al., 2014; John et al., 2014; Ding et al., 2015; Li et al., 2015; Offermann et al., 2015; Covshoff et al., 2016; Rao et al., 2016), or among related C3 and C4 species (Bräutigam et al., 2011; Gowik et al., 2011; Chen et al., 2014; Külahoglu et al., 2014; Wang et al., 2014; Ding et al., 2015; Lauterbach et al., 2017; Schlüter et al., 2017; Dunning et al., 2019). These transcriptome data have been complemented by surveys of protein populations during C4 differentiation and in isolated BS and M chloroplasts (Majeran et al., 2005; Bräutigam et al., 2008; Majeran et al., 2008; Friso et al., 2010; Majeran et al., 2010).
The results from these studies support the view that transcriptional control plays the major role in determining patterns of gene expression during C4 differentiation and in mature BS and M cells. That said, evidence for post-transcriptional contributions emerged in several studies (Patel et al., 2006; John et al., 2014; Ponnala et al., 2014; Berry et al., 2016; Williams et al., 2016), yet translational regulation has barely been explored (Schlüter and Weber, 2020). Ribosome profiling (ribo-seq) provides the means to comprehensively address this issue. Ribo-seq uses deep sequencing to map and quantify ribosome-protected mRNA fragments (ribosome footprints [RFs]). Because average translation elongation rates are generally similar among mRNAs under a given condition, the normalized abundance of RFs is a widely accepted proxy for relative rates of protein synthesis (Brar and Weissman, 2015). Comparison of RF abundance with the abundance of the corresponding mRNA allows inferences about translational efficiencies on a genome-wide scale.
Previously, we used ribo-seq in conjunction with RNA-seq to analyze chloroplast gene expression in BS- and M-enriched leaf fractions in maize (Zea mays), a C4 species of the NADP-malic enzyme (NADP-ME) subtype (Chotewutmontri and Barkan, 2016). We found that differences in mRNA abundance largely account for differential expression of chloroplast genes in the two cell types, but differences in translational efficiency (TE) synergize with mRNA-level effects in some cases. We have now extended this analysis to cytosolic mRNAs. Similar to what we observed in chloroplasts, the differential expression of nuclear genes in BS and M cells results primarily from differences in mRNA abundance. However, our data identified a subset of mRNAs whose translational efficiencies are significantly different in the two cell types, and revealed mRNA sequence features and trans-factors that correlate with cell-type-dependent translation. Additionally, our results expand the set of genes known to be differentially expressed in maize BS and M cells, suggesting additional genes of potential relevance to C4 traits.
Results
Overview of ribo-seq and RNA-seq data collected from BS- and M-enriched leaf fractions
Analyses reported here used BS- and M-enriched fractions generated from the apical region of seedling leaves with a rapid mechanical fractionation method similar to that used in our prior study of chloroplast translatomes (Chotewutmontri and Barkan, 2016). Marker proteins for each cell type were highly enriched in these fractions (Figure 1A). RFs and RNA were purified from aliquots of the same BS and M preparations, in three biological replicates. The ribo-seq reads mapping to cytosolic mRNAs exhibit the expected characteristics of RFs: they map almost exclusively to protein-coding regions, their size distribution is heavily weighted to 29–30 nt, and their positions within open reading frames (ORFs) show strong 3-nt periodicity (Supplemental Figure S1, A–C). We used rRNA-depleted total RNA for RNA-seq to avoid the 3′-bias that can lead to false inferences about differential expression (Denton et al., 2017; Supplemental Figure S2, A and B). Correlation coefficients among replicates ranged from ∼0.93 to 0.98, with replicate datasets clustering together as expected (Supplemental Figure S1D).
Figure 1.

Enrichment of cell type-specific markers in BS and M fractions. A, Immunoblots showing abundance of marker proteins in BS and M fractions. Replicate blots were probed with the indicated antibodies. BS markers: NADP-dependent malic enzyme (NADP-ME), H subunit of the NDH-like complex (NdhH), Rubisco large subunit (RbcL). M markers: phosphoenolpyruvate carboxylase (PEPC), pyruvate orthophosphate dikinase (PPDK), D2 subunit of Photosystem II (D2). An image of one blot stained with Ponceau S is shown below. B, Abundance of RNA-seq and ribo-seq reads from BS and M marker genes. BS markers: NADP-ME (Zm00001d000316) and Rubisco small subunit (RBCS; Zm00001d052595). M markers: PEPC (Zm00001d046170) and PPDK (Zm00001d038163). RNA-seq data from Tausta et al. (2014) (leaf section 14 with 2 replicates) and Denton et al. (2017) (leaf section S1 with three replicates) are compared with our data. Each line symbol represents normalized reads for one gene (see color key at top) in one replicate. RPKM, reads per kilobase per million reads mapped to nuclear coding sequences. C, Abundance of RNA-seq and ribo-seq reads from epidermis and phloem marker genes. Data for additional marker genes are shown in Supplemental Figure S2C.
Four RNA-seq studies of maize BS and M fractions were reported previously (Li et al., 2010; Chang et al., 2012; Tausta et al., 2014; Denton et al., 2017), all of which used polyA-selected mRNA. We used the two most recent studies as points of comparison for our data. Tausta et al. (2014) generated BS- and M-enriched fractions by laser capture microdissection, whereas Denton et al. (2017) used a mechanical method that differed from ours. Both studies analyzed tissue slices at several positions along the leaf blade, which represent different stages along the pathway of photosynthetic differentiation. The apical sections were most analogous to our material, so we selected those data for our comparisons. To facilitate comparisons, we aligned both prior datasets to the current maize genome assembly (B73 RefGen_v4) and we calculated differential expression using the same analysis pipeline we used for our data.
Figure 1B compares the cell-type enrichment of sequence reads from four C4 marker genes in our data to those in the prior datasets. In comparison with the Denton data, our RNA-seq data exhibited greater enrichment of both M and BS markers. In comparison with the Tausta data, our RNA-seq data exhibited similar enrichment of M markers and less enrichment of BS markers. We expected that our mechanical fractionation method would result in contamination of the M and BS fractions with epidermal and vascular tissue, respectively, and analysis of epidermal and phloem markers confirmed this to be the case (Figure 1C; Supplemental Figure S2C). Despite the considerable differences in sample preparation and developmental stage, a core gene set associated with C4 photosynthesis (Schlüter and Weber, 2020) showed Pearson correlation coefficients of at least 0.85 among all three datasets (Supplemental Figure S3A).
Comparison of our ribo-seq and RNA-seq data (Supplemental Figure S3B) returned correlation coefficients of 0.72 and 0.89 for genome-wide and C4 gene comparisons, respectively. These values are considerably lower than those for replicate samples, suggesting some differences in TE in the two cell types. In addition, we compared our data to that from a proteomic study of material obtained with a similar mechanical fractionation method (Friso et al., 2010; Supplemental Figure S3C). Proteins that were quantified with higher confidence levels showed considerable correlation with our ribo-seq data. Furthermore, our ribo-seq data correlated better with the proteomic data than did our RNA-seq data (Supplemental Figure S3C), consistent with a role for translational control in maintaining the distinct proteomes in the two cell types.
Differences in translational efficiency contribute to the differential expression of genes in BS and M cells.
To detect mRNAs that experience differential translation in M and BS cells, we calculated differences in TE with XTAIL (Xiao et al., 2016). XTAIL reports the normalized ratio of ribo-seq to RNA-seq reads for each gene together with a false discovery rate (FDR) for differential translation. We drew conclusions about differential translation only for genes with an average of at least 100 RNA-seq reads mapped to coding sequences in both the BS and M fractions, as well as an average of at least 100 ribo-seq reads in either the BS or M fraction. 9,476 genes met these read-count criteria. The XTAIL output for these genes is provided in Supplemental Dataset S1. We defined a gene as being differentially translated using stringent criteria: greater than three-fold difference in TE between the two tissues and an FDR < 0.001. A total of 355 genes met these criteria (Figure 2A; Supplemental Dataset S2). A comparison of the ribo-seq and RNA-seq data (Figure 2B) parses these genes into several groups: those that are differentially expressed primarily due to differences in TE, those for which changes in RNA and TE synergize to amplify differential expression (RNA&TE), and those for which a change in TE offsets a change in RNA (buffering). Buffering of this type has been detected in many ribo-seq studies (McManus et al., 2014; Schafer et al., 2015; Oertlin et al., 2019). However, we are hesitant to draw conclusions about the buffering set because our RNA-seq data for those particular genes correlated poorly with data from prior RNA-seq studies (Supplemental Figure S4). In contrast, RNA-seq data for our TE and RNA&TE sets correlated well with those from prior studies (Supplemental Figure S4). Examples of read coverage plots for genes whose cell type-specific expression results primarily from a difference in TE are shown in Figure 2C with additional examples in Supplemental Figure S5.
