

SUMMARY
Ligands bind to an occluded orthosteric ligand-binding pocket within the nuclear receptor (NR) ligand-binding domain (LBD). Molecular simulations have revealed theoretical ligand entry/exit pathways to the orthosteric pocket; however, it remains unclear whether ligand binding proceeds through induced fit or conformational selection mechanisms. Here, using NMR spectroscopy, ITC, and SPR analysis, we provide evidence that structurally distinct agonists bind PPARγ via a two-step induced fit mechanism involving an initial fast kinetic step followed by a slow conformational change. The agonist encounter complex binding pose is suggested in crystal structures where ligands bind to a surface pore suggested as a ligand entry site in molecular simulations. Our findings suggest an activation mechanism for PPARγ where agonist binding occurs through an initial encounter complex followed by a transition of the ligand into the final binding pose within the orthosteric pocket inducing a transcriptionally active conformation.
eTOC Blurb
Shang and Kojetin use NMR and biophysical methods to study the binding mechanism of PPARγ agonist ligands. This study supports a model by which ligand binding to PPARγ occurs through an induced fit mechanism involving an initial encounter complex followed by a conformational change into the final bound state.
Graphical Abstract

INTRODUCTION
Nuclear receptors (NRs) comprise a superfamily of transcription factors that evolved to bind and functionally respond to endogenous small molecule ligands (Holzer et al., 2017). NRs contain a conserved domain organization including a central DNA-binding domain flanked by two regulatory regions, a disordered N-terminal activation function-1 (AF-1) domain and a C-terminal ligand-binding domain (LBD) containing the activation function-2 (AF-2) coregulator interaction surface. The LBD harbors the orthosteric ligand-binding pocket, an occluded region within the core of the NR LBD where endogenous and synthetic NR ligands bind and regulate NR activity. Ligand binding affects the conformation of the AF-2 surface and changes the binding affinity for chromatin remodeling transcriptional coregulator proteins resulting in activation or repression of gene transcription (Kojetin and Burris, 2013; Santos et al., 2011).
Crystal structures have defined static active and inactive/repressive conformations of NR LBDs bound to ligands that enable binding of transcriptional coactivator and corepressor proteins, respectively, by stabilizing specific conformations of the AF-2 helix 12 (Weikum et al., 2018). However, mechanistically it remains poorly understood how ligands engage the LBD and enter the orthosteric ligand-binding pocket—whether ligand binding occurs through conformational selection or induced fit mechanisms (Zhou, 2010). In the conformational selection scenario, ligand binding selectively binds to and selects a particular conformation that is populated within the dynamic LBD conformational ensemble. In the induced fit scenario, ligand binding occurs through an encounter complex and induces or pushes the LBD conformational ensemble into the final ligand-bound complex.
In NR LBD crystal structures, ligands bound to orthosteric pockets are occluded from solvent suggesting an induced fit binding mechanism. A recent review of molecular simulations on various NR LBDs identified six potential locations involved in ligand entry and exit pathways to the NR orthosteric pocket (Fischer and Smieško, 2019). Most molecular simulation studies focused on ligand egress or unbinding from the orthosteric pocket. However, coarse grained simulations on farnesoid X receptor (FXR) suggest ligand binding occurs through induced fit mechanism (Souza et al., 2020) via an orthosteric pocket entry site that was also observed in simulations of peroxisome proliferator-activated receptor gamma (PPARγ) (Aci-Sèche et al., 2011). Molecular simulations of ligand binding to steroid receptors including androgen receptor (AR), estrogen receptor alpha (ERα), glucocorticoid receptor (GR), mineralocorticoid receptor (MR), and progesterone receptor (PR) also suggest an induced fit mechanism (Edman et al., 2015; Grebner et al., 2017), although the site of ligand entry into the orthosteric pocket is different than FXR (Souza et al., 2020) and PPARγ (Aci-Sèche et al., 2011). Indeed, ligand binding to nuclear receptors is often described to induce an active conformation (Bruning et al., 2007, 2010; Huang et al., 2010; Okafor et al., 2019; Rastinejad et al., 2013; Sladek, 2011; Weikum et al., 2018). However, there is evidence from NMR studies on PPARγ that in the absence of ligand the apo-LBD exchanges between transcriptionally active and transcriptionally inactive/repressive conformations (Shang et al., 2020) suggesting a role for conformational selection in the ligand binding mechanism of NR agonists, which are thought to stabilize an active conformation from a dynamic ensemble of active and inactive/repressive conformations (Batista and Martínez, 2015; Bruning et al., 2010; Chrisman et al., 2018; Cronet et al., 2001; Hughes et al., 2012; Johnson et al., 2000; Köhler et al., 2020; Lu et al., 2006; Merk et al., 2019; Shang et al., 2019; Singarapu et al., 2011). Taken together, these observations stem from the ability of the ligand-bound NR LBD to exert specific functions such as coactivator interaction and transcription, but not directly on the mechanism of ligand binding to the orthosteric pocket.
Here, we use protein NMR chemical shift perturbation (CSP)- analysis, which provides atomic resolution structural insight into conformational selection and induced fit binding mechanisms (Kovrigin, 2012), to study the mechanism of agonist binding to PPARγ. NMR CSP analysis suggests that agonist binding to the PPARγ LBD occurs through an induced fit mechanism with two steps: an initial fast kinetic step followed by a slow conformational change step. The fast and slow binding steps are further suggested by association rates from fitted surface plasmon resonance (SPR) data. Temperature-dependent isothermal titration calorimetry (ITC) heat capacity further support a ligand binding mechanism that involves a conformational change after binding. Crystal structures reveal that ligands can bind to a surface pore in the PPARγ LBD, a site implicated as a putative ligand entry site in molecular simulations. We discuss the implication of our findings within the context of our recent NMR study revealing helix 12 occupies the orthosteric ligand-binding pocket in apo-PPARγ (Shang et al., 2020) that, when taken together with this current study, describes a more complete mechanism by which agonist binding induces activation of PPARγ.
RESULTS
Biophysical analysis of agonist ligand binding
To study the mechanism of ligand binding to PPARγ, we used a previously reported high affinity synthetic PPARγ agonist called GW1929 (Figure 1A) (Henke et al., 1998) that displays a 100 pM inhibitory binding constant (Ki) (Figure 1B). We collected 2D [1H-15N]-TROSY-HSQC NMR spectra of 15N-PPARγ LBD in the absence and presence of increasing substoichiometric molar concentrations of GW1929 (Figure S1). For a simple two-state ligand binding mechanism, also called a “lock-and-key” or U model (Kovrigin, 2012), titration of a high affinity ligand with mid-picomolar affinity in principle should result in NMR chemical shift perturbations that occur in slow exchange on the NMR time scale. The intensity of the ligand-free/apo-protein signal would be expected to decrease during the titration while the ligand-bound/holo-protein signal increases. By comparison, titration of a low affinity ligand should cause chemical shift perturbations that occur in fast exchange, causing the apo-protein signal to shift towards the holo-protein signal.
Figure 1. Affinity characterization of GW1929.

(A) Chemical structure of GW1929. (B) TR-FRET fluorescent tracer ligand displacement assay measuring the inhibition constant (Ki) of GW1929 binding to the PPARγ LBD. Data are represented as mean ± SEM (n=3).
During the GW1929 titration, a mixture of fast and slow exchange NMR CSP profiles are observed (Figure 2A) for residues structurally dispersed throughout the PPARγ LBD (Figure 2B). The ligand-free apo-peaks disappear in fast exchange, and the ligand-bound holo-peaks are populated in slow exchange. The mixture of fast and slow exchange suggests that GW1929 binding to PPARγ LBD occurs via a three-state mechanism (Kovrigin, 2012) where the protein or ligand either isomerizes between two conformations or undergoes monomer-dimer equilibrium in the absence or presence of ligand. Small-angle X-ray scattering (SAXS), dynamic light scattering (DLS), size exclusion chromatography (SEC), and NMR data indicate that the apo- and ligand-bound PPARγ LBD is monomeric (Bernardes et al., 2012; Johnson et al., 2000), ruling out a potential contribution from protein dimerization. Furthermore, GW1929 does not contain a racemizable chiral center that would result in an enantiomeric ligand isomerization mixture and it is not likely that GW1929 forms a dimer, although these features would not contribute to the three-state protein-observed exchange mechanism (Kovrigin, 2012). This leaves two protein isomerization scenarios that would show a combination of fast and slow exchange components: ligand binding that occurs via conformational selection or induced fit. In the conformational selection scenario, the protein undergoes slow isomerization and the ligand binds with fast kinetics to a sparsely populated conformation, causing the apo-peak to disappear in slow exchange and the appearance of a holo-peak in fast exchange; this is opposite of what we see in the GW1929 titration data. However, these data are consistent with an induced fit scenario where the ligand binds to the protein via a fast kinetic initial step that causes the apo-peak to disappear in fast exchange, which is followed by a slow step where the initial ligand-protein complex changes conformation into a more tightly bound conformation that causes the holo-peak to appear in slow exchange.
