

Abstract
Aldose reductase (AR) is an NADPH-dependent aldo-keto reductase that has been shown to be involved in the pathogenesis of several blinding diseases such as uveitis, diabetic retinopathy (DR) and cataract. However, possible mechanisms linking the action of AR to these diseases are not well understood. As DR and cataract are among the leading causes of blindness in the world, there is an urgent need to explore therapeutic strategies to prevent or delay their onset. Studies with AR inhibitors and gene-targeted mice have demonstrated that the action of AR is also linked to cancer onset and progression. In this review we examine possible mechanisms that relate AR to molecular signaling cascades and thus explain why AR inhibition is an effective strategy against colon cancer as well as diseases of the eye such as uveitis, cataract, and retinopathy.
Keywords: aldose reductase, inflammation, diabetes, cancer, inhibitor, cataract, retinopathy, nephropathy, neuropathy
1. Introduction
Aldo-keto reductases (AKRs) are a superfamily of enzymes involved in phase 1 metabolism of carbonyl substrates such as sugars, lipid aldehydes, keto-steroids and keto-prostaglandins [1–4]. The AKR superfamily contains 16 families (AKR1–16) [5] based on their high sequence similarity and common protein fold structure [AKR website: http://www.med.upenn.edu/akr/]. Enzymes within this family share many catalytic and structural properties. As a group, AKRs are nicotinamide adenine dinucleotide (phosphate) (NAD(P)H)-dependent oxidoreductases and are expressed as 34–37 kDa polypeptides [6].
The AKR family 1 includes: AKR1A (aldehyde reductases) [7], AKR1B (aldose reductases) [8], AKR1C (hydroxysteroid dehydrogenases) [9], AKR1D (steroid 5β-reductases) [9], and AKR1E (1,5-anhydro-D-fructose reductase) [10]. Among enzymes in the AKR family 1, AKR1B is the most well-studied with well over 6,000 reports published in PubMed (as of September 2016). Three AKR1B subfamily enzymes include AKR1B1 (human aldose reductase, HAR), AKR1B10 (human small intestine-like aldose reductase, HSIR) and AKR1B15 that are all encoded by genes localized to chromosome 7 [11] see Table 1. Since it is so well-studied among aldo-keto reductases, this review will focus primarily on recent studies on the role of aldose reductase (AR, AKR1B1) in human health, cancer, and ocular disease.
Table 1.
The table for human aldo-keto reductase family 1B
| Gene | Protein | Also known as | Chromosomal localization |
|---|---|---|---|
| AKR1B1 | Aldose reductase | AR, ALR2 | 7q35 |
| AKR1B10 | Small intestine-like aldose reductase | HSIR, ALR1 | 7q33 |
| AKR1B15 | Aldo-keto reductase family 1, member B15 | AKR1B10L | 7q33 |
2. Function of AR in cellular responses
2.1. Inflammation
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is a transcription factor that controls gene expression affecting cellular processes such as cell cycle regulation, apoptosis [12], and activation of genes involved with inflammation [13]. In unstimulated cells, NF-κB is localized mainly in the cytoplasm [14]. Following cellular activation, NF-κB translocates into the nucleus and becomes acetylated by histone acetyltransferase (HATs) including CREB-binding protein (CBP) or its homolog p300 [15, 16]. Studies have shown that acetylation enhances both DNA-binding ability and transcriptional activity of NF-κB [17]. Removal of acetyl groups, catalyzed by a family of deacetylating proteins, can also influence the activity of transcription factors. SIRT1 (silent mating type information regulation 2 homolog) 1, the sirtuin 1 protein in mammals, is an NAD+-dependent deacetylase that has been known to inhibit NF-κB transcription signaling by removing acetylation from its subunit RelA/p65 at lysine 310 [18]. Therefore, activity of SIRT1 plays a crucial role in regulating inflammatory responses by influencing the acetylation, and thus the activation state of NF-κB, the key transcription factor controlling expression of proinflammatory genes.
Pioneering studies by Ramana and Srivastava and their colleagues demonstrated a possible connection between AR and NF-κB activation when they showed that ARIs suppress the endotoxin-induced activation of NF-κB in macrophages [19]. Ramana and Srivastava have studied the intersection of AR and NF-κB by focusing on the role of AR in the NADPH-dependent detoxification of reactive aldehydes [20, 21]. AR has been shown to play a role in the reduction of toxic aldehydes such as 4-hydroxy-trans-2-nonenal (HNE) and its glutathione adducts (GS-HNE). Under oxidative stress conditions characterized by increased levels of lipid peroxidation, AR converts HNE and GS-NHE to 1,4-dihydroxynonene (DHN) and GS-DHN, respectively [22]. GS-DHN is the predominant metabolite derived from HNE due to the high reactivity of the aldehyde with glutathione. GS-DHN is a potent activator of the phospholipase C (PLC)/protein kinase C (PKC)/NF-κB pathway [23, 24]. Therefore, several studies indicated that AR can play an important role in the activation of the PLC/PKC/NF-κB cascade: AR inhibition suppresses these signaling events [23, 25–27]. In ocular models of endotoxin-induced uveitis, injection of LPS induces NF-κB activation in the anterior or posterior chambers of the eye [28, 29]. Clinically, topical corticosteroids, which act by suppressing NF-κB signaling [30], represent the primary treatment strategy for patients with noninfectious anterior uveitis [31, 32].
2.2. Intersection of glucose metabolism and inflammatory signaling
Hyperacetylation of NF-κB is well known to drive expression of inflammatory signaling genes [33]. Among many factors, high glucose has been shown to cause increased acetylation of NF-κB [34]. We hypothesize that the polyol pathway may provide a functional link between glucose metabolism, protein acetylation, and NF-κB activation. In the first step of the polyol pathway, AR catalyzes the NADPH-dependent conversion of glucose to sorbitol, which is then converted to fructose by the NAD+-dependent sorbitol dehydrogenase (SDH) [35]. Thus, accelerated glucose flux through the polyol pathway results in a reduction in the NAD+/NADH. Consequently, lower levels of NAD+ could be expected to attenuate the NAD+-dependent deacetylase activity of sirtuin-1 (Sirt-1), resulting in higher levels of NF-κB acetylation and therefore higher activity of Sirt-1 as an activator of inflammatory gene transcription. Thus, the imbalance between NAD+/NADH provides a plausible linkage between polyol pathway activation and accumulation of acetyl-NF-κB, leading to an inflammatory phenotype (Fig. 1). We hypothesize that blockade of the poylol pathway by inhibition of either AR or SDH would prevent the high glucose-induced redox imbalance and thereby support the higher deacetylase activity of sirtulin 1. This would result in reduced levels of activated NF-κB and thereby lower expression of inflammatory genes.
Fig. 1.
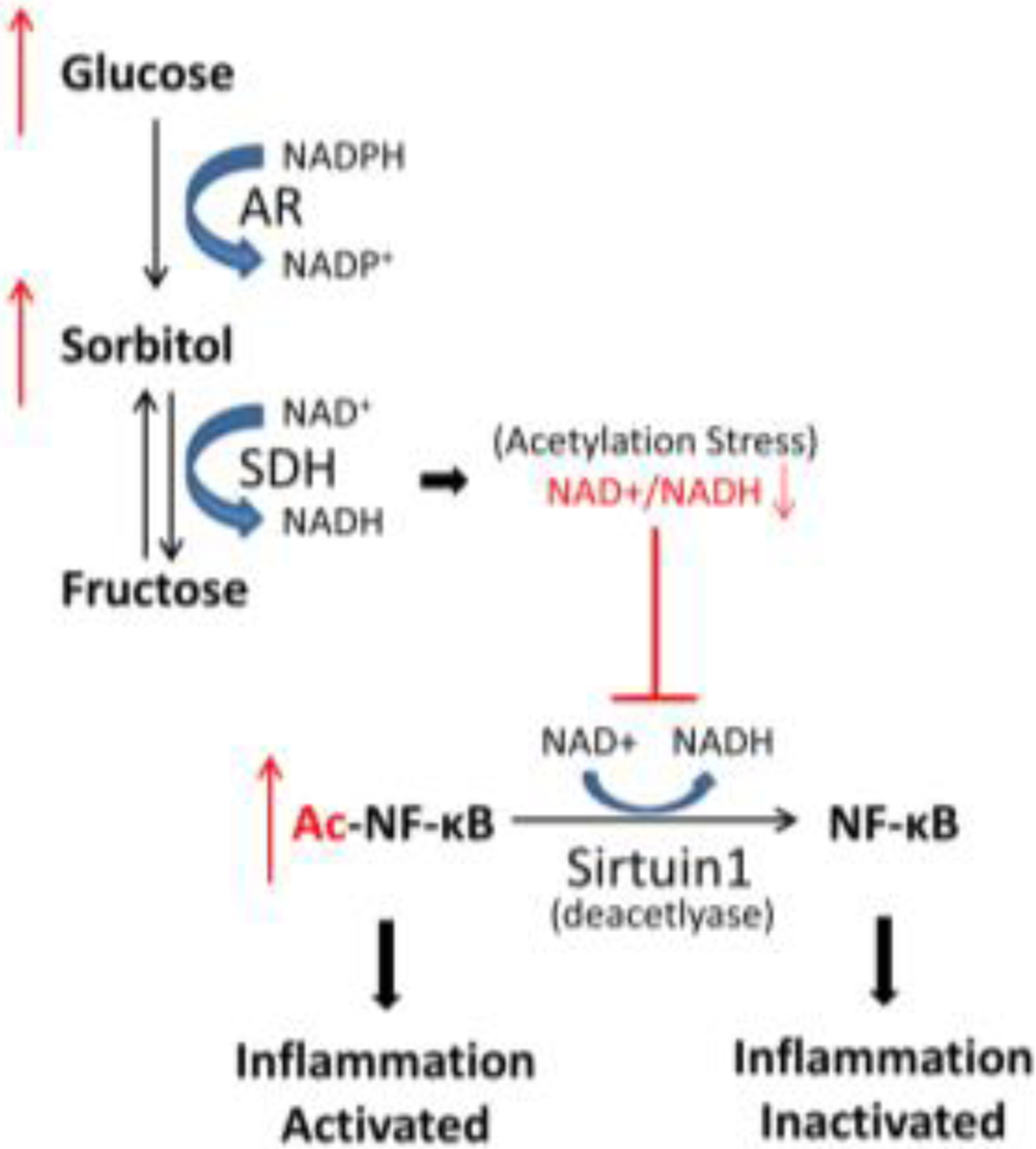
Linkage between polyol pathway activation and induction of inflammatory signaling. Glucose metabolism through the polyol pathway results in a reduced NAD+/NADH ratio, which lowers the ability of Sirt1 to deacetylate NF-κB.
Our studies are in concordance with those mentioned previously by Ramana and Srivastava [36–38], demonstrating that AR inhibition prevents endotoxin-induced inflammation in the eye [39, 40]. However, further studies will be needed to elucidate whether AR inhibition protects against the inflammatory response through its control of lipid derived aldehydes or alternatively through its influence on NAD+/NADH ratios and acetylation status of transcription factors such as NF-κB. It is also possible that AR is involved in the inflammatory response via both pathways.
2.3. Oxidative stress
Hyperglycemia is a leading cause of oxidative stress in diabetic organs such as heart, kidney and eye. Pathways of hyperglycemia-induced oxidative stress include polyol pathway, mitochondrial electron transport system, protein kinase C (PKC) and advanced glycation end products (AGEs) (Fig. 2) [23]. During hyperglycemia, increased flux of AR-mediated reduction of glucose into sorbitol in a NADPH-dependent reaction was observed [41, 42]. NADPH plays reductive roles in metabolic steps, such as detoxification of reactive oxygen species (ROS) by the glutathione reductase/glutathione peroxidase system [43]. Therefore, excessive utilization of NADPH by the polyol pathway could compromise the ability of cells to protect themselves from oxidative stress.
Fig. 2.
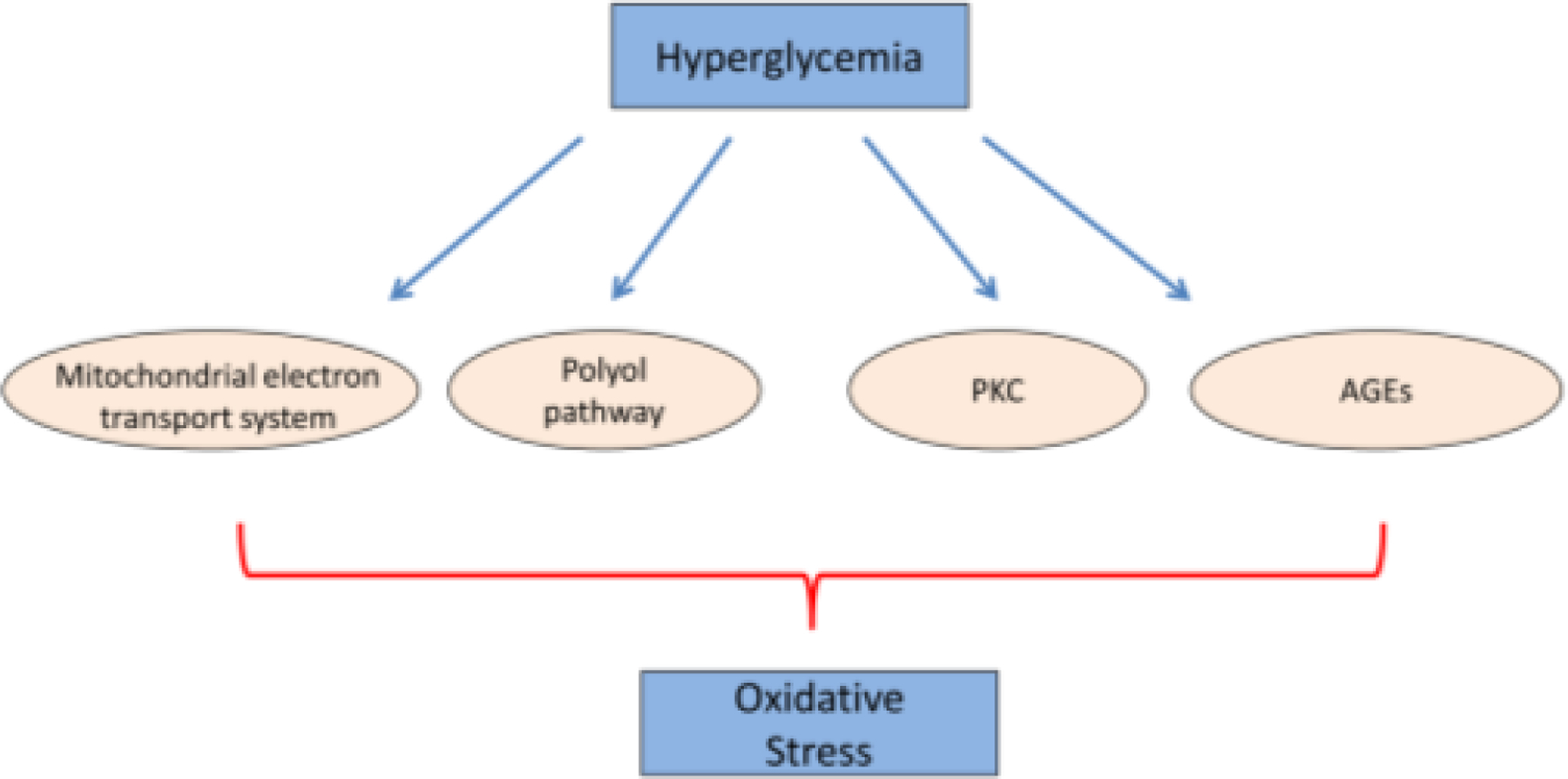
Hyperglycemia-induced oxidative stress is contributed by mitochondrial electron transport system, polyol pathway, PKC and AGEs.