Figure 2.

Overview of genes exhibiting differential translation in BS and M fractions. A, Volcano plot of TE and FDR values. Each symbol represents one gene. Genes exhibiting preferential translation in M or BS fractions are shown in blue or red, respectively. Dashed lines indicate the FDR and fold-change cutoffs we used to define differentially translated genes. B, Scatter plot comparing RNA-seq to ribo-seq BS/M ratios. Each symbol represents one gene. Gene sets are labeled according to the dominant feature underlying differential expression (TE, RNA, or both). Dashed lines indicate the fold-change cutoffs we used to categorize the genes. C, Examples of RNA-seq and RF read coverage for genes whose differential expression results primarily from differential translation. Normalized read coverage in BS or M fractions is shown above or below the line, respectively. Coverage was normalized by million reads mapped to nuclear coding sequences. Each line represents one replicate. The re-analyzed values of published RNA-seq data are shown for comparison. Additional examples are shown in Supplemental Figure S5. D, Heatmaps showing BS/M values for genes exhibiting differential translation. Prior RNA-seq (Tausta et al. (2014) leaf section 14 and Denton et al. (2017) leaf section S1) and proteome (Friso et al. (2010)) data are shown for comparison. The previous RNA-seq data were re-aligned with our pipeline and analyzed with DESeq2; genes that did not meet our read count cutoffs are in gray. The expanded heat map to the right provides information for genes that are of particular relevance to C4 photosynthesis. Asterisks mark transcription factors (TFs) for which differential TE plays the dominant role in dictating cell type-specific expression that were not reported as differentially expressed in RNA-seq studies. Values are provided in Supplemental Dataset S2.
The data for translationally regulated genes of particular relevance to C4 physiology are shown in an expansion of the heat map in Figure 2D. These include (1) “C4” genes (Schlüter and Weber, 2020) involved in photorespiration, the Calvin-Benson cycle, C4 metabolism, redox regulation, and nitrogen and sulfur assimilation; (2) membrane transporters, subunits of photosystem II (PSII), and subunits of the NADH dehydrogenase-like complex (NDH) that are strongly enriched in one cell type or the other (Majeran and van Wijk, 2009); and (3) genes encoding transcription factors (TFs), which could potentially contribute to the establishment of cell type-specific transcriptomes. Roughly, one-third of these genes are regulated primarily at the level of TE, and the remainder by a combination of RNA abundance and TE. The latter set includes the gene encoding ribose-5-phosphate isomerase (RPI), which had been suggested to be under translational control based on a comparison of Setaria and maize transcriptome data (John et al., 2014). We detected three TFs that are differentially expressed primarily due to a difference in TE, two of which (Trihelix TF28 and NAC TF40) had not been detected as differentially expressed in transcriptome studies (Figure 2D, asterisks).
Features of untranslated regions that correlate with differential translation in BS and M cells
The rate of translation initiation is influenced by various features of mRNA untranslated regions (UTRs), including upstream ORFs (uORFs), start codon sequence and sequence context, RNA structure, and binding sites of translational regulators. To gain insight into the basis for differences in TE between BS and M cells, we compared UTR features among mRNAs exhibiting cell type-dependent translation to those that do not. RNAs that are translated preferentially in the BS fraction show a tendency toward UTRs that are shorter and more GC-rich than those in the control set, particularly in the 5′-UTR (Figure 3A). We identified six sequence motifs that are enriched in UTRs of mRNAs that are preferentially translated in one cell type or the other (Figure 3B; Supplemental Dataset S3): three in 5′-UTRs of the BS set, one in 3′-UTRs of the BS set, and one each in 5′- and 3′-UTRs of the M set. Out of the 182 genes whose cell type-dependent translation contributes to differential expression (greater than three-fold difference in TE), 78 harbor at least one of these motifs and 20 harbor more than one (Figure 3C). Enriched sequence motifs of this nature are candidates for binding sites of translational regulators. Few sequence-specific RNA-binding proteins have been identified in plants that regulate the translation of specific mRNAs. However, members of the PUF (Pumilio/FBF) family are attractive candidates because they are known to regulate translation and RNA stability by binding specific UTRs in animals and fungi (Goldstrohm et al., 2018; Wang et al., 2018), and the PUF family is unusually large in plants (Dedow and Bailey-Serres, 2019; Joshna et al., 2020). Furthermore, the UGUGCCG motif that is enriched in 3′-UTRs of the BS set (Figure 3B) resembles the consensus UGUR core binding site for canonical PUF proteins. With this in mind, we examined expression of genes encoding PUF proteins in BS and M fractions in our ribo-seq and RNA-seq data and in prior RNA-seq studies (Figure 3D). We identified several PUF-encoding genes that are expressed preferentially in one cell type or the other in at least one dataset (Figure 3D). A particularly intriguing example is Zm00001d023781, whose expression is highly enriched in BS cells across multiple datasets. Furthermore, the expression of this gene along the seedling leaf blade peaks in the zone of active chloroplast biogenesis (http://bar.utoronto.ca/efp_maize/cgi-bin/efpWeb.cgi; Li et al., 2010; Wang et al., 2014), consistent with a role in regulating chloroplast differentiation. These results highlight Zm00001d023781 as an attractive candidate for future investigation with regard to the post-transcriptional control of BS and M differentiation.
Figure 3.
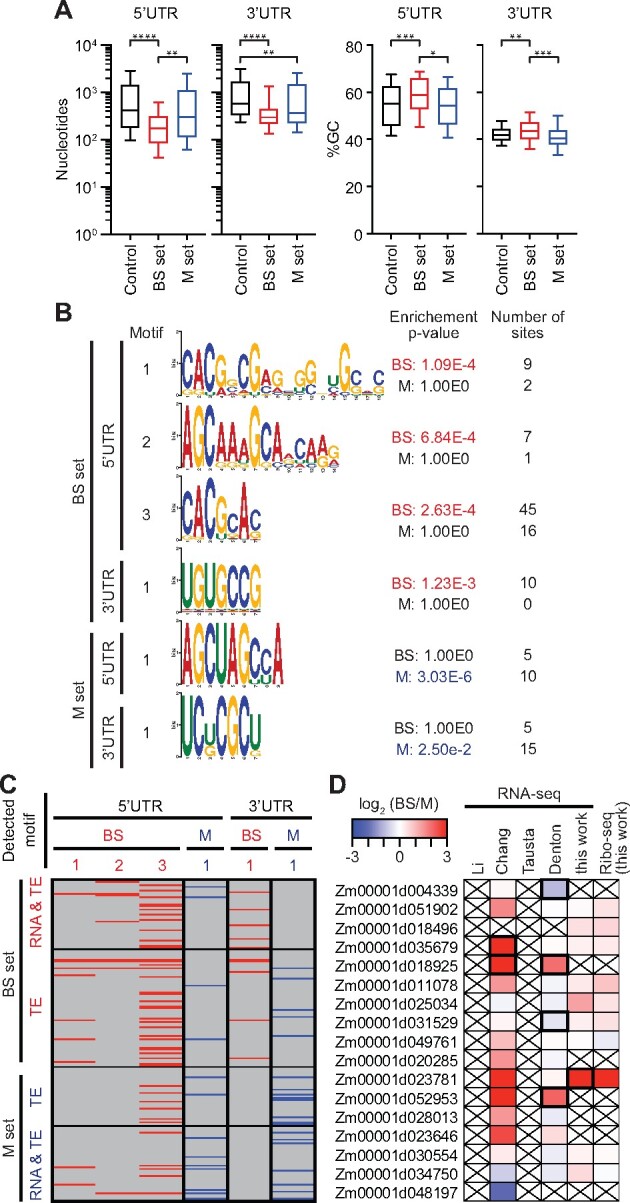
Characteristics of UTRs of differentially translated genes. Differentially translated genes used for these analyses are the TE and RNA&TE sets from Figure 2B (|log2(BS/M TE)| > 1.585, FDR < 0.001 excluding buffering sets, BS set n = 107 and M set n = 75). The control set was defined as |log2 (BS/M) TE| ≤ 0.585 (n = 5280). A, UTR length and GC content. Horizontal lines show median values, boxes show 25–75th percentile, and whiskers indicate 10–90th percentiles. Brackets show significant P-values from Dunn’s multiple comparison test of the means. *P = 0.01; **P < 0.005; ***P = 0.002, and ****P < 0.0001. B, Enriched motifs in UTRs of differentially translated genes. Enriched motifs in the data set indicated to the left were identified with STREME (Bailey, 2021) and those matching known DNA motifs were removed. Motifs reported by AME (McLeay and Bailey, 2010) as significantly enriched (P < 0.05) are shown in red or blue font. The enrichment P-values and number of sites for the nonenriched data are shown for comparison. C, Presence of enriched motifs in differentially translated genes. Gene sets are taken from Figure 2B. Motifs are identified at top via the motif numbers shown in (B). D, Differential expression of PUF genes. Values from prior RNA-seq datasets come from Li et al. (2010), Chang et al. (2012), Tausta et al. (2014) section 14, and Denton et al. (2017) section S1. Genes reported with high confidence as differentially expressed in prior studies or in our data [|log2(BS/M)| > 1, FDR < 0.001] are bordered in black. Crossed boxes indicate absence of data or read counts below cutoffs.
