

Abstract
Objectives
SARS‐CoV‐2 infection induces virus‐reactive memory B cells expressing unmutated antibodies, which hints at their emergence from naïve B cells. Yet, the dynamics of virus‐specific naïve B cells and their impact on immunity and immunopathology remain unclear.
Methods
We longitudinally profiled SARS‐CoV‐2‐specific B‐cell responses in 25 moderate‐to‐severe COVID‐19 patients by high‐dimensional flow cytometry and isotyping and subtyping ELISA. We also explored the relationship of B‐cell responses to SARS‐CoV‐2 with the activation of effector and regulatory cells from the innate or adaptive immune system.
Results
We found a virus‐specific antibody response with a broad spectrum of classes and subclasses during acute infection, which evolved into an IgG1‐dominated response during convalescence. Acute infection was associated with increased mature B‐cell progenitors in the circulation and the unexpected expansion of virus‐targeting naïve‐like B cells. The latter further augmented during convalescence together with virus‐specific memory B cells. In addition to a transitory increase in tissue‐homing CXCR3+ plasmablasts and extrafollicular memory B cells, most COVID‐19 patients showed persistent activation of CD4+ and CD8+ T cells along with transient or long‐lasting changes of key innate immune cells. Remarkably, virus‐specific antibodies and the frequency of naïve B cells were among the major variables defining distinct immune signatures associated with disease severity and inflammation.
Conclusion
Aside from providing new insights into the complexity of the immune response to SARS‐CoV‐2, our findings indicate that the de novo recruitment of mature B‐cell precursors into the periphery may be central to the induction of antiviral immunity.
Keywords: adaptive immunity, antibodies, B‐cell memory, COVID‐19, naïve B cells
We longitudinally studied moderate‐to‐severe COVID‐19 patients to dissect SARS‐CoV‐2‐specific B cell responses overtime. We found a broad virus‐specific antibody response during acute infection, which evolved into an IgG1‐dominated response. Moreover, we identified a persistent expansion of virus‐targeting naïve‐like and memory B cells in COVID‐19 patients, as well as unique immune signatures that associated with disease severity and inflammation.

Introduction
To date, the rapidly spreading severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) has infected about 200 million people, resulting in more than four million deaths worldwide.1 Infection with SARS‐CoV‐2 causes the coronavirus disease 2019 (COVID‐19), which is characterised by a wide variety of clinical manifestations that range from asymptomatic to acute respiratory distress syndrome (ARDS), multiorgan failure and death.2 Although such diversity in disease pathogenesis is partially explained by the patient’s comorbidities and genetic and socio‐demographic factors, severe manifestations of the disease are strongly associated with dysregulated immune responses.3, 4 Immune dysregulation in severe COVID‐19 patients is characterised by delayed and impaired type I interferon responses that associate with failure to control primary infection.5, 6 The resulting aberrant activation of innate immune cells leads to an exacerbated release of pro‐inflammatory cytokines, causing systemic inflammation and tissue damage.7 Interestingly, interferon signalling and hyperinflammation may associate with autoimmunity. Indeed, severe COVID‐19 patients develop autoantibodies against immunomodulatory proteins, including antibodies against type I interferon.8, 9, 10
Besides playing a role in immunopathogenesis, the host immune response is a major determinant of recovery and immune protection through the development of durable SARS‐CoV‐2‐specific T‐ and B‐cell responses. Several studies have documented the early activation of CD4+ and CD8+ T cells following SARS‐CoV‐2 infection and the generation of long‐lasting virus‐specific T‐cell responses required for immune protection.11, 12, 13, 14 In the early response to SARS‐CoV‐2, infected individuals generate antibodies against the viral nucleocapsid (NP) and spike (S) proteins. About 90% of COVID‐19 patients produce detectable neutralising antibody responses against the receptor‐binding domain (RBD) of the viral S protein, which persist for up to 8 months.15, 16, 17 Early humoral responses are driven by the transient expansion of antibody‐secreting plasmablasts (PBs). During convalescence, humoral memory is sustained by somatically mutated memory switched B cells and long‐lived plasma cells.14, 16, 18, 19 Of note, recent studies identified convergent antibody responses to SARS‐CoV‐2 by B cells with preferential immunoglobulin heavy chain variable‐joining (IGHV‐J) gene usage and minimal somatic hypermutation.18, 20, 21, 22, 23, 24, 25 These findings suggest that humoral protection involves SARS‐CoV‐2 recognition by naïve B cells with little or no antigen‐driven affinity maturation required.21, 26
In spite of our growing understanding of SARS‐CoV‐2 infection, both the kinetics and composition of virus‐specific B‐cell responses remain poorly understood. In particular, the dynamics of virus‐reactive naïve B cells and their role in immune protection and immunopathology are unclear. In addition, the temporal trajectories of innate and adaptive immune responses to SARS‐CoV‐2 and their functional relationship remain elusive. A better understanding of these facets of SARS‐CoV‐2 infection may help in the evaluation of the protective effects afforded by individual vaccination programmes.
Here, we longitudinally profiled global and virus‐specific B‐cell responses from a cohort of moderate‐to‐severe COVID‐19 patients at different stages of SARS‐CoV‐2 infection. We also explored the relationship of B‐cell responses to SARS‐CoV‐2 with the activation of effector and regulatory cells from the innate or adaptive immune system. We identified specific properties of immune response dynamics from COVID‐19 patients and unique immune trajectories that associate with disease severity and inflammation. We also found some evidence of a novel mechanism adopted by the host immune system to fight against SARS‐CoV‐2. This mechanism may involve the continuous peripheral recruitment of early mature B‐cell precursors to enhance viral recognition by the naïve B‐cell repertoire.
Results
SARS‐CoV‐2 triggers a broad antibody response in the early stage of infection and a robust and long‐lasting RBD‐specific IgG1 response
To dissect the dynamics of humoral immune responses to SARS‐CoV‐2, we collected blood and serum samples from a cohort of 25 hospitalised COVID‐19 patients in acute phase of infection (COVT1). Of these patients, 24% (n = 6; darker symbols in Figures) were admitted to intensive care unit (ICU). On average, COVT1 samples were collected 11 days post‐symptom onset (PSO). A follow‐up blood sample was drawn from 20 out of 25 patients (COVT2), including 4 ICU patients, averaging 70 days PSO (Figure 1a). Blood and serum samples were also collected from healthy controls (n = 21). Aside from being a major risk factor for fatal outcome in COVID‐19 patients, age profoundly influences the immune system of healthy individuals. To minimise these age‐related effects, we mostly selected patients younger than 65 (median age = 51) and included age‐matched healthy controls (median age = 50). A summary of demographic and clinical data of all the individuals included in this study is provided in Supplementary table 1.
Figure 1.
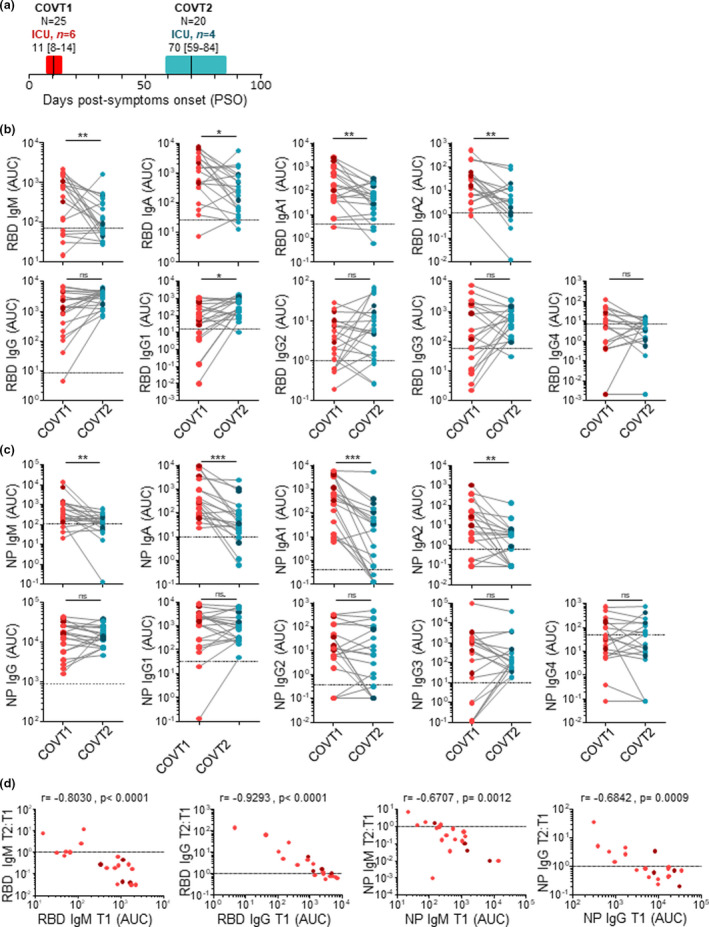
SARS‐CoV‐2 induces a broad antibody response in the early stage of infection and a long‐lasting IgG response. (a) Schematic diagram of the study timeline and cohort characteristics. Range of days post‐symptom onset (PSO) is indicated with a box, and median is indicated with a line for COVID‐19 patients in the acute (COVT1) and convalescent (COVT2) phase of infection. (b) Area under the curve (AUC) for each of the RBD‐specific and (c) NP‐specific antibody classes and subclasses analysed from COVT1 and COVT2 sera samples. Sera from healthy controls (HCs) were analysed in parallel to establish negative threshold values defined as the HC AUC mean plus 2 times the standard deviation of the mean. Data are presented as paired individual dots. Dashed line indicates negative threshold. The Wilcoxon matched‐pairs test was used to compare COVT1 with COVT2 in b and c. (d) Relative change in antibody levels between COVT1 and COVT2 plotted against the corresponding antibody levels at COVT1. r represents Spearman’s rank‐order correlation. (a–d) Dark‐coloured dots show ICU patients. ns, non‐significant (P > 0.05), *P < 0.05, **P < 0.01 and ***P < 0.001. COVT1, n = 20; COVT2, n = 20.
To elucidate whether COVID‐19 patients can mount a broad and long‐lasting humoral response to SARS‐CoV‐2, we performed isotype‐specific (IgM, IgA and IgG) and subtype‐specific (IgA1, IgA2, IgG1, IgG2, IgG3 and IgG4) enzyme‐linked immunosorbent assays (ELISAs) that quantified antibody responses to the RBD of the spike protein or NP from SARS‐CoV‐2. Compared with healthy controls, sera from COVID‐19 patients in acute infection (COVT1) and convalescence (COVT2) showed significantly higher titres of all RBD‐ and NP‐specific antibody classes and subclasses analysed (Figure 1b, c). According to our longitudinal study and as previously reported,18, 27 RBD‐ and NP‐specific IgM and IgA titres significantly decreased two months PSO (Figure 1b, c). Interestingly, both IgA1 and IgA2 subclasses contributed to the decline in virus‐specific IgA, suggesting a limited involvement of mucosal immunity during convalescence. Unlike RBD‐ and NP‐specific IgM and IgA, virus‐specific IgG titres were maintained over the first two months PSO (Figure 1b, c). Remarkably, RBD‐specific IgG1 was the only IgG subclass that significantly increased over time (Figure 1b).
