

Abstract
Effective antiviral immunity requires generation of T and B lymphocytes expressing the transcription factor T-bet, a regulator of type 1 inflammatory responses. Using T-bet expression as an endogenous marker for cells participating in a type 1 response, we report coordinated interactions of T-bet-expressing T and B lymphocytes based on their dynamic co-localization at the T cell zone and B follicle boundary (T-B boundary) and germinal centers (GC) during lung influenza infection. We demonstrate that the assembly of this circuit takes place in distinct anatomical niches within the draining lymph node, guided by CXCR3 that enables positioning of TH1 cells at the T-B boundary. The encounter of B and TH1 cells at the T-B boundary enables IFNγ produced by the latter to induce IgG2c class switching. Within GCs, T-bet+ TFH cells represent a specialized stable sub-lineage required for GC growth, but dispensable for IgG2c class switching. Our studies show that during respiratory viral infection, T-bet-expressing T and B lymphocytes form a circuit assembled in a spatiotemporally controlled manner that acts as a functional unit enabling a robust and coherent humoral response tailored for optimal antiviral immunity.
One-sentence summary
T-bet-expressing T and B lymphocytes form a spatiotemporal circuit that enables optimal antiviral humoral immunity.
Introduction
The immune system of higher organisms is dependent on a combination of prepositioned sentinels and circulating cells that traffic to secondary lymphoid organs, spleen, LNs (LN) and Peyer’s patches for tissue surveillance and defense against pathogens (1). Adaptive immune cells, T and B lymphocytes, are spatially segregated in the secondary lymphoid organs to the T cell zone (T zone) and B follicle, respectively. Yet cognate interactions between antigen-specific B cells and CD4+ T cells are required for long-lasting protective humoral responses to and vaccination against infectious agents, processes that include immunoglobulin (Ig) class switching and antibody affinity maturation (2). Beyond the well-known partitioning of secondary lymphoid organs into the T zone and B follicles, distinct positioning of different cell types within discrete areas of secondary lymphoid organs has been shown to contribute to processes ranging from pathogen containment to helper T cell differentiation, highlighting a role for cellular positioning and compartmentalized intercellular interactions in the course of the immune response (3-8).
After ingesting a pathogen or its products in infected tissues, dendritic cells (DCs) migrate to the T zones of tissue-draining LN where they activate, or prime, rare recirculating naïve antigen-specific T cells expressing cognate T cell receptors (TCR) (1). During priming, TCR and co-stimulatory receptor signaling along with cytokines released by DCs and other cells elicit differentiation of distinct effector CD4+ T cell types: TH1, TH2, and TH17, defined by expression of the transcription factors T-bet, GATA3, and RORγt, respectively (9). These specialized effector T cells are tailored to coordinate and mediate protective cellular and humoral immunity commensurate to the class of the invading pathogen. Thus, LNs also serve as the setting where T cells are exposed to cues specialized for the establishment of pathogen-specific effector programs. The transcription factors that define these programs not only induce the expression of distinct sets of effector molecules, but also of certain chemokines, chemokine receptors and adhesion molecules that allow migration of effector cells to sites of inflammation (10-12).
These observations raise the possibility that coordinated positioning of pathogen-specific lymphocytes in discrete locations within the LN enables their cooperative interactions leading to concordant expression of an appropriate specific pro-inflammatory effector program, culminating in protective immunity. Viral infections elicit type 1 immunity, which necessitates inter-lineage cell cooperation for the deployment of a spectrum of effector mechanisms, including antiviral mediators, cytotoxic T cells and NK cells, and neutralizing antibodies that collectively promote viral clearance. Expression of the transcription factor T-bet, known to orchestrate type 1 immunity, is induced in several innate and adaptive immune cell types, including CD4+ T cells and B cells (13, 14). While T-bet-expressing B cells have been implicated in autoantibody production in lupus, other studies have also suggested that T-bet expression in B cells plays a role during antiviral responses to acute and chronic infections: promoting class switching to IgG2a (IgG2c in C57Bl/6 mice), their differentiation into antibody-secreting cells and memory B cells, and viral clearance (15-20). It is noteworthy that class switching to IgG2a/IgG2c dominates antiviral B cell responses and has been linked to effective viral clearance (21-23).
While different DC subsets have been suggested to prime CD4+ and CD8+ T cells in discrete areas within the secondary lymphoid organs (6, 7, 24, 25), it remains unknown whether and how spatial and temporal localization of different helper T cell subsets post-priming contributes to the coordination of humoral responses during the course of infection. In this regard, the T cell zone and B follicle boundary (T-B boundary) and the interfollicular zones of the secondary lymphoid organs are of a particular interest, as these sites can serve as potential areas of encounters between activated antigen-specific B cells and CD4+ T cells (26, 27). These interactions are thought to precede B cell entry into germinal centers (GC), where clonal expansion, somatic hypermutation, affinity maturation as well as development of B cell memory and long-lived plasma cells occurs (28). Besides the aforementioned niches, CD4+ T cells that differentiate into T follicular helper (TFH ) cells enter the B cell follicle where they provide support to GC B cells (2). In addition to TH1 and B cells, T-bet expression in TFH cells has also been described, yet a role of T-bet expressing TFH cells in isotype switching or GC development is unknown (29-31).
Broad expression of T-bet among T and B cell lineages raises the possibility of this transcription factor having a major role in coordinating type 1 immune responses by inducing expression of receptors and effector molecules that facilitate interaction and cooperation between pertinent cell types. T-bet expression in different T and B lymphocyte lineages has been proposed to support mutually independent functionality of individual T-bet-expressing lymphocyte subtypes acting in a modular manner to facilitate different aspects of humoral immune response to infection or vaccination (30) . Here, we sought to test the alternative conception that during viral infection, distinct types of T-bet-expressing T and B lymphocytes form a functional circuit assembled to promote antiviral humoral immunity. Through the use of mice carrying a fluorescent reporter and Cre recombinase expressed under the control of the endogenous Tbx21 locus we were able to analyze the localization and genetically manipulate T-bet-expressing lymphocyte lineages during viral infection. We find coordinated T-bet expression in key lymphocyte lineages—TH1 cells, TFH cells and GC B cells—localizing to distinct regions within the draining lymph node. Furthermore, the disruption of the T-bet+ T-B cell circuit by ablating its individual cellular component or disrupting their localization hinders the coherency and magnitude of the influenza antibody response.
Results
T-bet+ CD4+ T cells show distinct intranodal localization during influenza infection.
We first sought to investigate the spatial distribution of T-bet-expressing lymphocytes in lung-draining mediastinal LNs during the course of intranasal influenza virus infection in mice carrying the previously characterized Tbx21tdTomato-T2Acre knock-in allele (Tbx21-Cre), which faithfully reports endogenous T-bet expression in all major immune cell subsets (32). We analyzed the number of T-bet-expressing effector CD4+ T cells (RFP+CD4+CD44+ cells) at the T-B boundary (50μm area between the T cell zone and the B follicles), the deep T zone (T cell zone minus T-B boundary), the B follicles, the interfollicular zone (between B follicles), the medulla and the subcapsullar sinus at day 0, 7, 14 and 21 after infection with the PR8 influenza A virus strain (Fig. S1). While the number of total and influenza nucleoprotein (NP)-specific RFP+CD4+ T cells increased over 21 days of infection (Fig. 1, A and B, Fig. S1C and D), they showed distinct time-dependent localization patterns across the deep T zone, the T-B boundary and the B follicles (Fig. 1,C and D and Fig. S1A and B). At day 7, a large proportion of RFP+CD4+ T cells was found in the deep T zone, which decreased at later time points (Fig. 1, C and D). In contrast, the proportion of RFP+CD4+ T cells at the T-B boundary remained relatively steady throughout the time course of the response with a notable increase at day 14 when 45% of all RFP+CD4+ cells were found at the T-B boundary followed by their increasing accumulation in the B follicles between day 14 and 21 (Fig. 1, C to E). The enrichment of CD4+RFP+ T cells at the boundary of the B follicle was consistent with the possibility of T-bet-expressing T and B lymphocytes forming a spatiotemporal circuit.
Figure 1. T-bet+ CD4+ T cells show distinct temporal intranodal localization during influenza infection.

Tbx21-Cre mice were intranasally infected with 50 TCID50 influenza (PR8). Mediastinal lymph nodes were collected on indicated time points and analyzed by flow cytometry and confocal microscopy. (A and B) Cell numbers of (A) CD4+CD44+RFP+ and (B) NP+CD4+CD44+RFP+ T cells. (C) Representative confocal micrographs of indicated lymph node areas at designated time points during infection. Scale bars, 10 μm. (D) Percentage of CD4+CD44+RFP+ T cells in indicated lymph node areas. (E) Percent of CD4+CD44+RFP+ cells of T-zone found in T-B boundary (T-B boundary / (deep T zone + T-B boundary) x 100). (A, B and D) The data are shown as mean ± s.e.m. (E) Each point represents an individual mouse with mean ± s.e.m. All data are representative of ≥ 2 experiments, n ≥ 3 mice per group.
T-bet+ B cells dominate GC and are required for virus-specific IgG response to influenza infection.
As expected from previous reports, we found that influenza infection induced T-bet expression in B cells (Fig. S2A). We first detected RFP+ B cells in the B follicles and in germinal centers (GC) (Fig. 1C, Fig. 2A). To more carefully characterize virus-specific T-bet-expressing B cell responses by flow cytometry, we utilized an influenza hemagglutinin (HA) probe with a structure-based modification (33). A sizeable population of HA-specific GC B cells (HA+B220+GL7+Fas+) was detected by day 14 in the mediastinal LNs (Fig. 2B and Fig. S2B) with over 80% of the bulk population of GC B cells and HA-specific GC B cells expressing RFP (Fig. 2, C and F). Among GC B cells, RFP expression was enriched and was the highest on a per cell basis in HA-specific GC cells that underwent IgG2c class switching compared to those that had switched to IgG1 (Fig. 2, D and E). These observations were consistent with previous reports showing that T-bet regulates switching to IgG2c in GC B cells (15-20). To test if the induction of T-bet in GC B cells is specific to type 1 responses, we infected Tbx21-Cre mice with Nippostongylus brasiliensis, a pathogenic helminth inducing type 2 immunity that also infects the lung. Despite stimulating a potent GC B cell response in the mediastinal LNs, RFP expression in GC B cells was not induced by infection with N. brasiliensis, suggesting that T-bet expression in GC B cells is not part of a general GC B cell program (Fig. S2C).