uORFs often regulate the translation of main ORFs (mORFs) in eukaryotic mRNAs (Hinnebusch et al., 2016). To address the relevance of uORFs to cell type-dependent translation, we expanded the set of differentially translated genes to include those that exhibit greater than two-fold differences in TE (P < 0.001). This resulted in 335 genes (excluding the buffering set), which we inspected for RFs upstream of their annotated start codon. We considered uORFs beginning with AUG as well as those with the noncanonical start codons CUG and ACG (van der Horst et al., 2019). Thirty-nine genes fit these criteria (Supplemental Dataset S4): 5 had only uORFs beginning with AUG, 28 had uORFs beginning with CUG or ACG, 6 had both AUG and non-AUG uORFs. Nine genes had overlapping uORFs and 17 genes had uORFs that overlap mORFs.
Comparison of uORF and mORF ribosome occupancies (ribo-seq/RNA-seq) in BS and M fractions revealed three types of relationship (Figure 4A; Supplemental Dataset S4): uORF translation was inversely related to mORF translation, suggesting a repressive effect of the uORF; uORF translation was positively correlated with mORF translation, suggesting an activating effect of the uORF; and uORF and mORF translation changed independently of one another. Positive correlations between non-AUG uORF and mORF translation in plants have been reported previously (Li and Liu, 2020). Figure 4B shows examples of normalized ribo-seq read coverage from two genes in each category. Interestingly, our uORF data suggest a bias toward translation of non-AUG start codons in BS cells (Figure 4C). However, a deeper analysis of start codon usage will be needed to draw firm conclusions. In yeast, the translation initiation factors eIF1 and eIF5A promote translation from AUG codons while eIF5 antagonizes this function, thereby favoring non-AUG initiation (Hinnebusch et al., 2016; Eisenberg et al., 2020). We, therefore, considered the possibility that differing ratios of these factors in BS and M cells underlie the differences in AUG versus non-AUG uORF initiation suggested by our data. In fact, the ratio of eIF1/eIF5A to eIF5 expression is elevated in BS cells (Figure 4D), running counter to the hypothesized relationship with non-AUG initiation. In particular, expression of one eIF1 paralog (Zm00001d021668) and two eIF5A paralogs (Zm00001d006760 and Zm00001d022042) is BS-enriched in our ribo-seq data (Figure 4D) and in many prior RNA-seq datasets (Supplemental Figure S6), suggesting that these paralogs may play noncanonical roles that favor non-AUG initiations.
Figure 4.

Translation of uORFs of differentially translated genes. The genes used for this analysis had |log2 (BS/M) TE| > 1 and FDR < 0.001, excluding the buffering sets. Ribo-seq read counts exclude ribosomes bound to the mORF start codon. RPKM, reads per kilobase per million reads mapped to nuclear coding sequences. A, TE (ribo-seq/RNA-seq RPKM) of uORFs and mORFs in BS and M fractions. Genes are grouped based on whether differences in TE of the uORF and mORF in BS and M fractions are correlated, anti-correlated, or independent. Thirty-nine differentially translated genes had ribosomes in uORFs; data are shown for the 29 genes with uORFs that begin with either AUG or non-AUG codons (but not both) and for which the relationship between uORF and mORF translation was reproducible. Data for the other ten genes are summarized in Supplemental Dataset S4. *CHUP1 is the only gene in (A) with AUG uORFs. The means ± sd from three replicates are shown. B, Examples of normalized ribo-seq read coverage from the 5′-regions of differentially translated uORF-containing genes. Gray shading indicates coverage from each replicate such that overlapping coverage from different replicates appears as a darker shade. Coverage in BS or M fractions is shown above or below the line, respectively. Values were normalized by million ribo-seq reads mapped to nuclear coding sequence and by RNA-seq RPKM for the mORF. Normalized P-site occupancies (sum of replicates) are shown in black (scale to the right). Nucleotide and three-frame translation tracks are shown above gene models. The green and red boxes in the translation tracks indicate AUG start and stop codons, respectively. Zm and LOC indicate gene models from Gramene and GenBank, respectively. C, TE (ribo-seq/RNA-seq) of uORFs with AUG or non-AUG start codons in BS and M fractions. Each symbol represents one gene. Values are the mean ± sd from three replicates. D, Expression of eIF1, eIF5, and eIF5A in BS and M fractions based on ribo-seq data (sum of three replicates).
Global analysis of BS and M translatomes expands the known set of differentially expressed genes
The abundance of RFs mapping to a gene reflects both mRNA abundance and TE. This is widely used as a proxy for relative rates of protein synthesis among genes, and there is considerable evidence that this is, with rare exceptions, a valid assumption (Brar and Weissman, 2015). Therefore, ribo-seq can provide a more accurate picture of differential expression than RNA-seq. With that in mind, we analyzed our ribo-seq data with DESeq2 to catalog genes that are differentially expressed in BS and M cells. We limited the analysis to the 13,182 genes having an average of at least 100 ribo-seq reads mapped to coding sequences in at least one of the two tissue types (Supplemental Dataset S5). We defined differentially expressed genes as having |log2 (BS/M)| > 1 and FDR < 0.001. With these criteria, we detected 1,345 and 993 genes that are preferentially expressed in the BS and M fractions, respectively (Figure 5A; Supplemental Dataset S6 and S7). Analysis of our RNA-seq data using the same criteria produced 14,476 genes that met our read count cutoffs, of which 1,111 and 1,457 genes are preferentially expressed in the BS and M fractions, respectively. We then compared the ribo-seq results to those from previous RNA-seq studies of maize BS and M fractions (Li et al., 2010; Chang et al., 2012; Tausta et al., 2014; Denton et al., 2017). Approximately 30% of the genes reported as differentially expressed in our ribo-seq analysis were not reported to be differentially expressed in prior RNA-seq studies (Supplemental Dataset S6 and S7; “unique” sets in Figure 5A). These “unique” gene sets showed similar functional-enrichment patterns as the complete sets of BS- and M-enriched genes (Figure 5B; Supplemental Dataset S8), instilling confidence that many of these genes are, in fact, differentially expressed in BS and M cells. The unique set includes many TFs (Figure 5C), as discussed below.
Figure 5.