We further studied the dynamics of the humoral response in COVID‐19 throughout the study period by plotting the relative change in antibody levels between COVT1 and COVT2 against the corresponding antibody levels at COVT1. This analysis revealed that the magnitude of relative changes in virus‐specific IgM or IgG titres directly correlated with their initial levels at COVT1. Indeed, patients with initial higher virus‐specific IgM or lower IgG titres showed a greater relative decrease or increase, respectively (Figure 1d). This resulted in much less variance in virus‐specific IgM or IgG titres among COVT2 samples (Figure 1b, c). Thus, SARS‐CoV‐2 infection triggers a broad antibody response in terms of antibody classes and subclasses in early stages and an expansion of an RBD‐specific and IgG1‐dominated response during convalescence.
COVID‐19 is associated with transitory expansion of circulating plasmablasts and naïve B cells during the acute phase of infection
To further explore the dynamics of the B‐cell response to SARS‐CoV‐2, we analysed circulating B cells from healthy controls and COVID‐19 patients in acute infection and convalescence by high‐dimensional spectral flow cytometry (Supplementary table 2). As reported by others,28 the frequency of CD19+ B cells within live peripheral blood mononuclear cells (PBMCs) was significantly higher in COVT1 than in healthy controls or COVT2 (Figure 2a). T‐distributed stochastic neighbour embedding (tSNE) defined the major B‐cell populations (Figure 2b), the contribution of each group of samples to these populations (Figure 2c) and the differential expression of multiple surface B‐cell proteins within these populations (Supplementary figure 1a). In parallel, we queried the data by traditional gating (Supplementary figures 1b, 2a and 3a). This analysis revealed that, for most COVID‐19 patients, the acute response to SARS‐CoV‐2 was dominated by a transitory expansion of CD38++CD10−CD27high PBs (Figure 2b–e), the majority of which expressed HLA‐DR+ (Figure 2f). Next, the analysis of antibody classes and subclasses confirmed the induction of unswitched and IgA1 or IgG class‐switched PBs soon after infection (Figure 2g and Supplementary figure 1c). We then assessed the homing potential of these PBs through the analysis of CXCR3 and CXCR4 chemokine receptors, which guide PBs to inflamed tissues or bone marrow, respectively.29 We found that the majority of PBs from COVT1 had a CXCR3+CXCR4− phenotype, suggesting their targeted migration into inflamed tissues (Figure 2h, i). Compared with healthy controls and COVT2, COVT1 samples also showed a significantly increased proportion of CXCR3+CXCR4+ PBs (Figure 2h, j).
Figure 2.
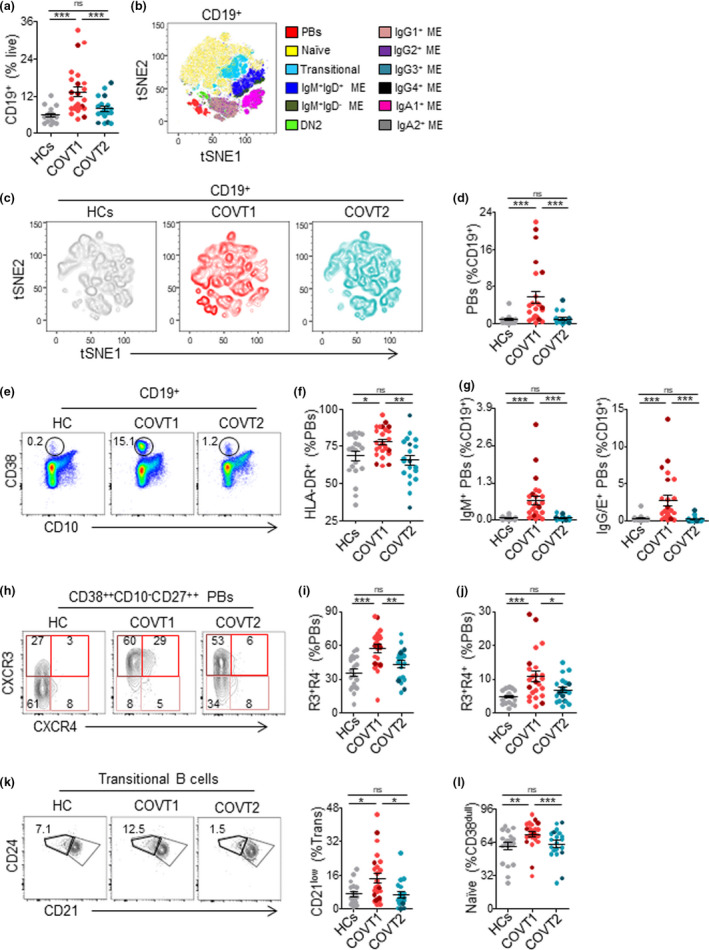
Deep profiling of B‐cell subsets in COVID‐19 patients reveals transitory expansion of circulating plasmablasts, immature transitional and naïve B cells during the acute phase of infection. (a) Frequency of CD19+ B cells from live peripheral blood mononuclear cells (PBMCs) in healthy controls (HCs) and COVT1‐COVT2. (b) Merged tSNE projection of CD19+ B cells for HCs (n = 11), COVT1 (n = 16) and COVT2 (n = 16) samples concatenated, with main B‐cell populations indicated by colour. PBs, plasmablasts. ME, memory. DN2, CD38dullCD10−IgD−CD27−CD21−CD11c+ double‐negative type 2. (c) tSNE projections of CD19+ B cells for each sample group. (d) Frequency of CD38++CD10−CD27+ plasmablasts (PBs) from total CD19+ B cells and (e) representative dot plots in HCs, COVT1 and COVT2. Numbers indicate percentages in the drawn gates. (f) Frequency of HLA‐DR+ from total circulating CD38++CD10−CD27+ PBs in HCs, COVT1 and COVT2. (g) Frequencies of IgM+ PBs (left) and IgG/E+ PBs (right) from total CD19+ B cells. (h) Representative flow cytometric plots of CXCR3−CXCR4− (R3−R4−), CXCR3−CXCR4+ (R3−R4+), CXCR3+CXCR4+ (R3+R4+) and CXCR3+CXCR4− (R3+R4−) PBs. Numbers indicate percentages in the drawn gates. (i) Frequency of CXCR3+CXCR4− cells within total circulating PBs. (j) Frequency of CXCR3+CXCR4+ cells within total circulating PBs. (k) Representative flow cytometric plots (left) and frequency (right) of immature cells (CD38intCD10+IgD+CD27−CD21low) from total transitional B cells, gated with thick black line. Numbers indicate percentages in the drawn gates. (l) Frequency of naïve B cells (CD19+CD38intCD27−IgD+) from CD19+CD38dull B cells in each group of samples. Data are presented as individual dots. Bars represent mean ± standard error mean (SEM). Dark‐coloured dots show ICU patients. The two‐tailed Mann–Whitney U‐test was performed to compare HCs with COVT1 and HCs with COVT2. The Wilcoxon matched‐pairs test was performed to compare COVT1 with COVT2. ns, non‐significant (P > 0.05), *P < 0.05, **P < 0.01 and ***P < 0.001. Unless mentioned otherwise, HCs, n = 19; COVT1, n = 25; COVT2, n = 20.
The tSNE projections also highlighted prominent differences in the transitional and naïve B‐cell compartments among healthy controls, COVT1 and COVT2 (Figure 2b, c and Supplementary figure 1a). Using conventional gating strategies (Supplementary figure 2a) and pairwise longitudinal comparisons, we identified an increased proportion of immature CD21low transitional B cells (Figure 2k) and a highly significant transient expansion of naïve B cells during the acute phase of infection (Figure 2l). Furthermore, our data revealed a positive correlation between the frequency of naïve B cells and the frequency of CD19+ B cells (Supplementary figure 2b), which implies that naïve B‐cell expansion could account for the increased frequency of CD19+ B cells. These changes likely reflect an increased inflammation‐dependent mobilisation of developing B cells from the bone marrow into the periphery, a phenomenon also observed in other infections.30 Moreover, compared with similar cells from healthy controls, naïve B cells from COVT1 exhibited significantly lower expression of CD21, HLA‐DR, IgD and the chemokine receptors CCR6, CXCR5 and CXCR3 but not CCR7 (Supplementary figure 2c–i). Thus, aside from promoting a transient induction of CXCR3+ unswitched and class‐switched PBs, infection‐associated inflammation may induce the mobilisation of precursors of mature B cells together with the transient expansion of naïve B cells with an atypical phenotype.
SARS‐CoV‐2 infection drives transient increase in extrafollicular memory switched B cells and long‐lasting expansion of IgG1 memory B cells
To gain insight into the dynamics of B‐cell memory (ME) responses to SARS‐CoV‐2, we analysed the frequency of different antigen‐experienced B‐cell populations. First, we found that the frequency of ME B cells within total CD19+ B cells was significantly lower in acute COVT1 than in COVT2 and healthy controls (Supplementary figure 3a and Figure 3a), presumably because of the transient expansion of newly formed PBs and naïve B cells. Unlike PBs but similar to naïve B cells, the frequency of CXCR3+ ME B cells diminished during the acute phase of infection (Figure 3b), suggesting a possible impairment in their recruitment into inflamed tissues and lymphoid follicles.31 Within the ME B‐cell compartment, we observed a significant and persistent reduction in the proportion of IgM+IgD+CD27+ unswitched ME B cells in COVID‐19 patients (Figure 3c) and a progressive expansion of IgG1‐expressing ME B cells (Figure 3d). Interestingly, the frequency of unswitched ME B cells positively correlated with the serum concentration of inflammatory marker ferritin (Figure 3e) and with NP‐specific IgM titres (Figure 3f). Moreover, COVID‐19 patients with higher plasma concentration of ferritin and other markers of inflammation (data not shown) also showed a lower frequency of IgG1 ME B cells during acute infection (Figure 3g). Remarkably, ME B cells expressing other IgG subclasses did not increase after infection (Supplementary figure 3b). These results may reflect the consolidation of efficient antiviral IgG1‐mediated immunity in less severe patients.
Figure 3.
