

SUMMARY
Astrocytic contributions to neuroinflammation are widely implicated in disease, but they remain incompletely explored. We assess medial prefrontal cortex (PFC) and visual cortex (VCX) astrocyte and whole-tissue gene expression changes in mice following peripherally induced neuroinflammation triggered by a systemic bacterial endotoxin, lipopolysaccharide, which produces sickness-related behaviors, including anhedonia. Neuroinflammation-mediated behavioral changes and astrocyte-specific gene expression alterations peak when anhedonia is greatest and then reverse to normal. Notably, region-specific molecular identities of PFC and VCX astrocytes are largely maintained during reactivity changes. Gene pathway analyses reveal alterations of diverse cell signaling pathways, including changes in cell-cell interactions of multiple cell types that may underlie the central effects of neuroinflammation. Certain astrocyte molecular signatures accompanying neuroinflammation are shared with changes reported in Alzheimer’s disease and mouse models. However, we find no evidence of altered neuronal survival or function in the PFC even when neuroinflammation-induced astrocyte reactivity and behavioral changes are significant.
Graphical Abstract

In brief
Astrocytic contributions to neuroinflammation are widely implicated in disease, but they remain incompletely explored. Diaz-Castro et al. report medial prefrontal cortex (PFC) and visual cortex (VCX) astrocyte and whole-tissue gene expression changes, as well as PFC neuronal properties, in mice following peripherally induced neuroinflammation triggered by a systemic bacterial endotoxin, lipopolysaccharide.
INTRODUCTION
Neuroinflammation is a complex multicellular response that accompanies a variety of disorders, including neurological and psychiatric diseases (Giovannoni and Quintana, 2020; Linnerbauer et al., 2020). Genetic, neuropathological, and cellular studies link immune cell dysfunction to several neurological and psychiatric disorders (Barnes et al., 2017; Ferrer, 2017; Hammond et al., 2019; Hickman et al., 2018; Lai et al., 2017; Matthews, 2019; Perez-Nievas and Serrano-Pozo, 2018; Pimenova et al., 2018; Pouget, 2018; Prinz and Priller, 2014; Syed et al., 2018). In many disorders involving neuroinflammation, it has proven problematic to separate correlation from causation, and thus the extent to which neuroinflammation accompanies brain diseases or is directly causal for disease-related phenotypes remains unclear in most cases. There is a need to understand how neuroinflammation affects relevant cell types at molecular and functional levels in brain nuclei known to contribute to brain disorders and thereby begin to explore the significance of neuroinflammation to disease-related phenotypes.
Astrocytes are among the main cell types that respond to, and contribute to, neuroinflammatory cues in the CNS (Bennett and Molofsky, 2019; Sofroniew, 2020). Astrocytes are the most numerous type of glia and tile the entire CNS. They intimately contact most, if not all, brain cell types and the vasculature. Astrocytes participate in the formation and function of the blood-brain barrier, provide metabolic support to neurons, are involved in synapse formation and removal, and regulate neuronal activity by buffering ions and neurotransmitters, as well as through regulated mechanisms that include the release of neuroactive substances (Barres, 2008; Khakh and Sofroniew, 2015; Nagai et al., 2021; Yu et al., 2020a). In addition, astrocytes are altered in all forms of brain disease, injury, and trauma, and are therefore proposed to contribute in a reactive or causative manner to disease states (Burda and Sofroniew, 2014; Giovannoni and Quintana, 2020; Linnerbauer et al., 2020). Much has been learned about basic mechanisms, but exploration of how astrocytes contribute to neurological disease is still in its infancy. In this study, we sought to understand astrocyte alterations accompanying neuroinflammation induced by the bacterial endotoxin lipopolysaccharide (LPS), which is associated with quantifiable behavioral changes in mice (Bhattacharya et al., 2018). We focused on anhedonia as a behavioral readout because this phenotype is strongly associated with psychiatric and dementia-related disorders such as depression and Alzheimer’s disease (AD), and it is readily observed and reliably quantified in mice as a sickness-related phenotype (De Fruyt et al., 2020; Scheggi et al., 2018).
We performed a series of functional evaluations, immunohistochemistry (IHC), behavioral assessments, and RNA sequencing (RNA-seq) in the prefrontal cortex (PFC) of mice injected with a single peripheral dose of LPS to mimic antigen presentation and neuroinflammation (Bhattacharya et al., 2018; Zamanian et al., 2012). As the main integrator of the decision-making circuits of the limbic system, the PFC becomes dysfunctional in both psychiatric and dementia-related disorders and when symptoms such as anhedonia are observed (Dafsari and Jessen, 2020; Elahi and Miller, 2017; Haber et al., 2020; Russo and Nestler, 2013). Astrocyte-specific and whole-tissue RNA-seq was performed during the development and recovery of anhedonia to identify molecular changes within astrocytes (Chai et al., 2017; Diaz-Castro et al., 2019; Srinivasan et al., 2016; Yu et al., 2018, 2020b). In addition, we compared astrocyte gene expression data from PFC to those from visual cortex (VCX) in order to evaluate how neuroinflammation affected astrocyte regional transcriptional identity. This is an emerging topic, because astrocytes are considered brain region specific (Ben Haim and Rowitch, 2017; Khakh and Deneen, 2019). However, the effect of neuroinflammation on region specificity has not been assessed. We also assessed bulk tissue gene expression changes in response to peripherally induced neuroinflammation and identified cell communication pathways that were altered in the PFC. Finally, we performed electrophysiological evaluations to determine how measurable astrocyte reactivity that accompanied neuroinflammation affected the functional properties of astrocytes and neurons in the PFC.
RESULTS
Neuroinflammation-induced anhedonia and PFC transcriptional alterations
To study the effects of inflammation on the CNS, we adapted a peripherally induced neuroinflammation model (NIM). We injected a single dose of LPS (5 mg/kg) intraperitoneally (i.p.) (Bhattacharya et al., 2018); this dose is identical to that used previously (Zamanian et al., 2012). For all of the experiments reported, control mice were handled equivalently and received an injection of PBS (Figure 1A). Subsequently, general health status and a focused set of behaviors were assessed during 3 days. The mice were weighed, locomotor activity was assessed using open field tests (OFTs), and anhedonia was evaluated daily with the sucrose preference test (SPT) (Scheggi et al., 2018) (Figures 1A–1D). We found that weight loss occurred after LPS injections and lasted until day 3 (Figure 1B, n = 21–22 mice). Locomotor activity was significantly reduced on day 1 (Figure 1C, n = 16). In contrast, sucrose preference was significantly reduced in the NIM mice for up to 2 days after LPS, returning back to control levels on day 3 (Figure 1D, n = 11 cages, 24 mice). To investigate whether the phenotypes induced by LPS fluctuated over time, we assessed a group of mice for 20 days after LPS or PBS administration and found that they remained stable after recovery (Figures S1A–S1E, n = 8 mice).
Figure 1. LPS-induced anhedonia, sickness behavior, and overall inflammatory responses.

(A) Timeline for the neuroinflammation model (NIM) and its behavioral assessments.
(B) Weight of control and LPS-injected animals before injection (0 days) and 1, 2, and 3 days after injection. ANOVA F (1.50, 61.74) = 11.84, p = 0.0002. n = 21–22 mice per group.
(C) Time immobile during 6 min of open field test 1,2, and 3 days after injection. Baseline was generated with non-injected animals. ANOVA F (1.73, 51.86) = 12.39, p < 0.0001. n = 16 mice per group. (D) On the left of the graph, the bottle preference is shown when the mice are given the choice between two bottles of water 2 days before the injection (baseline). On the right, the sucrose preference test 1, 2, and 3 days after injection is shown. ANOVA F (1.72, 34.46) = 7.30, p = 0.0034. n = 11 cages, 24 mice per group.
(E) One day after LPS or PBS i.p. injection, whole-tissue PFC RNA-seq detected 1,517 differentially expressed genes (DEGs) between NIM and control, with FDR < 0.05 and FPKM (fragments per kilobase of transcript per million mapped reads) > 1. n = 4 mice per group.
(F) Principal component analysis of the top 2,000 most variable genes.
(G) NIM versus control log2 ratio of known inflammatory molecules.
(H) Top 20 pathways altered in NIM PFC astrocytes, identified by Ingenuity Pathway Analysis (IPA).
Closed circles in graphs indicate mean ± SEM. In some cases, the error bars representing SEM are smaller than the symbol used for the mean. The statistical analyses for (B)–(D) were performed with two-way ANOVA for repeated measures. Time treatment interaction F and p values are reported above. **p < 0.01 > 0.001, ***p < 0.001 after a Tukey post hoc analysis for the interaction between time and treatment. IPA Z score indicates whether the pathway is predicted to be inhibited (blue) or activated (red). In some cases, activation or inhibition cannot be predicted (gray).
Next, to evaluate whether peripheral LPS did indeed cause inflammatory responses in the PFC, we performed bulk RNA-seq for four NIM and four control mice 1 day after LPS or PBS. 1,517 differentially expressed genes (DEGs) were found between NIM and control PFC 1 day after LPS injections (Figure 1E). Principal component analysis (PCA) separation of the two treatments by component 1 corroborated clear transcriptional differences between NIM and control samples (Figure 1F). Within the DEGs, we found upregulation of several inflammatory molecules (Figure 1G), and Ingenuity Pathway Analysis (IPA) showed that 15 of the top 20 altered pathways in the PFC were related to inflammatory responses (Figure 1H). Thus, peripheral administration of LPS induced anhedonia and was associated with clear neuroinflammatory transcriptional profiles in the PFC (Figure 1).
RiboTag to assess PFC and VCX astrocyte-specific transcriptional changes
To evaluate PFC astrocyte gene expression changes during neuroinflammation, we performed astrocyte-specific RNA-seq by crossing RiboTag mice (Sanz et al., 2009) to astrocyte-specific Aldh1l1-cre/ERT2 mice (Chai et al., 2017; Srinivasan et al., 2016). Bigenic male and female progeny were injected with tamoxifen at 6 weeks of age to induce the expression of Rpl22HA (RiboTag) specifically in astrocytes (Figures S2A–S2E). Three weeks later, the mice were injected with LPS (or PBS) to trigger neuroinflammation. Whole-tissue (INPUT) and astrocyte-specific RNA (IP) was extracted for PFC as well as VCX and sequenced (Figure S2F). We performed such analyses 1, 2, and 14 days after induction of neuroinflammation. The astrocyte-specific IP fraction was replete with known astrocyte markers and depleted of markers of other cells for PFC and VCX datasets at all three time points (Figures S2G–S2J). Thus, we could reliably assess astrocyte-specific gene expression 1, 2, and 14 days after the induction of neuroinflammation.
Impact of neuroinflammation on PFC and VCX astrocyte transcriptional identity
To address how transcriptional changes were related to the behavioral alterations reported in Figures 1 and S1, we analyzed RNA-seq of INPUT and IP samples at three time points in the NIM and control mice. We focused on day 1 when both locomotor dysfunction and anhedonia were observed, day 2 when locomotor activity was normal, but anhedonia persisted, and day 14 when there were no significant differences between the NIM and control groups in the measures we assessed (Figure 1; Figure S1). PFC and VCX gene expression was analyzed to determine whether differences existed between these two functionally distinct cortical areas; PFC becomes dysfunctional in both psychiatric and dementia-related disorders and when symptoms such as anhedonia are observed, while VCX has not been associated with such phenotypes. Furthermore, we reasoned that assessing PFC and VCX would allow us to determine whether neuroinflammation affected astrocytes in these two cortical areas similarly or separably. Initial assessment of the transcriptional differences between IP samples showed that the NIM group was separated from controls by the first principal component at days 1 and 2, but not at day 14 (Figures 2A–2C). PCA also revealed clear transcriptional differences between astrocytes from PFC and VCX, implying diversity between these areas under control conditions that also persisted following neuroinflammation at day 1, 2, and 14 (Figure 2A–2C). Accordingly, we found 1,218 DEGs between PFC and VCX regions in control conditions (Figure 2D) and 1,176 DEGs in NIM conditions (Figure 2E). To evaluate the genes more directly, we compared the PFC-enriched genes (yellow in Figures 2D and 2E) from control and NIM samples. We found that more than half of the PFC-enriched genes did not change their region specificity following neuroinflammation (Figure 2F), and thus they appeared intrinsic to these astrocytes irrespective of the neuroinflammatory insult. A similar trend was observed for the VCX-enriched genes (green in Figures 2D and 2E), implying that neuroinflammation does not markedly cause astrocytes to lose their PFC and VCX regional molecular identities (Figure 2G). The top 20 PFC and VCX-enriched genes are listed in Figures 2H and 2I, respectively. Some top PFC-enriched genes were transcription factors (Zic3 and Zic1) or involved in cell growth and proliferation (Greb1, Acvr1c, Zic1, and Nrn1). We had previously identified μ-crystallin as a protein enriched in striatal astrocytes (Chai et al., 2017). Interestingly, another crystallin (Crybb1) was identified as enriched in PFC versus VCX astrocytes. Crybb1 has been linked to anxiety and schizophrenia (Spadaro et al., 2015). Several of the VCX-enriched genes were related to the formation and function of the extracellular matrix (Colq, Fgfbp1, Spp1, and Egfl6). Cell adhesion molecules were found in both PFC and VCX astrocytes; for example, Bves was enriched in PFC, and Cdh1 was enriched in VCX.