Comparison of translatomes in BS and M fractions. A, Overview of ribo-seq data. 13,182 genes met the read-count cutoff used in this analysis (average of ≥100 reads in at least one of the two fractions). The BS- and M-enriched gene sets [|log2 (BS/M)| > 1 and FDR < 0.001] were compared to genes that had been reported to be differentially expressed in previous RNA-seq studies: Chang et al. (2012), Li et al. (2010), Denton et al. (2017) section S1, and Tausta et al. (2014) section 14. The “conflict gene model” sets consist of genes that cannot be compared among studies because of conflicting gene models. The “agree” sets include genes that were reported by at least one prior RNA-seq study to be preferentially expressed in the same tissue type as in our ribo-seq data. The “unique” sets are genes reported by our ribo-seq data as differentially expressed that were not reported as differentially expressed in prior studies. B, Functional enrichment analysis of genes identified as differentially expressed in our ribo-seq data that had not been reported as differentially expressed in previous studies. Heatmaps display the term enrichment z-scores when comparing BS- and M-enriched gene sets to the set of all expressed genes. Data for the complete BS- and M-enriched sets (BS and M) are compared with data for the unique sets described in (A). HC, hydroxycinnamic acid. C, Heatmap of log2 (BS/M) values of ribo-seq data for TF genes that exhibited BS- or M-enriched RF abundance. Values from prior RNA-seq studies are shown for comparison. Genes are ranked based on the number of reports concluding that the gene is differentially expressed (# report, left). Genes not reported as differentially expressed are in gray. The TFs that were identified in a cross-species transcriptome analysis by Huang and Brutnell (2016) are marked in the right column. TFs reported to be differentially expressed among all studies are listed in Supplemental Dataset S10. IAA10*, Aux/IAA TF10 also known as ZmIAA7.
Three photosynthetic complexes harboring plastid-encoded subunits accumulate differentially in BS and M cells: Rubisco and the NDH complex accumulate preferentially in BS cells, whereas PSII accumulates preferentially in M cells. The biogenesis of each complex involves a large number of nuclear genes, some of which are involved in complex assembly and others in the expression of plastid-encoded subunits. Differential expression data for genes that function specifically in the biogenesis of these complexes (Supplemental Figure S7; Supplemental Dataset S9) show that many of them are preferentially expressed in the same cell type as the cognate complex. Furthermore, three genes encoding proteins that activate translation of the chloroplast psbA mRNA (HCF173, HCF244, and OHP2; Schult et al., 2007; Link et al., 2012; Chotewutmontri et al., 2020) are expressed preferentially in M cells. This can account for the preferential translation of psbA mRNA in M cells we had reported previously (Chotewutmontri and Barkan, 2016).
Differential expression of TFs and microRNAs in BS and M fractions
The distinct transcriptomes of BS and M cells are presumably maintained, at least in part, by cell-type-specific regulatory molecules, including TFs and microRNAs. We identified genes encoding 114 and 37 TFs that were enriched in BS and M translatomes, respectively (Supplemental Dataset S10). Approximately one-third of these had not been reported as differentially expressed in prior RNA-seq studies. One interesting example is a BS-enriched member of the Aux/IAA family of auxin-regulated TFs (Gene ID Zm00001d041416; IAA10 in Figure 5C), which also emerged as relevant to C4 biology based on evolutionary inferences (Huang et al., 2017). Aux/IAA proteins govern diverse developmental processes including vascular patterning (Zhang et al., 2014; Luo et al., 2018). These observations suggest the possibility that this particular Aux/IAA protein regulates C4 vascular patterning in the maize leaf. Only one-fifth of these TFs were reported as differentially expressed across all studies (Figure 5C). The low concordance among studies is likely due to a variety of factors, including varying sequencing depth, varying criteria used to identify differentially expressed genes, and imperfectly matched developmental stages. In addition, cell-type-dependent translation accounted for the differential expression of two of the TFs detected uniquely in our set (Trihelix TF28 and NAC TF40 in Figure 2D). Expression of Golden2-like 1 (GLK1), a TF required specifically for M-cell differentiation in maize (Wang et al., 2013a) was M-enriched in our data as in prior RNA-seq studies (Figure 5C).
MicroRNAs regulate mRNA stability and translation. To address whether cell-type-specific microRNA expression accounts for the differential expression of any genes in BS and M cells, we sequenced small RNAs in the same RNA preparations used for RNA-seq. DESeq2 returned six differentially expressed microRNAs [|log2 (BS/M)| > 1 and FDR < 0.05] (Supplemental Dataset S11), representing two microRNA families: MIR160 and MIR166 (Supplemental Figure S8). Five members of the MIR160 gene family encode identical mature microRNAs, and these are strongly enriched in our BS fraction (Figure 6, A and B). One member of the MIR166 family (zma-MIR166a; Supplemental Figure S8) is enriched in our M fraction (Figure 6, A and B).
Figure 6.

Differential expression of microRNAs and their TF targets in BS and M fractions. A, Differential expression of microRNAs from sRNA-seq analysis of BS and M fractions. Six genes identified by DESeq2 are shown [|log2 (BS/M)| > 1 and FDR < 0.05]. The mature zma-MIR160 microRNAs have identical sequences, so sRNA-seq cannot distinguish them. B, Normalized read coverage of genes encoding zma-MIR160a and zma-MIR166a. Coverage was normalized by million reads mapped to all microRNA precursors. Single sequence reads spanned the full length of each microRNA, resulting in uniform coverage across their length. Each line represents one replicate, with reads from the BS and M fractions shown above and below the horizontal line, respectively. Each microRNA precursor (diagrammed at bottom) folds into a hairpin via the pairing of complementary regions (gray). One strand in the duplex becomes mature microRNA and the other is typically degraded. An asterisk marks the degraded strand identified by miRBase (http://www.mirbase.org/). C, Cell-type-specific expression of mRNA targets of differentially expressed microRNAs. The mRNA targets of MIR166 and MIR160 were recovered from the DPMIND database (Fei et al., 2018). The original log2 (BS/M) values reported in prior RNA-seq datasets are summarized. Genes reported as differentially expressed are bordered in black. Crossed boxes indicate absence of data. ARFTF, auxin-responsive factor TF; HD-ZIP III, Class III homeodomain leucine zipper; RLD1/2, Rolled leaf1/2; HB69, homeobox-transcription factor69.
We then used the DPMIND database (Fei et al., 2018) to identify mRNA targets of these microRNAs. Zma-MIR160, which is enriched in our BS fraction, targets mRNAs encoding three auxin-responsive factor (ARF) TFs. RNA-seq data show that expression of one of these, ARFTF15, is enriched in the M fraction (Figure 6C), supporting a role for zma-MIR160 in directing ARFTF15 mRNA decay in BS cells. The M-enriched microRNA zma-MIR166 targets mRNAs encoding three Class III homeodomain leucine zipper (HD-ZIP III) TFs (Juarez et al., 2004; Fei et al., 2018). RNA-seq data reported by Chang et al. (2012) indicate BS enrichment for all three of these mRNA targets, and our RNA-seq data support this for one of them (Rolled leaf1 [RLD1]; Figure 6C). Zma-MIR166 is known to control spatial expression of RLD1 in maize leaf primordia (Juarez et al., 2004). Our data suggest further that zma-MIR166 regulates RLD1 in differentiated leaf tissue.
Discussion
Numerous transcriptome studies have sought to identify genes whose differential expression is relevant to programming C4 traits (Wang et al., 2016; Schlüter and Weber, 2020). However, the contribution of translational regulation has been largely unexplored (Schlüter and Weber, 2020). Our study addressed this gap through analysis of BS and M translatomes in maize. Our results show that translational regulation contributes to the differential expression of genes in maize BS and M cells and provide clues about underlying mechanisms. Our data also add to the set of genes known to be differentially expressed in the two cell types, including additional regulatory genes of potential relevance to the establishment or maintenance of C4 traits.
A conservative interpretation of our data identified 182 genes for which differential translation makes a strong contribution (greater than three-fold difference in TE) to their differential expression in BS and M cells (Supplemental Dataset S2). These include several genes encoding TFs and numerous genes encoding proteins involved in C4 photosynthesis, including one (the gene encoding RPI) that had previously been suggested to be under translational control (John et al., 2014; Figure 2D). Our previous study of chloroplast translatomes in BS and M fractions showed that controls at the level of translation and mRNA abundance often synergize to produce robust differential expression (Chotewutmontri and Barkan, 2016). Results here indicate that a similar theme holds true for many nuclear genes. The relative contributions of controls at the level of RNA abundance and translation vary and, in some cases, translational regulation plays the major role (Figure 2B).