In the secondary part of the polyol pathway, sorbitol is converted to fructose by SDH resulting in decreased ratio of NAD+/NADH. Elevation of cytosolic NADH/ NAD+ ratio leads to induction of ROS via mitochondrial NADH dependent pathway [44]. Increased NADH could also enhance the synthesis of diacylglycerol (DAG), which activates PKC and subsequently induces oxidative stress via PKC-dependent activation of NAD(P)H oxidase [45].
In addition to pyridine cofactor imbalances, AGEs resulting from hyperglycemia also contribute to oxidative stress [46]. AGEs are formed by nonenzymatic glycation reactions involving addition of a carbohydrate to a protein under high glucose conditions typical of diabetic individuals [46]. A study using glucose and fructose in comparison of the ability of forming AGEs showed that fructose forms AGE-BSA much faster than glucose [47]. This observation indicates that elevation of fructose from increased flux of the polyol pathway is another contributor for oxidative stress. Collectively, reduction of the ratio of NADPH/NADP+ and NAD+/NADH, and induction of AGEs are the major causes of oxidative stress in hyperglycemic environment (Fig. 3).
Fig. 3.
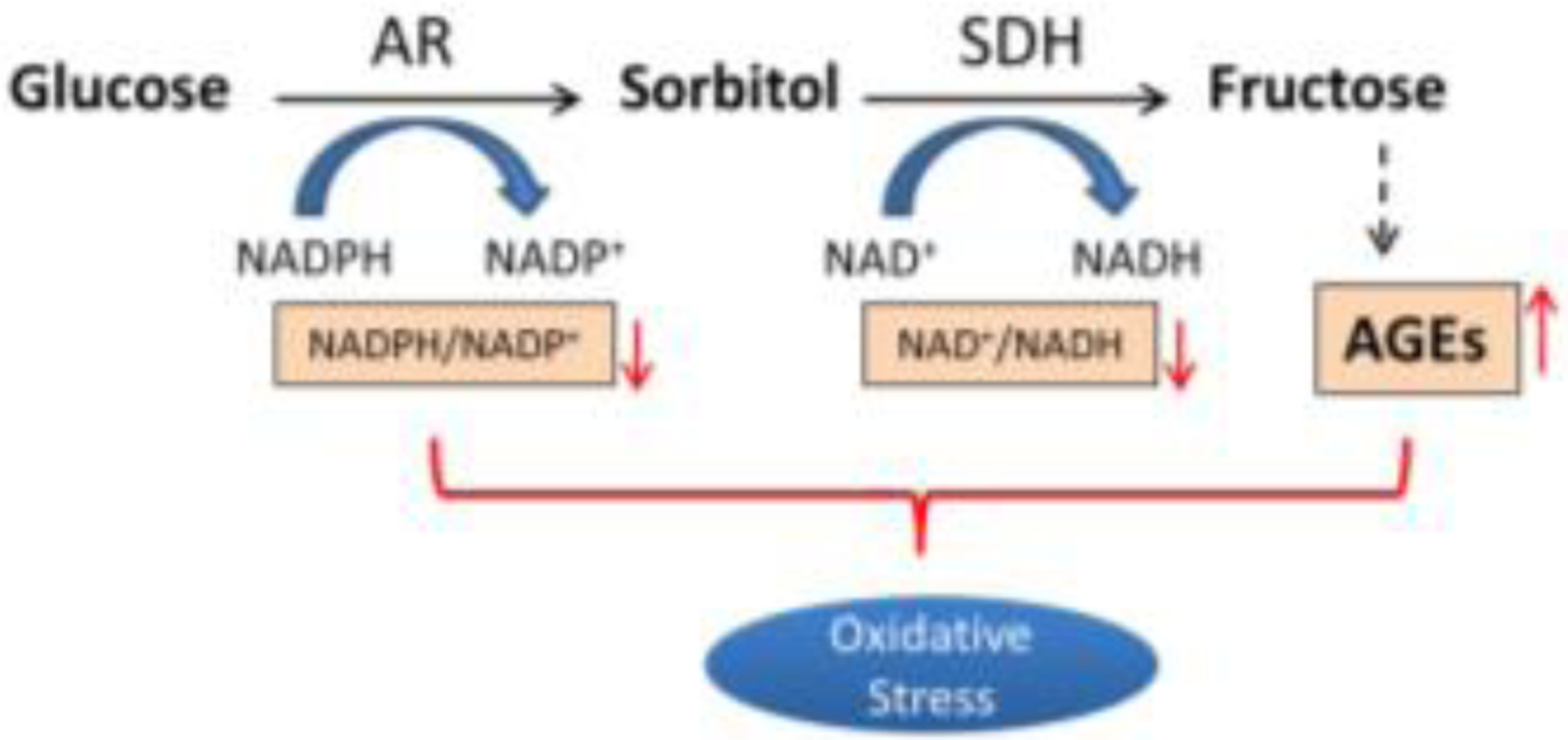
Increased flux of polyol pathway initiates oxidative stress by reducing the ratio of NADPH/NADP+ and NAD+/NADH, and inducing AGEs production.
3. AR and Complications of Diabetes and Chronic Hyperglycemia
Many studies indicate that AR is implicated in inflammatory responses in immune cells [36–40], in heart [48], in kidney [49, 50] and in the eye [39, 51–53]. Additionally, AR is a major factor that causes a variety of diabetic complications such as autonomic neuropathy [54–58], ischemic myocardial injury [59–68] cardiomyopathy [48, 69, 70], nephropathy [71–73], cataract [74–79] and retinopathy [80–84] see Table 2. Pharmacological inhibition of AR or genetic deficiency in animal models with targeted disruption of the AR gene (AR knock out) brings about protection against these complications of diabetes and therefore provide a valuable tool for investigating pathogenesis caused by endotoxin or diabetic hyperglycemia. In this review, we will focus on AR inhibitors’ effects on the major complications of diabetes mellitus.
Table 2.
The table for the role of AKR1B1 in health and disease.
| Organ | Associated disease |
|---|---|
| Heart | Myocardial ischemic injury |
| Cardiomyopathy | |
| Kidney | Diabeitc Nephropathy |
| Acute Kidney Injury | |
| Nerve | Diabetic peripheral neuropathy |
| Cardiac autonomic neuropathy | |
| Eye | Uveitis |
| Diabetic cataract | |
| Diabetic retinopathy | |
| Posterior Capsular Opacification |
3.1. Heart
3.1.1. Myocardial ischemic injury
Myocardial ischemia is a heart disease caused by lack of oxygen to cardiac muscle, usually due to blockage of blood vessels. Diabetic patients have high incidence of cardiovascular disease and myocardial infarction [85, 86]. In diabetic patients, more sorbitol accumulation and a decrease of NADPH, from a flux of AR, have been considered a causal factor in cardiac dysfunction [56, 87]. Many AR inhibitors such as Zopolrestat [59–61, 64], Tolrestat [63, 67] and Sorbinil [63, 67] have been reported effectively against myocardial ischemic injury in mice [64], rat [59, 60, 67] and rabbit [61, 63] models in both diabetic and non-diabetic conditions. To further understand the influence of AR in the heart, Ramasamy [62] and Bhatnagar [66] groups conducted experiments to measure AR activity and kinetic properties between normal and ischemic heart. They both found that ischemia increases myocardial AR activity, with higher Kcat and Vmax due to oxidative stress, without affecting Km [62]. In addition, the expression and activity of AR was significantly higher in aged hearts than young ones in a rat model [68]. Treating aged rats with AR inhibitor reduced ischemic injury and improved cardiac function in aged hearts [68]. Therefore, there is still a need for developing AR inhibitors on myocardial ischemia therapy.
3.1.2. Cardiomyopathy
Cardiomyopathy is degeneration of the myocardium, which causes severe cardiac failure and arrhythmia [88]. Studies showed that AR activation induces oxidative stress [39, 80] which could further trigger the NF-κB pathway [36, 38, 48]. The NF-κB pathway is involved in inflammatory condition which contributes to cardiovascular dysfunction [89]. Sakamoto and Sugamoto observed the upregulation of AR-like gene in heart of cardiomyopathic rodent [70]. Ramana and colleagues also reported that AR inhibition is capable of preventing endotoxin-induced cardiomyopathy [48]. In addition, AR inhibition is able to prevent acute hyperglycemia-induced cardiac contractile dysfunction by reducing oxidative stress [69]. Taken together, observations above indicate that blockade of NF-κB pathway and oxidative stress through AR suppression could be a therapeutic strategy for preventing cardiomyopathy.
3.2. Kidney
3.2.1. Nephropathy
Diabetes has high influences in kidney complications and approximately 30% of diabetic patients have diabetic nephropathy, which is a leading cause of kidney failure in US [90, 91]. A strong immunohistochemical staining of AR was found in diabetic individuals, when compared to non-diabetic individuals, indicating that the level of AR is increased in the kidney of diabetics [92]. Supportive studies reported that high glucose in blood or cultured condition induces AR expression that leads to renal sorbitol accumulation [93–95], reactive oxygen species (ROS) production [96, 97] and inflammation [96] in nephropathy kidney or renal cells culture. Thus, a variety of AR inhibitors had been utilized for treating nephropathy by inhibiting the polyol pathway [73] but showed no influences on kidney weight, body weight or blood glucose [72]. In vivo studies utilizing AR null mice showed that genetic ablation of AR plays a strong protective role in preventing diabetic nephropathy [71]; however, the AR deletion in kidney came with abnormal functioning of the inner medulla [98]. Kidneys from diabetic patients also highly express TGF-β [99], which is an inducer of epithelial-mesenchymal-transition (EMT) [100]. Evidence from human mesangial cells showed that AR inhibition prevents transforming growth factor-beta 1 (TGF-β1)-induced fibronectin expression [101], which is an EMT marker that leads to kidney fibrosis. Another study conducted in renal proximal tubular cells showed that AR inhibition attenuated hyperglycemia-induced fibronectin elevation [93]. Collectively, AR inhibition could be a therapeutic strategy by preventing ROS production and EMT marker expression in patients of nephropathy.
3.2.2. Acute kidney injury
Acute kidney injury (AKI), also called acute renal failure (ARF), is a rapid onset loss of kidney function that may arise from an intense inflammatory process. In AKI mouse model, endotoxin injection increases the levels of blood urea nitrogen, creatinine and cytokines which cause vacuolar degeneration, apoptosis of renal tubular cells and immune cells infiltration [50]. Pretreatment with Fidarestat, an ARI, was able to ameliorate LPS-induced AKI by reducing inflammation and increase survival rate [49, 50]. The polyol pathway has recently been implicated in ischemia/reperfusion tissue injury. Hindlimb ischemia in mice revealed accumulation of sorbitol and fructose in ischemic muscles accompanying secretion of TNF-α and IL-6 in serum, which led to AKI [49]. Treatment with the AR inhibitor was effective at suppressing inflammatory reactions and renal failure [49]. These results suggest that AR inhibition may be a potential therapeutic strategy for treatment of AKI.
3.3. Peripheral Nervous System Disorders
3.3.1. Diabetic peripheral neuropathy
Diabetic peripheral neuropathy (DPN) is nerve damage that affects the 50% of patients with both Type 1 and Type 2 diabetes [102] and causes loss of sensation in the arms, hands, legs and feet [103]. DPN is considered one of the most painful complications affecting diabetic patients [104]. Patients usually feel painful prickling, burning, electrical, sharp, or jabbing sensation [105]. Assessing sensory nerve conduction velocity (SNCV) and motor nerve conduction velocity (MNCV) allows clinicians to determine the degree of neuronal activity or damage [106]. Measurement of F-wave latency is the most common procedure to be diagnose peripheral neuropathy [107]. The efficacy of treatment on DPN is determined by using electrophysiological measurements of three key nerves—median motor, tibial motor, and median sensory nerves. A variety of strategies have been explored for management of DPN including the use of oriental medicine and natural products [108]. Conventional treatments of DPN include the commonly known analgesic drug such as non-steroidal anti-inflammatory drugs (NSAIDs) and opiates [109, 110]. Among the medicine in the management of DPN, tramadol is often used as a second-line analgesic. Tramadol is a semisynthetic opium-derived compound that binds to μ and δ opioid receptors and interferes the re-uptake of serotonin and norepinephrine [111, 112]. However, clinical use of tramadol is not recommended due to the high risk of epileptic seizures and psychiatric disorders. Other analgesic, in particular opium-derived, also has revealed physical and psychological side effects [113]. Therefore, alternative medicine is still an urgent need for management of DPN. Many natural compounds have been reported in the management of DPN. These compounds include phenolic compounds [114], cannabinoids [115, 116], vanilloids [117, 118] and essential fatty acid [119].
In the diabetic patient, AR is overexpressed in a variety of organs/tissues, particular in peripheral nerve [92], suggesting a possible link between AR and DPN. Sorbitol is the metabolite of glucose converted by AR in polyol pathway. Oka and Kato reported that increased accumulation of sorbitol results in the decrease of myo-inositol in the peripheral nerve [120]. Downregulatoin of myo-inositol subsequcetly results in lower Na+, K+ -ATPase activity, which is important for nerve conduction [120]. Therefore, blocking sorbitol accumulation by inhibiting AR polyol pathway is a strategy being considered for DPN treatment. Genetic studies support this concept, as AR knockout mice appear to be protected from delayed motor nerve conduction velocity [71]. Pharmacological inhibition of AR also showed encouraging or convincing results for clinical use. Epalrestat has been approved for use in the treatment of diabetic neuropathy in Japan [121]. Several clinical trials with epalrestat showed that 150 mg/day improves MNCV and SNCV, and subjective symptoms in patients with DPN [122, 123]. Two additional AR inhibitors, Fidarestat [124–126] and Rainrestat [124, 127, 128], also provide encouraging experimental and/or clinincal results in treatment of DPN and diabetic sensorimotor polyneuropathy (DSP), respectively. With these promising results of AR inhibitors, more about their efficacy and safety will need to be investigated to promote them in the U.S. market. Therefore, the natural products with lower cytotoxicity are potential candidates in AR inhibitor development for DPN treatment.
3.3.2. Cardiac autonomic neuropathy
Cardiac autonomic neuropathy (CAN) is characterized by dysfunction in the cardiac autonomic nerves causing dysregulation of heart rate. CAN results in higher incidence of heart failure and sudden death in diabetic patients [129]. In diabetic animal models, sorbitol accumulation from polyol pathway has been considered a major contributor to diabetic neuropathy [130]. Since it is considered an effective AR inhibitor, Sorbinil was used as a therapeutic agent in diabetic patients exhibiting improvement in CAN symptoms [54–56]. Other AR inhibitors such as Epalrestat [131–133], Ponalrestat [134–136] and Tolrestat [137, 138] were also reported as therapeutic agents against CAN. In 1981, Kline et al. developed a useful method entitled I-123 Meta-Iodobenzylguanidin (MIBG) to facilitate imaging the myocardium. This method provides quantitative information of heart rate [139] and is a useful tool in investigating diabetic autonomic disorder in patients [139]. Using MIBG, AR inhibition was observed to alleviate CAN progression in diabetic rats [57] and patients [58]. As a result, AR inhibitors might be a promising treatment for CAN. Thus, development of novel, effective, and nontoxic AR inhibitors is still necessary for slowing the progression of diabetic autonomic neuropathy [140].