Figure 2. Cortical region-specific and neuroinflammation time-specific transcriptional alterations in astrocytes.

(A–C) Principal component analysis (top 1,000 most variable genes) of NIM and control astrocyte RNA, from PFC and VCX, 1 (A), 2 (B) and 14 days (C) after LPS or PBS injection.
(D) Number of astrocyte DEGs between 1-day control PFC and VCX (FDR < 0.05, FPKM > 1).
(E) Number of astrocyte DEGs between 1-day NIM PFC and VCX (FDR < 0.05, FPKM > 1).
(F) Overlap of PFC-enriched genes (compared to VCX) between 1-day control and NIM conditions.
(G) Overlap of VCX-enriched genes (compared to PFC) between 1-day control and NIM conditions.
(H) Top 20 PFC-enriched genes when compared to VCX in both 1-day control and NIM conditions (see F).
(I) Top 20 VCX-enriched genes when compared to PFC in both 1-day control and NIM conditions (see G).
(J and K) Number of PFC (J) or VCX (K) NIM versus control DEGs (FDR < 0.05, FPKM > 1) in astrocytes at 1, 2, and 14 days.
(L) Overlap of 1-day NIM versus control DEGs between PFC and VCX (FDR < 0.05, FPKM > 1).
(M) Overlap of NIM versus control astrocyte DEGs (FDR < 0.05, FPKM > 1) between PFC and VCX at 1 and 2 days.
(N) Top 20 NIM versus control upregulated (top) and downregulated (bottom) genes that are common across 1-day PFC, 1-day VCX, 2-day PFC, and 2-day VCX astrocytes.
(O) Top 20 NIM versus control upregulated (top) and downregulated (bottom) genes that are uniquely altered (FDR < 0.05, FPKM > 1) in 1-day PFC astrocytes.
For the whole figure, n = 3–4 biological samples per group; each biological sample was generated from the tissue of two mice.
We next focused on PFC and VCX astrocyte gene expression changes in response to neuroinflammation (Figures 2J and 2K). Both PFC and VCX astrocytes showed large transcriptional responses at day 1, with >3,000 DEGs. This number was reduced to <1,000 DEGs at day 2, and remarkably, no significant gene expression changes were observed at day 14 (with a false discovery rate [FDR] < 0.05 threshold). Since we observed that PFC and VCX astrocytes were transcriptionally separable, we tested whether their responses to neuroinflammation were also different. Focusing on day 1, we found that from the 3026 and 3221 astrocyte DEGs in PFC and VCX respectively, 2114 genes were shared between the two regions (Figure 2L). Of these 2,114 genes, 269 DEGs maintained their differences at day 2 (Figures 2M and 2N). Upregulated genes in this highly conserved group of genes are mostly involved in inflammatory/immune responses, and the downregulated genes were related to metabolism (Figure 2N; Figure S3B). Notably, up to one third of the significantly DEGs caused by neuroinflammation were specific to the brain region (Figures 2L and 2M). Broadly, these data demonstrate that astrocytes from different brain areas show diverse responses to the same stimulus and underscore the necessity of drawing conclusions based on evaluations of defined brain areas, rather than mixing between brain regions. These data add support to the emerging view that astrocytes are separable between brain areas and respond in a context-specific manner (Chai et al., 2017; Ben Haim and Rowitch, 2017; Khakh and Deneen, 2019; Yu et al., 2020b).
In the day 1 dataset, 854 genes were unique to the PFC (Figure 2M); of these, the top 20 upregulated and downregulated DEGs are shown in Figure 2O. Evaluation of these genes shows that region- and time-specific DEGs were involved in a wide variety of functions (Figure S3). Within the region-specific pathways, the ones from day 1 in PFC astrocytes stood out because of the consistent inhibition of signaling pathways involved in astrocyte communication with other cells (Figure S3C, blue boxes next to each pathway). Moreover, specific analyses of known functions performed by astrocytes suggested potential alterations of K+ homeostasis (Figure S4A), Ca2+ signaling (Figure S4B), gap junctions (Figure S4C), as well as synapse morphogenesis and removal (Figure S4D). Taken together, the aforementioned RNA-seq data reveal astrocyte diversity that was dependent on the brain area and the time from induction of neuroinflammation. In particular, the data show quantitatively that NIM presented profound changes in gene expression at day 1, and that these were completely reversible by day 14. Furthermore, even though neuroinflammation evoked strong changes in gene expression, astrocytes maintained many PFC versus VCX regional gene expression differences.
Exploring brain cell type contributions to neuroinflammation
The encompassing mechanisms by which a peripheral stimulus such as LPS results in changed behavior are unknown, but they likely involve multiple cell types. We next used the INPUT RNA-seq data, which represents all cells, to explore this aspect. Recalling observations with astrocyte-specific data (Figure 2), the first two principal components of the PCA plot separated control from NIM mice and PFC from VCX at day 1 and day 2. At day 14, there was no separation between NIM and control, but there was for PFC and VCX as observed in controls (Figures 3A–3C). Almost 2,000 DEGs between NIM and control mice were detected at day 1 for PFC and VCX, but this value fell to ~400 DEGs at day 2, and to zero at day 14 (Figures 3D and 3E). In order to assign these DEGs to their likely cell types, we took advantage of recently published single-cell RNA (scRNA)-seq data that identified 14 cell types in the mouse PFC (Saunders et al., 2018) (Figure 3F). Using that dataset (http://dropviz.org/) we generated a list of enriched genes for each of the 14 cell types and determined which were differentially expressed between NIM and control mice (Figures 3G and 3H; Figure S4E and S4F). Based on the overlap with the scRNA-seq data, 515 of the 1,868 PFC DEGs at day 1 could be mapped to specific cell types (Figure 3G). Most downregulated genes mapped to neurons, followed by astrocytes as the second largest cell type. In contrast, the upregulated genes mapped to microglia and endothelial cells. For the day 2 time point, 178 of the 412 PFC DEGs were assigned to specific cell types (Figure 3H). Only 13 were downregulated and most mapped to astrocytes. Two thirds of the upregulated genes at day 2 mapped to microglia, followed by endothelial cells. To evaluate how many of the DEGs at day 2 were shared with those at day 1, we calculated the overlap between them based on cell type (Figures 3I and 3J). Thus, 62%–64% of the DEGs observed at day 2 for microglia, endothelial cells, and astrocytes were already significantly differentially expressed on day 1 (neurons and oligodendrocytes displayed only two DEGs on day 2). Interestingly, all cell types showed a strong decrease in the number of DEGs from day 1 to 2, with the exception of microglia (Figures 3J and 3K). By mapping our INPUT RNA-seq data onto cell types known to exist in the PFC, our analysis demonstrates that neuroinflammation involves a multicellular response that wanes from day 1 to day 2 in most cells (including astrocytes) except microglia.
Figure 3. Assessment of PFC cell-specific responses during neuroinflammation from input RNA-seq.

(A–C) Principal component analysis (top 2,000 most variable genes) of NIM and control input RNA, from PFC and VCX, 1 (A), 2 (B) and 14 days (C) after LPS/PBS injection.
(D–E) Number of PFC (D) or VCX (E) NIM versus control DEGs (FDR < 0.05, FPKM > 1) in input samples at 1, 2, and 14 days.
(F) tSNE (t-distributed stochastic neighbor embedding) plot of the mouse frontal cortex main cell types identified with single cell RNA (scRNA)-seq from a published dataset (http://dropviz.org; Saunders et al., 2018).
(G) Pie charts indicating the proportion of 1-day input downregulated (left) and upregulated (right) genes that are enriched in each cell type listed in the legend below. The numbers in each section of the pie chart indicate the number of DEGs for that cell type.
(H) Same as in (G) but for 2-day input samples.
(I) Overlap of all cell-enriched NIM versus control DEGs between 1 and 2 days.
(J) Overlap of microglia, endothelial, projection neuron (P. Neuron), astrocyte, and oligodendrocyte NIM versus control DEGs between 1 and 2 days.
(K) Bar graph representing the 2 day/1 day ratio of the number of DEGs for all cell-enriched genes (input), microglia (M), endothelial cells (E), projection neurons (P-N), astrocytes (A), and oligodendrocytes (O).
For the whole figure, n = 3–4 biological samples per group; each biological sample was generated from tissue of two mice.
Brain cell type responses and cell-cell interactions during neuroinflammation
We used IPA to explore the pathways that were altered in microglia, endothelial cells, neurons, astrocytes, and oligodendrocytes (Figures 4A–4O). Microglial pathways indicated direct engagement of these cells in immune responses and phagocytosis (Figures 4A and 4B). Endothelial cells were involved in inflammatory and vascular signaling (Figures 4D and 4E). Projection neuron-altered pathways related to stress responses, analgesia, and neuronal function (Figures 4G and 4H). Pathways affected in astrocytes were mostly metabolic (Figures 4J and 4K). Oligodendrocytes underwent metabolic and other diverse changes (Figures 4M and 4N). Comparison of the pathways identified at day 1 and day 2 for each cell type showed that even though the DEGs at each stage vary (Figure 3J), microglia and endothelial cells maintain their functional roles at these time points (Figures 4C and 4F), whereas other cells do so to a lesser degree (Figures 4I, 4L, and 4O).
Figure 4. PFC cell-specific altered pathways and cell-cell interaction mechanisms.
