

Abstract
The farnesyltransferase inhibitor, Lonafarnib, reduces tau inclusions and associated atrophy in familial tauopathy models through activation of autophagy, mediated by the inhibition of farnesylation of the Ras GTPase, Rhes. While hinting at a role of Rhes in tau aggregation, it is unclear how translatable these results are for sporadic forms of tauopathy. We examined histological slides of allocortex and neocortex from multiple postmortem cases in five different tauopathies, FTLD-TDP, and healthy controls using immunofluorescence for Rhes, several tau post-translational modifications, and phospho-TDP-43. Single nucleus RNA data suggest that Rhes is found in all cortical neuron subpopulations but not in glia. Histologic investigation showed that nearly all neurons in control brains display a pattern of diffuse cytoplasmic Rhes positivity. However, in the presence of abnormal tau, but not abnormal TDP-43, the patterns of neuronal cytoplasmic Rhes tend to present as either punctiform or entirely absent. This observation reinforces the relevance of findings that link Rhes changes and tau pathology from the in vivo and in vitro models of tauopathy. The results here support a potential clinical application of Lonafarnib to tauopathies.
Keywords: Tauopathies, Neuropathology, Alzheimer’s disease, Farnesyltransferase, Human, Autopsy
INTRODUCTION
Despite being the most frequent underlying cause of dementia, tauopathies remain without effective treatments and are increasing in prevalence [8, 24]. Tauopathies encompass many distinct neuropathological entities that feature the accumulation of abnormally folded tau protein followed by associated neuron and synapse loss. Alzheimer’s disease (AD) is the most common tauopathy and features both 3- and 4-repeat tau inclusions. Among the sporadic tauopathies, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and argyrophilic grain disease (AGD) show 4-repeat tau inclusions predominantly while Pick’s disease (PiD) features 3-repeat tau inclusions. Although the overwhelming majority of tauopathies occur sporadically, more than 50 mutations in MAPT lead to abnormal tau accumulation and different expression patterns of frontotemporal lobar degeneration (FTLD) in an autosomal dominant pattern of inheritance [22, 30, 31]. These mutations are the basis for biomedical research in tauopathies, as they can be incorporated into transgenic cell lines or in vivo systems that model tauopathies; however, it is unclear how generalizable findings from these familial models are for the more common sporadic tauopathies.
Immunotherapies have been the most common class of drugs brought to clinical trials for tauopathies; however, they have, thus far, produced lackluster results [11, 33]. Other approaches have attempted to modulate post-translational modifications of tau [18, 20, 33, 36] or reduce MAPT mRNA expression using antisense oligonucleotides [4]. The well-documented dysfunction in autophagy across tauopathies [2, 32] establishes autophagy modulation as an attractive strategy to enhance clearance of aggregated tau across the broad spectrum of tauopathies [2, 15]. These efforts have shown some promise in preclinical settings and may become effective treatment strategies for tauopathies.
Farnesyltransferase inhibitors were initially developed for cancer treatment by acting on farnesyl protein transferase, a necessary protein CaaX farnesylation catalyst, to inhibit Ras oncoproteins [13]. A pharmacological class effect of farnesyltransferase inhibitors is enhanced autophagy [25]. The farnesyltransferase inhibitor, Lonafarnib, was successfully used in vitro and in mouse models of familial tauopathy to reduce the burden of tau inclusions by enhancing autophagic activity [10]. Specifically, Lonafarnib acts through the Ras guanosine triphosphatase, Ras homolog enriched in striatum (Rhes) to activate autophagic degradation of tau aggregates. Rhes may suppress autophagy by binding to and activating the mammalian target of rapamycin (mTOR) [29]. Through sequestration of Beclin-1 from inhibitory binding to Bcl-2, Rhes can also activate autophagy independent of mTOR [19]. When farnesylation is blocked by an inhibitor such as Lonafarnib, Rhes dissociates from membranes, allowing it to sequester Beclin-1 and upregulate autophagy [10].
Although promising, these model-based observations’ translatability to treating tauopathies using a farnesyltransferase inhibitor, particularly in the case of sporadic tauopathies, is unclear. Furthermore, little is known about the pattern of Rhes expression in human neurons, particularly outside the striatum and in the context of neurodegenerative conditions.
Rhes is encoded by the gene RASD2 and was thought to be predominantly expressed in the striatum [10, 34]. Some studies suggest that Rhes expression in specific neuronal subpopulations is the basis of selective vulnerability to Huntington’s disease, a genetic neurodegenerative condition causing profound striatal and mild cortical neuronal loss [9]. However, tauopathies show only mild striatal involvement, with significant involvement of cortical and other subcortical structures. The evidence for Rhes expression in human cortical neurons is scant with just one study, to our knowledge, showing scattered expression of RASD2 mRNA throughout the cortices of humans and mouse models using autoradiography [34].
An in-depth understanding of Rhes expression, localization within neurons, and disease-related changes in cortical regions vulnerable to tauopathies, if any, is a necessary step to inform whether Lonafarnib should be explored as a treatment for tauopathies. To fill this gap, we used multiplex immunofluorescence and single nucleus RNA sequencing (snRNA-seq) in human brain tissue from healthy controls (HC) and a broad range of sporadic tauopathies to characterize and measure phenotype changes in Rhes and its relationship to the accumulation of tau aggregates.
MATERIALS AND METHODS
Participants and Neuropathological Assessment
Cases were sourced from the Neurodegenerative Disease Brain Bank at the University of California, San Francisco Memory and Aging Center. Consent for brain donation was obtained from subjects or next of kin following a protocol approved by the Institutional Review Board of the University of California, San Francisco.
Upon autopsy, the brains were slabbed fresh into coronal slabs and fixed in 4% neutral buffered formalin or frozen within 24 hours of death. Slabs undergoing formalin fixation were transferred to phosphate buffered saline-azide after 72-hours of fixation and stored at 4°C. A standardized set of 26 tissue blocks from neurodegenerative regions of interest are dissected from these slabs and embedded into paraffin blocks. Immunohistochemical and basic histological stains were used to assess neurodegenerative lesions for diagnoses using antibodies for phospho-Ser202 Tau (CP13, Mouse, 1:250, courtesy of Peter Davies), TDP-43 (Rabbit, 1:2000, 10782-2-AP, Proteintech, Chicago, IL), β-amyloid (Mouse, 1:500, MAB5206, Millipore, Billerica, MA), and α-Synuclein (Mouse, 1:5000, LB509, courtesy of J. Trojanowski and V. Lee). A final neuropathologic diagnosis was obtained after consensus (LTG, SS, and WWS) using currently accepted guidelines [16, 17, 21].
For the immunohistochemical component of this study, we selected 37 cases encompassing five HC cases, 25 sporadic tauopathy (AD, PSP, CBD, PiD, and AGD) cases, and seven cases of FTLD with TDP-43 inclusions (FTLD-TDP Type A and Type B). Half of the cases for the tauopathy analysis were female, albeit with an uneven distribution of sexes between groups, with a mean (sd) age of death of 73 (10) years (Table 1). A majority (71%) of cases for the TDP-43 analysis were female, with a mean (sd) age of death of 11.8 (7.4) years (Table 2). Inclusion criteria included: lack of more than one primary or contributing neuropathological diagnosis, lack of an Axis I psychiatric disorder, lack of non-dementia neurological disorder, lack of gross non-degenerative structural neuropathology, and a postmortem interval below 24 hours. HC cases were free of any clinical record of cognitive decline, neurological, or a non-incidental neuropathological diagnosis.
Table 1:
Demographics and case characteristics for analysis of tauopathies.
| Diagnosis | n | Age (mean, sd) | Female (n, %) | PMI (mean, sd) | CDR (median, IQR) | ABC Score |
|---|---|---|---|---|---|---|
| HCa | 5 | 78, 10 | 5, 100% | 8.2, 2.4 | 0, 0 |
A0B1C0 (1) A1B1C0 (2) A1B1C1 (1) A2B0C0 (1) |
| AD | 5 | 66, 5 | 2, 40% | 8.6, 0.9 | 3, 0 | A3B3C3 (5) |
| AGD | 5 | 84, 12 | 1, 20% | 8.7, 4.1 | 0.5, 0 | A?B1C1 (1) A?B2C1 (1) A0B2C0 (1) A1B2C0 (2) |
| CBD | 5 | 66, 4 | 2, 40% | 9.3, 2.7 | 1, 2.5 | A1B1C0 (5) |
| PiD | 5 | 68, 8 | 2, 40% | 10.2, 3.9 | 3, 0 |
A0B0C0 (1) A1B0C0 (1) A1B1C0 (2) A1B0C1 (1) |
| PSP | 5 | 76, 10 | 3, 60% | 11.1, 4.9 | 0.5, 0 |
A0B0C0 (1) A0B1C0 (3) A1B1C0 (1) |
| Total | 30 | 73, 10 | 15, 50% | 9.3, 3.3 | 0.75, 2.5 |
A0B0C0 (2) A0B1C0 (4) A0B2C0 (1) A1B0C0 (1) A1B0C1 (1) A1B1C0 (10) A1B1C1 (1) A1B2C0 (2) A2B0C0 (1) A3B3C3 (5) A?B1C1 (1) A?B2C1 (1) |
One of the HC cases had inclusions of α-synuclein in the form of Lewy bodies in the brain stem; however, none noted in the cortex.
