

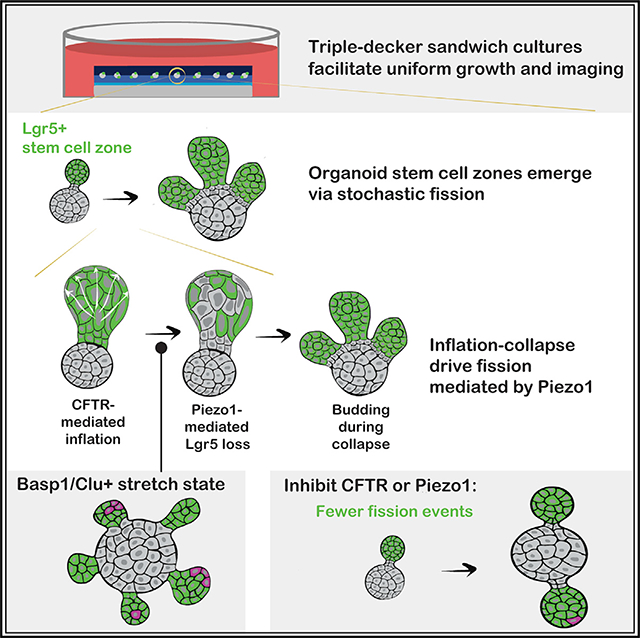
SUMMARY
How stem cells self-organize to form structured tissues is an unsolved problem. Intestinal organoids offer a model of self-organization as they generate stem cell zones (SCZs) of typical size even without a spatially structured environment. Here we examine processes governing the size of SCZs. We improve the viability and homogeneity of intestinal organoid cultures to enable long-term time-lapse imaging of multiple organoids in parallel. We find that SCZs are shaped by fission events under strong control of ion channel-mediated inflation and mechanosensitive Piezo-family channels. Fission occurs through stereotyped modes of dynamic behavior that differ in their coordination of budding and differentiation. Imaging and single-cell transcriptomics show that inflation drives acute stem cell differentiation and induces a stretch-responsive cell state characterized by large transcriptional changes, including upregulation of Piezo1. Our results reveal an intrinsic capacity of the intestinal epithelium to self-organize by modulating and then responding to its mechanical state.
In brief
Intestinal organoids contain stem cell zones (SCZs) that resemble the crypt base in vivo. We studied SCZ patterning by live imaging and perturbing organoids in “sandwich” cultures. We found that SCZs undergo Piezo-dependent fission driven by inflation and collapse of the organoid lumen, and inflation induces a stretch-associated cell state.
Graphical Abstract
INTRODUCTION
In homeostasis, stem and progenitor cells divide to compensate for differentiation and cell loss. How cells coordinate these processes is central to understanding tissue physiology and disease (Donati and Watt, 2015; Simons and Clevers, 2011). Two broad classes of mechanisms coordinate cell behaviors in tissues. First, cells are regulated externally by a structured micro-environment (a “niche”) through mechanical and biochemical signaling (Scadden, 2006; Spradling et al., 2008). Second, tissues can self-organize spontaneously even in the absence of structured cues from the microenvironment (Sasai, 2013; Shahbazi et al., 2019). A goal of stem cell biology is to characterize these intrinsic, niche-independent programs of tissues.
The intestinal epithelium is a stereotypical self-renewing tissue that offers a model to study self-organization and niche control (Clevers, 2013). In most mammals, the intestinal epithelium undergoes near-complete turnover every 3–5 days (Barker, 2014; Leblond, 1981). Supporting this turnover are equipotent Lgr5+ intestinal stem cells (ISCs) (Barker et al., 2007; Lopez-Garcia et al., 2010; Snippert et al., 2010), which cluster in stem cell zones (SCZs) at the bases of invaginated crypts of Lieberkühn (Barker et al., 2007; Bjerknes and Cheng, 1981a, 1981b; Clevers, 2013). ISCs differentiate into several cell types that emerge from crypts into villi and form the luminal surface of the intestine. In homeostasis, cell division at the crypt base precisely balances cell loss at the villus tip. An extrinsic niche formed by the underlying non-epithelial mesenchyme supports these behaviors; as cells migrate up the crypt-villus axis, they lose mitogenic stimulation and become exposed to signals that promote differentiation (Meran et al., 2017; Spit et al., 2018).
ISCs also generate components of their own niche, including progeny that produce Wnt and Notch ligands (Farin et al., 2012; Sato et al., 2011b). They exhibit an intrinsic capacity to self-organize a structured tissue, as observed in organoid cultures (Sato et al., 2009). Even a single ISC can give rise to a highly structured multicellular epithelium, with Lgr5+ SCZs localized to the tips of crypt-like buds with characteristic physiological size (Sato et al., 2009). Although organoid formation requires exposure to growth factors, in culture these factors are added to media, where they diffuse and impose no spatial pattern. Therefore, intestinal organoids offer an opportunity to study self-organization of tissue stem cells. Our central question here is how an epithelium in isolation can generate and maintain SCZs with typical numbers of stem cells.
In the intestinal epithelium, self-organization requires signaling through the epidermal growth factor (EGF), Notch, and Wnt cascades (Farin et al., 2016; Sato et al., 2011a; Serra et al., 2019) and establishment of EphB-Ephrin boundaries (Batlle et al., 2002). Mechanical patterning is also thought to play a key role in crypt formation, with compressive stresses in the epithelial monolayer driving buckling and emergence of a typical tissue curvature and length scale (Buske et al., 2012; Drasdo, 2000; Dunn et al., 2012; Hannezo et al., 2011; Langlands et al., 2016; Nelson et al., 2013; Pin et al., 2015). In vivo, buckling drives epithelial folding (Shyer et al., 2015) and constriction (Sumigray et al., 2018), which occur during development or shortly after birth (Trier and Moxey, 1979). Post-natally, SCZ buckling is also thought to drive fission of crypts into two or more daughter crypts (Baker et al., 2014; Cummins et al., 2008). Crypt fission occurs physiologically and complements crypt hyperplasia as a mode of intestinal growth, but it is accelerated abnormally in colorectal adenomas and carcinomas (Baker et al., 2014; Berlanga-Acosta et al., 2001; Pin et al., 2015). Crypt fission occurs frequently in organoids; thus, it could help set the size and morphology of SCZs.