Figure 2. NMR analysis of GW1929 binding to PPARγ LBD.

(A) Snapshots of 2D [1H,15N]-TROSY-HSQC NMR spectra of 15N-labeled PPARγ LBD in the absence or presence of increasing concentrations of GW1929. (B) Structural locations of the residues highlighted in the NMR analysis shown in (A). See also Figure S1.
Heat capacity (ΔCp) analysis of ITC data collected at different temperatures can be used to qualitatively assess whether ligand binding occurs without a conformational change (lock-and-key model; no or small ΔCp; e.g., > −0.2 kcal mol−1 K−1) or with a conformational change (conformational selection or induced fit model; moderate or large ΔCp; e.g., < −0.4 kcal mol−1 K−1) (Vega et al., 2016). In cases of ligand binding associated with a conformational change, a temperature dependent curvature of the ΔH vs. temperature plot can suggest binding via a conformational selection mechanism, whereas a linear relationship can suggest an induced fit mechanism. We performed isothermal titration calorimetry (ITC) to experiments measuring the thermodynamic parameters of GW1929 binding to PPARγ LBD at several temperatures (Figure S2). GW1929 binding to PPARγ LBD reveals a linear coupling relationship with a ΔCp of −0.55 kcal mol−1 K−1 (Figure 3A), suggestive of an induced fit binding mechanism whereby a conformational change occurs after GW1929 binding. These ITC data are consistent with the NMR CSP data indicating an induced fit binding mechanism. However, one caveat to this analysis is that we are unable to perform ITC experiments at temperatures above 40°C due to thermal unfolding of the PPARγ LBD; thus, it is not possible to completely rule out that curvature in the ΔH vs. temperature plot could occur at higher temperatures.
Figure 3. ITC and SPR analysis of GW1929 binding to PPARγ LBD.

(A) Heat capacity (ΔCp) analysis from temperature-dependent ITC titrations of GW1929 into PPARγ LBD. Data are represented as a fitted ΔH and ΔG values from the ITC analysis (n=1). (B) SPR analysis of GW1929 binding to PPARγ LBD fit with a two-step binding equation accounting for a conformational change after binding model. (C) A two-state induced fit binding model consistent with the NMR CSP titration, ITC, and SPR data with the SPR rate constants shown from the fit of the data in (B). See also Figure S2.
The induced fit binding mechanism suggested by the NMR CSP titration data indicates that the initial ligand binding step involves fast kinetics. We performed SPR experiments to measure the kinetics of GW1929 binding to the PPARγ LBD (Figure 3B). The SPR sensorgram profiles qualitatively reveal that GW1929 binds or associates with the PPARγ LBD (Kon or Ka) and dissociates (Koff or Kd) with fast kinetic rates. Other published SPR studies of various structurally distinct synthetic PPARγ ligands similarly reveal fast kinetic rates of association and dissociation to the PPARγ LBD (Choi et al., 2014; Jang et al., 2019; Jin et al., 2016; Kratochvil et al., 2019; Li et al., 2009; Lu et al., 2011; van Marrewijk et al., 2016; Vasaturo et al., 2017; Waku et al., 2010; Xie et al., 2015; Ye et al., 2006; Yue et al., 2005), suggesting that a fast initial binding step may be a common mechanism of ligand binding to the PPARγ. Given the two-step fast and slow exchange binding process observed by NMR CSP analysis, we fit the GW1929 SPR sensorgram data using a two-step kinetic model that accounts for an initial binding event followed by a conformational change to extract association and dissociation rates of binding for each step. The SPR data fitting provides fast Ka1 and slow Ka2 association rates (Figure 3C) that are consistent with NMR CSP profiles when considering exchange regimes of fast (>103 s−1) and slow (<1 s−1) NMR-observed binding events, respectively (Kleckner and Foster, 2011). Taken together, the SPR and ITC data provide support to the NMR data, all of which suggest that GW1929 binds to PPARγ via a two-step induced fit mechanism that includes a fast initial kinetic binding step followed by a slow conformational change.
Crystal structures reveal the putative ligand entry site to the orthosteric pocket
A common method for obtaining ligand-bound crystal structures of PPARγ LBD is to grow apo-protein crystals and then perform a ligand soak to obtain the ligand-bound complex (Frkic and Bruning, 2019). This procedure is premised on the idea that the path of ligand entry into the orthosteric pocket is accessible to solvent channels within the crystal lattice. To visualize the putative ligand entry site, we grew crystals of apo-PPARγ LBD and solved the structure at 2.27 Å (Table 1). Two chains are present in the asymmetric unit (Figure S3) with different helix 12 conformations that are stabilized by a crystal artifact. Helix 12 in the chain A molecule adopts an active conformation and helix 12 in a chain B adopts a non-active conformation because it interacts with the AF-2 surface of a symmetry related chain A molecule. The structure reveals a putative orthosteric pocket entry site: a solvent accessible pore is formed by a surface consisting of helix 3, the β-sheet, and the Ω-loop (Figure 4A). This region was also suggested by molecular dynamics simulations as a putative ligand entry and exit site to the occluded orthosteric ligand-binding pocket in the PPARγ LBD (Aci-Sèche et al., 2011; Genest et al., 2008).
Table 1.
X-ray crystallography data collection and refinement statistics.
| PPARγ LBD-Y473E- GW1929 | PPARγ LBDGW1929 | PPARγ LBD-Y473E- Darglitazone | PPARγ LBDY473E | PPARγ LBD-Apo Delipidated | |
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | C 1 2 1 | C 1 2 1 | C 1 2 1 | C 1 2 1 | C 1 2 1 |
| Cell dimensions | |||||
| a, b, c (Å) | 92.70, 61.62, 118.66 | 92.84, 62.13, 119.12 | 93.07, 61.59, 120.53 | 92.64, 61.75, 118.78 | 93.04, 62.10, 118.85 |
| α, β, γ (°) | 90, 102.44, 90 | 90, 102.16, 90 | 90, 102.16, 90 | 90, 101.93, 90 | 90, 102.22, 90 |
| Resolution | 36.15–2.15 (2.23–2.15) | 51.27–2.07 (2.14–2.07) | 33.41–2.40 (2.49–2.40) | 33.01–2.3 (2.38–2.3) | 58.08–2.27 (2.35–2.27) |
| Rmerge | 0.0134 (0.196) | 0.0511 (1.374) | 0.0245 (0.362) | 0.0163 (0.360) | 0.0367 (0.341) |
| I / σ(I) | 22.00 (3.49) | 13.59 (1.22) | 13.59 (2.06) | 19.42 (1.94) | 8.43 (1.58) |
| CC1/2 in highest shell | 0.934 | 0.727 | 0.774 | 0.877 | 0.862 |
| Completeness (%) | 98.19 (98.62) | 98.75 (97.88) | 98.16 (98.76) | 97.89 (94.32) | 99.50 (97.78) |
| Redundancy | 2.0 (2.0) | 6.4 (6.4) | 2.0 (2.0) | 2.0 (2.0) | 2.0 (1.9) |
| Refinement | |||||
| Resolution (Å) | 2.15 | 2.07 | 2.40 | 2.30 | 2.27 |
| No. of unique reflections | 35158 | 40606 | 25861 | 28880 | 30849 |
| Rwork/Rfree (%) | 23.0/26.5 | 25.3/28.6 | 22.2/27.3 | 22.4/27.5 | 22.3/26.7 |
| No. of atoms | |||||
| Protein | 3999 | 4012 | 4075 | 4050 | 4122 |
| Water | 90 | 335 | 205 | 122 | 270 |
| B-factors | |||||
| Protein | 58.40 | 29.54 | 32.78 | 54.96 | 31.34 |
| Ligand | 81.00 | 53.92 | 43.47 | n/a | n/a |
| Water | 54.06 | 31.37 | 31.70 | 50.39 | 31.84 |
| Root mean square deviations | |||||
| Bond lengths (Å) | 0.016 | 0.010 | 0.010 | 0.010 | 0.011 |
| Bond angles (°) | 1.30 | 1.12 | 1.18 | 1.08 | 1.18 |
| Ramachandran favored (%) | 96.91 | 96.92 | 96.96 | 97.76 | 96.61 |
| Ramachandran outliers (%) | 0.82 | 0.41 | 0.40 | 0.20 | 0.40 |
Values in parentheses indicate highest resolution shell.
Figure 4. Ligand soaking into apo-PPARγ LBD crystals reveals an orthosteric pocket ligand entry pathway.