Abbreviations: HC, Healthy Controls; AD, Alzheimer’s disease; AGD, Argyrophilic Grain Disease; CBD, Corticobasal Degeneration; PiD, Pick’s disease; PSP, Progressive Supranuclear Palsy; PMI, Postmortem Interval; CDR, Clinical Dementia Rating
Table 2:
Demographics and case characteristics for analysis of TDP-43.
| Diagnosis | n | Age (mean, sd) | Female (n, %) | PMI (mean, sd) | Clinical syndrome | ABC Score |
|---|---|---|---|---|---|---|
| FTLD-TDP Type A | 3 | 63, 3.6 | 3, 100% | 12.4, 4.1 | CBS (2) bvFTD (1) |
A0B0C0 (1) A1B1C0 (1) A1B1C1 (1) |
| FTLD-TDP Type B | 4 | 59, 3.7 | 2, 50% | 11.5, 9.8 | bvFTD (2) bvFTD-ALS (1) nfvPPA-ALS (1) |
A0B0C0 (3) A0B1C0 (1) |
| Total | 7 | 60, 4 | 5, 71% | 11.8, 7.4 | bvFTD (3) CBS (2) bvFTD-ALS (1) nfvPPA-ALS (1) |
A0B0C0 (4) A0B1C0 (1) A1B1C0 (1) A1B1C1 (1) |
Abbreviations: FTLD, Frontotemporal lobar degeneration; TDP, TAR DNA-binding protein; PMI, Postmortem Interval; CBS, Corticobasal syndrome; bvFTD, Behavioral variant frontotemporal dementia; ALS, Amyotrophic Lateral Sclerosis; nfvPPA, non-fluent variant primary progressive aphasia
Tissue processing, immunohistochemistry, and microscopy
Analysis of Rhes cytoplasmic neuronal distribution in healthy controls and tauopathies
Thirty cases encompassing HC and tauopathies were used in this analysis. The paraffin block containing the hippocampal formation at the level of the lateral genicular body was sectioned at 8 μm for all 30 cases. The paraffin block containing the superior frontal gyrus (SFG) was also sectioned at 8 μm for all cases, excluding the five AGD cases (as AGD lacks inclusions in the SFG) for a final n=25 sections. Multiplex immunofluorescence staining included antibodies for phospho-Ser396/404 tau (PHF-1), Rhes, and NeuN (a pan-neuronal marker). Supplementary Methods 1.1 describe the staining protocol in detail.
To establish criteria for analyzing Rhes in cortical neurons, we qualitatively examined the cytoplasmic neuronal distribution of Rhes signal. Based on consensus (AJE and LTG), we defined three different intraneuronal Rhes distribution patterns: Diffuse, Punctiform, and Absent. To further understand the intraneuronal location of Rhes, we used multiplex immunofluorescence against MAP2, Rhes, and NeuN (See Supplementary Methods 2) in sections cut from the hippocampal formation cut at the level of the genicular body from two tauopathy cases (one 64-year-old female with AD and one 69-year-old female with PiD). Next, we quantified digital images of our sections in ImageJ, classifying and counting neurons based on the pattern of Rhes cytoplasmic distribution and the presence of PHF-1 tau inclusions. Methodological details are described in Supplementary Methods 1.2.
Analysis of Rhes cytoplasmic neuronal distribution in FTLD-TDP
We selected cortical areas representing regions with a moderate-to-severe pathological burden in FTLD-TDP. Paraffin blocks representing the inferior frontal gyrus (IFG) were cut at 8 μm from three FTLD-TDP Type A cases. Similarly, we cut sections from paraffin blocks representing the middle frontal gyrus (MFG) of four FTLD-TDP Type B cases. Multiplex immunofluorescence staining included antibodies against phospho-TDP-43, Rhes, and NeuN (see Supplementary Methods 3.1). Next, neurons were quantified based on the Rhes pattern and presence of phospho-TDP-43 inclusions (see Supplementary Methods 3.2). We did not include FTLD-TDP Type C cases because this subtype rarely shows cytoplasmic neuronal inclusions in neocortical areas.
Analysis of the association between Rhes neuronal patterns association and tau post-translational modifications
To examine if the association between tau inclusions and changes in Rhes neuronal patterns vary among inclusions featuring different post-translational modifications, in addition to the analysis using PHF-1 labeled phospho-tau, we sectioned paraffin blocks containing the hippocampal formation at the level of the lateral genicular body from two AD cases (one 59-year-old male, one 64-year-old female). These sections were immunostained with antibodies against Rhes, NeuN, and a variety of abnormal tau forms: AT100 (phospho-Thr212/Ser214 tau), MC1 (conformational tau changes), 1f3c (acetyl-Lys274 tau), and Tau-C3 (ΔAsp421 Tau). As in previous analyses, neurons were quantified based on Rhes subtype and the presence of tau inclusions. Supplementary Methods 4.1 and 4.2 describe the staining, imaging, and quantification protocols in detail.
Cell type specificity for Rhes
Rhes has been extensively described in human striatal neurons, but its presence in other neuron groups or glia cells has been less explored. To close this gap, we first analyzed snRNA-seq data generated from an additional cohort of ten postmortem brain samples representing HC and individuals across the neuropathological spectrum of AD, initially published in Leng et al. [14]. This dataset is also available on Synapse (ID: syn21788402). This dataset represents single-cell gene expression information and metadata from the EC, labeled as “EC_allCells.rds”, and SFG (Brodmann area 8), labeled as “SFG_allCells.rds”. Although the dataset includes different cases than those used in our immunohistochemical analyses, the brain regions examined are similar. The log-scaled counts per million (CPM) of RASD2 transcripts in cells from the cell type subpopulations identified in the entorhinal cortex (EC) and SFG of Braak stage 0 cases were used to assess expression patterns. Next, the mean log CPM of RASD2 transcripts within groups 2 and 4 of excitatory EC neurons [14] was taken for each case (n=10) and compared between Braak stages 0, II, and VI [1] using a one-way ANOVA. These findings were validated in independent datasets containing other brain areas using an online tool (brainrnaseq.org) [37].
Cell-type specificity for Rhes was further studied histologically, focusing on neurons and astrocytes. To illustrate the lack of Rhes in astrocytes, we immunostained the hippocampal formation cut at the level of the genicular body of an 82-year-old HC female. Supplementary Methods 5 describes this staining protocol in detail.
Statistical analysis
To examine differences in the relative proportions of neuron populations belonging to each phenotype between diagnoses, the proportion of neurons belonging to each Rhes pattern was computed as the number of neurons featuring each of the three phenotypes divided by the total number of neurons counted per case. Wilcoxon-Mann-Whitney tests were used to test the null hypothesis that no significant difference exists in the proportion of total neurons counted that present as Diffuse Rhes between HC and each disease in pairwise comparisons.
The proportions of neurons positive and negative for PHF-1 inclusions per Rhes phenotype were computed for each region of interest (EC, Cornu Ammonis 1, and SFG) to test for differences in PHF-1 tau inclusion burden within each neuronal Rhes phenotype population. Kruskal-Wallis rank-sum tests were used to test the null hypothesis that there was no significant difference in the burden of PHF-1 tau inclusions between the three neuronal Rhes phenotypes in each disease. At a group-level, combining all cases, one-way ANOVA’s were used to test the null hypothesis that there was no significant difference in the burden of PHF-1 tau inclusions between the three neuronal Rhes phenotypes in the EC, Cornu Ammonis 1 (CA1), or SFG. For both Kruskal-Wallis rank-sum tests and one-way ANOVA’s, cases without a computed proportion (i.e., those with a denominator of 0) were omitted from the analysis. We repeated the same analysis for the FTLD-TDP cases, classifying inclusions based on labeling for phospho-TDP-43 instead of PHF-1 positive tau.
The analyses involving other post-translational modifications of tau did not involve statistical tests as they were exploratory. Instead, the percentage of neurons positive for each post-translational modification classified as each Rhes phenotype was computed and averaged between the two cases.
The alpha level for all analyses was set to 0.05. Statistical analysis was conducted in the software R, and graphs were produced with the R package ggplot2.
RESULTS
Rhes is widespread in cortical neurons of healthy controls, presenting in a diffuse granular cytoplasmic distribution
Several studies have suggested that Rhes expression is mostly limited to the striatum, thus the name Rhes. We detected Rhes signal in neurons throughout the allocortex (Figure 1a, Supplementary Figure 1) and one neocortical area using immunofluorescence. In HC, we found Rhes signal in over 95% of EC and over 99% of CA1 and SFG neurons. In these HC, most neurons present Rhes as a cytoplasmic granular distribution extending into the dendrite (Figure 1b–i).
Figure 1:
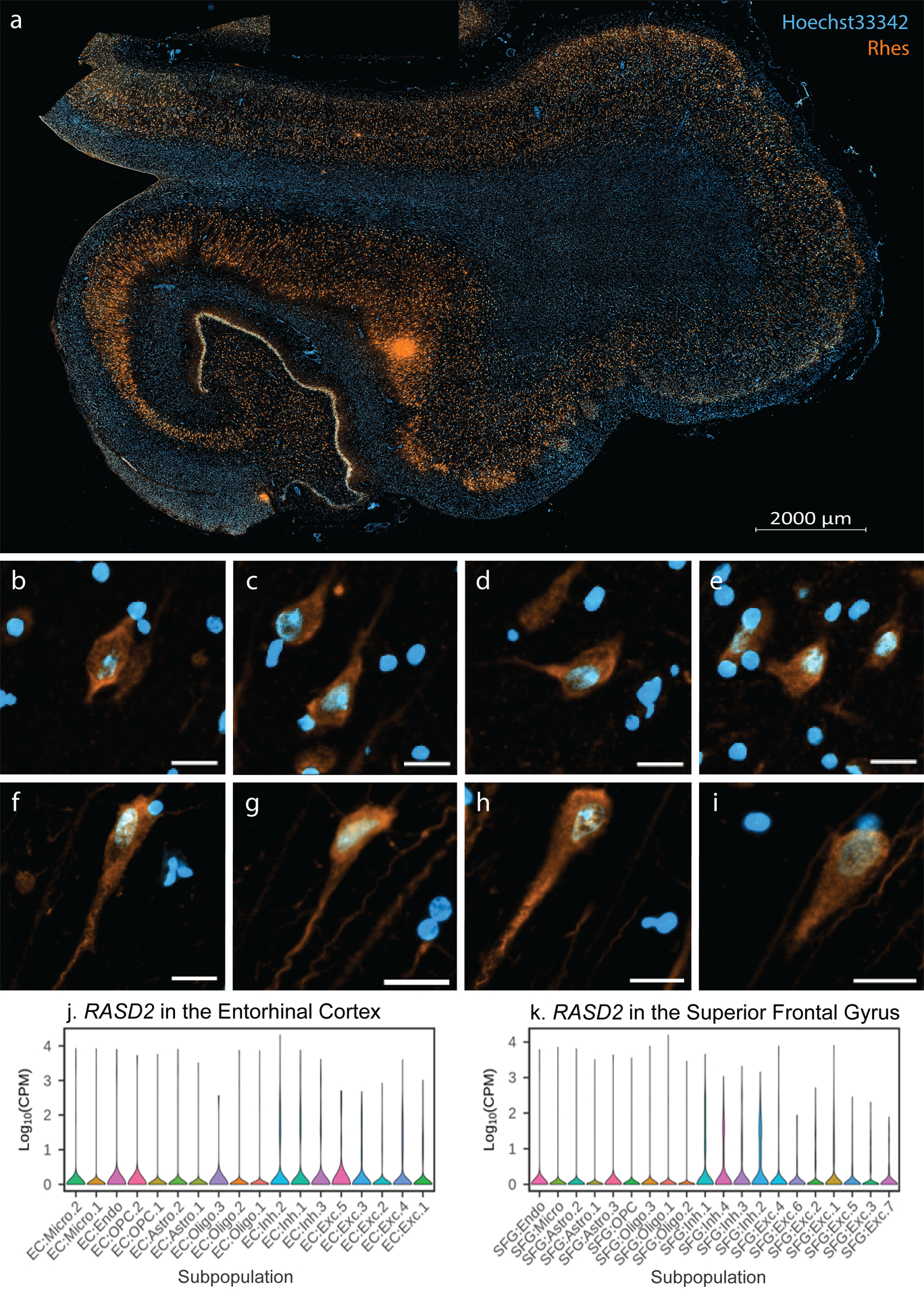
Rhes (orange) is ubiquitously expressed throughout the human hippocampal formation of an 82-year-old female free of any neuropathological diagnosis (a). See Supplementary Figure 1 for the overlapping NeuN+ distribution. In the entorhinal cortex (b-e) and cornu ammonis 1 (f-i) neurons, Rhes has a cytoplasmic granular appearance (Hoechst 33342 in blue), extending into proximal segments of neuronal processes (Diffuse Phenotype). All scale bars B-I correspond to 20 μm. (j-k) Violin plots of RASD2 transcript levels in different cell type subpopulations in the entorhinal cortex (EC) and superior frontal gyrus (SFG) from Leng et al. 2021. Cell type abbreviations: Exc, excitatory neurons; Oligo, oligodendrocytes; Astro, astrocytes; Inh, inhibitory neurons; OPC, oligodendrocyte precursor cells; Micro, microglia; Endo, endothelial cells.