We used intestinal organoid cultures to explore the mechanism by which ISCs are able to spontaneously generate multicellular SCZs with physiological sizes. For this, we developed a culture system that enabled high-resolution, long-term imaging and perturbation of fluorescently labeled intestinal organoids. From the initial results we identified stochastic SCZ fission as the primary determinant of organoid morphology. Using live imaging, perturbation, and single-cell transcriptomics, we then clarified cellular and molecular events associated with and necessary for crypt fission.
RESULTS
A “triple-decker” hydrogel sandwich culture for organoid imaging and molecular perturbation
We optimized organoid cultures for uniform growth and long-term time-lapse imaging. Organoids are commonly cultured in hemispherical hydrogel droplets deposited on multi-well plates and overlaid with medium (Figure 1A). Although these “dome” cultures are easy to prepare, they are not optimal for imaging because organoids grow in distinct focal planes at different distances from a microscope objective. Organoids can also occupy non-uniform mechanical and chemical environments that lead to location-dependent variation in growth. These sources of variation reduce the number of analyzable organoids per well, and they can obscure perturbation phenotypes. Imaging far from a coverslip also demands increased light exposure, which can lead to phototoxicity during long-term imaging.
Figure 1. Triple-decker hydrogel sandwiches for uniform organoid culture and imaging.
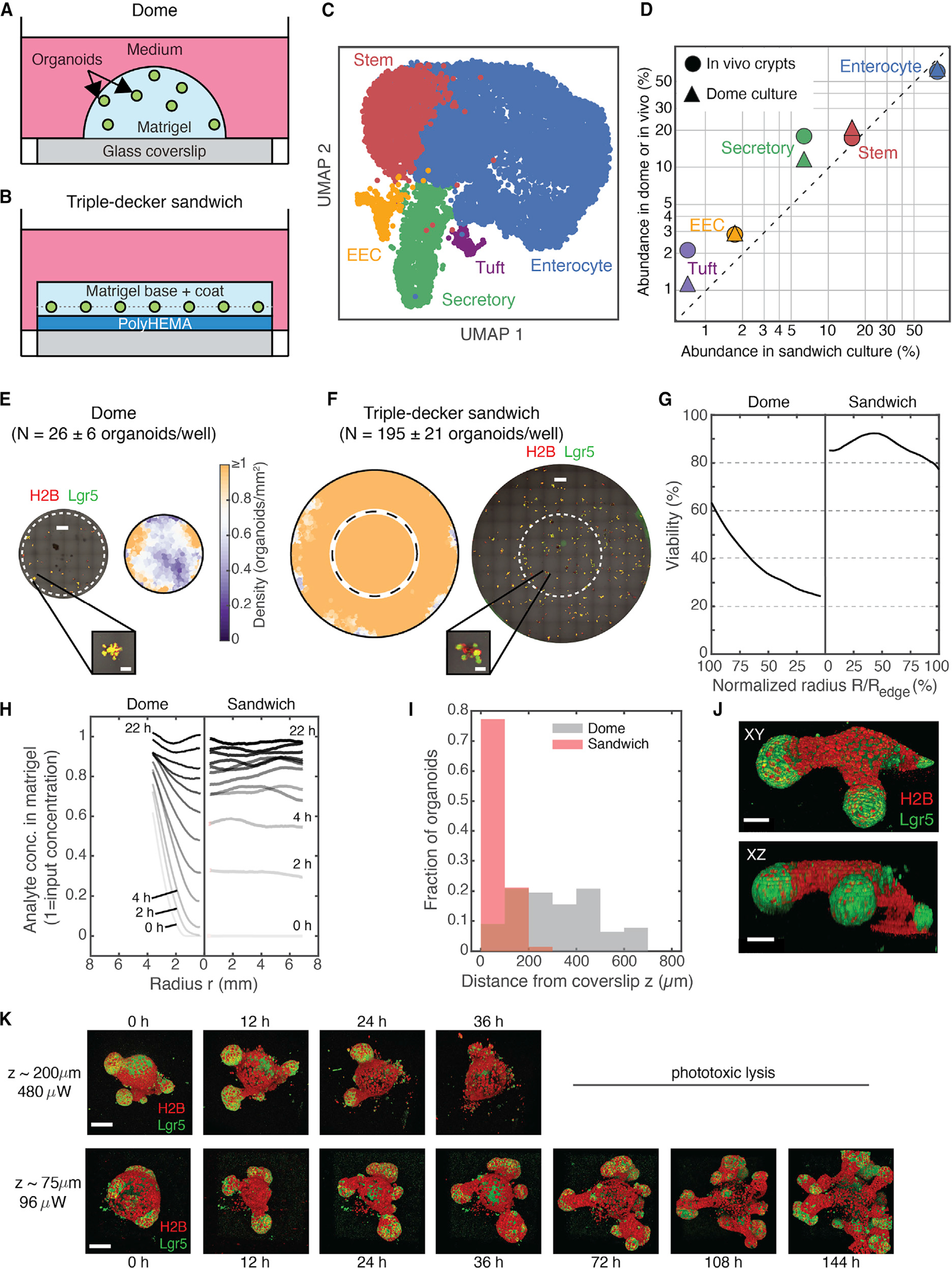
(A and B) Configuration of organoid cultures in Matrigel droplets (domes) compared with sandwich cultures consisting of a passivating polyHEMA layer and Matrigel layers.
