

Abstract
Myocardial toxicity and drug resistance caused by drug efflux are major limitations of doxorubicin (Dox)-based chemotherapy. Dox structure modification could be used to develop conjugates with an improved biological profile, such as antiproliferative activity and higher cellular retention. Thus, Dox thiol conjugates, Dox thiol (Dox-SH), thiol-reactive Dox-SS-pyridine (SS = disulfide), and a Dox-SS-cell-penetrating cyclic peptide, Dox-SS-[C(WR)4K], were synthesized. Dox was reacted with Trauťs reagent to generate Dox-SH. The thiol group was activated by the reaction with dithiodipyridine to afford the corresponding Dox-SS-Pyridine (Dox-SS-Pyr). A cyclic cell-penetrating peptide containing a cysteine residue [C(WR)4K] was prepared using Fmoc solid-phase strategy. Dox-SS-Py was reacted with the free sulfhydryl of cysteine in [C(WR)4K] to generate Dox-SS-[C(WR)4K] as a Dox-cyclic peptide conjugate. Cytotoxicity of the compounds was examined in human embryonic kidney (HEK-293), human ovarian cancer (SKOV-3), human fibrosarcoma (HT-1080), and human leukemia (CCRF-CEM) cells. Dox-SH and Dox-SS-pyridine were found to have significantly higher or comparable cytotoxicity when compared to Dox in HEK-293, HT-1080, and CCRF-CEM cells after 24 h and 72 incubation, presumably because of higher activity and retention of the compounds in these cells. Furthermore, Dox-SS-[C(WR)4K] showed significantly higher cytotoxic activity in HEK-293, HT-1080, and SKOV-3 cells when compared with Dox after 72 h incubation. Dox-SS-Pyr exhibited higher cellular uptake than Dox-SS-[C(WR)4K] in HT-1080 and HEK-293 cells as shown by flow cytometry. Fluorescence microscopy exhibited that Dox-SS-Pyr, Dox-SH, and Dox-SS-[C(WR)4K] localized in the nucleus as shown in four cell lines, HT-1080, SKOV-3, MDA-MB-468, and MCF-7. Of note, Dox-SS-[C(WR)4K] was significantly less toxic in mouse myoblast cells compared to Dox at the same concentration. Further mechanistic study demonstrated that the level of intracellular reactive oxygen species (ROS) in myoblast cells exposed to Dox-SS-[C(WR)4K] was reduced in comparison of Dox when co-treated with FeCl2. These data indicate that Dox-SH, Dox-SS-Pyr, and Dox-SS-[C(WR)4K] have the potential to be further examined as Dox alternatives and anticancer agents.
Keywords: Anticancer, Cyclic peptide, Disulfide, Doxorubicin, Cellular uptake, Thiol, Cardiotoxicity
1. Introduction
Cancer remains among the leading causes of morbidity and mortality around the world. Approximately 17.5 million new cases and 8.7 million cancer-related deaths were reported in 2015. During the next two decades, the number of new cases is estimated to rise by 70% [1]. Consequently, there is an urgent need to synthesize new potent and less toxic chemotherapeutic agents.
One of the major limitations of cancer chemotherapy treatment is the development of tumor resistance to the conventional chemotherapy. Tumors generally develop chemoresistance to repeated treatment with one type of anticancer agent and then often become resistant to similar or completely different drugs, which is called multidrug resistance (MDR). There are two main classes of membrane transporter proteins which influence the pharmacokinetics of drugs in cells, adenosine triphosphate-binding cassette (ABC) and solute carrier (SLC) transporters, and their changes lead to MDR in tumors. In this regard, several hypotheses have been proposed to account for the phenomenon of drug resistance, including alteration of the target protein, decreased membrane permeability and drug metabolism, and/or efflux pumping [2–4].
Doxorubicin (Dox) has been one of the most effective anthracycline antibiotics with a broad anti-tumor spectrum since more than 50 years ago, as mono or in combination therapy for the treatment of a variety of tumors, including solid tumors, soft tissue sarcomas, and hematological malignancies [5–9]. Over the years, a variety of different mechanisms has been proposed for the cytostatic and cytotoxic of Dox activity. However, the mechanism of actions is uncertain and has long been the subject of considerable controversy [10]. The main mechanism is the inhibition of topoisomerase II, critical to DNA function, causing DNA damage by intercalation the DNA double helix [11,12]. Unfortunately, the use of Dox has been limited clinically, due to its pharmacokinetics properties, such as rapid distribution, excretion and low bioavailability, high volume of distribution, and short half-life [13–15]. Moreover, a high cumulative dose of Dox leads to dose-dependent side effects, such as cumulative cardiotoxicity, nephrotoxicity, and extravasation [16–18]. Furthermore, Dox is not used commonly for the treatment a number of tumors [19,20] due to efflux pumping, such as ovarian carcinoma cells, leading to extrusion of Dox, keeping intracellular drug concentration below a cell-killing limit [21] that could be related to the overexpression of energy-dependent drug efflux pump protein transporter, such as P-glycoprotein (P-gp).
Carrier-mediated drug delivery has emerged as a powerful methodology for optimizing cellular uptake, improving the efficacy, and reducing the cytotoxicity of anticancer drugs [22]. In this regard, drug delivery systems are viewed as one of the main pillars to overcome MDR, through P-gp inhibition consequently, blocking efflux pump [23–26]. The prodrug strategy, in which different substances can be attached to the molecule to modify its chemical structure, has gained much attention in Dox delivery [27–29]. Several conjugation methods have been used to improve Dox delivery including polymeric nanoparticles [30], polymeric micelles [31], liposomes [32], lipids [33], dendrimers [34], and peptides [35–37]. However, the release of the drug from the carrier is based primarily on reactions catalyzed by endogenous physiological factors such as reduction [38], low pH [39], and hydrolytic enzymes [40] and still presents a challenge. In this regard, researchers are trying to incorporate Glutathione (GSH)-responsive moieties to impart stimuli-responsive properties [41–43].
We have previously reported that cyclic peptides containing 4 arginine and 4 tryptophan residues can act as a cell-penetrating peptide and molecular transporter [44–46]. Cyclic [W(RW)4]-Dox conjugate containing an ester linker between the cyclic peptide-linker and Dox exhibited 3.6-fold higher cellular uptake when compared with Dox alone in ovarian cancer cells after 24 h incubation, and 99% of the conjugate was hydrolyzed to release Dox intracellularly after 72 h [47].
The objective of this study was to generate Dox thiol derivatives for improving the Dox biological profile, such as antiproliferative activity, cellular uptake, nuclear delivery, and retention. Herein, a cell-penetrating cyclic peptide composed of arginine (R) and tryptophan (W) was designed and conjugated with Dox via disulfide bridge to construct a smart drug delivery system Dox-SS-[C(WR)4K]. The conjugate was evaluated for the antiproliferative activity in different cancer cell lines, cellular uptake, and retention. We hypothesized that Dox conjugation through the disulfide bridge with a cell-penetrating cyclic peptide would be more efficient in antiproliferative activity against cancer cells that have a high concentration of glutathione activity compared to the normal cells [48]. The disulfide bridge is expected to be advantageous because they can be cleaved easily by disulfide exchange with intracellular thiol such as glutathione. Furthermore, we also evaluated the antiproliferative activity of Dox thiol derivatives, Dox-SH and Dox-SS-Pyridine (Dox-SS-Pyr), as potential Dox alternatives in comparison with Dox-SS-[C(WR)4K] and Dox.
2. Results and discussion
2.1. Chemistry
2.1.1. Synthesis of cyclic peptide [C(WR)4K]
Cyclic peptide was composed of positively-charged arginine (R) and hydrophobic tryptophan (W), in addition to β-Alanine (A), lysine (K) and cysteine (C) residues and was synthesized by using a solid phase methodology according to the previously reported procedure [49]. The thiol group in cysteine moiety was used for the conjugation to the thiol-containing anticancer drug. The peptide sequence was designed to have lysine and β-Ala moieties as the spacer to provide a free NH2 group with less steric hindrance.
As shown in Scheme 1, the linear protected peptide (Fmoc-R(Pbf = 2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl) C(Trt; trityl)R(Pbf)W(Boc = t-butoxycarbonyl)R(Pbf)W(Boc)K(β-A(Boc))W(Boc)R(Pbf)W(Boc)) was assembled on acid-labile chlorotrityl resin, followed by removing the last Fmoc (fluorenylmethyloxycarbonyl) group on the N-terminal by piperidine (20% v/v, DMF). The peptidyl-resin was washed, dried, and the resin was removed by a mild acidic cocktail cleavage (DCM/TFE/AcOH) [50] to afford the linear protected peptide with free amino and carboxylic groups. The peptide was cyclized using 1-hydroxy-7-azabenzotriazole (HOAt) and N,N'-diisopropylcarbodiimide (DIC) as coupling reagents under anhydrous conditions, followed by side chain deprotection to generate the targeted peptide[C(WR)4K]. The structure of the cyclic peptide was confirmed by MALDI-TOF/TOF mass spectrometry, showing a peak of 1675.4534 Da (Supporting Information), corresponding to [M+5H]+.
Scheme 1.
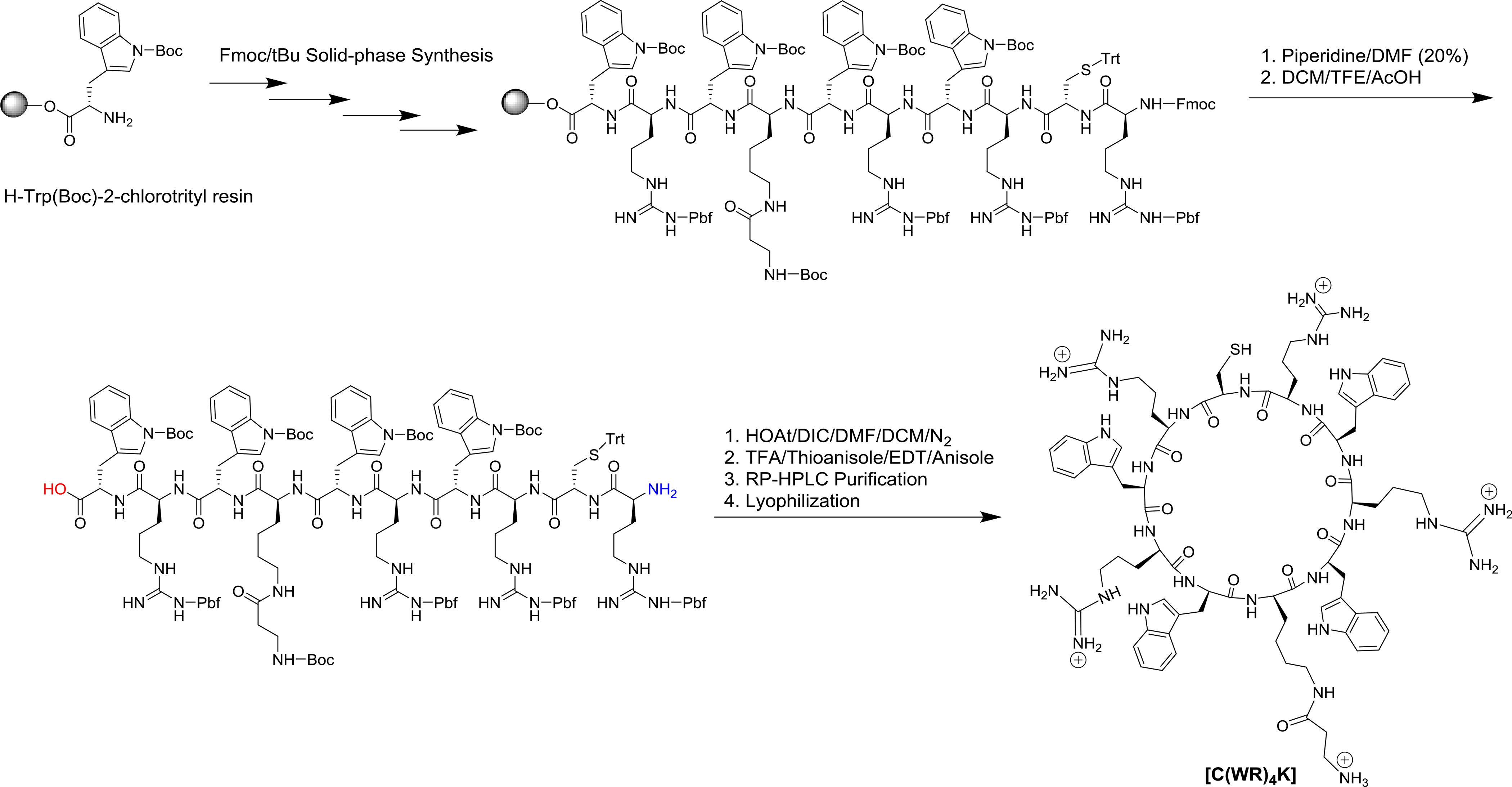
Synthesis of Cyclic Peptide [C(WR)4K].