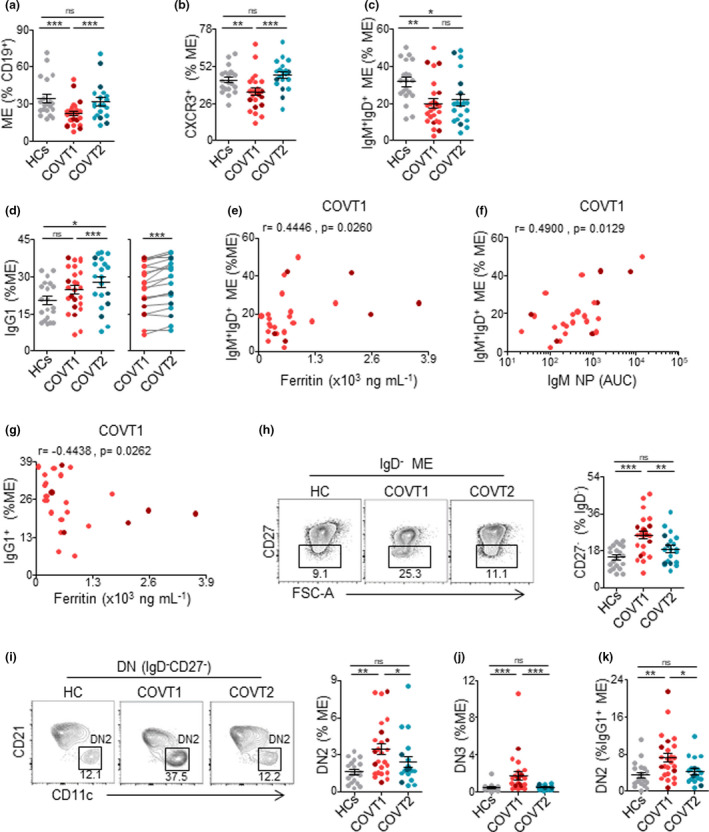
COVID‐19 is associated with temporary expansion of extrafollicular switched memory B cells, long‐lasting contraction of IgM+IgD+CD27+ memory B cells and late increase in IgG1+ memory B cells. (a) Frequency of memory (ME) B cells defined as depicted in Supplementary figure 3 from total CD19+ B cells in HCs, COVT1 and COVT2. (b) Frequency of CXCR3+ cells from total ME B cells in HCs, COVT1 and COVT2. (c) Frequency of CD38dullCD10−IgM+IgD+CD27+ ME B cells within total ME B cells in HCs, COVT1 and COVT2. (d) Frequency of IgG1+ ME B cells within total ME B cells in HCs, COVT1 and COVT2 (left) and paired analysis in COVT1 (n = 20) and COVT2 (n = 20, right). (e) Spearman’s correlation analysis of ferritin levels in COVT1 plotted against CD38dullCD10−IgM+IgD+CD27+ ME B cells within total ME B cells in COVT1. (f) Spearman’s correlation analysis of NP‐specific IgM antibody titres in COVT1 plotted against CD38dullCD10−IgM+IgD+CD27+ ME B cells within total ME B cells in COVT1. (g) Spearman’s correlation analysis of IgG1 ME B cells within total ME B cells in COVT1 plotted against ferritin levels in COVT1. (e–g) r represents Spearman's rank‐order correlation. (h) Representative flow cytometric plots and frequency of DN memory B cells defined as CD38dullCD10−IgD−CD27− out of total IgD− ME B cells. Numbers indicate percentages in the drawn gates. (i) Representative flow cytometric plots and frequency of extrafollicular DN2 ME B cells defined as CD38dullCD10−IgD−CD27−CD21−CD11c+ out of total ME B cells. Numbers indicate percentages in the drawn gates. (j) Frequency of DN3 ME B cells defined as CD38dullCD10−IgD−CD27−CD21−CD11c− out of total ME B cells. (k) Frequency of DN2 ME B cells from total IgG1+ ME B cells. Data are presented as individual dots. Dark‐coloured dots show ICU patients. Error bars represent mean ± SEM. The two‐tailed Mann–Whitney U‐test was performed to compare HCs with COVT1 and HCs with COVT2. The Wilcoxon matched‐pairs test was performed to compare COVT1 with COVT2. ns, non‐significant (P > 0.05), *P < 0.05, **P < 0.01 and ***P < 0.001. Unless mentioned otherwise, HCs, n = 19; COVT1, n = 25; COVT2, n = 20.
In agreement with findings published previously,32 we categorised IgD−CD27− double‐negative (DN) ME B cells as early activated CD27−CD21+CD11c− ME or DN1, extrafollicular CD27−CD21− CD11c+ PB precursors or DN2, and DN3 CD27−CD21−CD11c− B cells (Supplementary figure 3c). The analysis of these ME B‐cell subsets revealed a significant and transient increase in the frequency of CD27−IgD− DN cells in COVT1 compared with COVT2 and healthy controls (Figure 3h), as reported by others.28 Among DN B cells, DN2 cells are recognised as primed precursors of antibody‐secreting cells that differentiate from newly activated naïve B cells through an extrafollicular pathway. In contrast, DN3 has unknown origin and function.28 First, we quantified the frequency of DN2 cells within different ME B‐cell subsets distinguished on the basis of their expressed isotype or subclass. We found a significantly increased proportion of DN2 in ME B cells expressing IgG3, IgG1 and, to a lesser extent, IgA1 (Supplementary figure 3d). The frequencies of extrafollicular ME DN2 B cells and DN3 cells were higher in COVT1 than in COVT2 and healthy controls (Figure 3i, j). Interestingly, the most significant increase in DN2 was observed within the IgG1+ ME B‐cell subset (Figure 3k). Thus, SARS‐CoV‐2 infection induces the transient expansion of extrafollicular IgG1+ ME B cells, which is followed by a long‐lasting IgG1+ ME B‐cell response.
SARS‐CoV‐2 induces RBD‐specific PBs during acute infection followed by sustained expansion of circulating RBD‐specific naïve and memory switched B cells in convalescence
Next, a fluorescently labelled recombinant RBD probe was used to identify SARS‐CoV‐2‐specific B cells capable of producing potentially neutralising antibodies (Supplementary figure 4a). We confirmed the specificity of our assay by comparing our results with those obtained using a double discrimination strategy via the inclusion of two fluorescently labelled RBD probes (Supplementary figure 4b). Antigen‐binding CD19+ B cells were further characterised according to the expression of CD27, CD21, CD11c, HLA‐DR, IgM, IgD, IgA and the Ig light chain λ (Supplementary figure 4c and Supplementary table 3). In agreement with published studies,14, 16, 17 SARS‐CoV‐2 infection induced a rapid increase in the frequency of RBD‐specific CD19+ B cells at COVT1 that persisted 2 months after viral exposure (Figure 4a). Interestingly, the phenotypic characterisation of these antigen‐binding B cells revealed significant changes in virus‐specific B‐cell populations when the acute phase was compared to the convalescent phase of infection (Figure 4b and Supplementary figure 4d). Consistent with the observed expansion of PBs, COVT1 showed an increased frequency of RBD‐specific CD27highCD21− PBs compared with healthy controls and COVT2 (Figure 4c and data not shown). Remarkably, virtually 100% of them expressed HLA‐DR (Figure 4d).
Figure 4.
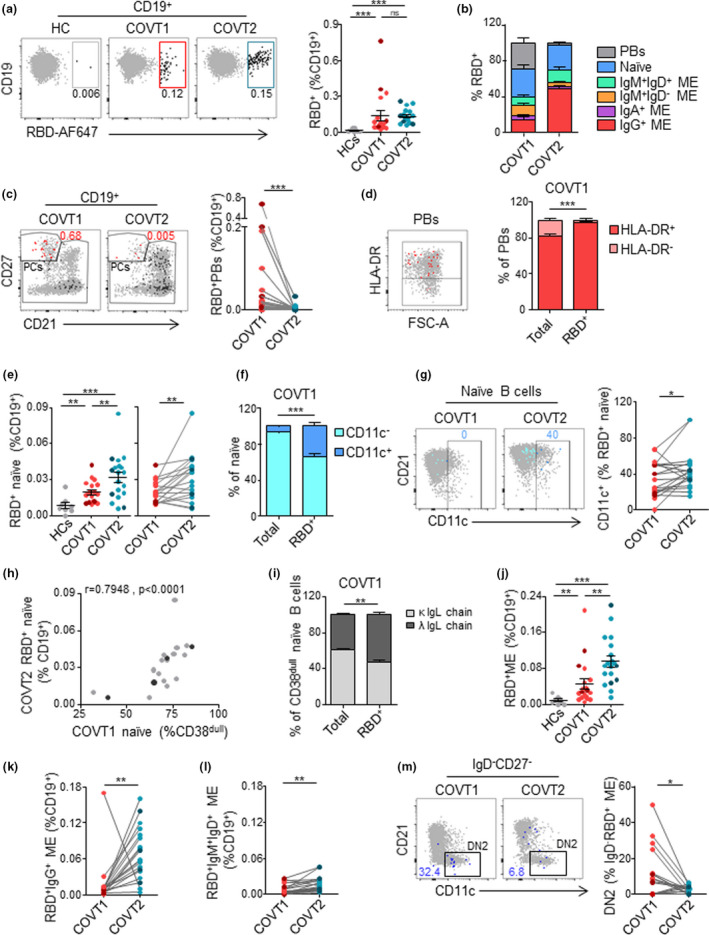
SARS‐CoV‐2 induces a temporal enrichment of RBD‐specific plasmablasts in the early stage of infection and a sustained expansion of RBD‐specific naïve and memory B‐cell subsets. (a) Representative flow cytometric plots of CD19+ RBD‐specific B cells (left) and frequency of RBD+ cells within total CD19+ B cells (right) from each group of samples. Numbers indicate the percentage of RBD‐specific cells within total CD19+ B cells. (b) Relative percentage of B‐cell subsets within total RBD+ CD19+ B cells in HCs, COVT1 and COVT2. (c) Representative flow cytometric plots and paired analysis of the frequency of RBD‐specific PBs (CD19+RBD+CD27++CD21−) within total CD19+ B cells. In the flow cytometric plot, red large dots represent RBD‐specific PBs and numbers indicate percentage. (d) Representative flow cytometric plot and relative percentage of total and RBD‐specific PBs expressing HLA‐DR in COVT1. In the flow cytometric plot, red large dots represent RBD‐specific PBs expressing HLA‐DR. (e) Frequency and paired analysis of RBD‐specific naïve B cells (CD19+RBD+CD27−IgD+) within total CD19+ in HCs, COVT1 and COVT2. (f) Relative percentage of total or RBD‐specific naïve B cells expressing CD11c in COVT1 samples. (g) Representative flow cytometric plots and paired analysis of the frequency of CD11c+ cells from RBD‐specific naïve B cells. Numbers indicate percentage of RBD‐specific CD11c+ naïve cells in the drawn gates. (h) Spearman’s correlation analysis of naïve B cells in COVT1 and RBD+ naïve B cells within CD19+ cells in COVT2. r represents Spearman’s rank‐order correlation. (i) Relative percentage of total and RBD+ naïve B cells expressing lambda or kappa light chains in COVT1. (j) Frequency of RBD‐specific ME B cells defined as non‐PB CD19+RBD+IgD− from total CD19+ B cells in HCs, COVT1 and COVT2. (k) Paired analysis of the frequency of RBD‐specific IgG+ ME B cells defined as IgA−IgM−IgD− within total CD19+ in COVT1 and COVT2. (l) Paired analysis of the frequency of RBD‐specific IgM+IgD+ ME B cells defined as non‐PB CD19+CD27+IgM+IgD+ within total CD19+ in COVT1 and COVT2. (m) Representative flow cytometric plots and paired analysis of the frequency of DN2 ME B cells (non‐PB CD19+RBD+CD27−IgD−CD11c+) from total IgD−RBD+ ME B cells. Numbers indicate percentages of RBD‐specific DN2 ME in the drawn gates. Dark‐coloured dots show ICU patients. Bars represent mean ± SEM. The two‐tailed Mann–Whitney U‐test was performed to compare HCs with COVT1 and HCs with COVT2. The Wilcoxon matched‐pairs test was performed to compare COVT1 with COVT2. ns, non‐significant (P > 0.05), *P < 0.05, **P < 0.01 and ***P < 0.001. HCs, n = 11; COVT1, n = 19; COVT2, n = 19.
