

Abstract
Background
The modification 6‐methyladenine (m6A) is the most common type in RNA methylation. Our study aims to explore the bioinformatic analysis of m6A in endometrial cancer.
Methods
The expression of 23 m6A RNA methylation regulators was compared through The Cancer Genome Atlas (TCGA) database among 406 endometrial tissue and 19 normal tissue samples. The Wilcoxon test was applied to compare the relationship between the clinicopathological characteristics and expression. Cox regressions were performed to identify the prognostic factors associated with overall survival. Gene ontology (GO) and Gene Set Enrichment Analysis (GSEA) were performed to evaluate the potential pathways.
Results
YTHDF2, HNRNPA2B1, HDRNPA2B1, YTHDF1, FMR1, IGF2BP3, METTL13, RBM15B, IGF2BP1, YTHDF3, YTHDC1, ZC3H13 IGF2BP2, KIAA1429, METTL14, RBMX, FTO, ALKBH5, and METTL16 were significantly abnormally expressed in endometrial cancer tissue samples. Both univariate and multivariate Cox regression analyses indicated that age, grade, and risk score were independent risk factors. High expression of FTO was associated with worse overall survival.
Conclusion
M6A RNA methylation regulators play vital role in endometrial cancer.
Keywords: bioinformatic analysis, endometrial cancer, methylation
The heatmap shows the expression levels of three 6RNA regulators in high and low‐risk groups (A). Univariate (B) and multivariate (C) Cox regression analyses indicated that ages, grades, and risk scores were relevant to prognosis.

1. INTRODUCTION
In 2013, there were an estimated 49,560 uterine cancer cases and 8,190 deaths1; by 2018, there were an estimated 63,230 endometrial cancer cases and 11,350 deaths.2, 3 Both the incidences and mortality of endometrial cancer in the United States have been rapidly rising in recent years. It has been suggested that these increases are due in part to declining rates of hysterectomy for benign causes.4
More than 150 types of chemical modifications have been identified in RNA molecules,5, 6 and methylation accounts for 66% of known RNA modifications.7 The RNA modification 6‐methyladenine (m6A) was first identified in 19758 and was the most prevalent type.9, 10 RNA modifications were identified in nearly every aspect of the mRNA life cycle, as well as in various cellular, developmental, and cancer processes.5, 11 The effects from m6A are determined by three homologous factors, including “writers” (including METTL3, METTL14, METTL16, WTAP, KIAA1429, ZC3H13, RBM15, and RBM15B), “readers” (including YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3, HNRNPC, FMR1, LRPPRC, HNRNPA2B1, IGFBP1, IGFBP2, IGFBP3, and RBMX), and “erasers” (including FTO and ALKBH5.12, 13, 14 Although RNA modifications have been mostly identified over the past decade, the biological functions and clinical value of endometrial cancer remain unclear.
In this article, we explore the function and therapeutic advances of m6A modifications in endometrial cancer and attempt to find potential biological molecules for targeted therapy. The cancer genome mapping (TCGA) database was used to compare the expression of 23 m6A RNA methylation regulators between endometrial cancer tissues and normal tissues. Gene Set Enrichment Analysis (GSEA) was used to explore the potential mechanisms of methylation regulators.
2. MATERIALS AND METHODS
2.1. Datasets and study cohort
RNA‐seq transcriptome data and clinical data from 406 endometrial cancer patients and 19 normal tissue samples were downloaded from TCGA database (http://www.cancergenome.nih.gov/). All data were normalized by the expectation‐maximization method and transformed into Sample IDs by Perl. Incomplete survival information from patients was excluded.23 m6A RNA methylation regulators were selected for subsequent bioinformatics analysis, including ALKBH5, FTO, HNRNPC, HNRNPA2B1, KIAA1429, METTL14, METTL3, METTL16, RBM15, RBMX, RBM15B, WTAP, YTHDC1, YTHDC2, YTHDF1, YTHDF2, ZC3H13, IGF2BP1, IGF2BP2, IGF2BP3, FMR1, and LRPPRC.