Figure 2. T-bet expression in GC B cells during influenza infection.

Tbx21-Cre mice were intranasally infected with 50 TCID50 influenza (PR8). Mediastinal lymph nodes were collected on indicated time points and analyzed by flow cytometry and confocal microscopy. (A) Representative confocal micrograph of day 14 infected or control mediastinal lymph node. Scale bars, 50 μm. T: T zone; B: B follicle; GC: germinal center. (B) Numbers of HA+ GC B cells (HA+B220+GL7+Fas+). (C) Percent RFP+ of GC B cells (B220+GL7+Fas+) and HA+ GC B cells (B220+GL7+Fas+HA+). (D) Percent RFP+ of IgG1+ GC B cells and IgG2c+ GC B cells on PBS (Ctrl) and day 21 after infection (Infected). (E) Mean fluorescence intensity (MFI) of RFP in IgG1+ GC B cells IgG2c+ GC B cells on PBS (Ctrl) and day 21 of infection (Infected). (F) Representative confocal micrograph of germinal center on day 14 of influenza infected mediastinal lymph node. Scale bars, 50 μm. (B and C) The data are shown as mean ± s.e.m. (D and E) Each point represents an individual mouse with mean ± s.e.m. All data are representative of ≥ 2 experiments, n ≥ 3 mice per group. Two-tailed t-test (***P < 0.001, **P < 0.01, and **P < 0.05).
To test the contribution of T-bet-expressing B cells to the humoral antiviral response, we generated Pax5f/fTbx21-Cre mice, which enabled selective ablation of T-bet-expressing B cells upon deletion of Pax5, a B cell lineage specifying transcription factor required for B cell differentiation and maintenance (34, 35). Pax5f/fTbx21-Cre mice showed no changes in overall B cell numbers, but a complete loss of total IgG2c serum titers compared to littermate controls, with no significant change in other isotypes (Fig. S2, D and E). However, we observed a severe decrease in the frequency and number of both bulk and HA-specific GC B cells induced by influenza infection in these mice (Fig. 3, A to C). This was at least partially due to a decrease in the IgG2c GC B cell population as reflected by the markedly reduced frequency of IgG2c+ cells within the total GC B cell pool (Fig. 3D). Consistent with a decrease in the GC population, we observed that influenza-specific total IgG and IgG2c antibody titers were substantially reduced in Pax5f/fTbx21-Cre mice compared to littermate controls (Fig. 3, E and F). These data show that the loss of T-bet+ B cells severely impaired virus-specific IgG production due to a loss of GC B cells.
Figure 3. T-bet+ B cells are required for anti-viral IgG production.
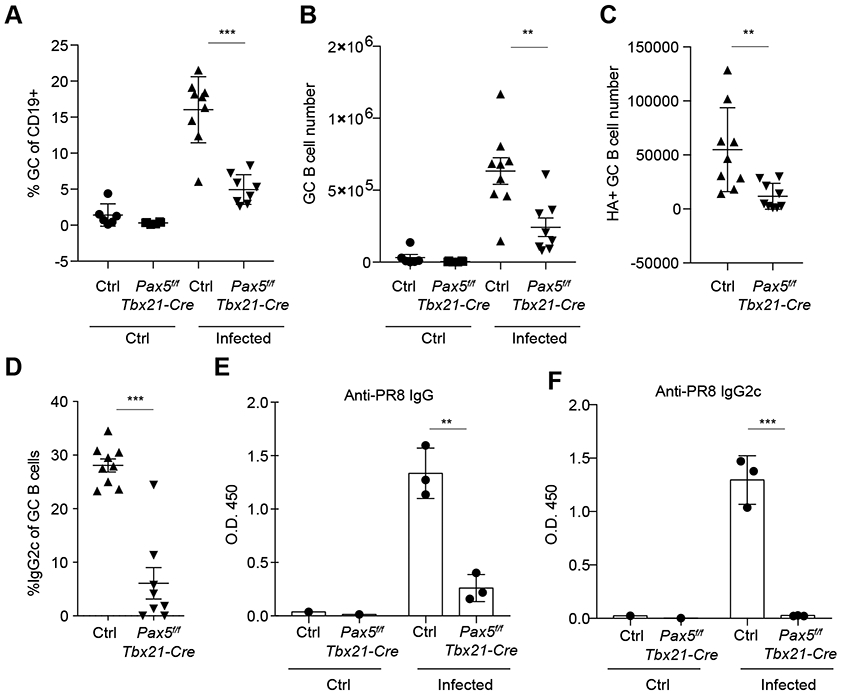
Pax5f/fTbx21-Cre and littermate controls (Pax5wt/fTbx21-Cre and Pax5wt/wtTbx21-Cre or Pax5wt/fTbx21-Cre) were intranasally infected with 50 TCID50 influenza (PR8) and analyzed 21 days after infection. (A) Frequency of GC B cells (GL7+Fas+) of total B cells. (B and C) Numbers of GC B cells (B220+GL7+Fas+) (B) and HA+ GC B cells (B220+GL7+Fas+HA+) (C). (D) Frequency of IgG2c+ of GC B cells in infected mice (B220+GL7+Fas+IgG2c+). (E and F) Anti-influenza antibodies were analyzed in serum by ELISA 21 days after influenza infection. Anti-influenza total IgG (E) and IgG2c (F). Each point represents an individual mouse with mean ± s.e.m. All data are representative of ≥ 2 experiments, n ≥ 3 mice per group. Two-tailed t-test (***P < 0.001, and **P < 0.01).
CXCR3 signaling is required for T-bet+ CD4+ T cell enrichment at the T-B boundary.
As Ig class switch recombination has been recently shown to initiate in B cells outside GC and T-bet is required for IgG2c switching, we reasoned that B cells may be exposed to signals at the T-B boundary which promote T-bet expression, IgG2c class switching and GC B cell differentiation (36). Given that T-bet-expressing GC B cells are essential for antiviral antibody production, we next asked whether the observed co-localization of T-bet-expressing RFP+ T cells and B cells at the T-B boundary was required for antiviral humoral response (Fig. 2A). T-bet directly regulates a set of genes important for TH1 cell function, including Cxcr3, encoding the chemokine receptor for the interferon-inducible chemokines CXCL9, CXCL10, and CXCL11 (10, 13, 37), of which only CXCL9 and CXCL10 are expressed in C57BL/6 mice (38). CXCR3 expression is required for positioning of effector CD4+ T cells to interfollicular peripheral T zone of the LN (39). Using immunofluorescence, we observed that in contrast to its low levels in T zone, CXCL9 was abundant in the B follicles throughout the course of infection with the difference peaking at day 14 (2.4-fold), coinciding with the maximum accumulation of T-bet-expressing CD4+ T cells at the T-B boundary (Fig. 4, A and B, Fig. S3A, and Fig. 1E). In contrast, CXCL10 was present at similarly low levels in the T zone and B follicle with its levels increasing in the B follicle after day 14 (Fig. 4, A and B and Fig. S3B). This was consistent with the idea of CXCR3-mediated recruitment of T-bet+ T cells to the T-B boundary and B follicle.
Figure 4. CXCR3 signaling is required for T-bet+ CD4+ T cell enrichment at the T-B boundary.
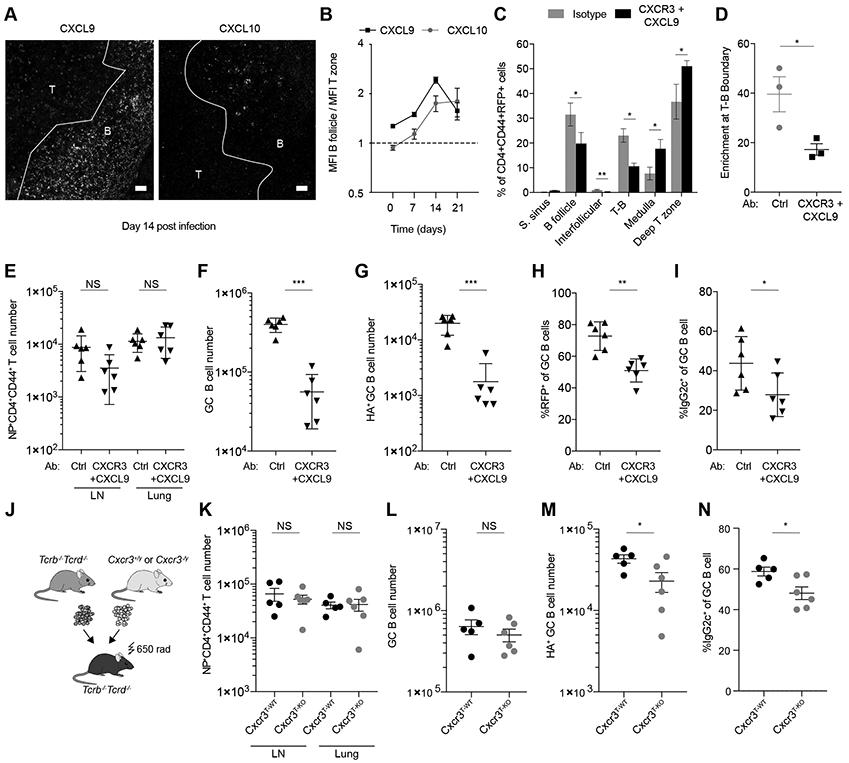
(A and B) Tbx21-Cre mice were intranasally infected with 50 TCID50 influenza (PR8). CXCL9 and CXCL10 were analyzed by immunofluorescence and confocal microscopy in mediastinal lymph nodes. (A) Representative confocal micrographs on day 14 after infection. Scale bars, 20 μm. T: T zone; B: B follicle. (B) Mean fluorescence intensity of CXCL9 and CXCL10 was quantified in T-zone and B follicles. Plot shows ratio of mean fluorescence intensity in B follicle over mean fluoresce intensity in T-zone at indicated time points after infection. (C to I) Tbx21-Cre mice were intranasally infected with 50 TCID50 influenza (PR8) and treated with CXCR3 and CXCL9 blocking antibodies or isotype controls. Mediastinal lymph nodes were collected at day 14 and analyzed by confocal microscopy and flow cytometry. (C) Quantification of percent of CD4+CD44+RFP+ T cells in indicated lymph node areas. (D) Percent of CD4+CD44+RFP+ cells of T-zone found in T-B boundary (T-B boundary / (deep T-zone + T-B boundary) x 100). (E) Numbers of NP+CD4+CD44+ T cells in mediastinal lymph nodes and lung. (F) Numbers of GC B cells (B220+GL7+Fas+). (G) Numbers of HA+ GC B cells (HA+B220+GL7+Fas+). (H) Percent RFP+ of GC B cells (B220+GL7+Fas+). (i) Percent IgG2c+ of GC B cells (B220+GL7+Fas+). (J to N) Lethally irradiated Tcrb−/−Tcrd−/− mice were reconstituted with bone marrow cells from Tcrb−/−Tcrd−/− mice and either Cxcr3+/y or CxCr3−/y mice mixed at a 4:1 ratio. After reconstitution mice were infected with 50 TCID50 influenza (PR8) and analyzed 14 days post-infection. (J) Schematic of bone marrow chimera generation. (K) Numbers of NP tetramer+CD4+CD44+ T cells in mediastinal lymph nodes and lung. (L) Numbers of GC B cells (B220+GL7+Fas+). (M) Numbers of HA+ GC B cells (B220+GL7+Fas+HA+). (N) Percent IgG2c+ of GC B cells (B220+GL7+Fas+). (E to N) Intravascular labeling was performed by injecting fluorophore-conjugated CD45 antibody i.v., labeled cells were gated out in this analysis. (B and C) The data are shown as mean ± s.e.m. (D to N) Each point represents an individual mouse with mean ± s.e.m. All data are representative of ≥ 2 experiments, n ≥ 3 mice per group. Two-tailed t-test (***P < 0.001, **P < 0.01, *P< 0.05, and NS = not significant).