In addition, RBD‐targeting IgM+IgD+CD27− naïve B cells increased progressively in COVID‐19 patients (Figure 4e). These cells were enriched in CD11c+ early activated B cells in COVT1 and even more in COVT2 (Figure 4f, g). Of note, the frequency of RBD‐specific naïve B cells during convalescence strongly correlated with the frequency of total naïve B cells in the acute phase of infection (Figure 4h), suggesting that the early mobilisation of developing B cells to the periphery and the expansion of naïve B cells could contribute to humoral defence by increasing germline RBD‐specific B cells. To elucidate the mechanisms leading to the sustained enrichment of RBD‐targeting naïve B cells, we measured the plasma concentration of IL‐7, a cytokine involved in B and T lymphopoiesis.33 Compared with healthy controls, COVID‐19 patients showed more IL‐7 during acute infection (Supplementary figure 4e) and its concentration correlated with the frequency of RBD‐specific naïve B cells in COVT2 (Supplementary figure 4f). Additionally, RBD‐specific naïve B cells expressed more Igλ than total naïve B cells (Figure 4i). Given that Igλ light chain has a more varied structural repertoire than Igκ,34 Igλ enrichment may provide the host with the advantage of developing a broader humoral response to SARS‐CoV‐2.
We then analysed whether RBD‐specific ME B cells were induced and maintained throughout the study’s time course. The number of RBD+ ME B cells in COVT1 was significantly greater than in healthy controls and increased even further in COVT2 (Figure 4j). Similarly, there was a sustained increase in the number of RBD‐specific IgG (IgM−IgD−IgA−) class‐switched ME and, to a lesser extent, in RBD‐specific IgM+IgD+CD27+ unswitched ME B cells (Figure 4k, l). The RBD‐specific ME compartment included only a few IgA+ and unswitched IgM+IgD− ME cells, and these subsets did not increase overtime (Supplementary figure 4g). Consistent with our characterisation of B‐cell subsets, RBD‐specific extrafollicular ME B cells, or DN2, expanded during the acute phase of infection, but their frequency dramatically decreased in convalescence (Figure 4m). Of note, enrichment of Igλ usage was also observed in RBD‐specific ME B cells and in IgA+ PBs (Supplementary figure 4h, i).
To assess the persistency of virus‐specific ME B‐cell responses, RBD‐targeting B‐cell subsets were analysed by flow cytometry in a small cohort of non‐hospitalised convalescent individuals 3 months (COVT3) and 6 months (COVT6) PSO (Supplementary figure 5). In agreement with previous reports,14, 17, 18 RBD‐specific B cells persisted up to 6 months after infection (Supplementary figure 5a) and mainly consisted of IgG+ ME B cells (Supplementary figure 5b). Accordingly, RBD‐specific IgG and, to a lesser extent, IgA were significantly higher in sera from COVT3 and COVT6 than in healthy controls (Supplementary figure 5c). Thus, SARS‐CoV‐2 infection promotes a rapid and transient induction of RBD‐specific PBs and extrafollicular ME B cells, as well as a long‐lasting expansion of RBD‐targeting activated naïve B cells and IgG class‐switched ME B cells.
COVID‐19 is associated with a sustained activation of CD4+ and CD8+ T cells
To further characterise the dynamics of immune responses over time in COVID‐19 patients, we simultaneously analysed by multicolour spectral flow cytometry the phenotypic landscape of other circulating lymphoid and myeloid populations using an in‐house‐developed 23‐colour antibody panel (Supplementary table 4). The projection of the data for CD19−CD3+ cells in tSNE space allowed the definition of the main T‐cell subsets, the contribution of each group of samples to T‐cell populations and the expression patterns of relevant T‐cell surface proteins (Figure 5a, b and Supplementary Figure 6a). In parallel, we queried the data by traditional gating (Supplementary figure 6b–d). As reported previously,12, 35 the acute phase of infection was characterised by a striking reduction in the frequency of CD3+ T cells (Figure 5c), which was driven by a decrease in the frequency of CD4+ and, to an even larger extent, CD8+ T cells (Supplementary figure 7a, b). Compared with healthy controls, convalescent individuals still had a significantly decreased frequency of CD3+ cells (Figure 5c) and in particular of CD4+ T helper cells (Supplementary figure 7a), which suggested their persistent loss from the periphery. We then evaluated how SARS‐CoV‐2 infection associated with temporary or long‐lasting perturbations in both CD4 and CD8 T‐cell compartments. Interestingly, the frequency of naïve T cells within total CD4+ T cells was significantly lower in acutely infected patients than in age‐matched healthy controls and further declined during convalescence (Figure 5d). This reduction was associated with a prominent expansion of activated CD38+HLA‐DR+ CD4+ T cells during acute infection and, to a lesser extent, in convalescence (Figure 5e), as observed in other viral infections.36, 37
Figure 5.

COVID‐19 is associated with long‐lasting contraction of naïve T‐cell compartment, CD4+ and CD8+ T‐cell activation, and expansion of circulating T follicular helper cells. (a) Merged tSNE projection of CD3+ cells for healthy controls (HCs; n = 21), COVT1 (n = 25) and COVT2 (n = 20) donors concatenated, with main T‐cell populations indicated by colour. CM, central memory. EM, effector memory. cTfh, circulating T follicular helper T cells. (b) tSNE projections of CD3+ T cells for each sample group. (c) Frequency of CD3+ T cells within live lymphocytes. (d) Frequency of naïve CD4+ T cells (CD27+CD45RA+CCR7+) from total CD4+ T cells. (e) Representative flow cytometric plots (left) and frequency of CD38+HLA‐DR+‐activated CD4+ T cells within total CD4+ T cells. Numbers indicate percentages in the drawn gates. (f) Frequency of circulating cTfh cells (non‐naïve CXCR5+PD‐1+) within total CD4+ T cells. (g) Frequency of activated cTfh cells (CD38+ICOS+) within total cTfh cells. (h) Frequency of naïve CD8+ T cells (CD27+CD45RA+CCR7+) within total CD8+ T cells. (i) Frequency of CM CD8+ (CD27+CD45RA−CCR7+) and (j) EM3 (CD27−CD45RA−CCR7−) T‐cell subsets within total CD8+ T cells. (k) Representative flow cytometric plots and frequency of CD38+HLA‐DR+‐activated CD8+ T cells within total CD8+ T cells. Numbers indicate percentages in the drawn gates. (l) Spearman’s correlation analysis of activated CD4+ T and CD8+ T cells in all samples analysed. r represents Spearman's rank‐order correlation. (c–k) Data are presented as individual dots. Dark‐coloured dots show ICU patients. Bars represent mean ± SEM. The two‐tailed Mann–Whitney U‐test was performed to compare HCs with COVT1 and HCs with COVT2. The Wilcoxon matched‐pairs test was performed to compare COVT1 with COVT2. ns, non‐significant (P > 0.05), *P < 0.05, **P < 0.01 and ***P < 0.001. HCs, n = 21; COVT1, n = 25; COVT2, n = 20.
We then analysed the frequency of circulating T follicular helper (cTfh) cells. Tfh cells are a specialised subset of CD4+ T cells that provide cognate help to antigen‐specific B cells in the germinal centre (GC) to initiate and maintain humoral immune responses.38 These cells include a circulating counterpart, cTfh cells, which co‐express PD‐1 and CXCR5 as their GC‐based equivalents do (Supplementary figure 6c). In general, cTfh cells can be used as a surrogate to evaluate the Tfh cell activity in lymphoid tissues.39 Of note, activated CD38+ICOS+ cTfh cells likely reflect a recent exit from the GC immediately after antigen encounter.40 The frequency of cTfh cells was significantly higher in COVT2 patients than in healthy controls and COVT1 patients, which is consistent with a model of antigen persistency and long‐lasting GC reaction previously suggested by others22 (Figure 5f). Interestingly, recently activated CD38+ICOS+ cTfh cells were more profoundly increased during the acute phase, which probably reflects the peak of GC responses in the early phase of the infection (Figure 5g).
We further explored whether SARS‐CoV‐2 infection promoted perturbations of other CD4+ helper T‐cell subsets, including central memory (CM) T cells, effector memory (EM) 1, 2 and 3 T cells, terminally differentiated effector memory (EMRA) T cells, T regulatory (Treg) cells and T helper 1 (Th1), Th1/17, Th2 and Th17 cells (Supplementary figure 6b). We detected an increase in the frequency of Treg cells during the acute phase of infection and a small but significant increase in the frequencies of Th1 and Th1/17 cells in COVT2 compared with COVT1 upon pairwise comparison, whereas Th2 cells showed a decreased frequency over time (Supplementary figure 7c).
COVID‐19 was also associated with several changes in the CD8+ T‐cell compartment. We observed a significantly reduced frequency of naïve CD8+ T cells during convalescence and an expansion of CM and EM3 CD8+ T cells (Figure 5h–j). However, our analysis showed no differences in the frequency of EM1, EM2 and EMRA CD8+ T cells between groups (Supplementary figure 7d). We also found that activated CD38+HLA‐DR+CD8+ T cells strongly expanded during the acute phase of infection and still persisted at a higher frequency during convalescence compared with healthy controls (Figure 5k). As expected, patients with a greater increase in activated CD4+ T cells also showed a higher frequency of activated CD8+ T cells (Figure 5l). Thus, SARS‐CoV‐2 causes a marked T‐cell loss during the acute phase of infection, which is associated with long‐lasting and coordinated activation of CD4+ and CD8+ T cells and expansion and activation of professional B‐cell‐helping T cells.
SARS‐CoV‐2 infection drives profound changes in the innate immune compartment
Acute SARS‐CoV‐2 infection triggers alterations in circulating innate immune cell subsets, some of which have been associated with COVID‐19 severity.41 Nevertheless, how these perturbations persist during convalescence after viral clearance is unclear. We combined global high‐dimensional mapping via tSNE of CD19−CD3− cells with traditional gating analysis to study significant alterations in the phenotype and frequency of peripheral blood innate immune cell subsets in COVT1 and COVT2 compared with healthy controls (Figure 6a, b and Supplementary figure 8a, b). Although the frequency of total circulating CD11c+HLA‐DR+ myeloid cells among live PBMCs in COVT1 and COVT2 was comparable to that of healthy controls (Supplementary figure 8c), we observed significant differences in the phenotype and subset composition of these myeloid cells. We initially explored the expression of the activation‐induced molecule CD38 and HLA‐DR on circulating myeloid cells. We found a striking induction of CD38 and a reduction in the expression of HLA‐DR during acute infection (Figure 6c, d). Of note, the decrease in HLA‐DR may interfere with proper antigen presentation and has been directly linked to an immunosuppressive phenotype of monocytes during sepsis.5 Indeed, HLA‐DR levels strongly correlated with disease severity defined according to the patient’s oxygen requirement (Supplementary figure 8d). However, these changes were only transient, as CD38 and HLA‐DR returned to their homeostatic expression levels in the convalescent phase (Figure 6c, d).