(C) UMAP representation of scRNA-seq (13,062 cells × 30,005 genes) of dome and sandwich (n = 6 wells each) cultures, colored by annotated cell type. EEC, enteroendocrine.
(D) The fraction of cells classified to each cell type in sandwich cultures compared with dome cultures and freshly isolated intestinal crypts.
(E and F) Representative tiled, flattened images of mouse intestinal organoids cultured in domes or sandwiches alongside smoothed density heatmaps (average of 3 replicate wells). Dashed circles indicate the circumference of dome culture, overlaid to show the difference in available imaging area. Representative organoids are magnified. Scale bars, 1 mm and 200 μm (inset). Here and elsewhere: green, Lgr5-DTR-EGFP; red, H2B-mCherry.
(G) The fraction of organoids surviving after 5 days in culture (viability), plotted against distance from the center of the dome/sandwich.
(H) Fluorescence intensity time series of an analyte (dextran) as a function of radius from the center of the dome/sandwich. Other molecules are shown in Figure S1.
(I) Histograms of organoid heights. Sandwich cultures (red) align organoids with the coverslip (n = 599 from 3 wells); organoids in domes (gray) are distributed over hundreds of micrometers (n = 68 organoids from 3 wells).
(J) Triple-decker sandwiches enable full-thickness structures to be resolved by confocal microscopy. Shown are orthographic projections of an organoid with two SCZs. Scale bar, 30 μm.
(K) 3D fluorescence time-lapse imaging using a laser-scanning confocal microscope (15-min time interval; Video S1) is phototoxic at 200 μm from the coverslip but not at the 75-μm distance typical for sandwich cultures. Scale bar, 100 μm.
Several groups have reconfigured organoid cultures to improve tissue uniformity and accessibility to imaging. Organoids can be confined between a Transwell insert and a glass coverslip (Saarela et al., 2017) or “sandwiched” between two hydrogel layers, restricting them to a narrow z plane (Debnath et al., 2003; Jaffe et al., 2008). Usually the base layer of hydrogel is very thin to maximize image quality. Existing sandwich methods are effective when imaging slow-growing structures like mammary organoids (Debnath et al., 2003; Gajewska and McNally, 2017; Lee et al., 2007; Mroue and Bissell, 2013) and bronchospheres (Hild and Jaffe, 2016). However, organoids undergoing rapid expansion, such as intestinal organoids, penetrate the base layer of a sandwich and revert to 2D growth on a coverslip.
To overcome this limitation, we prepared triple-decker hydrogel sandwiches that facilitate high-quality imaging and long-term 3D organoid growth. A triple-decker sandwich consists of three polymer layers (Figure 1B): (1) a base coat of poly(2-hydroxyethyl methacrylate) (polyHEMA), (2) a thin overlay of Matrigel onto which cells are deposited, and (3) a top coat of Matrigel. The base layer of polyHEMA prevents cells from contacting and spreading onto the coverslip over multiple days in culture, enabling the growth of viable, multi-budded organoids with Lgr5+ stem cells located at the tips of the buds (Figures S1A and S1B). Using single-cell RNA sequencing (scRNA-seq) (Figures 1C, S1C, and S1D) and immunofluorescence for canonical cell type markers (Figure S1E), we established that organoids in triple-decker sandwich cultures contain the same cell types as organoids in dome cultures, including stem, enterocyte, secretory, enteroendocrine, and tuft cells (log-abundance Pearson correlation R > 0.99; Figure 1D), and they do so in similar proportions.
Organoids in triple-decker sandwiches grow more uniformly and are more viable than in domes. After 6 days in culture, domes were densest on their peripheries and sparsest at their centers (Figure 1E), whereas in triple-decker sandwiches, organoids were present at uniform density across the well (≥1 organoid/mm2, n = 3 replicates) (Figure 1F). “Dead zones” at the centers of domes emerged from preferential survival along the edges, not from inhomogeneity in the initial plating density (Figure 1G). At the edge of a dome, 60% of organoids deposited initially survived to day 6, and viability dropped to less than 30% at the center of a dome. In contrast, viability consistently exceeded 80% in triple-decker sandwiches regardless of spatial position. Likewise, organoids in triple-decker sandwiches also exhibit more uniform and reproducible growth (Figure S1F); organoid volumes varied with spatial position in domes but not in sandwiches.
These differences in organoid viability and growth may reflect better access to diffusible nutrients, gases, and growth factors in sandwich cultures, as seen by tracking the diffusion of fluorescently labeled molecules (immunoglobulin G [IgG; 150 kDa], dextran [3 kDa], and phenol red [0.3 kDa]) into Matrigel domes and sandwiches (Figures 1H and S1G). A concentration gradient between the center and edge of a dome equilibrated with a characteristic time constant T = 19 ± 4 h for IgG (time for the center concentration to reach 80% or greater of the edge concentration, mean ± SD across replicate domes) compared with T = 3 ± 1 h in a sandwich.
As with other sandwich cultures, triple-decker sandwiches enable high-resolution and high-throughput imaging by aligning organoids close to the coverslip (Figure 1I). Organoids in triple-decker sandwiches were confined to a narrow band (z = 78 ± 39 μm from the coverslip [mean ± SD; n = 586 organoids from 3 technical replicates]). Organoids in domes were located much farther from the coverslip, and their z positions were far more variable (z = 317 ± 167 μm [mean ± SD; n = 77 organoids from 3 technical replicates]). This 4-fold reduction in the average distance from organoid to objective reduces scattering and spherical aberration (Ross et al., 2014), allowing imaging of full-thickness Lgr5+ buds (Figure 1J).