To corroborate the findings, we analyzed snRNA-seq data from the EC (an allocortical region) and SFG (a neocortical region) from a different cohort [14]. Within the Braak stage 0 cases (n=3), we detected RASD2 transcripts in all excitatory and inhibitory neuronal populations of these two regions (Figure 1j–k). Moreover, these results were validated in independent datasets using the online tool at brainrnaseq.org [37] and immunohistologically (Supplementary Figure 2). Both neuropathological and snRNA-seq data indicate that glia lack Rhes (Figure 2, Supplementary Figure 2).
Figure 2:
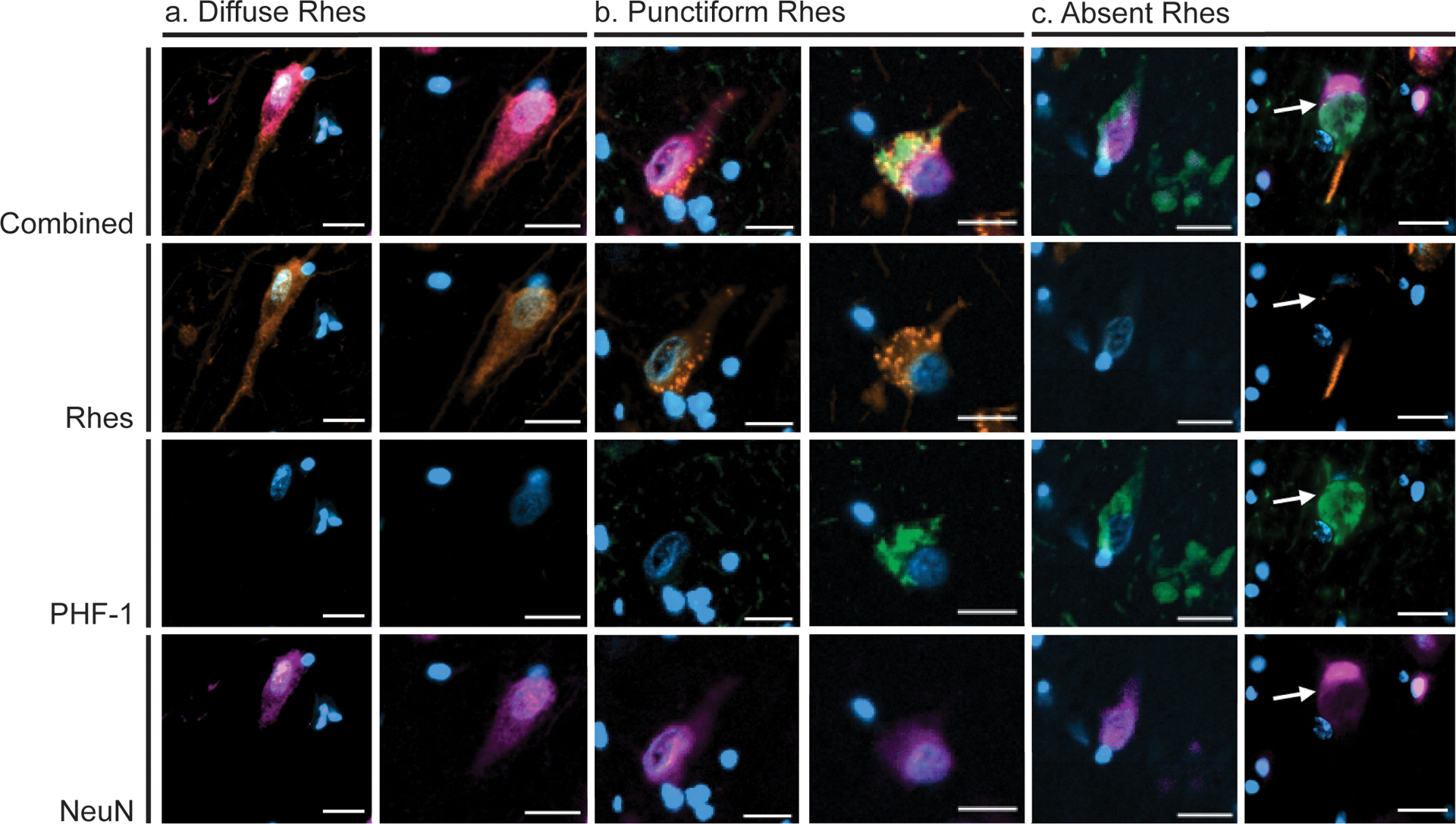
Diffuse Phenotype (orange) neurons (a) feature diffuse Rhes staining in the cell body extending into the apical dendrite. Punctiform Phenotype neurons (c) feature punctiform Rhes signal in the cell body and often co-occur with PHF-1 (pink) positive tau inclusions. Absent Phenotype neurons (c) feature clearance of Rhes from the cell body and often co-occur with PHF-1 positive tau inclusions. Blue, DAPI; Orange, Rhes; Green, PHF-1; Pink, NeuN. All scale bars correspond to 20 μm.
In tauopathies, neurons present as three phenotypes of Rhes cytoplasmic distribution
We qualitatively examined neurons in the EC, CA1, and SFG of all 30 HC and tauopathy cases (Table 1) and defined three distinct phenotypes of neuronal cytoplasmic Rhes distribution. The ‘Diffuse’ type features granular Rhes staining in the cell body extending into the dendrite (Figure 2a). The ‘Punctiform’ type has large cytoplasmic Rhes+ puncta with a variable granular background, extending into the dendrite (Figure 2b). The ‘Absent’ type features an absence of Rhes staining in the cell body and variable positivity in the dendrite (Figure 2c).
To confirm that Rhes is expressed in the dendrites, histological slides of the human hippocampal formation from AD and PiD cases underwent multiplex immunostaining with Rhes, MAP2, and NeuN. We observed that Rhes overlaps with MAP2 signal in the neuronal processes of all three subtypes, indicating that it is present in dendrites (Figure 3, Supplementary Figure 3).