To test if CXCR3 is required for the migration of T-bet+CD4+ T cells to the boundary, we blocked CXCR3-ligand interactions with a combination of CXCR3 and CXCL9 blocking antibodies, as the CXCR3 antibody blocks CXCL10, but not CXCL9 signaling (40). While CXCR3 blockade markedly perturbed the distribution of RFP+CD4+ T cells, resulting in their preferential accumulation in the T zone and a pronounced decrease at the T-B boundary and in the B follicles (Fig. 4, C and D and Fig. S4, A and B), it did not affect the overall numbers of NP-specific effector CD4+ T cells in the lungs and draining LNs of infected mice (Fig. 4E). The diminished localization of RFP+CD4+ T cells at the T-B boundary in these mice was accompanied by an impaired B cell response reflected by diminished numbers of total and HA-specific GC B cells (Fig. 4, F and G). Furthermore, the RFP+ frequency of the remaining GC B cells was lower compared to control mice (Fig. 4H). Consistent with a decrease in RFP+ GC B cells, the proportion of IgG2c+ GC B cells were also decreased (Fig. 4I). These data suggest that the localization of T-bet+ T cells at the T-B boundary is required to induce proper responses of B cells, likely by providing them with cues for T-bet induction and entry into GCs, and by ensuring a pool of activated and pre-positioned cells that can differentiate into T follicular helper cells (TFH) and access the B follicle.
Next, we assessed a requirement for CXCR3 expression by T cells for GC formation and IgG2c switching. We generated mixed bone marrow (BM) chimeras by transferring BM precursors from either CXCR3-deficient or -sufficient Cxcr3−/y or Cxcr3+/y littermate mice combined with BM precursors from T cell-deficient Tcrb−/−Tcrd−/− mice at a 1:4 ratio into irradiated Tcrb−/−Tcrd−/− recipients (Fig. 4J). These mice (referred as Cxcr3T-KO or Cxcr3T-WT) harbor only CXCR3-deficient or -sufficient T cells derived from the Cxcr3−/y or Cxcr3wt/y BM cells, respectively, while the rest of hematopoietic cells can be derived from the Cxcr3-sufficient mixed donor (Fig. S4C). Following influenza infection, Cxcr3T-KO and Cxcr3T-WT mice showed comparable overall NP-specific CD4+ T cell numbers in the draining LNs and total number of GC B cells (Fig. 4, K and L). However, HA-specific GC B cells and the proportion of IgG2c+ GC B cells were decreased in Cxcr3T-KO compared to Cxcr3T-WT controls (Fig. 4, M and N).
To test a requirement for CXCR3 expression in B cells, we generated chimeric Cxcr3B-KO or Cxcr3B-WT mice by transferring either Cxcr3−/y or Cxcr3+/WT mixed with muMt−/− BM precursors, respectively, into irradiated CD45.1 hosts at a 1:4 ratio (Fig. S5A). These mice harbor only CXCR3-deficient or -sufficient B cells derived from the Cxcr3−/y or Cxcr3wt/y BM cells, respectively, while the rest of hematopoietic cells can be derived from the Cxcr3-sufficient mixed donor (Fig. S5F). In contrast to T cell-intrinsic CXCR3 deficiency, Cxcr3B-KO and Cxcr3B-WT mice showed no difference in the numbers of total and HA-specific GC B cells or frequency of IgG2c+ B cells following influenza infection (Fig. S5, B to E). These results suggest a cell-intrinsic role for CXCR3 expression in T cells, but not B cells, in inducing efficient virus-specific IgG2c humoral responses. A lesser effect of T cell-restricted CXCR3 deficiency on GC responses compared to that observed upon acute CXCR3 blockade treatment may be due to a partial compensation of the genetic defect by other TH1 chemokine receptors.
IFNγ production by CD4+ T cells is required for class switch recombination to IgG2c.
The requirement for CXCR3 expression by T cells for their proper localization to the T-B boundary and for switching to IgG2c by GC B cells suggests that signals derived from T-bet-expressing T cells plays a crucial role in driving T-bet expression in B cells after activation. A likely candidate for such a signal is IFNγ, as STAT1 activation has been shown to be required for T-bet expression in B cells in lupus-prone B6.Sle1b mice and IFNγ–induced T-bet expression in B cells in vitro (41, 42). IFNγ is produced by a wide range of lymphoid cell types in the LN during the course of viral infection including NK cells, NKT cells, γδT cells, ILC1 and CD4+ and CD8+ T cells. The location of specific IFNγ-producing cells likely plays a critical role in ensuring targeted delivery of IFNγ, as they inhabit different niches and their mislocalization results in defective IFNγ production (43, 44). Given that CD4+T-bet+ T cells are enriched at the T-B boundary, we hypothesized that they serve as key producers of the IFNγ required to locally drive T-bet expression in B cells and subsequent switching to IgG2c. Consistent with this possibility, we observed higher numbers and proportion of B cells expressing phosphorylated STAT1 (pSTAT1) outside of GCs, suggesting that the induction of T-bet expression in B cells likely occurs prior to their entry into the GC (Fig. 5, A to C).
Figure 5. IFNγ derived from CD4+ T cells is required for class switch recombination to IgG2c.

(A to C) Tbx21-Cre mice were intranasally infected with 50 TCID50 influenza (PR8). Mediastinal lymph nodes were collected at day 14 post infection and analyzed by confocal microscopy. (A) Representative confocal micrographs. Scale bars, 10 μm. T: T zone; B: B follicle; GC: germinal center. (B) Number of pSTAT1+B220+ cells inside and outside GCs. (C) Frequency of pSTAT+ of B cells (B220+) inside and outside GCs. (D to J) Ifngf/fCD4CreERT2/wt (IfngΔ) and Ifngf/wtCD4CreERT2/wt littermate controls (Ctrl) were gavaged with tamoxifen at 3 and 1 days prior to intranasal infection with 50 TCID50 influenza (PR8) and analyzed 14 days after infection. (D) Diagram of tamoxifen (TAM) regimen and infection. (E to I) Numbers of CD4+CD44+ and CD8+CD44+ T cells (E), NP+CD4+CD44+ T cells (F), CD4+CD44+PD-1HiCXCR5Hi T cells (G), GC B cells (B220+GL7+Fas+) (H), and HA+ GC B cells (B220+GL7+Fas+HA+) (I) in mediastinal LN. (J) Percent IgG2c+ of GC B cells (B220+GL7+Fas+) in mediastinal LN. (B to J) Each point represents an individual mouse with mean ± s.e.m. All data are representative of ≥ 2 experiments, n ≥ 4 mice per group. Two-tailed t-test (***P < 0.001, *P< 0.05, and NS = not significant).
To test the effect of IFNγ derived specifically from CD4+ T cells on GC responses, we induced acute ablation of a conditional Ifng allele in CD4+ T cells upon administration of tamoxifen to IfngfloxCd4Cre-ERT2 mice (Fig. 5D, and Fig. S6, A and B). While ablation of Ifng in CD4 T cells during influenza infection did not affect the number of effector CD4+ and CD8+ T cells, TFH cells or GC B cells (Fig. 5, E to I), it did cause a marked reduction in the proportion on GC B cells switching to IgG2c compared to control mice (Fig. 5J). The decrease in IgG2c switching was independent from a defect in the generation of T-bet+ TFH cells as we observed no difference in the frequency of T-bet+Bcl6+ T cells or in the level of T-bet expression on a per cell basis by TFH cells (Fig. S6, C and D). These data are consistent with a prominent role for locally produced IFNγ by CD4+ T cells within the LN and suggest that it is required for the coherency of the humoral response, as other cells abundant in the T zone that produce IFNγ, most prominently CD8+ T cells, fail to compensate for its loss in CD4+ T cells.
T-bet+ TFH cells facilitate GC formation but are dispensable for IgG2c isotype switching during influenza infection.