Figure 6.
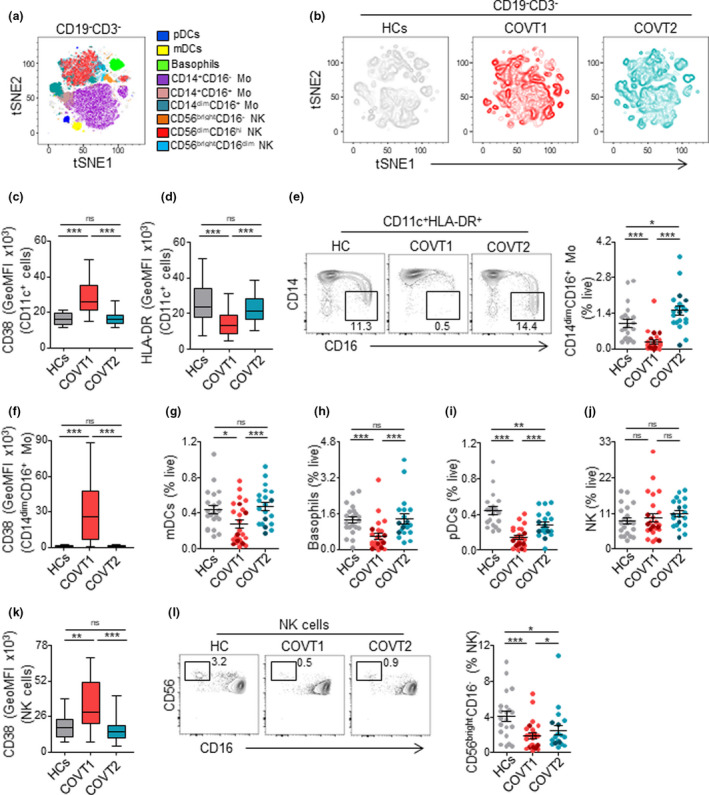
SARS‐CoV‐2 infection associates with transitory and long‐lasting changes in the innate immune compartment. (a) Merged tSNE projection of CD19−CD3− cells for healthy controls (HCs; n = 21), COVT1 (n = 25) and COVT2 (n = 20) samples concatenated and overlaid, with main innate immune cell populations indicated by colour. pDCs, plasmacytoid dendritic cells. mDCs, myeloid dendritic cells. Mo, monocytes. NK, natural killer cells. (b) tSNE projections of CD19−CD3− innate immune cells for each sample group. (c) Geometric mean fluorescence intensity (GeoMFI) of CD38 and (d) HLA‐DR expression in CD19−CD3−CD11c+ myeloid cells from HCs, COVT1 and COVT2. (e) Representative flow cytometric plots and frequency of non‐classical CD14dimCD16+ Mo as a proportion of live cells. Numbers indicate percentages in the drawn gates. (f) GeoMFI of CD38 in CD14dimCD16+ Mo. (g) Frequency of mDCs, (h) basophils, (i) pDCs and (j) NK cells within total live cells. (k) GeoMFI of CD38 in NK cells. (l) Representative flow cytometric plots and frequency of CD56brightCD16− NK cells. Numbers indicate percentages in the drawn gates. Data are presented as individual dots. Dark‐coloured plots show ICU patients. Bars represent mean ± SEM. Two‐tailed Mann–Whitney U‐test was performed to compare HCs with COVT1 and HCs with COVT2. The Wilcoxon matched‐pairs test was performed to compare COVT1 with COVT2 (c, d, f and k). Data are presented in boxplots. Box boundaries represent the 1st and 3rd quartile of the distribution, while the centre line represents the 2nd quartile (median). Whiskers go down to the smallest value and up to the largest. ns, non‐significant (P > 0.05), *P < 0.05, **P < 0.01 and ***P < 0.001. HCs, n = 21; COVT1, n = 25; COVT2, n = 20.
We then analysed the frequency of CD14+CD16− conventional monocytes, CD14+CD16+ intermediate monocytes and CD14dimCD16+ non‐conventional monocytes. Classical CD14+CD16− monocytes remained unaltered, whereas intermediate CD14+CD16+ monocytes showed a higher frequency in COVT2 than in COVT1 and healthy controls (Supplementary figure 8e, f). Furthermore, we observed a robust but transient decrease in the frequency of non‐conventional CD14dimCD16+ monocytes (Figure 6e), which appeared to be strongly activated during acute infection (Figure 6f). Non‐conventional monocytes have a patrolling function and contribute to the antiviral response,42 which may point to their selective recruitment to the inflamed lung upon FcγRIIIA‐mediated activation by virus–IgG immune complexes. Accordingly, non‐conventional monocytes have been shown to be enriched in the lungs of critical COVID‐19 patients.43 The frequency of circulating myeloid dendritic cells (mDCs) and basophils was also strongly reduced in COVT1 compared with healthy controls, but both returned to homeostatic levels two months PSO (Figure 6g, h). Interestingly, we detected increased CD25 expression on basophils from COVT1 compared with COVT2, which suggests basophil activation during the acute phase of infection (Supplementary figure 8g).
Finally, we analysed the frequency of circulating plasmacytoid DCs (pDCs) and natural killer (NK) cells, two subsets of the innate immune system that play crucial protective roles in viral infections. As reported previously,44 the frequency of pDCs significantly decreased during acute infection (Figure 6i). Remarkably, convalescent individuals maintained significantly lower frequency of pDCs compared with healthy controls, even after viral clearance (Figure 6i), suggesting persistent impaired type I IFN responses in infected individuals.
Although our data did not reveal any significant changes in the frequencies of natural killer (NK) cells within live cells, the analysis of CD38 expression indicated an activated phenotype during the acute phase of the infection (Figure 6j, k). Moreover, the characterisation of NK cell subsets based on the relative expression of CD56 and CD16 showed a persistent decrease in the frequency of circulating immature CD56brightCD16− cells and an increase in the proportion of naturally cytotoxic CD56dimCD16high cells within total NK cells (Figure 6l and Supplementary figure 8h). Thus, SARS‐CoV‐2 infection drives profound changes in the circulating innate immune compartment, some of which persist after viral clearance during convalescence.
High‐dimensional analysis reveals coordinated immune responses and their association with disease severity
The immune system is composed of layered defence mechanisms of increasing specificity that protect the host from infection. To investigate the putative associations between innate and adaptive immune trajectories developed upon SARS‐CoV‐2 infection, we performed pairwise correlations across 41 variables that were significantly different among the groups analysed, including virus‐specific antibody titres measured by ELISA and selected immune parameters identified by high‐dimensional flow cytometry (Supplementary data file 1). We found that the global antibody response to SARS‐CoV‐2 positively correlated with the frequency of circulating PBs, extrafollicular DN2 ME B cells, activated CD4+ T cells, cTfh cells and CD8+ T cells. It also positively correlated with CD38 expression on circulating myeloid cells, which suggests a close coordination between innate and adaptive immune responses. However, the level of virus‐specific antibody titres, the percentage of circulating PBs and the frequency of CD19+ cells inversely correlated with the frequency of CD3+ T cells, non‐conventional monocytes and pDCs, which suggests virus‐induced interference with different components of both adaptive and innate immune responses in a fraction of COVID‐19 patients (Figure 7a). Next, principal component analysis (PCA) was performed to examine the general distribution of healthy controls and infected individuals during acute and convalescent stage according to these immune parameters. The analysis revealed that patients during acute infection were notably more distant from healthy controls than the same patients during convalescence in PCA space (Figure 7b). Virus‐specific IgA titres, as well as the frequency of IgG and IgM PBs, activated CD4 T cells and pDCs, were among the variables that mostly contributed to the observed clustering in PC1 space. The frequency of naïve B cells and, to a lesser extent, IgM+IgD+ ME B cells, IgA1 PBs and NP‐specific IgM titres was the immune features that mostly contributed to the clustering in PC2 space (Figure 7c, d). We then used the first two principal components of the PCA to compute the Euclidean distance of all COVT1 to the centroid of the healthy controls. Remarkably, we found a positive correlation between this distance and disease severity defined according to the patient’s oxygen requirement and serum concentration of ferritin and lactate dehydrogenase during acute infection (Figure 7e). Of note, the same analysis performed computing the Euclidean distance of COVT2 samples to the centroid of the healthy controls did not show equivalent correlation patterns (Supplementary figure 9), which suggests that immunological recovery trajectories in convalescence occur independently of disease severity status during acute infection. Thus, cross‐data set correlation and principal component analyses reveal coordinated immune responses in COVID‐19 patients and immune parameters associated with disease severity and inflammation.
Figure 7.

High‐dimensional analysis of all variables studied reveals coordinated immune responses and their association with disease severity (a) Spearman’s correlation mapping of indicated parameters for healthy controls (HCs), COVT1 and COVT2. Spearman’s rank correlation coefficient (ρ) was indicated by heat scale; Spearman’s P‐value significance levels were corrected using the Benjamini–Hochberg method significance (*P < 0.05, **P < 0.01 and ***P < 0.001). PBs, plasmablasts. R3, CXCR3. R4, CXCR4. ME, memory. cTfh, circulating T follicular helper cells. CM, central memory. EM3, effector memory type 3. pDCs, plasmacytoid dendritic cells. mDCs, myeloid dendritic cells. Baso, basophils. NK, natural killer cells. trans, transitional B cells. (b) Principal component analysis (PCA) based on antibody titres, frequencies and GeoMFI of all measured markers (centred and scaled). RBD‐specific parameters were excluded from the analysis. The first component explains 25% of the variation, whereas the second component explains 12.3% of the variation. Each dot represents a donor, and each colour represents a donor group. Dark‐coloured dots show ICU patients. Confidence ellipses for each group are plotted (confidence level set to 95%). (c) Variable correlation plot showing eigenvector‐based coordinates of the top 20 variables in the two‐dimensional space defined by the first two principal components. The relative position of the clinical variables reflects their relationship (positive correlated variables point to the same side of the plot; negative correlated variables point to opposite sides of the plot), while the length of the arrow is proportional to their contribution to the principal components. (d) Relative contribution of the top 10 variables to PC1 (top panel) and PC2 (bottom panel). (a–d) HCs, n = 16; COVT1, n = 25; COVT2, n = 20. (e) Spearman’s correlation analysis of oxygen flow, ferritin and lactate dehydrogenase (LDH) levels in COVID‐19 patients at COVT1 plotted against the Euclidean distance computed using the two first principal components from the PCA of each COVT1 patient to the centroid of the healthy control (HC) group. The HC centroid is computed by averaging PC1 and PC2 for all HCs. Data are presented as individual dots. Dark‐coloured dots show ICU patients. r represents Spearman’s rank‐order correlation.