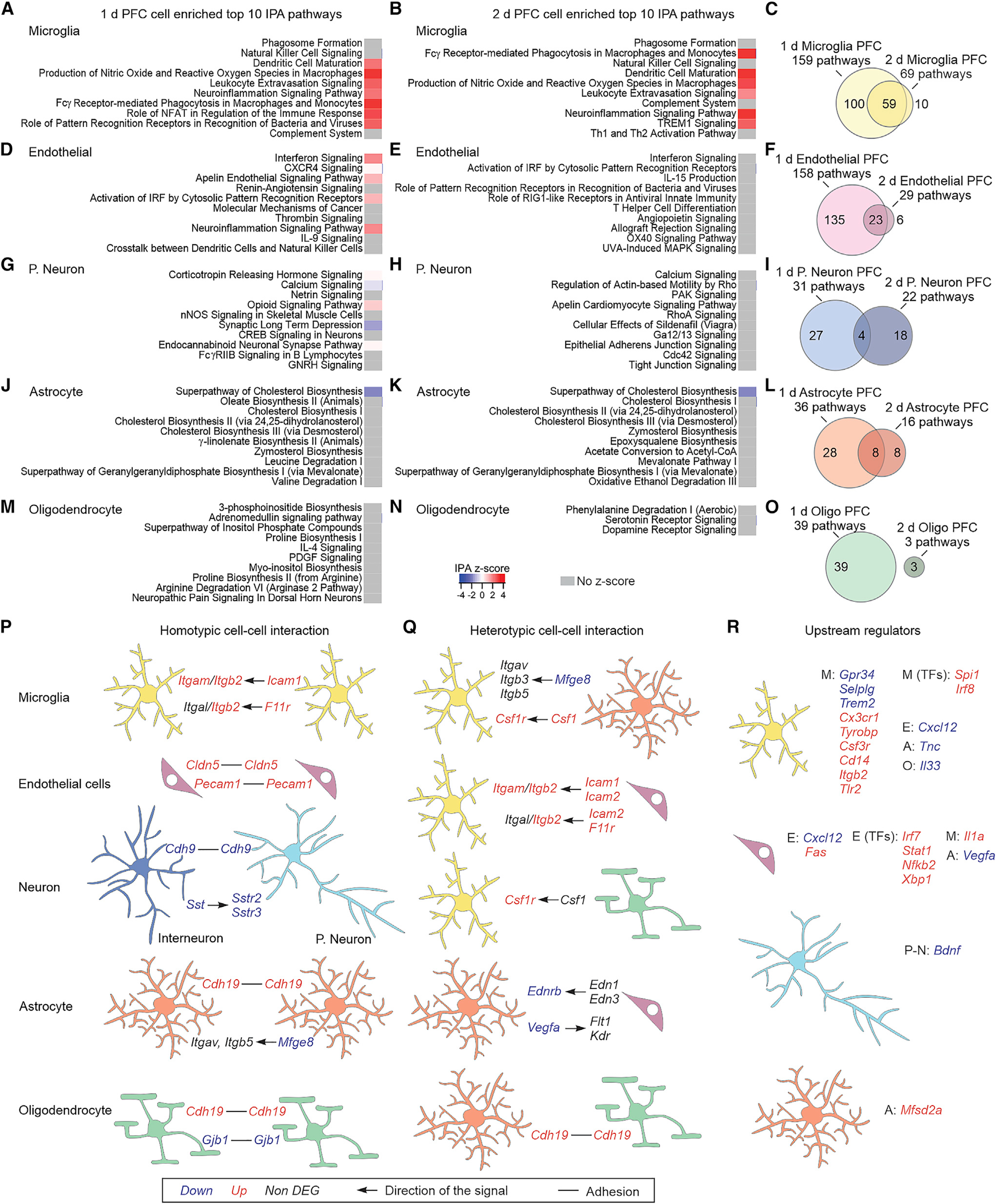
(A and B) Top 10 IPA pathways identified for the microglia-enriched DEGs at 1 day (A) or 2 days (B).
(C) Overlap of the identified IPA pathways from microglia-enriched DEGs between 1 and 2 days.
(D–O) Same as (A)–(C) but for endothelial cells (D–F), projection neurons (G–I), astrocytes (J–L), or oligodendrocytes (M–O).
(P and Q) Homotypic (P) and heterotypic (Q) adhesion and ligand-receptor cell-cell interaction mechanisms that are altered in NIM. The arrows indicate the directionality of the signal, e.g., ligand → receptor.
(R) Differentially expressed upstream regulators of microglia, endothelial cells, projection neurons, and astrocyte DEGs. The upstream regulators could be receptors, ligands, or transcription factors (TFs) that are expressed in the same cell, or in the neighboring ones: microglia (M), endothelial cells (E), projection neurons (P-N), astrocytes (A), or dendrocytes (O).
For the whole figure, n = 3–4 biological samples per group; each biological sample was generated from tissue of two mice.
We observed that many pathways overlapped between cell types (Figures S4G and S4H), which may suggest synergistic or reciprocal cell-cell interaction mechanisms, in which, for instance, a receptor involved in a specific pathway is expressed in one cell type, and its ligand in another. We explored this possibility by focusing on common pathways between cell types and sought known molecular interactions that were enriched in specific cell types. We thus identified potential homotypic cell-cell interaction mechanisms that were altered during neuroinflammation (Figure 4P). For example, several cell adhesion molecules were upregulated in NIM when compared to controls: Cldn5 and Pecam1 in endothelial cells or Cdh19 in astrocytes and oligodendrocytes. Integrin pathways, with different combinations of ligands and receptors depending on the cell type, were affected in microglia and astrocytes. Interestingly, both Sst within interneurons and its receptors Sstr2 and Sstr3 in projection neurons were downregulated. Somatostatin interneuron deficiency has been proposed as a mechanism involved in depression (Fee et al., 2017), and somatostatin infusion induces antidepressant-like effects (Engin and Treit, 2009). Heterotypic interactions were also found to be changed (Figure 4Q). We identified several upregulated pathways whereby astrocytes, endothelial cells, and oligodendrocytes produced ligands that signaled to microglia. For example, Csf1r in microglia was found to be upregulated in NIM, while its ligand Csf1 was enriched in astrocytes and potentially oligodendrocytes (Saunders et al., 2018). Csf1r has been identified to be essential for microglia survival (Elmore et al., 2014). Integrin heterotypic communication was also detected between microglia and astrocytes or endothelial cells. We next used IPA to identify predicted upstream regulators that may influence the expression of the DEGs in different cell types (Figure 4R).
Comparison of neuroinflammatory PFC transcriptional profiles with AD
Astrocyte neuroinflammatory phenotypes may have roles in neuropsychiatric and neurodegenerative diseases (Liddelow and Barres, 2017; Perez-Nievas and Serrano-Pozo, 2018; Sofroniew, 2015; Wang et al., 2017). Available evidence indicates that reactive astrocytes can have diverse phenotypes that vary depending on age, species, type of injury, and disease stage (Escartin et al., 2021). The possibility that there are specific astrocyte changes that are conserved across different conditions remains incompletely explored, but this could have important implications for disease treatment strategies. In addition, growing evidence suggests that circulating molecules derived from the gut or oral microbiome, including LPS, have the potential to influence neuroinflammation in neurodegenerative conditions (Tremlett et al., 2017). We therefore compared our NIM data to recent post-mortem human PFC single nuclei RNA (snRNA)-seq from patients with AD and from a mouse model of AD (Habib et al., 2020; Mathys et al., 2019) in order to assess to what extent the astrocyte neuroinflammatory responses we observed in our acute NIM were also observed within chronic disease.
Mathys et al. (2019) used snRNA-seq to assess PFC cell transcriptional alterations in 24 patients with AD and 24 controls. 514 DEGs were identified in astrocytes (Figure S5A). From these, 28 were also altered at day 1 and 2 in NIM PFC astrocytes (Figure S5B). Furthermore, half of them changed in the same direction in both AD and NIM. The top common upregulated gene was Csf1, the gene that encodes the cytokine ligand of the essential microglial receptor Csf1r (Elmore et al., 2014), and one of the genes identified by our heterotypic cell-cell interaction analysis (Figure 4Q). Common upregulated genes also included Gfap and the inflammatory mediators Tnfrsf1a and Cd81. Several of the genes downregulated in the three datasets were involved with lipid metabolism, e.g., Acaca, Fdft1, Sqle, and Acat2 (Figure S5B). 165 DEGs were altered in both AD and day 1 NIM PFC astrocytes (Figures S5B, S5C, S6A, and S6B). Of these, 56% changed in the same direction in both datasets, with most of them being downregulated (Figure S5C). Unexpectedly, the three biological processes identified with gene ontology analyses were not related to inflammatory processes, but instead they included neuron projection morphogenesis, modulation of synaptic transmission, and cellular component organization (Figure S5D). Several of the genes in these pathways encode cell adhesion molecules (Cdh11, Cntnap2, and Nrxn1), receptors or ligands (Ephb1, Cck, Ptn, Grid1, Grm3, and Tnfrsf1a), or cytoskeleton related proteins (Dst, Gfap, Fam107a, Mapt, and Tubb2b) (Figure S5D). 34 DEGs were common between AD and day 2 NIM astrocytes (Figures S5B, S5E, S6A, and S6C); 61% of them changed in the same direction (Figure S5E). Gene Ontology analysis of these emphasized that cholesterol synthesis is downregulated in both AD and NIM (Figure S5F).
We also evaluated how similar PFC astrocyte NIM transcriptomes were to those from the 5xFAD AD mouse model. For this we used scREAD (Jiang et al., 2020) to identify PFC astrocyte DEGs from the recently published 5xFAD dataset (Habib et al., 2020). Two mouse ages were analyzed, that is, 7 and 10 months old. We found 131 (7 months old) and 63 (10 months old) DEGs in 5xFAD mice when compared to wild-type (WT) controls (Figure S5G). Although the numbers of DEGs differed, 42% of the astrocyte 5xFAD DEGs were shared with day 1 NIM astrocytes (Figure S5H), and half of those changed in the same direction. Seven DEGs were found to be shared between 7-month 5xFAD day 1 and day 2 NIM astrocytes (Figure S5H). At 10 months, 5xFAD astrocytes showed a very similar overlap with day 1 NIM (43%). However, the proportion of genes that changed in the same direction was 85% (Figure S5I). Seven of these genes were shared also with day 2 NIM astrocytes (Figure S5I). To assess to what extent the common astrocyte genes between 7- and 10-month 5xFAD were shared with the astrocyte DEGs from human AD samples, we made comparisons among datasets (Figure S5J). Six DEG were common across all datasets: Plce1, which encodes a protein that catalyzes the production of diacylglycerol and inositol trisphosphate; Gfap, encoding for an astrocyte intermediate filament protein; Cd81, involved in immune response; Sdc4, encoding a protein that could facilitate exosome biogenesis; Nhsl1, of unknown function; and Grm3, which encodes for one of the main astrocyte glutamate G-protein-coupled receptors (Figure S5J). Interestingly, the direction of the differential expression in these six shared genes is also observed in the disease-associated astrocytes described with snRNA-seq analyses (Habib et al., 2020).
To summarize, we compared transcriptional changes in PFC astrocytes after LPS-induced neuroinflammation to astrocyte transcriptional changes in the equivalent brain region of patients with AD and in the 5xFAD mouse model. The common genes identified were mainly involved in immune responses, reduced interactions with neurons, and reduced lipid synthesis.
Effects of neuroinflammation on astrocyte phenotypes
It has been proposed that LPS activates microglial release of cytokines that convert astrocytes into a neurotoxic “A1” type that can kill surrounding neurons (Liddelow et al., 2017). We next asked whether PFC and VCX astrocytes preferentially expressed the proposed “pan reactive,” A1, and A2 marker genes in NIM mice relative to controls (Liddelow et al., 2017; Zamanian et al., 2012). The majority of pan reactive and A1 genes were upregulated at day 1 in the NIM mice for both PFC and VCX. The number of upregulated genes decreased at day 2, and these transcriptional changes were fully recovered by day 14 when no significant differences were observed (Figure 5A). Upregulation of some proposed A2 genes was also observed on day 1 in both PFC and VCX astrocytes (Figure 5A), which is consistent with overall astrocyte reactivity. It is noteworthy that the functions of these proposed correlative marker genes are largely unknown and therefore the functional relevance of their presence or absence remains largely uncertain. We also performed IHC for GFAP. In accord with the gene expression data, upregulation of GFAP was observed in astrocytes of PFC and VCX (Figure 5C–5D), while the number of S100β-positive astrocytes or Iba1-expressing microglia did not change (Figures 5B and 5E–5H). Although the IHC data need to be interpreted cautiously, together with RNA-seq they nonetheless suggest the existence of neuroinflammatory signatures in whole tissue (Figures 1E–1H and 5A–5D; Figure S3).
Figure 5. Astrocyte reactivity in PFC and VCX astrocytes.

(A) NIM versus control RNA-seq differential expression of the top astrocyte reactivity genes in 1-, 2-, and 14-day PFC and VCX astrocytes. In the heatmaps, red indicates upregulation and blue indicates downregulation when comparing NIM versus control. An asterisk indicates the genes that were differentially expressed with FDR < 0.05. n = 3–4 biological samples per group; each biological sample was generated from tissue of two mice.
(B) Representative immunostaining of S100β in PFC brain slices.
(C and D) Representative images from GFAP staining and area % coverage of GFAP in control and NIM PFC (C) or VCX (D). n = 9 mice per group in (C) and 4 mice per group in (D).
(E and F) Representative images from S100β staining and number of S100β-positive cells per mm2 in control and NIM PFC (E) or VCX (F). n = 3–4 mice per group.