3.4. Ocular Disorders
3.4.1. Uveitis
Uveitis is an ocular inflammatory disease of the uvea, the middle layer of the eye that consists of the iris (anterior uveitis), ciliary body (intermediate uveitis) and choroid (posterior uveitis), that contributes about 10 to 20% legal blindness per year [82]. Therefore, the aim of uveitis treatment is to prevent inflammatory responses. Topical eye drops or oral administration of glucocorticoid steroids is the most common treatment for uveitis [141]. In animal studies, lipopolysaccharide (LPS) is commonly used for induction of experimental uveitis, or so-called endotoxin-induced uveitis (EIU) [39, 51, 52, 142, 143]. LPS injection results in induction of TNF-α [51, 52, 142], ROS [51, 142], cyclooxygenase-2 (COX-2) [51, 52, 143], inducible nitric oxide synthase (iNOS) [51, 52, 143] and NF-κB activation [51, 143] in rodent eyes. Experimental autoimmune uveoretinitis (EAU) is another model for the investigation of ocular inflammatory response [52, 144]. Within these two uveitis models, secretion of proinflammatory cytokines is thought to play an important role resulting in damage to ocular tissue [52, 142]. Regarding the effects of AR on inflammatory responses, many studies using macrophages demonstrated that pharmacological inhibition or genetic ablation of AR attenuates LPS-induced cytokines secretion, oxidative stress and cell migration by suppressing MMP-9 and NF-κB activation [36–40]. In addition, studies also reported that downregulation of AR by enzymatic activity or gene expression is capable of preventing experimental models of EIU or EAU [39, 51, 52]. In the eye, retinal microglia (RMG) are one of the major immune cells that participate in surveillance in retinal environment. While they are typically located in the inner and outer plexiform layers in a healthy condition [145, 146], in uveitis they become activated [147] leading to morphological transformation [142] and migration into the outer nuclear layer (photoreceptor layer) where they secrete cytokines and peroxynitrites [144, 146]. In vitro studies using primary cells showed that RMG can be activated by LPS exposure [40, 148] and such activation can be suppressed by addition of an AR inhibitor or in mice lacking the functional allele of the AR gene [40]. For these reasons, we propose that AR could be a therapeutic target for uveitis.
3.4.2. Diabetic cataract
In 2010, it was estimated that around 285 million people worldwide had diabetes [149]. There is an estimation that around 552 million people, which is one in ten, will have diabetes by 2030 [150]. Many studies have shown that diabetes is associated with higher prevalence of cataracts, which remains a major cause of blindness in the world [151–153]. Tight glycemic control is known to reduce the risk of cataracts in subjects with type 2 diabetes [154]. Among the factors thought to induce lens opacification, oxidative damage is thought to be a major mechanism in the onset or progression of diabetic cataract (DC) [155]. Thus, researchers observed that the use of dietary antioxidant alleviates the cataract progression [156, 157]. Galactose is another substrate metabolized by AR and results in accumulation of galactitol, which also causes cataract formation. Studies on the anterior part of eyes showed that AR activation plays a key role in DC formation [158, 159]. AR inhibitors prevent cataract formation in streptozotocin (STZ)-diabetic animal models [77, 78] and galactose-fed rats [76]. Other than the effect of sorbitol, fructose metabolized from glucose in AR polyol pathway is another precursor that initiates production of AGE [79], which contributes to cataractous lenses of human subjects with diabetes [74]. Therefore, there is still an urgent need for ARI development. Since AR expression is low in wildtype mice, even in diabetes, Lee and colleagues generated the human AR expressing mice to accelerate cataractogenesis for the sugar cataract study [159]. We recently generated human AR transgenic (AR-Tg) mice that can shorten the time for DC formation by STZ injection [160], thus providing a useful laboratory model for studying DC formation and prevention. The role for AR in DC formation was further substantiated when it was observed that a lower level of diabetic cataract formed in AR null mice compared to wild type [161].
3.4.3. Diabetic retinopathy
Diabetic retinopathy (DR) is one of the major complications of diabetes and has become the leading cause of blindness in people of working age in the past century [75, 162]. Clinical features of DR are macular edema, retinal ischemia, retinal hemorrhages and microaneurysms, formation of intraretinal microvascular abnormalities, growth of neovascular vessels onto the retina, and retinal detachment [163]. Patients with DR experience a decline of visual acuity that affects many activities of daily living. Among the factors causing DR, vascular endothelial growth factor (VEGF) is considered a major one that leads to neovascularization in retina [164, 165]. Animal studies have also shown a correlation between elevated VEGF and diabetic vasculopathy [166]. Clinical trials have shown that use of the VEGF neutralizing antibodies by intravitreal injection improves visual acuity [167, 168]. Although anti-VEGF therapy has revolutionized management of DR, this procedure requires repeated injections, often monthly for two years, and may lead to impaired survival of neuronal and vascular cells [169]. Thus, alternative strategies are being sought to offset VEGF-driven pathology in the diabetic retina, such as reduction of VEGF protein production. In animal studies, genetic ablation of VEGF in Muller cells reveals the important role of VEGF production in retinopathy [170, 171]. Genetically predisposed diabetic mice (db/db) carrying a mutation in leptin receptor are type 2 diabetic models for investigation of DR [172–174]. Elevation of VEGF in the diabetic retina is decreased when the AR null mutation is introduced into db/db mice, which further prevents blood-retinal barrier (BRB) breakdown and apoptosis in retina [175]. Deletion of AR also prevents mice from streptozotocin-induced DR by inhibiting retinal capillary degeneration and superoxide generation [83]. Reduction of AR activity using inhibitors helps to normalize VEGF levels [166], suppressing VEGF-induced tube formation in retinal endothelial cells [84] and alleviating hyperglycemia-induced damage in retinal pigment epithelial cells [80].
Considering the source of VEGF, Muller cells are believed to be the major immune cells that secrete VEGF in the diabetic eye [81]. However, our current unpublished studies show that genetic ablation of AR reduces hypoxia-induced VEGF secretion by attenuating COX-2 expression in retinal microglia (RMG) indicating that RMG could be another source of VEGF in retina (Fig. 5).
Fig. 5.
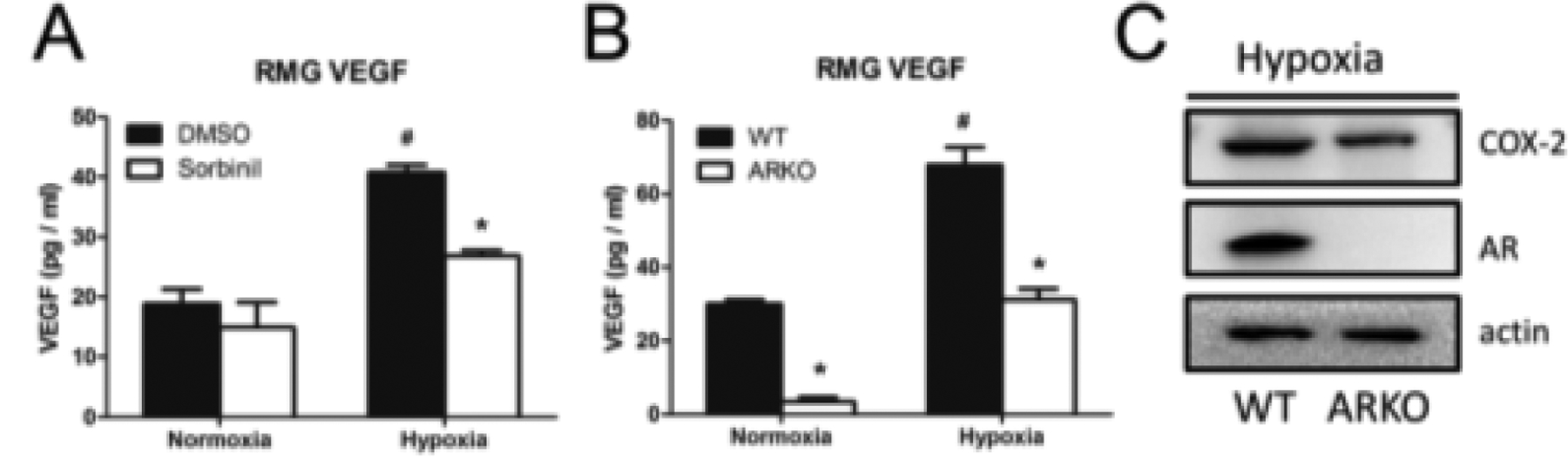
AR inhibition or knockdown prevents hypoxia-induced VEFG secretion by reducing COX-2 expression. RMG were treated with 10 μM Sorbinil (A) or isolated from ARKO mice (B) in normoxia or hypoxia (1% O2) condition for 24 h. Secreted VEGF was measured by using mouse VEGF detection ELISA kit. Hypoxia-induced COX-2 expression in wild type (WT) or ARKO RMG was probed by using Western blot (C). Data shown are means ± SEM (N = 3). *P, #P < 0.05.
Other than anti-VEGF therapy, intravitreal injection of steroids is also used for treatment of diabetic macular edema [176]. Steroids are well-known to reduce inflammatory responses by suppressing NF-κB pathway [177, 178]. In the clinic, patients treated with intravitreal injections of steroids such as triamcinolone and dexamethasone showed improvement in diabetic macular edema by reducing central macular thickness [179]. However, these treatments have the side effects of causing cataracts. Nevertheless, suppression of NF-κB pathway is an effective strategy for prevention of onset and/or progression of DR which can be a blinding disease if left untreated.
Systemic inflammation is considered to be an intrinsic response to diabetes [180]. Inflammatory cytokines like interleukin-1β (IL-1β) and TNF-α are increased in the vitreous of patients with DR [181]. Increased TNF-α in retina leads to retinal vascular permeability [182], microglia activation [183] and induction of apoptotic protein markers [183] in retina. Collective data suggested that anti-inflammatory treatment with glucocorticoids [184] or minocycline [183] attenuates severity of retinopathy and helps to restore the BRB. Muller cells and RMG are thought to contribute to inflammatory responses in retina [163]. Our previous study showed that downregulation of AR reduces inflammatory responses in RMG [40] suggesting AR inhibition plays an alternative role in preventing DR by suppressing inflammatory responses in the diabetic eye.
Advanced glycation end-products (AGEs) have been shown to induce VEFG production [81] and matrix metalloproteinases (MMPs) [185] in the diabetic retina. Induction of MMPs alters the BRB by initiating morphological changes in retinal endothelial cells [185], which is often observed in DR. Amadori-glycated protein is the precursor to AGEs [46]. Patients with diabetes have increase of Amadori-glycated albumin (AGA) in serum [186] which correlates with higher risk of retinopathy [187]. Animal studies also showed the increase of AGA in the diabetic retina [188]. Inflammation in diabetic individuals could be induced by AGEs in kidney via NF-κB pathway [189] or by AGA in retina through Mitogen-activated protein kinases (MAPK) pathway [188]. Previous studies showed that AR inhibition or genetic deficiency suppresses the NF-κB [36–38] and MAPK pathways [39]. Therefore, it would be an interesting question whether AR mediates AGE or AGA-induced inflammatory responses in diabetic retina. Recent studies have shown that RMG were activated in the presence of AGE [190, 191] and AGA [188], and follow the induction of TNF-α. Our current unpublished data observed that AR inhibition suppresses AGA-induced TNF-α secretion and cell migration in RMG suggesting that AR is involved in AGA-mediated DR (Fig. 4).
Fig. 4.
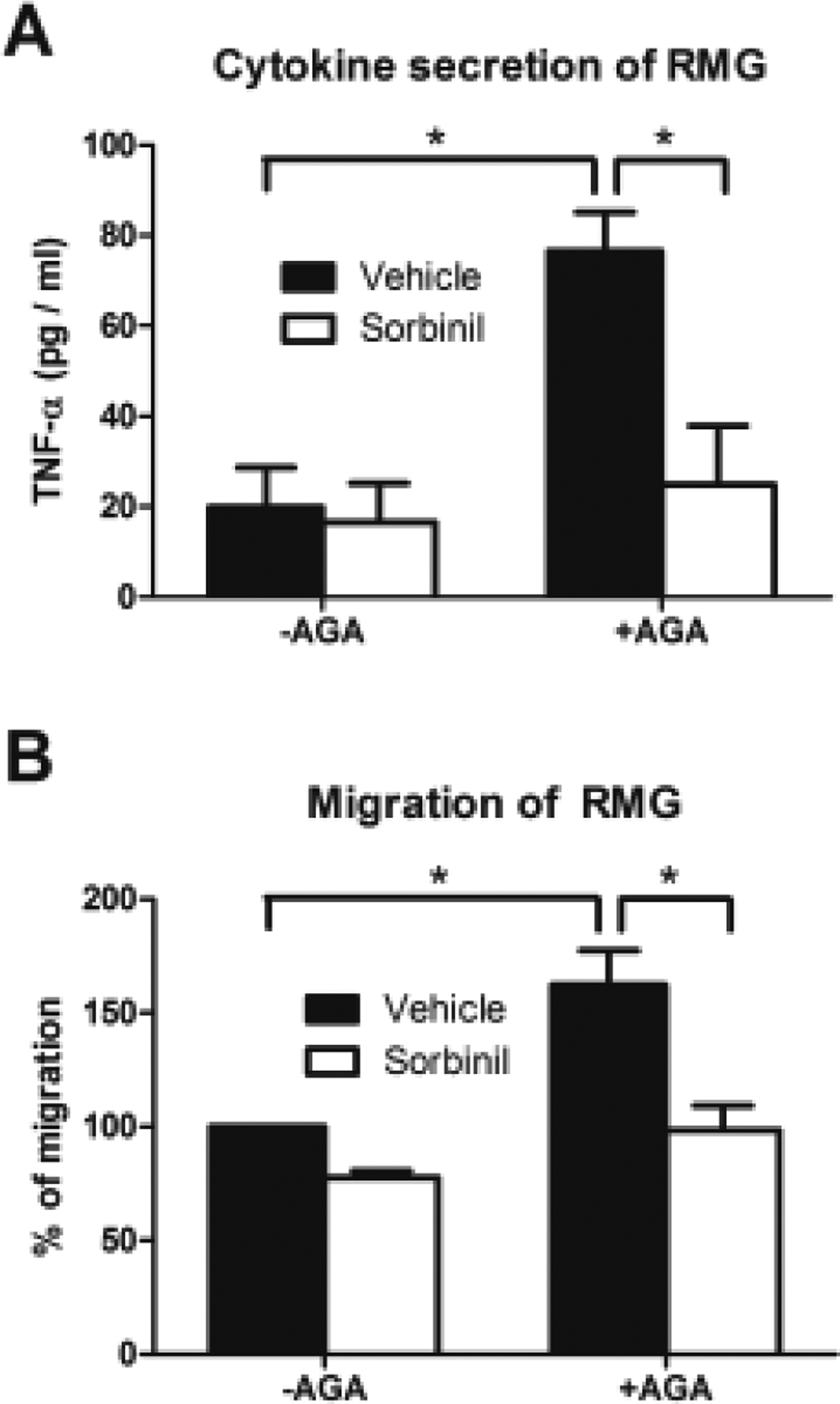
Effect of ARI on AGA-induced cytokine secretion and migration in RMG. RMG were treated with vehicle or Sorbinil (10 μM) in the absence or presence of AGA (500 μg/ml) for 24 h for TNF-α secretion (A) or migration assay (B). Secreted TNF- α was measured by using mouse TNF-α detection ELISA kit. The migration assay was conducted by using transwell method. Data shown are means ± SEM (N = 3). *P < 0.05.