Discussion
A better understanding of immune changes occurring during COVID‐19 progression and recovery is required to predict disease outcome and development of long‐lasting immune protection. The vast majority of studies so far have focused on the immune profiling of elderly COVID‐19 patients, who have an intrinsic risk to develop severe disease because of ageing of the immune system and potential underlying health conditions. To circumvent this bias, we selected hospitalised patients younger than 65 and compared their immune parameters with equivalent parameters from age‐matched healthy controls. This approach provided new insights into the breadth and kinetics of virus‐specific B‐cell responses to SARS‐CoV‐2 and on the immunological landscape of not aged COVID‐19 patients ranging from moderate to severe.
During acute SARS‐CoV‐2 infection, the presence of virus‐specific antibodies with highly diversified Fc receptor‐associated effector functions is consistent with an early involvement of multiple systemic and mucosal mechanisms of humoral protection, which presumably include immune pathways emerging from the gut.45 The observed decline in RBD‐ and NP‐specific IgM, IgA1 and IgA2 and the progressive increase in RBD‐specific IgG1 during convalescence may reflect two temporal phases of humoral immunity to SARS‐CoV‐2: an initial stage characterised by a rapid release of poorly mutated and highly diversified antibody classes and subclasses, largely produced by short‐lived PBs through extrafollicular pathways, followed by the emergence of high‐affinity IgG released by long‐lived plasma cells presumably emerging from GCs. Indeed, the analysis of virus‐specific B‐cell subset dynamics showed a transient increase in circulating RBD‐specific HLA‐DR+ PBs and extrafollicular DN B cells in acute infection, followed by the waning of early non‐GC‐derived B‐cell responses and sustained expansion of virus‐specific IgG‐dominated B‐cell memory. This breadth and kinetics of humoral responses may be set in place to consolidate robust antibody responses aimed at achieving complete viral clearance and long‐lasting immune protection. Indeed, immune inhibitory signals generated by systemically produced monomeric IgA via the FcαRI receptor could interfere with the clearance of IgG‐opsonised viral particles by phagocytes at a later phase of the infection. When engaged by monomeric forms of systemic IgA, the FcαRI receptor predominantly deploys inhibitory ITIM‐mediated signals to phagocytes and NK cells.46
Another possible explanation for the reduction in virus‐specific IgA1 and IgA2 two months PSO is the relocation of IgA‐secreting PBs to mucosal inflamed tissues, including the upper respiratory tract. Indeed, acutely infected patients showed increased proportion of CXCR3+CXCR4− PBs that are programmed to migrate to inflamed tissues rather than bone marrow niches. Consistently, two earlier reports described elevated SARS‐CoV‐2‐specific IgA antibodies in nasal fluids, tears and saliva of infected individuals.27, 47 However, a recent study failed to detect PBs in the lung of deceased COVID‐19 patients.48
Overall, our data are in agreement with recent reports showing detectable S‐specific IgG and neutralising antibodies at 6–8 months PSO and rapid decline in circulating IgA titres.14, 15, 16, 17, 18 Moreover, we provided additional information on the dynamics of SARS‐CoV‐2‐specific IgG subclasses in different phases of the infection. Compared with healthy controls, COVID‐19 patients showed higher titres of all RBD‐ and NP‐specific IgG subclasses, including poorly reported IgG2 and IgG4 subclasses, during acute infection and convalescence. Yet, only RBD‐specific IgG1 increased over time, presumably because this antibody mostly originated from delayed GC responses. Finally, the magnitude and kinetics of IgG subclasses were highly heterogeneous between infected individuals. Thus, more studies with larger cohorts and later time points are required to explore the contribution of each IgG subclass to immune protection and recovery trajectories.
Recent reports have shown sustained development of virus‐targeting ME B cells expressing IgG and a continuous evolution of the virus‐specific antibody repertoire over time,14, 16, 17 indicating that SARS‐CoV‐2 infection promotes long‐lasting GC‐derived humoral responses. Similarly, we detected a sustained increase in RBD‐specific IgG ME B cells that lasted for 6 months PSO. In addition, our analysis revealed that SARS‐CoV‐2 infection profoundly shaped the overall B‐cell memory compartment, promoting a sustained increase in the IgG1+ ME subset and a persistent contraction of unswitched CD27+IgM+IgD+ ME B cells. Loss of IgM+ ME B cells in COVID‐19 patients has been previously described and associated with increased mortality and superimposed infections.49 However, in our cohort of COVID‐19 patients, the frequency of CD27+IgM+IgD+ ME B cells positively correlated with several markers of inflammation and NP‐specific IgM titres. These results suggest that, in a subset of patients, humoral responses might diverge towards less efficient and non‐neutralising NP‐targeting antibodies. These antibodies may arise from the reactivation of B‐cell subsets primed during previous seasonal coronavirus infections and therefore expressing cross‐reactive but poorly protective IgM.
The mapping of global and SARS‐CoV‐2‐specific B‐cell subsets in COVID‐19 patients younger than 65 years also revealed some striking changes in mature B‐cell precursors. The increased frequency in CD21low transitional and naïve B cells in COVT1 patients compared with age‐matched healthy controls and COVT2 patients was consistent with the mobilisation of B‐cell precursors to the periphery following virus‐induced inflammation.30 In the presence of massive immune sensing of viral particles, the immune system may be evolutionary programmed to recruit mature B‐cell precursors from the bone marrow into the periphery as a ‘last‐ditch’ defence against invading virions. Multiple inflammation‐induced cytokines, including IL‐7, may contribute to this process.33
In agreement with this hypothesis, we documented a significant expansion of RBD‐specific naïve B cells enriched in Igλ that were largely activated and more numerous in COVID‐19 patients with increased serum IL‐7 and expanded naïve B cells. Acute infection was also characterised by an increased proportion of immature CD21low transitional B cells and, as recently reported,25 an increase in serum BAFF, an homeostatic regulator of peripheral B lymphopoiesis. Altogether, these results support a scenario where the de novo recruitment of mature B‐cell precursors to the periphery promotes the emergence of B cells expressing virus‐reactive specificities constitutively engraved in the germline repertoire.
Consistent with this interpretation, stereotypic neutralising antibodies with no or very few somatic mutations have been recently described in both the naïve and ME B‐cell repertoires from COVID‐19 patients.20, 21, 22, 23, 24, 25 Antibodies expressed by naïve and transitional B cells have notoriously different structural properties compared with mature antibodies expressed by GC‐selected B cells. Compared with the latter, naïve and transitional B cells express antibodies that have a generally longer H‐CDR3 in addition to increased autoreactivity and polyreactivity.50 These properties may be central to the initial recognition and neutralisation of the RBD‐containing S protein on SARS‐CoV‐2. Antigen‐activated naïve and transitional B cells could then transit through the GC to increase their affinity for viral antigens while redeeming their potential harmful autoreactivity. Therefore, the expansion of a naïve B‐cell repertoire expressing BCRs reactive to a wider range of neutralising epitopes across the RBD of the S antigen may confer a functional advantage with respect to the initiation of protective antibody responses to SARS‐CoV‐2. However, the mobilisation of B‐cell precursors to the periphery could also promote the emergence of pathogenic antibody responses to self‐antigens because of defective central tolerance.30, 50 Indeed, the frequency of naïve B cells was a key variable to define distinct immune signatures associated with disease severity and inflammation in COVID‐19 patients. More studies are needed to evaluate longitudinal changes in the naïve Ig repertoire upon infection and their putative correlation with the onset of autoimmunity.
To further characterise the coordination of humoral immunity with other arms of the immune system in COVID‐19 patients, we analysed other circulating lymphoid and myeloid populations. Consistent with recent studies,12, 51 we showed robust activation of CD4+ and CD8+ T cells, including cTfh cells, in acutely infected patients and a strong correlation of T‐cell activation with several hallmarks of humoral responses. Our longitudinal analysis additionally revealed residual activation of the T‐cell pool and progressive expansion of cTfh, Th1 and Th1/17 CD4+ T cells and subsets of memory CD8+ T cells during convalescence. These results suggest the continuous evolution of adaptive immunity to SARS‐CoV‐2 and are consistent with viral persistence in mucosal reservoirs or peripheral lymph nodes.18
In agreement with earlier reports,52 severe COVID‐19 patients with increased inflammation also showed augmented T‐cell activation and humoral responses during acute infection. This observation may be explained by the presence of higher viral loads or prolonged periods of active viral replication in severe patients. However, a pathogenic role of adaptive immunity cannot be completely ruled out.
The role of innate immune responses in COVID‐19 pathogenesis has been extensively described.41 SARS‐CoV‐2 infection induces delayed and dysregulated IFN responses, thereby impairing proper priming of adaptive immune responses. In concordance with recent literature,44, 53 our data revealed a dramatic decrease in pDCs in acute infection that was marginally restored during convalescence. Although the depletion of circulating pDCs during acute infection could be suggestive of a targeted recruitment into virally infected tissues, their persistent lower frequency during convalescence argues in favor of a massive pDC apoptosis because of their activation through IFN signalling.54 Moreover, being pDCs the main source of type I IFN, their loss may be linked to the aberrant interferon responses displayed by severe COVID‐19 patients. Indeed, our study indicated that pDC depletion contributed to the definition of distinct immune signatures that correlated with poor prognosis and inflammation.
Moreover, as reported by others,5, 55 we found that acute SARS‐CoV‐2 infection markedly reduced HLA‐DR expression on circulating myeloid cells and depleted non‐conventional monocytes from circulation. Furthermore, our data revealed a negative correlation between HLA‐DR expression and disease severity. Indeed, such decrease in HLA‐DR expression has been recently associated with defective antigen presentation driven by an excessive release of IL‐6.5 Finally, we found persistent changes in the circulating NK cell pool of patients infected with SARS‐CoV‐2. Our data are in line with previous reports suggesting a dysfunctional/exhausted phenotype of NK cells in COVID‐19 patients as a consequence of hyperinflammation.56
In summary, our in‐depth characterisation of SARS‐CoV‐2‐specific B‐cell responses revealed a previously unappreciated expansion of virus‐specific naïve‐like B cells over time, perhaps through the continuous mobilisation of mature B‐cell precursors to the periphery. Moreover, our results consolidated previous findings on the immune response dynamics occurring in COVID‐19 patients, showing both transient and long‐lasting changes associated with disease severity and development of immune memory.