(G and H) Representative images from Iba1 staining and number of Iba1-positive cells per mm2 in control and NIM PFC (G) or VCX (H). n = 4 mice per group. In the scatterplots, open circles are raw data with closed circles indicating mean ± SEM. In some cases, the error bars representing SEM are smaller than the symbol used for the mean. Scale bars, 50 μm. Note that in (C), the number of mice is greater for the GFAP evaluations. This is because all of the experiments shown in this figure were not performed in parallel, and thus we included GFAP in all such evaluations as an internal reference, which meant its n number was higher.
Since RNA-seq data identified significant changes in astrocyte transcriptional profiles, we performed a series of experiments under identical conditions to assess functional alterations that accompany reactive astrocytes in NIM versus control mice. Using intracellular lucifer yellow iontophoresis we assessed astrocyte morphology (Figures 6A–6E), and with a whole-cell patch clamp we evaluated astrocyte resting membrane potentials, membrane conductance, current waveforms, macroscopic current-voltage relationships, and Ba2+-sensitive currents (Figures 6F–6J). Using imaging of intracellular Ca2+ signals, we assessed spontaneous Ca2+ signaling in astrocyte somata, branches, and processes, as well as G protein-coupled receptor (GPCR)-mediated Ca2+ signals evoked by ATP and phenylephrine, which activate P2Y and α1 adrenoceptors on astrocytes (Figures 6K–6M; Table S1). Using patch clamp-mediated intracellular loading with biocytin, we monitored gap-junctional dye coupling between astrocytes (Figure S7). From all of these assessments, we detected an ~25% decrease in the astrocyte branch and process volumes following neuroinflammation (Figure 6D) and modest alterations in amplitude, half-width, and frequency of spontaneous Ca2+ signals (Table S1). Astrocyte electrophysiological properties were essentially normal in NIM mice, implying that core functions such as K+ buffering and homeostasis were not significantly altered (Figures 6F–6J). Overall, our data show that PFC astrocytes display strong changes in gene expression when assessed agnostically, as well as some significant functional alterations.
Figure 6. Functional and morphological alterations of PFC astrocytes during neuroinflammation.
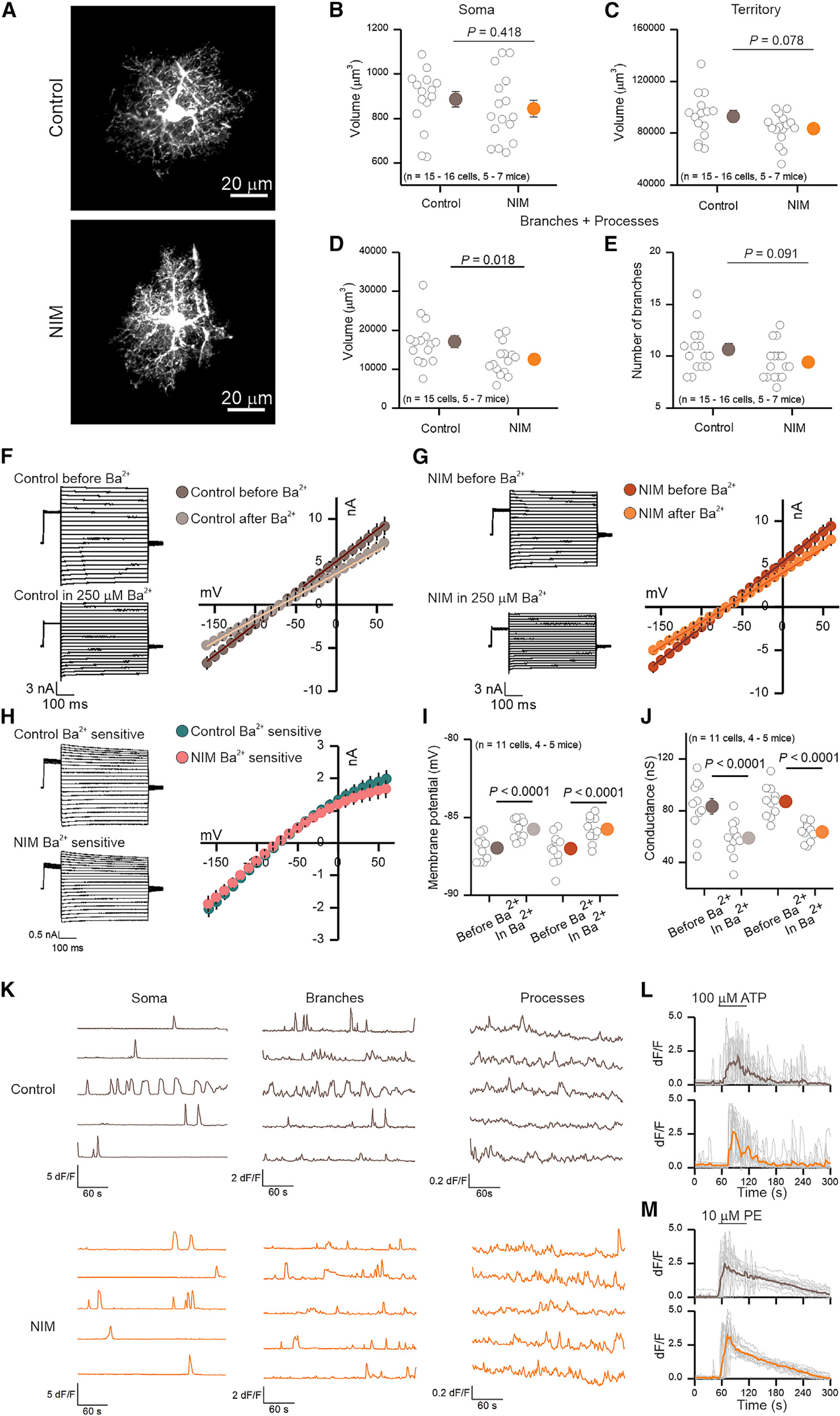
(A) Representative images of 2-day control and NIM PFC astrocytes filled by lucifer yellow iontophoresis.
(B–D) Volume of soma (B), territory (C), and branches + processes (D) of 2-day control and NIM astrocytes. Cell compartment volumes were calculated with Imaris software.
(E) Number of branches that ramify from the soma of 2-day control and NIM astrocytes. n = 15–16 cells from five to seven mice per group.
(F and G) Whole-cell voltage clamp was performed on 2-day control (F) and NIM (G) astrocytes before and in the presence of 250 μM Ba2+. On the left, representative waveforms for total and Ba2+ insensitive currents are shown. On the right, average current-voltage relationships are shown.
(H) On the left, representative waveforms for control and NIM Ba2+-sensitive currents are shown. On the right, average current-voltage relationships for control and NIM Ba2+-sensitive currents are shown.
(I) Membrane potential of control and NIM astrocytes before and during Ba2+.
(J) Conductance of control and NIM astrocytes before and during Ba2+. n = 11 cells from four to five mice per group.
(K) Representative traces of 2-day control and NIM spontaneous Ca2+ signals (in 300 nM TTX) in soma, branches, and processes. The amplitude of Ca2+ signals is higher in control somas and branches than in NIM; however, Ca2+ signals are more frequent in the processes of NIM than in control (Table S1).
(L and M) Single (gray) and average (brown or orange) traces of astrocyte soma and branch Ca2+ increase in response to 1-min exposure to 100 μM ATP (L) or 10 μM phenylephrine (PE) (M).
In the scatterplots, open circles are raw data, with closed circles indicating mean ± SEM. In some cases, the error bars representing SEM are smaller than the symbol used for the mean. Scale bars, 20 μm.
Functional assessments of PFC neurons following neuroinflammation
To test whether astrocyte-reactive phenotypes in NIM had an effect on the survival of neurons, we performed IHC with the neuronal marker NeuN. No differences in neuron numbers were found between control and NIM in PFC or VCX (Figures 7A–7C). We further assessed potential alterations in neuronal excitability by performing whole-cell patch clamp of PFC pyramidal neurons of layers 2/3 (Figures 7D–7U). Remarkably, no differences between NIM and control were observed in membrane potential (Vm), membrane resistance (Rm), or the minimum current necessary to produce an action potential (rheobase) (Figures 7D–7I). Furthermore, no untoward issues were noted when recording from neurons from NIM mice, and the cells looked healthy in both NIM and control conditions. We next recorded and quantified spontaneous excitatory postsynaptic currents (sEPSCs; in 10 μM bicuculline). Neither the amplitude nor the frequency of the events was different between the experimental conditions (Figures 7J–7O). Action potential independent miniature EPSCs (mEPSCs; in 10 μM bicuculline + 300 nM tetrodotoxin [TTX]), however, were slightly increased in frequency in the NIM neurons. There were no differences in mEPSC amplitude (Figure 7P–U). All in all, PFC pyramidal neurons were ostensibly normal with no evidence of dysfunction or neurotoxicity under conditions when astrocyte reactivity genes proposed to be associated with A1 phenotypes were significantly elevated.
Figure 7. Neuronal and synaptic properties of PFC pyramidal neurons during neuroinflammation.
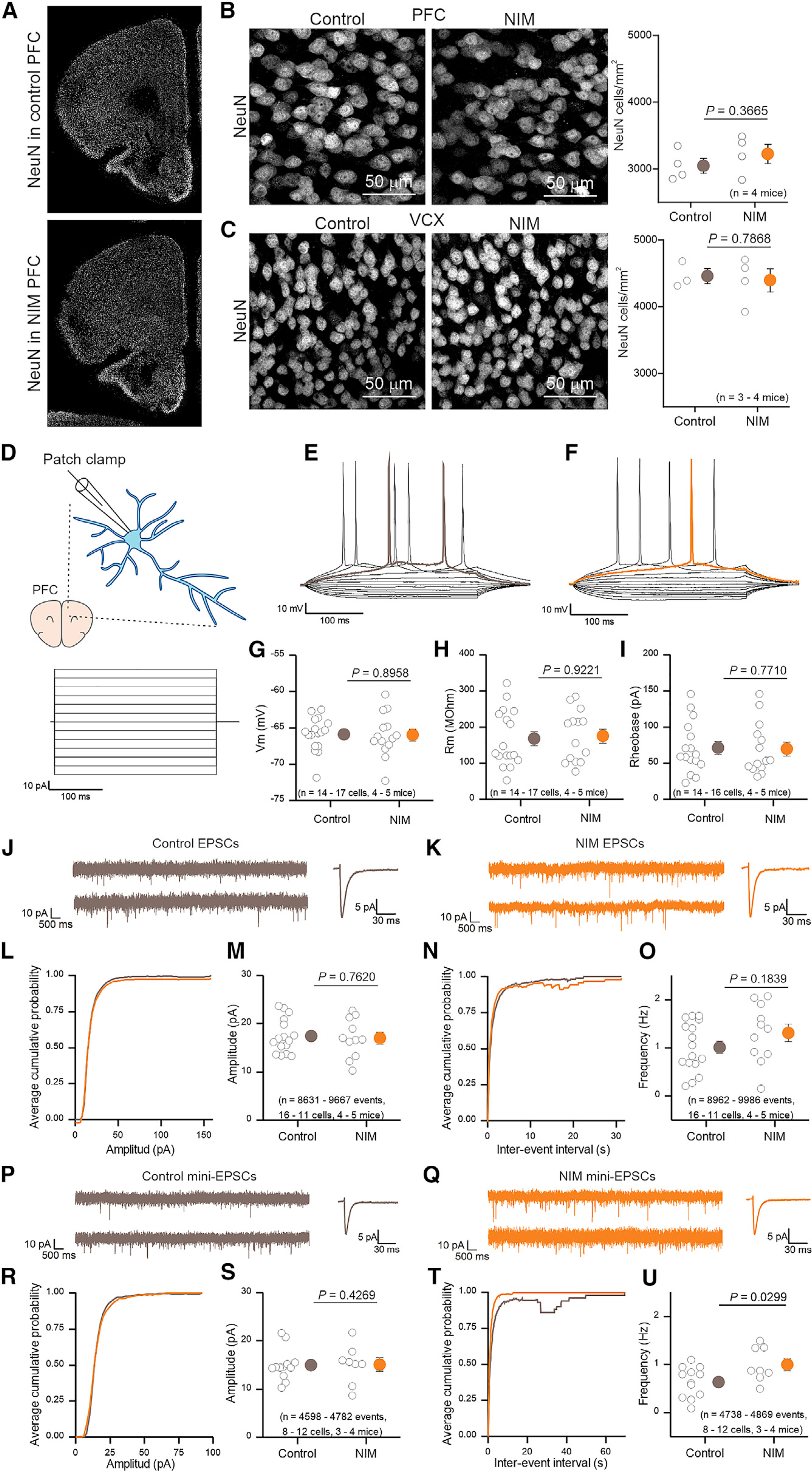
(A) Representative immunostaining of NeuN in PFC brain slices.