Ribo-seq can provide a more accurate view of differential expression than RNA-seq because it accounts for differences in both mRNA abundance and TE. In fact, our ribo-seq data provide evidence for the differential expression of many more genes than had been detected in prior RNA-seq studies (Figure 5; Supplemental Dataset S6 and S7). Of particular interest are candidates for cell-type-specific regulatory molecules. For example, we detected 44 new TF candidates (Supplemental Dataset S10) including one (IAA10) that had been implicated in C4 differentiation on the basis of a cross-species selection scan (Huang et al., 2017), and two (NAC TF40 and THX28) that are expressed preferentially in the BS fraction due primarily to differential translation. Furthermore, we present evidence that the differential expression of two microRNAs underlies the cell-type-enriched expression of several TFs: zma-MIR166a and zma-MIR160 are preferentially expressed in M and BS fractions, respectively, correlating with expression of their mRNA targets encoding TFs RLD1 and ARF proteins, respectively, in the opposite cell type (Figure 6). Interestingly, a gain-of-function Rld1 mutant that is no longer controlled by zma-MIR166 exhibited normal vasculature in leaf primordia (Juarez et al., 2004) but failed to increase vascular bundle density and lacked photosynthetic BS and M cells in mature leaf (Nelson et al., 2002). These results together with our findings suggest that zma-MIR166-mediated inhibition of RLD1 expression in mature M cells is important to maintain high vein density and differentiated BS/M cells, both of which are important C4 traits (Ermakova et al., 2020).
Our results also provide clues about the molecular basis for the differential translation events reported here. Our microRNA-seq data did not detect differentially expressed microRNAs that target translationally regulated mRNAs, suggesting that microRNAs do not contribute to translational control in this context. Our data hint, however, that differential use of AUG versus non-AUG initiation codons in uORFs is relevant to the cell-type-dependent translation of several genes (Figure 4C) and that non-AUG initiations are favored in BS cells. It is intriguing in this regard that specific eIF1 and eIF5A paralogs, two general translation factors that function in start codon selection (Nanda et al., 2009; Manjunath et al., 2019), exhibit BS-enriched expression (Figure 4D). A method called Translation Initiation Site Profiling offers more robust detection of functional start codons than does ribo-seq, and revealed a shift to use of non-AUG start codons during yeast meiosis (Eisenberg et al., 2020). Analysis of the BS and M translatomes with this assay could address whether translational reprogramming of this type affects BS and M proteomes in biologically meaningful ways. In addition, sequence motifs that are enriched in UTRs of translationally regulated mRNAs (Figure 3B) are candidate binding sites for sequence-specific RNA binding proteins. The motif UGUGCCG enriched in 3′-UTRs of mRNAs that exhibit preferential translation in BS cells (Figure 3B) resembles the consensus binding site of PUF proteins, which regulate the translation of specific mRNAs in animals and fungi. Furthermore, the expression of one PUF-encoding gene is strongly enriched in the BS fraction (Figure 3D), implicating this gene in the post-transcriptional control of gene expression in BS cells. PUF proteins typically repress translation, but examples of activation have been reported (Goldstrohm et al., 2018; Wang et al., 2018). Functional analysis of the candidate cis-elements and trans-factors to emerge from this study is an attractive direction for future investigation.
Materials and methods
Preparation of BS and M fractions
We prepared BS and M fractions by using a minor modification of the rapid mechanical procedure described previously (Chotewutmontri and Barkan, 2016). In brief, maize (Z. mays inbred line B73) was grown for 13 d under cycles of 12-h light (300 μmol m−2s−1) at 31°C and 12 h darkness at 22°C. The apical one-third of the second and third leaves to emerge were harvested 2 h into the light cycle. The M fraction was taken as the supernatant following gentle grinding of fresh tissue in a mortar and pestle as described before (Chotewutmontri and Barkan, 2016). However, differing from our previous procedure which used the residual material from the M preparation as the source of the BS fraction, we prepared the BS fraction from separate tissue aliquots as the tissue remaining after several rounds of gentle grinding and rinsing to remove released cells. This fractionation procedure took roughly 3 min for M fractions and 10 min for BS fractions, at which point samples were flash frozen in liquid nitrogen.
Ribo-seq, RNA-seq, and sRNA-seq libraries
RFs and total RNA were prepared from the same lysates in three biological replicates as described previously (Chotewutmontri and Barkan, 2016). Ribosome protected fragments from ∼20 to ∼40 nt were selected for ribo-seq library preparation, and libraries were prepared with the NEXTflex Small RNA Sequencing Kit v2 (Bioo Scientific, Austin, TX, USA) in combination with rRNA depletion with customized biotinylated antisense oligonucleotides (Chotewutmontri et al., 2018). RNA-seq libraries were prepared from total RNA after rRNA depletion with the Ribo-Zero rRNA Removal Kit (Plant Leaf; Illumina, San Diego, CA, USA) using the NEXTflex Rapid Directional qRNA-Seq Kit (Bioo Scientific). To catalog microRNAs, the same RNA samples were analyzed by sRNA-seq with the same kit used to sequence RFs. The ribo-seq, RNA-seq, and microRNA libraries were sequenced using a HiSeq 4000 (Illumina) in single-read mode with read lengths of 100 nt.
Sequence read processing
Ribo-seq and RNA-seq data were processed as described previously (Chotewutmontri and Barkan, 2020) with minor modifications. In brief, adapter sequences were trimmed using cutadapt (Martin, 2011). For ribo-seq, only trimmed reads between 18 and 40 nt were retained for alignments. Reads were aligned sequentially to the maize chloroplast genome (GenBank accession X86563), the maize mitochondrial genome (B73 RefGen_v4 assembly release 38), and the maize nuclear genome (B73 RefGen_v4 assembly release 38) using STAR version 2.5.3a (Dobin et al., 2013). Read splicing was restricted to annotated splice junctions. Read counting was performed using featureCounts from Subread package version 1.6.0 (Liao et al., 2014), counting only those reads that mapped to a unique site. Ribo-seq read counts for nuclear and organellar genes excluded the first 25 and 10 nt of each ORF, respectively, to avoid counting ribosome pileups at start codons. RNA-seq read counts used full-length ORFs. Read counts are provided in Supplemental Dataset S12.
RF length distribution, 3-nt periodicity, and metagene analysis were performed using in-house Perl scripts. Comparison among replicates was evaluated with Pearson correlation and hierarchical clustering using log10 reads per kilobase per million reads mapped to nuclear coding sequences (RPKM) values. Transcript coverage and 3′-bias were analyzed using Picard Tool version 2.6.0 (http://broadinstitute.github.io/picard/). Samtools (Li et al., 2009) was used to extract gene-specific read coverages. P-site assignment of ribo-seq reads was inferred from the RF size-dependent placements of footprint 5′-ends to the P-site location, which was observed from metagene analysis of reads mapping to start codons in the data discussed here.
MicroRNA data were trimmed with cutadapt (Martin, 2011). Trimmed reads between 15 and 45 nucleotides were retained and aligned to the B73 RefGen_v4 assembly using STAR version 2.5.3a (Dobin et al., 2013). For multi-mapped reads, STAR parameters were set to distribute each multi-mapped read randomly to one of the mapped locations (–outMultimapperOrder Random –outSAMmultNmax 1). Read counting was performed using featureCounts (Liao et al., 2014) with maize microRNA annotation release 22 (mirbase.org), where counting included multi-mapped reads. The read counts are provided in Supplemental Dataset S11.
Differential expression analysis
Differential expression analysis based on RNA-seq, ribo-seq, or microRNA data was performed using DESeq2 (Love et al., 2014). Outputs are reported in Supplemental Dataset S5, Supplemental Dataset S11 and Supplemental Dataset S13. Genes were reported as differentially expressed if they showed an average of at least 100 reads in at least one of the tissue types, a |log2 BS/M| > 1 and FDR < 0.001 for RNA-seq and ribo-seq or FDR < 0.05 for microRNA data.
For analysis of differential TE, we used XTAIL (Xiao et al., 2016). Only genes with an average of at least 100 RNA-seq reads in both tissue types and an average of at least 100 ribo-seq reads in at least one of the tissue types were reported. Genes were reported as differentially translated if they showed |log2(BS/M) TE| > 1.585 with FDR < 0.001. The XTAIL output is provided in Supplemental Dataset S1.