2.1.2. Synthesis of Dox-SH, Dox-SS-Pyr
Moreover, Dox was modified by reacting with a bifunctional cross linking agent. The amine group of Dox was reacted with 2-iminothiolane hydrochloride (Trauťs reagent) under optimal conditions of basic medium to generate Dox-SH, containing an amidine with a free sulfhydryl group, which was activated by reacting with pyridyl disulfide (Pyr-SS-Pyr) (SS = disulfide) in acidic medium, affording the corresponding more reactive Dox-SS-Pyr containing a disulfide bridge with extrusion of pyridine-2-thione as depicted in Scheme 2. This type of chemical modification for Dox permitted the conjugation with the thiol group in the cysteine - of the cyclic peptide.
Scheme 2.
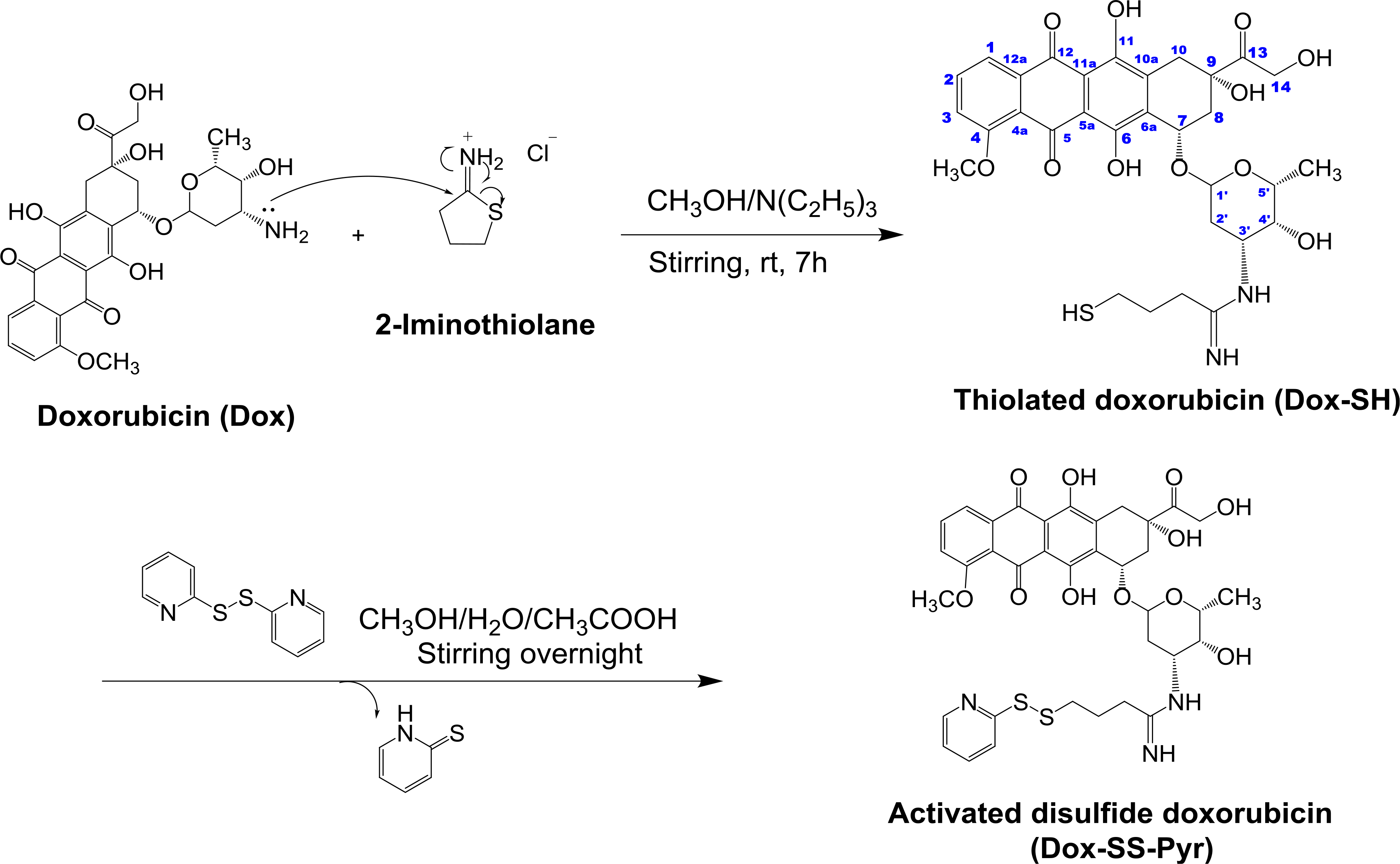
Synthesis of Dox-SH and Dox-SS-Pyr.
2.1.3. Synthesis of Dox-SS-[C(WR)4K]
Cyclic peptide [C(WR)4K] was conjugated with Dox-activated disulfide (Dox-SS-Pyr) via thiolate-disulfide interchange, using water as a polar protic solvent (Scheme 3). Mass spectra showed 2315.3719 Da corresponding to [M+2H]+, in addition to 1919.0574 (extrusion tetracyclic anthracycline aglycone) and 1672.0049 (the cysteinyl peptide) as fragments (Supporting Information, 1D) since disulfide peptide can be specifically fragmented at S-S bond during MALDI-MS as shown previously for other disulfide compounds [51]. The Dox-SS-[C(WR)4K] product was purified by reverse-phase preparative HPLC (RP-HPLC) on a C18 column and lyophilized. The purity of the product was determined by analytical HPLC (Supporting Information, 3D).
Scheme 3.
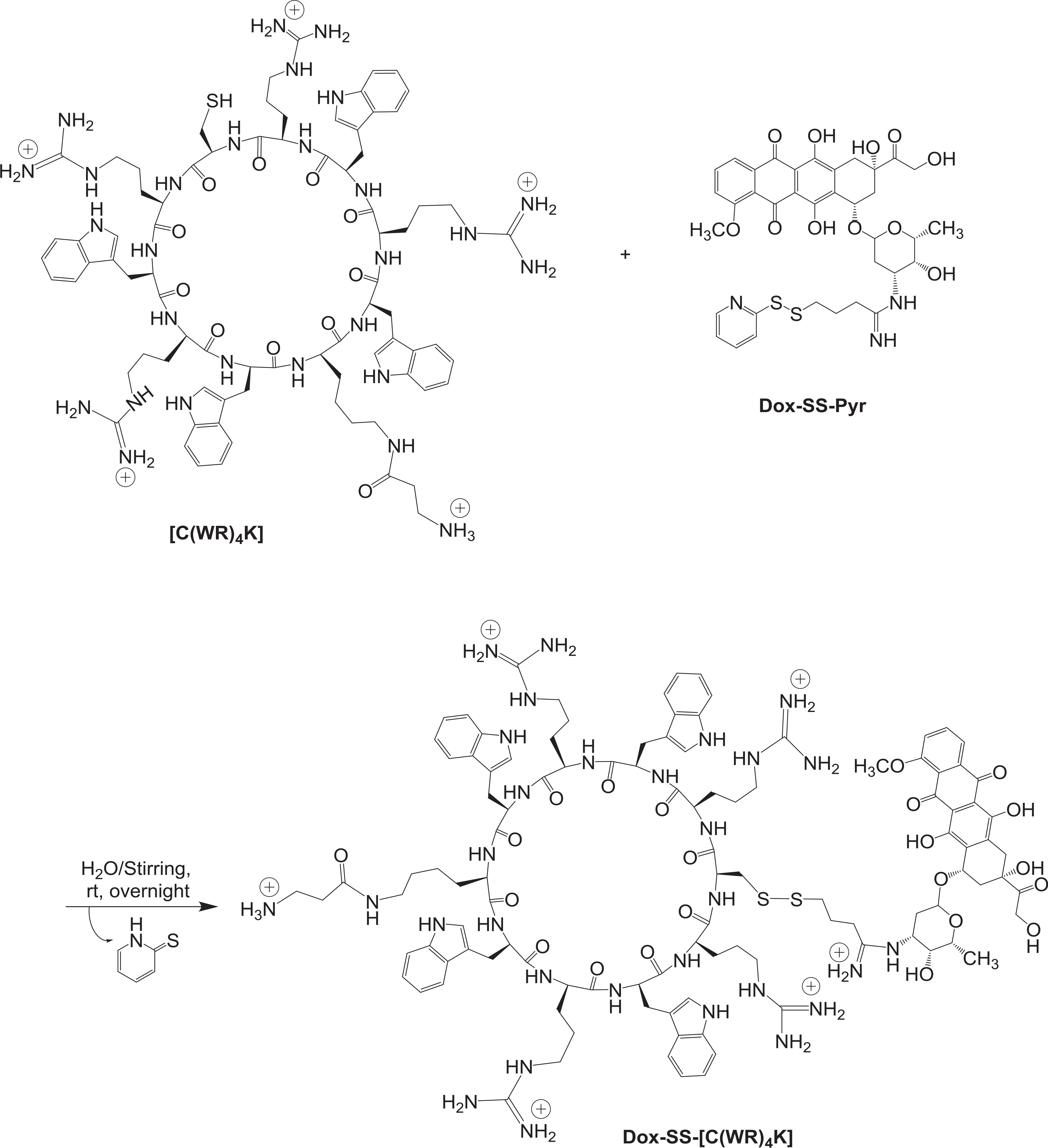
Synthesis Dox-SS-[C(WR)4K].