2.2. Bioinformatic analysis
Limma package was used to explore the expressions of m6A RNA methylation regulators among endometrial cancer using R v3.6.1. The 23 m6A RNA methylation regulators were compared to the expressions between tumor and normal tissues through heatmap diagram and vioplot diagram. Corrplot was conducted to analyze the correlations between m6A RNA methylation regulators. Forestplot was conducted to select potential prognostic regulators for LASSO Cox regression. Univariate and multivariate Cox regression analyses were conducted to evaluate the risk scores. We chose three m6A RNA methylation regulators to compare the paired expressions between normal tissues and endometrial cancer tissues. The levels of expression for the three m6A RNA methylation regulators were also compared according to grade statuses. The high‐ and low‐expression groups were compared with prognostic condition. Gene ontology (GO) was conducted with the R packages. Gene Set Enrichment Analysis (GSEA) was performed to study the functions of the three m6A RNA methylation regulators by using “kegg.v7.1 symbol.gmt” as a gene set database.
2.3. Statistical analysis
The Wilcoxon test was applied to explore the differences in expression of m6A RNA methylation regulators between endometrial cancer patients and normal tissues. The relationship between the clinicopathological variables and risk scores was compared through the chi‐square test. The median risk scores were applied to classify patients into low‐risk and high‐risk groups. Univariate and multivariate Cox regression analyses were conducted to evaluate the relationship between clinicopathological variables and risk scores. The t test was conducted to compare the differences between low‐risk and high‐risk groups. The significant statistics were considered with p < 0.05. The fold change<−2 (p < 0.05) or fold change >2 (p < 0.05) was applied to select the genes for GO analysis.
3. RESULTS
3.1. Expression of m6A RNA methylation regulators
The levels of expression for the 23 m6A RNA methylation regulators were compared between 406 endometrial cancer patients and 19 normal tissue samples in the TCGA database. Compared to normal tissues, endometrial cancer patients possess greater expressions of YTHDF2, HNRNPA2B1, HDRNPA2B1, YTHDF1, FMR1, IGF2BP3, METTL13, RBM15B, and IGF2BP1 and low expression of YTHDF3, YTHDC1, ZC3H13, IGF2BP2, KIAA1429, METTL14, RBMX, FTO, ALKBH5, and METTL16 (Figure 1A,B). YTHDF3 and KIAA1429 (correlation score of 0.66) showed the most significant positive correlation according to the Spearman correlation analysis (Figure 1C).
FIGURE 1.
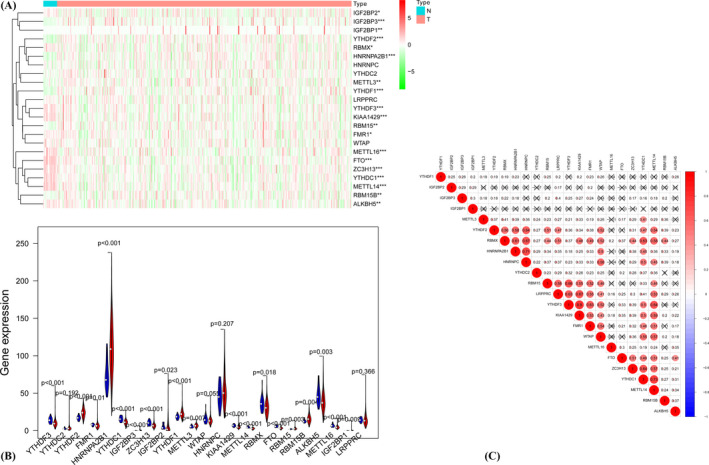
Expression heatmap of 23 m6A RNA methylation regulations in tumor tissue and normal tissue. *p < 0.05, **p < 0.01,***p < 0.001 (A). Violet plots of 23 regulation factors with the comparison between tumor tissue and normal issue (B). Spearman's correlation analysis of m6A RNA methylation regulators (C)
3.2. Prognostic role of m6A RNA methylation regulators
We performed the univariate Cox regression analysis to evaluate the prognostic values of 23 m6A RNA methylation regulators (Figure 2A). To select the most significant m6A prognostic regulators, we set the criteria as p < 0.1. FTO, RBMX, and YTHDF1 were selected for the LASSO Cox, and the coefficients were calculated to separate patients into low‐risk and high‐risk groups. The high‐risk group showed worse survival chances than the low‐risk group (Figure 2B).
FIGURE 2.