(A) Crystal structure of apo-PPARγ LBD reveals a surface pocket with access to the orthosteric ligand-binding pocket. (B,C) Soaking of GW1929 (B) or darglitazone (C) into preformed apo-PPARγ LBD crystals reveals two ligand binding poses, one within the orthosteric pocket in chain A (blue ligand) and a second at the orthosteric pocket ligand entry site (pink ligand). Insets show ligand 2Fo-Fc maps contoured at 1σ, and key structural elements are labeled. See also Figures S3–S4.
We soaked GW1929 into preformed apo-PPARγ LBD crystals and solved the structure to 2.07 Å (Table 1). In chain A where helix 12 is stabilized in an active conformation, GW1929 adopts a binding mode within the orthosteric ligand-binding pocket as typical of PPARγ agonists (Figure 4B). However, in chain B where helix 12 adopts a crystal contact-induced non-active helix 12 conformation, GW1929 bound to the putative orthosteric pocket entry site. Other ligand-bound PPARγ LBD crystal structures obtained from soaking ligand into apo-protein crystals have similarly revealed a ligand bound to this pocket entrance in chain B molecules (Figure S4) (Bae et al., 2016; Hopkins et al., 2006; Jang et al., 2017, 2019; Laghezza et al., 2018; Li et al., 2008; Mosure et al., 2019; Shang et al., 2019), including a crystal structure we previously solved of darglitazone-bound PPARγ LBD (Figure 4C). These crystallography observations provide support that this solvent accessible pore at the helix 3/β-sheet/Ω-loop surface constitutes the ligand entry site to the PPARγ orthosteric pocket.
Induced fit as a general PPARγ ligand binding mechanism
The data above raise a question as to whether other structurally distinct synthetic PPARγ agonists bind using an induced fit mechanism. To address this we used NMR to study the binding mechanism of a different full agonist, darglitazone, and a partial agonist, MRL24 (Figure 5A), which display Ki values at or below 1 nM in a ligand displacement assay (Figure 5B). We collected 2D [1H-15N]-TROSY-HSQC NMR spectra of 15N-PPARγ LBD in the absence and presence of substoichiometric molar concentrations of the high affinity agonists. Similar to the NMR titration of GW1929, we observed a mixture of fast and slow exchange NMR CSP profiles upon titration of darglitazone (Figure 5C, Figure S5) and MRL24 (Figure 5C, Figure S5) associated with the disappearance of ligand-free apo-peaks (fast exchange) and appearance of ligand-bound holo-peaks (slow exchange). Taken together with the GW1929 studies, these data indicate that the induced fit binding mechanism may be a general ligand binding mechanism that is not an artifact associated with any one specific PPARγ ligand scaffold.
Figure 5. NMR analysis of two structurally distinct ligands binding to PPARγ LBD.

(A) Chemical structures of darglitazone and MRL24. (B) TR-FRET fluorescent tracer ligand displacement assay measuring the inhibition constant (Ki) of darglitazone and MRL24 binding to the PPARγ LBD. Data are represented as mean ± SEM (n=3). (C) Snapshots of 2D [1H,15N]-TROSY-HSQC NMR spectra of 15N-labeled PPARγ LBD in the absence or presence of increasing concentrations of darglitazone or MRL24. See also Figure S5.
A helix 12 mutant impairs full agonist-induced function but not binding mechanism
When bound to the orthosteric pocket, full agonists such as darglitazone and GW1929 form a hydrogen bond with the side chain hydroxyl group of residue Y473 on helix 12. This tyrosine residue is thought to be critical for binding affinity and transcriptional efficacy of full agonists, but not partial agonists that do not interact with Y473 and display lower levels of PPARγ-mediated transcription (Einstein et al., 2008). The crystal structures of wild-type PPARγ LBD with the non-active helix 12 conformation observed in chain B provide one glimpse into a ligand encounter complex when helix 12 does not adopt an active conformation that points the Y473 side chain into the orthosteric pocket. To further determine how Y473 impacts the binding mechanism of darglitazone and GW1929, we generated a mutant [Y473E]-PPARγ LBD construct that we hypothesized would impact the ligand binding model in both chains A and B. In a TR-FRET coregulator interaction assay, the Y473E mutation significantly decreases the darglitazone EC50 and efficacy for increasing interaction of the TRAP220/MED1 coactivator peptide, and GW1929 shows essentially no concentration-dependent effect on the interaction (Figure 6A).
Figure 6. Ligand binding analysis to [Y473E]-PPARγ LBD.
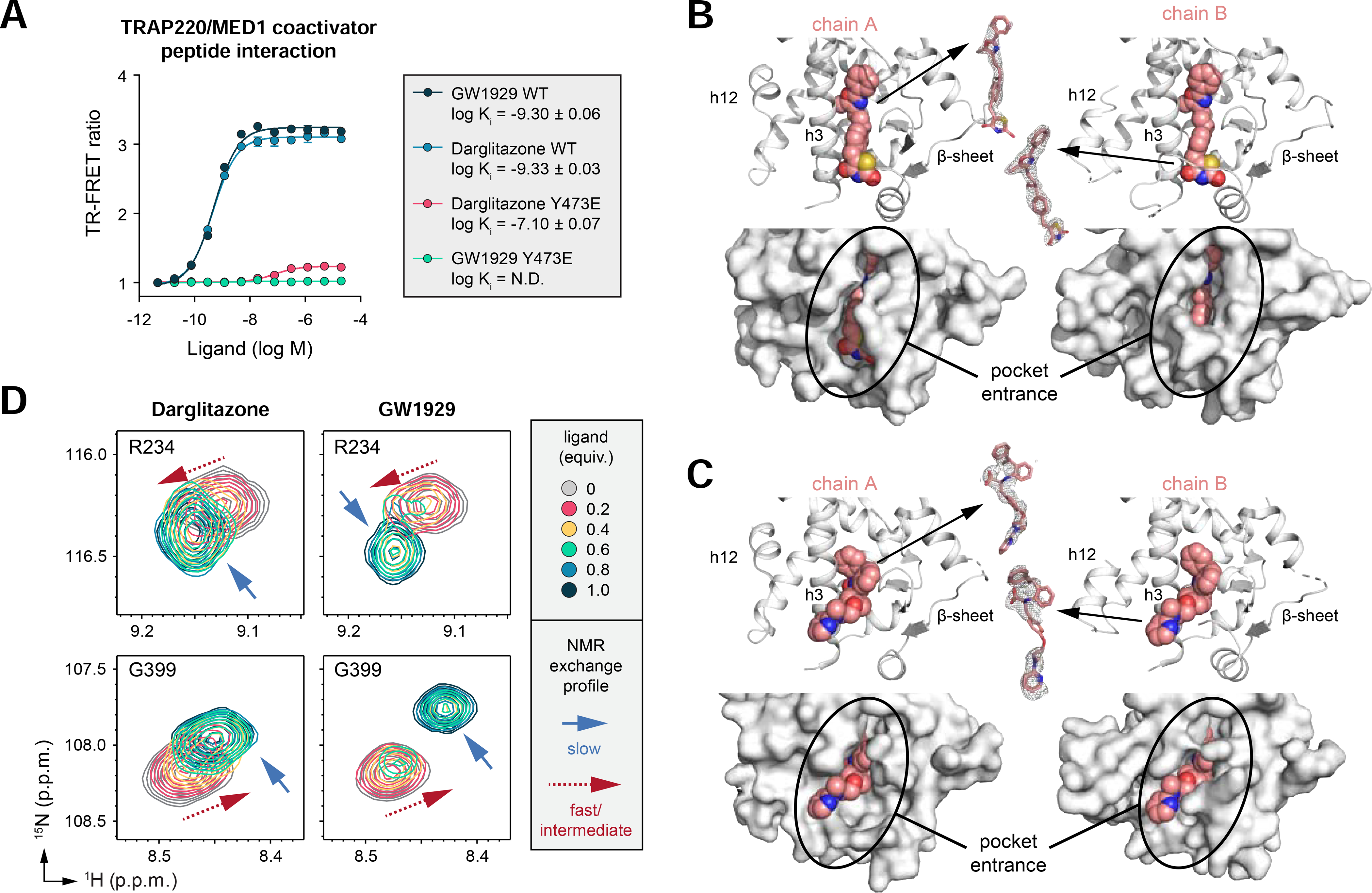
(A) TR-FRET coregulator interaction assay characterizing the activity of two full agonists, darglitazone and GW1929, on the interaction of a peptide derived from the TRAP220/MED1 coactivator protein to either wild-type (WT) PPARγ LBD or [Y473E]-PPARγ LBD. Data are presented as mean ± SEM (n = 3). (B,C) Soaking of darglitazone (B) or GW1929 (C) into preformed apo-[Y473E]-PPARγ LBD crystals reveals the ligands bind only to the orthosteric pocket ligand entry site in both chain A and B (pink ligand). Insets show ligand 2Fo-Fc maps contoured at 1σ, and key structural elements are labeled. (D) Snapshots of 2D [1H,15N]-TROSY-HSQC NMR spectra of 15N-labeled [Y473E]-PPARγ LBD in the absence or presence of increasing concentrations of darglitazone or GW1929. See also Figures S6–S8.