2.2. Biological activities
Dox is commonly used in the treatment of leukemia, multiple lymphoma, thyroid, ovarian, lung, and breast cancers. Thus, antiproliferative activity of Dox-SH, Dox-SS-Pyr, and the conjugate were compared with Dox at a concentration of 5 μM in human leukemia cell line (CCRF-CEM, ATCC No. CCL-119) [6], human ovarian cancer cells (SKOV-3, ATCC No. HTB-77), human fibrosarcoma cells (HT-1080, ATCC No. CRL 12012) [7,52], and human embryonic kidney cells (HEK-293, ATCC No. CRL-1573) [53,54] (Figs. 1 and 2). We chose different cancer cell lines (e.g. CCRF-CEM, SKOV-3, HT-1080) and normal human embryonic kidney cells (HEK-293) to test both cytotoxicity and antiproliferative activity of synthesized compounds versus Dox.
Fig. 1.
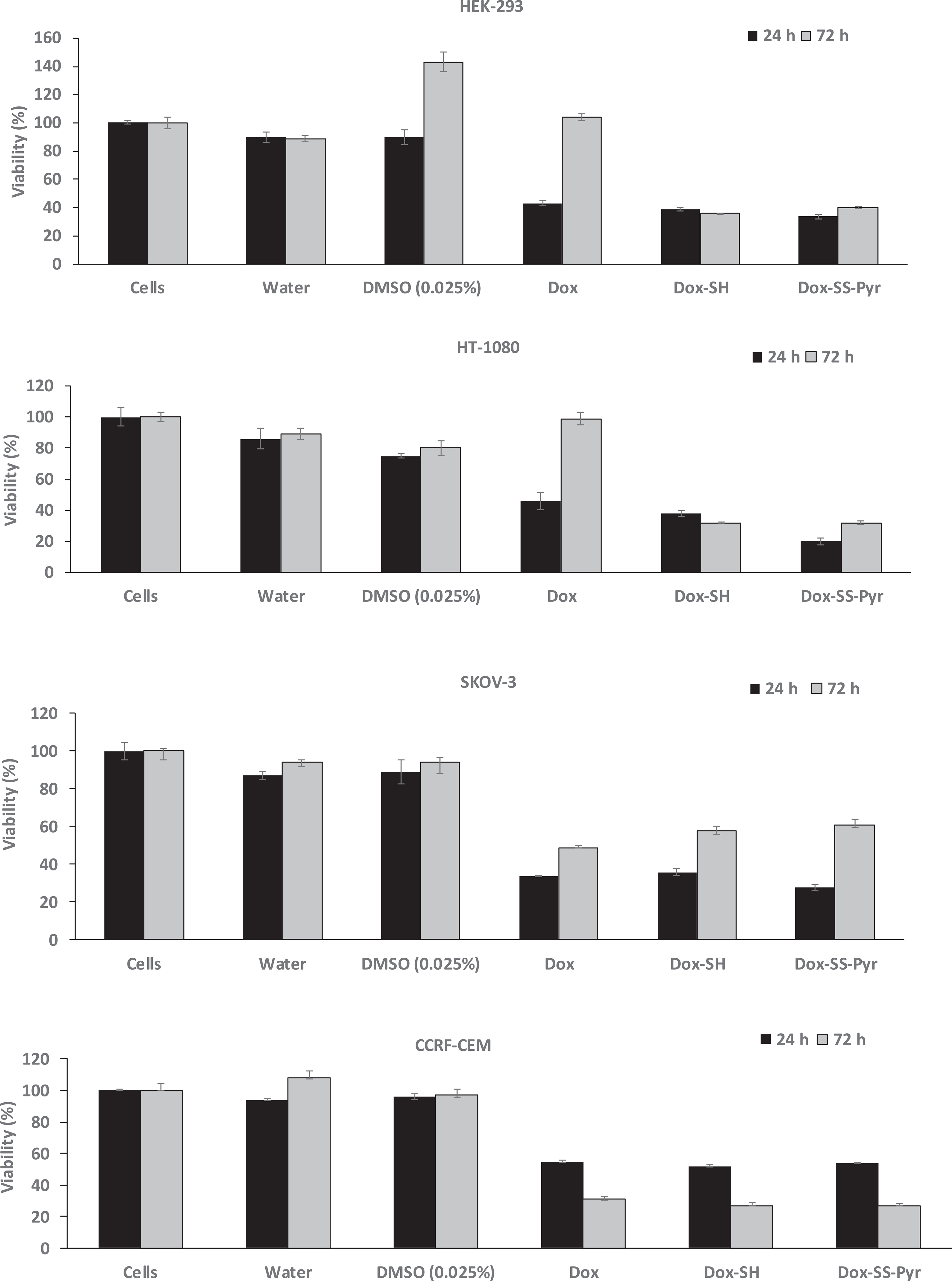
Inhibition of cells by Dox-SH (5 μM), Dox-SS-Pyr (5 μM), and Dox (5 μM) after 24 h and 72 h incubation. The results are shown as the percentage of the control cells that have no compound (set at 100%). All the experiments were performed in triplicate (±SD).
Fig. 2.
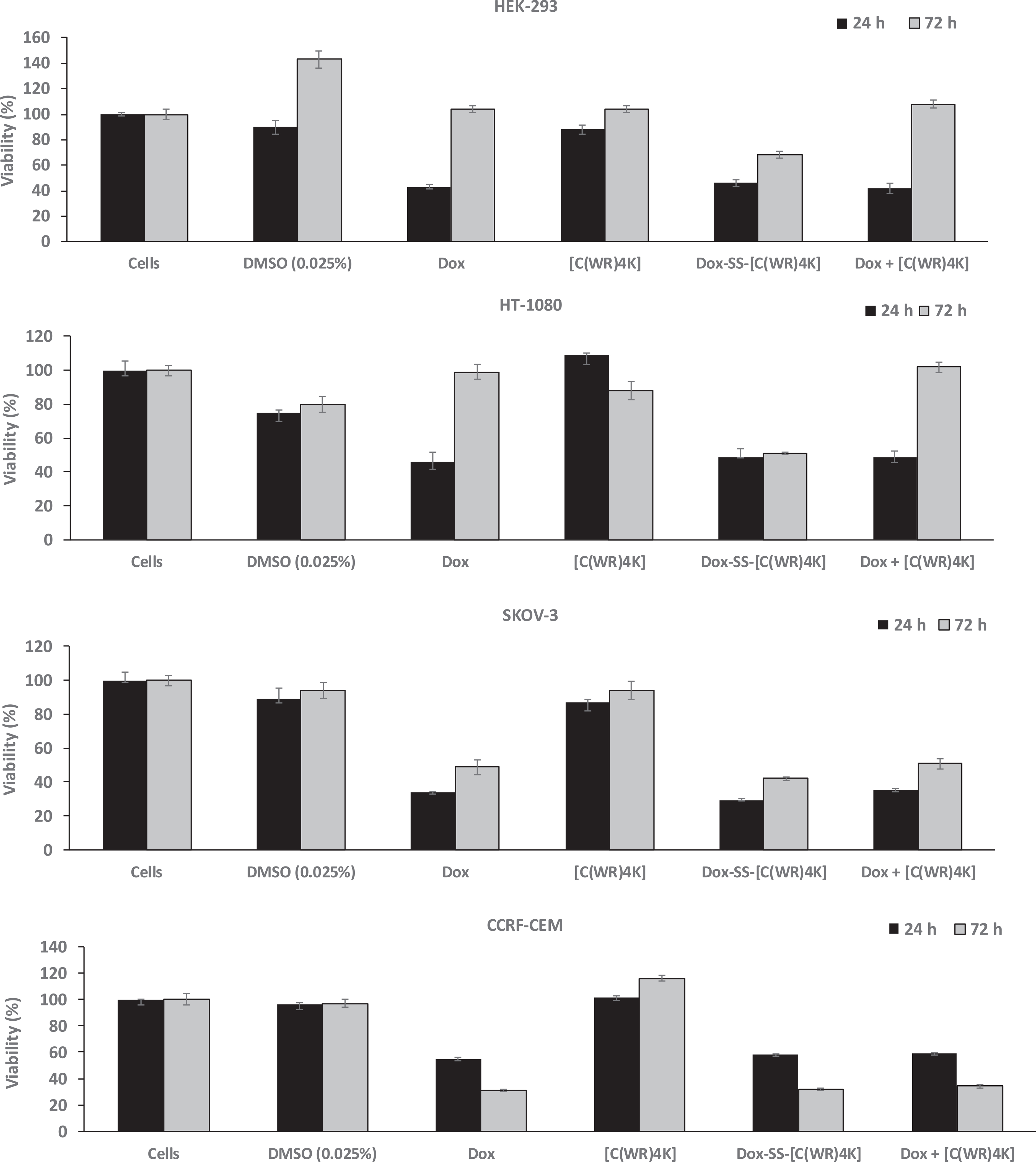
Inhibition of cells by Dox-SS-[C(WR)4K] (5 μM), a physical mixture of Dox, and [C(WR)4K] (1:1 ratio based on M.W., and Dox (5 μM) after 24 h and 72 h incubation. The results are shown as the percentage of the control cells that have no compound (set at 100%). All the experiments were performed in triplicate (±SD).
Fig. 1 shows the antiproliferative activity of Dox versus Dox-SS-Pyr and Dox-SH. Dox-SS-Pyr inhibited the cell proliferation of HEK-293 (66%), HT-1080 (80%), and SKOV-3 (72%), and CCRF-CEM cells (46%) after 24 h incubation while Dox alone exhibited inhibition the cell proliferation of HEK-293 (57%), HT-1080 (66%), and SKOV-3 (76%), and CCRF-CEM cells (45%). Thus, Dox-SS-Pyr showed significantly higher cytotoxicity than Dox in HEK-293 and HT-1080 when compared with Dox while showed comparable activity against and SKOV-3 cells CCRF-CEM cells after 24 h incubation.
After 72 h incubation, Dox-SS-Pyr inhibited the cell proliferation of HEK-293 (60%), HT-1080 (68%), and SKOV-3 (39%), and CCRF-CEM cells (73%). Dox alone did not exhibit inhibition the cell proliferation of HEK-293 and HT-1080, while inhibited the proliferation of SKOV-3 (51%) and CCRF-CEM cells (69%). Thus, Dox-SS-Pyr was more cytotoxic than Dox in all cell lines except SKOV-3 after 72 h incubation when compared with Dox. The antiproliferative activities of the compounds in HEK-293, HT-1080, and SKOV-3 were found to be less after 72 h, possibly due to the efflux mechanism. However, this effect was more obvious in HEK-293 and HT-1080 where Dox did not show any significant cytotoxicity. Thus, Dox-SS-Pyr enhances the intracellular retention of Dox in HEK-293 and HT-1080 cells while Dox had no effect.
Dox-SH inhibited the cell proliferation of HEK-293 (61% and 64%), HT-1080 (62% and 68%), and SKOV-3 (64% and 42%), and CCRF-CEM cells (48% and 73%) after 24 h and 72 h incubation, respectively, suggesting slightly higher inhibition and retention in all cells in longer incubation time except SKOV-3 cells. Furthermore, Dox-SH was found to be more cytotoxic in all cell lines except SKOV-3 cells when compared with Dox alone after 24 h and 72 h incubation. Dox-SH was less cytotoxic in HEK-293, HT-1080, and SKOV-3 cells when compared with Dox-SS-Pyr after 24 h incubation, but showed comparable activity after 72 h incubation. These data indicate Dox-SH and Dox-SS-Pyr have significantly higher or comparable antiproliferative activity when compared to Dox in HEK-293, HT-1080, and CCRF-CEM cells after 24 h and 72 incubation, presumably because of higher activity and retention of the compounds in these cells. We speculate Dox-SS-Pyr hydrolyzes and releases Dox-SH slowly in the presence of intracellular thiol groups, such as glutathione. Hence, Dox-SH and Dox-SS-Pyr have comparable activity after 72 h.