Univariate Cox regression analysis showed the p value, hazard ratio (HR), and 95% confidence interval (CI) with prognostic value of 23 m6A RNA methylation regulators (A). Kaplan‐Meier curve showed the overall survival of high‐ and low‐risk group according to the risk score (B)
3.3. Clinicopathological features of the three m6A RNA methylation regulators
The clinicopathological characteristics of the three m6A RNA methylation regulators vary differently according to the heat map (Figure 3A). The ages, grades, and risk scores were relevant to prognosis according to both univariate and multivariate Cox regression analyses (Figure 3B,C).
FIGURE 3.
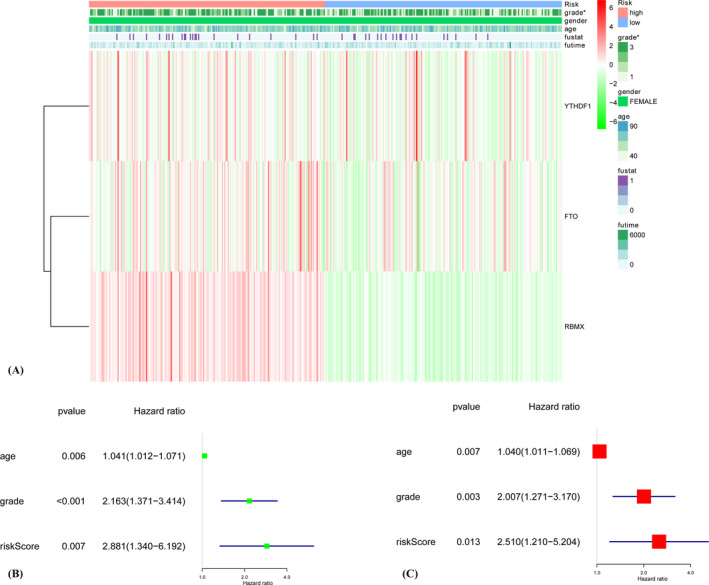
The heatmap shows the expression levels of three m6A RNA regulators in high‐ and low‐risk groups (A). The association between clinicopathological factors and overall survival of breast cancer patients through univariate (B) and multivariate (C) Cox regression analyses
The consequences of expression for FTO (Figure 4A) and YTHDF1 (Figure 4C) were consistent with the situation in the paired comparison. The levels of expression for FTO were not significantly correlated with grades (Figure 4D), while the patients with high grades tended to have high expressions of RBMX (Figure 4E) and YTHDF1 (Figure 4F). Survival analysis indicated that high FTO expression was associated with worse overall survival (Figure 4G). The survival differences between the low‐expression and high‐expression groups of RBMX and YTHDF1 were not detected.
FIGURE 4.
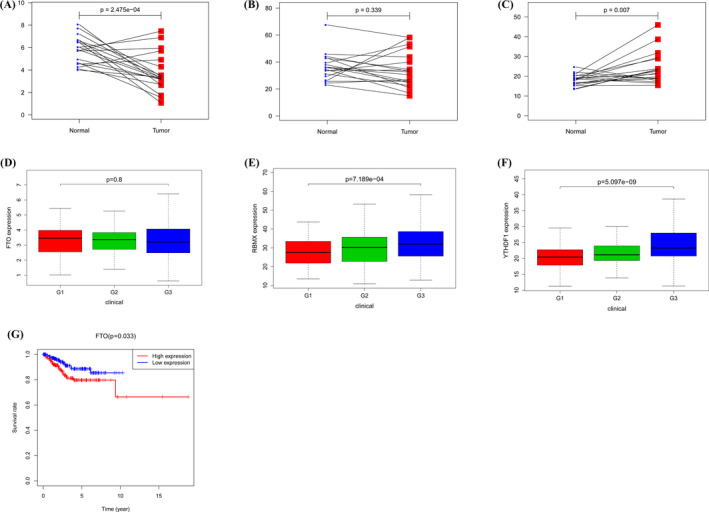
The results of expression of three m6A RNA regulators in paired breast cancer and normal specimens (A–C). C: Differential expression of three m6A RNA regulators in different grades (D–F). Kaplan‐Meier curves showed the prognostic value of FTO on overall survival (G)
GO analysis showed that these m6A RNA methylation regulators worked on regulating the mRNA metabolic process, RNA catabolic process, mRNA stability, and so on (Figure 5). The GSEA analysis was conducted to identify associated KEGG pathways. We selected significantly enriched signaling pathways based on their normalized enrichment score (NES) and normalized p value. FTO was enriched in TGF‐β signaling pathway (NES = 2.33, p < 0.001), ERBB signaling pathway (NES = 2.18, p = 0.005), WNT signaling pathway (NES = 2.34, p = 0.002), and pathways in cancer (NES = 2.07, p = 0.006). FTO was down‐regulated in oxidative phosphorylation (NES = −2.13, p = 0.010). RBMX was enriched in the cell cycle (NES = 2.45, p < 0.001), ERBB signaling pathway (NES = 2.43, p < 0.001), homologous recombination (NES = 2.29, p < 0.001), and ubiquitin‐mediated proteolysis (NES = 2.55, p < 0.001). YTHDF1 was enriched in cell cycle (NES = 2.42, p < 0.001), endocytosis (NES = 2.28, p < 0.001), ubiquitin‐mediated proteolysis (NES = 2.51, p < 0.001), and ERBB signaling pathway (NES = 2.40, p < 0.001)(Figure 6).