We crystallized [Y473E]-PPARγ LBD under the same conditions used to generate apo-PPARγ LBD crystals and solved the structure at 2.30 Å (Table 1). Overall, the [Y473E]-PPARγ LBD structure is highly similar to the apo-PPARγ LBD structure; chain A and B in both structures adopt an active and inactive conformation with Cα R.M.S.D values of 0.36 Å and 0.40 Å to the wild-type chain A and B conformers, respectively. We also solved structures after soaking darglitazone (Figure 6B) and GW1929 (Figure 6C) into preformed [Y473E]-PPARγ LBD crystals to 2.40 Å and 2.15 Å (Table 1), respectively. Ligand density is present at the ligand entry site but not in the orthosteric pocket in both chain A and B, indicating the Y473 side chain may be necessary for transition of the ligands into the final high affinity orthosteric binding pose within the crystals.
Although the TR-FRET and crystallography data indicate that the Y473E mutant inhibits or weakens GW1929 and darglitazone binding to the orthosteric pocket, 2D [1H-15N]-TROSY-HSQC NMR spectra of 15N-[Y473E]-PPARγ LBD titrated with substoichiometric molar concentrations of darglitazone and GW1929 (Figure S6) reveals the ligands indeed bind to [Y473E]-PPARγ LBD. Moreover, NMR CSP analysis of the titration series reveals a mixture of fast and slow exchange indicating that binding of darglitazone and GW1929 (Figure 6D) proceeds through an induced fit mechanism to [Y473E]-PPARγ LBD similar to the binding mechanism to wild-type PPARγ LBD. The slow exchange characteristic of the NMR titration profile suggests that the ligands bind to [Y473E]-PPARγ LBD with a reasonably high affinity, which we confirmed for GW1929 using ITC (Figure S7). Overlay of NMR spectra of wild-type or Y473E mutant 15N-PPARγ LBD bound to 1 equivalent of darglitazone or GW1929 (Figure S8) look very similar with two notable exceptions. First, NMR peaks corresponding to residues in the AF-2 surface, in particular helix 12 but also helix 3–5 and other nearby structural elements, are present in wild-type spectra but missing in the Y473E mutant spectra due to dynamics on the μs-ms intermediate exchange NMR time scale. Second, NMR peaks corresponding to residues within or nearby the AF-2 surface display chemical shift perturbations, likely due to the fact that helix 12 is dynamic and samples multiple conformations in the Y473E mutant but in wild-type PPARγ LBD it is stabilized in an active conformation via hydrogen bond formation between the ligand and the Y473 hydroxyl group.
Taken together, the Y473E ligand binding data are consistent with a dynamic activation model (Kojetin and Burris, 2013) whereby PPARγ agonism is associated with ligand-induced stabilization of helix 12, which is dynamic on the μs-ms time scale in apo-PPARγ (Chrisman et al., 2018; Hughes et al., 2012; Johnson et al., 2000; Shang et al., 2019). The NMR data indicate that darglitazone and GW1929 binding to [Y473E]-PPARγ LBD stabilizes the dynamics of most of the orthosteric ligand-binding pocket, likely by binding to the orthosteric pocket in solution though not in the crystallized form. However, because the ligands do not hydrogen bond to the side chain of Y473E, helix 12 remains dynamic, which could explain the relatively flat ligand dose response curve in the TR-FRET assay despite their ability to bind with relatively high affinity. It is also possible that the addition of a coactivator peptide forces the [Y473E]-PPARγ LBD into an active AF-2 helix 12 conformation resulting in a clash between the glutamic acid side chain and the acid headgroup of GW1929 in the orthosteric binding pose. An electrostatic clash in the [Y473E]-PPARγ LBD crystals, or lack of the Y473 side chain in chain B of wild-type PPARγ crystals, could also explain why the ligands do not crystallize with an orthosteric binding pose. However, the NMR and ITC data indicate that the Y473E mutant does not prevent the ligands from binding to the orthosteric pocket in solution with high affinity.
DISCUSSION
There is evidence that the functional activity of ligand-bound NRs is associated with a shift in the dynamic LBD conformational ensemble from a ground state to an active state. For example, in the absence of ligand, the PPARγ LBD is conformationally dynamic, samples multiple conformation, and binding of an agonist stabilizes the LBD in an active conformation to a degree correlated with the activity of the ligand (Chrisman et al., 2018; Hughes et al., 2012; Johnson et al., 2000; Shang et al., 2019). The association between ligand-bound NR LBD conformation and graded function is evidence for conformational selection in the mechanism of NR ligand activity (Bruning et al., 2010; Changeux and Edelstein, 2011; Cronet et al., 2001; Köhler et al., 2020; Lu et al., 2006; Okafor et al., 2019; Singarapu et al., 2011). However, these studies only address the functional mechanism of the ligand-bound state; they do not address the mechanism of ligand binding. In this study, we directly probed the mechanism of ligand binding to PPARγ using NMR CSP analysis, a powerful method for studying binding equilibria at atomic resolution that can differentiate conformational selection and induced fit binding mechanisms (Kovrigin, 2012).
Our NMR data indicate that agonists bind to the PPARγ orthosteric pocket via a two-state induced fit mechanism. The first step is characterized by a fast binding event with an exchange rate of ~105 s−1, which is supported by SPR data and likely occurs at a surface pore formed by helix 3, the β-sheet, and the Ω-loop. This is a site where ligands can bind in the non-active conformation PPARγ LBD crystal structures as well as our Y473E mutant crystal structures, and it is the ligand entry site suggested in unbiased coarse-grained molecular simulations of FXR (Souza et al., 2020) and targeted simulations of PPARγ (Aci-Sèche et al., 2011). Molecular simulations of other NRs have suggested other ligand entry sites including a conserved surface in steroid receptors at the intersection of helix 3, 7, and 11 (Edman et al., 2015; Grebner et al., 2017), suggesting that certain classes of NRs may use different ligand entry sites to the orthosteric pocket. The ligands bound to the entry site to the orthosteric pocket represent a likely snapshot of the induced fit ligand encounter complex.
The second slower step detected by our NMR studies is associated with a conformational change after the initial fast binding event, which is supported by ITC heat capacity and SPR analysis. This step likely represents transition of the ligand from the initial encounter complex at the surface pore ligand entry site to the final bound conformation within the orthosteric pocket. Molecular simulation studies of ligand binding to FXR (Souza et al., 2020) and PPARγ (Aci-Sèche et al., 2011) support this conformational change step. In the study on PPARγ, binding of an agonist called GW0072 was described to rotate on itself during a transition from its initial binding pose to the final bound conformation. This is conceptually similar to the crystallized PPARγ ligand binding modes we describe here. In the structures of darglitazone bound to the pocket entry site, the thiazolidinedione (TZD) headgroup is solvent exposed—if this represents an initial encounter complex binding pose that is populated in solution, the TZD headgroup would need to swing around helix 3 through a rotation point at the intersection of helix 3 and 5 to migrate to the final orthosteric binding pose. In the structures of GW1929 bound to the pocket entry site, the acid headgroup points into the pocket and if this also represents an initial encounter complex binding pose that is populated in solution it would need to flip ~180° during the transition into the final orthosteric binding pose. The unbiased coarse-grained simulations of obeticholic acid binding to FXR revealed a similar multi-step mechanism with the final step associated with a rearrangement of the FXR LBD and a transition of the ligand to the inner binding pose within the orthosteric pocket (Souza et al., 2020).