We found numbers of T-bet-expressing RFP+CD4+ T cells within the B follicles progressively increased over the course of the response to infection while numbers of RFP− effector CD4+ T cells remained low and largely unchanged (Fig. 6A). These observations were consistent with pronounced increases in the frequency and number of RFP+ TFH cells during the course of influenza infection (Fig. 6, B and C, and Fig. S7A). In contrast, infection with N. brasiliensis did not induce RFP expression in TFH cells, suggesting sustained T-bet expression in TFH cells is not a universal feature of response to infection (Fig. S7B). While T-bet expression in TFH cells has been described previously, its stability in TFH cells has remained controversial with both stable or transient expression of T-bet observed in different models of infection and vaccination (29-31). Thus, we assessed the stability of T-bet expression in TFH cells using Tbx21tdTomato-T2A-creERT2 mice harboring the Rosa26Lox-STOP-Lox-YFP recombination reporter (32). These mice were infected with influenza and provided with a single dose of tamoxifen by oral gavage 7 days after infection. One week after tamoxifen administration, the vast majority of YFP-tagged TFH cells continued to express RFP (Fig. 6D), in agreement with the aforementioned progressive increase in RFP+, but not RFP− CD4+ T cells in the B follicles over the course of infection suggesting that T-bet+ TFH cells represent a stable specialized cell subset.
Figure 6. T-bet+ TFH cells are induced during viral infection are a stable population functionally distinct from T-bet− TFH.

(A to C) Tbx21-Cre mice were intranasally infected with 50 TCID50 influenza (PR8). Mediastinal lymph nodes were collected on indicated time points and analyzed by confocal microscopy and flow cytometry. (A) Cell numbers of CD4+CD44+RFP+ and CD4+CD44+RFP− in B cell follicles. (B) Percent RFP+ of CD4+CD44+CXCR5HiPD-1Hi T cells analyzed by confocal microscopy. (C) Numbers of NP tetramer+CD4+CD44+CXCR5HiPD-1HiRFP+ T cells. (D) Tbx21tdTomato-T2A-creERT2Rosa26Lox-STOP-Lox-YFP mice were intranasally infected with 50 TCID50 influenza (PR8); 7 days after infection mice were gavaged with 1 dose of tamoxifen. Mice were analyzed on day 14 after infection. Percent RFP+ of YFP+CD4+CD44+CXCR5HiPD-1Hi T cells. (E and F) Tbx21-Cre; Bcl6-YFP/Bcl6WT; Foxp3Thy1.1 mice were intranasally infected with 50 TCID50 influenza (PR8). Mediastinal lymph nodes were collected on day 14. Indicated cell types were sorted and analyzed by RNA-seq. (E) Heatmap showing k-means clustering of genes differentially expressed in any pairwise comparison. Values are Z-score normalized by row. (F) Expression patterns of select IFN and IL-12 signaling, TFH signature and cell migration-related genes. (A to C) The data are shown as mean ± s.e.m. (D) Each point represents an individual mouse with mean ± s.e.m. All data are representative of ≥ 2 experiments, n ≥ 3 mice per group. (E and F) RNA-seq analysis of gene expression was performed using three biological replicates.
To gain further insight into shared and distinct features of TH1, T-bet+TFH and T-bet−TFH cells, we performed RNA sequencing (RNA-seq) of these cell subsets isolated from Tbx21tdTomato-T2Acre:Bcl6-YFP:Foxp3Thy1.1 reporter mice on day 14 of influenza infection (Fig. S7E). The corresponding populations were defined as Thy1.1−RFP+YFP− (TH1), Thy1.1−RFP+YFP+ (T-bet+ TFH), and Thy1.1−RFP−YFP+ (T-bet− TFH) cells. We identified clusters of genes uniquely, differentially, and commonly expressed by the 3 populations (Fig. 6E, Supplementary Table 1). TFH gene signature, including heightened expression of Bcl6, Tox2 and Trim8, was shared by RFP+TFH and RFP−TFH cells, but not TH1 cells (clusters 1 and 4) (Fig. 6F). We also found gene sets with shared expression between TH1 and RFP+TFH cells compared to RFP−TFH cells (clusters 2 and 3); notably, these included genes linked to type II (IFNγ) and type I (IFNα) and the IL-12 signaling pathways such as Ifng, Irf1 and Cxcl10 (Fig. 6F). This was consistent with the observed higher capacity of T-bet+ TFH cells to produce IFNγ upon re-stimulation compared to T-bet− TFH cells (Fig. S7C). Also of note, we found that a number of genes in cluster 5, which were expressed in both TFH subsets albeit higher in T-bet+ TFH cells, but not in TH1 cells, have been implicated in TFH function; this set included canonical TFH expressed genes Cxcr5, Pdcd1, and Il21 (Fig. 6F) (2). Moreover, we examined the expression of transcripts for migratory and homing molecules that could offer clues to differential intranodal localization of T-bet+ TFH and TH1 cells. While transcript levels of Cxcr3 and Gpr183 that have been linked to peripheral T zone or outer B follicle localization, respectively, were comparable in TH1 and RFP+TFH cells, CCR7 and PSGL1 (SelpIg) linked to T zone localization were expressed relatively higher in TH1 compared to RFP+TFH cells (Fig. 6F) (45-47). Conversely, Cxcr5 and Cxcr4 genes encoding receptors promoting entry into the B cell follicle and GCs were expressed at higher levels in T-bet+ TFH cells (Fig. 6F) (48, 49). These data are consistent with a scenario where T-bet-induced expression of CXCR3 enables localization of TH1 cells to the T-B boundary following CXCL9 and CXCL10 gradients, whereas Cxcr5 or Ccr7 and Selplg expression pattern may help guide these T-bet+ CD4+ T cell subsets to the B cell follicle (TFH) or retain them in the T zone (TH1). This model was consistent with the localization of T-bet+ TFH cells observed exclusively within the GCs in the B cell follicles (Fig. S7D).
To explore the role of T-bet+ TFH cells in virus-specific B cell responses, we generated Bcl6f/fTbx21-Cre mice, in which T-bet expression leads to deletion of Bcl6 and loss of these cells, which rely on Bcl6 for their differentiation and function (50-52). As both GC B cells and TFH cells express T-bet during influenza infection, mixed chimeras were generated by transferring BM cells from Bcl6f/fTbx21-Cre or control littermates and Tcrb−/−Tcrd−/− BM cells mixed at a 1:4 ratio into irradiated Tcrb−/−Tcrd−/−recipients (Fig. 7A). The resulting experimental (Bcl6f/fTbx21-Cre) and control (Bcl6+/+Tbx21-Cre) chimeras harbored Bcl6-sufficient T-bet-expressing B cells and either Bcl6-deficient or -sufficient T-bet+ T cells, respectively. Consistent with the observed T-bet expression by a large fraction of TFH cells, Bcl6f/fTbx21-Cre BM chimeras showed a 4.7-fold reduction in the number of TFH cells 14 days after influenza infection but no reduction in the total number of NP-specific effector CD4+ T cells in comparison to control group (Fig. 7, B to D). Accordingly, Bcl6f/fTbx21-Cre BM chimeras had reduced frequency and number of GC B cells compared to control BM chimeras in response to influenza infection (Fig. 7, E to G). However, we did not observe a defect in isotype switching as the proportion of GC B cells switched to IgG2c in mice lacking T-bet+ TFH cells remained unchanged compared to control mice (Fig. 7H). These results suggest that while the magnitude of the GC response to influenza virus is dependent on T-bet+TFH cells, isotype switching to IgG2c does not require T-bet+TFH cells. These observations support a model where temporally coordinated CXCR3-dependent migration of T-bet+ CD4+ T cells to the T-B boundary generates a localized pool of IFNγ that specifically promotes T-bet expression and subsequent class switching to IgG2c in B cells, which seed an appropriate virus-specific GC response whose magnitude is scaled by T-bet-expressing TFH cells (Fig. S8).
Figure 7. T-bet+ TFH cells are required for GC robustness, but dispensable for switching to IgG2c.
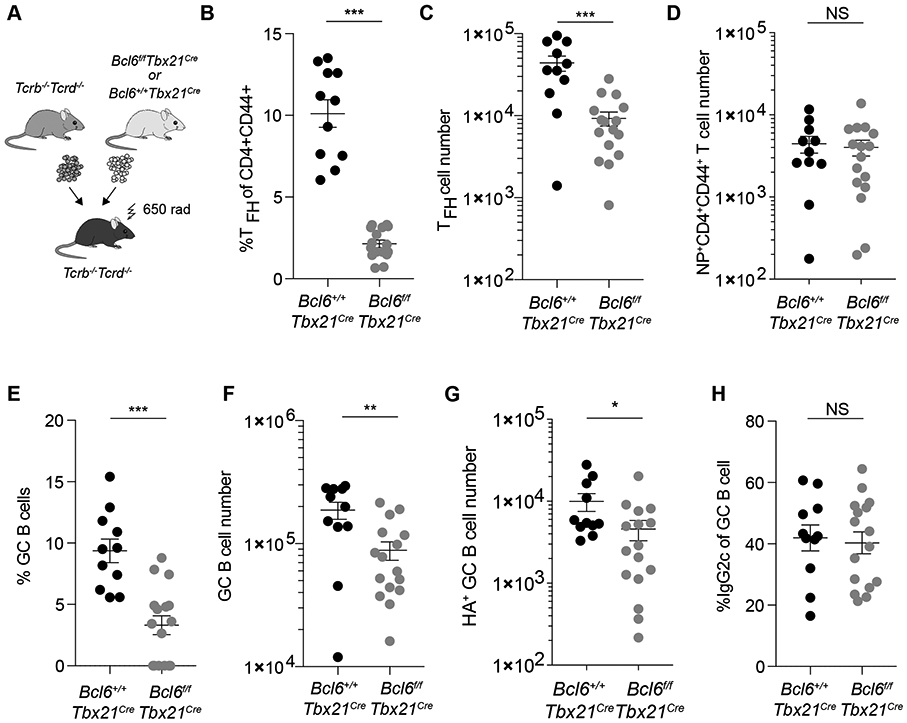
Lethally irradiated Tcrb−/−Tcrd−/− mice were reconstituted with bone marrow cells from Tcrb−/−Tcrd−/− mice and either Bcl6+/+Tbx21-Cre or Bcl6f/fTbx21-Cre mice mixed at a 4:1 ratio. After reconstitution mice were infected with 50 TCID50 influenza (PR8). Mediastinal lymph nodes were and analyzed 14 days post-infection. (A) Schematic for bone marrow chimera generation. (B and C) Frequencies (B) and numbers (C) of CD4+CD44+PD-1HiCXCR5Hi T cells. (D) Numbers of NP tetramer+CD4+CD44+ T cells. (E) Percent of GL7+Fas+ of total B cells. (F) Numbers of GC B cells (B220+GL7+Fas+). (G) Numbers of HA+ GC B cells (B220+GL7+Fas+HA+). (H) Percent IgG2c+ of GC B cells (B220+GL7+Fas+). (B to H) Each point represents an individual mouse with mean ± s.e.m. All data are representative of ≥ 2 experiments, n ≥ 11 mice per group. Two-tailed t-test (***P < 0.001, **P < 0.01, *P< 0.05, and NS = not significant).