Fig. 2 shows the antiproliferative activity of the cyclic peptide [C(WR)4K], Dox-SS-[C(WR)4K], and the physical mixture of Dox and [C(WR)4K] versus Dox. In general, cyclic peptide [C(WR)4K] alone did not exhibit any significant cytotoxicity against HT-1080 and CCRF-CEM cells after 24 h incubation and against HEK-293 and CCRF-CEM cells after 72 h incubation. The compound inhibited the proliferation of HEK-293 (12%) and SKOV-3 cells (13%) after 24 h incubation and HT-1080 (12%) and SKOV-3 (6%) after 72 h incubation. Thus, the contribution of the cyclic peptide in overall cytotoxicity in the conjugate is minimal. While Dox-SS-[C(WR)4K] exhibited slightly higher inhibition of proliferation in SKOV-3 cells when compared with Dox (71% versus 66%) after 24 h incubation, comparable cytotoxicity was observed when two compounds were compared in HEK-293, HT-1080, and CCRF-CEM cells. The physical mixture of Dox and [C(WR)4K] also exhibited comparable cytotoxicity with Dox in all cell lines after 24 h incubation.
On the other hand, Dox-SS-[C(WR)4K] inhibited the proliferation of HEK-293 (32%), HT-1080 (49%), SKOV-3 (58%), and CCRF-CEM cells (68%) after 72 h incubation while Dox alone did not inhibit the cell proliferation of HEK-293 and HT-1080 cells and exhibited antiproliferative activity in SKOV-3 (51%) and CCRF-CEM cells (69%). Thus, Dox-SS-[C(WR)4K] showed comparatively higher cytotoxicity in HEK-293, HT-1080, and SKOV-3 cells when compared with Dox after 72 h incubation, suggesting that the conjugate enhanced the retention of Dox in these cells presumably by slowly releasing Dox and reducing the efflux.
The physical mixture of Dox and [C(WR)4K] was found to show no inhibition in HEK-293 and HT-1080 cells after 72 h incubation similar to Dox but showed comparable activity with Dox in SKOV-3 and CCRF-CEM cells. These data indicate that the physical mixture has no advantage compared to Dox, and the cytotoxicity of the physical mixture is due to the presence of free Dox. Furthermore, the cyclic peptide does not improve the delivery or retention of Dox when used in the physical mixture form with Dox. On the other hand, Dox-SS-[C(WR)4K] is more cytotoxic than Dox in HEK-293, HT-1080, and SKOV-3 cells after 72 h incubation, suggesting the potential of the conjugate to improve the cytotoxicity and retention of Dox.
Comparison of Dox-SS-[C(WR)4K] with Dox-SS-Pyr (Figs. 1 and 2) revealed that the conjugate was significantly less cytotoxic than Dox-SS-Pyr in HEK-293 (54% versus 66%), HT-1080 (51% versus 80%) and showed comparable activity against SKOV-3 (71% versus 72%), and CCRF-CEM (42% versus 46%), respectively, after 24 h incubation. Similarly the Dox-SS-[C(WR)4K] was less cytotoxic than Dox-SS-Pyr in HEK-293 (32% versus 60%), HT-1080 (49% versus 68%), and CCRF-CEM (68% versus 73%) cells after 72 h incubation, but was more cytotoxic against SKOV-3 (58% versus 39%), respectively. These data suggest that Dox-SS-Pyr has higher antiproliferative activity than the conjugate in all cell lines after 24 h incubation and in HEK-293, HT-1080, and CCRF-CEM cells after 72 h incubation.
Flow cytometry was used to determine the cellular uptake of the conjugate Dox-SS-[C(WR)4K] versus Dox-SS-Pyr in HEK-293, HT-1080, SKOV-3, and CCRF-CEM cells (Fig. 3). Cellular uptake studies indicated that Dox-SS-Pyr had higher cellular uptake in HT-1080, HEK-293, and CCRF-CEM cells when compared with Dox-SS-[C(WR)4K]. Dox and Dox-SS-[C(WR)4K] showed a similar pattern and had higher cellular uptake in SKOV-3 and HT-1080 cells.
Fig. 3.
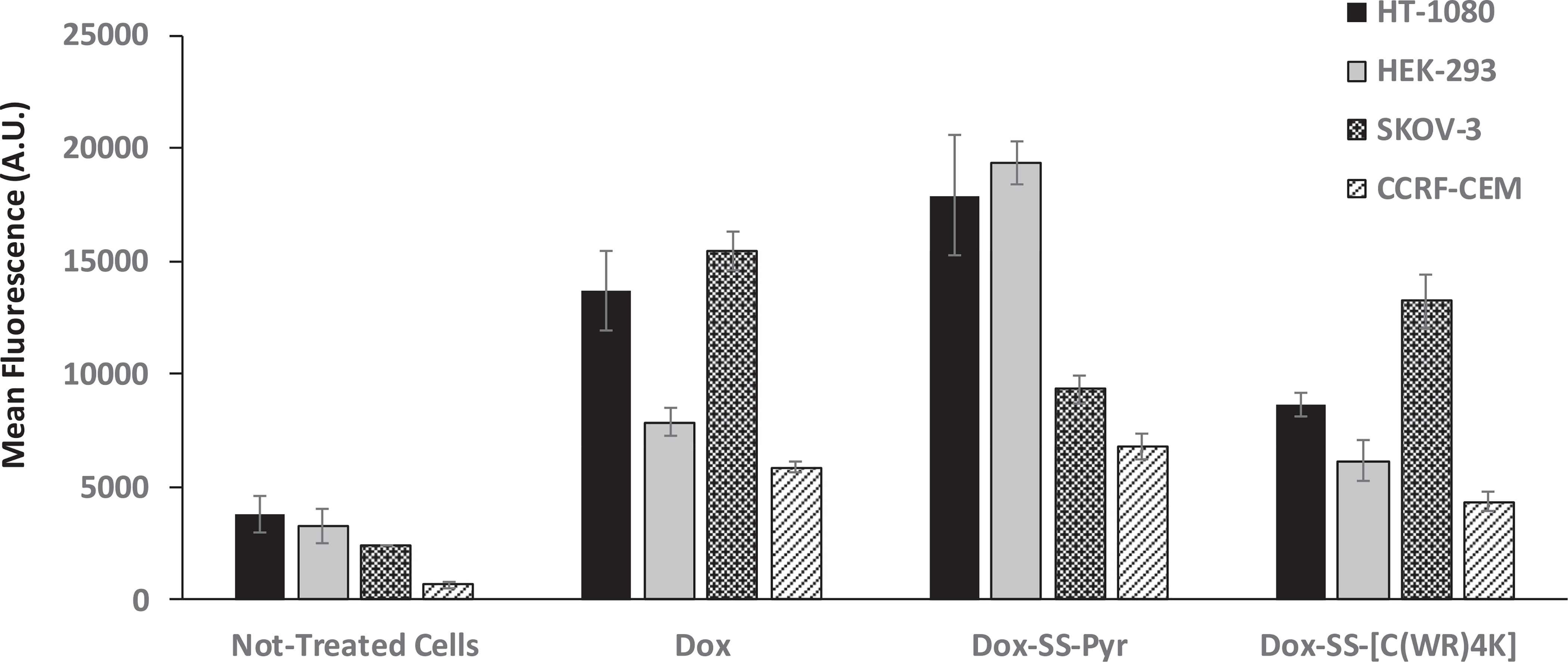
Flow cytometry analysis (FACS) analysis of cellular uptake assays of Dox (5 μM), Dox-SS-Pyr (50 μM) and Dox-SS-[C(WR)4K] (50 μM) in HT-1080, HEK-293, SKOV-3, and CCRF-CEM cells after 3 h of incubation (mean ± SD).
Based on the cytotoxicity and flow cytometry data Dox-SH, Dox-SS-Pyr, and Dox-SS-[C(WR)4K] were selected for fluorescence microscopy studies. Fluorescence microscopy was used to evaluate the cellular uptake and cellular localization in HT-1080, SKOV-3, human breast adenocarcinoma (MDA-MB-468, ATCC No. HTB-132), and human breast adenocarcinoma cell (MCF-7, ATCC No. HTB-22). Figs. 4 and 5, and S1 show localization of all the compounds after 1 h and 24 h in the nucleus in HT-1080, MDA-MB-468, and MCF-7 cells. Dox-SH and Dox-SS-Pyr exhibited much higher intensity when compared with Dox as shown in Figs. 4 and 5, and Figures S1 and S2 (Supporting Information).
Fig. 4.
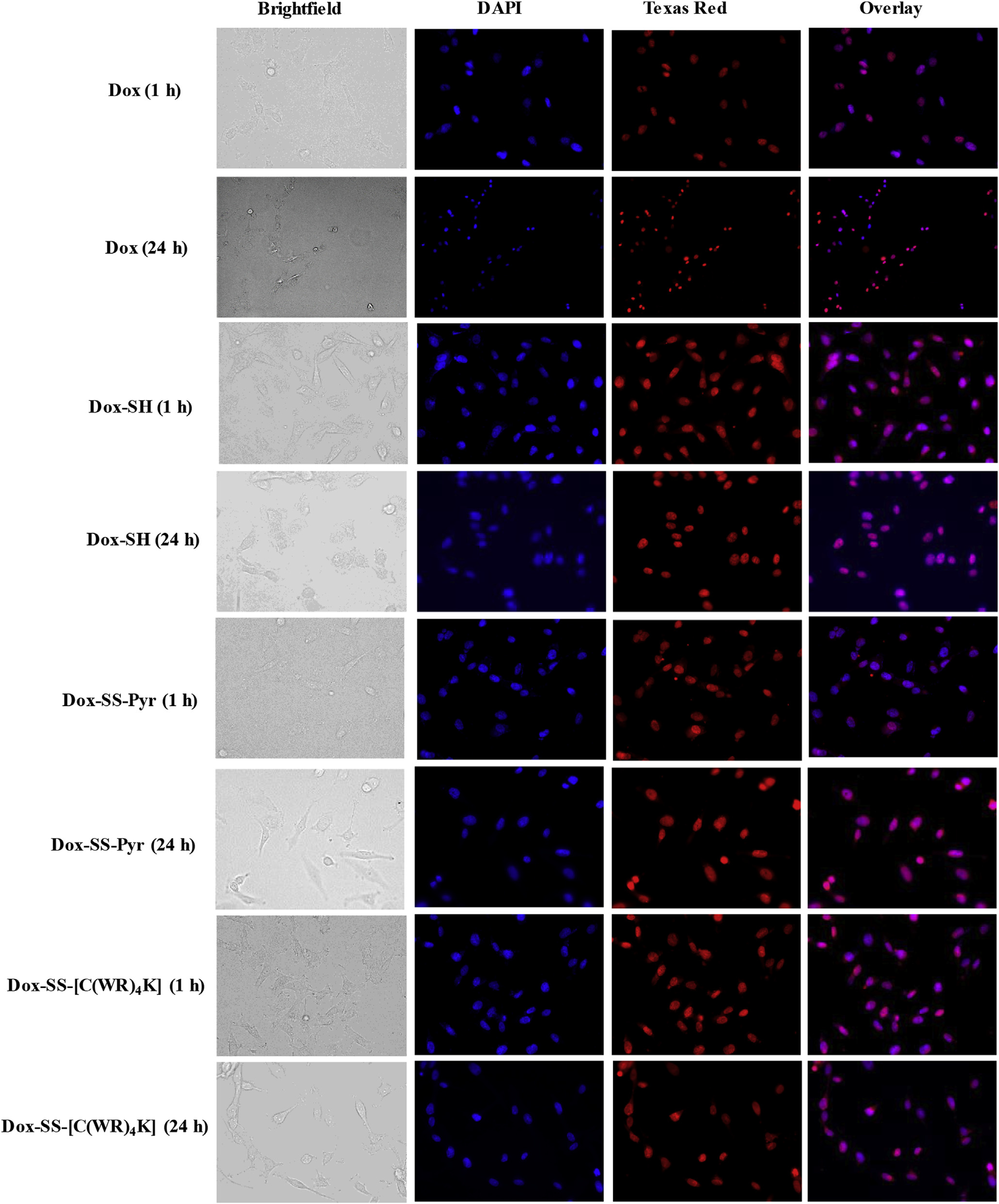
Fluorescence microscopy images of Dox (5 μM), Dox-SH (5 μM), Dox-SS-Pyr (5 μM), and Dox-SS-[C(WR)4K] (5 μM) uptake in HT-1080 cells after 1 h and 24 h. Cells treated with the compound for 1 h or 24 h. Red represents the fluorescence of Dox. To identify the nucleus, the cells were dyed with DAPI.