Our findings extend a model for the molecular mechanism of agonist binding and activation of PPARγ (Figure 7). In the absence of ligand, several regions of apo-PPARγ LBD exchanges between multiple conformations on the microsecond-millisecond (μs-ms) time scale including the orthosteric ligand-binding pocket and the AF-2 helix 12 (Johnson et al., 2000). One of these conformations was assumed to be the active NR LBD conformation that has been captured in most NR crystal structures. Using paramagnetic relaxation enhancement (PRE) NMR, we recently demonstrated for apo-PPARγ LBD that the μs-ms time scale dynamics is caused by exchange of the AF-2 helix 12 in and out of the orthosteric ligand-binding pocket, which are associated with the transcriptionally repressive and active conformations (Shang et al., 2020). The PRE measurements were particularly robust for the transcriptionally repressive helix 12 conformation within the orthosteric pocket. When considered with our findings here, this suggests that there may be a competition between the repressive helix 12 conformation within the orthosteric pocket and the transition of the ligand from the encounter complex bound at the entry site to the final binding pose in the orthosteric pocket. It is possible that this transition pushes helix 12 out of the repressive conformation within the orthosteric pocket to a solvent-exposed active conformation, a signature of what might be considered a true induced fit binding mechanism. In this case, the agonist-bound transcriptionally active conformation may not be significantly populated in the apo-LBD conformational ensemble. Alternatively, the binding mechanism could involve a mixture of induced fit binding and conformational selection where helix 12 may adopt an active-like conformation prior to the ligand transitioning to the final binding pose in the orthosteric pocket if the time scale by which helix 12 exchanges out of the pocket is not rate limiting (Zhou, 2010). Other NMR studies have similarly found that the orthosteric pocket and helix 12 in other apo-NR LBDs are dynamic on the μs-ms time scale including FXR, PPARα, retinoid X receptor alpha (RXRα), and vitamin D receptor (VDR) (Cronet et al., 2001; Lu et al., 2006; Merk et al., 2019; Singarapu et al., 2011). Thus, the ligand binding mechanism we describe here for PPARγ may occur for other NRs as well, a hypothesis that could be tested by NMR and biophysical studies as demonstrated in this study.
Figure 7. Schematic model of agonist binding to a dynamic apo-PPARγ LBD conformational ensemble via a two-step process involving an initial fast step followed by a slow conformational change step.

In the absence of ligand, helix 12 in apo-PPARγ LBD exchanges between a transcriptionally repressive conformation within the orthosteric ligand-binding pocket and a solvent exposed active conformation. Agonist binding to the ligand entry site via an encounter complex could occur to either of these conformations. In the pure induced fit scenario, agonist binding to the repressive LBD conformation would push helix 12 into an active conformation. In the mixed induced fit & conformational selection scenario, ligand binding to the active LBD conformation would facilitate transition into the final ligand binding pose. Structures displayed include PDB code 6ONJ (active conformation; chain A), 6ONI (repressive conformation; chain B), and 6DGL (darglitazone ligand; chain B). Black arrows denote processes supported by experimental studies (NMR, SPR, and ITC heat capacity analysis); grey arrows denote other steps that are part of the binding kinetic pathways.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents may be directed to and will be fulfilled by the Lead Contact, Douglas J. Kojetin (dkojetin@scripps.edu).
Materials Availability
Plasmids generated in this study may be requested from the Lead Contact.
Data and Code Availability
The atomic coordinates of PPARγ LBD-WT-Apo Delipidated, PPARγ LBD/GW1929 complex, PPARγ LBD-Y473E, PPARγ LBD-Y473E/darglitazone complex and PPARγ LBD-Y473E/GW1929 complex reported in this paper are deposited to the Protein Data Bank (PDB) under accession codes 6VZO, 6VZL, 6VZN, 6VZM and 7JQG, respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial Strains Used for Protein Expression
Wild-type or Y473E mutant human PPARγ LBD (UniProt P37231; residues 203–477, isoform 1 numbering) protein was expressed from a pET46 Ek/LIC vector (Novagen) as a Tobacco Etch Virus (TEV)-cleavable N-terminal Hexa(6x)His-tagged fusion protein in Escherichia coli BL21(DE3) cells using autoinduction ZY media (grown at 22°C; harvested after overnight growth), or M9 minimal media supplemented with 15NH4Cl (induced with 0.4 mM isopropyl β-d-1-thiogalactopyranoside at O.D. ~0.8; grown at 18°C; harvested after overnight growth). The Y473E PPARγ LBD mutant was generated with site directed mutagenesis using the following forward primer and the corresponding reverse primer: 5’-CTGCAGGAGATCGAAAAGGACTTGTAC-3’.
METHOD DETAILS
Ligands and other reagents
Darglitazone and GW1929 were obtained from Cayman Chemical, MRL24 was obtained from BioVision; ligands were dissolved in DMSO-d6 at 20 mM stocks. Peptides of LXXLL-containing motif from human TRAP220/MED1 (UniProt Q15648; residues 638–656; NTKNHPMLMNLLKDNPAQD) with an N-terminal FITC label, a six-carbon linker (Ahx), and an amidated C-terminus for stability was synthesized by LifeTein.
Protein expression and purification
Proteins were purified using Ni-NTA affinity chromatography, in some cases a second Ni-NTA affinity chromatography step after cleavage of the 6xHis-tag by TEV protease (at a ratio of 1 mg TEV protease : 50 mg PPARγ LBD), and finally gel filtration chromatography. Purified protein was concentrated to 10 mg/mL in a buffer consisting of 20 mM potassium phosphate (pH 7.4), 50 mM potassium chloride, 5 mM tris(2-carboxyethyl)phosphine (TCEP), and 0.5 mM ethylenediaminetetraacetic acid (EDTA). Purified proteins were verified by SDS-PAGE as >95% pure. Delipidated protein was obtained by denaturation using a chloroform/methanol lipid extraction method and refolding using a fast dilution/dialysis procedure followed by size exclusion chromatography (Armstrong et al., 2014).
TR-FRET assays
The time-resolved fluorescence resonance energy transfer (TR-FRET) assays were performed in black 384-well plates (Greiner). Ligand stocks were prepared via serial dilution in DMSO and added to wells. For the TR-FRET ligand displacement assay, each well (23 μL total volume) contained 1 nM 6xHis-tagged PPARγ LBD protein, 1 nM LanthaScreen Elite Tb-anti-HIS antibody (Thermo Fisher), and 5 nM Fluormone Pan-PPAR Green fluorescent tracer ligand (Invitrogen) in the same buffer. For the coregulator interaction assay, each well (23 μL total volume) contained 4 nM 6xHis-tagged PPARγ LBD, 1 nM LanthaScreen Elite Tb-anti-HIS antibody (Thermo Fisher), and 400 nM FITC-labeled TRAP220 or NCoR peptide in a buffer consisting of 20mM potassium phosphate (pH 8), 50 mM potassium chloride, 5 mM TCEP, and 0.005% Tween 20. Plates were incubated at 25°C for 1 h and read using Synergy Neo plate reader (BioTek). The Tb donor was excited at 340 nm, its emission was monitored at 492 nm, and the acceptor FITC emission was measured at 520 nm. Data were analyzed using GraphPad Prism. TR-FRET coregulator data were fit to sigmoidal dose response curve equation, and ligand displacement data were fit to the “one site – Fit Ki” binding equation to obtain Ki values using the published binding affinity of Fluormone Pan-PPAR Green (2.8 nM; Invitrogen PV4894 info sheet).
NMR spectroscopy
Two dimensional (2D) [1H,15N]-transverse relaxation optimized spectroscopy (TROSY) heteronuclear single quantum coherence (HSQC) data were collected on a Bruker 700 MHz NMR spectrometer equipped with a QCI cryoprobe at 298K using Topspin 3.0 (Bruker). Samples contained 200 μM 15N-PPARγ LBD at increasing ligand concentrations (0, 0.2, 0.4, 0.6, 0.8, and 1.0 equivalents); a DMSO control experiment (1.0 equivalent of vehicle) revealed no significant perturbations. Data were processed using NMRFx Processor (Norris et al., 2016) and analyzed using NMRViewJ and NMRFx Analyst (Johnson, 2018). Backbone NMR chemical shift assignments for GW1929-bound PPARγ LBD were previously reported (Lu et al., 2008).
Surface plasmon resonance
Surface plasmon resonance (SPR) measurements were performed on a Biacore X100 instrument (GE Healthcare). 6xHis-tagged PPARγ LBD protein was immobilized on an NTA sensor chip (GE Healthcare), which includes a reference flow cell for background subtraction, at a density not exceeding 2,000 RU using reagents supplied by NTA reagent kit (GE Healthcare). Measurements were performed in a buffer (1X HBS-P+ buffer) containing 10 mM HEPES, 150 mM NaCl and 0.05% (v/v) Surfactant P20 at a flow rate of 10 μL/min. For kinetic and affinity measurements, GW1929 was diluted in 1X HBS-P+ buffer and injected at 8 concentrations with one duplicate. The NTA sensor chip was regenerated between each GW1929 concentration measurement by stripping Ni2+ using EDTA and recharging with NiCl2. SPR sensorgrams were fit using Biacore Insight Evaluation Software (Cytiva/GE) to a two-state kinetic model that accounts for a conformational change associated with ligand binding.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were carried out on a iTC200 calorimeter (MicroCal/GE/Malvern) using the iTC200 software (v 1.24.2) for instrument control and data acquisition. GW1929 (present in the syringe at 300 μM) was diluted in a buffer consisting of 20 mM potassium phosphate (pH 7.4), 50 mM potassium chloride, 5 mM TCEP, 0.5 mM EDTA, and 0.17% DMSO. Wild-type or Y473E PPARγ LBD protein (present in the sample cell at 30 μM) was diluted in the same buffer. Titrations were performed with a 0.4 μL pre-injection followed by nineteen 2 μL injections. Mixing was carried out at five temperatures (10°C, 18°C, 25°C, 30°C, 37°C) for wild-type PPARγ LBD or 25°C for [Y473E]-PPARγ LBD with reference power and rotational stirring set at 5 μcal s−1 and 1200 rpm, respectively. Data analysis was performed using software packages NITPIC, SEDPHAT, and GUSSI (Brautigam et al., 2016; Houtman et al., 2007; Keller et al., 2012; Zhao et al., 2015).