(B and C) Representative images from NeuN staining and number of NeuN-positive cells per mm2 in control and NIM PFC (B) or VCX (C). n = 4 mice per group.
(D) Whole-cell current clamp was performed in 2-day NIM and control PFC pyramidal neurons. 10-pA steps were applied during 300 ms.
(E and F) Representative voltage waveforms from control (E) and NIM (F). Rheobase trace is highlighted in brown for control and orange for NIM.
(G–I) Control and NIM pyramidal neuron membrane potential (G), membrane resistance (H), and rheobase (I). n = 14–17 cells from four to five mice per group.
(J and K) Representative EPSC (10 μM bicuculline) current clamp recording and single EPSC example from control (J) and NIM (K) PFC pyramidal neurons.
(L) Average cumulative probability plot for EPSC amplitude.
(M) EPSC average amplitude for 2-day control and NIM PFC pyramidal neurons. n = 8,631–9,667 events from 11–16 cells from four to five mice per group.
(N) Average cumulative probability plot for EPSC inter-event intervals.
(O) EPSCs frequency for 2-day control and NIM PFC pyramidal neurons in a 10-min recording. n = 8,962–9,986 events from 11–16 cells from four to five mice per group.
(P and Q) Representative mini-EPSC (10 μM bicuculline and 300 nM TTX) current clamp recording and single mini-EPSC example from control (P) and NIM (Q) PFC pyramidal neurons.
(R) Average cumulative probability plot for mini-EPSC amplitude.
(S) Mini-EPSC average amplitude for 2-day control and NIM PFC pyramidal neurons. n = 4,598–4,782 events from 8–12 cells from three to four mice per group.
(T) Average cumulative probability plot for mini-EPSC inter-event intervals.
(U) Mini-EPSCs frequency for 2-day control and NIM PFC pyramidal neurons in a 10-min recording. N = 4,738–4,869 events from 8–12 cells from three to four mice per group.
In the scatterplots, open circles are raw data with closed circles indicating mean ± SEM. In some cases, the error bars representing SEM are smaller than the symbol used for the mean. Scale bars, 20 μm.
DISCUSSION
Neuroinflammation has long been associated with CNS disorders and is emerging as a target for treatment, but many open questions remain at the interface between the immune system and the brain. In this study, we used a peripherally induced NIM to explore the molecular and functional profiles of astrocytes at specific behaviorally defined stages. We examined three time points: day 1, when the mice displayed locomotor dysfunction and anhedonia; day 2, when locomotor activity was normal, but anhedonia persisted; and day 14, when there were no detectable differences between the NIM and controls. We performed astrocyte-specific and whole-tissue RNA-seq from PFC and VCX at each time point and comment on the major conclusions that can be drawn.
First, astrocyte-specific RNA-seq experiments showed transcriptional diversity between PFC and VCX astrocytes in control conditions as well as during neuroinflammation at days 1 and 2 after LPS. Notably, up to one third of DEGs between NIM and control mice were specific to the cortical region, and region-specific molecular identities of PFC and VCX astrocytes were largely maintained during the response to neuroinflammation, indicating that astrocyte regional identity can be preserved in spite of reactive changes. Second, RNA-seq analyses suggested that, in addition to activation of immune signaling in astrocytes, astrocyte pathways involved in homeostatic roles such cholesterol metabolism, cytoskeleton dynamics, gap junctions, K+ and Ca2+ transport, synapse plasticity, and signaling to other cells were significantly affected in the PFC. Third, whole-tissue RNA-seq analysis of the cell type-enriched markers revealed that microglia and brain endothelial cells were the main responders in NIM in both PFC and VCX. Fourth, astrocyte gene expression changes observed in NIM were completely reversible, and no significant differences persisted at day 14 after LPS. Fifth, modest overlap was found between NIM and human or mouse AD in terms of PFC astrocyte gene expression changes. An increase in astrocyte transcriptome inflammatory profiles and a decrease in neuron supportive functions were found as common phenotypes. Sixth, in NIM, although astrocytes upregulated a set of genes related to astrocyte reactivity, these changes did not exert detectable detrimental effects on neuronal health or survival, indicating that astrocyte reactivity can be associated with maintenance of essential neural homeostatic functions. These findings add to steadily growing evidence that astrocytes play central roles in CNS innate immune responses (Farina et al., 2007; Sofroniew, 2020) and we think they will help in the interpretation and design of studies aimed at disentangling the effects of neuroinflammation on the CNS.
Rodent models of neuroinflammation display behaviors such as anhedonia that have been associated with depression (Bhattacharya et al., 2018; Scheggi et al., 2018; Yirmiya, 1996). Behavioral phenotypes in these models typically present in two phases. The initial phase dominated by sickness behavior is followed by a second phase in which sickness phenotypes wane, but depressive-like behaviors remain (Dantzer et al., 2008). Similarly, we describe how NIM mice showed reduced motility on the first day and anhedonia, measured as a lack of preference for sucrose, for up to 2 days. To better understand how neuroinflammation affected astrocytes during the sickness (day 1), anhedonia-like (day 2), and recovery (day 14) phases we performed astrocyte RNA-seq from two cortical regions: the PFC, which is relevant to anhedonia and cognitive decline in many CNS diseases, and VCX, an area engaged in different visual circuits and behaviors. In parallel to the severity of the NIM phenotypes, the highest number of DEGs in both PFC and VCX were detected at day 1, followed by day 2. Remarkably, no DEGs were identified at day 14, indicating that astrocyte transcriptional changes associated with acute neuroinflammation may be a completely reversible phenomenon.
Transcriptionally different astrocyte populations have been observed between and within brain regions (Ben Haim and Rowitch, 2017; Khakh and Deneen, 2019). We compared PFC and VCX astrocyte transcriptomes to assess whether the response of astrocytes from two different brain circuits to the same stimulus was equivalent or region-dependent. As expected, the transcriptomes of both PFC and VCX astrocytes changed during neuroinflammation. Interestingly, around 60% of the region-specific genes were common between control and NIM, indicating that a considerable proportion of identity-defining gene expression in these two areas was preserved during neuroinflammation. When we focused on DEGs between the NIM versus control in each brain region, we discovered that one third were region specific. Our data show that astrocytes from distinct regions respond differently to the same stimulus, which merits further mechanistic work. This is potentially an important question with relevance to regional susceptibility of CNS regions to specific diseases. Furthermore, this result underscores the need for considerable caution when analyzing gene expression data that are available in various resources and published studies: the impact of regional variation (Ben Haim and Rowitch, 2017; Khakh and Deneen, 2019), highly context-specific responses (Yu et al., 2020b), and of variable responses to the same stimulus that is dependent on the brain region, as shown here, need to be thoughtfully accounted for in order to make meaningful conclusions. To what extent these region-specific responses are due to cell-autonomous mechanisms or to the variability of response of surrounding cells needs further exploration.
Our gene pathway analyses evinced prominent alterations of cell signaling pathways in PFC NIM astrocytes. We evaluated cell-cell interaction mechanisms during neuroinflammation. We identified the brain cell type-enriched genes of the frontal and posterior cortices (Saunders et al., 2018). We then asked which of those genes changed their expression in our whole-tissue (INPUT) samples in response to neuroinflammation. The cell type with the most upregulated genes was microglia followed by endothelial cells. Both microglia and endothelial cells also displayed clear activation of immune signaling. Interestingly, our cell-cell interaction analysis identified several ligand-receptor pairs that may signal from endothelial cells to microglia and astrocytes. Since endothelial cells are the first barrier between the blood and the brain at the neurovascular unit, our data suggest that these cells may translate peripheral inflammatory signals into the brain by directly signaling to astrocytes and microglia. This is a somewhat unexplored area and our data and analyses will be valuable to design hypothesis-driven experiments that will contribute to the understanding of such mechanisms.
We also explored functional alterations of astrocytes. Despite the large transcriptional response of astrocytes to neuroinflammation, we detected more subtle differences at the functional level. We emphasize that the topic of astrocyte functional changes should be revisited in the future when tools are available to reliably assess a greater number of metrics, because our data do not rule out the existence of functional changes that we could not assess carefully in our experiments with the available methods. In addition, transcriptional changes could differentially influence particular subcellular compartments (e.g., processes near synapses) that we cannot assess with the current tools. Moreover, we do not know to what extent subtle differences can impact astrocyte roles and their interaction with other cells. In these regards, the metrics to assess astrocyte function are limited in relation to the large number of genes that can be assessed with the high sensitivity of RNA-seq. Notwithstanding the limitations of the current tools, our data suggest that astrocytes may be able to maintain tissue homeostasis and function even during profound gene expression changes associated with reactivity. In this eventuality, our data have important and potentially broad implications for studies where gene expression data alone are interpreted to invoke altered astrocyte functions in disease settings, especially in the case of postmortem human tissue.
It has been proposed, based primarily on in vitro observations, that LPS-induced signaling leads to release of C1q, tumor necrosis factor (TNF), and Il1α by microglia, which in turn convert astrocytes into neurotoxic A1 astrocytes, which could lead to neuronal death in mouse models and human neurodegenerative disease (Liddelow et al., 2017). In this study, we administered the same LPS dose using i.p. injection as originally used to identify a set of genes proposed to characterize this reactive A1 phenotype (Zamanian et al., 2012). We confirmed that most of the proposed pan-reactive and A1 genes were upregulated 1 day after LPS injection in both PFC and VCX. However, no neuronal death or neuronal electrophysiological dysfunction was observed in the PFC. Recently, the original propositions have been changed and it has been suggested that for reactive astrocytes to become neurotoxic, neuronal injury, such as axotomy of retinal ganglion neurons, must precede the astrocyte transformation to proposed A1 states (Guttenplan et al., 2020). Our studies suggest caution in concluding that neurons would be dysfunctional based simply on the appearance of published proposed correlative “neurotoxic” gene expression profiles within astrocytes. Additional studies are needed to better understand the nuances of proposed protective and detrimental roles of reactive astrocytes in pathology (Escartin et al., 2021).
Chronic neuroinflammation is considered a major potential contributor to AD and other neurodegenerative diseases. This neuroinflammation may derive not only from CNS-intrinsic responses to degenerative changes, but may be exacerbated by peripherally derived molecules from gut or oral microbiome, such as LPS (Long and Holtzman, 2019; Tremlett et al., 2017). We therefore compared the PFC astrocyte transcriptional changes we observed after acute peripherally induced neuroinflammation to the changes observed in PFC astrocytes from chronic human AD and a mouse model of AD. Although the overlap between datasets was modest, some common changes between NIM, AD, and other disease datasets were observed, such as downregulation of cholesterol synthesis pathways and reduction in pathways associated support of neurons and synapses (Boisvert et al., 2018; Diaz-Castro et al., 2019; Itoh et al., 2018; Mathys et al., 2019). These findings help understand how astrocyte changes associated with neuroinflammation can impact surrounding cells and thereby on disease processes as well. The precise contribution of neuroinflammation to pathophysiology in diseases such as AD needs further work; although neuroinflammation may occur, it remains to be fully elucidated whether this is phenotypically causal to disease.