Fig. 5.

Fluorescence microscopy images of Dox (5 μM), Dox-SH (5 μM), Dox-SS-Pyr (5 μM), and Dox-SS-[C(WR)4K] (5 μM) uptake in MDA-MB-468 cells after 1 h and 24 h. Cells treated with the compound for 1 h or 24 h. Red represents the fluorescence of Dox. To identify the nucleus, the cells were dyed with DAPI.
Dox-SS-[C(WR)4K] and Dox was localized in the nucleus after 1 h and 24 h in SKOV-3 cells while Dox-SH and Dox-SS-Pyr were distributed mostly outside the nucleus after 1 h. After 24 h, all the compounds were mostly localized in the nucleus (Figure S2), suggesting the slow nuclear localization of Dox-SH and Dox-SS-Pyr in SKOV-3 cells. These data indicate that Dox-SH, Dox-SS-Pyr and Dox-SS-[C(WR)4K] can be localized in the nucleus that allows DNA targeting by potentially released Dox or the intact compound.
2.3. Cytotoxicity in mouse myoblast cells
The clinical use of Dox is greatly limited by the risk of developing severe cardiomyopathy and congestive heart failure. In order to investigate the potential cardiotoxicity of Dox-SS-[C(WR)4K] in comparison to Dox, a mouse myoblast cell line (C2C12, ATCC No. CRL 1772) was used in the study. Cells were maintained in DMEM (ATCC No. 30–2002) containing 10% fetal bovine serum at 37 °C. After incubation with Dox or Dox-SS-[C(WR)4K] for 72 h, cell viability was determined by MTT colorimetric analysis as described before [55].
As shown in Fig. 6, at m5M concentration, Dox treatment significantly reduced the cell viability to 28.6% of control, while in cells exposed to Dox-SS-[C(WR)4K], the cell survival rate was significantly increased (71% of control, p < 0.05 compared to Dox alone), indicating that Dox-SS-[C(WR)4K] may exhibit less cardiotoxicity in comparison to Dox.
Fig. 6.
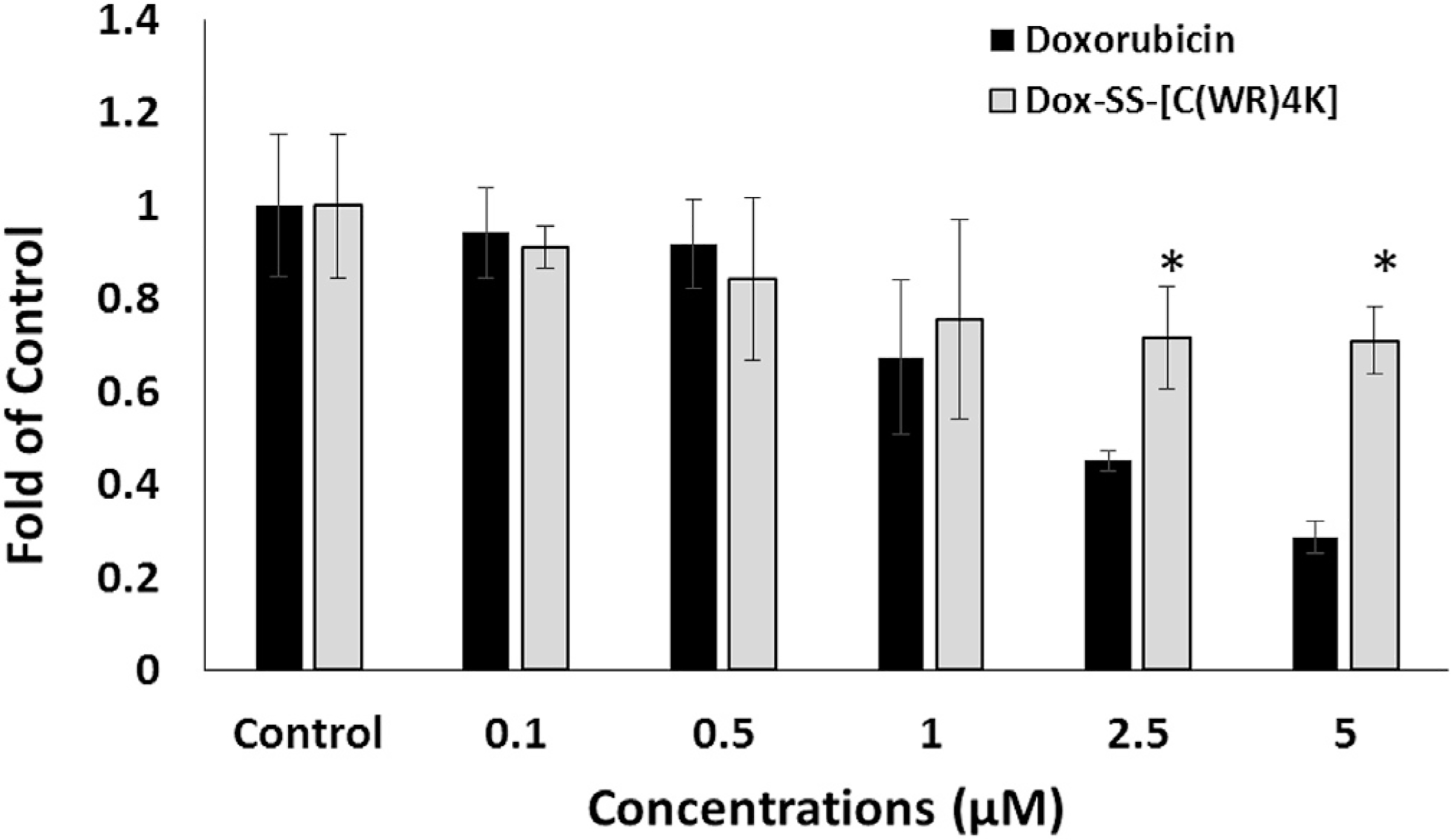
Dox-SS-[C(WR)4K] in mouse myoblast cells after 72 h incubation compared to that of Dox. The results are shown as the folds of the control cells that have no compound (set at 100%). All the experiments were performed in triplicate (mean ± SD). *, p < 0.05 compared to doxorubicin.
Oxidative stress has been well documented as the primary cause of anthracycline-induced cardiotoxicity (AIC) [56], which is exacerbated by iron overload [56,57]. To further determine the effect of Dox-SS-[C(WR)4K] on reactive oxygen spices (ROS), we conducted flow cytometry analysis using a cell-permeant fluorogenic reagent 2',7'-dichlorodihydrofluorescein diacetate (H2DCFDA) (ThermoFisher Scientific, Cat. No. D399), which can detect intracellular reactive oxygen intermediates.
Cultured C2C12 cells in a 6 well plate were treated with Fe2+ (10 μM) in the absence or presence of Dox or Dox-SS-[C(WR)4K] (0.5 μM) for 72 h. Once the incubation period was complete, cells were collected and stained with H2DCFDA (10 μM) to detect intracellular ROS levels [55].
As shown in Fig. 7, FeCl2 treatment increased the intracellular ROS levels to 2.5-fold of control, while the levels of ROS in cells exposed to Dox or Dox-SS-[C(WR)4K] alone at low concentration (0.5 mM) were not elevated in comparison to that of control cells (1.09-fold and 1.05-fold of control respectively). Notably, the coincubation of Dox with Fe2+ increased ROS levels higher to 3.48-folds of control; however, induction in Fe2+-treated cells was not observed in the presence of Dox-SS-[C(WR)4K]. The distinct pattern of effects on ROS generation between Dox and Dox-SS-[C(WR)4K] may explain, at least partially, the reduced cytotoxicity of Dox-SS-[C(WR)4K] in C2C12 cells. Further study is warranted to define the underlying mechanisms.
Fig. 7.
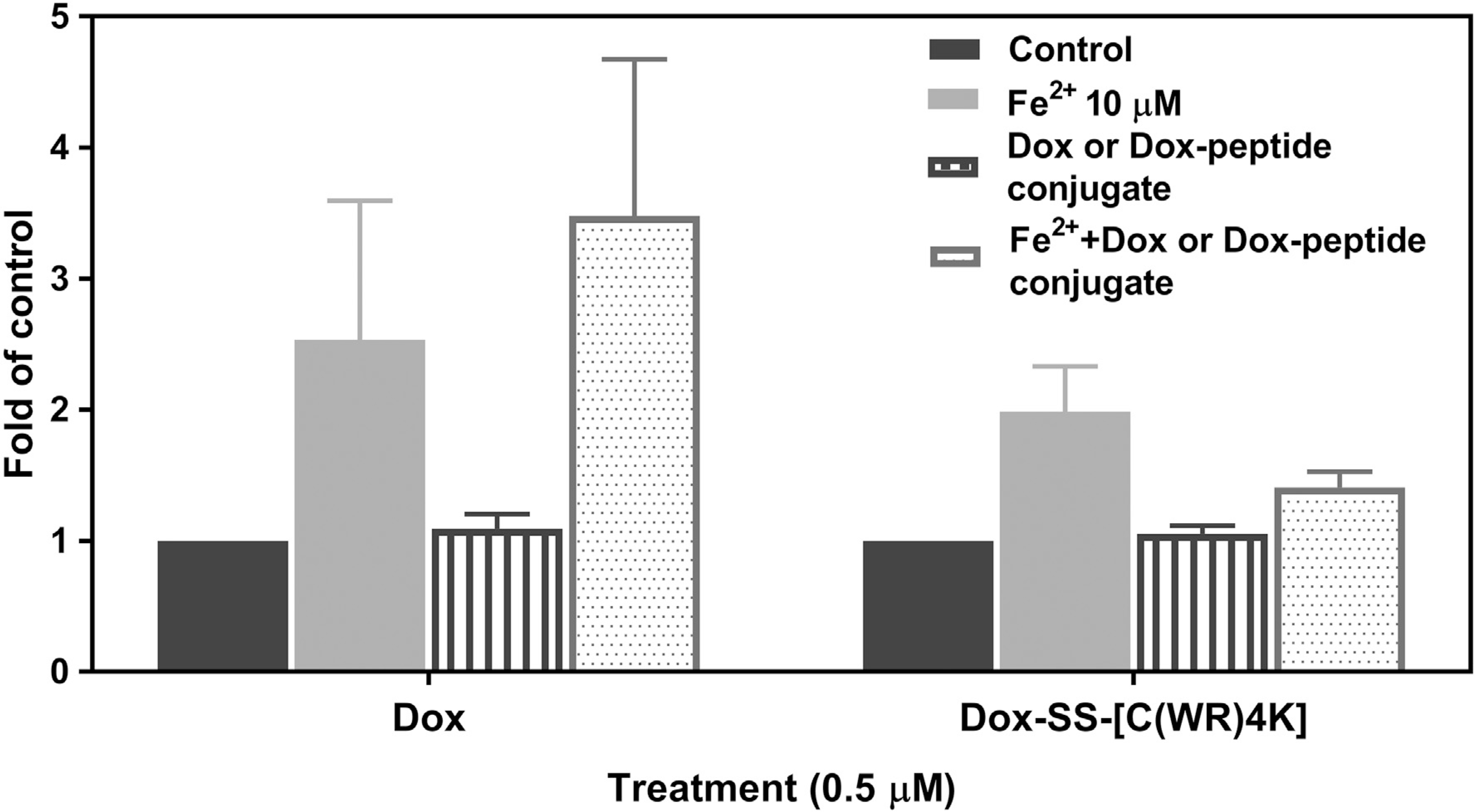
Flow cytometry analysis (FACS) of intracellular ROS levels using H2DCFDA staining in C2C12 cells. Cells were treated with Fe2+ (10 μM) in the absence or presence of Dox or Dox-SS-[C(WR)4K] (0.5 μM) for 72 h before staining with H2DCFDA (10 μM). Data is represented as the fold of control and each experiment was repeated two times.