UTR sequence motif analysis
UTRs from genes that are differentially translated (107 genes in BS set and 75 genes in M set; |log2(BS/M) TE| > 1.585 excluding the buffering genes) were compared with one another and with a set of nondifferentially translated genes (Control set; 5280 genes; |log2(BS/M) TE| ≤ 0.585). The UTR sequences were downloaded from the BioMart of Ensembl Plants release 50 (https://plants.ensembl.org/). Missing UTR sequences for genes in the BS and M sets were manually extracted from GenBank. Analyses used the longest annotated UTR sequences for each gene. Differences in UTR lengths and GC content were analyzed with a nonparametric one-way analysis of variance test, the Kruskal–Wallis Test, followed by Dunn’s multiple comparisons test for all possible pairs using Prism 8 (GraphPad, San Diego, CA, USA). Motif discovery was performed using STREME (Bailey, 2021) with a motif width of 4–25 nt for the BS or M sets against the Control set. Motifs with P < 0.05 were retained. To remove known DNA motifs and identify known RNA motifs, the motifs were searched against three DNA and RNA motif datasets (JASPAR CORE, CISBP-RNA Z. mays and Arabidopsis DAP motifs) using TOMTOM (Gupta et al., 2007) based on Euclidean distance. No known RNA motifs were detected. DNA motifs that produced any match with a P ˂ 0.07 were removed. Motif enrichment analysis was performed using AME (McLeay and Bailey, 2010) for the BS or M sets against the Control set. FIMO (Grant et al., 2011) was used to find motif positions in UTR sequences and only the top score FIMO sites corresponding to the number of sites found by AME were used. The motif analysis data are provided in Supplemental Dataset S3.
uORF analysis
Genes with |log2(BS/M) TE| > 1 and FDR < 0.001 excluding the buffering genes (n = 335) were screened manually for ribo-seq reads in the 5′-UTR. P-site assignments and identification of uORFs beginning with AUG, CUG, or ACG were determined for those genes with substantial ribo-seq reads in the UTR. Genes that show P-site coverage in predicted uORFs are reported in Supplemental Dataset S4. uORF read count was performed using featureCounts (Liao et al., 2014), counting only those reads that mapped to a unique site and excluding the 25-nt region upstream of the main ORF to avoid counting ribosome pileups at start codons. The total “TE” of all uORFs in an mRNA was calculated from the normalized ribo-seq abundance (number of ribo-seq reads mapped to any uORF of the gene per total uORF kilobase per million reads mapped to nuclear coding sequences) divided by the normalized mRNA abundance (mORF RNA-seq RPKM).
Functional-enrichment analysis
The enrichment analysis was performed with MapMan (Thimm et al., 2004) as described (Zones et al., 2015), using maize X4.2 functional assignments and Mapman terms Levels 1–3. In brief, a set of genes showing an average ribo-seq RPKM value >1 in at least one tissue type (18,980 genes) was used to calculate background distribution of each MapMan term among the expressed gene set. To evaluate a pair of BS and M datasets of sizes m and n genes, a total of 10,000 permutations by random sampling without replacement for pair sets of m and n genes from the expressed gene pool were performed. The term occurrences in these 10,000 permutated dataset pairs produced the mean and standard deviation (sd) for each term occurrence in the background. The observed term occurrence in each dataset together with the mean and sd of the term occurrence in the background from the corresponding permutation set were used to calculate z-score and P-value. The adjusted P-values or FDR values were calculated using the Benjamini–Hochberg method in R as described previously (Zones et al., 2015). The output is provided in Supplemental Dataset S8.
Reanalysis of external data
Previously published RNA-seq analyses of maize BS and M fractions used noncurrent maize genome assemblies and gene models. For comparison to our data, we re-analyzed the +9 cm data (also called section 14) from Tausta et al. (2014) and the slice 1 data from Denton et al. (2017) using our pipeline. To compare our ribo-seq data to proteome data from Friso et al. (2010), those data were matched to B73 version 4 gene identifiers using the gene ID history (gramene.org). For comparison to our differential translatomes in Figures 3E, 5, and 6; Supplemental Figures S6 and S7, the originally reported BS/M values were compiled from the BS and M data from Li et al. (2010), Chang et al. (2012), the section 14 data from Tausta et al. (2014), and the slice 1 data from Denton et al. (2017). The values reported with older gene IDs were matched using the gene ID history (gramene.org).
Antibodies
Antibodies to NdhH and PPDK were generously provided by Tsuyoshi Endo (Kyoto University) and Kazuko Aoyagi (University of California, Berkeley), respectively. Antibodies to PEPC, malic enzyme, and Rubisco large subunit (RbcL) were gifts of William Taylor (University of California, Berkeley). The antibody to the D2 subunit of PSII was obtained from Agrisera.
Accession numbers
The ribo-seq and RNA-seq data were deposited at the NCBI Sequence Read Archive (SRA) with accession number PRJNA667075. RNA-seq data from Tausta et al. (2014) and Denton et al. (2017) were downloaded from SRA (accession numbers SRP035577 and SRP052802, respectively). Alignments of reads to the maize chloroplast genome used Genbank accession X86563. B73 RefGen_v4 assembly with annotation release 38 (gramene.org) was used for alignments to the nuclear and mitochondrial genomes.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1 Characteristics of ribo-seq and RNA-seq data from BS and M fractions.
Supplemental Figure S2 Comparison of RNA-seq data characteristics with those from prior studies.
Supplemental Figure S3 Comparison of RNA-seq, ribo-seq, and proteome data collected for maize BS and M leaf fractions.
Supplemental Figure S4 Comparison of our RNA-seq and prior RNA-seq data for genes exhibiting preferential translation in M or BS fractions.
Supplemental Figure S5 Normalized read coverage of translationally regulated genes.
Supplemental Figure S6 Differential expression of eIF1, eIF5, and eIF5A genes.
Supplemental Figure S7 Differential expression of biogenesis factors for PSII, Rubisco, and the NDH complex.
Supplemental Figure S8 Alignment of microRNA sequences of zma-MIR160 and zma-MIR166 families.
Supplemental Dataset S1 . XTAIL output and related data.
Supplemental Dataset S2 . Genes reported by XTAIL as having a greater than three-fold difference in TE between BS and M fractions.
Supplemental Dataset S3 . Sequence motif enrichment data.
Supplemental Dataset S4 . Data on uORF translation for differentially translated genes.
Supplemental Dataset S5 . DESeq2 output of ribo-seq data.
Supplemental Dataset S6 . BS-enriched genes in ribo-seq data.
Supplemental Dataset S7 . M-enriched genes in ribo-seq data.
Supplemental Dataset S8 . Enrichment of MapMan terms in gene sets that are differentially expressed in BS versus M fractions based on ribo-seq data.
Supplemental Dataset S9 . Differential expression of nucleus-encoded proteins involved in expression and assembly of Rubisco, NDH-like, and PSII complexes.
Supplemental Dataset S10 . Data for TF genes that are differentially expressed based on ribo-seq data.
Supplemental Dataset S11 . MicroRNA data.
Supplemental Dataset S12 . Ribo-seq and RNA-seq read counts, including re-analyzed data from Denton S1 and Tausta Sec 14 samples.
Supplemental Dataset S13. DESeq2 outputs of RNA-seq data, including re-analyzed data from Denton S1 and Tausta Sec 14 samples.
Supplementary Material
Acknowledgments
We wish to thank Rosalind Williams-Carrier for assistance with sample preparation and for comments on the manuscript, Susan Belcher for comments on the manuscript, the University of Oregon Genomics and Cell Characterization Core Facility for Illumina sequencing, and the University of Oregon Research Advanced Computing Services for access to the high-performance computer Talapas.
Funding
This work was supported by grant IOS-1339130 to A.B. from the US National Science Foundation.
Conflict of interest statement. None declared.
Conceived and designed the experiments: P.C. and A.B. Performed the experiments: P.C. Analyzed the data: P.C. and A.B. Contributed reagents/materials/analysis tools: P.C. Wrote the paper: P.C. and A.B.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plphys/pages/general-instructions) is: Alice Barkan (abarkan@uoregon.edu).