Overall, our observations are consistent with the emerging consensus on astrocyte reactivity as a complex non-binary response and that astrocyte reactivity phenotypes are not only region-dependent, but also tailored to the age, disease, and stage of the disorder (Escartin et al., 2021). Our findings and gene expression data provide a rich source of information to formulate and test specific hypotheses in relation to the influence of neuroinflammation on brain function. For example, are there causative relationships between PFC-specific astrocyte NIM changes and the concomitant anhedonia we observed? Are the region-specific astrocyte responses to neuroinflammation due to cell-autonomous or non-cell-autonomous mechanisms? How does loss of astrocyte homeostatic support affect neuron circuit function and can this be a target for disease treatment? How do other cells and the communication between them contribute to the initiation and maintenance of the brain neuroinflammatory response? What restoration mechanisms allow for a full reversal of the transcriptional changes in NIM, are they protective in chronic disease, and could they be exploited? The availability of improved experimental tools and approaches to study astrocytes (Yu et al., 2020a) should allow such questions to be addressed with confidence in the years ahead.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| rabbit anti-GFAP (1:1000) | Agilent | Cat# Z0334, RRID:AB_10013382 |
| rabbit anti-S100β (1:1000) | Abcam | Cat# ab41548, RRID:AB_956280 |
| mouse anti-S100β (1:1000) | Sigma-Aldrich | Cat# S2532, RRID:AB_477499 |
| mouse anti-HA antibody | Biolegend | Cat# 901514, RRID:AB_2565336 |
| rabbit anti-NeuN (1:2000) | Cell Signaling Technology | Cat# 12943, RRID:AB_2630395 |
| rabbit anti-Iba1 (1:1000) | Wako | Cat# 019-19741, RRID:AB_839504 |
| Alexa goat anti-mouse 488 (1:1000) | Invitrogen | Cat# A10680 |
| Alexa goat anti-rabbit 546 (1:1000) | Invitrogen | Cat# A11010 |
| Streptavidin conjugated Alexa 488 (1:250) | Molecular Probes | Cat# S32354, RRID:AB_2315383 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Lipopolysaccharide | Sigma-Aldrich | Cat# L5024 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Sucrose | Sigma-Aldrich | Cat# S7903 |
| 10% buffered formalin | Fisher | Cat# SF100-20 |
| 10% NP-40 | Roche | Cat# 11 332 473 001 |
| 1M DTT | Sigma-Aldrich | Cat# 646563-10X0.5 ml |
| Protease inhibitor | Sigma-Aldrich | Cat# P8340-5ml |
| RNAsin | Promega | Cat# N2115 |
| Cyclohexamide | Sigma-Aldrich | Cat# C7698 |
| Heparin | Sigma-Aldrich | Cat# H3393 |
| Pierce A/G magnetic beads | Pierce | Cat# 88803 |
| Lucifer yellow CH di-Lithium salt | Sigma-Aldrich | Cat# L0259 |
| Biocytin | Tocris | Cat# 3349 |
| Carbenoxolone | Tocris | Cat# 3096 |
| Bicuculline | Sigma-Aldrich | Cat# 14340 |
| TTX | Cayman Chemical Company | Cat# 14964 |
| Phenylephrine | Tocris | Cat# 2838 |
| ATP disodium salt | Tocris | Cat# 3245 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq data | Gene Expression Omnibus | GSE135110 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mouse | JAX | RRID: IMSR_JAX: 000664 |
| B6N.129-Rpl22tm1.1Psam/J mice | JAX | RRID: IMSR_JAX: 011029 |
| Aldh1/1-cre/ERT2 mice | In house, now at JAX | RRID: IMSR_JAX: 031008 |
| Ai95 mice | JAX | RRID: IMSR_JAX: 024105 |
|
| ||
| Software and algorithms | ||
|
| ||
| Axon pCLAMP10 | Molecular devices | RRID:SCR_011323 |
| Ingenuity Pathway Analysis | QIAGEN | RRID:SCR_008653 |
| Panther | http://www.pantherdb.org/ | RRID:SCR_004869 |
| Fiji | NIH | RRID:SCR_002285 |
| ClampFit10.7 | Molecular devices | RRID:SCR_011323 |
| Graphpad Instat 3 | Graphpad | RRID:SCR_000306 |
| Imaris Version 7.6.5. | Bitplane | RRID:SCR_007370 |
| OriginPro | Origin lab | RRID:SCR_014212 |
|
| ||
| Other | ||
|
| ||
| RNeasy Plus Micro | QIAGEN | Cat# 74034 |
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: Baljit S. Khakh (bkhakh@mednet.ucla.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data S1 contains the annotated RNA-seq data and all the raw data have been submitted to the Gene Expression Omnibus (GEO) with accession ID GSE135110. Other data are available upon reasonable request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Most animal experiments were conducted in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Chancellor’s Animal Research Committee at the University of California, Los Angeles. A subset of the animal experiments conformed to national and institutional guidelines including the Animals [Scientific Procedures Act] 1986 (UK), and the Council Directive 2010/63EU of the European Parliament and the Council of 22 September 2010 on the protection of animals used for scientific purposes, and had full Home Office ethical approval. All mice were housed with food and water available ad libitum in a 12 hour light/dark environment. All animals were sacrificed during the light cycle, and none were involved in previous studies.
Experiments were performed using 8–10 week old C57BL/6J (RRID: IMSR_JAX: 000664) mice. For astrocyte specific RNA-seq experiments, B6N.129-Rpl22tm1.1Psam/J (RRID: IMSR_JAX: 011029) (Sanz et al., 2009) were bred with Aldh1l1-cre/ERT2 mice (Srinivasan et al., 2016) from an in-house colony (RRID: IMSR_JAX: 031008); male and female homozygous knock-in hemizygous transgenic mice were used. For Ca2+ imaging Ai95 (RRID: IMSR_JAX: 024105) were bred with Aldh1l1-cre/ERT2 mice. RNA-seq experiments were performed with male and female mice, all other experiments were performed with female mice. All mice were young adults between 8 and 10 weeks old at the time of LPS injection.
METHOD DETAILS
Drug administration
Lipopolysaccharide (Sigma Cat# L5024) from E. coli was diluted to 1 mg/ml in sterile PBS and stored in aliquots at −80°C. Eight to ten week old mice received one single injection of 5 mg/kg at 11 AM. Control mice received an equivalent volume of PBS. Mice were weighed every day from the day of injection until sacrifice. Tamoxifen (Sigma Cat# T5648, 20 mg/ml) was administered intraperitoneally for five consecutive days at 100 mg/kg body weight to 6–7 week old Aldh1l1-cre/ERT2 × RiboTag mice (Rpl22tm1.1Psam). Mice were used 2–3 weeks after the last tamoxifen injection.
Behavioral assessments
The behavioral assessments shown in this manuscript were all performed in female mice. Similar results are observed in male mice that receive the same treatment (data not shown).
Sucrose preference test
One week before LPS injections, mice underwent a 3 day baseline test for sucrose preference. Each cage received two bottles filled with water ad libitum. The following day, one bottle was replaced by 1% sucrose solution (Sigma Cat# S7903). On day 3, that bottle was replaced by water. Consumption of each liquid was measured. One day prior to LPS injection, each cage received a fresh bottle of water and 1% sucrose solution, which were weighed every day until sacrifice. The bottle containing sucrose was maintained in the same position for the duration of the experiment. Sucrose preference was calculated by the following formula: 100*weight of the liquid drunk from the sucrose bottle/weight of the liquid drunk from the two bottles.
Open field test (OFT)
Mice injected with PBS, LPS (5 mg/kg) or those that were uninjected (as control for effects of the injection on locomotor activity), were placed in plastic containers measuring 30.5 × 18 × 25.5 cm inside a dimly lit room. Mice underwent OFT 6 hours after the injection and on days one, two, three, ten, and 20 at 5 PM. Mice were allowed to acclimate for 3 min, and then were recorded for the following 6 min. To eliminate ambient noise, a white noise generator was used (San Diego Instruments). AnyMaze video analysis software was used to quantify OFT data.
Transcardial perfusions
For transcardial perfusion, mice were euthanized with i.p. injected pentobarbitol and perfused with 10% buffered formalin (Fisher Cat# SF100–20). Briefly, once all reflexes subsided, the abdominal cavity was opened. The animal was perfused with 6 mL of 0.1 M phosphate buffered saline (PBS) containing 10 units/ml of heparin, followed by 60 mL 10% buffered formalin. After gentle removal from the skull, the brain was post fixed in 10% buffered formalin overnight at 4°C. The tissue was cryoprotected in 30% sucrose PBS solution the following day for at least 48 hours at 4°C until use.
Immunohistochemistry (IHC)
All immunohistochemistry studies were performed with tissue from female mice. Coronal sections (40 μm) of formalin perfused brains were prepared using a cryostat microtome (Leica) and kept in 0.05 M PBS, 250 mM sucrose, 7 mM MgCl2, 50% Glycerol at −20°C. For IHC, sections were washed twice in 0.1 M PBS for 10 min, once in 1M HCl, and then incubated in blocking solution containing 10% normal goat serum (NDS or NGS) in 0.1 M PBS with 0.05% Triton® X-100 for 1 hr at room temperature with agitation. Sections were subsequently incubated in primary antibodies diluted in 0.1 M PBS with 0.05% Triton® X-100 overnight at 4°C with agitation. The following primary antibodies were used: rabbit anti-GFAP (1:1000; Agilent Cat# Z0334, RRID:AB_10013382), rabbit anti-S100β (1:1000; Abcam Cat# ab41548, RRID:AB_956280), mouse anti- S100β (Sigma-Aldrich Cat# S2532, RRID:AB_477499), mouse anti-HA (1:1000; BioLegend Cat# 901514, RRID:AB_2565336), rabbit anti-NeuN (1:2000; Cell Signaling Technology Cat# 12943, RRID:AB_2630395), rabbit anti-Iba1 (1:1000, Wako Cat# 019–19741, RRID:AB_839504). The next day the sections were washed three times in 0.1 M PBS for 10 min each before incubation at room temperature for 2 hr with secondary antibodies diluted in blocking solution. The following secondary antibodies were used: goat anti-mouse 488 (1:1000, Invitrogen Cat# A10680), goat anti-rabbit 546 (1:1000, Invitrogen Cat# A11010), streptavidin conjugated Alexa 488 (1:250, Molecular Probes Cat# S32354, RRID:AB_2315383). The sections were rinsed two times in 0.1 M PBS for 10 min each, and once in 0.1 M PBS with 0.2% Triton® X-100 and 140 ng/μl Dapi for 10 min. The sections were mounted on microscope slides in fluoromount-G. Fluorescent images were taken using either UplanSApo 20X 0.85 NA oil immersion objective lens on a commercial confocal laser-scanning microscope (Fluoview V1000; Olympus), a 25X 0.8 NA oil immersion lens on a Zeiss LSM900 confocal laser-scanning microscope, or a Zeiss Axioplan 2 with FluoArc pseudo-confocal. The settings were the same within each experiment.
RNA-seq of mouse whole-tissue and astrocyte transcriptomes
Mice were anesthetized with isoflurane and decapitated. The brain was quickly extracted and cut into 2 mm coronal slices using a rodent brain matrix (Electron Microscopy Sciences). Slices were placed in ice cold PBS and PFC and VCX were further dissected and collected for RNA extraction. Whole tissue or astrocyte specific RNA was used for RNA-seq studies. For whole tissue PFC, female C57/BL6J mice were used. The dissected tissue was homogenized and RNA was purified following the RNeasy kit instructions (RNeasy Plus Micro QIAGEN Cat# 74034). For determining the astrocyte transcriptomes, Ribotag mice crossed with Aldh1l1-cre/ERT2 mice were used. Four biological replicates per condition were collected. Two of them were harvested from male, and the other two from female mice. To obtain enough RNA, each biological replicate contained PFC from two mice. Briefly, freshly dissected tissues were collected and individually homogenized in 1 mL of homogenization buffer (50 mM Tris pH 7.4, 100 mM KCl, 12 mM MgCl2, 1% NP-40, 1 mM Dithiothreitol (DTT), 1X Protease inhibitors, 200 U/ml RNAsin, 100 μg/ml Cyclohexamide, 1 mg/ml Heparin). RNA was extracted from 20% of cleared lysate as input (200 μl). The remaining lysate (800 μl) was incubated with 5 μl of mouse anti-HA antibody (Biolegend Cat# 901514) with rocking for 4 hours at 4°C followed by addition of 200 μl of magnetic beads (Pierce Cat# 88803) and overnight incubation with rocking at 4°C. The beads were washed three times in high salt solution (50 mM Tris pH 7.4, 300 mM KCl, 12 mM MgCl2, 1% NP-40, 1 mM Dithiothreitol (DTT), 100 μg/ml Cyclohexamide). RNA was purified from the IP and corresponding input samples (RNeasy Plus Micro QIAGEN Cat# 74034).