3. Conclusions
Dox thiol conjugates, thiolated doxorubicin (Dox-SH), thiol-reactive Dox-SS-Pyr, and a Dox-SS-cell-penetrating cyclic peptide, Dox-SS-[C(WR)4K] were synthesized and purified by using RP-HPLC. Dox-SS-Pyr showed significantly higher antiproliferative activity than Dox in HEK-293, HT-1080, and SKOV-3 cells when compared with Dox which showed comparable activity against CCRF-CEM cells after 24 h incubation. Dox-SS-[C(WR)4K] was found to be more cytotoxic in HEK-293, HT-1080, and SKOV-3 cells when compared with Dox after 72 h incubation. Dox-SS-Pyr exhibited higher cellular uptake in HT-1080, HEK-293, and CCRF-CEM cells as shown by flow cytometry when compared with Dox-SS-[C(WR)4K]. Dox-SS-Pyr, Dox-SH, and Dox-SS-[C(WR)4K] showed high cellular uptake and were localized mostly in the nucleus as shown consistently in four different cancer cell lines by fluorescence microscopy studies. Our study further demonstrated that Dox-SS-[C(WR)4K] exhibited less cytotoxicity in mouse myoblast cells in comparison to Dox. Intracellular ROS levels were elevated by the co-treatment of Dox and Fe2+, but such induction was not evident in cells exposed to Dox-SS-[C(WR)4K] and Fe2+. These data indicate that these compounds can be considered as Dox alternatives and anticancer agents, which may exhibit less cardiotoxicity in comparison to Dox.
4. Experimental section
4.1. Materials and methods
The resin, Fmoc-protected amino acid building blocks, and chemical reagents were purchased from AAPPTec. All solvents used for peptide synthesis or HPLC solvents were obtained from Sigma-Aldrich, USA and used without further purification. Peptide purification was carried out using Shimadzu RP-HPLC system, C18 column (19 cm × 250 mm), and the purity was confirmed by analytical HPLC, using water (0.1% TFA) as eluent A and acetonitrile (0.1% TFA) as eluent B. NMR spectra were recorded on a Bruker Avance III HDTM 400 NMR spectrometer using DMSO-d6 as the solvent and TMS as internal reference. Mass spectra were obtained by a Bruker Impact 11, UHR-qTOF and a Bruker Daltonics Autoflex MALDI-TOF, with α-cyano-4-hydroxycinnamic Acid (CHCA) as the matrix (15 mg/mL).
4.2. Chemistry
4.2.1. Synthesis cyclic peptide [C(WR)4K]
Cyclic peptide [C(WR)4K] was synthesized by Fmoc/tBu solid-phase peptide synthesis strategy. The coupling and activating reagents were N,N,N',N'-tetramethyl-O-(1H-benzotriazol-1-yl)uro-niumhexafluorophosphate (HBTU) and N,N-diisopropylethylamine (DIPEA), respectively, in anhydrous N,N-dimethylformamide (DMF). Fmoc was deprotected using piperidine in DMF (20% v/v). The H-Trp(Boc)-2-chlorotrityl resin (811 mg, 0.30 mmol, 0.37 mmol/g) was swelled in DMF (10 mL) for 10 min with shaking and mixing using N2. The excess of DMF was filtered off. The swelling and filtration steps were repeated two times before the coupling reactions. The solvent was drained, followed by coupling with Fmoc-Arg(Pbf)-OH (583 mg, 0.90 mmol, 3 equiv.) using HBTU (341 mg, 0.90 mmol, 3 equiv.) and DIPEA (313 μL, 1.80 mmol, 6 equiv.) in anhydrous DMF (5 mL) for 1 h. The resin was washed three times using DMF (3 × 10 mL), followed by deprotection of Fmoc group by previously prepared piperidine solution in DMF (20 mL, 20% v/v). The resin was washed with DMF three times (3 × 10 mL) to be ready for the next coupling. The subsequent amino acids, Fmoc-Trp(Boc)-OH (473 mg, 0.90 mmol, 3 equiv.), Dde-Lys(Fmoc)-OH (479 mg, 0.90 mmol, 3 equiv.), Fmoc-Trp(Boc)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Cys(Trt)-OH (527 mg, 0.90 mmol) and Fmoc-Arg(Pbf)-OH were coupled in the same manner. The Dde (1-(4,4-dimethyl-2,6-dioxocyclohex-1-ylidene)ethyl) group of Dde-Lys (Fmoc)-OH was removed using hydrazine hydrate in DMF (2% v/v, 20 mL, 2 × 5 min) before coupling with Boc-β-Ala-OH (170 mg, 0.90 mmol, 3 equiv.). The deprotection of Fmoc groups was achieved with the piperidine solution. The side-chain protected peptide was cleaved from the resin by shaking the resin with a mixture of dichloromethane (DCM)/2,2,2-trifluoroethanol (TFE)/acetic acid (AcOH) (50 mL, 7:2:1, v/v/v) for 1 h. The resin was filtered off, and the solution was evaporated under vacuum, using hexane to remove acetic acid azeotropically. The residue was dried overnight under vacuum. The crude linear protected peptide was used directly for cyclization. The crude peptide was dissolved in a diluted solution of DMF/DCM (240 mL, 4:1 v/v) under inert gas, using HOAt (122 mg, 0.90 mmol) and DIC (139 μl, 0.90 mmol) as coupling reagents. The mixture was stirred overnight at rt. After cyclization, the solvent was evaporated and cleavage of the side-chain protecting groups was carried out by using trifluoroacetic acid (TFA)/thioanisole/1,2-ethanedithiol (EDT)/anisole (90:5:3:2, v/v/v/v, 10 mL) for 2 h. The crude deprotected cyclic peptide was precipitated with addition of cold diethyl ether treatment followed by centrifugation, providing the targeted cyclic peptide, which was purified by using RP-HPLC (reversed-phase high-performance liquid chromatography). The fraction was evaporated and lyophilized to afford the powdered cyclic peptide. MALDI-TOF (m/z) [C80H110N28O11S]: calcd, 1670.8630; found, 1675.4534 [M + 5H]+.
4.2.2. Synthesis thiolated doxorubicin (Dox-SH)
Dox (50 mg, 0.09 mmol) and 2-iminothiolane hydrochloride (12 mg, 0.09 mmol) were dissolved in methanol (40 mL) followed by addition of a catalytic amount of triethylamine (TEA) and stirred at rt for 7 h. After evaporation of the solution, the crude product was washed multiple times with diethyl ether affording 45 mg (70%) of Dox-SH. 1H NMR (DMSO-d6 + D2O, 400 MHz, δ ppm): 7.90–7.83 (m, 2H, ArC1H, ArC2H), 7.62–7.60 (m, 1H, ArC3H), 5.26 (t, J = 12.0 Hz, 1H, H-1'), 4.95–4.86 (m, 1H, H-14), 4.50–4.59 (m, 3H, H-14 and H-7), 4.15 (d, J = 8.0 Hz, 1H, H-50), 3.95 (d, J = 12.0 Hz, 3H, OCH3), 3.55–3.50 (m, 1H, H-4'), 3.39–3.30 (m, 1H, H-3'), 2.80–2.96 (m, 2H, H-10), 244–2.32 (m, 2H, CH2SH), 2.17–1.42 (m, 8H, CH2 (H-8), HSCH2CH2, CH2C=NH, CH2 (H-2'), 1.16–1.10 (br S, 3H, CH3); 13CNMR (DMSO-d6 + D2O, 100 MHz, δ ppm): 214.39 (C13 = O), 187.07 (C12 = O),186.52 (C5=O), 166.53 (C-4), 161.33 (C=NH), 156.16 (C-6), 154..62 (C-11), 137.13 (C-6a), 135.40 (C-10a), 135.06 (C-12a), 134.51 (C-2), 120.33 (C-4a), 120.23 (C-1), 119.76 (C-3), 111.30 (C-5a), 110.93 (C-11a), 99.81 (C-1'), 75.73 (C-9), 75.37 (C-7), 70.35 (C-4'), 66.75 (C-5'), 64.25 (C-14), 57.16 (C4-OCH3), 47.06 (C3'), 36.77 (C-8), 32.57 (C-10), 32.46 (C-2'), 28.56 (CH2C(NH) = NH), 27.53 (CH2SH), 26.82 (CH2CH2SH), 17.21 (C-5'-CH3); HR-MS (ESI-qTOF) (m/z) [C31H36N2O11S]: calcd, 644.2040; found 645.4678 [M+H]+.
4.2.3. Synthesis of pyridyl doxorubicin (Dox-SS-Pyr)
Thiolated Dox (100 mg, 0.15 mmol) and 2,2'-dithiodipyridine (33 mg, 0.15 mmol) were dissolved in methanol (40 mL) followed by adding a catalytic amount of acetic acid (AcOH). The mixture was stirred overnight at room temperature. After evaporation of the solution, the precipitated product was crystallized from diethyl ether, affording 85 mg (73%) of Dox-SS-Pyr. 1H NMR (DMSO-d6, 400 MHz, δ ppm): 14.08 (s, 1H, OH Phenolic C-6), 13.28 (s, 1H, OH Phenolic C-11), 8.47–7.28 (m, 9H, NH and ArH), 5.37–5.31 (m, 2H, CH2OH), 5.04–5.01 (m, 2H, OH Sugar and OH cyclohexane), 4.61–4.55 (m, 1H, OH, CH2OH), 3.98 (s, 3H, OCH3), 1.61–1.57 (m, 17H, CH2 and CH of sugar, cyclohexane and thiolane), 1.14 (d, J = 6.8 Hz, 3H, CH3); 13C NMR (DMSO-d6, 100 MHz, δ ppm): 207.80 (C13 = O),186.90 (C12 = O), 185.70 (C5=O), 165.09 (C-4), 161.09 (C=NH), 158.90, 157.33, 154.39, 149.72, 149.61, 138.17, 137.82, 136.38, 134.20, 121.82, 121.21, 119.55, 119.28, 112.83, 110.63, 110.43 (16 Aromatic carbons), 99.26, 74.87, 68.93, 64.30, 63.06, 48.66, 38.88, 36.60, 32.56, 31.29, 28.20, 26.43 (12C, CH2 and CH of sugar, cyclohexane and thiolane), 63.06 (CH2OH), 56.60 (OCH3), 16.89 (CH3); HR-MS (ESI-qTOF) (m/z) [C36H39N3O11S2]: calcd, 753.2026; found 754.4053 [M+H]+.