FIGURE 5.
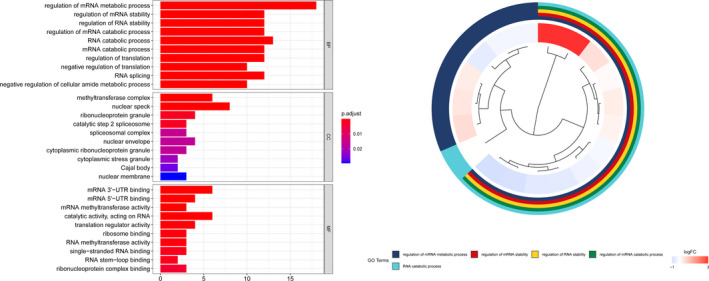
Gene Ontology analysis of the differential expression genes
FIGURE 6.
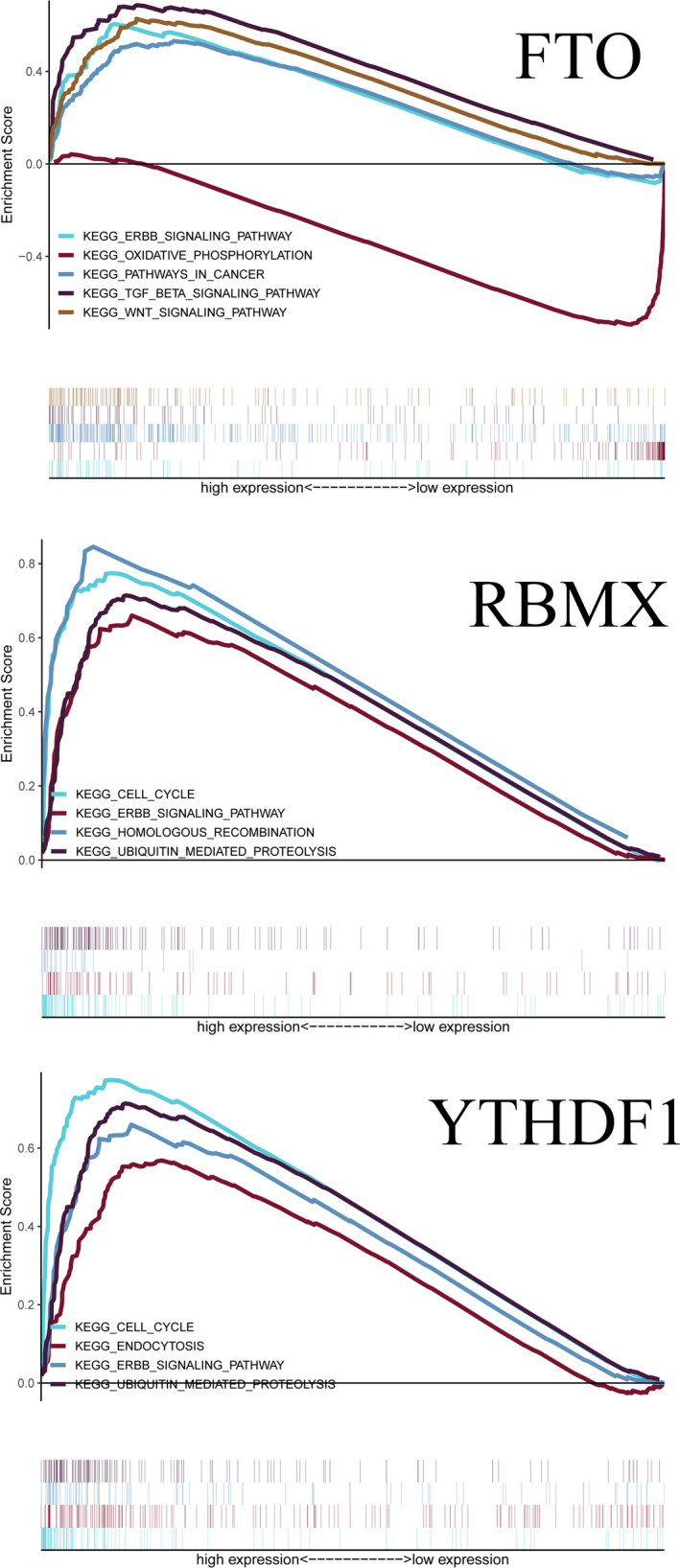
Enrichment plots from gene set enrichment analysis
4. DISCUSSION
Endometrial cancer is one of the most common gynecologic tumors.15 m6A RNA methylation is an important post‐transcriptional factor for tumor occurrence and development.10 Database‐based bioinformatic analysis helps find potential biological molecules for targeted therapy. Therefore, we conducted this analysis to explore potential molecular mechanisms between RNA methylation and endometrial cancer.