Crystallization, structure determination, and structural analysis
Crystals of wild-type and Y473E PPARγ LBD were obtained after 3–8 days at 22°C by sitting-drop vapor diffusion against 50 μL of well solution using 96-well format crystallization plates. The crystallization drops contained 1 μL of protein/ligand sample mixed with 1 μL of reservoir solution containing 0.1 M MOPS (pH 7.6) and 0.8 M sodium citrate. Crystals for the darglitazone and GW1929-PPARγ LBD complexes were obtained by soaking ligands (1.5 mM in reservoir solution containing 5% DMSO) into preformed apo-PPARγ LBD crystals for about 1 week. All crystals were flash-frozen in liquid nitrogen before data collection. Data collection for the wild-type apo-refolded and GW1929-bound structures were collected at Berkeley Center for Structural Biology beamline 5.0.3; the apo-Y473E mutant and GW1929 or darglitazone-bound Y473E mutant structures were collected at SLAC National Accelerator Laboratory/Stanford Synchrotron Radiation Lightsource (SSRL) beamline 12–2. Data were processed, integrated, and scaled with the programs Mosflm (Battye et al., 2011) and Scala in CCP4 (Winn et al., 2011). The structure was solved by molecular replacement using the program Phaser (McCoy et al., 2007) that is implemented in the PHENIX package (Adams et al., 2011) using previously published PPARγ LBD structure (PDB code 1PRG) (Nolte et al., 1998) as the search model. The structure was refined using PHENIX with several cycles of interactive model rebuilding in Coot (Emsley and Cowtan, 2004). The align command in PyMOL (Schrödinger) was used to calculate Cα R.M.S.D. values with the number of alignment refinement cycles set to 0.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism (v8 or v9) software was used to determine average values, standard errors, and standard deviations. For each figure, the number of experimental replicates and other information relevant for assessing the accuracy and precision of the analysis are included in the accompanying legend.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| LanthaScreen Elite Tb-anti-His Antibody | Thermo Fisher Scientific | Cat: PV5863 |
| Bacterial and Virus Strains | ||
| Escherichia coli BL21(DE3) cells | Invitrogen | Cat: C600003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ammonium Chloride, 99%15N | Cambridge Isotope Labs | Cat: NLM-467 |
| Deuterium Oxide, 99.9% D | Sigma-Aldrich | Cat: 151882 |
| Dimethyl Sulfoxide-d6, 99.9% D | Cambridge Isotope Labs | Cat: DLM-10 |
| FITC-NH-NTKNHPMLMNLLKDNPAQD | LifeTein, LLC | N/A |
| HiLoad 16/600 Superdex 75 pg column | GE Healthcare Life Sciences | Cat: 28989333 |
| HisTrap FF column | GE Healthcare Life Sciences | Cat: 17525501 |
| NTA Reagent Kit | GE Healthcare Life Sciences | Cat: 28995043 |
| HBS-P+ Buffer | GE Healthcare Life Sciences | Cat: BR100827 |
| Isopropyl-β-D-thiogalactoside | Gold Biotechnology | Cat: I2481C |
| TCEP (Tris (2-Carboxyethyl) phosphine Hydrochloride) | Gold Biotechnology | Cat: TCEP25 |
| Sodium citrate tribasic dihydrate | Hampton Research | Cat: HR2–549 |
| MOPS sodium salt | Sigma-Aldrich | Cat: M9381 |
| Imidazole | Sigma-Aldrich | Cat: 792527 |
| Potassium Chloride | Sigma-Aldrich | Cat: 746436 |
| Potassium phosphate monobasic | Sigma-Aldrich | Cat: 795488 |
| Potassium phosphate dibasic | Sigma-Aldrich | Cat: P3786 |
| Trizma Base | Sigma-Aldrich | Cat: 93352 |
| Ethylenediaminetetraacetic acid disodium dihydrate | Sigma-Aldrich | Cat: E9884 |
| Sodium Chloride | Sigma-Aldrich | Cat: 746398 |
| Sodium phosphate dibasic | Sigma-Aldrich | Cat: S5136 |
| Calcium chloride dihydrate | Sigma-Aldrich | Cat: 223506 |
| Manganese (II) chloride tetrahydrate | Sigma-Aldrich | Cat: 221279 |
| Copper (II) Chloride Dihydrate | Sigma-Aldrich | Cat: 22201–1 |
| Tryptone | Fisher Scientific | Cat: BP1421–2 |
| Yeast Extract | Fisher Scientific | Cat: BP1422–2 |
| D-Glucose | Fisher Scientific | Cat: FLBP350–1 |
| Magnesium sulfate | Fisher Scientific | Cat: BP213–1 |
| Ampicillin | Fisher Scientific | Cat: BP1760 |
| Ferrous sulfate heptahydrate | Fisher Scientific | Cat: I146–500 |
| Cobalt (II) chloride hexahydrate | Acros Organic | Cat: 7791–13-1 |
| Zinc sulfate heptahydrate | EMD Millipore | Cat: ZX0065–1 |
| Fluormone Pan-PPAR Green | Invitrogen | Cat: PV4894 |
| Darglitazone | Cayman Chemical | Cat: 17681 |
| GW1929 | Cayman Chemical | Cat: 13689 |
| MRL24 | BioVision | Cat: 2764–5 |
| Human PPARγ LBD WT, residues 203–477; isoform 1 protein | This paper | N/A |
| Human PPARγ LBD Y473E mutant protein | This paper | N/A |
| Deposited Data | ||
| Human PPARγ LBD in Complex with Darglitazone crystal structure | (Shang et al., 2019) | PDB: 6DGL |
| Human PPARγ LBD-Apo Delipidated crystal structure | This paper | PDB: 6VZO |
| Human PPARγ LBD in Complex with GW1929 crystal structure | This paper | PDB: 6VZL |
| Human PPARγ LBD-Y473E crystal structure | This paper | PDB: 6VZN |
| Human PPARγ LBD-Y473E in Complex with Darglitazone crystal structure | This paper | PDB: 6VZM |
| Human PPARγ LBD-Y473E in Complex with GW1929 crystal structure | This paper | PDB: 7JQG |
| Human PPARγ LBD Bound to GW1929 NMR chemical shift assignments | (Lu et al., 2008) | BMRB: 15518 |
| Oligonucleotides | ||
| Y473E mutagenesis forward primer: 5’-CTGCAGGAGATCGAAAAGGACTTGTAC-3’ | IDT technologies | N/A |
| Y473E mutagenesis reverse primer: 5’-GTACAAGTCCTTTTCGATCTCCTGCAG-3’ | IDT technologies | N/A |
| Recombinant DNA | ||
| Plasmid: pET-46 Ek/LIC | Novagen | Cat#71335 |
| pET46-PPARγ LBD WT | (Hughes et al., 2012) | N/A |
| pET46-PPARγ LBD Y473E | This paper | N/A |
| Software and Algorithms | ||
| NMRViewJ | OneMoon Scientific, Inc. (Johnson 2018) | https://nmrfx.org/nmrfx/nmrviewj |
| NMRFx Analyst | OneMoon Scientific, Inc. (Norris et al. 2016; Johnson 2018) | https://nmrfx.org/nmrfx/analyst |
| Topspin | Bruker Biospin | https://www.bruker.com/products/mr/nmr/software/topspin.html |
| GraphPad Prism | GraphPad Software Inc. | http://www.graphpad.com/scientific-software/prism/ |
| PyMOL | Schrödinger | https://pymol.org/2/ |
| PHENIX | (Adams et al., 2011) | http://www.phenix-online.org/ |
| CCP4 | (Winn et al., 2011) | http://www.ccp4.ac.uk/ |
| COOT | (Emsley et al., 2004) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| iTC200 software | MicroCal Inc. | v 1.24.2 |
| NITPIC | (Keller et al., 2012) | http://biophysics.swmed.edu/MBR/software.htm |
| GUSSI | (Brautigam et al., 2016) | http://biophysics.swmed.edu/MBR/software.htm |
| SEDPHAT | (Houtman et al., 2007) | http://www.analyticalultracentrifugation.com/sedphat/ |
HIGHLIGHTS.