4.2.4. Synthesis of cyclic peptide attached doxorubicin (Dox-SS[C(WR)4K])
A cyclic peptide ([C(WR)4K], 30 mg, 0.01 mmol) and the activated Dox (Dox-SS-Pyr, 11 mg, 0.01 mmol) were dissolved in methanol/water mixture (1:4, v/v, 4 mL). The reaction mixture was stirred under nitrogen overnight at rt. After completion of the reaction as monitored by MALDI mass, the solution was concentrated using rotatory evaporator and subjected to RP-HPLC for purification, followed by analytical HPLC to confirm the purity. MALDI-TOF (m/z) [C111H144N30O22S2]: calcd, 2313.0513; found 2315.3719 [M + 2H]+.
4.3. Cell culture
Human embryonic kidney cells (HEK-293, ATCC No. CRL-1573), human ovarian cancer cells (SKOV-3, ATCC No. HTB-77), human fibrosarcoma cells (HT-1080, ATCC No. CRL-12012), human leukemia cell line (CCRF-CEM, ATCC No. CCL-119), human breast adenocarcinoma (MDA-MB-468, ATCC No. HTB-132), human breast adenocarcinoma cell (MCF-7, ATCC No. HTB-22), and mouse myoblast cells (C2C12, ATCC No. CRL-1772) were purchased from American Type Culture Collection. The cells were grown on 75 cm2 cell culture flasks with RPMI-16 medium for CCRF. DMEM for MDA-MB-468, MCF-7, and C2C12, and EMEM for SKOV-3, HT-1080, and HEK-293, supplemented with 10% fetal bovine serum (FBS), and 1% penicillin-streptomycin solution (10,000 units of penicillin and 10 mg of streptomycin in 0.9% NaCl) in a humidified atmosphere of 5% CO2, 95% air at 37 °C.
4.4. Viability assay
HEK-293 (5000 cells), SKOV-3 (5000 cells), HT-1080 (5000 cells), CCRF-CEM cells (50,000 cells), and C2C12 (10,000 cells) were seeded in 0.1 mL per well in 96-well plates 24 h prior to the experiment. Cells were treated with 5 μM of Dox, Dox-SH, Dox-SS-Pyr, or Dox-SS-[C(WR)4K]. Water and DMSO (0.025%) were used as negative control. Plates were incubated for 24 or 72 h at 37 °C in a humidified atmosphere of 5% CO2. Before adding MTT or MTS reagent, the medium of SKOV-3, HT-1080, and HEK-293 was replaced with fresh medium. Cell viability was then determined by measuring the fluorescence intensity of the formazan product at 595 nm (MTT) or 490 nm (MTS) using a SpectraMax M2 microplate spectrophotometer. The percentage of cell survival was calculated as [(OD value of cells treated with compounds) – (OD value of culture medium)]/[(OD value of control cells) – (OD value of culture medium)] × 100.
4.4.1. Flow cytometry
HEK-293, SKOV-3, HT-1080, and CCRF-CEM cells were taken in 6-well plates, 5 × 104 cells/well for adherent cells and 5 × 105 for CCRF-CEM as non-adherent cells in serum-free medium and incubated overnight. The next day, Dox (5 μM), Dox-SS-Pyr (50 μM), or and Dox-SS-[C(WR)4K] (50 μM) were added to the wells. The cells with no treatment were used as control groups. The plates were incubated for 3 h at 37 °C. After 3 h incubation at 37 °C, the medium containing the drugs treated with HEK-293, SKOV-3, and HT-1080 cells was removed, and the cells were washed once with Dulbecco's phosphate-buffered saline (DPBS). Then cells were detached with 0.25% trypsin/EDTA (0.53 mM) for 5 min, neutralized with serum free fresh medium, and centrifuged at 800 rpm for 5 min to remove any artificial surface association and to detect only intracellular uptake. For CCRF-CEM cells, the cells were collected after 3 h and centrifuged at 800 rpm for 5 min. To wash the cells, the pellets of all cell lines were resuspended in DPBS and centrifuged twice. Finally, the cells were resuspended in flow cytometry buffer and analyzed by flow cytometry (FACSCalibur: BD) using TEXAS RED channel and CellQuest software (BD FACSVerse). The data presented were based on the mean fluorescence signal for 10,000 cells collected. All assays were performed in triplicate.
4.4.2. Detection of intracellular reactive oxygen species (ROS)
C2C12 cells were seeded in 6-well plates in DMEM medium supplemented with 10% FBS and incubated with 0.5% CO2 at 37 °C for 24 h prior to treatments. Cells were then incubated with 10 μM Fe2+ with or without 0.5 νM of Dox or Dox-SS-[C(WR)4K] in serum-free DMEM for 72 h. After treatments, cells were collected and stained with H2DCFDA (10 μM) in Hank's Balanced Salt Solution (HBSS) for 30 min at 37 °C followed by flow cytometry analysis. The mean fluorescence signal for 5000 cells is represented here as the fold of control. Each experiment was repeated twice.
4.5. Fluorescence microscopy
SKOV-3, HT-1080, MDA-MB-468, and MCF-7 cells were seeded on each chamber of the μ-Slide VI0.4 plates (ibidi cat. # 80604). 1 × 104 cells in 30 μL medium were injected to each chamber and incubated for 60 min to fix the cells. After this time, 60 μL of supplied medium was added to each column and incubated overnight in humidified atmosphere of 95% air, 0.5% CO2 at 37 °C. The next day, the medium was removed and a solution of 120 mL of fresh supplied medium contain final concentration of 5 mM of Dox, Dox-SH, Dox-SS-Pyr, or Dox-SS-[C(WR)4K] were added to each chamber and incubated for 1 and 24 h. Cells without treatment were considered as the control group. After each treatment time, the medium was removed, and chambers were washed 2 times with 100 μL DPBS, fixed with 100 μL of 4% PFA (paraformaldehyde) for 30 min and washed again with DPBS for 2–3 times. Since Dox is fluorescent, no dye was added. To identify nucleus, the cells were dyed with 50 μL DAPI (300 nM) for 5–10 min and washed with DPBS 2 times. One drop of mounting solution was added to each chamber. Plates were kept in the dark by rolling in aluminum foil and kept at room temperature for further analysis by Keyence Fluorescent microscope.
Supplementary Material
Acknowledgments
The authors would like to thank the Egyptian Cultural Affairs and Mission Sector, Egypt and Chapman University School of Pharmacy and Chapman University Office of Undergraduate Research and Creative Activity (OURCA), USA for their support. Research reported in this publication was also supported by the National Cancer Institute of the National Institutes of Health, USA under Award Number K08CA179084 (Sun Yang). We also thank support of Dr. Innokentiy Maslennikov for assisting in performing NMR spectroscopy.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2018.10.042.
References
- [1]. http://www.who.int/mediacentre/factsheets/fs297/en/.
- [2].Gottesman MM, Mechanisms of drug resistance, Annu. Rev. Med. 53 (2002) 615–627. [DOI] [PubMed] [Google Scholar]
- [3].Longely B, Johnston PG, Molecular mechanisms of drug resistance, J. Pathol. 205 (2005) 275–292. [DOI] [PubMed] [Google Scholar]
- [4].Wu Q, Yang Z, Nie Y, Shi Y, Fan D, Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches, Cancer Lett. 347 (2014) 159–166. [DOI] [PubMed] [Google Scholar]
- [5].Murphy GP, Lawrence W, Lenhard RE (Eds.), American Society Textbook of Clinical Oncology, second ed., American Cancer Society, Atlanta, GA, 1995. [Google Scholar]
- [6].Wiernik PH, Dutcher JP, Clinical importance of anthracyclines in the treatment of acute myeloid leukemia, Leukemia 6 (1992) 67–69. [PubMed] [Google Scholar]
- [7].Vincenzi B, Frezza M, Santini D, Tonini G, Kim H, New therapies in soft tissue sarcoma, Expet Opin. Emerg. Drugs 15 (2010) 237–248. [DOI] [PubMed] [Google Scholar]
- [8].Kim SH, Kim JH, Lethal effect of adriamycin on the division cycle of HeLa cells, 1972, Cancer Res. 32 (1972) 323–325. [PubMed] [Google Scholar]
- [9].Momparler L, Karon M, Siegel E, Avila F, Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells, Cancer Res. 36 (1976) 2891–2895. [PubMed] [Google Scholar]
- [10].Gewirtz D, A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin, Bio. Chem. Pharmacol. 57 (1999) 727–741. [DOI] [PubMed] [Google Scholar]
- [11].Capranico GI, Kohn KW, Pommier Y, Local sequence requirements for DNA cleavage by mammalian topoisomerase II in the presence of doxorubicin, Nucleic Acids Res. 18 (1990) 6611–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12] (a).Quigley J, Wang H, Ughetto G, Van der Marel G, Van Boom H, Rich A, Molecular structure of an anticancer drug-DNA complex: daunomycin plus d(CpGpTpApCpG), Proc. Natl. Acad. Sci. U.S.A. 77 (1980) 7204–7208; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Patel J, Kozlowski A, Rice A, Hydrogen bonding, overlap geometry and sequence specificity in anthracycline antitumor antibiotic. DNA complexes in solution, Proc. Natl. Acad. Sci. U.S.A. 78 (1981) 3333–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Raoul L, Heresbach D, Bretagne F, Ferrer B, Duvauferrier R, Bourguet P, Messner M, Gosselin M, Chemoembolization of hepatocellular carcinomas a study of the biodistribution and pharmacokinetics of doxorubicin, Cancer 70 (1992) 585–590. [DOI] [PubMed] [Google Scholar]
- [14].Rahman A, Carmichael D, Harris M, Roh K, Comparative pharmacokinetics of free doxorubicin and doxorubicin entrapped in cardiolipin liposomes, Cancer Res. 46 (1986) 2295–2299. [PubMed] [Google Scholar]
- [15].(a) Chhikara S, Jean N, Mandal D, Kumar A, Parang K, Fatty acyl amide derivatives of doxorubicin: synthesis and in vitro anticancer activities, Eur. J. Med. Chem. 46 (2011) 2037–2042; [DOI] [PubMed] [Google Scholar]; (b) Chhikara S, Mandal D, Parang K, Synthesis, anticancer activities and cellular uptake studies of lipophilic derivatives of doxorubicin succinate, J. Med. Chem 55 (2012) 1500–1510. [DOI] [PubMed] [Google Scholar]
- [16].Takemura G, Fujiwara H, Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management, Prog. Cardiovasc. Dis. 49 (2007) 330–352. [DOI] [PubMed] [Google Scholar]