First, the expressions of the 23 m6A RNA methylation regulators were compared between endometrial cancer tissues and normal tissues. YTHDF2, HDRNPA2B1, YTHDF1, FMR1, IGF2BP3, METTL13, RBM15B, and IGF2BP1 were up‐regulated when endometrial cancer was present, while YTHDF3, YTHDC1, ZC3H13, IGF2BP2, KIAA1429, METTL14, RBMX, FTO, ALKBH5, and METTL16 were down‐regulated. According to the LASSO Cox regression, FTO, RBMX, and YTHDF1 were selected to construct a risk signature that assigned patients into low‐risk and high‐risk groups. We found that the high‐risk group had shorter survival times when compared with the low‐risk group. Both univariate and multivariate Cox regressions showed that the age and grade were associated with prognosis. However, the pathological stage and stage TNM were not available from TCGA, so we were limited to discussing. Increased expression of RBMX and YTHDF1 correlated significantly with the high grade status. Up‐regulated FTO was correlated with poor prognosis when endometrial cancer was present. To verify our results, The Human Protein Atlas (http://www.proteinatlas.org/) and GEO database (https:// www.ncbi.nlm.nih.gov/geo/) were applied to compare regulator expressions between endometrial cancer tissues and normal tissues. In the Human Protein Atlas database, FTO and RBMX were down‐regulated in endometrial cancer when compared to normal tissues (Supplementary Figure). This aligned with our results. However, YTHDF1 was not available in the Human Protein Atlas database. The dataset GSE17025 contains 91 endometrial cancer tissue samples and 12 normal tissue samples. The heatmap showed that RBMX was down‐regulated, while YTHDF1 was up‐regulated when endometrial cancer was present (Supplementary Figure). However, the expression of FTO was not found to be any different between tumor tissue samples and normal tissue samples.