Proximity to the coverslip also facilitated long-term live imaging. Controlling for the signal-to-noise ratio (SNR), imaging a typical organoid in a triple-decker sandwich required 80% less laser power than for an organoid three times as far from the coverslip (Figure 1K). We were able to capture large, high-resolution z stacks of entire intestinal organoids every 15 min for over 1 week with minimal phototoxicity (Figure 1K; Videos S1 and S2), resolving cell membranes and tracking nuclei through mitosis (Figure S1H). Later we carried out imaging every 6 min. In contrast, intestinal organoids positioned farther above the coverslip showed signs of photodamage within 24 h and lost most of their Lgr5+ stem cells after 36 h of imaging at the same frequency (Figure 1K). The improved SNR also enabled use of dry microscope objectives as well as high numerical aperture (NA) water immersion objectives to image organoids (Figure S1I). Therefore, triple-decker sandwiches make it possible to capture high-resolution videos of organoids in multiple wells in a single experiment.
Organoid stem cell niche sizes emerge from constitutive growth and stochastic fission
To investigate how physiological SCZ size (10–20 Lgr5+ cells; Snippert et al., 2010) emerges spontaneously in organoids, we identified the processes shaping SCZ size. SCZs could grow by merging with each other, but we only rarely observed SCZ fusion (Video S3). Small Lgr5+ SCZs could also appear from Lgr5− cells, but we observed no de novo Lgr5 expression except during an early transient period after plating, as reported previously (Serra et al., 2019). This left us to consider three processes shaping SCZ size distribution: self-renewing ISC divisions that expand SCZs, ISC differentiation that contracts SCZs, and SCZ fission events that generate smaller SCZs from larger ones.
Growth and fragmentation processes could be regulated to set SCZ size in several ways (Figure 2A): ISCs could tune their division or differentiation rates (“growth control”), SCZs could become unstable and undergo fission above the physiological size, or both forms of control could occur. It is also possible that there is no intrinsic control of SCZ size; if so, then a typical SCZ size could still emerge from uncontrolled growth and stochastic fission, albeit with significant variability in SCZ size about the average. To distinguish these possibilities, we recorded growth and fission events of SCZs by imaging live Lgr5-DTR-EGFP/H2B-mCherry organoids.
Figure 2. ISCs in organoid cultures lack intrinsic size control.
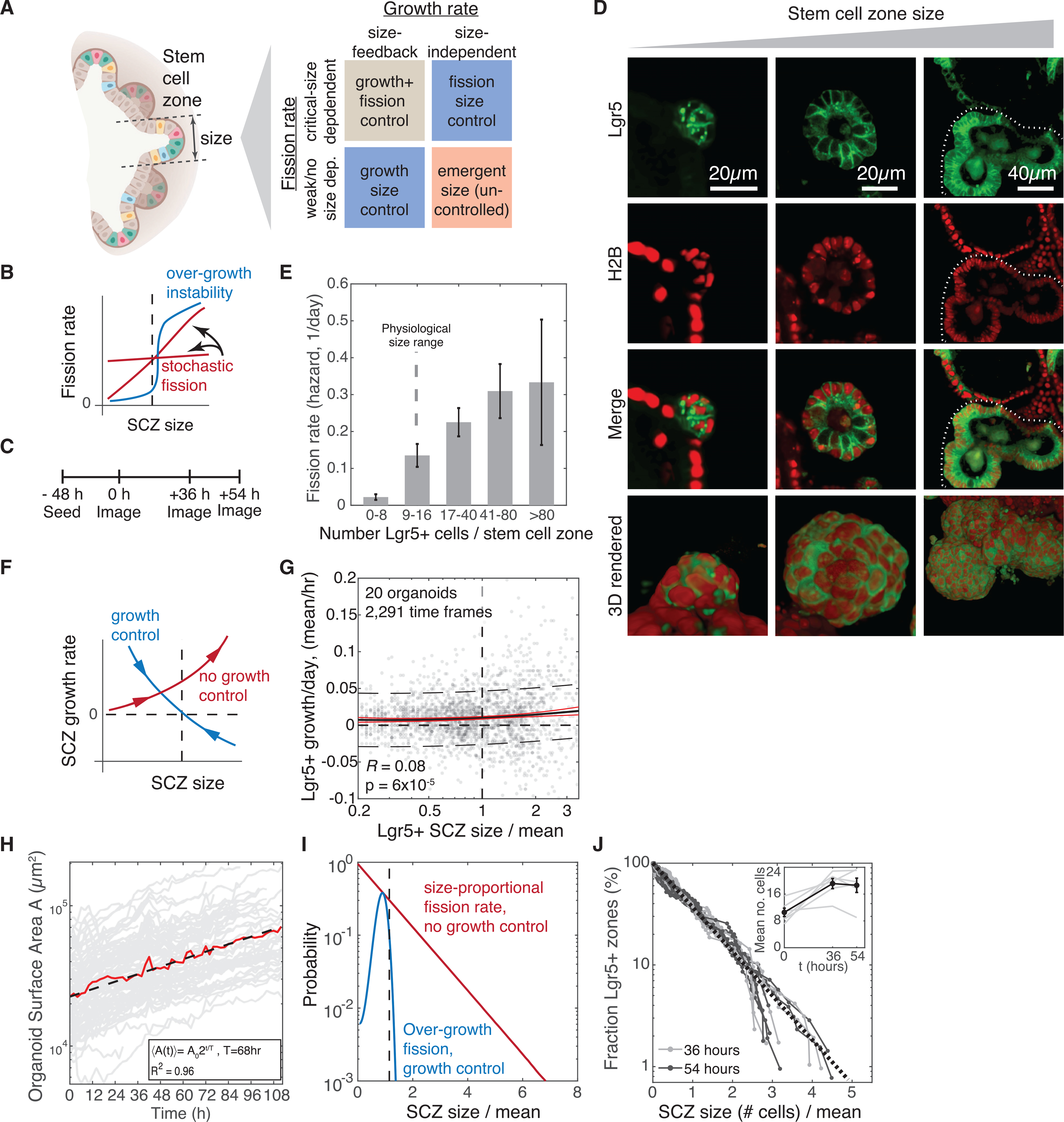
(A) Classes of mechanisms by which Lgr5+ stem cell zones (SCZs) in culture could spontaneously acquire characteristic mean size: by reducing growth rates of large SCZs, triggering fission of large SCZs, or both.