References
- Aubry S, Kelly S, Kumpers BM, Smith-Unna RD, Hibberd JM (2014) Deep evolutionary comparison of gene expression identifies parallel recruitment of trans-factors in two independent origins of C4 photosynthesis. PLoS Genet 10: e1004365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL (2021) STREME: accurate and versatile sequence motif discovery. Bioinformatics btab203; https://doi.org/10.1093/bioinformatics/btab203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry JO, Mure CM, Yerramsetty P (2016) Regulation of Rubisco gene expression in C4 plants. Curr Opin Plant Biol 31: 23–28 [DOI] [PubMed] [Google Scholar]
- Brar GA, Weissman JS (2015) Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol 16: 651–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräutigam A, Hoffmann-Benning S, Weber AP (2008) Comparative proteomics of chloroplast envelopes from C3 and C4 plants reveals specific adaptations of the plastid envelope to C4 photosynthesis and candidate proteins required for maintaining C4 metabolite fluxes. Plant Physiol 148: 568–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bräutigam A, Kajala K, Wullenweber J, Sommer M, Gagneul D, Weber KL, Carr KM, Gowik U, Mass J, Lercher MJ, et al. (2011) An mRNA blueprint for C4 photosynthesis derived from comparative transcriptomics of closely related C3 and C4 species. Plant Physiol 155: 142–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YM, Liu WY, Shih AC, Shen MN, Lu CH, Lu MY, Yang HW, Wang TY, Chen SC, Chen SM, et al. (2012) Characterizing regulatory and functional differentiation between maize mesophyll and bundle sheath cells by transcriptomic analysis. Plant Physiol 160: 165–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Zhu XG, Lin Y (2014) Major alterations in transcript profiles between C3-C4 and C4 photosynthesis of an amphibious species Eleocharis baldwinii. Plant Mol Biol 86: 93–110 [DOI] [PubMed] [Google Scholar]
- Chotewutmontri P, Barkan A (2016) Dynamics of chloroplast translation during chloroplast differentiation in maize. PLoS Genet 12: e1006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotewutmontri P, Barkan A (2020) Light-induced psbA translation in plants is triggered by photosystem II damage via an assembly-linked autoregulatory circuit. Proc Natl Acad Sci USA 117: 21775–21784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chotewutmontri P, Stiffler N, Watkins KP, Barkan A (2018) Ribosome profiling in maize. Methods Mol Biol 1676: 165–183 [DOI] [PubMed] [Google Scholar]
- Chotewutmontri P, Williams-Carrier R, Barkan A (2020) Exploring the link between photosystem II assembly and translation of the chloroplast psbA mRNA. Plants 9: pii: E152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covshoff S, Szecowka M, Hughes TE, Smith-Unna R, Kelly S, Bailey KJ, Sage TL, Pachebat JA, Leegood R, Hibberd JM (2016) C4 photosynthesis in the rice paddy: insights from the noxious weed Echinochloa glabrescens. Plant Physiol 170: 57–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedow LK, Bailey-Serres J (2019) Searching for a match: structure, function and application of sequence-specific RNA-binding proteins. Plant Cell Physiol 60: 1927–1938 [DOI] [PubMed] [Google Scholar]
- Denton AK, Mass J, Kulahoglu C, Lercher MJ, Bräutigam A, Weber AP (2017) Freeze-quenched maize mesophyll and bundle sheath separation uncovers bias in previous tissue-specific RNA-Seq data. J Exp Bot 68: 147–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Weissmann S, Wang M, Du B, Huang L, Wang L, Tu X, Zhong S, Myers C, Brutnell TP, et al. (2015) Identification of photosynthesis-associated C4 candidate genes through comparative leaf gradient transcriptome in multiple lineages of C3 and C4 species. PLoS One 10: e0140629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning LT, Moreno-Villena JJ, Lundgren MR, Dionora J, Salazar P, Adams C, Nyirenda F, Olofsson JK, Mapaura A, Grundy IM, et al. (2019) Key changes in gene expression identified for different stages of C4 evolution in Alloteropsis semialata. J Exp Bot 70: 3255–3268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg AR, Higdon AL, Hollerer I, Fields AP, Jungreis I, Diamond PD, Kellis M, Jovanovic M, Brar GA (2020) Translation initiation site profiling reveals widespread synthesis of non-AUG-initiated protein isoforms in yeast. Cell Syst 11: 145–160 e145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermakova M, Danila FR, Furbank RT, von Caemmerer S (2020) On the road to C4 rice: advances and perspectives. Plant J 101: 940–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei Y, Wang R, Li H, Liu S, Zhang H, Huang J (2018) DPMIND: degradome-based plant miRNA-target interaction and network database. Bioinformatics 34: 1618–1620 [DOI] [PubMed] [Google Scholar]
- Friso G, Majeran W, Huang M, Sun Q, van Wijk KJ (2010) Reconstruction of metabolic pathways, protein expression, and homeostasis machineries across maize bundle sheath and mesophyll chloroplasts: large-scale quantitative proteomics using the first maize genome assembly. Plant Physiol 152: 1219–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstrohm AC, Hall TMT, McKenney KM (2018) Post-transcriptional regulatory functions of mammalian pumilio proteins. Trends Genet 34: 972–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowik U, Brautigam A, Weber KL, Weber AP, Westhoff P (2011) Evolution of C4 photosynthesis in the genus Flaveria: how many and which genes does it take to make C4? Plant Cell 23: 2087–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS (2007) Quantifying similarity between motifs. Genome Biol 8: R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch M (1987) C4 photosynthesis: a unique blend of modified biochemistry, anatomy and ultrastructure. BBA Rev Bioenergetic 895: 81–106 [Google Scholar]
- Hinnebusch AG, Ivanov IP, Sonenberg N (2016) Translational control by 5'-untranslated regions of eukaryotic mRNAs. Science 352: 1413–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, , Brutnell TP (2016) A synthesis of transcriptomic surveys to dissect the genetic basis of C4 photosynthesis. Curr Opin Plant Biol 31: 91–99 [DOI] [PubMed] [Google Scholar]
- Huang P, Studer AJ, Schnable JC, Kellogg EA, Brutnell TP (2017) Cross species selection scans identify components of C4 photosynthesis in the grasses. J Exp Bot 68: 127–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John CR, Smith-Unna RD, Woodfield H, Covshoff S, Hibberd JM (2014) Evolutionary convergence of cell-specific gene expression in independent lineages of C4 grasses. Plant Physiol 165: 62–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshna CR, Saha P, Atugala D, Chua G, Muench DG (2020) Plant PUF RNA-binding proteins: A wealth of diversity for post-transcriptional gene regulation. Plant Sci 297: 110505. [DOI] [PubMed] [Google Scholar]
- Juarez MT, Kui JS, Thomas J, Heller BA, Timmermans MC (2004) microRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature 428: 84–88 [DOI] [PubMed] [Google Scholar]
- Külahoglu C, Denton AK, Sommer M, Mass J, Schliesky S, Wrobel TJ, Berckmans B, Gongora-Castillo E, Buell CR, Simon R, et al. (2014) Comparative transcriptome atlases reveal altered gene expression modules between two Cleomaceae C3 and C4 plant species. Plant Cell 26: 3243–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterbach M, Schmidt H, Billakurthi K, Hankeln T, Westhoff P, Gowik U, Kadereit G (2017) De novo transcriptome assembly and comparison of C3, C3-C4, and C4 species of tribe Salsoleae (Chenopodiaceae). Front Plant Sci 8: 1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, Kebrom TH, Provart N, Patel R, Myers CR, et al. (2010) The developmental dynamics of the maize leaf transcriptome. Nat Genet 42: 1060–1067 [DOI] [PubMed] [Google Scholar]