YTHDF1 was overexpressed in ovarian cancer, colorectal cancer, and hepatocellular carcinoma.16, 17, 18 In ovarian cancer cells, YTHDF1 was crucial for proliferation and metastasis. YTHDF1‐deficient mice showed an elevated antigen‐specific CD8+ T cell antitumor response, which means YTHDF1 may play a vital role in anticancer immunotherapies.19 Moreover, cell cycle, endocytosis, ubiquitin‐mediated proteolysis, and ERBB signaling pathway may be the potential pathway regulated by YTHDF1 in endometrial cancer. RBMX is a small protein gene encoded by paralogs on the mammalian Y chromosome and other chromosomes20 and was overexpressed in hepatocellular carcinoma.21 The viability and proliferation of hepatocellular carcinoma cells exhibited a positive relationship to the expression of RBMX. RBMX promotes hepatocellular carcinoma development and sorafenib resistance.21 This indicated that RBMX was a potential therapeutic target in cancer. According to GESA analysis, cell cycle, homologous recombination, ubiquitin‐mediated proteolysis, and ERBB signaling pathway may be the potential pathway regulated by RBMX in endometrial cancer. Obesity is a risk factor of endometrial cancer.22 Epidemiological investigations attributed to approximately 40% of cases due to physical inactivity and being overweight.23 FTO has been associated with obesity,24, 25 and Frayling et al.24 identified 10 single‐nucleotide polymorphisms (SNPs) in the FTO gene (represented by rs9939609). The 16% of adults who are homozygous for the at‐risk allele weighed about 3 kilograms more and had 1.67 times the chances to be obese when compared with those that did not inherit the at‐risk allele. Fischer et al.26 found that FTO was involved in energy homeostasis by controlling energy expenditure. There is no clear molecular mechanism through which FTO affects obesity‐mediated tumorigenesis and tumor progression. However, FTO seems to increase the risk of cancer by promoting obesity.27 As the prognostic m6A RNA methylation regulator, FTO has been identified as an oncogene in endometrial cancer, breast cancer, and pancreatic cancer.28, 29, 30 Niu et al.30 found that high levels of FTO were crucially associated with lower survival rates in breast cancer patients, and FTO could promote breast cancer cell proliferation, colony formation, and metastasis through epigenetically down‐regulating BNIP3 in vitro and in vivo. Accumulated adipocytes partly contributed to the increased production of estrogen, which induced FTO nuclear accumulation via the mTOR signaling pathway.29 Zhang et al.31 discovered that FTO could enhance endometrial carcinoma cell proliferation and invasions by activating the PI3K/AKT and MPAK signal pathways. Zhang et al. found that FTO may promote metastasis by activating the WNT signaling pathway in endometrial cancer.32 FTO overexpression is also involved in chemo‐radiotherapy resistance of cervical squamous cell carcinoma through reducing m6A levels in its mRNA transcripts and in turn increasing ERCC1.33 M6A RNA methylation is complex, and how FTO regulates cancer cells is still in the initial stages and warrants further research. We found that cell cycle, endocytosis, ubiquitin‐mediated proteolysis, and ERBB signaling pathway may be the potential pathway regulated by FTO in endometrial cancer.
In our study, we conducted bioinformatic analysis for 23 m6A RNA methylation regulators in endometrial cancer. We found FTO was a prognostic regulator in endometrial cancer, which was in accordance with in vitro and in vivo experiments. There are some limitations in this study. FTO seems to increase the risk of endometrial cancer by promoting obesity. However, FTO shows diverse characteristics in people from different regions. In our study, all endometrial cancer information was obtained from the TCGA database, and the patients are primarily Americans. Endometrial cancer patients from other regions remain to be further verified with additional evidence. Second, our study is purely bioinformatic analysis; future experimental and clinical data remain to be further confirmed.
In conclusion, our study explored the expressions, prognostic values, GO, and GSEA analyses of the m6A RNA methylation regulators in endometrial cancer. Among the 23 m6A RNA methylation regulators, FTO was associated with prognosis for endometrial cancer. FTO is a promising prognostic biomarker in endometrial cancer, and the mechanisms remain to be explored.
Supporting information
Figure S1
Zhang Y, Yang Y. Effects of m6A RNA methylation regulators on endometrial cancer. J Clin Lab Anal. 2021;35:e23942. 10.1002/jcla.23942
DATA AVAILABILITY STATEMENT
All data generated or analyzed during this study are included in this article.