3.4.4. Posterior capsule opacification
Posterior capsule opacification (PCO), which is the complication after cataract surgery [192], results from abnormal proliferation and migration of lens epithelia cells (LECs) [193] in the central posterior capsule resulting in degraded visual acuity. LECs undergo differentiation from an epithelial to a myofibroblast phenotype [100] and matrix contraction [194], which further leads to opacification. TGF-β overexpression in Tg mice led to morphological changes in lens that resembled PCO in human [195]. In the process of PCO, TGF-β plays an important role in developing epithelial-mesenchymal-transition (EMT) [100], resulting in expression of EMT related markers such as α-smooth muscle actin (α-SMA) [194], and forming cells with a spindled-shaped myofibroblastic morphology [100]. TGF-β also induces MMPs such as MMP-2 and -9, which has been demonstrated that can be induced under mechanical trauma of cataract surgery [194]. Therefore, suppression of EMT and MMPs activation could lead to prevention of PCO. Previous works showed that AR inhibition suppresses LPS or high glucose-induced MMP-9 activation [40, 196, 197]. Kidney studies further reported that TGF-β-induced MMPs and EMT activations were attenuated by AR inhibitor treatment in renal cells [101, 198]. Yadav and colleagues reported a study using pig capsule that AR inhibitor were shown to reduce LECs proliferation and expression EMT markers [199]. SMAD signaling pathway has been identified as playing a critical downstream role in TGF-β-mediated signaling [200]. TGF-β interaction with its receptor leads to SMADS phosphorylation and subsequent translocation to the nucleus to trigger EMT process [201]. Recently, an encouraging study in LECs showed that AR inhibition suppresses TGF-β2-induced SMADs phosphorylation and its downstream regulatory pathways including cell migration, EMT initiation and MMPs activation [202]. A novel function of AR was reported, involving AR interaction with SMADs. This novel model offers a possible mechanism to explain how AR inhibitor treatment suppresses SMADs activation [202]. This finding indicated the AR is required to facilitate TGF-β/SMADs pathway and AR inhibitor disrupts AR-SMADs interaction. Accordingly, AR could be a therapeutic target for PCO.
4. Summary and future directions
4.1. N ovel AR inhibito rs
In the view of AR inhibitor and ocular inflammation, we determined whether β-glucogallin (BGG), isolated from Indian gooseberry, is an efficacious AR inhibitor. We observed that BGG is a potential anti-inflammatory agent against endotoxin-induced uveitis in an experimental mouse model. BGG not only alleviated inflammatory responses in macrophages but also suppressed infiltration of immune cells into the eye [39]. However, our extensive research showed the instability of BGG in thermal acidic condition [203]. We observed that the ester linkage in BGG (glycosyl 1-ester) is labile in aqueous solution. Thus, our collaborators Dr. Daniel LaBarbera and his lab designed β-glucogallin amide (BGA), a derivative of BGG produced by replacing the ester with an amide linkage to join the gallic acid and glucose ring. BGA demonstrated similar inhibitory activity in vitro and ex vivo but much better stability under thermal acidic condition [203]. With compatible activity and greatly improved stability, BGA holds a promise to be an attractive therapeutic lead toward the treatment of ocular inflammation. Further structure-based drug design is presently ongoing to improve the pharmacological profile of BGA, and more sophisticated animal models will be used to test BGA efficacy in vivo.
Our studies of BGG were motivated by strong animal study results which showed that this AR inhibitor can prevent complications of diabetes mellitus. Clinical trials of many AR inhibitors have been unsuccessful in part due to toxicity from their metabolic breakdown products. In our studies of the effect of AR inhibition on inflammation associated with endotoxin-induced uveitis, we included Sorbinil as a positive AR inhibitor control. Sorbinil is no longer considered a candidate for human therapy because previous clinical studies showed that microsomal metabolites of Sorbinil are cytotoxic [204]. Similarly, other AR inhibitors such as Imirestat [205], Tolrestat [206, 207] and Zoporestat [207] also failed in clinical trials due to liver and/or renal toxicity. So far, only Epalrestat has been shown to be safe and efficacious against diabetic peripheral neuropathy and is now marketed in Japan [121, 207]. Therefore, the encouraging results of BGG and preliminary data from BGA provide a new direction for development of AR inhibitors based on a novel pharmacophore structurally unrelated to previously failed AR inhibitors. Since BGG is abundant in many fruits consumed by humans (gooseberry, rhubarb), it is likely that it will not cause liver and renal complications.
In the study of DR, we wanted to know whether BGG is capable of preventing diabetic complications. We developed hyperglycemic condition using high glucose medium on ARPE-19 cells. We found that BGG is an efficacious AR inhibitor reducing hyperglycemia-induced cell death, ROS production, ER stress and mitochondrial dysfunction [80]. Hyperglycemia also elevates the level of advanced glycation end products (AGEs) in the serum of diabetic patients [186]. A study on AGEs has been known to induce ocular inflammation by triggering RMG activation [148]. Our preliminary studies showed that AR inhibition attenuates amadori glycated albumin (AGA)-induced inflammatory responses in RMG (Fig. 4). The mechanism behind this finding remains unknown and should be studied. Since low cytotoxicity of BGG has been shown in cell line model [39], extensive studies of BGG in animal study are feasible. Diabetes shows higher risk in elevating VEGF in retina that causes neovascularization [164, 165]. Our preliminary showed that pharmacological inhibition or genetic ablation of AR prevents hypoxia-induced VEGF secretion from RMG (Fig. 5). Based on our previous study of ARI on RMG, We believe that BGG and BGA are a potential therapeutics for diabetic complication by preventing RMG activation in diabetic retina. However, to further apply BGA to next step, more pharmaceutical kinetics and toxicity experiments are necessary. Although BGA is derived from natural compound, the toxicity in vivo has not been studied. Animal toxicity study of BGA injection should be conducted before BGA’s further application. Since BGA was designed for its higher stability, in vivo pharmaceutical kinetics should also be studied in the future.
4.2. A role for AR in ocular inflammation
In the eye, RMG is one of the immune cells that normally reside in the inner retina. However, under inflammatory conditions, RMG can be found in higher numbers in the subretinal space between photoreceptor outer segments and the RPE. Activated RMG secrete chemokines and/or cytokines to damage neural and retinal cells in the eye. Therefore, regulation of RMG may be critical for preventing ocular inflammation. To investigate a more specific area of the eye, we conducted an ex vivo study to examine a potential role for AR in the response of RMG to endotoxin exposure. We observed that inflammatory responses in RMG following exposure to endotoxin were substantially suppressed when cells were treated with AR inhibitors or were genetically deficient for AR gene expression. These results demonstrated that pharmacological inhibition or genetic ablation of AR prevents endotoxin-induced inflammation in the retina by suppressing RMG activation [40]. An MMP-9 inhibitor, designed to inhibit gelatinase activity, was shown to prevent LPS-induced cell migration. By a different route, AR inhibition prevents cell migration by reducing MMP-9 protein expression. Therefore, one would expect additive effects of combined treatment with AR and MMP inhibitors on preventing cell migration. A preliminary animal study was extensively performed using CX3CR1 transgenic mice, a strain in which monocytes, including RMG, constitutively express green fluorescence protein and therefore are easily observed in ocular tissue sections. We observed that LPS injection triggers RMG activation and migration into inner and outer nuclear layers (Fig. 6). Co-injection with Sorbinil alleviated LPS-induced RMG activation and migration in the retina (Fig. 6) indicating that AR inhibition is valid in vivo to prevent ocular inflammation. In inflammation, TNF-α plays a robust role in causing apoptosis. We have previously shown that downregulation of AR by either pharmacological inhibition or genetic ablation reduces TNF-α secretion in RMG as well as apoptosis in co-cultured RPE cells [40]. Studies on the detection the level of TNF-α secretion and apoptosis in retina should be conducted in the future. Since we have AR null mice, genetic effects of AR on LPS-induced RMG activation would be a convincing experiment to confirm the role of AR in retinal inflammation. The study of RMG provides a therapeutic target in the retina for AR-associated ocular inflammatory diseases such as uveitis and retinopathy. In addition, Muller cells are the glial cells that have immune functions in the eye. AR was also reported to express in the Muller cells [208, 209]. Several studies showed that Muller cells are involved in uveitis, proliferative vitreoretinopathy (PVR) and DR [210–212]. Therefore, the effect of AR on Muller cells is an interesting study to be conducted in the future.
Fig. 6.
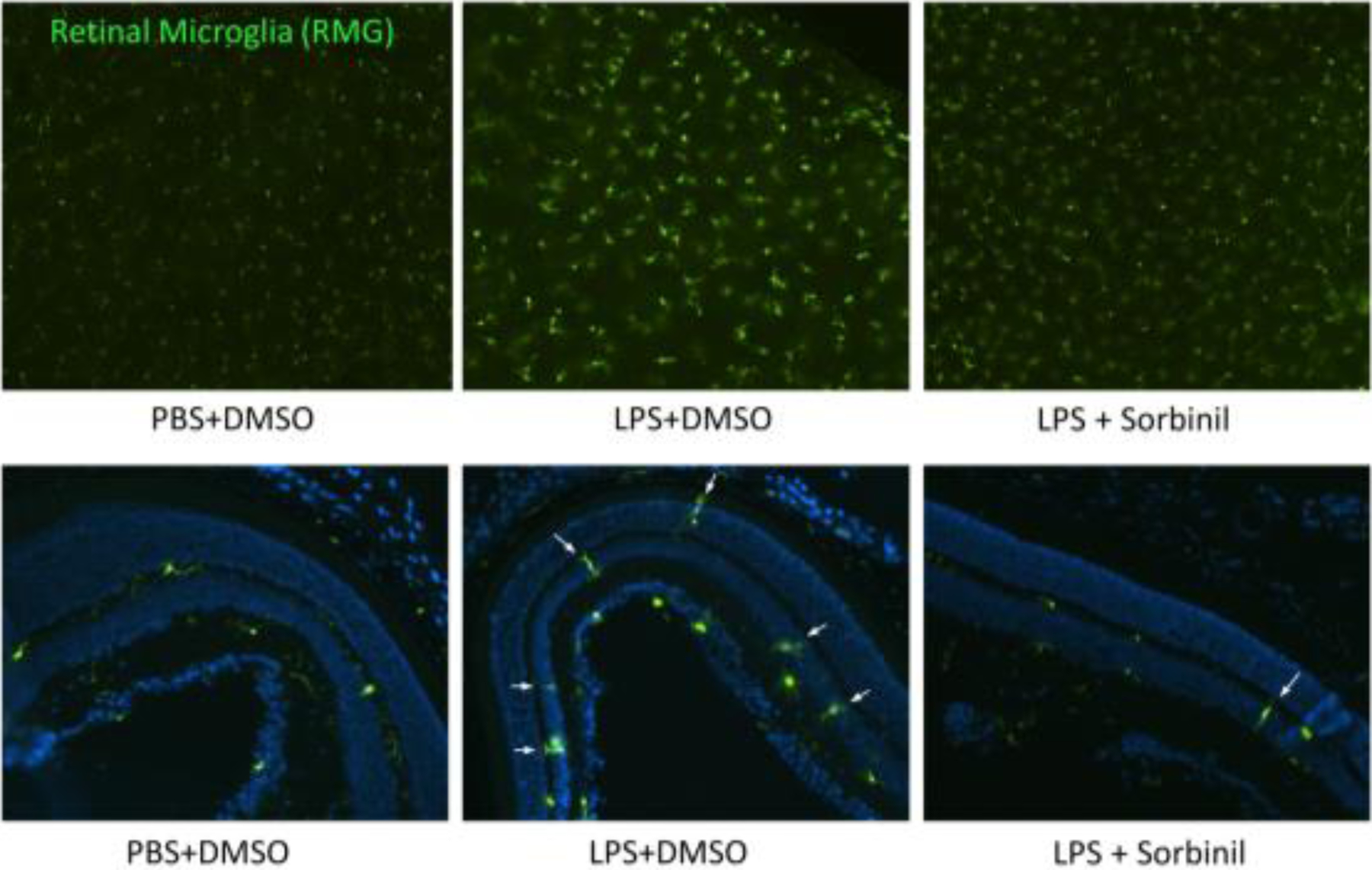
AR inhibition prevents RMG activation and migration under LPS exposure. Cx3Cr1 transgenic mice were injected with LPS (500 ug / mouse) for 24 h with or without Sorbinil co-injection. Green spots indicate RMG in retinas. White arrows indicate RMG migration into inner or outer nuclear layers. Figure adapted by Chang and Petrash from Biochem Biophys Res Commun. 2016; 473(2):565–571.
In the experimental uveitis model or hyperglycemic stress experiment, we treated the mice or cell line at the same time while uveitis or hyperglycemic stresses were being induced. However, in real practice, treatment is always applied after disease occurs. Based on this point, the efficacy of AR inhibitors after stressor such as LPS or hyperglycemia needs to be tested. Although we haven’t conducted experiments regarding this issue, administration of AR inhibitors after retinal inflammation or hyperglycemia would better mimic the treatment paradigm in uveitis or diabetes management. It is possible that the effect of post-treatment would be less effective than pre-treatment due to some possible irreversible changes that may have occurred or the disease may have gone to a more advanced stage that is less susceptible to ARI therapy.
We observed that AR inhibitors lowered the abundance of inflammatory cells in the vitreous of endotoxin-treated animals. One possibility is that inhibitors reduce expression of MMPs and thereby reduce the ability of cells to migrate toward their targets. Another possible explanation for reduced immune cell infiltration may relate to their ability to pass through the vascular endothelium. Adhesion molecules are cell surface receptors that facilitate the binding of immune cells to endothelial cells and penetration [213]. These adhesion molecules include intracellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule (VCAM). TNF-α is an inflammatory cytokine that mediates pathological endothelial changes which cause induction of adhesion molecule expression and vascular leakage at the site of inflammation [214]. Ramana and colleagues reported that AR inhibition prevents TNF-α-induced increases of ICAM-1 and VCAM in human umbilical vein endothelial cells (HUVECs) as well as decreases monocytes adhesion to these cells [215]. This observation may provide another explanation for the effect of AR inhibitors on prevention of inflammatory cells infiltration into the eye.
4.3. Studies of AR as a mediator of EMT during development of posterior capsule opacification
Regarding the effect of AR inhibitor on PCO development, we proposed a novel idea of AR with a noncatalytic function. We observed AR interacts with Smads in a NADPH-dependent pathway but in a manner not requiring enzymatic activity [202]. This is the first paper that reports a non-enzymatic function of AR. We also found that AR inhibition or genetic ablation prevents TGF-β2-induced Smads activation and expression of EMT markers, thus indicating a potential therapeutic strategy for prevention of PCO development. However, remaining unknown are the details of how AR interacts with Smads and whether there is a distinct interaction site. Sequence deletion could be used for understanding the possible site or domain. Since AR null mice are available, we can develop PCO in either wildtype or AR null mice to confirm the hypothesis in vivo. Sorbinil disruption of AR-Smads interaction could be resulted from an AR conformational change or actual blocking of a protein-protein interaction site. In this study, we only tested the noncatalytic function of AR using Sorbinil. However, whether other ARIs such as BGG also contribute similar effect is still unknown. Other ARIs may fail to disrupt AR-Smads interaction due to different results of conformational change or blocking site. Therefore, more studies on different ARIs on noncatalytic effect need to be conducted to elucidate the hypothesis.