(B) Schematic of a test for fission control by evaluating fission rates at different SCZ sizes. The dashed line indicates characteristic physiological size.
(C) Experimental schematic for measuring size-dependent SCZ fission rates. The same SCZs are tracked over 3 imaging time points to assess their initial size and whether they undergo fission. See Figure S2 and STAR Methods for SCZ size segmentation and calibration.
(D) Single z sections and 3D rendered projections of representative Lgr5+ SCZs varying from tens to several hundreds of cells. On the right, the dashed lines delineate the SCZ.
(E) Organoid SCZs are unstable to fission even at sizes that are normally stable in vivo (n = 74). Fission rates are calculated as the number of fission events of SCZs with initial size n1 < n < n2 prior to fission (indicated in the plot), divided by the recorded duration in which any organoid is of size n1 < n < n2. Error bars indicate SEM by error propagation of volume and sampling SEMs.
(F) Schematic of the test for SCZ size-dependent feedback control on net self-renewal rates of stem cells. Arrows indicate the mean dynamics associated with each case.
(G) Time series measurements of SCZ size in multiple organoids indicate a weak positive correlation between SCZ size and growth rate, ruling out intrinsic feedback growth control. n = 20 organoids, 2,291 time frames.
(H) The organoid surface area grows exponentially with a doubling time of 68 h, as determined by analyzing time series data from z stacks of H2B-mCherry-labeled organoids (n = 94).
(I) Predicted steady-state size distributions of SCZs under assumptions of size-dependent feedback (blue) or no size control of growth and fission rates (red). Curves show numerical PBE model solutions.
(J) Experimentally observed SCZ sizes are distributed exponentially at steady state, consistent with a model lacking growth and fission size control (n = 659 SCZs from 5 wells across 3 time points). Inset: average SCZ size.
First we examined whether small SCZs might grow stably but undergo fission above their typical physiological size (Figure 2B). We imaged z stacks of organoids at three consecutive time points (n = 5 wells; see schematic in Figure 2C). At any given time point, we found that SCZs in organoids varied considerably in size, containing as few as ~10 cells or as many as several hundred (Figure 2D). This wide distribution allowed us to assess how fission frequency varies with size. We measured the sizes of SCZs by segmenting Lgr5+ regions (Figures S2A–S2I) and scored whether each SCZ underwent fission. From these data (of 659 SCZs, 74 underwent fission), we observed that fission frequency does increase with size (Figure 2E), but at no size were SCZs intrinsically stable. The fission rate for SCZs of physiological size far exceeded the fission rate for mouse intestinal crypts in vivo (more than 10% per day in vitro for SCZs in organoids with 9–16 Lgr5+ cells versus less than 0.2% per day in vivo; Snippert et al., 2014; Lopez-Garcia et al., 2010). Thus, although small SCZs in vivo rarely undergo fission, this stability is not intrinsic to the epithelium.
Next we tested whether the typical size of SCZs might reflect self-tuning of ISC self-renewal or differentiation to modulate net growth. If so, then SCZs should expand when small and contract when large (Figure 2F). To assess this, we obtained volumetric time-lapse videos of 20 organoids over periods of 48–144 h with 1- to 2-h imaging intervals. We segmented the SCZs in each organoid and calculated changes in size between consecutive frames between fission events. We observed that SCZ growth rates were highly variable but tended, on average, to increase with size (Figure 2G; R = 0.08, p < 10−4 for zero correlation). This growth was consistent with our observation that the total epithelial surface area of organoids increases exponentially with time, doubling, on average, every 68 h (N = 94 organoids) (Figure 2H; Video S4). Further, immunostaining showed that SCZs contained a stable concentration of Paneth cells, which support ISC self-renewal, across all SCZ sizes (1 Paneth cell per 5 ISCs; Figure S2J). These measurements rule out size-dependent feedback on organoid SCZ growth rates. Because such size control must occur in vivo, we attribute it to extrinsic cues rather than an intrinsic program of the epithelium.
The absence of growth control and fission control suggests that, in organoids, the size of SCZs emerges from a constitutive ISC fate imbalance toward self-renewal and stochastic SCZ fission. If true, then this conclusion should allow prediction of the SCZ size distribution in culture; namely, it should be an exponential distribution (coefficient of variation [CV] of 100%). This prediction emerges from a solution to the population balance equation (PBE), which models how the sizes of particles evolve over time because of processes that grow, shrink, merge, and split them (Krapivsky et al., 2010; Rulands et al., 2018). In contrast, if active SCZ size control occurs, then the PBE predicts a narrower size distribution (CV < 100%) with a mode at the typical size (Figure 2I). The volume distributions we measured (N = 159, 183, and 224 SCZs, respectively, across 3 technical replicates) fit well to a simple exponential with no mode at the typical size and no change in mean size between 36–54 h (Figure 2J). The shape of the distribution and the live imaging results show that the intestinal epithelium does not encode a preferred SCZ size through intrinsic control of ISC self-renewal or SCZ fission rates.
SCZ fission correlates with inflation of the organoid lumen
Crypt fission is controlled tightly in vivo. It occurs rarely during adult homeostasis but is crucial for postnatal tissue growth and the response to large-scale tissue damage, and it is far more frequent in malignant disease (Snover et al., 2005). In organoid cultures, SCZ fission occurs frequently. Our results above clarified that the high rate of fission in organoids is not simply the product of excessive growth in culture because fission often occurs even in small SCZs at sizes that would be stable in vivo. This observation suggested that organoids might be used to uncover events that trigger fission.