Methods
Experimental model and subject details
Study cohort
Blood samples were collected from COVID‐19 hospitalised patients (n = 25) in acute phase of infection (COVT1) at the Hospital del Mar (Barcelona, Spain), with patient informed consent. Only patients with confirmed SARS‐CoV‐2 infection by reverse transcription–quantitative polymerase chain reaction (RT‐qPCR) of nasopharyngeal swab were included. All COVT1 samples were collected within the first 14 days following symptom onset, being the median time from symptom onset to blood testing 11 days [8–14]. A follow‐up blood sample was collected from 20 out of the 25 initial participants two months after first sample collection (COVT2), being the median time from symptom onset to COVT2 blood testing 70 days [59–84]. The median age of patients was 51 years. 52% were males. The median length of stay from hospitalisation to discharge was 16 days [5–90]. Clinical laboratory data were collected from the date closest to the first day of hospitalisation. Blood samples from age‐matched healthy donors (n = 21) were also collected. In addition, blood samples were obtained from 5 COVID‐19 convalescent individuals 3 and 6 months after symptom onset to assess the persistency of humoral responses. All research subjects were pseudonymised by replacing directly identifiable variables with an ID immediately when collecting data. Patients ID were not known to anyone outside the research group. Demographic and clinical data of patients included in the study are summarised in Supplementary table 1. All procedures followed were approved by the Ethical Committee for Clinical Investigation of the Institut Hospital del Mar d’Investigacions Mèdiques (Number 2020/9189/I).
Sample collection and processing
For all COVID‐19 patients and healthy volunteers, sera were collected from whole blood in silica‐treated tubes (BD Biosciences, Franklin Lakes, USA), heat‐inactivated at 56°C for 1 h and stored at −80°C prior to use. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood collected with EDTA anticoagulant via Ficoll–Paque Premium (Cytiva, Freiburg, Germany) following the manufacturer’s instructions. PBMCs were counted using Turk solution, resuspended in foetal bovine serum (FBS; Gibco, Riverside, MA, USA) with 10% dimethyl sulphoxide (DMSO; Sigma‐Aldrich, St. Louis, USA) and stored in liquid nitrogen prior to use.
Production of recombinant SARS‐CoV‐2 proteins
The pCAGGS RBD construct, encoding for the receptor‐binding domain of the SARS‐CoV‐2 Spike protein (amino acids 319–541 of the Spike protein) along with the signal peptide plus a hexahistidine tag, was provided by Dr Krammer (Mount Sinai School of Medicine, NY USA). The pLVX‐EF1alpha‐nCoV2019‐N‐2xStrep‐IRES‐Puro construct, encoding for the full‐length SARS‐CoV‐2 nucleocapsid protein (NP) fused to a double Strep‐tag at the C‐terminus, was a gift from Dr Krogan (University of California, San Francisco USA). Recombinant proteins were expressed in‐house in Expi293F human cells (Thermo Fisher, Waltham, MA, USA) by transfection of the cells with purified DNA and polyethylenimine (PEI). For secreted RBD proteins, cells were harvested 3 days post‐transfection and RBD‐containing supernatants were collected by centrifugation at 20000 g for 15 min. RBD proteins were purified in HiTrap Ni Columns in an automated fast protein liquid chromatography (FPLC; ÄKTA avant, Cytiva), concentrated through 10 kDa Amicon centrifugal filter units (Merck Millipore, Darmstadt, Germany) and resuspended in PBS. For NP, cell lysates from transfected cells were centrifuged at 20000 g for 20 min and the supernatant was loaded into a 5‐mL Strep‐Tactin XT column. The eluted nucleocapsid was concentrated through a 10 kDa Amicon centrifugal filter unit and purified with SEC (Sephadex 10/300) in PBS.
Enzyme‐linked immunosorbent assay (ELISA)
ELISAs performed in this study were adapted from previously established protocols.45 96‐well half‐area flat‐bottom high‐bind microplates (Corning, New York, USA) were coated overnight at 4°C with each respective recombinant viral protein at 2 µg mL−1 in PBS (30 µL per well) or with PBS alone. Plates were washed with PBS 0.05% Tween‐20 (PBS‐T) and blocked with blocking buffer (PBS containing 1.5% bovine serum albumin, BSA) for 2 h at RT. Serum samples were serially diluted in PBS‐T supplemented with 1% BSA and added to the viral protein‐ or PBS‐coated plates for 2 h at RT. After washing, plates were incubated with horseradish peroxidase (HRP)‐conjugated anti‐human Ig secondary antibodies diluted in PBS‐T supplemented with 1% BSA for 45 min at RT. Plates were washed 5 times with PBS‐T and developed with TMB substrate reagent set (BD bioscience) with development reaction stopped with 1 M H2SO4. Absorbance was measured at 450 nm on a microplate reader (Infinite 200 PRO, Tecan, Männedorf, Switzerland). To detect RBD‐specific and NP‐specific IgM, IgA and IgG, goat HRP‐conjugated anti‐human IgA and F(ab’)2 anti‐human IgG and IgM (Southern Biotech, Birmingham, USA) were used at a 1:4000 dilution. To measure RBD and NP‐specific IgG subclasses, HRP‐conjugated mouse anti‐human IgG1, IgG2, IgG3 and IgG4 (Southern Biotech) were used at a 1:3000 dilution. To detect SARS‐CoV‐2‐specific IgA1 and IgA2, HRP‐conjugated mouse anti‐human IgA1 or IgA2 (Southern Biotech) was used at a dilution of 1:2000 and 1:4000, respectively. To quantitate the level of each viral antigen‐specific antibody class or subclasses, optical density (OD) values were calculated after subtraction of background (OD450 of serum dilutions on PBS‐coated plates) and the area under the curve (AUC) derived from optical density measurements of six serial dilutions was determined using Prism 8 (GraphPad Software; San Diego, USA). AUC values below an established cut‐off were replaced by the AUC value of the cut‐off for plotting and calculation purposes. Negative threshold values were set using healthy control AUC levels plus 2 times the standard deviations of the mean.
The concentration of interleukin‐7 (IL‐7) was measured by a sandwich ELISA kit (PeproTech, London, UK) following the manufacturer’s instructions on 50 µL plasma samples. The levels of IL‐7 were expressed as pg mL−1 of plasma.
Spectral flow cytometry
For spectral flow cytometry, frozen PBMCs were thawed and resuspended in 2 mL of LIVE/DEAD Fixable Yellow Dead Cell Stain Kit (Thermo Fisher), incubated for 30 min at RT and stained with two different fluorophore‐conjugated antibody cocktails (Supplementary tables 2 and 4). For staining, a sequential approach was followed. Anti‐CCR7 antibody was added first and incubated for 10 min. Then, all other anti‐chemokine receptor antibodies (CXCR3, CXCR4, CXCR5, CCR4 and CCR6 for MIX3, and CXCR3 and CXCR4 for MIX 1) were added to the corresponding tubes and incubated for 10 min. After that, the remaining antibodies were added and incubated for 20 min (MIX 1) or 30 min (MIX 3). Stained cells resuspended in 4% paraformaldehyde in PBS, and incubated for 12 min in ice. Fixed cells were then washed, resuspended in PBS and acquired using the Aurora spectral analyser (Cytek Biosciences, Fremont, CA, USA). Data were analysed using FlowJo V10.6.2 software (TreeStar Inc., Ashland, USA).
Flow cytometric detection of SARS‐CoV‐2‐RBD‐specific B cells
1.5 million frozen PBMCs were thawed, centrifuged and resuspended in a 1:20 000 PBS dilution of LIVE/DEAD Fixable Yellow Dead Cell Stain Kit (Thermo Fisher) for 30 min at RT to exclude dead cells. Cells were then washed with PBS and incubated with 13.2 pmol SARS‐CoV‐2 RBD‐HIS Biotinylated Recombinant Protein (Sino Biological Inc., Beijing, China) diluted in 50 µL 1X PBS supplemented with 0.2% BSA and 2 mM EDTA for 30 min in ice. Cells were washed again and stained with the MIX 2 antibody cocktail (Supplementary table 3) plus 1.32 pmol Streptavidin Alexa Fluor 647 (Thermo Fisher) for 30 min. Stained cells were washed, fixed with 4% paraformaldehyde in PBS for 12 min and acquired with LSR Fortessa (BD Biosciences). Data were further analysed with FlowJo V10.6.2 software.
Alternatively, 6.6 pmol SARS‐CoV‐2 RBD‐HIS Biotinylated Recombinant Protein was incubated for 1 h in PBS with 0.94 pmol Streptavidin Alexa Fluor 647 and 0.94 pmol Streptavidin Alexa Fluor 488 (Thermo Fisher), separately. Meanwhile, 1.5 million frozen PBMCs were thawed, centrifuged and resuspended in 50 µL PBS. Next, cells were incubated in the same staining tube with the labelled RBD probes for 20 min in ice. Cells were then washed and stained with the MIX 2 antibody cocktail for 10 min using anti‐human IgA AmCyan instead of anti‐human IgA FITC (Supplementary table 3) and DAPI fluorescent dye (Sigma‐Aldrich). Stained cells were washed and resuspended in PBS 2% FBS and acquired on LSR Fortessa (BD Bioscience).
Data analysis and visualisation
t‐Distributed stochastic neighbour embedding (tSNE) analyses were performed with FlowJo V10.6.2 software. tSNE analysis was performed using equal sampling from each FCS file, with 1000 iterations, a perplexity of 30, a Barnes‐Hut gradient algorithm and exact KNN algorithm.
GraphPad Prism (version 8.0) and R (version 3.6.3; R Core Team (2019). R: A Language and Environment for Statistical Computing) were used to conduct statistical analyses. For each experiment, the type of statistical test, summary statistics and levels of significance were specified in the figures and corresponding legends. The correlation analysis was performed in R. We performed data imputation of the missing variables using predictive mean matching implemented in the mice package.57 Pairwise Spearman's rank correlations between 41 variables (including antibody titres measured by ELISA and immune parameters defined by high‐dimensional flow cytometry; Supplementary data file 1) for COVT1 (n = 25), COVT2 (n = 20) and healthy controls (n = 16) were calculated and visualised as a correlogram using R package corrplot (Taiyun Wei and Viliam Simko (2017). R package ‘corrplot’: Visualization of a Correlation Matrix). Spearman’s rank correlation coefficient (ρ) was indicated by heat scale; significance was indicated by *P < 0.05, **P < 0.01 and ***P < 0.001; FDR correction was performed using the Benjamini–Hochberg procedure at the FDR < 0.05 significance threshold. Principal component analysis (PCA) was used to identify the most important features from 41 variables (including antibody titres and immune parameters; Supplementary data file 1) using COVT1 (n = 25), COVT2 (n = 20) and healthy controls (n = 16). The PCA was conducted using the ‘prcomp’ function in R and visualised using the ‘factoextra’ package (Kassambara A, and Mundt F. factoextra: Extract and Visualize the Results of Multivariate Data Analyses, 2020).
Conflict of interest
The authors declare that they have no competing financial interests.
Author contributions
Leire de Campos‐Mata: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Resources; Visualization; Writing‐original draft; Writing‐review & editing. Sonia Tejedor Vaquero: Conceptualization; Data curation; Formal analysis; Methodology; Resources; Visualization; Writing‐review & editing. Roser Tachó‐Piñot: Investigation; Methodology; Writing‐review & editing. Janet Piñero: Data curation; Formal analysis; Investigation; Visualization. Emilie K Grasset: Data curation; Writing‐review & editing. Itziar Arrieta: Resources; Writing‐review & editing. Natalia Rodrigo Melero: Resources. Carlo Carolis: Resources. Juan P Horcajada: Investigation; Writing‐review & editing. Andrea Cerutti: Conceptualization; Investigation; Writing‐original draft; Writing‐review & editing. Judit Villar‐Garcia: Investigation; Project administration; Resources; Writing‐review & editing. Giuliana Magri: Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing‐original draft; Writing‐review & editing.