RNA concentration and quality were assessed with an Agilent 2100 Bioanalyzer. RNA samples with RNA integrity numbers (RIN) greater than seven (mean RIN 8.5) were processed with Ribo-Zero Gold kit (Epicenter, WI) to remove ribosomal RNA. Sequencing libraries were prepared using Illumina TruSeq RNA sample prep kits following the manufacturer’s protocol. After library preparation, amplified double-stranded cDNA was fragmented into 125 bp (Covaris-S2, Woburn, MA) DNA fragments, which were (200 ng) end-repaired to generate blunt ends with 5′- phosphates and 3′- hydroxyls and adapters ligated. The purified cDNA library products were evaluated using the Agilent Bioanalyzer (Santa Rosa, CA) and diluted to 10 nM for cluster generation in situ on the HiSeq paired-end flow cell using the CBot automated cluster generation system. Four separate sequencing experiments were performed, one for whole tissue PFC 1 d after injection, another one for IP and INPUT 1 d PFC and VCX, a third one for IP and INPUT 2 d PFC and VCX, and a fourth one for IP and INPUT 14 d PFC and VCX. Samples in each experiment were multiplexed into a single pool in order to avoid batch effects and 69 bp-paired-end sequencing was performed using an Illumina HiSeq 4000 sequencer (Illumina, San Diego, CA). A yield between 30 and 90 million reads was obtained per sample.
Quality control was performed on base qualities and nucleotide composition of sequences. Alignment to the M. musculus (mm10) refSeq (refFlat) reference gene annotation was performed using the STAR spliced read aligner with default parameters. Additional QC was performed after the alignment to examine: the degree of mismatch rate, mapping rate to the whole genome, repeats, chromosomes, key transcriptomic regions (exons, introns, UTRs, genes), insert sizes, AT/GC dropout, transcript coverage and GC bias. Total counts of read-fragments aligned to candidate gene regions were derived using the HTSeq program (https://www.huber.embl.de/users/anders/HTSeq/doc/overview.html) with mouse mm10 refSeq (refFlat table) as a reference and used as a basis for the quantification of gene expression. Only uniquely mapped reads were used for subsequent analyses. Four mice per condition were used; however, in the 2 d dataset two outlier samples were excluded, one LPS-PFC-IP, and another one LPS-VCX-INPUT. Differential expression analysis was conducted with R-project and the Bioconductor package limma-voom. RNA-seq data have been deposited within the Gene Expression Omnibus (GEO) repository (https://www.ncbi.nlm.nih.gov/geo), with accession number GSE135110.
Astrocyte intracellular lucifer yellow iontophoresis
This method for filling cells in fixed tissue was modified from published methods (Bushong et al., 2002; Chai et al., 2017). C57BL/6J female mice injected with PBS or LPS were euthanized two days after the injection with i.p. injected pentobarbitol overdose and transcardially perfused with 10 mL of 35°C Ringer’s Solution with 0.02% lidocaine followed by 50 mL of 10% buffered formalin (Fisher #SF100–20). Ringer’s Solution contains the following (in mM): 135 NaCl, 14.7 NaHCO3, 5 KCl, 1.25 Na2HPO4, 2 CaCl2, 1 MgCl2, and 11 D-glucose, bubbled with 95% O2 and 5% CO2. Brains were lightly post-fixed at room temperature for 1.5 hr and then washed three times in ice cold 0.1 M PBS for 10 min. 100 μm coronal sections were cut using Pelco Vibrotome 3000 and then placed in ice-cold PBS for the duration of the experiment. 10 mg Lucifer yellow CH di-Lithium salt (Sigma Cat# L0259) was dissolved in 1 mL 5 mM KCl solution and filtered prior to use. Sharp (200 MOhm) glass electrodes were pulled from Borosilicate glass capillaries with filament (O.D. 1.0 mm, I.D. 0.58 mm). Electrodes were gravity filled. Sections were transferred to a solution of room temperature PBS for filling. Astrocytes were identified using IR-DIC and then impaled with the sharp electrode. Lucifer yellow was injected into the cell by applying ~1 V for 10 minutes. The electrode was removed and the filled astrocyte was imaged using a confocal microscope (Fluoview V1000; Olympus), with UplanFL 40X water-immersion objective lens 0.8 NA and at a digital zoom of two to three and a 0.25 um confocal z-steps. Quantitative soma and branch + process volumes were generated using Imaris (Bitplane; RRID:SCR_007370) surface function. The astrocyte territory volume was measured from a low-intensity threshold reconstruction encompassing the cell volume and the space between its processes. The number of primary branches was counted visually.
Acute brain slice preparation for imaging and electrophysiology
Slice procedures have been described previously (Jiang et al., 2016; Shigetomi et al., 2013). Coronal striatal or hippocampal slices were prepared from 8–9 week old female mice two days after injection of PBS or LPS. Briefly, animals were deeply anesthetized with isoflurane and decapitated. The brains were placed and sliced in ice-cold modified artificial cerebrospinal fluid (aCSF) containing the following (in mM): 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 D-glucose, saturated with 95% O2 and 5% CO2. A vibratome (DSK-Zero1) was used to cut 300 μm brain sections. The slices were allowed to equilibrate for 30 min at 32–34°C in normal aCSF containing (in mM); 124 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 D-glucose continuously bubbled with 95% O2 and 5% CO2. Slices were then stored at 21–23°C in the same buffer. All experiments were performed within 4–6 hr of slicing.
Electrophysiological recording and assessment of cell coupling in brain slices
Slices were placed in the recording chamber and continuously perfused with 95% O2 and 5% CO2 bubbled normal aCSF. Cells were visualized with infrared optics on an upright microscope (Fluoview FV1000; Olympus). pCLAMP10 software (Molecular Devices; RRID:SCR_011323) and a Multi-Clamp 700B amplifier was used for electrophysiology (Molecular Devices). For recording from astrocytes and dye coupling experiments, currents were measured in whole-cell mode using pipettes with a typical resistance of 5.5 MΩ when filled with internal solution containing the following (in mM): 130 K-gluconate, 2 MgCl2, 10 HEPES, 5 EGTA, 2 Na-ATP, 0.5 CaCl2, with pH set to 7.3. Cells were recorded in normal aCSF and in aCSF + 250 μM Ba2+. For a subset of cells, 2 mg/ml biocytin (Tocris Cat# 3349) was added to the intracellular solution to examine gap junction coupling. Astrocytes were held in whole-cell mode for 20 min to allow biocytin to diffuse from the patched cell to other cells connected by gap junctions. In some cases 100 μM carbenoxolone (CBX; Tocris Cat# 3096) was added to the recording solution to block gap junctions. Brain slices were then rescued from the recording chamber for IHC.
For recording from pyramidal neurons, currents were measured in whole-cell mode using pipettes with a typical resistance of 5–6 MΩ when filled a K+ internal solution consisting of the following (in mM): 135 potassium gluconate, 5 KCl, 0.5 CaCl2, 5 HEPES, 5 EGTA, 2 Mg-ATP and 0.3 Na-GTP, pH 7.3 adjusted with KOH. In some cases, 2 mg/ml biocytin was added to the intracellular solution to subsequently visualize the patched neuron. Neurons were voltage-clamped at −70 mV unless otherwise stated. Excitatory postsynaptic potentials (EPSCs) were recorded in the presence of bicuculline (10 mM), and mini-EPSCs in bicuculline (Sigma Cat# 14340) (10 μM) and tetrodotoxin (TTX; Cayman Chemical Company Cat# 14964) (300 nM). ClampFit 10.7 software was used to analyze traces from astrocyte and neuronal recordings.
Intracellular Ca2+ imaging
Slice preparation was performed as above. Cells for all the experiments were imaged using a confocal microscope (Fluoview FV1000; Olympus) with a 40X water-immersion objective lens with a numerical aperture (NA) of 0.8. A 488 nm line of an Argon laser was used, with the intensity adjusted to 9% of the maximum output of 10 mW and 2–3 digital zoom. The emitted light pathway consisted of an emission high pass filter (505–525 nm) before the photomultiplier tube. Astrocyte somata were typically 25 μm below the slice surface and scanned at 1 frame per second for imaging sessions. Spontaneous signals were recorded in aCSF + 300 nM TTX. For evoked Ca2+ signals, slices were washed on 100 μM adenosine 5′ triphosphate (ATP; Tocris Cat# 3245) or 10 μM phenylephrine (PE; Tocris Cat# 2838) for 1 min.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq differential expression analyses
Statistical significance of the differential expression was determined at false discovery rate (FDR) < 0.05. In the cases in which an FPKM > 1 cutoff was used, we excluded the genes that had an FPKM < 1 in at least one of the samples being compared. Gene ontology analyses were performed using Ingenuity Pathway Analysis (IPA – QIAGEN Bioinformatics; RRID:SCR_008653) with P-value < 0.05 and PANTHER (RRID:SCR_004869) overrepresentation test with FDR < 0.05.
INPUT combined with scRNA-seq analysis
Mouse frontal cortex scRNA-seq gene expression data were obtained from http://dropviz.org/ (Saunders et al., 2018). We obtained a list of cell enriched genes per cell type identified in this dataset by comparing each cell type to the rest and selecting a “minimum fold ratio” of 2, “maximum P-value exponent” of −2, “min mean log amount in target” of 0.25 and a “max mean log amount in comp” of 2. We compared the list of cell enriched genes to our list of DEGs in INPUT 1 d and 2 d NIM versus control and assigned some of the DEGs to the cells in which they were enriched. To find cell-cell interaction molecules, we first identified the IPA pathways that were common between cell types. Within those, we looked for DEGs that encode for receptors, ligands or adhesion molecules. We further filtered these by keeping the ones that were enriched in less than two cells types based on our analysis of Saunders et.al. 2018. With this information, we identified the potential cell-cell interactions mechanisms that could be altered. For example, in the IPA “Neuroinflammation signaling pathway,” we found the receptor Csf1r is upregulated and enriched in microglia. We checked if its potential ligand were cell enriched, and found that its ligand Csf1, was enriched and also upregulated in astrocytes. We used IPA to identify cell enriched upstream regulators. To detect potential non-cell autonomous regulation, we compared the list of upstream regulators and selected the ones that were cell specific, based on our analysis of Saunders et al., 2018.
Comparison of NIM RNA-seq with human AD and AD mouse model snRNA-seq
A list of snRNA-seq cell type specific DEGs from human PFC of patients with AD (Mathys et al., 2019) was obtained from their respective manuscript publications. The DEGs that had a mouse ortholog were selected and compared to the DEGs of our mouse 1 d and 2 d PFC IP NIM versus control analysis. PANTHER gene ontology analysis was used to identify the biological processes that were represented by genes that changed in both human disease and mouse NIM. To compare to 5xFAD mouse model of AD, scREAD was used to analyze a PFC dataset and determine the astrocyte DEGs between 5xFAD and WT at seven and 10 months of age (FDR < 0.05, no fold change threshold). These genes were then compared to the DEGs of our mouse 1 d and 2 d PFC IP NIM versus control analysis.
IHC analysis
All images were analyzed with Fiji (NIH; RRID:SCR_002285). Cell counting was performed from maximum intensity projections using the Cell Counter plugin; only cells with somata completely within the region of interest (ROI) were counted. For signal area and intensity measurements, images were converted to 8-bit and then standardized ROIs were created and thresholded to eliminate background noise in experimental and control images. Two ROIs were analyzed per section (1 per hemisphere), and 3 sections were analyzed per animal in both PFC and VCX.
Calcium imaging analysis
Analyses of time-lapse image series were performed using Fiji (NIH). XY drift was corrected using a custom plugin for Fiji; cells with Z-drift were excluded from analyses. The data were analyzed essentially as previously reported (Yu et al., 2018). Time traces of fluorescence intensity were extracted from the soma, branch or process ROI signals in Fiji and presented as the change in fluorescence relative to the baseline (dF/F). A signal was declared as a Ca2+ transient if it exceeded the baseline by greater than twice the baseline noise. Peak amplitude, half-width, frequency, time of the first peak and area under the curve of Ca2+ signals were analyzed in ClampFit10.7 (Molecular Devices; RRID:SCR_011323).