Figure 3:
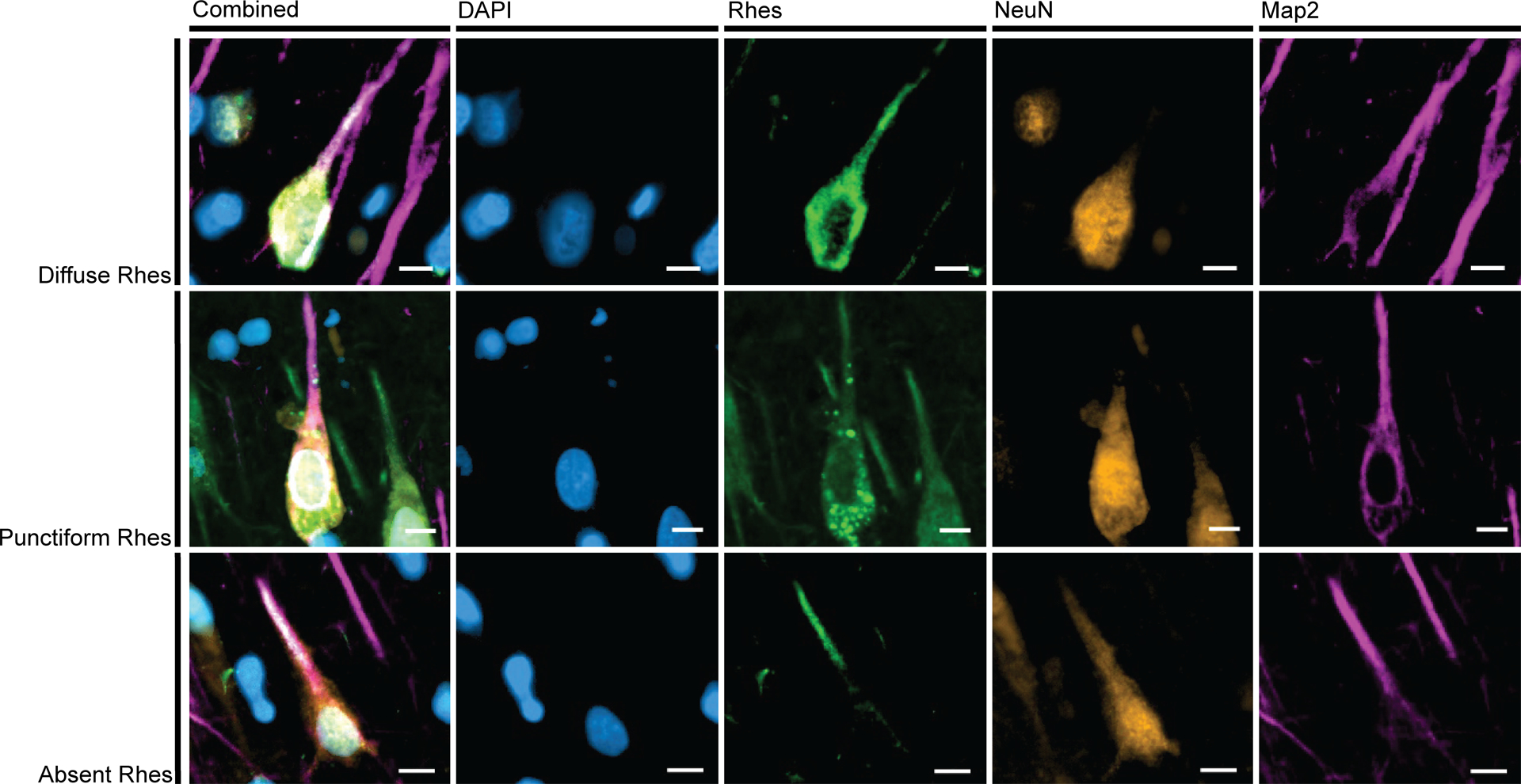
In all three Rhes Phenotypes, projections of Rhes (green) overlap with MAP2 (pink). This indicates that Rhes occurs in dendrites. Examples here from CA1 of a 64-year-old female with AD (A3B3C3). Scale bars correspond to 10 μm.
The relative abundance of Rhes neuronal phenotypes in AD and other tauopathies
In HC, 94.4% of neurons in the EC, 97.6% of neurons in CA1, and 99.8% of neurons in the SFG showed a diffuse Rhes phenotype, whereas tauopathy cases had a higher representation of Punctiform and Absent phenotypes (Figure 4). A one-way ANOVA showed that the proportion of neurons with a Diffuse phenotype was significantly different between each of the tauopathies and HC in the EC (p = 0.002), CA1 (p < 0.001), and the SFG (p < 0.001). This finding suggests that in tauopathies, intraneuronal Rhes assumes a punctiform presentation or is cleared from the neuronal cytoplasm in affected neurons.
Figure 4:
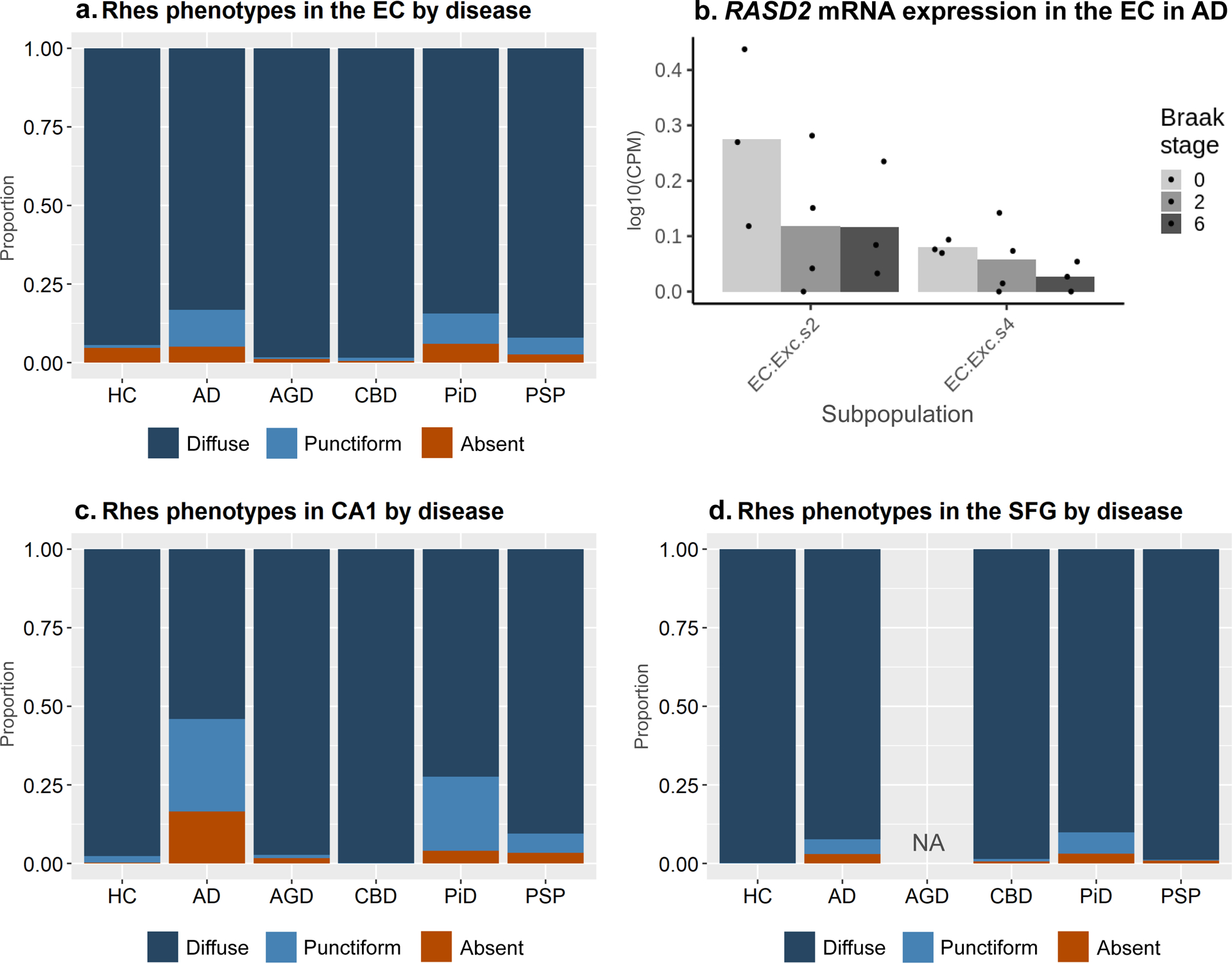
The relative proportions of Rhes phenotypes across tauopathies in the entorhinal cortex (A), CA1 (C), and SFG (D). Diffuse Phenotype is the dominant phenotype, and its relative proportion decreases in tauopathies, most notably in AD, PiD, and PSP. Single-nucleus RNA expression data show a negative trend in the quantity of RASD2 with increasing Braak stages transcripts within two selectively vulnerable excitatory neuron populations, but the groups do not statistically significantly differ from one another (C).
AD and PiD showed the highest proportion of neurons with Punctiform or Absent Rhes phenotypes (Figure 4a, c, d, Table 3). Notably, in AGD, a four-repeat tauopathy featuring scarce to moderate numbers of cytoplasmic neuronal inclusions in the EC and CA1, the proportion of neurons with diffuse Rhes phenotypes did not show a significant change compared to HC. Similarly, CBD and PSP, 4-repeat tauopathies with mild neuronal pathology, showed no significant differences in the proportion of neurons with Diffuse Rhes phenotypes compared to HC in the EC, CA1, and SFG.
Table 3:
Proportion (mean, sd) of counted neuron population classified as each Rhes phenotype and with PHF-1 inclusions by region.
| Diagnosis | Diffuse Rhes | Punctiform Rhes | Absent Rhes | PHF-1 + | |
|---|---|---|---|---|---|
| a) Entorhinal Cortex | Total (n=30) | 0.918, 0.086 | 0.048, 0.062 | 0.034, 0.048 | 0.108, 0.12 |
| HC (n=5) | 0.944, 0.103 | 0.008, 0.010 | 0.048, 0.094 | 0.061, 0.106 | |
| AD (n=5) | 0.832, 0.068 | 0.117, 0.061 | 0.051, 0.033 | 0.195, 0.075 | |
| AGD (n=5) | 0.983, 0.019 | 0.004, 0.006 | 0.013, 0.018 | 0.013, 0.017 | |
| CBD (n=5) | 0.984, 0.009 | 0.010, 0.011 | 0.006, 0.004 | 0.015, 0.006 | |
| PiD (n=5) | 0.843, 0.065 | 0.096, 0.053 | 0.061, 0.050 | 0.277, 0.107 | |
| PSP (n=5) | 0.920, 0.075 | 0.053, 0.079 | 0.027, 0.034 | 0.084, 0.071 | |
| b) Cornu Ammonis 1 | Total (n=30) | 0.852, 0.195 | 0.104, 0.136 | 0.044, 0.067 | 0.178, 0.24 |
| HC (n=5) | 0.976, 0.029 | 0.020, 0.028 | 0.004, 0.006 | 0.027, 0.041 | |
| AD (n=5) | 0.540, 0.082 | 0.295, 0.077 | 0.165, 0.016 | 0.441, 0.089 | |
| AGD (n=5) | 0.972, 0.027 | 0.010, 0.007 | 0.017, 0.022 | 0.025, 0.026 | |
| CBD (n=5) | 0.998, 0.003 | 0.002, 0.003 | 0, 0 | 0.013, 0.013 | |
| PiD (n=5) | 0.723, 0.139 | 0.236, 0.112 | 0.040, 0.044 | 0.489, 0.287 | |
| PSP (n=5) | 0.904, 0.191 | 0.061, 0.114 | 0.035, 0.078 | 0.071, 0.112 | |
| c) Superior Frontal Gyrus | Total (n=25) | 0.959, 0.05 | 0.025, 0.036 | 0.016, 0.018 | 0.061, 0.08 |
| HC (n=5) | 0.998, 0.001 | 0.002, 0.002 | 0, 0.001 | 0, 0 | |
| AD (n=5) | 0.923, 0.056 | 0.046, 0.046 | 0.031, 0.02 | 0.118, 0.091 | |
| CBD (n=5) | 0.985, 0.009 | 0.008, 0.004 | 0.007, 0.005 | 0.022, 0.018 | |
| PiD (n=5) | 0.901, 0.046 | 0.068, 0.03 | 0.031, 0.019 | 0.148, 0.084 | |
| PSP (n=5) | 0.988, 0.005 | 0.003, 0.002 | 0.009, 0.007 | 0.016, 0.006 |
Abbreviations: HC, Healthy Controls; AD, Alzheimer’s disease; AGD, Argyrophilic Grain Disease; CBD, Corticobasal Degeneration; PiD, Pick’s disease; PSP, Progressive Supranuclear Palsy
Changes in Rhes subtypes correlate with the presence of intraneuronal tau aggregates
Seeing that there is a possible correlation between abnormal tau accumulation in neurons and Rhes cytoplasmic distribution changes, we examined how well the presence of PHF-1 positive tau inclusions associated with Rhes phenotype.