Discussion
For elaboration of antibody responses, the secondary lymphoid organ’s microanatomy must support effective communication among distinct lymphocyte types. Our studies reveal the spatiotemporal connectivity between T-bet-expressing T and B cells, which forms a specialized dynamic circuit in the lung draining lymph nodes required for type 1 humoral immune response to a respiratory viral infection. We demonstrate that the assembly of this circuit takes place in distinct anatomical niches guided by a type 1 response-linked chemokine (CXCL9/CXCL10) gradient, which enables time-dependent positioning of TH1 cells at the T-B boundary (Fig. 1, D and E and Fig 4, A and B). The close position of T-bet+ B cells and TH1 cells at the T-B boundary suggests that IFNγ produced by the latter induces T-bet expression in B cells at this particular site. This notion is consistent with the coordinated contemporaneous appearance of T-bet+ B and T cells, the prominently enriched phospho-STAT1 expression in B cells at the T-B boundary, and the requirement of CD4+ T cell-derived IFNγ for IgG2c class switching (Figure 2A, Figure 5, A and J). It remains unknown whether antigenic specificity of T-bet-expressing CD4+ T cells is contributing to their enrichment at the T-B boundary as we analyzed total CD4+CD44+RFP+ T cells.
Genetic ablation of individual cellular components of the T-bet lymphocyte circuit - T-bet+ B cells and T-bet+ TFH cells - confirmed their roles in the downstream functional output as we observed a marked decline in overall GC response and loss of virus-specific IgG production, especially IgG2c - the isotype known to provide protective humoral immunity against viral infections and confer optimal viral clearance (Fig. 3, A to F, and Fig 5, E to H) (53). The impaired responses observed upon selective ablation of T-bet+ B cells were concordant with earlier reports of B cell-restricted T-bet deficiency resulting in a shift from IgG2c to IgG1 production (16, 17). Taken alone, these results cannot distinguish between independent roles of different T-bet-expressing lymphocyte lineages in humoral response vs. their roles as components of a circuit acting as a minimal functional unit. However, the latter possibility is strongly supported by the impaired GC and anti-viral IgG responses observed upon selective uncoupling of this circuit using genetic and antibody-based interference with the spatial positioning of its principal initiating component - T-bet+ CD4+ T cells - assuming that CXCR3 signaling in these cells does not have additional roles other than their chemotactic guidance (Fig. 4, C to N). Importantly, the impediment in the functional output of the circuit was not due to diminished generation of TH1 cells, whose localization at T-B boundary was impeded but overall numbers were unaffected (Fig. 4, E and K). A more pronounced decline in virus-specific GC B cells and IgG2c class-switch observed upon acute antibody induced blockage of CXCR3 signaling in comparison to T cell-restricted CXCR3 deficiency was likely due to a compensation by other TH1-linked chemokine receptors.
In opposition to previous reports of a transient T-bet expression in TFH cells (29), our fate-mapping studies revealed its stable expression, suggesting that T-bet+ TFH cells represent a specialized stable TFH sub-lineage (Fig. 6D). As we found enrichment of CD4+CD44+T-bet+ T cells at the T-B boundary, it was tempting to assume that some of them are capable of differentiation into TFH cells and migration to GCs. However, we failed to observe Bcl6 expression by T-bet+ CD4+ T cells at the T-B boundary, whereas T-bet+ Bcl6+ TFH cells were readily detectable within GCs (Fig. S7D). While this observation does not formally rule out the possibility that some of the TH1 cells at the T-B boundary may give rise to ‘immature” TFH cells with a very short dwell time at this site, it seems more likely that T-bet+ TFH cells are generated in the deep T zone and migrate rapidly to promote GC expansion. This possibility is in agreement with the reported parallel emergence of TFH and TH1 cells from a common transitional precursor or from separate ones observed early in the immune response (31, 54-56). Therefore, the observed decrease in the numbers of TFH cells upon CXCR3 signaling blockage was likely a consequence of the marked decrease in GC size and numbers.
We found that ablation of T-bet+ TFH cells resulted in overall decreased GC numbers (Fig. 7, E to G). Interestingly, the proportion of B cells that switch to IgG2c was unaffected within the numerically diminished GC B cell population (Fig. 7H). Thus, TH1 cells providing IFNγ at the T-B boundary most likely induce IgG2c class switch in activated B cells preceding their arrival at the GC, whereas T-bet+ TFH cells within the GC control their expansion. This scenario is consistent with recent studies showing that isotype class switching occurs most frequently outside of GCs (36). Thus, contrary to a previous suggestion that T-bet expression in B cells and TFH cells independently promotes IgG2c class switching (30), our studies demonstrate that the signals required for Ig switching during type 1 response are provided by Bcl6-negative TH1, but not by TFH cells. These findings suggest a division of labor between these two specialized T-bet-expressing CD4+ T cell subtypes during humoral response to viral infection.
Although the mechanisms underlying differential distribution of TH1 and T-bet+ TFH cells remain to be explored, RNA-seq analysis showed differential expression of multiple chemokine and other receptors by these cell subsets. While CXCR3 and GPR183 promoting peripheral T zone or outer B follicle localization, respectively, were not differently expressed in TH1 and TFH cells, genes encoding CCR7 (Ccr7) and PSGL1 (Selplg) linked to T zone localization were present at a higher level in TH1 compared to T-bet+ TFH cells (Fig. 6F) (39, 45-47, 57). Conversely, genes encoding receptors linked to cell entry into the B cell follicle and GC such as Cxcr5 and Cxcr4 were expressed at higher levels in T-bet+ TFH cells (Fig. 6F) (48, 49). Differential expression of Cxcr5, Cxcr4, Ccr7 and Selplg genes may support the migration of T-bet+ TFH cells to B cell follicle and account for the continuing residence of TH1 cells at the T-B boundary or even return to the T zone.
The observed circuitry of T-bet-expressing lymphocytes we uncovered may also play a role in the induction of long-lasting B cell memory responses, as T-bet expression has been recently linked to B cells with memory potential (18, 58). In addition, T-bet expression has been shown in a B cell subset that accumulates with age and is linked to autoantibody production in a murine model of systemic lupus erythematosus (59, 60). Therefore, understanding protective and pathogenic T-bet+ cell circuits may lead to development of therapeutic interventions that promote their assembly and function for enhanced and long-lasting protective B cell responses to vaccines and infectious agents, or block them in the case of autoimmune responses.
In conclusion, our studies demonstrated coordinated assembly of a distinct spatiotemporal circuit of T-bet-expressing lymphocytes in specific anatomical niches in response to viral infection. The assembly of this circuit in the reactive lymph node draining a virus-infected tissue is enabled by a TH1-linked chemokine gradient. Furthermore, we demonstrate the importance of this circuit in coherent response to infection in relation to the type of antibody produced and the magnitude of the response. Our studies suggest that in semblance of neuronal circuits, a circuit of T-bet-expressing lymphocytes acts as a functional unit enabling the transmission of information between its cellular components to ensure a robust and coherent humoral response tailored for optimal protective immunity against viral infection.
Materials and Methods
Study Design
The goal of this study was to explore whether distinct types of T-bet-expressing CD4+ T and B lymphocytes form a spatiotemporal circuit to promote robust and coherent antiviral humoral immunity. Through the use of mice carrying a fluorescent reporter and Cre recombinase expressed under the control of the endogenous Tbx21 locus we were able to analyze the dynamic localization and genetically manipulate T-bet-expressing lymphocyte lineages during viral infection. In addition, we used genetic approaches combined with mixed bone marrow transfers to inactivate or eliminate distinct cellular components of the circuit to assess their contribution to its overall output.
Mice
Tbx21tdTomato-T2Acre and Tbx21tdTomato-T2A-creERT2, Foxp3Thy1.1, Cd4CreERT2 and Bcl6-YFP mice were described previously (32, 61-63). Pax5f, Cxcr3− (Stock ID: 005796), Tcrb−, Tcrd−, muMt−, Bcl6f, CD45.1, and Rosa26Lox-STOP-Lox-YFP mice were purchased from Jackson Laboratories (35, 64-69). Targeting of the murine Ifng locus was performed in C57BL/6N-A/a (Agouti; JM8A3.N1) ES cells by the EUCOMM (European Conditional Mouse Mutagenesis Program) consortium. Obtained clones were screened by Southern blot to confirm specific recombination and integration of the Ifngtm1a(EUCOMM)Hmgu allele in which exons 2 and 3 of murine Ifng were flanked by LoxP sites and an FRT-flanked gene trap cassette encoding βgalactosidase (lacZ) and neomycin resistance (neo) were introduced to generate a “conditional-ready” (floxed) Infg locus. Positive clones were injected into C57BL/6 blastocysts and resulting progeny were bred to confirm germ-line transmission. Excision of lacZ and neo was obtained by breeding with flip recombinase (Flp) transgenic mice resulting in germ-line–targeted Ifngf/f animals that were subsequently bred to C57BL/6 mice for loss of the Flp allele. Mice were 6-10 weeks old at the time of analysis, except for bone marrow chimeras, which were 22-26 weeks old at the time of analysis. Male and female mice were used depending on availability, as sex did not appear to have a major impact on the measurements made. In all cases mice were compared to littermate controls. Generation and treatments of mice were performed under protocol 08-10-023 approved by the Sloan Kettering Institute (SKI) Institutional Animal Care and Use Committee. All mouse strains were maintained in the SKI animal facility in specific pathogen free (SPF) conditions in accordance with institutional guidelines and ethical regulations.