- Li Y, Ma X, Zhao J, Xu J, Shi J, Zhu XG, Zhao Y, Zhang H (2015) Developmental genetic mechanisms of C4 syndrome based on transcriptome analysis of C3 cotyledons and C4 assimilating shoots in Haloxylon ammodendron. PLoS One 10: e0117175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, Liu MJ (2020) Prevalence of alternative AUG and non-AUG translation initiators and their regulatory effects across plants. Genome Res 30: 1418–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) Feature counts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Link S, Engelmann K, Meierhoff K, Westhoff P (2012) The atypical short-chain dehydrogenases HCF173 and HCF244 are jointly involved in translational initiation of the psbA mRNA of Arabidopsis. Plant Physiol 160: 2202–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WY, Chang YM, Chen SC, Lu CH, Wu YH, Lu MY, Chen DR, Shih AC, Sheue CR, Huang HC et al. (2013) Anatomical and transcriptional dynamics of maize embryonic leaves during seed germination. Proc Natl Acad Sci USA 110: 3979–3984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Zhou JJ, Zhang JZ (2018) Aux/IAA gene family in plants: molecular structure, regulation, and function. Int J Mol Sci 19: 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeran W, Cai Y, Sun Q, van Wijk KJ (2005) Functional differentiation of bundle sheath and mesophyll maize chloroplasts determined by comparative proteomics. Plant Cell 17: 3111–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeran W, Friso G, Ponnala L, Connolly B, Huang M, Reidel E, Zhang C, Asakura Y, Bhuiyan NH, Sun Q, et al. (2010) Structural and metabolic transitions of C4 leaf development and differentiation defined by microscopy and quantitative proteomics in maize. Plant Cell 22: 3509–3542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeran W, van Wijk KJ (2009) Cell-type-specific differentiation of chloroplasts in C4 plants. Trends Plant Sci 14: 100–109 [DOI] [PubMed] [Google Scholar]
- Majeran W, Zybailov B, Ytterberg AJ, Dunsmore J, Sun Q, van Wijk KJ (2008) Consequences of C4 differentiation for chloroplast membrane proteomes in maize mesophyll and bundle sheath cells. Mol Cell Proteomics 7: 1609–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunath H, Zhang H, Rehfeld F, Han J, Chang TC, Mendell JT (2019) Suppression of ribosomal pausing by eIF5A is necessary to maintain the fidelity of start codon selection. Cell Rep 29: 3134–3146 e3136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17: 10–12 [Google Scholar]
- Mattiello L, Riano-Pachon DM, Martins MC, da Cruz LP, Bassi D, Marchiori PE, Ribeiro RV, Labate MT, Labate CA, Menossi M (2015) Physiological and transcriptional analyses of developmental stages along sugarcane leaf. BMC Plant Biol 15: 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeay RC, Bailey TL (2010) Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11: 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CJ, May GE, Spealman P, Shteyman A (2014) Ribosome profiling reveals post-transcriptional buffering of divergent gene expression in yeast. Genome Res 24: 422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda JS, Cheung YN, Takacs JE, Martin-Marcos P, Saini AK, Hinnebusch AG, Lorsch JR (2009. ) eIF1 controls multiple steps in start codon recognition during eukaryotic translation initiation. J Mol Biol 394: 268–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JM, Lane B, Freeling M (2002) Expression of a mutant maize gene in the ventral leaf epidermis is sufficient to signal a switch of the leaf's dorsoventral axis. Development 129: 4581–4589 [DOI] [PubMed] [Google Scholar]
- Oertlin C, Lorent J, Murie C, Furic L, Topisirovic I, Larsson O (2019) Generally applicable transcriptome-wide analysis of translation using anota2seq. Nucleic Acids Res 47: e70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offermann S, Friso G, Doroshenk KA, Sun Q, Sharpe RM, Okita TW, Wimmer D, Edwards GE, van Wijk KJ (2015) Developmental and subcellular organization of single-cell C(4) photosynthesis in bienertia sinuspersici determined by large-scale proteomics and cDNA assembly from 454 DNA sequencing. J Proteome Res 14: 2090–2108 [DOI] [PubMed] [Google Scholar]
- Patel M, Siegel AJ, Berry JO (2006) Untranslated regions of FbRbcS1 mRNA mediate bundle sheath cell-specific gene expression in leaves of a C4 plant. J Biol Chem 281: 25485–25491 [DOI] [PubMed] [Google Scholar]
- Pick TR, Brautigam A, Schluter U, Denton AK, Colmsee C, Scholz U, Fahnenstich H, Pieruschka R, Rascher U, Sonnewald U, et al. (2011) Systems analysis of a maize leaf developmental gradient redefines the current C4 model and provides candidates for regulation. Plant Cell 23: 4208–4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnala L, Wang Y, Sun Q, van Wijk KJ (2014) Correlation of mRNA and protein abundance in the developing maize leaf. Plant J 78: 424–440 [DOI] [PubMed] [Google Scholar]
- Rao X, Lu N, Li G, Nakashima J, Tang Y, Dixon RA (2016) Comparative cell-specific transcriptomics reveals differentiation of C4 photosynthesis pathways in switchgrass and other C4 lineages. J Exp Bot 67: 1649–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage RF, Christin PA, Edwards EJ (2011) The C(4) plant lineages of planet Earth. J Exp Bot 62: 3155–3169 [DOI] [PubMed] [Google Scholar]
- Schafer S, Adami E, Heinig M, Rodrigues KEC, Kreuchwig F, Silhavy J, van Heesch S, Simaite D, Rajewsky N, Cuppen E, et al. (2015) Translational regulation shapes the molecular landscape of complex disease phenotypes. Nat Commun 6: 7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter U, Bräutigam A, Gowik U, Melzer M, Christin PA, Kurz S, Mettler-Altmann T, Weber AP (2017) Photosynthesis in C3-C4 intermediate Moricandia species. J Exp Bot 68: 191–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter U, Weber APM (2020) Regulation and evolution of C4 photosynthesis. Annu Rev Plant Biol 71: 183–215 [DOI] [PubMed] [Google Scholar]
- Schult K, Meierhoff K, Paradies S, Toller T, Wolff P, Westhoff P (2007) The nuclear-encoded factor HCF173 is involved in the initiation of translation of the psbA mRNA in Arabidopsis thaliana. Plant Cell 19: 1329–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova OV, Hughes TE, Langdale JA (2018) Understanding the genetic basis of C4 Kranz anatomy with a view to engineering C3 crops. Annu Rev Genet 52: 249–270 [DOI] [PubMed] [Google Scholar]
- Tausta SL, Li P, Si Y, Gandotra N, Liu P, Sun Q, Brutnell TP, Nelson T (2014) Developmental dynamics of Kranz cell transcriptional specificity in maize leaf reveals early onset of C4-related processes. J Exp Bot 65: 3543–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O, Blasing O, Gibon Y, Nagel A, Meyer S, Kruger P, Selbig J, Muller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37: 914–939 [DOI] [PubMed] [Google Scholar]
- van der Horst S, Snel B, Hanson J, Smeekens S (2019) Novel pipeline identifies new upstream ORFs and non-AUG initiating main ORFs with conserved amino acid sequences in the 5' leader of mRNAs in Arabidopsis thaliana. RNA 25: 292–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Czedik-Eysenberg A, Mertz RA, Si Y, Tohge T, Nunes-Nesi A, Arrivault S, Dedow LK, Bryant DW, Zhou W, et al. (2014) Comparative analyses of C(4) and C(3) photosynthesis in developing leaves of maize and rice. Nat Biotechnol 32: 1158–1165 [DOI] [PubMed] [Google Scholar]
- Wang M, Oge L, Perez-Garcia MD, Hamama L, Sakr S (2018) The PUF protein family: overview on PUF RNA targets, biological functions, and post transcriptional regulation. Int J Mol Sci 19: 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Fouracre J, Kelly S, Karki S, Gowik U, Aubry S, Shaw MK, Westhoff P, Slamet-Loedin IH, Quick WP, et al. (2013a) Evolution of GOLDEN2-LIKE gene function in C(3) and C (4) plants. Planta 237: 481–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Kelly S, Fouracre JP, Langdale JA (2013b) Genome-wide transcript analysis of early maize leaf development reveals gene cohorts associated with the differentiation of C4 Kranz anatomy. Plant J 75: 656–670 [DOI] [PubMed] [Google Scholar]
- Wang P, Vlad D, Langdale JA (2016) Finding the genes to build C4 rice. Curr Opin Plant Biol 31: 44–50 [DOI] [PubMed] [Google Scholar]
- Williams BP, Burgess SJ, Reyna-Llorens I, Knerova J, Aubry S, Stanley S, Hibberd JM (2016) An untranslated cis-element regulates the accumulation of multiple C4 enzymes in Gynandropsis gynandra mesophyll cells. Plant Cell 28: 454–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Zou Q, Liu Y, Yang X (2016) Genome-wide assessment of differential translations with ribosome profiling data. Nat Commun 7: 11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Paschold A, Marcon C, Liu S, Tai H, Nestler J, Yeh CT, Opitz N, Lanz C, Schnable PS, et al. (2014) The Aux/IAA gene rum1 involved in seminal and lateral root formation controls vascular patterning in maize (Zea mays L.) primary roots. J Exp Bot 65: 4919–4930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zones JM, Blaby IK, Merchant SS, Umen JG (2015) High-resolution profiling of a synchronized diurnal transcriptome from Chlamydomonas reinhardtii reveals continuous cell and metabolic differentiation. Plant Cell 27: 2743–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.