In slides immunostained for Rhes, PHF-1, and NeuN, we counted a total 26,357 neurons (8,121 in the EC, 5,636 in CA1, and 12,600 in the SFG). Only a mean (sd) of 4.3% (0.7%) Diffuse Type neurons in the EC, 7.4% (17.1%) in CA1, and 2.8% (3.8%) in the SFG were PHF-1 positive. Conversely, a mean (sd) of 91.9% (18.8%) EC, 93.6% (14.9%) CA1, and 85.1% (35.7%) Absent Type SFG neurons harbored PHF-1 positive inclusions. Punctiform type neurons showed a more balanced distribution of PFH-1 positivity with a mean (sd) 72.6% (32.3%) in the EC, 84.1% (25.0%) in CA1, and 56.7% (39.5%) in the SFG (Figure 6, Table 5).
Figure 6:
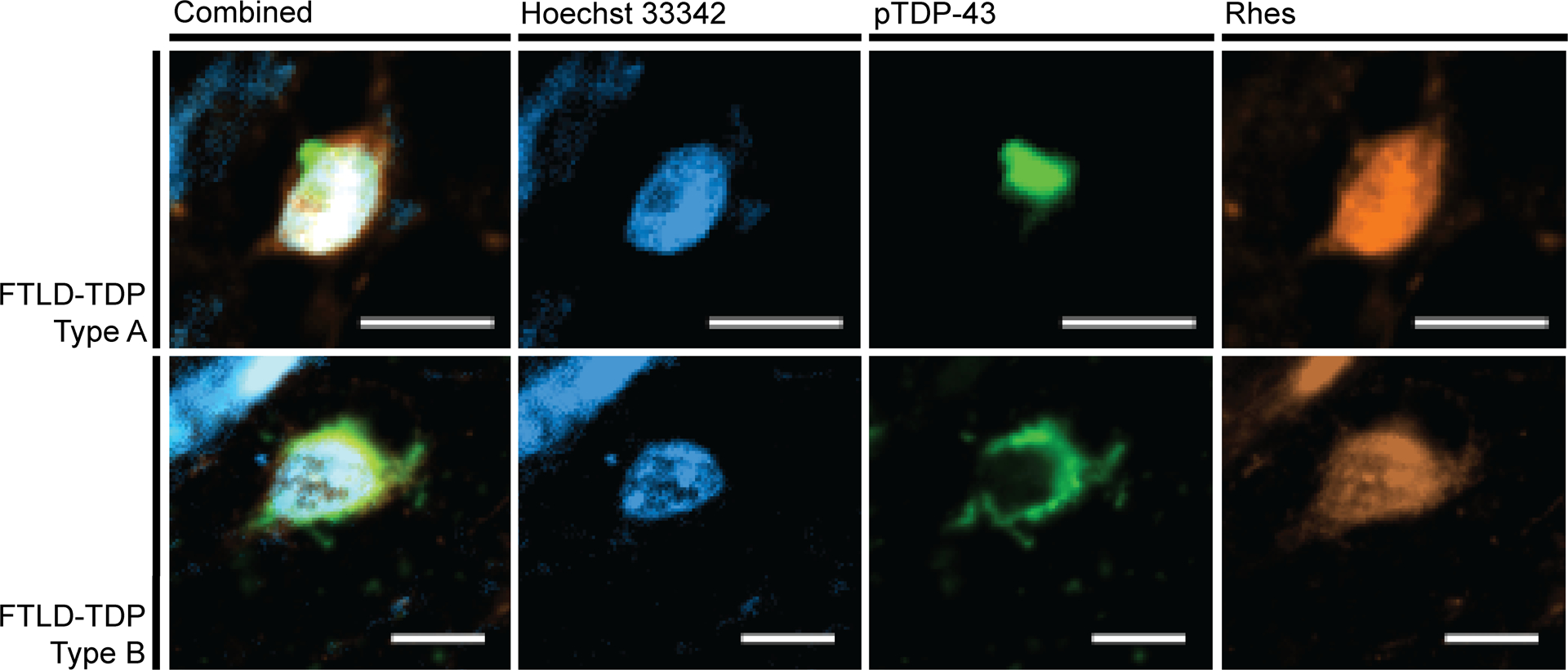
Neurons positive for pTDP-43 (green) proteinopathy display a Diffuse Rhes (orange) phenotype. The FTLD-TDP Type A images come from the inferior frontal gyrus of a 66-year-old female with behavioral variant frontotemporal dementia. The FTLD-TDP Type B images come from the middle frontal gyrus of a 57-year-old male with behavioral variant frontotemporal dementia and amyotrophic lateral sclerosis. Scale bar represents 10 μm.
Table 5:
Proportions of tau inclusion positive CA1 neurons classified as each Rhes phenotype for different post-translational modifications of tau.
| Antibody | Epitope | Diffuse Rhes | Punctiform Rhes | Absent Rhes |
|---|---|---|---|---|
| PHF-1a | phospho-Ser396, Ser404 Tau | 5.99% | 61.08% | 32.93% |
| 1f3c | acetyl-Lys274 Tau | 10.42% | 29.17% | 60.42% |
| AT100 | phospho-Thr212, Ser214 Tau | 8.46% | 55.38% | 36.15% |
| Tau-C3 | ΔAsp421 Tau | 4.11% | 35.62% | 60.27% |
| MC1 | Conformational tau | 5.69% | 60.98% | 33.33% |
While calculated from the same two AD cases as the other tau post-translational modifications, PHF-1 was calculated from one large 1×1mm counting frame instead of the three 650×650μm counting frames used for the other post-translational modifications.
The proportion of neurons with PHF-1 positive tau inclusions significantly differ between the three neuronal Rhes phenotypes (p < 0.0001 for EC, CA1, and SFG). Kruskal-Wallis tests showed that this pattern is statistically significant (p < 0.05) in the EC for AD, CBD, PiD, and PSP individually; all tauopathies individually for CA1; and AD, PiD, and PSP for the SFG. When not statistically significant for an individual tauopathy, the patterns generally followed the same trend.
The CA1 sector of AD cases showed the highest proportion of Punctiform and Absent Rhes phenotypes. To examine if the correlation between neuronal Rhes phenotypes and tau inclusions differ across various tau post-translational modifications, we immunostained CA1 of two AD cases (59-year-old male and 64-year-old female) against Rhes, NeuN, and several post-translational modifications of tau (Supplementary Figure 4): phospho-Thr212/Ser214 Tau (AT100), acetyl-Lys274 Tau (1f3c), truncated ΔAsp421 Tau (Tau-C3), or conformationally-changed tau (MC1). Overall, the correlation between these different tau post-translational modifications and Rhes phenotypes followed the same patterns seen for PHF-1. We sought to identify tau post-translational modifications that better discriminated Rhes types than PHF-1 but did not identify such a post-translational modification (Table 6).
Table 6:
TDP-43: Proportions (mean, sd) of counted neuron population classified as each Rhes phenotype and with pTDP-43 inclusions.
| Diagnosis | Diffuse Rhes | Punctiform Rhes | Absent Rhes | pTDP-43 + |
|---|---|---|---|---|
| FTLD-TDP Type A (IFG, n=3) | 1, 0 | 0, 0 | 0, 0 | 0.025, 0.027 |
| FTLD-TDP Type B (MFG, n=4) | 1, 0 | 0, 0 | 0, 0 | 0.032, 0.015 |
| Total (n=7) | 1, 0 | 0, 0 | 0, 0 | 0.028, 0.021 |
TDP-43, TAR DNA-binding protein 43; FTLD, Frontotemporal lobar degeneration; IFG, Inferior Frontal Gyrus; MFG, Middle Frontal Gyrus
TDP-43 pathological change is not associated with Rhes neuronal phenotype
To examine if abnormal Rhes phenotype also associates with phospho-TDP-43 inclusions, we examined cortical sections with moderate or severe TDP-43 pathological changes from three FTLD-TDP Type A cases and four FTLD-TDP Type B cases. We failed to identify any neurons with Punctiform or Absent Rhes phenotypes (Table 4, Figure 5).