The process of SCZ fission entails changes in SCZ cell composition and in 3D morphology. Contiguous Lgr5+ regions of the epithelium split into distinct domains separated by differentiated Lgr5− cells, and the initial SCZ breaks into two or more spherical buds. The order of these cellular and morphological changes is not yet clear. Several studies have proposed that SCZ fission may coincide with growth-mediated compression of the epithelium that causes the crypt base to buckle or invaginate (Hannezo et al., 2011; Langlands et al., 2016; Pin et al., 2015). Supporting this view, static images of crypts in vivo show invaginations of the Lgr5+ epithelium between Paneth cell-rich domains (Langlands et al., 2016). However, we noticed that SCZs in organoids are frequently subject to stretching (Figure 3A) and that Lgr5− cells appear to partition Lgr5+ domains during stretching. Thus, SCZ fission may occur prior to compression of the epithelium (Figure 3B). We examined the temporal relationship between epithelial stretching, compression, and SCZ fission.
Figure 3. SCZ fission correlates with inflation of the organoid lumen.
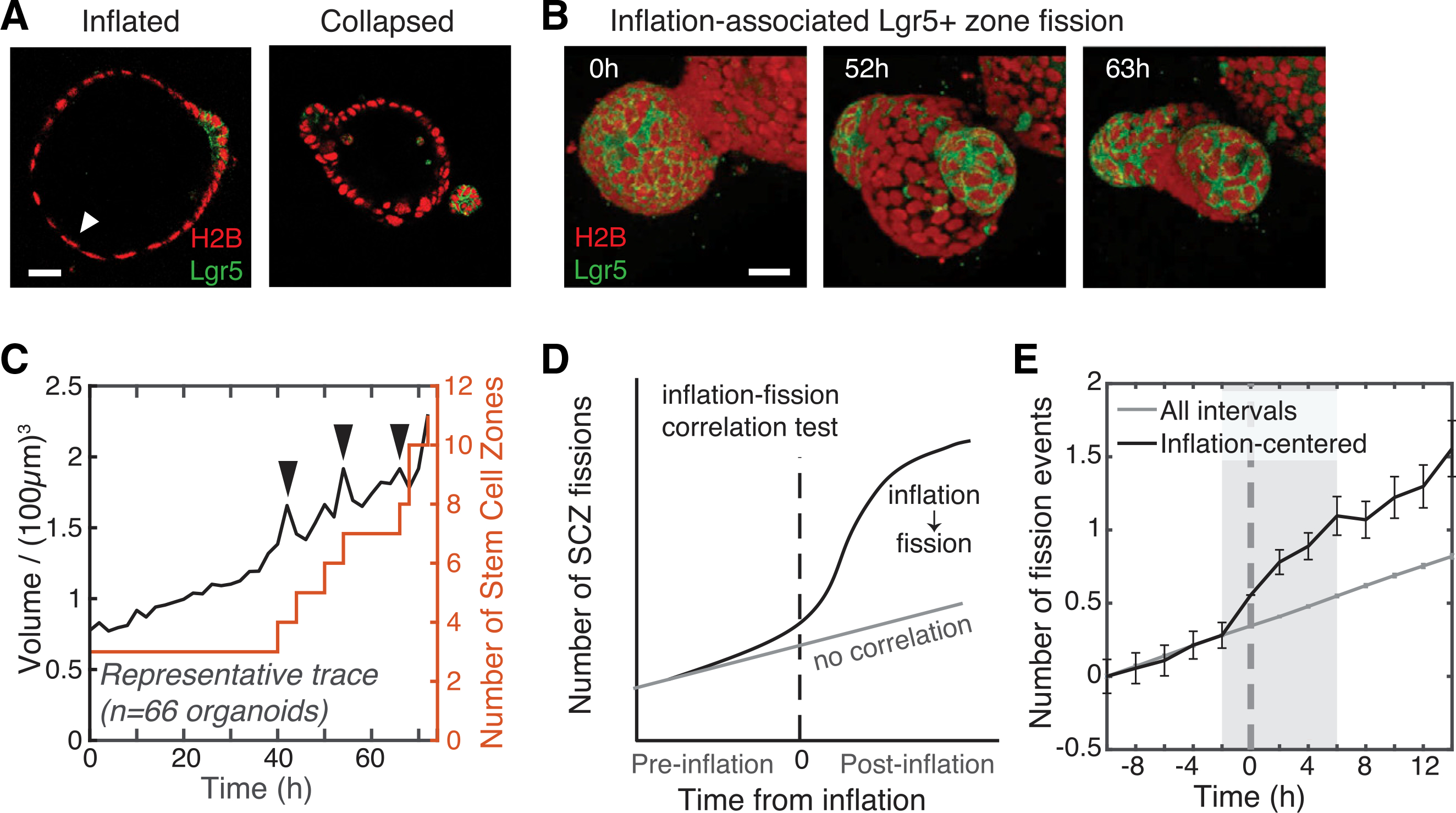
(A) Micrographs of inflated and collapsed organoids. Lgr5− cells in inflated organoids (white arrow) are squamous like. Scale bar, 40 μm.
(B) Time series images showing a contiguous Lgr5+ region splitting in two as the bud inflates (52 h). Scale bar, 30 μm.
(C) Representative time series data of segmented organoid volume (black) and SCZ number (red) used to test for correlation between inflation-collapse and fission. The organoid was imaged every 2 h for 72 h. Black arrows indicate autodetected inflation-collapse events.
(D) Schematic of the test for temporal correlation of inflation and fission. If correlated, then the number of SCZ fission events should increase immediately after inflation compared with all intervals of the same duration (gray, no correlation).