Most of the published studies surrounding Sorbinil as an anti-inflammatory agent implicitly presume its efficacy derives from inhibition of AR catalytic activity. In our PCO study, we showed that a catalytically inactive mutant of AR was able to facilitate TGF-β2-induced Smad activation, demonstrating that the ability of Sorbinil to downregulate Smad activation was unrelated to its ability to inhibit AR catalysis. Therefore, the effect of Sorbinil can be segregated into its effects on catalytic and noncatalytic functions of AR. Previous studies reported that AR plays a catalytic role in reduction of aldehydes during lipid peroxidation pathway following NF-κB activation [20, 21] and AR inhibitors such as Sorbinil suppress this pathway [23, 25–27]. However, the noncatalytic role of AR in NF-κB activation has not been studied to date. Going forward, it would be possible to address this question by transfecting AR null cells with wildtype AR (wtAR) or an active site mutant AR (mutAR) and treat the cells with or without AR inhibitors (Sorbinil or BGG) under LPS exposure. If LPS induces the same level of NF-κB activation in mutAR group compared to wtAR group, one could conclude that AR facilitates NF-κB activation in a noncatalytic fashion. Similarly, if Sorbinil treatment prevented NF-κB activation in both groups, it would be consistent with the notion that an AR interaction domain overlapping the ARI binding site is important for NF-κB activation in a manner similar to our findings with AR and Smad activation.
AR has been studied extensively as a catalyst that results in sorbitol accumulation and contributes to pathogenesis of diabetic complications. Our current pharmacological studies confirm the importance of AR through its role as a regulator of glucose metabolism through the polyol pathway (catalytic function). In addition, we have compelling results which point to AR as a component of the TGF-β signaling pathway (noncatalytic function). It will be exciting to see the results of future studies which aim to further clarify the mechanism linking AR to TGF-β signaling and possible therapeutic strategies to prevent TGF-β-associated disease.
5 References
- 1.Bohren KM, Bullock B, Wermuth B, Gabbay KH. The aldo-keto reductase superfamily. cDNAs and deduced amino acid sequences of human aldehyde and aldose reductases. The Journal of biological chemistry 1989; 264(16):9547–51. [PubMed] [Google Scholar]
- 2.Iwata N, Inazu N, Satoh T. The purification and characterization of NADPH-dependent carbonyl reductase from rat ovary. Progress in clinical and biological research 1989; 290:307–21. [PubMed] [Google Scholar]
- 3.Penning TM, Burczynski ME, Jez JM, Hung CF, Lin HK, Ma H, Moore M, Palackal N, Ratnam K. Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. The Biochemical journal 2000; 351(Pt 1):67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Komoto J, Yamada T, Watanabe K, Takusagawa F. Crystal structure of human prostaglandin F synthase (AKR1C3). Biochemistry 2004; 43(8):2188–98. [DOI] [PubMed] [Google Scholar]
- 5.Hyndman D, Bauman DR, Heredia VV, Penning TM. The aldo-keto reductase superfamily homepage. Chemico-biological interactions 2003; 143–144:621–31. [DOI] [PubMed] [Google Scholar]
- 6.Jez JM, Bennett MJ, Schlegel BP, Lewis M, Penning TM. Comparative anatomy of the aldo-keto reductase superfamily. The Biochemical journal 1997; 326 (Pt 3):625–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barski OA, Gabbay KH, Bohren KM. Characterization of the human aldehyde reductase gene and promoter. Genomics 1999; 60(2):188–98. [DOI] [PubMed] [Google Scholar]
- 8.Hayman S, Kinoshita JH. Isolation and Properties of Lens Aldose Reductase. The Journal of biological chemistry 1965; 240:877–82. [PubMed] [Google Scholar]
- 9.Bauman DR, Steckelbroeck S, Penning TM. The roles of aldo-keto reductases in steroid hormone action. Drug news & perspectives 2004; 17(9):563–78. [DOI] [PubMed] [Google Scholar]
- 10.Sakuma M, Kametani S, Akanuma H. Purification and some properties of a hepatic NADPH-dependent reductase that specifically acts on 1,5-anhydro-D-fructose. Journal of biochemistry 1998; 123(1):189–93. [DOI] [PubMed] [Google Scholar]
- 11.Penning TM. The aldo-keto reductases (AKRs): Overview. Chemico-biological interactions 2015; 234:236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nature immunology 2002; 3(3)221–7. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunological reviews 2006; 210:171–86. [DOI] [PubMed] [Google Scholar]
- 14.Baldwin AS Jr. The NF-kappa B and I kappa B proteins: new discoveries and insights. Annual review of immunology 1996; 14:649–83. [DOI] [PubMed] [Google Scholar]
- 15.Sheppard KA, Phelps KM, Williams AJ, Thanos D, Glass CK, Rosenfeld MG, Gerritsen ME, Collins T. Nuclear integration of glucocorticoid receptor and nuclear factor-kappaB signaling by CREB-binding protein and steroid receptor coactivator-1. The Journal of biological chemistry 1998; 273(45):29291–4. [DOI] [PubMed] [Google Scholar]
- 16.Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, Thanos D, Rosenfeld MG, Glass CK, Collins T. Transcriptional activation by NF-kappaB requires multiple coactivators. Molecular and cellular biology 1999; 19(9):6367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. The EMBO journal 2002; 21(23):6539–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. The EMBO journal 2004; 23(12):2369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramana KV, Fadl AA, Tammali R, Reddy ABM, Chopra AK, Srivastava SK. Aldose Reductase Mediates the Lipopolysaccharide-induced Release of Inflammatory Mediators in RAW264.7 Murine Macrophages. Journal Of Biological Chemistry 2006; 281(44):33019–29. [DOI] [PubMed] [Google Scholar]
- 20.Srivastava S, Dixit BL, Cai J, Sharma S, Hurst HE, Bhatnagar A, Srivastava SK. Metabolism of lipid peroxidation product, 4-hydroxynonenal (HNE) in rat erythrocytes: role of aldose reductase. Free radical biology & medicine 2000; 29(7):642–51. [DOI] [PubMed] [Google Scholar]
- 21.Vander Jagt DL, Kolb NS, Vander Jagt TJ, Chino J, Martinez FJ, Hunsaker LA, Royer RE. Substrate specificity of human aldose reductase: identification of 4-hydroxynonenal as an endogenous substrate. Biochimica et biophysica acta 1995; 1249(2):117–26. [DOI] [PubMed] [Google Scholar]
- 22.Maccari R, Ottana R. Targeting aldose reductase for the treatment of diabetes complications and inflammatory diseases: new insights and future directions. Journal of medicinal chemistry 2015; 58(5):2047–67. [DOI] [PubMed] [Google Scholar]
- 23.Srivastava SK, Ramana KV, Bhatnagar A. Role of aldose reductase and oxidative damage in diabetes and the consequent potential for therapeutic options. Endocrine reviews 2005; 26(3):380–92. [DOI] [PubMed] [Google Scholar]
- 24.Ramana KV. Aldose reductase: New Insights for an Old Enzyme. Biomolecular concepts 2011; 2(1–2):103–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramana KV, Friedrich B, Srivastava S, Bhatnagar A, Srivastava SK. Activation of nuclear factor-kappaB by hyperglycemia in vascular smooth muscle cells is regulated by aldose reductase. Diabetes 2004; 53(11):2910–20. [DOI] [PubMed] [Google Scholar]
- 26.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocrine reviews 2002; 23(5):599–622. [DOI] [PubMed] [Google Scholar]
- 27.Ramana KV, Friedrich B, Tammali R, West MB, Bhatnagar A, Srivastava SK. Requirement of aldose reductase for the hyperglycemic activation of protein kinase C and formation of diacylglycerol in vascular smooth muscle cells. Diabetes 2005; 54(3):818–29. [DOI] [PubMed] [Google Scholar]
- 28.Shen W, Gao Y, Lu B, Zhang Q, Hu Y, Chen Y. Negatively regulating TLR4/NF-kappaB signaling via PPARalpha in endotoxin-induced uveitis. Biochimica et biophysica acta 2014; 1842(7):1109–20. [DOI] [PubMed] [Google Scholar]
- 29.Kalariya NM, Shoeb M, Ansari NH, Srivastava SK, Ramana KV. Antidiabetic drug metformin suppresses endotoxin-induced uveitis in rats. Investigative ophthalmology & visual science 2012; 53(7):3431–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lustig MJ, Cunningham ET Jr. Use of immunosuppressive agents in uveitis. Current opinion in ophthalmology 2003; 14(6):399–412. [DOI] [PubMed] [Google Scholar]
- 31.Cunningham ET Jr., Wender JD. Practical approach to the use of corticosteroids in patients with uveitis. Canadian journal of ophthalmology Journal canadien d’ophtalmologie 2010; 45(4):352–8. [DOI] [PubMed] [Google Scholar]
- 32.Sadiq MA, Agarwal A, Hassan M, Afridi R, Sarwar S, Soliman MK, Do DV, Nguyen QD. Therapies in Development for Non-infectious Uveitis. Current molecular medicine 2015. [DOI] [PubMed] [Google Scholar]
- 33.Vermeulen L, De Wilde G, Notebaert S, Vanden Berghe W, Haegeman G. Regulation of the transcriptional activity of the nuclear factor-kappaB p65 subunit. Biochemical pharmacology 2002; 64(5–6):963–70. [DOI] [PubMed] [Google Scholar]
- 34.Ceolotto G, De Kreutzenberg SV, Cattelan A, Fabricio AS, Squarcina E, Gion M, Semplicini A, Fadini GP, Avogaro A. Sirtuin 1 stabilization by HuR represses TNF-alpha- and glucose-induced E-selectin release and endothelial cell adhesiveness in vitro: relevance to human metabolic syndrome. Clinical science 2014; 127(7):449–61. [DOI] [PubMed] [Google Scholar]
- 35.Petrash JM. All in the family: aldose reductase and closely related aldo-keto reductases. Cellular and molecular life sciences : CMLS 2004; 61(7–8):737–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramana KV, Fadl AA, Tammali R, Reddy AB, Chopra AK, Srivastava SK. Aldose reductase mediates the lipopolysaccharide-induced release of inflammatory mediators in RAW264.7 murine macrophages. The Journal of biological chemistry 2006; 281(44):33019–29. [DOI] [PubMed] [Google Scholar]
- 37.Ramana KV, Reddy AB, Tammali R, Srivastava SK. Aldose reductase mediates endotoxin-induced production of nitric oxide and cytotoxicity in murine macrophages. Free radical biology & medicine 2007; 42(8):1290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramana KV, Srivastava SK. Mediation of aldose reductase in lipopolysaccharide-induced inflammatory signals in mouse peritoneal macrophages. Cytokine 2006; 36(3–4):115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang KC, Lafin B, Ponder J, Enzsoly A, Nemeth J, Labarbera DV, Petrash JM. Beta-glucogallin reduces the expression of lipopolysaccharide-induced inflammatory markers by inhibition of aldose reductase in murine macrophages and ocular tissues. Chemico-biological interactions 2013; 202(1–3):283–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang KC, Ponder J, Labarbera DV, Petrash JM. Aldose reductase inhibition prevents endotoxin-induced inflammatory responses in retinal microglia. Invest Ophthalmol Vis Sci 2014; 55(5)2853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhatnagar A, Srivastava SK. Aldose reductase: congenial and injurious profiles of an enigmatic enzyme. Biochemical medicine and metabolic biology 1992; 48(2):91–121. [DOI] [PubMed] [Google Scholar]
- 42.Sheetz MJ, King GL. Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. Jama 2002; 288(20):2579–88. [DOI] [PubMed] [Google Scholar]
- 43.Lou MF. Redox regulation in the lens. Progress in retinal and eye research 2003; 22(5):657–82. [DOI] [PubMed] [Google Scholar]
- 44.Vedantham S, Ananthakrishnan R, Schmidt AM, Ramasamy R. Aldose reductase, oxidative stress and diabetic cardiovascular complications. Cardiovascular & hematological agents in medicinal chemistry 2012; 10(3):234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koya D, King GL. Protein kinase C activation and the development of diabetic complications. Diabetes 1998; 47(6):859–66. [DOI] [PubMed] [Google Scholar]
- 46.Kandarakis SA, Piperi C, Topouzis F, Papavassiliou AG. Emerging role of advanced glycation-end products (AGEs) in the pathobiology of eye diseases. Progress in retinal and eye research 2014; 42:85–102. [DOI] [PubMed] [Google Scholar]
- 47.Sadowska-Bartosz I, Galiniak S, Bartosz G. Kinetics of glycoxidation of bovine serum albumin by glucose, fructose and ribose and its prevention by food components. Molecules 2014; 19(11):18828–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramana KV, Willis MS, White MD, Horton JW, DiMaio JM, Srivastava D, Bhatnagar A, Srivastava SK. Endotoxin-induced cardiomyopathy and systemic inflammation in mice is prevented by aldose reductase inhibition. Circulation 2006; 114(17):1838–46. [DOI] [PubMed] [Google Scholar]
- 49.Yagihashi S, Mizukami H, Ogasawara S, Yamagishi S, Nukada H, Kato N, Hibi C, Chung S, Chung S. The role of the polyol pathway in acute kidney injury caused by hindlimb ischaemia in mice. The Journal of pathology 2010; 220(5):530–41. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi K, Mizukami H, Kamata K, Inaba W, Kato N, Hibi C, Yagihashi S. Amelioration of acute kidney injury in lipopolysaccharide-induced systemic inflammatory response syndrome by an aldose reductase inhibitor, fidarestat. PloS one 2012; 7(1):e30134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yadav UC, Srivastava SK, Ramana KV. Aldose reductase inhibition prevents endotoxin-induced uveitis in rats. Investigative ophthalmology & visual science 2007; 48(10):4634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yadav UC, Shoeb M, Srivastava SK, Ramana KV. Aldose reductase deficiency protects from autoimmune- and endotoxin-induced uveitis in mice. Investigative ophthalmology & visual science 2011; 52(11)8076–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Filippo C, Zippo MV, Maisto R, Trotta MC, Siniscalco D, Ferraro B, Ferraraccio F, La Motta C, Sartini S, Cosconati S, Novellino E, Gesualdo C, Simonelli F, Rossi S, D’Amico M. Inhibition of ocular aldose reductase by a new benzofuroxane derivative ameliorates rat endotoxic uveitis. Mediators of inflammation 2014; 2014:857958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jaspan JB, Towle VL, Maselli R, Herold K. Clinical studies with an aldose reductase inhibitor in the autonomic and somatic neuropathies of diabetes. Metabolism: clinical and experimental 1986; 35(4 Suppl 1):83–92. [DOI] [PubMed] [Google Scholar]