Table 4:
Proportions (mean, sd) of each Rhes phenotype with PHF-1+ tau inclusions by region.
| Diffuse Rhes | Punctiform Rhes | Absent Rhes | ||||||
|---|---|---|---|---|---|---|---|---|
| Dx | PHF-1+ | PHF-1− | PHF-1+ | PHF-1− | PHF-1+ | PHF-1− | p-valuea | |
| a) Entorhinal cortex | Total (n=30) | 0.043, 0.073 | 0.957, 0.073 | 0.726, 0.323 | 0.274, 0.323 | 0.919, 0.188 | 0.081, 0.188 | < 0.0001b |
| HC (n=5) | 0.01, 0.011 | 0.99, 0.011 | 0.556, 0.509 | 0.444, 0.509 | 0.889, 0.192 | 0.111, 0.192 | 0.0737 | |
| AD (n=5) | 0.074, 0.036 | 0.926, 0.036 | 0.688, 0.187 | 0.312, 0.187 | 0.96, 0.089 | 0.04, 0.089 | 0.0024 | |
| AGD (n=5) | 0.006, 0.009 | 0.994, 0.009 | 0.444, 0.509 | 0.556, 0.509 | 0.587, 0.361 | 0.413, 0.361 | 0.0693 | |
| CBD (n=5) | 0.004, 0.006 | 0.996, 0.006 | 0.675, 0.395 | 0.325, 0.395 | 1, 0 | 0, 0 | 0.0061 | |
| PiD (n=5) | 0.154, 0.118 | 0.846, 0.118 | 0.913, 0.092 | 0.087, 0.092 | 0.98, 0.045 | 0.02, 0.045 | 0.0049 | |
| PSP (n=5) | 0.008, 0.009 | 0.992, 0.009 | 0.927, 0.12 | 0.073, 0.12 | 1, 0 | 0, 0 | 0.0103 | |
| b) Cornu Ammonis 1 | Total (n=30) | 0.074, 0.171 | 0.926, 0.171 | 0.841, 0.25 | 0.159, 0.25 | 0.936, 0.149 | 0.064, 0.149 | < 0.0001b |
| HC (n=5) | 0.01, 0.018 | 0.99, 0.018 | 0.508, 0.45 | 0.492, 0.45 | 1, 0 | 0, 0 | 0.0586 | |
| AD (n=5) | 0.067, 0.102 | 0.933, 0.102 | 0.833, 0.078 | 0.167, 0.078 | 0.992, 0.017 | 0.008, 0.017 | 0.0016 | |
| AGD (n=5) | 0.001, 0.003 | 0.999, 0.003 | 0.917, 0.167 | 0.083, 0.167 | 0.833, 0.289 | 0.167, 0.289 | 0.0107 | |
| CBD (n=5) | 0.011, 0.013 | 0.989, 0.013 | 1, 0 | 0, 0 | NAc | NAc | 0.0486 | |
| PiD (n=5) | 0.345, 0.294 | 0.655, 0.294 | 0.965, 0.052 | 0.035, 0.052 | 0.894, 0.157 | 0.106, 0.157 | 0.0145 | |
| PSP (n=5) | 0.011, 0.018 | 0.989, 0.018 | 0.778, 0.385 | 0.222, 0.385 | 1, 0 | 0, 0 | 0.0392 | |
| c) Superior frontal gyrus | Total (n=25) | 0.028, 0.038 | 0.972, 0.038 | 0.567, 0.395 | 0.433, 0.395 | 0.851, 0.357 | 0.149, 0.357 | < 0.0001b |
| HC (n=5) | 0, 0 | 1, 0 | 0, 0 | 1, 0 | 0, 0 | 1, 0 | NAd | |
| AD (n=5) | 0.056, 0.047 | 0.944, 0.047 | 0.764, 0.12 | 0.236, 0.12 | 0.8, 0.447 | 0.2, 0.447 | 0.0365 | |
| CBD (n=5) | 0.014, 0.013 | 0.986, 0.013 | 0.413, 0.433 | 0.587, 0.433 | 0.775, 0.437 | 0.225, 0.437 | 0.1765 | |
| PiD (n=5) | 0.065, 0.043 | 0.935, 0.043 | 0.816, 0.215 | 0.184, 0.215 | 1, 0 | 0, 0 | 0.0023 | |
| PSP (n=5) | 0.005, 0.003 | 0.995, 0.003 | 0.625, 0.479 | 0.375, 0.479 | 1, 0 | 0, 0 | 0.0163 | |
p-value computed by a Kruskal-Wallis rank sum test comparing the PHF-1+ proportions in each Rhes phenotype for each disease.
p-value for all diseases combined computed using a one-way ANOVA comparing the PHF-1+ proportions in each Rhes phenotype
No CA1 neurons with the absent Rhes phenotype were detected in CBD cases
The Kruskal-Wallis rank sum test cannot estimate the significant of data lacking variance
Abbreviations: HC, Healthy Controls; AD, Alzheimer’s disease; AGD, Argyrophilic Grain Disease; CBD, Corticobasal Degeneration; PiD, Pick’s disease; PSP, Progressive Supranuclear Palsy
Figure 5:
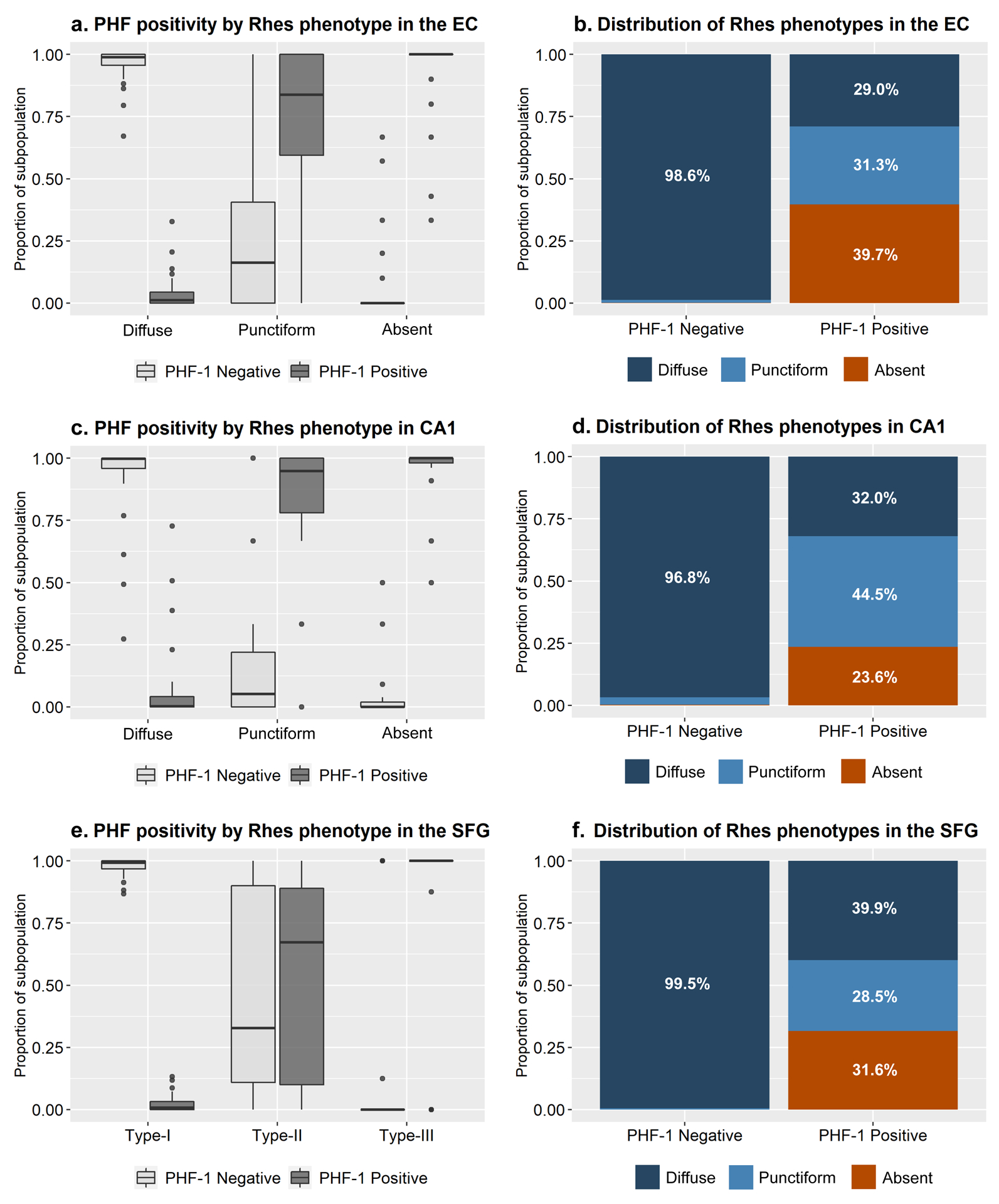
Entorhinal cortex neurons with Diffuse Rhes Phenotype are predominantly negative for p-tau (PHF-1), while neurons with Absent Rhes Phenotype are predominantly PHF-1 positive (a). While neurons with Punctiform Rhes Phenotype appear to be roughly split between PHF-1 positive and negative neurons in the entorhinal cortex, they make up roughly a third of the PHF-1 positive neurons, indicating a tendency to co-occur with tau inclusions (b). The majority of the PHF-1 positive neurons in the entorhinal cortex, either have Punctiform or Absent Rhes Phenotype. A similar correlation occurs in the CA1 and the superior frontal gyrus (c-f).
RASD2 mRNA levels in selectively vulnerable excitatory neurons of the EC
Recent work using snRNA-seq [14] has identified two subpopulations of excitatory neurons in the EC that show a higher vulnerability to AD changes than neighboring excitatory neuron populations. Here, we used this same dataset, featuring three cases at Braak stage 0, four at Braak stage II, and three at Braak stage VI, to test if RASD2 mRNA levels change in these two vulnerable neuronal populations across AD progression. We computed the mean log10(CPM) transcript levels for each case. For each of the two subpopulations, the mean log10(CPM) of RASD2 transcripts was low (< 0.5). We failed to detect significant differences in RASD2 transcript levels among the three Braak stage groups for both subpopulations (EC:Exc.s2 at p=0.286 and EC:Exc.s4 at p=0.404); however, we noted a negative trend with increasing Braak stages (Figure 4b).
DISCUSSION
As an abbreviation for Ras homolog enriched in striatum, Rhes has been thought to be a striatal-predominant protein. Here, we showed that Rhes is expressed throughout the allocortex and neocortex, confirming other studies suggesting that Rhes expression extends beyond striatal neurons [10, 34]. Rhes is widespread in cortical neurons and typically presents with a diffuse cytoplasmic distribution (referred to as the Diffuse type) in over 94% of the EC, CA1, and SFG neurons in HC, suggesting that such distribution represents the standard (i.e., healthy) neuronal phenotype. Noticeably, sporadic tauopathies feature an increased representation of neurons with a cytoplasmic distribution of Rhes presenting as punctiform in cell bodies (Punctiform type) or entirely without Rhes in cell bodies (Absent type) in the three areas examined (the EC, CA1, and SFG).