(E) A correlation test (n = 66 organoids) shows that the SCZ fission rate increases significantly above the baseline rate in an 8-h window around inflation events, with maximum fission rate 6 h after peak inflation. Error bars indicate SEM.
The intestinal organoid epithelium undergoes stretching and compression as its lumen inflates and deflates. Inflation often stretches the epithelial monolayer to the point where cells appear squamous with flattened nuclei (Figure 3A). An entire organoid can inflate. In some cases, compartmentalized inflation occurs when a single bud lumen is obstructed (Video S5). Eventually an inflated structure collapses when luminal fluid escapes through rupture of the epithelial monolayer, shunting into a different part of the organoid, or possibly through paracellular leakage. Collapse relieves stress on the epithelial wall and restores cells to a columnar shape. We observed cells shed into the lumen at a higher rate after luminal collapse (Video S6), reminiscent of compression-induced delamination (Eisenhoffer et al., 2012). Cycles of inflation and collapse occurred across medium conditions, culture configurations, and genotypes as well as in videos published by others (Fordham et al., 2013; Miura and Suzuki, 2017; Sato et al., 2011a; Schwank et al., 2013).
To test whether inflation-collapse events correlate with SCZ fission, we carried out tiled 3D time-lapse imaging of Lgr5-DTR-EGFP/H2B-mCherry organoids in sandwich culture to follow large fields of organoids for over 100 h at 2-h intervals. We scored the number of SCZs over time in a subset of the organoids (177 total organoids, 66 of which were analyzed) and computationally segmented whole organoid volumes (Video S4; Figure 3C). Hereafter, we score a “fission event” as partitioning of Lgr5+ domains rather than a change in tissue curvature. Inflation-collapse events appeared as sawtooth-shaped volume pulses (Figure 3C, black arrows). Fission events appeared as stair-step increases in SCZ numbers. Across all organoids (in total, 184 fission events and 99 inflation-collapse events), inflation-collapse and SCZ fission were correlated significantly in time; SCZ fission occurred at double the baseline rate in an 8-h window around inflation-collapse events (Figures 3D and 3E). SCZ fission was most likely to occur 6 h after peak inflation, indicating that luminal inflation most often preceded SCZ fission (Figure 3E).
Qualitative differences between SCZ fission with or without luminal inflation
To determine the order of luminal inflation, collapse, SCZ fission and buckling, we examined SCZ fission events using high-magnification time series. In the absence of inflation, we observed that epithelial tissue buckled first and the SCZ partitioned only subsequently, with Lgr5− cells appearing at sites of negative curvature (Figure 4A; Video S7). This mode of fission resembles the prevailing model for crypt fission (Langlands et al., 2016). In the presence of inflation, the order of events was reversed (Figure 4B; Video S8): Lgr5+ cells partitioned into separate patches during inflation. Only after luminal collapse did the epithelium invaginate, with nascent buds emerging from the sites of separate Lgr5+ patches. These observations argue that buckling is not required to drive ISC differentiation and SCZ fission. Hereafter, we refer to these two modes of fission (inflation-independent and inflation-dependent) as type I and type II fission, respectively.
Figure 4. Stereotyped SCZ fission dynamics in intestinal organoids.
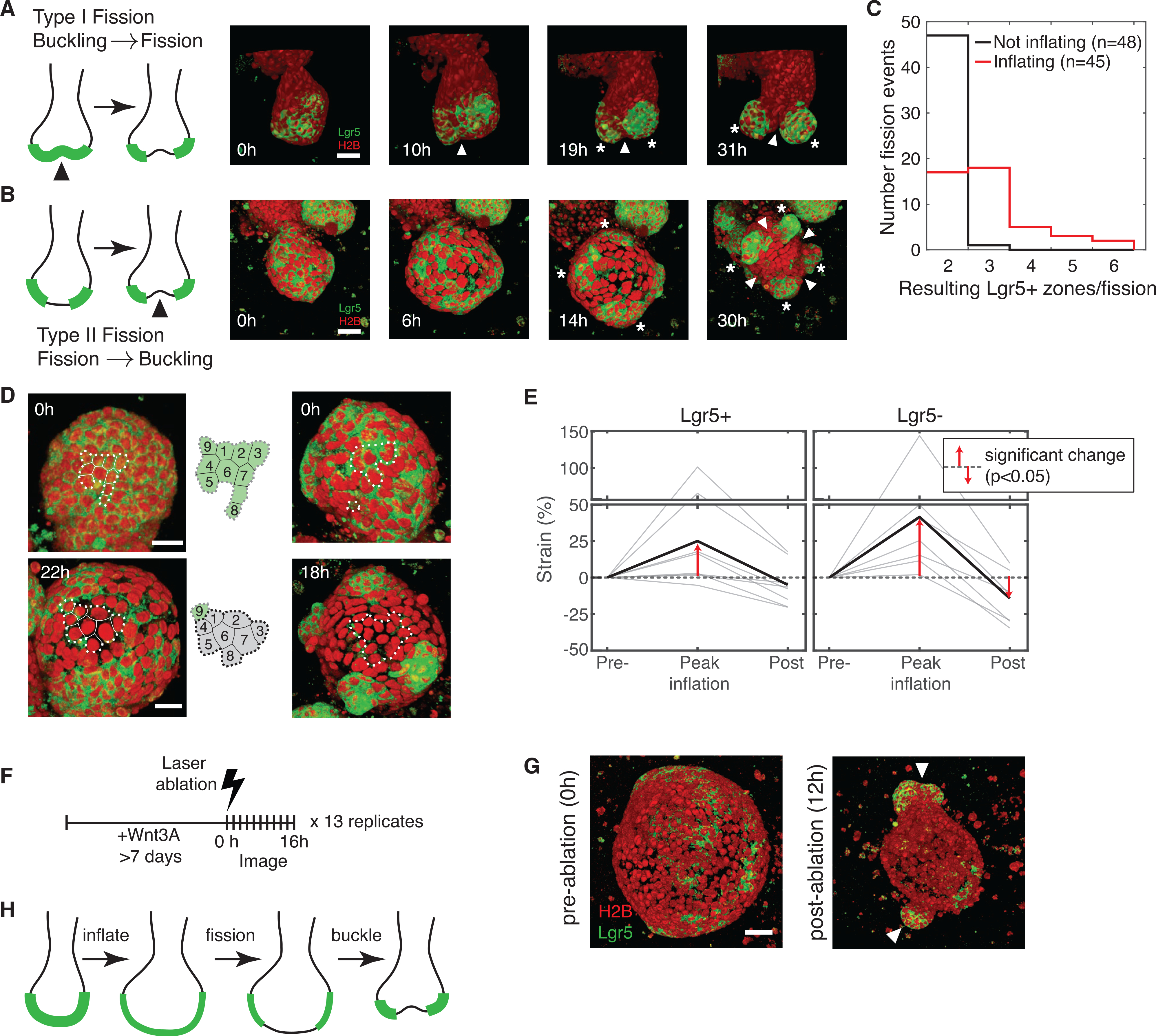
(A) Fission sequence (type I) by tissue buckling and invagination (white arrows) preceding Lgr5+ stem cell differentiation and fragmentation of SCZs (white asterisks). Red, H2B; green: Lgr5.