- 55.Green A, Jaspan J, Kavin H, Chung S, Schoenberg H. Influence of long-term aldose reductase inhibitor therapy on autonomic dysfunction of urinary bladder, stomach and cardiovascular systems in diabetic patients. Diabetes research and clinical practice 1987; 4(1):67–75. [DOI] [PubMed] [Google Scholar]
- 56.Roy TM, Broadstone VL, Peterson HR, Snider HL, Cyrus J, Fell R, Rothchild AH, Samols E, Pfeifer MA. The effect of an aldose reductase inhibitor on cardiovascular performance in patients with diabetes mellitus. Diabetes research and clinical practice 1990; 10(1):91–7. [DOI] [PubMed] [Google Scholar]
- 57.Kurata C, Okayama K, Wakabayashi Y, Shouda S, Mikami T, Tawarahara K, Sugiyama T. Cardiac sympathetic neuropathy and effects of aldose reductase inhibitor in streptozotocin-induced diabetic rats. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 1997; 38(11):1677–80. [PubMed] [Google Scholar]
- 58.Utsunomiya K, Narabayashi I, Tamura K, Nakatani Y, Saika Y, Onishi S, Kariyone S. Effects of aldose reductase inhibitor and vitamin B12 on myocardial uptake of iodine-123 metaiodobenzylguanidine in patients with non-insulin-dependent diabetes mellitus. European journal of nuclear medicine 1998; 25(12):1643–8. [DOI] [PubMed] [Google Scholar]
- 59.Ramasamy R, Oates PJ, Schaefer S. Aldose reductase inhibition protects diabetic and nondiabetic rat hearts from ischemic injury. Diabetes 1997; 46(2):292–300. [DOI] [PubMed] [Google Scholar]
- 60.Ramasamy R, Liu H, Oates PJ, Schaefer S. Attenuation of ischemia induced increases in sodium and calcium by the aldose reductase inhibitor zopolrestat. Cardiovascular research 1999; 42(1):130–9. [DOI] [PubMed] [Google Scholar]
- 61.Tracey WR, Magee WP, Ellery CA, MacAndrew JT, Smith AH, Knight DR, Oates PJ. Aldose reductase inhibition alone or combined with an adenosine A(3) agonist reduces ischemic myocardial injury. American journal of physiology Heart and circulatory physiology 2000; 279(4)H1447–52. [DOI] [PubMed] [Google Scholar]
- 62.Hwang YC, Sato S, Tsai JY, Yan S, Bakr S, Zhang H, Oates PJ, Ramasamy R. Aldose reductase activation is a key component of myocardial response to ischemia. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2002; 16(2):243–5. [DOI] [PubMed] [Google Scholar]
- 63.Shinmura K, Bolli R, Liu SQ, Tang XL, Kodani E, Xuan YT, Srivastava S, Bhatnagar A. Aldose reductase is an obligatory mediator of the late phase of ischemic preconditioning. Circulation research 2002; 91(3):240–6. [DOI] [PubMed] [Google Scholar]
- 64.Hwang YC, Kaneko M, Bakr S, Liao H, Lu Y, Lewis ER, Yan S, Ii S, Itakura M, Rui L, Skopicki H, Homma S, Schmidt AM, Oates PJ, Szabolcs M, Ramasamy R. Central role for aldose reductase pathway in myocardial ischemic injury. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2004; 18(11):1192–9. [DOI] [PubMed] [Google Scholar]
- 65.Hwang YC, Shaw S, Kaneko M, Redd H, Marrero MB, Ramasamy R. Aldose reductase pathway mediates JAK-STAT signaling: a novel axis in myocardial ischemic injury. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2005; 19(7):795–7. [DOI] [PubMed] [Google Scholar]
- 66.Kaiserova K, Srivastava S, Hoetker JD, Awe SO, Tang XL, Cai J, Bhatnagar A. Redox activation of aldose reductase in the ischemic heart. The Journal of biological chemistry 2006; 281(22):15110–20. [DOI] [PubMed] [Google Scholar]
- 67.Kaiserova K, Tang XL, Srivastava S, Bhatnagar A. Role of nitric oxide in regulating aldose reductase activation in the ischemic heart. The Journal of biological chemistry 2008; 283(14):9101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ananthakrishnan R, Li Q, Gomes T, Schmidt AM, Ramasamy R. Aldose reductase pathway contributes to vulnerability of aging myocardium to ischemic injury. Experimental gerontology 2011; 46(9):762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tang WH, Cheng WT, Kravtsov GM, Tong XY, Hou XY, Chung SK, Chung SS. Cardiac contractile dysfunction during acute hyperglycemia due to impairment of SERCA by polyol pathway-mediated oxidative stress. American journal of physiology Cell physiology 2010; 299(3):C643–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sakamoto A, Sugamoto Y. Identification of a novel aldose reductase-like gene upregulated in the failing heart of cardiomyopathic hamster. Molecular and cellular biochemistry 2011; 353(1–2):275–81. [DOI] [PubMed] [Google Scholar]
- 71.Ho EC, Lam KS, Chen YS, Yip JC, Arvindakshan M, Yamagishi S, Yagihashi S, Oates PJ, Ellery CA, Chung SS, Chung SK. Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes 2006; 55(7):1946–53. [DOI] [PubMed] [Google Scholar]
- 72.Itagaki I, Shimizu K, Kamanaka Y, Ebata K, Kikkawa R, Haneda M, Shigeta Y. The effect of an aldose reductase inhibitor (Epalrestat) on diabetic nephropathy in rats. Diabetes research and clinical practice 1994; 25(3):147–54. [DOI] [PubMed] [Google Scholar]
- 73.Oates PJ. Aldose reductase inhibitors and diabetic kidney disease. Current opinion in investigational drugs 2010; 11(4):402–17. [PubMed] [Google Scholar]
- 74.Ahmed N, Thornalley PJ, Dawczynski J, Franke S, Strobel J, Stein G, Haik GM. Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins. Investigative ophthalmology & visual science 2003; 44(12):5287–92. [DOI] [PubMed] [Google Scholar]
- 75.Congdon NG, Friedman DS, Lietman T. Important causes of visual impairment in the world today. Jama 2003; 290(15):2057–60. [DOI] [PubMed] [Google Scholar]
- 76.Kador PF, Randazzo J, Babb T, Koushik K, Takamura Y, Zhu W, Blessing K, Kompella UB. Topical aldose reductase inhibitor formulations for effective lens drug delivery in a rat model for sugar cataracts. J Ocul Pharmacol Ther 2007; 23(2):116–23. [DOI] [PubMed] [Google Scholar]
- 77.Kawakubo K, Mori A, Sakamoto K, Nakahara T, Ishii K. GP-1447, an inhibitor of aldose reductase, prevents the progression of diabetic cataract in rats. Biological & pharmaceutical bulletin 2012; 35(6)866–72. [DOI] [PubMed] [Google Scholar]
- 78.Matsumoto T, Ono Y, Kuromiya A, Toyosawa K, Ueda Y, Bril V. Long-term treatment with ranirestat (AS-3201), a potent aldose reductase inhibitor, suppresses diabetic neuropathy and cataract formation in rats. Journal of pharmacological sciences 2008; 107(3):340–8. [DOI] [PubMed] [Google Scholar]
- 79.Schalkwijk CG, Stehouwer CD, van Hinsbergh VW. Fructose-mediated non-enzymatic glycation: sweet coupling or bad modification. Diabetes/metabolism research and reviews 2004; 20(5):369–82. [DOI] [PubMed] [Google Scholar]
- 80.Chang KC, Snow A, LaBarbera DV, Petrash JM. Aldose reductase inhibition alleviates hyperglycemic effects on human retinal pigment epithelial cells. Chem Biol Interact 2015; 234:254–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hirata C, Nakano K, Nakamura N, Kitagawa Y, Shigeta H, Hasegawa G, Ogata M, Ikeda T, Sawa H, Nakamura K, Ienaga K, Obayashi H, Kondo M. Advanced glycation end products induce expression of vascular endothelial growth factor by retinal Muller cells. Biochemical and biophysical research communications 1997; 236(3):712–5. [DOI] [PubMed] [Google Scholar]
- 82.Nguyen QD, Hatef E, Kayen B, Macahilig CP, Ibrahim M, Wang J, Shaikh O, Bodaghi B. A cross-sectional study of the current treatment patterns in noninfectious uveitis among specialists in the United States. Ophthalmology 2011; 118(1):184–90. [DOI] [PubMed] [Google Scholar]
- 83.Tang J, Du Y, Petrash JM, Sheibani N, Kern TS. Deletion of aldose reductase from mice inhibits diabetes-induced retinal capillary degeneration and superoxide generation. PloS one 2013; 8(4):e62081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yadav UC, Srivastava SK, Ramana KV. Prevention of VEGF-induced growth and tube formation in human retinal endothelial cells by aldose reductase inhibition. Journal of diabetes and its complications 2012; 26(5):369–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lehto S, Pyorala K, Miettinen H, Ronnemaa T, Palomaki P, Tuomilehto J, Laakso M. Myocardial infarct size and mortality in patients with non-insulin-dependent diabetes mellitus. Journal of internal medicine 1994; 236(3):291–7. [DOI] [PubMed] [Google Scholar]
- 86.Stone GW, Grines CL, Browne KF, Marco J, Rothbaum D, O’Keefe J, Hartzler GO, Overlie P, Donohue B, Chelliah N, et al. Predictors of in-hospital and 6-month outcome after acute myocardial infarction in the reperfusion era: the Primary Angioplasty in Myocardial Infarction (PAMI) trail. Journal of the American College of Cardiology 1995; 25(2):370–7. [DOI] [PubMed] [Google Scholar]
- 87.Greene DA, Lattimer SA, Sima AA. Sorbitol, phosphoinositides, and sodium-potassium-ATPase in the pathogenesis of diabetic complications. The New England journal of medicine 1987; 316(10):599–606. [DOI] [PubMed] [Google Scholar]
- 88.Braunwald E, Bristow MR. Congestive heart failure: fifty years of progress. Circulation 2000; 102(20 Suppl 4):IV14–23. [DOI] [PubMed] [Google Scholar]
- 89.Tiwari S, Ndisang JF. The role of obesity in cardiomyopathy and nephropathy. Current pharmaceutical design 2014; 20(9):1409–17. [DOI] [PubMed] [Google Scholar]
- 90.Fioretto P, Caramori ML, Mauer M. The kidney in diabetes: dynamic pathways of injury and repair. The Camillo Golgi Lecture 2007. Diabetologia 2008; 51(8):1347–55. [DOI] [PubMed] [Google Scholar]
- 91.Diabetes C, Complications Trial/Epidemiology of Diabetes I, Complications Research G, Nathan DM, Zinman B, Cleary PA, Backlund JY, Genuth S, Miller R, Orchard TJ. Modern-day clinical course of type 1 diabetes mellitus after 30 years’ duration: the diabetes control and complications trial/epidemiology of diabetes interventions and complications and Pittsburgh epidemiology of diabetes complications experience (1983–2005). Archives of internal medicine 2009; 169(14):1307–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kasajima H, Yamagishi S, Sugai S, Yagihashi N, Yagihashi S. Enhanced in situ expression of aldose reductase in peripheral nerve and renal glomeruli in diabetic patients. Virchows Archiv : an international journal of pathology 2001; 439(1):46–54. [DOI] [PubMed] [Google Scholar]
- 93.Morrisey K, Steadman R, Williams JD, Phillips AO. Renal proximal tubular cell fibronectin accumulation in response to glucose is polyol pathway dependent. Kidney international 1999; 55(6):2548–72. [DOI] [PubMed] [Google Scholar]
- 94.Gabbay KH. The sorbitol pathway and the complications of diabetes. The New England journal of medicine 1973; 288(16)831–6. [DOI] [PubMed] [Google Scholar]
- 95.Kador PF, Robison WG Jr., Kinoshita JH. The pharmacology of aldose reductase inhibitors. Annual review of pharmacology and toxicology 1985; 25:691–714. [DOI] [PubMed] [Google Scholar]
- 96.Palsamy P, Subramanian S. Resveratrol protects diabetic kidney by attenuating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via Nrf2-Keap1 signaling. Biochimica et biophysica acta 2011; 1812(7):719–31. [DOI] [PubMed] [Google Scholar]
- 97.Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney Disease in diabetes. Diabetes 2008; 57(6):1446–54. [DOI] [PubMed] [Google Scholar]
- 98.Yang JY, Tam WY, Tam S, Guo H, Wu X, Li G, Chau JF, Klein JD, Chung SK, Sands JM, Chung SS. Genetic restoration of aldose reductase to the collecting tubules restores maturation of the urine concentrating mechanism. American journal of physiology Renal physiology 2006; 291(1)F186–95. [DOI] [PubMed] [Google Scholar]
- 99.Makino H, Kashihara N, Sugiyama H, Kanao K, Sekikawa T, Okamoto K, Maeshima Y, Ota Z, Nagai R. Phenotypic modulation of the mesangium reflected by contractile proteins in diabetes. Diabetes 1996; 45(4):488–95. [DOI] [PubMed] [Google Scholar]
- 100.de Iongh RU, Wederell E, Lovicu FJ, McAvoy JW. Transforming growth factor-beta-induced epithelial-mesenchymal transition in the lens: a model for cataract formation. Cells, tissues, organs 2005; 179(1–2):43–55. [DOI] [PubMed] [Google Scholar]
- 101.Huang P, Zhang Y, Jiang T, Zeng W, Zhang N. Aldose reductase is a potent regulator of TGF-beta1 induced expression of fibronectin in human mesangial cells. Molecular biology reports 2010; 37(7):3097–103. [DOI] [PubMed] [Google Scholar]
- 102.Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman R, Malik RA, Maser RE, Sosenko JM, Ziegler D, American Diabetes A. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes care 2005; 28(4):956–62. [DOI] [PubMed] [Google Scholar]
- 103.Jaspan JB. Taking control of diabetes. Hospital practice 1995; 30(10):55–62. [DOI] [PubMed] [Google Scholar]
- 104.Zatalia SR, Sanusi H. The role of antioxidants in the pathophysiology, complications, and management of diabetes mellitus. Acta medica Indonesiana 2013; 45(2):141–7. [PubMed] [Google Scholar]
- 105.Zochodne DW. Diabetes mellitus and the peripheral nervous system: manifestations and mechanisms. Muscle & nerve 2007; 36(2):144–66. [DOI] [PubMed] [Google Scholar]
- 106.Hotta N, Toyota T, Matsuoka K, Shigeta Y, Kikkawa R, Kaneko T, Takahashi A, Sugimura K, Koike Y, Ishii J, Sakamoto N, Group SNKDNS. Clinical efficacy of fidarestat, a novel aldose reductase inhibitor, for diabetic peripheral neuropathy: a 52-week multicenter placebo-controlled double-blind parallel group study. Diabetes care 2001; 24(10):1776–82. [DOI] [PubMed] [Google Scholar]
- 107.Pan H, Jian F, Lin J, Chen N, Zhang C, Zhang Z, Ding Z, Wang Y, Cui L, Kimura J. F-wave latencies in patients with diabetes mellitus. Muscle & nerve 2014; 49(6):804–8. [DOI] [PubMed] [Google Scholar]
- 108.Galuppo M, Giacoppo S, Bramanti P, Mazzon E. Use of natural compounds in the management of diabetic peripheral neuropathy. Molecules 2014; 19(3):2877–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Possidente CJ, Tandan R. A survey of treatment practices in diabetic peripheral neuropathy. Primary care diabetes 2009; 3(4):253–7. [DOI] [PubMed] [Google Scholar]
- 110.Tesfaye S, Vileikyte L, Rayman G, Sindrup SH, Perkins BA, Baconja M, Vinik AI, Boulton AJ, Toronto Expert Panel on Diabetic N. Painful diabetic peripheral neuropathy: consensus recommendations on diagnosis, assessment and management. Diabetes/metabolism research and reviews 2011; 27(7):629–38. [DOI] [PubMed] [Google Scholar]