We found a direct relationship between PHF-1 positive tau inclusions and changes in Rhes phenotype in all tauopathies. Neurons with Punctiform and Absent phenotypes were more likely to harbor tau inclusions than neurons with Diffuse Rhes. This observation suggests that the population-level decreases in the proportions of neurons with Diffuse Rhes in tauopathies are associated with either the tau inclusion itself or an upstream process affecting both tau aggregation and Rhes expression. While neurons with Diffuse Rhes feature few PHF-1 positive tau inclusions, Absent Rhes neurons are predominantly PHF-1 positive, suggesting that abnormal (i.e., diseased) neurons are cleared of Rhes in the soma. Meanwhile, punctiform Rhes appears to be an intermediate phenotype (Figure 7). Similar patterns were observed for other tau post-translational modifications in AD cases.
Figure 7:
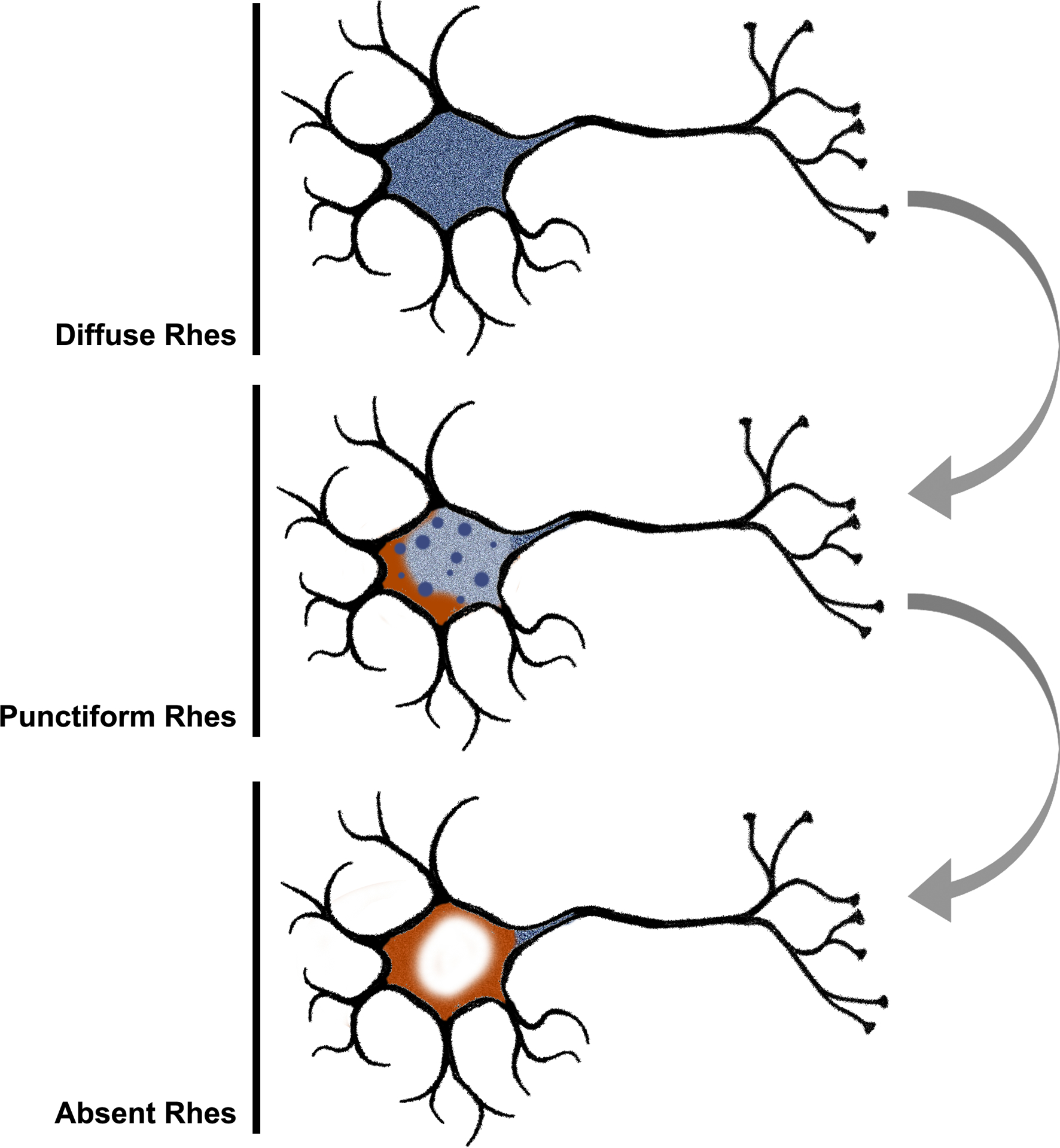
We propose an ordered manner in which the Rhes (blue) phenotypes relate to each other based on the collective evidence from this investigation. At baseline, Rhes is expressed throughout the cytoplasm extending into neuronal processes. As neuropathologic changes (orange) emerge, Rhes expression becomes dysregulated and appears punctiform within the cell body. Finally, Rhes is no longer expressed in the cell body of those neurons most affected by neuropathologic changes with some signal persisting in neuronal processes.
Given that the Absent phenotype is more common in AD than HC, we examined whether RADS2 mRNA expression levels decreased in the two excitatory neuronal subpopulations of the EC thought to be more vulnerable to tau inclusions and neuron loss than other excitatory neuronal subpopulations in AD [14]. Although we did not detect statistically significant changes in transcript levels of RASD2 between Braak stages, a negative trend emerged. Further work with larger RNA-sequencing datasets should be done to understand how the expression of RASD2 changes with disease progression in tauopathies.
This postmortem study in humans, together with in vivo and in vitro work done in familial tauopathy models [10], suggests the presence of a causal relationship between Rhes clearance and tau proteinopathic lesions. The lack of abnormal Rhes phenotypes (i.e., Punctiform or Absent types) associated with FTLD-TDP reinforces a unique mechanistic link between Rhes changes and accumulation of abnormal tau inclusions. Impaired autophagy is a well-established hallmark of neurodegenerative diseases, including tauopathies [2, 23, 26, 32]. Rhes has an established role in autophagy modulation [10, 19, 25], including its role in forming tunneling nanotube-like protrusions [28]. These protrusions are thought to facilitate the selective transfer of membrane vesicles and organelles, including lysosomes and endosomes, between cells. Taking our data together with the previous work on Rhes, the high co-incidence of Absent type neurons with PHF-1 positive inclusions may be explained by the established role of Rhes in autophagy modulation [10, 19, 25].
While our findings suggest a causal relationship between Rhes clearance and tau proteinopathic lesions, our study design precludes us from determining the direction of the association. Hernandez and colleagues [10] demonstrated that pathogenic MAPT mutations initially suppress age-related increases in Rhes, indicating that tau inclusions may drive neurons towards the Absent phenotype. Conversely, the abundance of Punctiform type neurons without PHF-1 positive tau inclusions suggests that changes in Rhes may precede tau accumulation.
Our observations provide further explanation for findings in Hernandez et al. [10]. They demonstrated that Lonafarnib effectively prevented the accumulation of tau proteinopathic lesions in model systems but was not effective in clearing existing tau aggregations. If end-stage neurons feature an absence of Rhes, they would lack the druggable target for Lonafarnib to act. A notable proportion of PHF-1 positive neurons (23.6–39.7%) had an absent Rhes phenotype in our sample, possibly explaining why Lonafarnib did not promote significant clearance of tau aggregations in model systems.
The strong association between neuronal tau inclusions and abnormal (i.e., Punctiform and Absent) Rhes phenotypes may explain why CBD and PSP showed only a minimal number of neurons with Rhes phenotypic changes. Most cortical tau aggregation in CBD and PSP occurs in glia [3], where we found little evidence of Rhes expression (Figure 1, Supplementary Figure 2). Despite this, the few cortical neurons with PHF-1 positive tau inclusions in PSP and CBD predominantly displayed either a Punctiform or Absent Rhes phenotype, as seen in AD and PiD. Similarly, AGD showed minimal changes in neuronal Rhes; however, AGD is thought to be a relatively benign tauopathy, with most tau deposits occurring in dendritic buttons and oligodendroglia [5, 27].
Caution should be taken in interpreting the results, as our investigation has limitations. Postmortem studies are cross-sectional by nature. As such, we cannot assess longitudinal changes in neuronal Rhes phenotypes in tauopathies and precisely how they mechanistically relate to tau. For this reason, our study should be interpreted with others using model systems [10]. Additionally, we have not yet identified sub-cellular structures that colocalize with the puncta observed in Punctiform Rhes, nor have we resolved the pathophysiologic changes that accompany the PHF-1 negative Punctiform neurons. Gaining a better mechanistic understanding of the Rhes neuronal phenotypes in the manner of Figure 6 would be essential for precise manipulation of Rhes as a therapeutic target. Finally, although we counted thousands of neurons, they only represented a fraction of the EC, CA1, and SFG and did not inform on other cortical regions affected by tauopathies. The primary outcomes here were agnostic to the neuropathological diagnosis. They focused on the relationship between tau proteinopathic inclusions and changes in neuronal Rhes phenotype in tauopathies as a whole class of diseases, to which we are sufficiently powered to make conclusions. However, we lacked the power to conduct an extensive investigation comparing each tauopathy.
This study also has several strengths. First, we used multiple independent datasets of RNA-seq data to validate our immunohistochemical findings. Additionally, we investigated well-characterized postmortem brain tissue of individuals with a range of sporadic tauopathies, making us well-positioned to examine the translatability of results drawn from model systems based on familial tauopathies. One limitation of in vivo and in vitro tauopathy models is the use of MAPT mutations. While also featuring tau aggregations, the specific pathophysiology and etiology of rare familial tauopathies have differences with the more common sporadic tauopathies studied here. It is infeasible to completely model the entire molecular and physiological milieu surrounding neurodegeneration in model systems. As such, it is unclear how generalizable the results of interventions in these model systems are. It is imperative to examine the conclusions of model systems in the context of human cases, as we have done here.
Lonafarnib is a well-tolerated drug with which there is existing clinical experience [6, 7, 12, 35]. Here, we show that the expression of a target of Lonafarnib, Rhes, is altered in tauopathies, reinforcing the conclusions drawn from model systems. Our findings here, in combination with the mechanistic work done by Hernandez and colleagues [10], lend support to the establishment of a clinical study of Lonafarnib for early intervention in tauopathies, including AD.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the UCSF Memory and Aging Center patients and their families for their contributions to this work. In particular, we thank those who have donated their brains to the Neurodegenerative Disease Brain Bank. We also thank the brain bank staff, without whom this work would not be possible. Additionally, we thank the Grinberg lab staff for their technical and administrative assistance with this work.