Influenza infection
Influenza A/Puerto Rico/8/34 (PR8) was grown in 10 day-old embryonated chicken eggs. For intranasal infections, mice were anesthetized with 2.5% isofluorane and nasally inoculated with 50 TCID50 of PR8 diluted in PBS. TCID50 values for PR8 stocks were determined by scoring cytopathic effects of infected Madin-Darby Canine Kidney (MDCK) cells and calculated using the Reed and Muench method.
Generation of bone marrow chimeric mice
Tcrb−/−Tcrd−/− and CD45.1 recipient mice were lethally irradiated with 650 Gy and 950 Gy, respectively. The following day, bone marrow was isolated from femurs and tibias of donor mice. 2×106 total bone marrow cells were transferred into recipient mice via retro-orbital injection.
Tamoxifen administration
40 mg/ml tamoxifen was dissolved in corn oil (Sigma-Aldrich). Mice were orally gavaged with one or two doses of 200μl tamoxifen emulsion.
CXCR3 blockade
Mice were injected intraperitoneally with 0.25 mg of anti-mouse CXCR3 (CD183) and anti-mouse CXCL9 (MIG) or hamster IgG (Catalogue number BE0091) (BioXCell) every other day for 14 days beginning on the day of infection.
Nippostrongylus brasiliensis infections
Nippostrongylus brasiliensis (Nb) was maintained by passage in 9 to 10 week-old male Wistar rats as previously described(70). Briefly, rats were injected subcutaneously (s.c.) with 7000 L3 Nb and stool was collected on days 6–9 post infection. Fecal pellets were mixed with 5×8 bone charcoal and incubated on moisten filter paper in Petri dishes at 26°C for 7 days. L3 larvae were recovered from the edge of the filter paper and the perimeter of the plates and extensively washed with PBS to eliminate contaminants before infection. Mice infections were carried out using a 23G needle at a concentration of 500 L3 Nb in 200 μL.
Influenza HA probe
The VRC 3687 plasmid for recombinant PR8 HA expression was a gift from Adrian McDermott to the Chaudhuri Lab. PR8 HA was expressed and purified as previously described(33). Briefly, 293F cells were cultured in Freestyle medium (ThermoFisher Scientific) in baffled flasks at 37°C and 8% CO2, and diluted to a concentration of 1 x 106 cells/mL prior to transfection. Cells were transfected with 100 μg of PR8 HA plasmid mixed with 400 μg of PEI MAX (Polysciences, Inc.) per 100mLs of culture, and cultured for 5 days. The supernatant was collected by centrifuging cells at 4000 x g, and loaded on a HisTrap excel column (GE) equilibrated in 20 mM sodium phosphate pH 7.4, 0.5 M NaCl. Columns were washed with 20 column volumes (CV) of wash buffer (20 mM sodium phosphate pH 7.4, 0.5 M NaCl, 10 mM imidazole), and eluted with 5 CV of elution buffer (20 mM sodium phosphate pH 7.4, 0.5 M NaCl, 500 mM imidazole). The elute was dialyzed into PBS, concentrated using an Amicon 100 kDa filter (Millipore), and loaded on a Superdex 200 increase 10/300 GL column (GE) equilibrated in PBS. Fractions corresponding to trimeric HA were pooled, concentrated and buffer exchanged into Tris pH 8.0 using an Amicon 100 kDa filter in preparation for biotinylation. Purified HA was biotinylated using a BirA500 biotin-protein ligase standard reaction kit (Avidity) according to the manufacturers protocol. Biotinylated HA was buffer exchanged into PBS and concentrated to 2 mgs/mL using an Amicon 100 kDa and stored at −80°C until use.
Microscopy
Confocal imaging was done using standard conditions. In brief, LN were excised, fixed for 2 hours at room temperature in 4% paraformaldehyde, and dehydrated at 4°C in 30% sucrose in PBS. Tissues were snap frozen in OCT compound (Sakura Tissue-Tek). 10 μm tissue sections were cut, fixed in acetone for 20 minutes at −20°C and dried overnight. Tissue sections were rehydrated in PBS for 10 minutes prior to staining. Staining was done in 10% donkey serum (Jackson ImmunoResearch) 0.3% Triton X-100 in 1x PBS overnight. Tissues were imaged in Slow Fade mounting media (Life Technologies). All images were acquired using LSM880 (Carl Zeiss) or SP8 (Leica) confocal microscopes with a 40X oil immersion objective. Images were processed and analyzed using ImageJ software (version 2.0.0-rc-54/1.51h; National Institutes of Health).
Cell isolation and Flow cytometry
Lymphocytes were isolated from mediastinal LN by mechanical disruption and filtration through a 100 μm cell strainer in Staining buffer (2%FBS, 1mM EDTA, 10 mM HEPES, 1x PBS). To label intra-vascular cells, mice were injected intravenously with 3 μg of anti-CD45 antibody. 3 minutes after injection mice were euthanized. Lung tissue was placed in 5mL snap-cap tubes (Eppendorf) in 3mL Wash buffer (1x RPMI 1640 w/ 2% FBS, 10mM HEPES buffer, 1% penicillin/streptomycin, 2mM L-glutamine) supplemented with 0.2U/mL collagenase A (Sigma) and 1U/mL DNase I (Sigma), along with three ¼ inch ceramic beads and shaken horizontally at 250RPM for 45 minutes at 37°C. Digested samples were then passed through a 100 μm strainer and centrifuged to remove collagenase solution. Samples were then treated with 1x ACK (155mM Ammonium Chloride, 10mM Potassium Bicarbonate, 100nM EDTA pH 7.2) to lyse red blood cells, and then washed by centrifugation in 40% Percoll™ (ThermoFisher) in 1x PBS to remove debris and enrich for lymphocytes. All flow cytometry staining was done in Staining buffer on ice. For CXCR5, cells were stained with purified rat anti-mouse CXCR5 antibody, followed by anti-rat biotin (5.5 μg/ml), and APC-Strepavidin, all in Staining buffer supplemented with 0.01% sodium azide. I-A(b) Influenza A NP tetramer (NIH Tetramer Core Facility) stain was performed in Complete media (1x RPMI 1640 w/ 5% FBS, 10mM HEPES buffer, 1% penicillin/streptomycin, 2mM L-glutamine) for 1 hour at room temperature. For staining cytokine production, cells were incubated in Complete RPMI supplemented with 50ng/mL Phorbol 12-myristate 13-acetate and 500ng/mL ionomycin with 1μg/mL brefeldin A and 2uM monensin to inhibit ER and Golgi transport for 3 hours at 37°C with 5% CO2. Cells were stained for intracellular antigens using the CytoFix/CytoPerm kit (BD Biosciences) according to manufacturer instructions, adjusted for 96-well staining (100μl fix, 100μl stain, 200μl washes). Cell counts were performed using 123eCount Beads (ThermoFisher). Samples were acquired on a LSR II (BD Biosciences) and analyzed using FlowJo software (BD Biosciences).
Antibodies
The following antibodies were used for flow cytometry and microscopy experiments: IgM (II/41), IgG1 (X56), CD45 (30-F11), CD19 (6D5), IgD (11-26c.2a), GL-7 (GL-7), CXCR3 (CXCR3-173), Fas (Jo2), IgG2c (goat polyclonal, Southern Biotech), TCRβ (H57-597), CD62L (MEL-14), CD4 (RM4-5), CD8 (53-6.7), CD44 (IM7), PD-1 (29F.1A12), CD90 (30-H12), CD45.1 (A20), CD45.2 (104), B220 (RA3-6B2), BCL6 (K112-91), LYVE1 (ALY7), CD169 (3D6.112), CXCR5 (2G8), IFNγ (XMG1.2), CXCL9 (11H1L14), CXCL10 (goat polyclonal, R&D Systems), Phospho-Stat1 (Tyr701) (58D6), DsRed (rabbit polyclonal, Takara; and goat polycloal, Mybiosource), CD16/36 (2.4G2), and Biotin-F(ab')2 Fragment Donkey Anti-rat (Jackson Immunoresearch). Avidin/Biotin Blocking Kit and biotinylated Peanut Agglutinin (PNA) were purchased from Vector Laboratories. Ghost Dye Red 780 was purchased from Tonbo Biosciences.
ELISA
Serum immunoglobulin concentrations were measured using SBA Clonotyping System-HRP per manufacturer instructions. IgE was quantified using biotinylated anti-IgE detection antibody (BD PharMingen) and streptavidin-conjugated HRP. ELISAs were read at OD 450 on a Synergy HTX instrument (BioTek). As previously described, for viral specific antibody in sera, PR8 virus was resuspended in PBS with 0.2% Triton X-100 and bound to the plate at 0.5 μg/ml in BBS buffer overnight at 4°C (71). Nonspecific binding was blocked by 1% bovine serum albumin in 1x PBS for 2 hours at 37°C. Serum samples were diluted in ELISA buffer and incubated on the plate for 1 hour at 4°C. Secondary antibodies against total IgG and IgG2c immunoglobulins conjugated to HRP (Southern Biotech) diluted in the ELISA buffer (1x PBS, 0.05% Tween 20) were used at the second step. Background obtained with the secondary antibody alone was subtracted.