- 111.Suehiro K, Funao T, Fujimoto Y, Yamada T, Mori T, Nishikawa K. Relationship between noradrenaline release in the locus coeruleus and antiallodynic efficacy of analgesics in rats with painful diabetic neuropathy. Life sciences 2013; 92(23):1138–44. [DOI] [PubMed] [Google Scholar]
- 112.Mehrpour O. Addiction and seizure ability of tramadol in high-risk patients. Indian journal of anaesthesia 2013; 57(1):86–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Aloisi AM, Buonocore M, Merlo L, Galandra C, Sotgiu A, Bacchella L, Ungaretti M, Demartini L, Bonezzi C. Chronic pain therapy and hypothalamic-pituitary-adrenal axis impairment. Psychoneuroendocrinology 2011; 36(7):1032–9. [DOI] [PubMed] [Google Scholar]
- 114.Bhanot A, Shri R. A comparative profile of methanol extracts of Allium cepa and Allium sativum in diabetic neuropathy in mice. Pharmacognosy research 2010; 2(6):374–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Selvarajah D, Gandhi R, Emery CJ, Tesfaye S. Randomized placebo-controlled double-blind clinical trial of cannabis-based medicinal product (Sativex) in painful diabetic neuropathy: depression is a major confounding factor. Diabetes care 2010; 33(1):128–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Snedecor SJ, Sudharshan L, Cappelleri JC, Sadosky A, Mehta S, Botteman M. Systematic review and meta-analysis of pharmacological therapies for painful diabetic peripheral neuropathy. Pain practice : the official journal of World Institute of Pain 2014; 14(2):167–84. [DOI] [PubMed] [Google Scholar]
- 117.Derry S, Sven-Rice A, Cole P, Tan T, Moore RA. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. The Cochrane database of systematic reviews 2013; 2:CD007393. [DOI] [PubMed] [Google Scholar]
- 118.Mou J, Paillard F, Turnbull B, Trudeau J, Stoker M, Katz NP. Efficacy of Qutenza(R) (capsaicin) 8% patch for neuropathic pain: a meta-analysis of the Qutenza Clinical Trials Database. Pain 2013; 154(9):1632–9. [DOI] [PubMed] [Google Scholar]
- 119.Khalil H Painful diabetic neuropathy management. International journal of evidence-based healthcare 2013; 11(1):77–9. [DOI] [PubMed] [Google Scholar]
- 120.Oka M, Kato N. Aldose reductase inhibitors. Journal of enzyme inhibition 2001; 16(6):465–73. [DOI] [PubMed] [Google Scholar]
- 121.Hotta N, Sakamoto N, Shigeta Y, Kikkawa R, Goto Y. Clinical investigation of epalrestat, an aldose reductase inhibitor, on diabetic neuropathy in Japan: multicenter study. Diabetic Neuropathy Study Group in Japan. Journal of diabetes and its complications 1996; 10(3):168–72. [DOI] [PubMed] [Google Scholar]
- 122.Goto Y, Hotta N, Shigeta Y, Sakamoto N, Kito S, Matsuoka K, Takahashi A, Kikkawa R, Sakuma A. A placebo-controlled double-blind study of epalrestat (ONO-2235) in patients with diabetic neuropathy. Diabetic medicine : a journal of the British Diabetic Association 1993; 10 Suppl 2:39S–43S. [DOI] [PubMed] [Google Scholar]
- 123.Uchida K, Kigoshi T, Nakano S, Ishii T, Kitazawa M, Morimoto S. Effect of 24 weeks of treatment with epalrestat, an aldose reductase inhibitor, on peripheral neuropathy in patients with non-insulin-dependent diabetes mellitus. Clinical therapeutics 1995; 17(3):460–6. [DOI] [PubMed] [Google Scholar]
- 124.Schemmel KE, Padiyara RS, D’Souza JJ. Aldose reductase inhibitors in the treatment of diabetic peripheral neuropathy: a review. Journal of diabetes and its complications 2010; 24(5):354–60. [DOI] [PubMed] [Google Scholar]
- 125.Stavniichuk R, Shevalye H, Hirooka H, Nadler JL, Obrosova IG. Interplay of sorbitol pathway of glucose metabolism, 12/15-lipoxygenase, and mitogen-activated protein kinases in the pathogenesis of diabetic peripheral neuropathy. Biochemical pharmacology 2012; 83(7):932–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hotta N, Yasuda K, Sumita Y, Sano T, Kakuta H, Nagashima M, Hayashi Y, Yamamoto M, Wakao T, Okuyama M, Kobayashi M, Mori K. Effects of a Novel Aldose Reductase Inhibitor, Fidarestat (SNK-860), on Vibration Perception Threshold and Subjective Symptoms in Patients with Diabetic Polyneuropathy : An Open-Label Pilot Study. Clinical drug investigation 2004; 24(11):671–80. [DOI] [PubMed] [Google Scholar]
- 127.Bril V, Hirose T, Tomioka S, Buchanan R, Ranirestat Study G. Ranirestat for the management of diabetic sensorimotor polyneuropathy. Diabetes care 2009; 32(7):1256–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bril V, Tomioka S, Buchanan RA, Perkins BA, m TSG. Reliability and validity of the modified Toronto Clinical Neuropathy Score in diabetic sensorimotor polyneuropathy. Diabetic medicine : a journal of the British Diabetic Association 2009; 26(3):240–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ewing DJ, Campbell IW, Clarke BF. Mortality in diabetic autonomic neuropathy. Lancet 1976; 1(7960):601–3. [DOI] [PubMed] [Google Scholar]
- 130.Greene DA, Lattimer S, Ulbrecht J, Carroll P. Glucose-induced alterations in nerve metabolism: current perspective on the pathogenesis of diabetic neuropathy and future directions for research and therapy. Diabetes care 1985; 8(3):290–9. [DOI] [PubMed] [Google Scholar]
- 131.Goto Y, Hotta N, Shigeta Y, Sakamoto N, Kikkawa R. Effects of an aldose reductase inhibitor, epalrestat, on diabetic neuropathy. Clinical benefit and indication for the drug assessed from the results of a placebo-controlled double-blind study. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 1995; 49(6):269–77. [DOI] [PubMed] [Google Scholar]
- 132.Hotta N, Akanuma Y, Kawamori R, Matsuoka K, Oka Y, Shichiri M, Toyota T, Nakashima M, Yoshimura I, Sakamoto N, Shigeta Y. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy: the 3-year, multicenter, comparative Aldose Reductase Inhibitor-Diabetes Complications Trial. Diabetes care 2006; 29(7):1538–44. [DOI] [PubMed] [Google Scholar]
- 133.Ikeda T, Iwata K, Tanaka Y. Long-term effect of epalrestat on cardiac autonomic neuropathy in subjects with non-insulin dependent diabetes mellitus. Diabetes research and clinical practice 1999; 43(3):193–8. [DOI] [PubMed] [Google Scholar]
- 134.Sundkvist G, Armstrong FM, Bradbury JE, Chaplin C, Ellis SH, Owens DR, Rosen I, Sonksen P. Peripheral and autonomic nerve function in 259 diabetic patients with peripheral neuropathy treated with ponalrestat (an aldose reductase inhibitor) or placebo for 18 months. United Kingdom/Scandinavian Ponalrestat Trial. Journal of diabetes and its complications 1992; 6(2):123–30. [DOI] [PubMed] [Google Scholar]
- 135.Ziegler D, Mayer P, Rathmann W, Gries FA. One-year treatment with the aldose reductase inhibitor, ponalrestat, in diabetic neuropathy. Diabetes research and clinical practice 1991; 14(1):63–73. [DOI] [PubMed] [Google Scholar]
- 136.Gill JS, Williams G, Ghatei MA, Hetreed AH, Mather HM, Bloom SR. Effect of the aldose reductase inhibitor, ponalrestat, on diabetic neuropathy. Diabete & metabolisme 1990; 16(4):296–302. [PubMed] [Google Scholar]
- 137.Giugliano D, Marfella R, Quatraro A, De Rosa N, Salvatore T, Cozzolino D, Ceriello A, Torella R. Tolrestat for mild diabetic neuropathy. A 52-week, randomized, placebo-controlled trial. Annals ofinternal medicine 1993; 118(1):7–11. [DOI] [PubMed] [Google Scholar]
- 138.Giugliano D, Acampora R, Marfella R, Di Maro G, De Rosa N, Misso L, Ceriello A, Quatraro A, D’Onofrio F. Tolrestat in the primary prevention of diabetic neuropathy. Diabetes care 1995; 18(4):536–41. [DOI] [PubMed] [Google Scholar]
- 139.Kline RC, Swanson DP, Wieland DM, Thrall JH, Gross MD, Pitt B, Beierwaltes WH. Myocardial imaging in man with I-123 meta-iodobenzylguanidine. Journal of nuclear medicine : official publication, Society of Nuclear Medicine 1981; 22(2):129–32. [PubMed] [Google Scholar]
- 140.Oates PJ. Aldose reductase, still a compelling target for diabetic neuropathy. Current drug targets 2008; 9(1):14–36. [DOI] [PubMed] [Google Scholar]
- 141.Pato E, Munoz-Fernandez S, Francisco F, Abad MA, Maese J, Ortiz A, Carmona L, Uveitis Working Group from Spanish Society of R. Systematic review on the effectiveness of immunosuppressants and biological therapies in the treatment of autoimmune posterior uveitis. Seminars in arthritis and rheumatism 2011; 40(4):314–23. [DOI] [PubMed] [Google Scholar]
- 142.El-Remessy AB, Tang Y, Zhu G, Matragoon S, Khalifa Y, Liu EK, Liu JY, Hanson E, Mian S, Fatteh N, Liou GI. Neuroprotective effects of cannabidiol in endotoxin-induced uveitis: critical role of p38 MAPK activation. Molecular vision 2008; 14:2190–203. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 143.Kalariya NM, Shoeb M, Reddy AB, Sawhney R, Ramana KV. Piceatannol suppresses endotoxin-induced ocular inflammation in rats. International immunopharmacology 2013; 17(2):439–46. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 144.Rao NA, Kimoto T, Zamir E, Giri R, Wang R, Ito S, Pararajasegaram G, Read RW, Wu GS. Pathogenic role of retinal microglia in experimental uveoretinitis. Investigative ophthalmology & visual science 2003; 44(1):22–31. [DOI] [PubMed] [Google Scholar]
- 145.Coorey NJ, Shen W, Chung SH, Zhu L, Gillies MC. The role of glia in retinal vascular disease. Clinical & experimental optometry : journal of the Australian Optometrical Association 2012; 95(3):266–81. [DOI] [PubMed] [Google Scholar]
- 146.Karlstetter M, Ebert S, Langmann T. Microglia in the healthy and degenerating retina: insights from novel mouse models. Immunobiology 2010; 215(9–10):685–91. [DOI] [PubMed] [Google Scholar]
- 147.Bousquet E, Zhao M, Ly A, Leroux Les Jardins G, Goldenberg B, Naud MC, Jonet L, Besson-Lescure B, Jaisser F, Farman N, De Kozak Y, Behar-Cohen F. The aldosterone-mineralocorticoid receptor pathway exerts anti-inflammatory effects in endotoxin-induced uveitis. PloS one 2012; 7(11):e49036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ma W, Zhao L, Fontainhas AM, Fariss RN, Wong WT. Microglia in the mouse retina alter the structure and function of retinal pigmented epithelial cells: a potential cellular interaction relevant to AMD. PloS one 2009; 4(11):e7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, Aboyans V, Abraham J, Ackerman I, Aggarwal R, Ahn SY, Ali MK, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Bahalim AN, Barker-Collo S, Barrero LH, Bartels DH, Basanez MG, Baxter A, Bell ML, Benjamin EJ, Bennett D, Bernabe E, Bhalla K, Bhandari B, Bikbov B, Bin Abdulhak A, Birbeck G, Black JA, Blencowe H, Blore JD, Blyth F, Bolliger I, Bonaventure A, Boufous S, Bourne R, Boussinesq M, Braithwaite T, Brayne C, Bridgett L, Brooker S, Brooks P, Brugha TS, Bryan-Hancock C, Bucello C, Buchbinder R, Buckle G, Budke CM, Burch M, Burney P, Burstein R, Calabria B, Campbell B, Canter CE, Carabin H, Carapetis J, Carmona L, Cella C, Charlson F, Chen H, Cheng AT, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahiya M, Dahodwala N, Damsere-Derry J, Danaei G, Davis A, De Leo D, Degenhardt L, Dellavalle R, Delossantos A, Denenberg J, Derrett S, Des Jarlais DC, Dharmaratne SD, Dherani M, Diaz-Torne C, Dolk H, Dorsey ER, Driscoll T, Duber H, Ebel B, Edmond K, Elbaz A, Ali SE, Erskine H, Erwin PJ, Espindola P, Ewoigbokhan SE, Farzadfar F, Feigin V, Felson DT, Ferrari A, Ferri CP, Fevre EM, Finucane MM, Flaxman S, Flood L, Foreman K, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabbe BJ, Gabriel SE, Gakidou E, Ganatra HA, Garcia B, Gaspari F, Gillum RF, Gmel G, Gosselin R, Grainger R, Groeger J, Guillemin F, Gunnell D, Gupta R, Haagsma J, Hagan H, Halasa YA, Hall W, Haring D, Haro JM, Harrison JE, Havmoeller R, Hay RJ, Higashi H, Hill C, Hoen B, Hoffman H, Hotez PJ, Hoy D, Huang JJ, Ibeanusi SE, Jacobsen KH, James SL, Jarvis D, Jasrasaria R, Jayaraman S, Johns N, Jonas JB, Karthikeyan G, Kassebaum N, Kawakami N, Keren A, Khoo JP, King CH, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lalloo R, Laslett LL, Lathlean T, Leasher JL, Lee YY, Leigh J, Lim SS, Limb E, Lin JK, Lipnick M, Lipshultz SE, Liu W, Loane M, Ohno SL, Lyons R, Ma J, Mabweijano J, MacIntyre MF, Malekzadeh R, Mallinger L, Manivannan S, Marcenes W, March L, Margolis DJ, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGill N, McGrath J, Medina-Mora ME, Meltzer M, Mensah GA, Merriman TR, Meyer AC, Miglioli V, Miller M, Miller TR, Mitchell PB, Mocumbi AO, Moffitt TE, Mokdad AA, Monasta L, Montico M, Moradi-Lakeh M, Moran A, Morawska L, Mori R, Murdoch ME, Mwaniki MK, Naidoo K, Nair MN, Naldi L, Narayan KM, Nelson PK, Nelson RG, Nevitt MC, Newton CR, Nolte S, Norman P, Norman R, O’Donnell M, O’Hanlon S, Olives C, Omer SB, Ortblad K, Osborne R, Ozgediz D, Page A, Pahari B, Pandian JD, Rivero AP, Patten SB, Pearce N, Padilla RP, Perez-Ruiz F, Perico N, Pesudovs K, Phillips D, Phillips MR, Pierce K, Pion S, Polanczyk GV, Polinder S, Pope CA 3rd, Popova S, Porrini E, Pourmalek F, Prince M, Pullan RL, Ramaiah KD, Ranganathan D, Razavi H, Regan M, Rehm JT, Rein DB, Remuzzi G, Richardson K, Rivara FP, Roberts T, Robinson C, De Leon FR, Ronfani L, Room R, Rosenfeld LC, Rushton L, Sacco RL, Saha S, Sampson U, Sanchez-Riera L, Sanman E, Schwebel DC, Scott JG, Segui-Gomez M, Shahraz S, Shepard DS, Shin H, Shivakoti R, Singh D, Singh GM, Singh JA, Singleton J, Sleet DA, Sliwa K, Smith E, Smith JL, Stapelberg NJ, Steer A, Steiner T, Stolk WA, Stovner LJ, Sudfeld C, Syed S, Tamburlini G, Tavakkoli M, Taylor HR, Taylor JA, Taylor WJ, Thomas B, Thomson WM, Thurston GD, Tleyjeh IM, Tonelli M, Towbin JA, Truelsen T, Tsilimbaris MK, Ubeda C, Undurraga EA, van der Werf MJ, van Os J, Vavilala MS, Venketasubramanian N, Wang M, Wang W, Watt K, Weatherall DJ, Weinstock MA, Weintraub R, Weisskopf MG, Weissman MM, White RA, Whiteford H, Wiersma ST, Wilkinson JD, Williams HC, Williams SR, Witt E, Wolfe F, Woolf AD, Wulf S, Yeh PH, Zaidi AK, Zheng ZJ, Zonies D, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; 380(9859):2163–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Webber S Internal Diabetes Federation. Diabetes Atlas 5th edition2011. [Google Scholar]
- 151.Klein BE, Klein R, Wang Q, Moss SE. Older-onset diabetes and lens opacities. The Beaver Dam Eye Study. Ophthalmic Epidemiol 1995; 2(1):49–55. [DOI] [PubMed] [Google Scholar]