Microscopy was done at the Cancer Research Lab Molecular Imaging Center at the University of California, Berkeley. The UC Berkeley Biological Faculty Research Fund provided financial support for the appropriate equipment. We thank Feather Ives and Holly Aaron, Ph.D., for their training and assistance.
This study was supported by National Institute on Aging grants K24AG053435, K08AG052648, R01AG062359, F30AG066418, and R56AG057528 as well as National Institute of Neurological Disorders and Stroke grant U54NS100717-04 with additional support from Institutional grants NIH P30 AG062422 and P01AG019724, the Rainwater Charitable Foundation, and the Larry L. Hillblom Foundation.
REFERENCES
- 1.Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: 239–259 Doi 10.1007/bf00308809 [DOI] [PubMed] [Google Scholar]
- 2.Caballero B, Wang Y, Diaz A, Tasset I, Juste YR, Stiller B, Mandelkow EM, Mandelkow E, Cuervo AM (2018) Interplay of pathogenic forms of human tau with different autophagic pathways. Aging Cell 17: Doi 10.1111/acel.12692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coughlin DG, Dickson DW, Josephs KA, Litvan I (2021) Progressive Supranuclear Palsy and Corticobasal Degeneration. Adv Exp Med Biol 1281: 151–176 Doi 10.1007/978-3-030-51140-1_11 [DOI] [PubMed] [Google Scholar]
- 4.DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, Shen T, Tran H, Nichols B et al. (2017) Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med 9: Doi 10.1126/scitranslmed.aag0481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrer I, Santpere G, van Leeuwen FW (2008) Argyrophilic grain disease. Brain 131: 1416–1432 Doi 10.1093/brain/awm305 [DOI] [PubMed] [Google Scholar]
- 6.Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD et al. (2012) Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 109: 16666–16671 Doi 10.1073/pnas.1202529109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon LB, Massaro J, D’Agostino RB Sr., Campbell SE, Brazier J, Brown WT, Kleinman ME, Kieran MW, Progeria Clinical Trials C (2014) Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation 130: 27–34 Doi 10.1161/CIRCULATIONAHA.113.008285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hebert LE, Weuve J, Scherr PA, Evans DA (2013) Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 80: 1778–1783 Doi 10.1212/WNL.0b013e31828726f5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heinsen H, Strik M, Bauer M, Luther K, Ulmar G, Gangnus D, Jungkunz G, Eisenmenger W, Gotz M (1994) Cortical and striatal neurone number in Huntington’s disease. Acta Neuropathol 88: 320–333 Doi 10.1007/bf00310376 [DOI] [PubMed] [Google Scholar]
- 10.Hernandez I, Luna G, Rauch JN, Reis SA, Giroux M, Karch CM, Boctor D, Sibih YE, Storm NJ, Diaz A et al. (2019) A farnesyltransferase inhibitor activates lysosomes and reduces tau pathology in mice with tauopathy. Sci Transl Med 11: Doi 10.1126/scitranslmed.aat3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hochgrafe K, Sydow A, Matenia D, Cadinu D, Konen S, Petrova O, Pickhardt M, Goll P, Morellini F, Mandelkow E et al. (2015) Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human Tau. Acta Neuropathol Commun 3: 25 Doi 10.1186/s40478-015-0204-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kieran MW, Packer RJ, Onar A, Blaney SM, Phillips P, Pollack IF, Geyer JR, Gururangan S, Banerjee A, Goldman S et al. (2007) Phase I and pharmacokinetic study of the oral farnesyltransferase inhibitor lonafarnib administered twice daily to pediatric patients with advanced central nervous system tumors using a modified continuous reassessment method: a Pediatric Brain Tumor Consortium Study. J Clin Oncol 25: 3137–3143 Doi 10.1200/JCO.2006.09.4243 [DOI] [PubMed] [Google Scholar]
- 13.Kohl NE, Mosser SD, deSolms SJ, Giuliani EA, Pompliano DL, Graham SL, Smith RL, Scolnick EM, Oliff A, Gibbs JB (1993) Selective inhibition of ras-dependent transformation by a farnesyltransferase inhibitor. Science 260: 1934–1937 Doi 10.1126/science.8316833 [DOI] [PubMed] [Google Scholar]
- 14.Leng K, Li E, Eser R, Piergies A, Sit R, Tan M, Neff N, Li SH, Rodriguez RD, Suemoto CK et al. (2021) Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat Neurosci: Doi 10.1038/s41593-020-00764-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez A, Lee SE, Wojta K, Ramos EM, Klein E, Chen J, Boxer AL, Gorno-Tempini ML, Geschwind DH, Schlotawa L et al. (2017) A152T tau allele causes neurodegeneration that can be ameliorated in a zebrafish model by autophagy induction. Brain 140: 1128–1146 Doi 10.1093/brain/awx005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122: 111–113 Doi 10.1007/s00401-011-0845-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK et al. (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65: 1863–1872 Doi 10.1212/01.wnl.0000187889.17253.b1 [DOI] [PubMed] [Google Scholar]
- 18.Mead E, Kestoras D, Gibson Y, Hamilton L, Goodson R, Jones S, Eversden S, Davies P, O’Neill M, Hutton M et al. (2016) Halting of Caspase Activity Protects Tau from MC1-Conformational Change and Aggregation. J Alzheimers Dis 54: 1521–1538 Doi 10.3233/JAD-150960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mealer RG, Murray AJ, Shahani N, Subramaniam S, Snyder SH (2014) Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy. J Biol Chem 289: 3547–3554 Doi 10.1074/jbc.M113.536912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA et al. (2015) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21: 1154–1162 Doi 10.1038/nm.3951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS et al. (2012) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123: 1–11 Doi 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munoz DG, Dickson DW, Bergeron C, Mackenzie IR, Delacourte A, Zhukareva V (2003) The neuropathology and biochemistry of frontotemporal dementia. Ann Neurol 54 Suppl 5: S24–28 Doi 10.1002/ana.10571 [DOI] [PubMed] [Google Scholar]
- 23.Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19: 983–997 Doi 10.1038/nm.3232 [DOI] [PubMed] [Google Scholar]
- 24.Onyike CU, Diehl-Schmid J (2013) The epidemiology of frontotemporal dementia. Int Rev Psychiatry 25: 130–137 Doi 10.3109/09540261.2013.776523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan J, Song E, Cheng C, Lee MH, Yeung SC (2009) Farnesyltransferase inhibitors-induced autophagy: alternative mechanisms? Autophagy 5: 129–131 Doi 10.4161/auto.5.1.7329 [DOI] [PubMed] [Google Scholar]
- 26.Piras A, Collin L, Gruninger F, Graff C, Ronnback A (2016) Autophagic and lysosomal defects in human tauopathies: analysis of postmortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun 4: 22 Doi 10.1186/s40478-016-0292-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez RD, Suemoto CK, Molina M, Nascimento CF, Leite RE, de Lucena Ferretti-Rebustini RE, Farfel JM, Heinsen H, Nitrini R, Ueda K et al. (2016) Argyrophilic Grain Disease: Demographics, Clinical, and Neuropathological Features From a Large Autopsy Study. J Neuropathol Exp Neurol 75: 628–635 Doi 10.1093/jnen/nlw034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramaniam S (2020) Rhes Tunnels: A Radical New Way of Communication in the Brain’s Striatum? Bioessays: e1900231 Doi 10.1002/bies.201900231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Subramaniam S, Napolitano F, Mealer RG, Kim S, Errico F, Barrow R, Shahani N, Tyagi R, Snyder SH, Usiello A (2011) Rhes, a striatal-enriched small G protein, mediates mTOR signaling and L-DOPA-induced dyskinesia. Nat Neurosci 15: 191–193 Doi 10.1038/nn.2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tacik P, Sanchez-Contreras M, Rademakers R, Dickson DW, Wszolek ZK (2016) Genetic Disorders with Tau Pathology: A Review of the Literature and Report of Two Patients with Tauopathy and Positive Family Histories. Neurodegener Dis 16: 12–21 Doi 10.1159/000440840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takada LT (2015) The Genetics of Monogenic Frontotemporal Dementia. Dement Neuropsychol 9: 219–229 Doi 10.1590/1980-57642015DN93000003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Theofilas P, Ehrenberg AJ, Nguy A, Thackrey JM, Dunlop S, Mejia MB, Alho AT, Paraizo Leite RE, Rodriguez RD, Suemoto CK et al. (2018) Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer’s disease Braak stages: a quantitative study in humans. Neurobiol Aging 61: 1–12 Doi 10.1016/j.neurobiolaging.2017.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.VandeVrede L, Boxer AL, Polydoro M (2020) Targeting tau: Clinical trials and novel therapeutic approaches. Neurosci Lett: 134919 Doi 10.1016/j.neulet.2020.134919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vitucci D, Di Giorgio A, Napolitano F, Pelosi B, Blasi G, Errico F, Attrotto MT, Gelao B, Fazio L, Taurisano P et al. (2016) Rasd2 Modulates Prefronto-Striatal Phenotypes in Humans and ‘Schizophrenia-Like Behaviors’ in Mice. Neuropsychopharmacology 41: 916–927 Doi 10.1038/npp.2015.228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yust-Katz S, Liu D, Yuan Y, Liu V, Kang S, Groves M, Puduvalli V, Levin V, Conrad C, Colman H et al. (2013) Phase 1/1b study of lonafarnib and temozolomide in patients with recurrent or temozolomide refractory glioblastoma. Cancer 119: 2747–2753 Doi 10.1002/cncr.28031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Hernandez I, Rei D, Mair W, Laha JK, Cornwell ME, Cuny GD, Tsai LH, Steen JA, Kosik KS (2013) Diaminothiazoles modify Tau phosphorylation and improve the tauopathy in mouse models. J Biol Chem 288: 22042–22056 Doi 10.1074/jbc.M112.436402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G et al. (2016) Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89: 37–53 Doi 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.