- [17].Ayla S, Seckin I, Tanriverdi G, Cengiz M, Eser M, Soner C, Oktem G, Doxorubicin-induced nephrotoxicity: protective effect of nicotinamide, Int. J. Cell Biol. (2011) 390238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kremer LC, Van Dalen EC, Offringa M, Ottenkamp J, Voute PA, Anthracycline-induced clinical heart failure in a cohort of 607 children: long-term follow-up study, J. Clin. Oncol. 19 (2001) 191–196. [DOI] [PubMed] [Google Scholar]
- [19].Kratz F, DOXO-EMCH (INNO-206): the first albumin-binding prodrug of doxorubicin to enter clinical trials, Expert Opin. Invest. Drugs 16 (2007) 855–866. [DOI] [PubMed] [Google Scholar]
- [20].Tang Y, McGoron AJ, Combined effects of laser-ICG photothermotherapy and doxorubicin chemotherapy on ovarian cancer cells, J. Photochem. Photobiol., B 97 (2009) 138–144. [DOI] [PubMed] [Google Scholar]
- [21].Seelig A, Gatlik-Landwojtowicz E, Inhibitors of multidrug efflux transporters: their membrane and protein interactions, Mini Rev. Med. Chem. 5 (2005) 135–151. [DOI] [PubMed] [Google Scholar]
- [22].Park W, Liposome-based drug delivery in breast cancer treatment, Breast Canc. Res. 4 (2002) 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kuppens IE, Witteveen EO, Jewell RC, Radema SA, Paul EM, Mangum SG, Beijnen JH, Voest EE, Schellens JH, A phase I, randomized, open-label, parallel-cohort, dose-finding study of elacridar (GF120918) and oral topotecan in cancer patients, Clin. Canc. Res. 13 (2007) 3276–3785. [DOI] [PubMed] [Google Scholar]
- [24].Pusztai L, Wagner P, Ibrahim N, Rivera E, Theriault R, Booser D, Symmans FW, Wong F, Blumenschein G, Fleming DR, Rouzier R, Boniface G, Hortobagyi GN, Phase II study of tariquidar, a selective P-glycoprotein inhibitor,in patients with chemotherapy-resistant, advanced breast carcinoma, Cancer 104 (2005) 682–691. [DOI] [PubMed] [Google Scholar]
- [25].Malingre M, Beihnen H, Rosing H, Koopman J, Jewell C, Paul M, Ten Bokkel W, Huinink W, Schellens H, Co-administration of GF120918 significantly increases the systemic exposure to oral paclitaxel in cancer patients, Br. J. Canc. 84 (2001) 42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kwak O, Lee SH, Lee GS, Kim MS, Ahn YG, Lee JH, Kim SW, Kim KH, Lee MG, Selective inhibition of MDR1 (ABCB1) by HM30181 increases oral bioavailability and therapeutic efficacy of paclitaxel, Eur. J. Pharmacol. 627 (2010) 92–98. [DOI] [PubMed] [Google Scholar]
- [27].Hay M, Wilson W, Denny W, Nitroarylmethylcarbamate prodrugs of doxorubicin for use with nitroreductase gene-directed enzyme prodrug therapy, Bioorg. Med. Chem. 13 (2005) 4043–4055. [DOI] [PubMed] [Google Scholar]
- [28].Pallerla S, Gauthier T, Sable R, Jois SD, Design of a doxorubicin-peptidomimetic conjugate that targets HER2-positive cancer cells, Eur. J. Med. Chem. 125 (2017) 914–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Singh B, Jang Y, Maharjan S, Kim HJ, Lee AY, Kim S, Gankhuyag N, Yang MS, Choi YJ, Cho MH, Cho CS, Combination therapy with doxorubicin-loaded galactosylated poly(ethyleneglycol)-lithocholic acid to suppress the tumor growth in an orthotopic mouse model of liver cancer, Biomaterials 116 (2017) 130–144. [DOI] [PubMed] [Google Scholar]
- [30].Cho K, Wang X, Nie S, Shin M, Therapeutic nanoparticles for drug delivery in cancer, Clin. Canc. Res. 14 (2008) 1310–1316. [DOI] [PubMed] [Google Scholar]
- [31].Xueqiong Z, Lianghong L, Chunfu L, Hua Z, Haoyuan S, Fuliang X, Tong Q, Jin Y, Cisplatin-crosslinked glutathione-sensitive micelles loaded with doxorubicin for combination and targeted therapy of tumors, Carbohydr. Polym. 155 (2017) 407–415. [DOI] [PubMed] [Google Scholar]
- [32].Ine L, Bart G, Joseph D, Stefaan C, Niek S, Design and evaluation of doxorubicin-containing microbubbles for ultrasound-triggered doxorubicin delivery: cytotoxicity and mechanisms involved, Mol. Ther. 181 (2010) 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ilbasmis-Tamer S, Unsal H, Tugcu-Demiroz F, Kalaycioglu GD, Degim IT, Aydogan N, Stimuli-responsive lipid nanotubes in gel formulations for the delivery of doxorubicin, Colloids Surf. B Biointerfaces 143 (2016) 406–414. [DOI] [PubMed] [Google Scholar]
- [34].Wen H, Lipeng Q, Lian C, Qing H, Yang L, Ziyang H, Dawei Ch, Lifang Ch, Redox and pH dual responsive poly(amidoamine) dendrimer-poly(ethylene glycol) conjugates for intracellular delivery of doxorubicin, Acta Biomater. 36 (2016) 241–253. [DOI] [PubMed] [Google Scholar]
- [35].Yang F, Yang Y, Xie Y, Cai S, Zhang H, Gong W, Wang Y, Mei G, PEGylated liposomes with NGR ligand and heat-activable cell-penetrating peptide-doxorubicin conjugate for tumor-specific therapy, Biomaterials 35 (2014) 4368–4381. [DOI] [PubMed] [Google Scholar]
- [36].Goksu C, Ayse O, Seren H, Gokhan G, Aykutlu D, Ayse T, Mustafa G, Local delivery of doxorubicin through supramolecular peptide amphiphile nano-fiber gels, Biomater. Sci. 5 (2017) 67–76. [DOI] [PubMed] [Google Scholar]
- [37].Ye X, Shin C, Liang L, He N, Yang C, 15 Years of ATTEMPTS: a macromolecular drug delivery system based on the CPP-mediated intracellular drug delivery and antibody targeting, J. Contr. Release 205 (2015) 58–69. [DOI] [PubMed] [Google Scholar]
- [38].Ojima I, Guided molecular missiles for tumor-targeting chemotherapy-case studies using the second-generation taxoids as war heads, Acc. Chem. Res. 41 (2008) 108–119. [DOI] [PubMed] [Google Scholar]
- [39].Feazell P, Nakayama-Ratchford N, Dai H, Lippard J, Soluble single-walled carbon nanotubes as longboat delivery systems for platinum (IV) anticancer drug design, J. Am. Chem. Soc. 129 (2007) 8438–8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dubowchik M, Walker A, Receptor-mediated and enzyme-dependent targeting of cytotoxic anticancer drugs, Pharmacol. Ther. 83 (1999) 67–123. [DOI] [PubMed] [Google Scholar]
- [41].Yu S, He C, Ding J, Cheng Y, Song W, Zhuang X, Chen X, pH and reduction dual responsive polyurethane triblock copolymers for efficient intracellular drug delivery, Soft Matter 9 (2013) 2637–2645. [Google Scholar]
- [42].Sun T, Zhang Y, Pang B, Hyun D, Yang M, Xia Y, Engineered nanoparticles for drug delivery in cancer therapy, Angew. Chem. Int. Ed. 53 (2014) 12320–12364. [DOI] [PubMed] [Google Scholar]
- [43].Wang H, Wang Y, Chen Y, Jin Q, Ji J, A biomimicPh-sensitive polymeric prodrug based on polycarbonate for intracellular drug delivery, Polym. Chem. 5 (2014) 854–861. [Google Scholar]
- [44].Shirazi AN, El-Sayed NS, Tiwari RK, Tavakoli K, Parang K, Cyclic peptide containing hydrophobic and positively charged residues as a drug delivery system for curcumin, Curr. Drug Deliv. 13 (2016) 409–417. [DOI] [PubMed] [Google Scholar]
- [45].Shirazi AN, Tiwari RK, Oh D, Banerjee A, Yadav A, Parang K, Efficient delivery of cell impermeable phosphopeptides by a cyclic peptide amphiphile containing tryptophan and arginine, Mol. Pharm. 10 (2013) 2008–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mandal D, Shirazi AN, Parang K, Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters, Angew. Chem. Int. Ed. 50 (2011) 9633–9637. [DOI] [PubMed] [Google Scholar]
- [47].Shirazi AN, Tiwari RK, Chhikara BS, Mandal D, Parang K, Design and evaluation of cell-penetrating peptide-doxorubicin conjugates as prodrugs, Mol. Pharm. 10 (2013) 488–499. [DOI] [PubMed] [Google Scholar]
- [48].Jaracz S, Chen J, Kuznetsova LV, Ojima I, Recent advances in tumor-targeting anticancer drug conjugates, Bioorg. Med. Chem. 13 (2005) 5043–5054. [DOI] [PubMed] [Google Scholar]
- [49].Oh D, Darwish Sh, Shirazi A, Tiwari R, Parang K, Amphiphilic bicyclic peptides as cellular delivery agents, ChemMedChem 9 (2014) 2449–2453. [DOI] [PubMed] [Google Scholar]
- [50].Barlos K, Gatos D, Kallitsis J, Papaphotiu G, Sotiriu P, Wenqing Y, Schäfer W, Darstellung geschützter peptid-fragmente unter einsatz substituierter triphenylmethyl-harze, Tetrahedron Lett. 30 (1989) 3943–3946. [Google Scholar]
- [51].Patterson SD, Katta V, Prompt fragmentation of disulfide-linked peptides during matrix-assisted laser desorption ionization mass spectrometry, Anal. Chem. 66 (1994) 3727–3732. [DOI] [PubMed] [Google Scholar]
- [52].Shi N, Gao W, Xiang B, Qi X, Enhancing cellular uptake of activable cell-penetrating peptideedoxorubicin conjugate by enzymatic cleavage, Int. J. Nanomed. 7 (2012) 1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Akan I, Akan S, Akca H, Savas B, Ozben T, N-acetylcysteine enhances multidrug resistance-associated protein I mediated doxorubicin resistance, Eur. J. Clin. Invest. 34 (2004) 683–689. [DOI] [PubMed] [Google Scholar]
- [54].Wei Y, Gao L, Wang L, Shi L, Wei E, Zhou B, Zhou L, Ge B, Polydopamine and peptide decorated doxorubicin-loaded mesoporous silica nanoparticles as a targeted drug delivery system for bladder cancer therapy, Drug Deliv. 42 (2017) 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yang Z, Misner B, Ji H, Poulos TL, Silverman RB, Meyskens FL, Yang S, Targeting nitric oxide signaling with nNOS inhibitors as a novel strategy for the therapy and prevention of human melanoma, Antioxid. Redox Signal 19 (2013) 433–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL, Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies, J. Mol. Cell Card. 52 (2012) 1213–1225. [DOI] [PubMed] [Google Scholar]
- [57].Gammella E, Maccarinelli F, Buratti P, Recalcati S, Cairo G, The role of iron in anthracyclin cardiotoxicity, Front. Pharmacol. 5 (2014) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.