- 152.Rowe NG, Mitchell PG, Cumming RG, Wans JJ. Diabetes, fasting blood glucose and age-related cataract: the Blue Mountains Eye Study. Ophthalmic epidemiology 2000; 7(2):103–14. [PubMed] [Google Scholar]
- 153.Leske MC, Wu SY, Hennis A, Connell AM, Hyman L, Schachat A. Diabetes, hypertension, and central obesity as cataract risk factors in a black population. The Barbados Eye Study. Ophthalmology 1999; 106(1):35–41. [DOI] [PubMed] [Google Scholar]
- 154.UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33).. Lancet 1998; 352(9131):837–53. [PubMed] [Google Scholar]
- 155.Spector A Review: Oxidative stress and disease. Journal of ocular pharmacology and therapeutics : the official journal of the Association for Ocular Pharmacology and Therapeutics 2000; 16(2):193–201. [DOI] [PubMed] [Google Scholar]
- 156.Kyselova Z, Stefek M, Bauer V. Pharmacological prevention of diabetic cataract. Journal of diabetes and its complications 2004; 18(2):129–40. [DOI] [PubMed] [Google Scholar]
- 157.Mares JA. High-dose antioxidant supplementation and cataract risk. Nutrition reviews 2004; 62(1)28–32. [DOI] [PubMed] [Google Scholar]
- 158.Kador PF. The role of aldose reductase in the development of diabetic complications. Medicinal research reviews 1988; 8(3):325–52. [DOI] [PubMed] [Google Scholar]
- 159.Lee AY, Chung SK, Chung SS. Demonstration that polyol accumulation is responsible for diabetic cataract by the use of transgenic mice expressing the aldose reductase gene in the lens. Proceedings of the National Academy of Sciences of the United States of America 1995; 92(7):2780–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Snow A, Shieh B, Chang KC, Pal A, Lenhart P, Ammar D, Ruzycki P, Palla S, Reddy GB, Petrash JM. Aldose reductase expression as a risk factor for cataract. Chemico-biological interactions 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chan AW, Ho YS, Chung SK, Chung SS. Synergistic effect of osmotic and oxidative stress in slow-developing cataract formation. Experimental eye research 2008; 87(5):454–61. [DOI] [PubMed] [Google Scholar]
- 162.Elwyn H Diabetic retinopathy. American journal of ophthalmology 1946; 29:591. [PubMed] [Google Scholar]
- 163.Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. The New England journal of medicine 2012; 366(13):1227–39. [DOI] [PubMed] [Google Scholar]
- 164.Campochiaro PA. Ocular neovascularization. Journal of molecular medicine 2013; 91(3):311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Costa PZ, Soares R. Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life sciences 2013; 92(22):1037–45. [DOI] [PubMed] [Google Scholar]
- 166.Tilton RG, Kawamura T, Chang KC, Ido Y, Bjercke RJ, Stephan CC, Brock TA, Williamson JR. Vascular dysfunction induced by elevated glucose levels in rats is mediated by vascular endothelial growth factor. The Journal of clinical investigation 1997; 99(9):2192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nguyen QD, Shah SM, Khwaja AA, Channa R, Hatef E, Do DV, Boyer D, Heier JS, Abraham P, Thach AB, Lit ES, Foster BS, Kruger E, Dugel P, Chang T, Das A, Ciulla TA, Pollack JS, Lim JI, Eliott D, Campochiaro PA, Group R-S. Two-year outcomes of the ranibizumab for edema of the mAcula in diabetes (READ-2) study. Ophthalmology 2010; 117(11):2146–51. [DOI] [PubMed] [Google Scholar]
- 168.Michaelides M, Kaines A, Hamilton RD, Fraser-Bell S, Rajendram R, Quhill F, Boos CJ, Xing W, Egan C, Peto T, Bunce C, Leslie RD, Hykin PG. A prospective randomized trial of intravitreal bevacizumab or laser therapy in the management of diabetic macular edema (BOLT study) 12-month data: report 2. Ophthalmology 2010; 117(6):1078–86 e2. [DOI] [PubMed] [Google Scholar]
- 169.Nishijima K, Ng YS, Zhong L, Bradley J, Schubert W, Jo N, Akita J, Samuelsson SJ, Robinson GS, Adamis AP, Shima DT. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. The American journal of pathology 2007; 171(1):53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Bai Y, Ma JX, Guo J, Wang J, Zhu M, Chen Y, Le YZ. Muller cell-derived VEGF is a significant contributor to retinal neovascularization. The Journal of pathology 2009; 219(4):446–54. [DOI] [PubMed] [Google Scholar]
- 171.Wang J, Xu X, Elliott MH, Zhu M, Le YZ. Muller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes 2010; 59(9):2297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Clements RS Jr., Robison WG Jr., Cohen MP. Anti-glycated albumin therapy ameliorates early retinal microvascular pathology in db/db mice. Journal of diabetes and its complications 1998; 12(1):28–33. [DOI] [PubMed] [Google Scholar]
- 173.Midena E, Segato T, Radin S, di Giorgio G, Meneghini F, Piermarocchi S, Belloni AS. Studies on the retina of the diabetic db/db mouse. I. Endothelial cell-pericyte ratio. Ophthalmic research 1989; 21(2):106–11. [DOI] [PubMed] [Google Scholar]
- 174.Tadayoni R, Paques M, Gaudric A, Vicaut E. Erythrocyte and leukocyte dynamics in the retinal capillaries of diabetic mice. Experimental eye research 2003; 77(4):497–504. [DOI] [PubMed] [Google Scholar]
- 175.Cheung AK, Fung MK, Lo AC, Lam TT, So KF, Chung SS, Chung SK. Aldose Reductase Deficiency Prevents Diabetes-Induced Blood-Retinal Barrier Breakdown, Apoptosis, and Glial Reactivation in the Retina of db/db Mice. Diabetes 2005; 54(11):3119–25. [DOI] [PubMed] [Google Scholar]
- 176.Grover D, Li TJ, Chong CC. Intravitreal steroids for macular edema in diabetes. The Cochrane database of systematic reviews 2008(1):CD005656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Payne AS, Freishtat RJ. Conserved steroid hormone homology converges on nuclear factor kappaB to modulate inflammation in asthma. Journal of investigative medicine : the official publication of the American Federation for Clinical Research 2012; 60(1):13–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cechin SR, Buchwald P. Effects of representative glucocorticoids on TNFalpha- and CD40L-induced NF-kappaB activation in sensor cells. Steroids 2014; 85:36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yumusak E, Buyuktortop N, Ornek K. Early results of dexamethasone implant, ranibizumab, and triamcinolone in macular edema due to branch retinal vein occlusion. European journal of ophthalmology 2015:0. [DOI] [PubMed] [Google Scholar]
- 180.Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006; 444(7121):860–7. [DOI] [PubMed] [Google Scholar]
- 181.Demircan N, Safran BG, Soylu M, Ozcan AA, Sizmaz S. Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye (London, England) 2006; 20(12):1366–9. [DOI] [PubMed] [Google Scholar]
- 182.Aveleira CA, Lin CM, Abcouwer SF, Ambrosio AF, Antonetti DA. TNF-alpha signals through PKCzeta/NF-kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 2010; 59(11):2872–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Krady JK, Basu A, Allen CM, Xu Y, LaNoue KF, Gardner TW, Levison SW. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes 2005; 54(5):1559–65. [DOI] [PubMed] [Google Scholar]
- 184.Diabetic Retinopathy Clinical Research N, Elman MJ, Aiello LP, Beck RW, Bressler NM, Bressler SB, Edwards AR, Ferris FL 3rd, Friedman SM, Glassman AR, Miller KM, Scott IU, Stockdale CR, Sun JK. Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmology 2010; 117(6):1064–77 e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Navaratna D, McGuire PG, Menicucci G, Das A. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes 2007; 56(9):2380–7. [DOI] [PubMed] [Google Scholar]
- 186.Sabbatini M, Sansone G, Uccello F, Giliberti A, Conte G, Andreucci VE. Early glycosylation products induce glomerular hyperfiltration in normal rats. Kidney international 1992; 42(4)875–81. [DOI] [PubMed] [Google Scholar]
- 187.Stitt AW. AGEs and diabetic retinopathy. Investigative ophthalmology & visual science 2010; 51(10):4867–74. [DOI] [PubMed] [Google Scholar]
- 188.Ibrahim AS, El-Remessy AB, Matragoon S, Zhang W, Patel Y, Khan S, Al-Gayyar MM, El-Shishtawy MM, Liou GI. Retinal microglial activation and inflammation induced by amadori-glycated albumin in a rat model of diabetes. Diabetes 2011; 60(4):1122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Heidland A, Sebekova K, Schinzel R. Advanced glycation end products and the progressive course of renal disease. American journal of kidney diseases : the official journal of the National Kidney Foundation 2001; 38(4 Suppl 1):S100–6. [DOI] [PubMed] [Google Scholar]
- 190.Dong N, Chang L, Wang B, Chu L. Retinal neuronal MCP-1 induced by AGEs stimulates TNF-alpha expression in rat microglia via p38, ERK, and NF-kappaB pathways. Molecular vision 2014; 20:616–28. [PMC free article] [PubMed] [Google Scholar]
- 191.Wang AL, Yu AC, He QH, Zhu X, Tso MO. AGEs mediated expression and secretion of TNF alpha in rat retinal microglia. Experimental eye research 2007; 84(5):905–13. [DOI] [PubMed] [Google Scholar]
- 192.Dewey S Posterior capsule opacification. Current opinion in ophthalmology 2006; 17(1):45–53. [DOI] [PubMed] [Google Scholar]
- 193.Wormstone IM. Posterior capsule opacification: a cell biological perspective. Experimental eye research 2002; 74(3):337–47. [DOI] [PubMed] [Google Scholar]
- 194.Wormstone IM, Tamiya S, Anderson I, Duncan G. TGF-beta2-induced matrix modification and cell transdifferentiation in the human lens capsular bag. Investigative ophthalmology & visual science 2002; 43(7):2301–8. [PubMed] [Google Scholar]
- 195.Hales AM, Schulz MW, Chamberlain CG, McAvoy JW. TGF-beta 1 induces lens cells to accumulate alpha-smooth muscle actin, a marker for subcapsular cataracts. Current eye research 1994; 13(12):885–90. [DOI] [PubMed] [Google Scholar]
- 196.Pladzyk A, Reddy AB, Yadav UC, Tammali R, Ramana KV, Srivastava SK. Inhibition of aldose reductase prevents lipopolysaccharide-induced inflammatory response in human lens epithelial cells. Investigative ophthalmology & visual science 2006; 47(12):5395–403. [DOI] [PubMed] [Google Scholar]
- 197.Reddy AB, Ramana KV, Srivastava S, Bhatnagar A, Srivastava SK. Aldose reductase regulates high glucose-induced ectodomain shedding of tumor necrosis factor (TNF)-alpha via protein kinase C-delta and TNF-alpha converting enzyme in vascular smooth muscle cells. Endocrinology 2009; 150(1):63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yoon J, Lee H, Chang HB, Choi H, Kim YS, Rho YK, Seong S, Choi DH, Park D, Ku B. DW1029M, a novel botanical drug candidate, inhibits advanced glycation end-product formation, rat lens aldose reductase activity, and TGF-beta1 signaling. American journal of physiology Renal physiology 2014; 306(10)F1161–70. [DOI] [PubMed] [Google Scholar]
- 199.Yadav UC, Ighani-Hosseinabad F, van Kuijk FJ, Srivastava SK, Ramana KV. Prevention of posterior capsular opacification through aldose reductase inhibition. Investigative ophthalmology & visual science 2009; 50(2):752–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003; 113(6):685–700. [DOI] [PubMed] [Google Scholar]
- 201.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nature genetics 2001; 29(2):117–29. [DOI] [PubMed] [Google Scholar]
- 202.Chang KC, Petrash JM. Aldose Reductase Mediates Transforming Growth Factor beta2 (TGF-beta2)-Induced Migration and Epithelial-To-Mesenchymal Transition of Lens-Derived Epithelial Cells. Invest Ophthalmol Vis Sci 2015; 56(8):4198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Li L, Chang KC, Zhou Y, Shieh B, Ponder J, Abraham AD, Ali H, Snow A, Petrash JM, LaBarbera DV. Design of an amide N-glycoside derivative of beta-glucogallin: a stable, potent, and specific inhibitor of aldose reductase. Journal of medicinal chemistry 2014; 57(1):71–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Riley RJ, Maggs JL, Lambert C, Kitteringham NR, Park BK. An in vitro study of the microsomal metabolism and cellular toxicity of phenytoin, sorbinil and mianserin. British journal of clinical pharmacology 1988; 26(5):577–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Suzen S, Buyukbingol E. Recent studies of aldose reductase enzyme inhibition for diabetic complications. Current medicinal chemistry 2003; 10(15):1329–52. [DOI] [PubMed] [Google Scholar]
- 206.Chalk C, Benstead TJ, Moore F. Aldose reductase inhibitors for the treatment of diabetic polyneuropathy. The Cochrane database of systematic reviews 2007(4):CD004572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Gabbay KH. Aldose reductase inhibition in the treatment of diabetic neuropathy: where are we in 2004? Current diabetes reports 2004; 4(6):405–8. [DOI] [PubMed] [Google Scholar]
- 208.Ludvigson MA, Sorenson RL. Immunohistochemical localization of aldose reductase. II. Rat eye and kidney. Diabetes 1980; 29(6):450–9. [DOI] [PubMed] [Google Scholar]
- 209.Chakrabarti S, Sima AA, Nakajima T, Yagihashi S, Greene DA. Aldose reductase in the BB rat: isolation, immunological identification and localization in the retina and peripheral nerve. Diabetologia 1987; 30(4):244–51. [DOI] [PubMed] [Google Scholar]
- 210.Guidry C The role of Muller cells in fibrocontractive retinal disorders. Progress in retinal and eye research 2005; 24(1):75–86. [DOI] [PubMed] [Google Scholar]
- 211.Wang JJ, Zhu M, Le YZ. Functions of Muller cell-derived vascular endothelial growth factor in diabetic retinopathy. World journal of diabetes 2015; 6(5):726–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Verwaerde C, Naud MC, Delanoye A, Wood M, Thillaye-Goldenberg B, Auriault C, de Kozak Y. Ocular transfer of retinal glial cells transduced ex vivo with adenovirus expressing viral IL-10 or CTLA4-Ig inhibits experimental autoimmune uveoretinitis. Gene therapy 2003; 10(23):1970–81. [DOI] [PubMed] [Google Scholar]
- 213.Anker SD, von Haehling S. Inflammatory mediators in chronic heart failure: an overview. Heart 2004; 90(4):464–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Experimental and molecular pathology 2001; 70(3):317–25. [DOI] [PubMed] [Google Scholar]
- 215.Ramana KV, Bhatnagar A, Srivastava SK. Inhibition of aldose reductase attenuates TNF-alpha-induced expression of adhesion molecules in endothelial cells. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2004; 18(11):1209–18. [DOI] [PubMed] [Google Scholar]