Biophysical data suggest PPARγ ligand binding occurs via an induced fit mechanism
NMR chemical shift perturbation data reveal a mixture of slow and fast exchange
SPR and ITC data support fast binding and slow conformational change steps
Crystal structures reveal a putative induced fit ligand encounter complex
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health (NIH) grant R01DK124870. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIDDK or NIH.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aci-Sèche S, Genest M, and Garnier N (2011). Ligand entry pathways in the ligand binding domain of PPARγ receptor. FEBS Lett. 585, 2599–2603. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Echols N, Headd JJ, Hung L-W, Jain S, Kapral GJ, Grosse Kunstleve RW, et al. (2011). The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong EH, Goswami D, Griffin PR, Noy N, and Ortlund EA (2014). Structural basis for ligand regulation of the fatty acid-binding protein 5, peroxisome proliferator-activated receptor β/δ (FABP5-PPARβ/δ) signaling pathway. J. Biol. Chem. 289, 14941–14954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae H, Jang JY, Choi S-S, Lee J-J, Kim H, Jo A, Lee K-J, Choi JH, Suh SW, and Park SB (2016). Mechanistic elucidation guided by covalent inhibitors for the development of anti-diabetic PPARγ ligands. Chem. Sci 7, 5523–5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista MRB, and Martínez L (2015). Conformational Diversity of the Helix 12 of the Ligand Binding Domain of PPARγ and Functional Implications. J. Phys. Chem. B 119, 15418–15429. [DOI] [PubMed] [Google Scholar]
- Battye TGG, Kontogiannis L, Johnson O, Powell HR, and Leslie AGW (2011). iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes A, Batista FAH, de Oliveira Neto M, Figueira ACM, Webb P, Saidemberg D, Palma MS, and Polikarpov I (2012). Low-resolution molecular models reveal the oligomeric state of the PPAR and the conformational organization of its domains in solution. PloS One 7, e31852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam CA, Zhao H, Vargas C, Keller S, and Schuck P (2016). Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat. Protoc. 11, 882–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JB, Chalmers MJ, Prasad S, Busby SA, Kamenecka TM, He Y, Nettles KW, and Griffin PR (2007). Partial agonists activate PPARgamma using a helix 12 independent mechanism. Struct. 15, 1258–1271. [DOI] [PubMed] [Google Scholar]
- Bruning JB, Parent AA, Gil G, Zhao M, Nowak J, Pace MC, Smith CL, Afonine PV, Adams PD, Katzenellenbogen JA, et al. (2010). Coupling of receptor conformation and ligand orientation determine graded activity. Nat. Chem. Biol 6, 837–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux J-P, and Edelstein S (2011). Conformational selection or induced fit? 50 years of debate resolved. F1000 Biol. Rep. 3, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S-S, Kim ES, Koh M, Lee S-J, Lim D, Yang YR, Jang H-J, Seo K-A, Min S-H, Lee IH, et al. (2014). A novel non-agonist peroxisome proliferator-activated receptor γ (PPARγ) ligand UHC1 blocks PPARγ phosphorylation by cyclin-dependent kinase 5 (CDK5) and improves insulin sensitivity. J. Biol. Chem 289, 26618–26629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrisman IM, Nemetchek MD, de Vera IMS, Shang J, Heidari Z, Long Y, Reyes-Caballero H, Galindo-Murillo R, Cheatham TE, Blayo A-L, et al. (2018). Defining a conformational ensemble that directs activation of PPARγ. Nat. Commun 9, 1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronet P, Petersen JF, Folmer R, Blomberg N, Sjöblom K, Karlsson U, Lindstedt EL, and Bamberg K (2001). Structure of the PPARalpha and -gamma ligand binding domain in complex with AZ 242; ligand selectivity and agonist activation in the PPAR family. Struct. 9, 699–706. [DOI] [PubMed] [Google Scholar]
- Edman K, Hosseini A, Bjursell MK, Aagaard A, Wissler L, Gunnarsson A, Kaminski T, Köhler C, Bäckström S, Jensen TJ, et al. (2015). Ligand Binding Mechanism in Steroid Receptors: From Conserved Plasticity to Differential Evolutionary Constraints. Struct. 23, 2280–2290. [DOI] [PubMed] [Google Scholar]
- Einstein M, Akiyama TE, Castriota GA, Wang CF, McKeever B, Mosley RT, Becker JW, Moller DE, Meinke PT, Wood HB, et al. (2008). The differential interactions of peroxisome proliferator-activated receptor gamma ligands with Tyr473 is a physical basis for their unique biological activities. Mol. Pharmacol 73, 62–74. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fischer A, and Smieško M (2019). Ligand Pathways in Nuclear Receptors. J. Chem. Inf. Model. 59, 3100–3109. [DOI] [PubMed] [Google Scholar]
- Frkic RL, and Bruning JB (2019). Obtaining Crystals of PPARγ Ligand Binding Domain Bound to Small Molecules. Methods Mol. Biol. Clifton NJ 1966, 253–260. [DOI] [PubMed] [Google Scholar]
- Genest D, Garnier N, Arrault A, Marot C, Morin-Allory L, and Genest M (2008). Ligand-escape pathways from the ligand-binding domain of PPARgamma receptor as probed by molecular dynamics simulations. Eur. Biophys. J. EBJ 37, 369–379. [DOI] [PubMed] [Google Scholar]
- Grebner C, Lecina D, Gil V, Ulander J, Hansson P, Dellsen A, Tyrchan C, Edman K, Hogner A, and Guallar V (2017). Exploring Binding Mechanisms in Nuclear Hormone Receptors by Monte Carlo and X-ray-derived Motions. Biophys. J 112, 1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henke BR, Blanchard SG, Brackeen MF, Brown KK, Cobb JE, Collins JL, Harrington WW, Hashim MA, Hull-Ryde EA, Kaldor I, et al. (1998). N-(2-Benzoylphenyl)-L-tyrosine PPARgamma agonists. 1. Discovery of a novel series of potent antihyperglycemic and antihyperlipidemic agents. J. Med. Chem 41, 5020–5036. [DOI] [PubMed] [Google Scholar]
- Holzer G, Markov GV, and Laudet V (2017). Evolution of Nuclear Receptors and Ligand Signaling: Toward a Soft Key-Lock Model? Curr. Top. Dev. Biol 125, 1–38. [DOI] [PubMed] [Google Scholar]
- Hopkins CR, O’neil SV, Laufersweiler MC, Wang Y, Pokross M, Mekel M, Evdokimov A, Walter R, Kontoyianni M, Petrey ME, et al. (2006). Design and synthesis of novel N-sulfonyl-2-indole carboxamides as potent PPAR-gamma binding agents with potential application to the treatment of osteoporosis. Bioorg. Med. Chem. Lett. 16, 5659–5663. [DOI] [PubMed] [Google Scholar]
- Houtman JCD, Brown PH, Bowden B, Yamaguchi H, Appella E, Samelson LE, and Schuck P (2007). Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. Publ. Protein Soc. 16, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Chandra V, and Rastinejad F (2010). Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu. Rev. Physiol. 72, 247–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TS, Chalmers MJ, Novick S, Kuruvilla DS, Chang MR, Kamenecka TM, Rance M, Johnson BA, Burris TP, Griffin PR, et al. (2012). Ligand and receptor dynamics contribute to the mechanism of graded PPARγ agonism. Struct. 20, 139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang JY, Koh M, Bae H, An DR, Im HN, Kim HS, Yoon JY, Yoon H-J, Han BW, Park SB, et al. (2017). Structural basis for differential activities of enantiomeric PPARγ agonists: Binding of S35 to the alternate site. Biochim. Biophys. Acta Proteins Proteomics 1865, 674–681. [DOI] [PubMed] [Google Scholar]
- Jang JY, Kim H-J, and Han BW (2019). Structural Basis for the Regulation of PPARγ Activity by Imatinib. Mol. Basel Switz. 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Han Y, Khadka DB, Zhao C, Lee KY, and Cho W-J (2016). Discovery of Isoquinolinoquinazolinones as a Novel Class of Potent PPARγ Antagonists with Anti-adipogenic Effects. Sci. Rep 6, 34661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA (2018). From Raw Data to Protein Backbone Chemical Shifts Using NMRFx Processing and NMRViewJ Analysis. Methods Mol. Biol. Clifton NJ 1688, 257–310. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Wilson EM, Li Y, Moller DE, Smith RG, and Zhou G (2000). Ligand-induced stabilization of PPARgamma monitored by NMR spectroscopy: implications for nuclear receptor activation. J. Mol. Biol 298, 187–194. [DOI] [PubMed] [Google Scholar]