Data availability statement
All data are available in the main text or the supplementary materials. Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author, Giuliana Magri (gmagri@imim.es).
Supporting information
Acknowledgments
We acknowledge the patients and the Parc de Salut Mar MARBiobanc (PT17/0015/0011) integrated in the Spanish National Biobanks Network from ISCIII for their collaboration. MARBiobanc’s work was supported by grants from Instituto de Salud Carlos III/FEDER (PT17/0015/0011) and the ‘Xarxa de Bancs de tumors’ sponsored by Pla Director d'Oncologia de Catalunya (XBTC). This study was supported by the COVID‐19 call grant from Generalitat de Catalunya, Department of Health (to GM and LDCM), and grant Miguel Servet Research Program (to GM).
References
- 1.WHO Coronavirus Disease (COVID‐19) Dashboard. Geneva, World Heal. Organ; 2020. https://covid19.who.int/ (accessed 12 August 2021).
- 2.Guan W, Ni Z, Hu Yet al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 2020; 382: 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID‐19: consider cytokine storm syndromes and immunosuppression. Lancet 2020; 395: 1033–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID‐19: immunity, inflammation and intervention. Nat Rev Immunol 2020; 20: 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giamarellos‐Bourboulis EJ, Netea MG, Rovina Net al. Complex immune dysregulation in COVID‐19 patients with severe respiratory failure. Cell Host Microbe 2020; 27: 992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hadjadj J, Yatim N, Barnabei Let al. Impaired type I interferon activity and inflammatory responses in severe COVID‐19 patients. Science 2020; 369: 718–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mangalmurti N, Hunter CA. Cytokine storms: understanding COVID‐19. Immunity 2020; 53: 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bastard P, Rosen LB, Zhang Qet al. Autoantibodies against type I IFNs in patients with life‐threatening COVID‐19. Science 2020; 370: eabd4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuo Y, Estes SK, Ali RAet al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID‐19. Sci Transl Med 2020; 12: eabd3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang EY, Mao T, Klein Jet al. Diverse functional autoantibodies in patients with COVID‐19. Nature 2021; 595: 283–288. [DOI] [PubMed] [Google Scholar]
- 11.Le Bert N, Tan AT, Kunasegaran Ket al. SARS‐CoV‐2‐specific T cell immunity in cases of COVID‐19 and SARS, and uninfected controls. Nature 2020; 584: 457–462. [DOI] [PubMed] [Google Scholar]
- 12.Mathew D, Giles JR, Baxter AEet al. Deep immune profiling of COVID‐19 patients reveals distinct immunotypes with therapeutic implications. Science 2020; 369: eabc8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peng Y, Mentzer AJ, Liu Get al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS‐CoV‐2 in UK convalescent individuals following COVID‐19. Nat Immunol 2020; 21: 1336–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dan JM, Mateus J, Kato Yet al. Immunological memory to SARS‐CoV‐2 assessed for up to 8 months after infection. Science 2021; 371: eabf4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wajnberg A, Amanat F, Firpo Aet al. Robust neutralizing antibodies to SARS‐CoV‐2 infection persist for months. Science 2020; 370: 1227–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rodda LB, Netland J, Shehata Let al. Functional SARS‐CoV‐2‐specific immune memory persists after mild COVID‐19. Cell 2021; 184: 169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartley GE, Edwards ESJ, Aui PMet al. Rapid generation of durable B cell memory to SARS‐CoV‐2 spike and nucleocapsid proteins in COVID‐19 and convalescence. Sci Immunol 2020; 5: eabf8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gaebler C, Wang Z, Lorenzi JCCet al. Evolution of antibody immunity to SARS‐CoV‐2. Nature 2021; 591: 639–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sokal A, Chappert P, Barba‐Spaeth Get al. Maturation and persistence of the anti‐SARS‐CoV‐2 memory B cell response. Cell 2021; 184: 1201–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreer C, Zehner M, Weber Tet al. Longitudinal isolation of potent near‐germline SARS‐CoV‐2‐neutralizing antibodies from COVID‐19 patients. Cell 2020; 182: 1663–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SI, Noh J, Kim Set al. Stereotypic neutralizing VH antibodies against SARS‐CoV‐2 spike protein receptor binding domain in patients with COVID‐19 and healthy individuals. Sci Transl Med 2021; 13: eabd6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robbiani DF, Gaebler C, Muecksch Fet al. Convergent antibody responses to SARS‐CoV‐2 in convalescent individuals. Nature 2020; 584: 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barnes CO, West AP, Huey‐Tubman KEet al. Structures of human antibodies bound to SARS‐CoV‐2 spike reveal common epitopes and recurrent features of antibodies. Cell 2020; 182: 828–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seydoux E, Homad LJ, Maccamy AJ, Pancera M, Mcguire AT, Stamatatos L. Analysis of a SARS‐CoV‐2‐infected individual reveals development of potent neutralizing antibodies with limited somatic mutation. Immunity 2020; 53: 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schultheiß C, Paschold L, Simnica Det al. Next‐Generation Sequencing of T and B Cell Receptor Repertoires from COVID‐19 Patients Showed Signatures Associated with Severity of Disease. Immunity 2020; 53: 442–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feldman J, Bals J, & Altomare CG et al. Naive human B cells engage the receptor binding domain of SARS‐CoV‐2, variants of concern, and related sarbecoviruses. bioRxiv 2021. 10.1101/2021.02.02.429458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sterlin D, Mathian A, Miyara Met al. IgA dominates the early neutralizing antibody response to SARS‐CoV‐2. Sci Transl Med 2020; 13: eabd2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woodruff MC, Ramonell RP, Nguyen DCet al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID‐19. Nat Immunol 2020; 21: 1506–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muehlinghaus G, Cigliano L, Huehn Set al. Regulation of CXCR3 and CXCR4 expression during terminal differentiation of memory B cells into plasma cells. Blood 2005; 105: 3965–3971. [DOI] [PubMed] [Google Scholar]
- 30.Cain D, Kondo M, Chen H, Kelsoe G. Effects of acute and chronic inflammation on B‐cell development and differentiation. J Invest Dermatol 2009; 129: 266–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 2011; 89: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanz I, Wei C, Jenks SAet al. Challenges and opportunities for consistent classification of human B cell and plasma cell populations. Front Immunol 2019; 10: 2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ceredig R, Rolink AG. The key role of IL‐7 in lymphopoiesis. Semin Immunol 2012; 24: 159–164. [DOI] [PubMed] [Google Scholar]
- 34.Chailyan A, Marcatili P, Cirillo D, Tramontano A. Structural repertoire of immunoglobulin λ light chains. Proteins 2011; 79: 1513–1524. [DOI] [PubMed] [Google Scholar]
- 35.Chen Z, John Wherry E. T cell responses in patients with COVID‐19. Nat Rev Immunol 2020; 20: 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilkinson TM, Li CKF, Chui CSCet al. Preexisting influenza‐specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med 2012; 18: 274–280. [DOI] [PubMed] [Google Scholar]
- 37.Miller JD, van der Most RG, Akondy RSet al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity 2008; 28: 710–722. [DOI] [PubMed] [Google Scholar]
- 38.Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity 2019; 50: 1132–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morita R, Schmitt N, Bentebibel SEet al. Human Blood CXCR5+CD4+ T Cells Are Counterparts of T Follicular Cells and Contain Specific Subsets that Differentially Support Antibody Secretion. Immunity 2011; 34: 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vella LA, Buggert M, Manne Set al. T follicular helper cells in human efferent lymph retain lymphoid characteristics. J Clin Invest 2019; 129: 3185–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schultze JL, Aschenbrenner AC. COVID‐19 and the human innate immune system. Cell 2021; 184: 1671–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Narasimhan PB, Marcovecchio P, Hamers AAJ, Hedrick CC. Nonclassical monocytes in health and disease. Ann Rev Immunol 2019; 37: 439–456. [DOI] [PubMed] [Google Scholar]
- 43.Sánchez‐Cerrillo I, Landete P, Aldave Bet al. COVID‐19 severity associates with pulmonary redistribution of CD1c+ DCs and inflammatory transitional and nonclassical monocytes. J Clin Invest 2020; 130: 6290–6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou R, To KKW, Wong YCet al. Acute SARS‐CoV‐2 infection impairs dendritic cell and T cell responses. Immunity 2020; 53: 864–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Campos Mata L, Piñero J,Vaquero STet al. SARS‐CoV‐2‐specific antibody profiles distinguish patients with moderate from severe COVID‐19. medRxiv 2020; 10.1101/2020.12.18.20248461. [DOI]
- 46.Pasquier B, Launay P, Kanamaru Yet al. Identification of FcαRI as an inhibitory receptor that controls inflammation: dual role of FcRγ ITAM. Immunity 2005; 22: 31–42. [DOI] [PubMed] [Google Scholar]
- 47.Cervia C, Nilsson J, Zurbuchen Yet al. Systemic and mucosal antibody responses specific to SARS‐CoV‐2 during mild versus severe COVID‐19. J Allergy Clin Immunol 2021; 147: 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferreira‐Gomes M, Kruglov A, Durek Pet al. SARS‐CoV‐2 in severe COVID‐19 induces a TGF‐β‐dominated chronic immune response that does not target itself. Nat Commun 2021; 12: 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lenti MV, Aronico N, Pellegrino Iet al. Depletion of circulating IgM memory B cells predicts unfavourable outcome in COVID‐19. Sci Rep 2020; 10: 20836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science 2003; 301: 1374–1377. [DOI] [PubMed] [Google Scholar]
- 51.Weiskopf D, Schmitz KS, Raadsen MPet al. Phenotype and kinetics of SARS‐CoV‐2‐specific T cells in COVID‐19 patients with acute respiratory distress syndrome. Sci Immunol 2020; 5: eabd2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brodin P. Immune determinants of COVID‐19 disease presentation and severity. Nat Med 2021; 27: 28–33. [DOI] [PubMed] [Google Scholar]
- 53.Kuri‐Cervantes L, Pampena MB, Meng Wet al. Comprehensive mapping of immune perturbations associated with severe COVID‐19. Sci Immunol 2020; 5: eabd7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J Exp Med 2011; 208: 2367–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schulte‐Schrepping J, Reusch N, Paclik Det al. Severe COVID‐19 Is marked by a dysregulated myeloid cell compartment. Cell 2020; 182: 1419–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Eeden C, Khan L, Osman MS, Tervaert JWC. Natural killer cell dysfunction and its role in COVID‐19. Int J Mol Sci 2020; 21: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Buuren S, Groothuis‐Oudshoorn K. mice: multivariate imputation by chained equations in R. J Stat Softw 2011; 45: 1–67. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials. Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author, Giuliana Magri (gmagri@imim.es).