General data analyses and statistics
Statistical tests were run in GraphPad Instat 3 (GraphPad, RRID:SCR_000306) and Origin 2020. Summary data are presented as mean ± s.e.m. Note that in some of the graphs, the bars representing the s.e.m. are smaller than the symbols used to represent the mean. For weight and behavioral studies, where repeated-measures were performed in the same mice and two drug treatments were being compared, two way ANOVA for repeated-measures were performed. When ANOVA had a p < 0.05, the Tukey post hoc test was used to determine if there were significant differences between groups. These analyses were performed with Origin 2020 software. For the rest of the comparisons, we determined within GraphPad Instat whether the data were normally distributed or not. If they were normally distributed, we used parametric tests. If the data were not normally distributed, we used non-parametric tests. Paired and unpaired Student’s two-tailed t tests (as appropriate) and two-tailed Mann–Whitney tests were used for most statistical analyses with significance declared at p < 0.05, but stated in each case with a precise P value. When the P value was less than 0.0001, it is stated as such to save space on the figure panels and text. N is defined as the numbers of cells or mice throughout on a case-by-case basis depending on the particular experiment; the unit of analysis is stated in each figure or figure legend. A statistical FDR value < 0.05 was used for all RNA-seq analyses. Where appropriate, key statistics are also reported in the text. No data points were excluded from any experiment.
Supplementary Material
Neuroinflammation causes reversible changes in mouse behavior and gene expression
Astrocytic region-specific signatures are maintained during neuroinflammation
Microglia and brain endothelial cells are the main responders
Neurons are largely normal during neuroinflammation-induced astrocyte reactivity
ACKNOWLEDGMENTS
Thanks to the UCLA Neuroscience Genomics Core for assistance with sequencing and to Fuying Gao for helping with RNA-seq data analysis. This work was supported by the National Institutes of Health (R35NS111583) and by the Ressler Family Foundation (to B.S.K.). B.-D.C. was supported by Khakh lab funds, the UCLA Depression Grand Challenge, and UK DRI. Collaborations between the B.S.K. and M.V.S. groups were supported by the UCLA Depression Grand Challenge. A.M.B. was supported by Sofroniew lab funds from the Dr. Miriam and Sheldon G. Adelson Medical Foundation and the UCLA Depression Grand Challenge. We acknowledge the NINDS Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691 to G.C.) and the Genetics, Genomics and Informatics Core of the Semel Institute of Neuroscience at UCLA (U54HD087101-01 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development).
Footnotes
DECLARATION OF INTERESTS
B.S.K. is a consultant for Third Rock Ventures. The remaining authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109508.
REFERENCES
- Barnes J, Mondelli V, and Pariante CM (2017). Genetic contributions of inflammation to depression. Neuropsychopharmacology 42, 81–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA (2008). The mystery and magic of glia: A perspective on their roles in health and disease. Neuron 60, 430–440. [DOI] [PubMed] [Google Scholar]
- Ben Haim L, and Rowitch DH (2017). Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci 18, 31–41. [DOI] [PubMed] [Google Scholar]
- Bennett FC, and Molofsky AV (2019). The immune system and psychiatric disease: A basic science perspective. Clin. Exp. Immunol 197, 294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Lord B, Grigoleit JS, He Y, Fraser I, Campbell SN, Taylor N, Aluisio L, O’Connor JC, Papp M, et al. (2018). Neuropsychopharmacology of JNJ-55308942: Evaluation of a clinical candidate targeting P2X7 ion channels in animal models of neuroinflammation and anhedonia. Neuropsychopharmacology 43, 2586–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert MM, Erikson GA, Shokhirev MN, and Allen NJ (2018). The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep 22, 269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, and Sofroniew MV (2014). Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, and Ellisman MH (2002). Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci 22, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, Cohn W, Rajendran PS, Vondriska TM, Whitelegge JP, et al. (2017). Neural circuit-specialized astrocytes: Transcriptomic, proteomic, morphological, and functional evidence. Neuron 95, 531–549.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafsari FS, and Jessen F (2020). Depression—An underrecognized target for prevention of dementia in Alzheimer’s disease. Transl. Psychiatry 10, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, and Kelley KW (2008). From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci 9, 46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Fruyt J, Sabbe B, and Demyttenaere K (2020). Anhedonia in depressive disorder: A narrative review. Psychopathology 53, 274–281. [DOI] [PubMed] [Google Scholar]
- Diaz-Castro B, Gangwani MR, Yu X, Coppola G, and Khakh BS (2019). Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med 11, eaaw8546. [DOI] [PubMed] [Google Scholar]
- Elahi FM, and Miller BL (2017). A clinicopathological approach to the diagnosis of dementia. Nat. Rev. Neurol 13, 457–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, and Green KN (2014). Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82, 380–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin E, and Treit D (2009). Anxiolytic and antidepressant actions of somatostatin: The role of sst2 and sst3 receptors. Psychopharmacology (Berl.) 206, 281–289. [DOI] [PubMed] [Google Scholar]
- Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A, et al. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci 24, 312–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina C, Aloisi F, and Meinl E (2007). Astrocytes are active players in cerebral innate immunity. Trends Immunol 28, 138–145. [DOI] [PubMed] [Google Scholar]
- Fee C, Banasr M, and Sibille E (2017). Somatostatin-positive gamma-aminobutyric acid interneuron deficits in depression: Cortical microcircuit and therapeutic perspectives. Biol. Psychiatry 82, 549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I (2017). Diversity of astroglial responses across human neurodegenerative disorders and brain aging. Brain Pathol 27, 645–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannoni F, and Quintana FJ (2020). The role of astrocytes in CNS inflammation. Trends Immunol 41, 805–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttenplan KA, Stafford BK, El-Danaf RN, Adler DI, Münch AE, Weigel MK, Huberman AD, and Liddelow SA (2020). Neurotoxic reactive astrocytes drive neuronal death after retinal injury. Cell Rep 31, 107776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Tang W, Choi EY, Yendiki A, Liu H, Jbabdi S, Versace A, and Phillips M (2020). Circuits, networks, and neuropsychiatric disease: Transitioning from anatomy to imaging. Biol. Psychiatry 87, 318–327. [DOI] [PubMed] [Google Scholar]
- Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, Green G, Dionne D, Nguyen L, Marshall JL, et al. (2020). Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci 23, 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Marsh SE, and Stevens B (2019). Immune signaling in neurodegeneration. Immunity 50, 955–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman S, Izzy S, Sen P, Morsett L, and El Khoury J (2018). Microglia in neurodegeneration. Nat. Neurosci 21, 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N, Itoh Y, Tassoni A, Ren E, Kaito M, Ohno A, Ao Y, Farkhondeh V, Johnsonbaugh H, Burda J, et al. (2018). Cell-specific and region-specific transcriptomics in the multiple sclerosis model: Focus on astrocytes. Proc. Natl. Acad. Sci. USA 115, E302–E309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R, Diaz-Castro B, Looger LL, and Khakh BS (2016). Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J. Neurosci 36, 3453–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Wang C, Qi R, Fu H, and Ma Q (2020). scREAD: A single-cell RNA-seq database for Alzheimer’s disease. iScience 23, 101769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, and Deneen B (2019). The emerging nature of astrocyte diversity. Annu. Rev. Neurosci 42, 187–207. [DOI] [PubMed] [Google Scholar]
- Khakh BS, and Sofroniew MV (2015). Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci 18, 942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai KSP, Liu CS, Rau A, Lanctôt KL, Köhler CA, Pakosh M, Carvalho AF, and Herrmann N (2017). Peripheral inflammatory markers in Alzheimer’s disease: A systematic review and meta-analysis of 175 studies. J. Neurol. Neurosurg. Psychiatry 88, 876–882. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, and Barres BA (2017). Reactive astrocytes: Production, function, and therapeutic potential. Immunity 46, 957–967. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnerbauer M, Wheeler MA, and Quintana FJ (2020). Astrocyte crosstalk in CNS Inflammation. Neuron 108, 608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JM, and Holtzman DM (2019). Alzheimer disease: An update on pathobiology and treatment strategies. Cell 179, 312–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, et al. (2019). Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews PM (2019). Chronic inflammation in multiple sclerosis—Seeing what was always there. Nat. Rev. Neurol 15, 582–593. [DOI] [PubMed] [Google Scholar]
- Nagai J, Yu X, Papouin T, Cheong E, Freeman MR, Monk KR, Hastings MH, Haydon PG, Rowitch D, Shaham S, and Khakh BS (2021). Behaviorally consequential astrocytic regulation of neural circuits. Neuron 109, 576–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Nievas BG, and Serrano-Pozo A (2018). Deciphering the astrocyte reaction in Alzheimer’s disease. Front. Aging Neurosci 10, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimenova AA, Raj T, and Goate AM (2018). Untangling genetic risk for Alzheimer’s Disease. Biol. Psychiatry 83, 300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouget JG (2018). The emerging immunogenetic architecture of schizophrenia. Schizophr. Bull. 44, 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M, and Priller J (2014). Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci 15, 300–312. [DOI] [PubMed] [Google Scholar]
- Russo SJ, and Nestler EJ (2013). The brain reward circuitry in mood disorders. Nat. Rev. Neurosci 14, 609–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, and Amieux PS (2009). Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. USA 106, 13939–13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174, 1015–1030.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheggi S, De Montis MG, and Gambarana C (2018). Making sense of rodent models of anhedonia. Int. J. Neuropsychopharmacol 21, 1049–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson-Weaver O, Kracun S, Xu J, Sofroniew MV, Ellisman MH, and Khakh BS (2013). Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J. Gen. Physiol 141, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV (2015). Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci 16, 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV (2020). Astrocyte reactivity: Subtypes, states, and functions in CNS innate immunity. Trends Immunol 41, 758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadaro PA, Flavell CR, Widagdo J, Ratnu VS, Troup M, Ragan C, Mattick JS, and Bredy TW (2015). Long noncoding RNA-directed epigenetic regulation of gene expression is associated with anxiety-like behavior in mice. Biol. Psychiatry 78, 848–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Lu T-Y, Chai H, Xu J, Huang BS, Golshani P, Coppola G, and Khakh BS (2016). New transgenic mouse lines for selectively targeting astrocytes and studying calcium signals in astrocyte processes in situ and in vivo. Neuron 92, 1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed SA, Beurel E, Loewenstein DA, Lowell JA, Craighead WE, Dunlop BW, Mayberg HS, Dhabhar F, Dietrich WD, Keane RW, et al. (2018). Defective inflammatory pathways in never-treated depressed patients are associated with poor treatment response. Neuron 99, 914–924.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremlett H, Bauer KC, Appel-Cresswell S, Finlay BB, and Waubant E (2017). The gut microbiome in human neurological disease: A review. Ann. Neurol 81, 369–382. [DOI] [PubMed] [Google Scholar]
- Wang Q, Jie W, Liu JH, Yang JM, and Gao TM (2017). An astroglial basis of major depressive disorder? An overview. Glia 65, 1227–1250. [DOI] [PubMed] [Google Scholar]
- Yirmiya R (1996). Endotoxin produces a depressive-like episode in rats. Brain Res 711, 163–174. [DOI] [PubMed] [Google Scholar]
- Yu X, Taylor AMW, Nagai J, Golshani P, Evans CJ, Coppola G, and Khakh BS (2018). Reducing astrocyte calcium signaling in vivo alters striatal microcircuits and causes repetitive behavior. Neuron 99, 1170–1187.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Nagai J, and Khakh BS (2020a). Improved tools to study astrocytes. Nat. Rev. Neurosci 21, 121–138. [DOI] [PubMed] [Google Scholar]
- Yu X, Nagai J, Marti-Solano M, Soto JS, Coppola G, Babu MM, and Khakh BS (2020b). Context-specific striatal astrocyte molecular responses are phenotypically exploitable. Neuron 108, 1146–1162.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, and Barres BA (2012). Genomic analysis of reactive astrogliosis. J. Neurosci 32, 6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data S1 contains the annotated RNA-seq data and all the raw data have been submitted to the Gene Expression Omnibus (GEO) with accession ID GSE135110. Other data are available upon reasonable request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.