REFERENCES
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11‐30. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7‐30. [DOI] [PubMed] [Google Scholar]
- 3.Brooks RA, et al. Current recommendations and recent progress in endometrial cancer. CA Cancer J Clin. 2019;69(4):258‐279. [DOI] [PubMed] [Google Scholar]
- 4.Luoto R, Raitanen J, Pukkala E, Anttila A. Effect of hysterectomy on incidence trends of endometrial and cervical cancer in Finland 1953–2010. Br J Cancer. 2004;90(9):1756‐1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187‐1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boccaletto P, Machnicka MA, Purta E, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303‐D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motorin Y, Helm M. RNA nucleotide methylation. Wiley Interdiscip Rev RNA. 2011;2(5):611‐631. [DOI] [PubMed] [Google Scholar]
- 8.Wei CM, Gershowitz A, Moss B. Methylated nucleotides block 5’ terminus of HeLa cell messenger RNA. Cell. 1975;4(4):379‐386. [DOI] [PubMed] [Google Scholar]
- 9.Liu N, Pan T. N6‐methyladenosine–encoded epitranscriptomics. Nat Struct Mol Biol. 2016;23(2):98‐102. [DOI] [PubMed] [Google Scholar]
- 10.Wang S, et al. Roles of RNA methylation by means of N(6)‐methyladenosine (m(6)A) in human cancers. Cancer Lett. 2017;408:112‐120. [DOI] [PubMed] [Google Scholar]
- 11.Yang J, et al. N6‐methyladenine RNA modification and cancer. Oncol Lett. 2020;20(2):1504‐1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28(6):616‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meyer KD, Jaffrey SR. Rethinking m(6)A Readers, Writers, And Erasers. Annu Rev Cell Dev Biol. 2017;33:319‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fu Y, Jia G, Pang X, et al. FTO‐mediated formation of N6‐hydroxymethyladenosine and N6‐formyladenosine in mammalian RNA. Nat Commun. 2013;4:1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7‐34. [DOI] [PubMed] [Google Scholar]
- 16.Liu T, et al. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 2020;48(7):3816‐3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishizawa Y, et al. Oncogene c‐Myc promotes epitranscriptome m(6)A reader YTHDF1 expression in colorectal cancer. Oncotarget. 2018;9(7):7476‐7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao X, et al. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomark. 2018;21(4):859‐868. [DOI] [PubMed] [Google Scholar]
- 19.Han D, et al. Anti‐tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019;566(7743):270‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elliott DJ, et al. RBMX family proteins connect the fields of nuclear RNA processing, disease and sex chromosome biology. Int J Biochem Cell Biol. 2019;108:1‐6. [DOI] [PubMed] [Google Scholar]
- 21.Song Y, et al. RBMX contributes to hepatocellular carcinoma progression and sorafenib resistance by specifically binding and stabilizing BLACAT1. Am J Cancer Res. 2020;10(11):3644‐3665. [PMC free article] [PubMed] [Google Scholar]
- 22.Schouten LJ, Goldbohm RA, van den Brandt PA. Anthropometry, physical activity, and endometrial cancer risk: results from the Netherlands Cohort Study. J Natl Cancer Inst. 2004;96(21):1635‐1638. [DOI] [PubMed] [Google Scholar]
- 23.Kaaks R, Lukanova A, Kurzer MS. Obesity, endogenous hormones, and endometrial cancer risk: a synthetic review. Cancer Epidemiol Biomarkers Prev. 2002;11(12):1531‐1543. [PubMed] [Google Scholar]
- 24.Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med. 2010;363(24):2339‐2350. [DOI] [PubMed] [Google Scholar]
- 26.Fischer J, et al. Inactivation of the Fto gene protects from obesity. Nature. 2009;458(7240):894‐898. [DOI] [PubMed] [Google Scholar]
- 27.Chen J, Du B. Novel positioning from obesity to cancer: FTO, an m(6)A RNA demethylase, regulates tumour progression. J Cancer Res Clin Oncol. 2019;145(1):19‐29. [DOI] [PubMed] [Google Scholar]
- 28.Huang X, Zhao J, Yang M, Li M, Zheng J. Association between FTO gene polymorphism (rs9939609 T/A) and cancer risk: a meta‐analysis. Eur J Cancer Care (Engl). 2017;26(5):e12464. [DOI] [PubMed] [Google Scholar]
- 29.Zhu Y, Shen J, Gao L, Feng Y. Estrogen promotes fat mass and obesity‐associated protein nuclear localization and enhances endometrial cancer cell proliferation via the mTOR signaling pathway. Oncol Rep. 2016;35(4):2391‐2397. [DOI] [PubMed] [Google Scholar]
- 30.Niu Y, Lin Z, Wan A. RNA N6‐methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Z, Zhou D, Lai Y, et al. Estrogen induces endometrial cancer cell proliferation and invasion by regulating the fat mass and obesity‐associated gene via PI3K/AKT and MAPK signaling pathways. Cancer Lett. 2012;319(1):89‐97. [DOI] [PubMed] [Google Scholar]
- 32.zhang L, Wan Y, Zhang Z. FTO demethylates m6A modifications in HOXB13 mRNA and promotes endometrial cancer metastasis by activating the WNT signalling pathway. RNA Biol. 2020;1‐14. 10.1080/15476286.2020.1841458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou S, Bai ZL, Xia D. FTO regulates the chemo‐radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting β‐catenin through mRNA demethylation. Mol Carcinog. 2018;57(5):590‐597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Data Availability Statement
All data generated or analyzed during this study are included in this article.