RNA-seq analysis
Cells from mediastinal lymph nodes were isolated from Tbx21tdTomato-T2Acre, Bcl6-YFP, Foxp3Thy1.1 reporter mice 14 days after influenza infection. Populations were FACS-sorted on an Aria II cell sorter (BD Bioscience). TH1 cells were defined as Thy1.1−RFP+YFP−; T-bet+TFH cells defined as Thy1.1−RFP+YFP+; and T-bet−TFH cells defined as Thy1.1−RFP−YFP+. Three replicates of each cell subset were generated. Cells were resuspended in Trizol™. RNA was extracted from Trizol™ and prepared for sequencing by the Integrated Genomics Core at Memorial Sloan Kettering Cancer Center. 10-30 million 50bp paired-end reads were acquired on an Illumina HiSeq 2000. RNA-sequencing reads were aligned to the reference mouse genome GRCm38 (72) using the STAR RNA-seq aligner (73) and local realignment was performed using the Genome Analysis Toolkit (GATK) (74). For each sample, raw count of reads per gene was measured using R, and DESeq2 R package (75) was used to perform differential gene expression analysis. A cutoff of 0.05 was set on the obtained p values (that were adjusted using Benjamini-Hochberg multiple testing correction) to identify differentially expressed genes for each comparison. The threshold to minimum average expression within samples of each comparison was set to 30 reads. DESeq2 package was used for PCA calculation of components as well. For clustering analysis we used k-means method by choosing 6 centers. The number represents the set of different trends we observed after a simple hierarchical clustering using Euclidean distance as metric.
Statistical analysis
All statistical analyses (excluding RNA-seq, described above) were performed using GraphPad Prism 6 software. Differences between individual groups were analyzed for statistical significance using the unpaired or paired two-tailed t-test. *, p ≥ 0.05; **, p ≥ 0.01; ***, p ≥ 0.001; NS, not significant. The number of mice used in each experiment to reach statistical significance was determined on the basis of preliminary data. No animals were excluded from the analyses. No methods of randomization were used to allocate animals into experimental groups. No blinding was used. Data met assumptions of statistical methods used and variance was similar between groups that were statistically compared.
Supplementary Material
Raw data (Excel file).
k-means Clustering analysis (Excel file).
Figure S1. Lymph node regions analysis using confocal microscopy.
Figure S2. T-bet expression in B cells during influenza infection and Nippostongylus brasiliensis infection.
Figure S3. in situ CXCL9 and CXCL10 during influenza infection.
Figure S4. Lymph node regions analysis with CXCR3 blockade and Cxcr3 deletion in T cells.
Figure S5. Cxcr3 deletion in B cells.
Figure S6. Ifng deletion in CD4 T cells.
Figure S7. T-bet expression in TFH cells during influenza infection and Nippostongylus brasiliensis infection.
Figure S8. Model of T-bet+ T-B cell circuit.
Acknowledgments
We thank N. Arpaia for generating Ifngf mice, A. Bravo and J. Verter for help with animal husbandry; A.M. Mujal for providing bones from Cxcr3+/y and Cxcr3−/y mice on a B6 background; S. Fujisawa and the staff at the Memorial Sloan Kettering Molecular Cytology Core facility for technical support and image analysis assistance; S. Dikiy for critical advice on the manuscript; and all the members of the Rudensky laboratory for technical input and discussion.
Funding
This work was supported by the National Institutes of Health/National Cancer Institute Cancer Center Support Grant P30 CA008748 (A.Y.R.), National Institutes of Health grant R01 AI034206 (A.Y.R.), Bristol Myers Squibb Fellowship from the Cancer Research Institute (A.M.), National Institutes of Health grant T32 CA009149 (A.M.), and the Ludwig Center at the Memorial Sloan Kettering Cancer Center. A.Y.R. is an investigator with the Howard Hughes Medical Institute.
Footnotes
Competing interests
The authors declare no competing interests.
Data and materials availability
The accession number for the RNA-seq datasets reported in this paper is GEO: GSE173605. The code used for image analysis are available via GitHub (https://github.com/mendozaalejandra/Image-analysis). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Ifngf mice are available under an MTA and requests for access should be directed to the corresponding authors.
References
- 1.von Andrian UH, Mempel TR, Homing and cellular traffic in lymph nodes. Nat Rev Immunol 3, 867–878 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Crotty S, T follicular helper cell differentiation, function, and roles in disease. Immunity 41, 529–542 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, van Rooijen N, Mempel TR, Whelan SP, von Andrian UH, Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature 450, 110–114 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Li J, Lu E, Yi T, Cyster JG, EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature 533, 110–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaya M, Barral P, Burbage M, Aggarwal S, Montaner B, Warren Navia A, Aid M, Tsui C, Maldonado P, Nair U, Ghneim K, Fallon PG, Sekaly RP, Barouch DH, Shalek AK, Bruckbauer A, Strid J, Batista FD, Initiation of Antiviral B Cell Immunity Relies on Innate Signals from Spatially Positioned NKT Cells. Cell 172, 517–533 e520 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, Kroczek RA, Kurts C, Garbi N, Barchet W, Iannacone M, Klauschen F, Kolanus W, Kaisho T, Colonna M, Germain RN, Kastenmuller W, CD8(+) T Cells Orchestrate pDC-XCR1(+) Dendritic Cell Spatial and Functional Cooperativity to Optimize Priming. Immunity 46, 205–219 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, Garbi N, Kaisho T, Germain RN, Kastenmuller W, Robust Anti-viral Immunity Requires Multiple Distinct T Cell-Dendritic Cell Interactions. Cell 162, 1322–1337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu P, Shih C, Qi H, Ephrin B1-mediated repulsion and signaling control germinal center T cell territoriality and function. Science 356, (2017). [DOI] [PubMed] [Google Scholar]
- 9.O'Shea JJ, Paul WE, Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327, 1098–1102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lord GM, Rao RM, Choe H, Sullivan BM, Lichtman AH, Luscinskas FW, Glimcher LH, T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood 106, 3432–3439 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chensue SW, Lukacs NW, Yang TY, Shang X, Frait KA, Kunkel SL, Kung T, Wiekowski MT, Hedrick JA, Cook DN, Zingoni A, Narula SK, Zlotnik A, Barrat FJ, O'Garra A, Napolitano M, Lira SA, Aberrant in vivo T helper type 2 cell response and impaired eosinophil recruitment in CC chemokine receptor 8 knockout mice. J Exp Med 193, 573–584 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, Yamaguchi T, Nomura T, Ito H, Nakamura T, Sakaguchi N, Sakaguchi S, Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med 204, 2803–2812 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH, A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Lazarevic V, Glimcher LH, Lord GM, T-bet: a bridge between innate and adaptive immunity. Nat Rev Immunol 13, 777–789 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng SL, Szabo SJ, Glimcher LH, T-bet regulates IgG class switching and pathogenic autoantibody production. Proc Natl Acad Sci U S A 99, 5545–5550 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang NS, McHeyzer-Williams LJ, Okitsu SL, Burris TP, Reiner SL, McHeyzer-Williams MG, Divergent transcriptional programming of class-specific B cell memory by T-bet and RORalpha. Nat Immunol 13, 604–611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barnett BE, Staupe RP, Odorizzi PM, Palko O, Tomov VT, Mahan AE, Gunn B, Chen D, Paley MA, Alter G, Reiner SL, Lauer GM, Teijaro JR, Wherry EJ, Cutting Edge: B Cell-Intrinsic T-bet Expression Is Required To Control Chronic Viral Infection. J Immunol 197, 1017–1022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson JL, Rosenthal RL, Knox JJ, Myles A, Naradikian MS, Madej J, Kostiv M, Rosenfeld AM, Meng W, Christensen SR, Hensley SE, Yewdell J, Canaday DH, Zhu J, McDermott AB, Dori Y, Itkin M, Wherry EJ, Pardi N, Weissman D, Naji A, Prak ETL, Betts MR, Cancro MP, The Transcription Factor T-bet Resolves Memory B Cell Subsets with Distinct Tissue Distributions and Antibody Specificities in Mice and Humans. Immunity, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu N, Ohnishi N, Ni L, Akira S, Bacon KB, CpG directly induces T-bet expression and inhibits IgG1 and IgE switching in B cells. Nat Immunol 4, 687–693 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Rubtsova K, Rubtsov AV, van Dyk LF, Kappler JW, Marrack P, T-box transcription factor T-bet, a key player in a unique type of B-cell activation essential for effective viral clearance. Proc Natl Acad Sci U S A 110, E3216–3224 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishizaka ST, Piacente P, Silva J, Mishkin EM, IgG subtype is correlated with efficiency of passive protection and effector function of anti-herpes simplex virus glycoprotein D monoclonal antibodies. J Infect Dis 172, 1108–1111 (1995). [DOI] [PubMed] [Google Scholar]
- 22.Markine-Goriaynoff D, Coutelier JP, Increased efficacy of the immunoglobulin G2a subclass in antibody-mediated protection against lactate dehydrogenase-elevating virus-induced polioencephalomyelitis revealed with switch mutants. J Virol 76, 432–435 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huber VC, McKeon RM, Brackin MN, Miller LA, Keating R, Brown SA, Makarova N, Perez DR, Macdonald GH, McCullers JA, Distinct contributions of vaccine-induced immunoglobulin G1 (IgG1) and IgG2a antibodies to protective immunity against influenza. Clin Vaccine Immunol 13, 981–990 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN, Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity 37, 364–376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, Haberman AM, Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 34, 947–960 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK, Visualization of specific B and T lymphocyte interactions in the lymph node. Science 281, 96–99 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, O'Garra A, Cahalan MD, Cyster JG, Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol 3, e150 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Victora GD, Nussenzweig MC, Germinal centers. Annu Rev Immunol 30, 429–457 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Fang D, Cui K, Mao K, Hu G, Li R, Zheng M, Riteau N, Reiner SL, Sher A, Zhao K, Zhu J, Transient T-bet expression functionally specifies a distinct T follicular helper subset. J Exp Med 215, 2705–2714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheikh AA, Cooper L, Feng M, Souza-Fonseca-Guimaraes F, Lafouresse F, Duckworth BC, Huntington ND, Moon JJ, Pellegrini M, Nutt SL, Belz GT, Good-Jacobson KL, Groom