- Keller S, Vargas C, Zhao H, Piszczek G, Brautigam CA, and Schuck P (2012). High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem 84, 5066–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner IR, and Foster MP (2011). An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta 1814, 942–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler C, Carlström G, Gunnarsson A, Weininger U, Tångefjord S, Ullah V, Lepistö M, Karlsson U, Papavoine T, Edman K, et al. (2020). Dynamic allosteric communication pathway directing differential activation of the glucocorticoid receptor. Sci. Adv 6, eabb5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojetin DJ, and Burris TP (2013). Small molecule modulation of nuclear receptor conformational dynamics: implications for function and drug discovery. Mol. Pharmacol 83, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovrigin EL (2012). NMR line shapes and multi-state binding equilibria. J. Biomol. NMR 53, 257–270. [DOI] [PubMed] [Google Scholar]
- Kratochvil I, Hofmann T, Rother S, Schlichting R, Moretti R, Scharnweber D, Hintze V, Escher BI, Meiler J, Kalkhof S, et al. (2019). Mono(2-ethylhexyl) phthalate (MEHP) and mono(2-ethyl-5-oxohexyl) phthalate (MEOHP) but not di(2-ethylhexyl) phthalate (DEHP) bind productively to the peroxisome proliferator-activated receptor γ. Rapid Commun. Mass Spectrom. RCM 33 Suppl 1, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laghezza A, Piemontese L, Cerchia C, Montanari R, Capelli D, Giudici M, Crestani M, Tortorella P, Peiretti F, Pochetti G, et al. (2018). Identification of the First PPARα/γ Dual Agonist Able To Bind to Canonical and Alternative Sites of PPARγ and To Inhibit Its Cdk5-Mediated Phosphorylation. J. Med. Chem 61, 8282–8298. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang Z, Furukawa N, Escaron P, Weiszmann J, Lee G, Lindstrom M, Liu J, Liu X, Xu H, et al. (2008). T2384, a novel antidiabetic agent with unique peroxisome proliferator-activated receptor gamma binding properties. J. Biol. Chem 283, 9168–9176. [DOI] [PubMed] [Google Scholar]
- Li Y, Li L, Chen J, Hu T, Huang J, Guo Y, Jiang H, and Shen X (2009). 7-Chloroarctinone-b as a new selective PPARgamma antagonist potently blocks adipocyte differentiation. Acta Pharmacol. Sin. 30, 1351–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Cistola DP, and Li E (2006). Analysis of ligand binding and protein dynamics of human retinoid X receptor alpha ligand-binding domain by nuclear magnetic resonance. Biochemistry 45, 1629–1639. [DOI] [PubMed] [Google Scholar]
- Lu J, Chen M, Stanley SE, and Li E (2008). Effect of heterodimer partner RXRalpha on PPARgamma activation function-2 helix in solution. Biochem. Biophys. Res. Commun 365, 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Che P, Zhang Y, Li H, Zou S, Zhu J, Deng J, Shen X, Jiang H, Li J, et al. (2011). HL005--a new selective PPARγ antagonist specifically inhibits the proliferation of MCF-7. J. Steroid Biochem. Mol. Biol. 124, 112–120. [DOI] [PubMed] [Google Scholar]
- van Marrewijk LM, Polyak SW, Hijnen M, Kuruvilla D, Chang MR, Shin Y, Kamenecka TM, Griffin PR, and Bruning JB (2016). SR2067 Reveals a Unique Kinetic and Structural Signature for PPARγ Partial Agonism. ACS Chem. Biol. 11, 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merk D, Sreeramulu S, Kudlinzki D, Saxena K, Linhard V, Gande SL, Hiller F, Lamers C, Nilsson E, Aagaard A, et al. (2019). Molecular tuning of farnesoid X receptor partial agonism. Nat. Commun 10, 2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosure SA, Shang J, Eberhardt J, Brust R, Zheng J, Griffin PR, Forli S, and Kojetin DJ (2019). Structural Basis of Altered Potency and Efficacy Displayed by a Major in Vivo Metabolite of the Antidiabetic PPARγ Drug Pioglitazone. J. Med. Chem 62, 2008–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, and Milburn MV (1998). Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 395, 137–143. [DOI] [PubMed] [Google Scholar]
- Norris M, Fetler B, Marchant J, and Johnson BA (2016). NMRFx Processor: a cross-platform NMR data processing program. J. Biomol. NMR 65, 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okafor CD, Colucci JK, and Ortlund EA (2019). Ligand-Induced Allosteric Effects Governing SR Signaling. Nucl. Recept. Res 6. [Google Scholar]
- Rastinejad F, Huang P, Chandra V, and Khorasanizadeh S (2013). Understanding nuclear receptor form and function using structural biology. J. Mol. Endocrinol 51, T1–T21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos GM, Fairall L, and Schwabe JWR (2011). Negative regulation by nuclear receptors: a plethora of mechanisms. Trends Endocrinol. Metab. TEM 22, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J, Brust R, Griffin PR, Kamenecka TM, and Kojetin DJ (2019). Quantitative structural assessment of graded receptor agonism. Proc. Natl. Acad. Sci. U. S. A 116, 22179–22188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J, Mosure SA, Zheng J, Brust R, Bass J, Nichols A, Solt LA, Griffin PR, and Kojetin DJ (2020). A molecular switch regulating transcriptional repression and activation of PPARγ. Nat. Commun 11, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singarapu KK, Zhu J, Tonelli M, Rao H, Assadi-Porter FM, Westler WM, DeLuca HF, and Markley JL (2011). Ligand-specific structural changes in the vitamin D receptor in solution. Biochemistry 50, 11025–11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek FM (2011). What are nuclear receptor ligands? Mol. Cell. Endocrinol 334, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza PCT, Thallmair S, Conflitti P, Ramírez-Palacios C, Alessandri R, Raniolo S, Limongelli V, and Marrink SJ (2020). Protein-ligand binding with the coarse-grained Martini model. Nat. Commun. 11, 3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasaturo M, Fiengo L, De Tommasi N, Sabatino L, Ziccardi P, Colantuoni V, Bruno M, Cerchia C, Novellino E, Lupo A, et al. (2017). A compound-based proteomic approach discloses 15-ketoatractyligenin methyl ester as a new PPARγ partial agonist with anti-proliferative ability. Sci. Rep 7, 41273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega S, Abian O, and Velazquez-Campoy A (2016). On the link between conformational changes, ligand binding and heat capacity. Biochim. Biophys. Acta 1860, 868–878. [DOI] [PubMed] [Google Scholar]
- Waku T, Shiraki T, Oyama T, Maebara K, Nakamori R, and Morikawa K (2010). The nuclear receptor PPARγ individually responds to serotonin- and fatty acid-metabolites. EMBO J. 29, 3395–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weikum ER, Liu X, and Ortlund EA (2018). The nuclear receptor superfamily: A structural perspective. Protein Sci. Publ. Protein Soc. 27, 1876–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Zhou X, Chen W, Long L, Li W, Yang X, Li S, and Wang L (2015). L312, a novel PPARγ ligand with potent anti-diabetic activity by selective regulation. Biochim. Biophys. Acta 1850, 62–72. [DOI] [PubMed] [Google Scholar]
- Ye F, Zhang Z-S, Luo H-B, Shen J-H, Chen K-X, Shen X, and Jiang H-L (2006). The dipeptide H-Trp-Glu-OH shows highly antagonistic activity against PPARgamma: bioassay with molecular modeling simulation. Chembiochem Eur. J. Chem. Biol. 7, 74–82. [DOI] [PubMed] [Google Scholar]
- Yue L, Ye F, Xu X, Shen J, Chen K, Shen X, and Jiang H (2005). The conserved residue Phe273(282) of PPARalpha(gamma), beyond the ligand-binding site, functions in binding affinity through solvation effect. Biochimie 87, 539–550. [DOI] [PubMed] [Google Scholar]
- Zhao H, Piszczek G, and Schuck P (2015). SEDPHAT--a platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 76, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H-X (2010). From induced fit to conformational selection: a continuum of binding mechanism controlled by the timescale of conformational transitions. Biophys. J 98, L15–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates of PPARγ LBD-WT-Apo Delipidated, PPARγ LBD/GW1929 complex, PPARγ LBD-Y473E, PPARγ LBD-Y473E/darglitazone complex and PPARγ LBD-Y473E/GW1929 complex reported in this paper are deposited to the Protein Data Bank (PDB) under accession codes 6VZO, 6VZL, 6VZN, 6VZM and 7JQG, respectively.