JR, Context-Dependent Role for T-bet in T Follicular Helper Differentiation and Germinal Center Function following Viral Infection. Cell Rep 28, 1758–1772 e1754 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakayamada S, Kanno Y, Takahashi H, Jankovic D, Lu KT, Johnson TA, Sun HW, Vahedi G, Hakim O, Handon R, Schwartzberg PL, Hager GL, O'Shea JJ, Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity 35, 919–931 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, Hoyos BE, Putintseva EV, Chaudhry A, Dikiy S, Fujisawa S, Chudakov DM, Treuting PM, Rudensky AY, Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 546, 421–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whittle JR, Wheatley AK, Wu L, Lingwood D, Kanekiyo M, Ma SS, Narpala SR, Yassine HM, Frank GM, Yewdell JW, Ledgerwood JE, Wei CJ, McDermott AB, Graham BS, Koup RA, Nabel GJ, Flow cytometry reveals that H5N1 vaccination elicits cross-reactive stem-directed antibodies from multiple Ig heavy-chain lineages. J Virol 88, 4047–4057 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nutt SL, Heavey B, Rolink AG, Busslinger M, Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 401, 556–562 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Horcher M, Souabni A, Busslinger M, Pax5/BSAP maintains the identity of B cells in late B lymphopoiesis. Immunity 14, 779–790 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, Ellyard J, Shen Q, Robert PA, Cappello J, Vohra H, Zhang Y, Nowosad CR, Schiepers A, Corcoran LM, Toellner KM, Polo JM, Meyer-Hermann M, Victora GD, Vinuesa CG, Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity 51, 337–350 e337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beima KM, Miazgowicz MM, Lewis MD, Yan PS, Huang TH, Weinmann AS, T-bet binding to newly identified target gene promoters is cell type-independent but results in variable context-dependent functional effects. J Biol Chem 281, 11992–12000 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Sierro F, Biben C, Martinez-Munoz L, Mellado M, Ransohoff RM, Li M, Woehl B, Leung H, Groom J, Batten M, Harvey RP, Martinez AC, Mackay CR, Mackay F, Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc Natl Acad Sci U S A 104, 14759–14764 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, von Andrian UH, Moon JJ, Mempel TR, Luster AD, CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 37, 1091–1103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uppaluri R, Sheehan KC, Wang L, Bui JD, Brotman JJ, Lu B, Gerard C, Hancock WW, Schreiber RD, Prolongation of cardiac and islet allograft survival by a blocking hamster anti-mouse CXCR3 monoclonal antibody. Transplantation 86, 137–147 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Domeier PP, Chodisetti SB, Soni C, Schell SL, Elias MJ, Wong EB, Cooper TK, Kitamura D, Rahman ZS, IFN-gamma receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med 213, 715–732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu W, Zhang JJ, Stat1-dependent synergistic activation of T-bet for IgG2a production during early stage of B cell activation. J Immunol 175, 7419–7424 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Kastenmuller W, Torabi-Parizi P, Subramanian N, Lammermann T, Germain RN, A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell 150, 1235–1248 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang V, Chaluvadi VS, Ramos-Perez WD, Mendoza A, Baeyens A, Rivera R, Chun J, Cammer M, Schwab SR, Gradients of the signaling lipid S1P in lymph nodes position natural killer cells and regulate their interferon-gamma response. Nat Immunol 18, 15–25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haynes NM, Allen CD, Lesley R, Ansel KM, Killeen N, Cyster JG, Role of CXCR5 and CCR7 in follicular Th cell positioning and appearance of a programmed cell death gene-1high germinal center-associated subpopulation. J Immunol 179, 5099–5108 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Okada T, Cyster JG, CC chemokine receptor 7 contributes to Gi-dependent T cell motility in the lymph node. J Immunol 178, 2973–2978 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Veerman KM, Williams MJ, Uchimura K, Singer MS, Merzaban JS, Naus S, Carlow DA, Owen P, Rivera-Nieves J, Rosen SD, Ziltener HJ, Interaction of the selectin ligand PSGL-1 with chemokines CCL21 and CCL19 facilitates efficient homing of T cells to secondary lymphoid organs. Nat Immunol 8, 532–539 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Ansel KM, McHeyzer-Williams LJ, Ngo VN, McHeyzer-Williams MG, Cyster JG, In vivo-activated CD4 T cells upregulate CXC chemokine receptor 5 and reprogram their response to lymphoid chemokines. J Exp Med 190, 1123–1134 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allen CD, Ansel KM, Low C, Lesley R, Tamamura H, Fujii N, Cyster JG, Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat Immunol 5, 943–952 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S, Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 325, 1006–1010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH, Dong C, Bcl6 mediates the development of T follicular helper cells. Science 325, 1001–1005 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, Srivastava M, Linterman M, Zheng L, Simpson N, Ellyard JI, Parish IA, Ma CS, Li QJ, Parish CR, Mackay CR, Vinuesa CG, The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity 31, 457–468 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Hangartner L, Zinkernagel RM, Hengartner H, Antiviral antibody responses: the two extremes of a wide spectrum. Nat Rev Immunol 6, 231–243 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, Huseby ES, Way SS, Jenkins MK, Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell 153, 785–796 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khatun A, Kasmani MY, Zander R, Schauder DM, Snook JP, Shen J, Wu X, Burns R, Chen YG, Lin CW, Williams MA, Cui W, Single-cell lineage mapping of a diverse virus-specific naive CD4 T cell repertoire. J Exp Med 218, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruterbusch M, Pruner KB, Shehata L, Pepper M, In Vivo CD4(+) T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Annu Rev Immunol 38, 705–725 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Baptista AP, Gola A, Huang Y, Milanez-Almeida P, Torabi-Parizi P, Urban JF Jr., Shapiro VS, Gerner MY, Germain RN, The Chemoattractant Receptor Ebi2 Drives Intranodal Naive CD4(+) T Cell Peripheralization to Promote Effective Adaptive Immunity. Immunity 50, 1188–1201 e1186 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Stone SL, Peel JN, Scharer CD, Risley CA, Chisolm DA, Schultz MD, Yu B, Ballesteros-Tato A, Wojciechowski W, Mousseau B, Misra RS, Hanidu A, Jiang H, Qi Z, Boss JM, Randall TD, Brodeur SR, Goldrath AW, Weinmann AS, Rosenberg AF, Lund FE, T-bet Transcription Factor Promotes Antibody-Secreting Cell Differentiation by Limiting the Inflammatory Effects of IFN-gamma on B Cells. Immunity 50, 1172–1187 e1177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hao Y, O'Neill P, Naradikian MS, Scholz JL, Cancro MP, A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 118, 1294–1304 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rubtsova K, Rubtsov AV, Thurman JM, Mennona JM, Kappler JW, Marrack P, B cells expressing the transcription factor T-bet drive lupus-like autoimmunity. J Clin Invest 127, 1392–1404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T, Okada T, Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity 34, 961–972 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Liston A, Nutsch KM, Farr AG, Lund JM, Rasmussen JP, Koni PA, Rudensky AY, Differentiation of regulatory Foxp3+ T cells in the thymic cortex. Proc Natl Acad Sci U S A 105, 11903–11908 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sledzinska A, Hemmers S, Mair F, Gorka O, Ruland J, Fairbairn L, Nissler A, Muller W, Waisman A, Becher B, Buch T, TGF-beta signalling is required for CD4(+) T cell homeostasis but dispensable for regulatory T cell function. PLoS Biol 11, e1001674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, Wang L, Ichikawa Y, Jaenisch R, Hooper ML, et al. , Mutations in T-cell antigen receptor genes alpha and beta block thymocyte development at different stages. Nature 360, 225–231 (1992). [DOI] [PubMed] [Google Scholar]
- 65.Itohara S, Mombaerts P, Lafaille J, Iacomini J, Nelson A, Clarke AR, Hooper ML, Farr A, Tonegawa S, T cell receptor delta gene mutant mice: independent generation of alpha beta T cells and programmed rearrangements of gamma delta TCR genes. Cell 72, 337–348 (1993). [DOI] [PubMed] [Google Scholar]
- 66.Kitamura D, Roes J, Kuhn R, Rajewsky K, A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature 350, 423–426 (1991). [DOI] [PubMed] [Google Scholar]
- 67.Hollister K, Kusam S, Wu H, Clegg N, Mondal A, Sawant DV, Dent AL, Insights into the role of Bcl6 in follicular Th cells using a new conditional mutant mouse model. J Immunol 191, 3705–3711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Janowska-Wieczorek A, Majka M, Kijowski J, Baj-Krzyworzeka M, Reca R, Turner AR, Ratajczak J, Emerson SG, Kowalska MA, Ratajczak MZ, Platelet-derived microparticles bind to hematopoietic stem/progenitor cells and enhance their engraftment. Blood 98, 3143–3149 (2001). [DOI] [PubMed] [Google Scholar]
- 69.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H, A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Camberis M, Le Gros G, Urban J Jr., Animal model of Nippostrongylus brasiliensis and Heligmosomoides polygyrus. Curr Protoc Immunol Chapter 19, Unit 19 12 (2003). [DOI] [PubMed] [Google Scholar]
- 71.Purdy A, Case L, Duvall M, Overstrom-Coleman M, Monnier N, Chervonsky A, Golovkina T, Unique resistance of I/LnJ mice to a retrovirus is due to sustained interferon gamma-dependent production of virus-neutralizing antibodies. J Exp Med 197, 233–243 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mouse Genome Sequencing C, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O'Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES, Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002). [DOI] [PubMed] [Google Scholar]
- 73.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA, The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Raw data (Excel file).
k-means Clustering analysis (Excel file).
Figure S1. Lymph node regions analysis using confocal microscopy.
Figure S2. T-bet expression in B cells during influenza infection and Nippostongylus brasiliensis infection.
Figure S3. in situ CXCL9 and CXCL10 during influenza infection.
Figure S4. Lymph node regions analysis with CXCR3 blockade and Cxcr3 deletion in T cells.
Figure S5. Cxcr3 deletion in B cells.
Figure S6. Ifng deletion in CD4 T cells.
Figure S7. T-bet expression in TFH cells during influenza infection and Nippostongylus brasiliensis infection.
Figure S8. Model of T-bet+ T-B cell circuit.