(B) Fission sequence (type II) with fragmentation of contiguous Lgr5+ SCZs preceding tissue buckling. The organoid bud appears inflated until 14 h. (A and B), arrows indicate invagination; asterisks indicate nascent SCZs. Scale bars, 20 μm.
(C) Number of nascent SCZs observed following fission from SCZs with inflated and non-inflated morphology.
(D) 40×-magnification and 6-min time lapse of organoid buds reveal that Lgr5+ cells differentiate in situ as organoid buds inflate. Scale bar, 40 μm. Dashed lines delineate a group of cells at early and late time points.
(E) The in-plane strain of the epithelium at three time points during luminal inflation and collapse. Gray, individual buds (n = 9); black, average. Strain is calculatedas the fractional change in mean inter-nuclear distance. Statistical significance by one-tailed t test.
(F and G) Laser ablation of cyst-like organoids grown in the presence of Wnt3A induces collapse, tissue buckling, bud formation, and localization of pre-existing Lgr5+ domains to nascent buds (white arrows). Scale bar, 50 μm.
(H) Summary of the model for type II SCZ fission.
Type I and II fission differed in their outcomes. In time series of organoid fission (cf. Figures 2C and 2E), we manually labeled each SCZ as inflated or collapsed based on the appearance of its lumen (a total of 93 fission events scored with similar frequencies of both types) and scored the number of nascent SCZs produced during a single fission event. Inflation-independent fission was largely binary, whereas inflation-associated fission was more prolific, with the majority of events producing 3 or more daughter SCZs (Figure 4C).
Cellular events during luminal inflation and collapse
To identify cellular events that lead to SCZ fission during inflation-collapse, we recorded high-magnification (403) time series of organoids at dense (6-min) intervals. SCZ fission requires a contiguous region of Lgr5+ cells to become separated by Lgr5− cells. These Lgr5− cells could emerge from ISCs differentiating in situ or from cell rearrangements enabling differentiated cells to flow into an SCZ. We found that stem cells differentiated in situ with little cell rearrangement (Figure 4D; Video S8). SCZ fission resulted from rapid loss of Lgr5 expression as cells were stretched during luminal inflation. ISCs can lose Lgr5 expression even when adjacent to a Paneth cell (seen in 6 of 8 videos), suggesting that epithelial stretching can override Paneth cell niche factors (Figure S3F). Loss of Lgr5 expression began, on average, 3.5 ± 1 h (mean ± SEM) after inflation onset (Figures S3A–S3C). Variation in the timing of Lgr5 loss (SD = 3.1 h) was explained in part by variation in initial epithelial stretching prior to inflation (Pearson R = −0.54 between timing of Lgr5 loss and the inter-nuclear distance prior to inflation; Figures S3D and S3E).
After SCZs partitioned, new buds containing fragments of the original SCZ emerged during luminal collapse. The mean inter-nuclear distance, a measure of stretching, increased during bud inflation; after collapse, only Lgr5+ regions returned to their pre-inflation density (5.0% ± 5.2% reduction in average inter-nuclear distance relative to baseline, p = 0.19). Lgr5− regions separating these domains showed a statistically significant increase in cell density (14.3% ± 6.7% reduction in average inter-nuclear distance relative to baseline, p = 0.038), indicating compression. Thus, budding is consistent with soft Lgr5− regions becoming compressed and buckling inward during collapse, leaving stiffer Lgr5+ regions to form nascent buds.
Based on these observations, we predicted that buds should form at the sites of pre-existing SCZs in cystic (bud-free) organoids when they are induced to collapse. Organoids grown in medium containing Wnt3A (WENR organoids) normally fail to form buds (Merker et al., 2016; Miyoshi and Stappenbeck, 2013; Mustata et al., 2013; Thalheim et al., 2018), but we found that they contain discrete patches of Lgr5+ cells akin to SCZs. We punctured WENR organoids by laser ablation and subsequently tracked changes in their morphology (Figures 4F and 4G; Video S9). Ablation was sufficient to cause luminal deflation, epithelial buckling, and rapid emergence (within 2 h after ablation) of budded outgrowths at the sites of Lgr5+ patches (N = 12 of 13 ablated organoids). Thus, Wnt3A organoids may lack buds only because they remain consistently inflated. These results clarify cellular events in type II SCZ fission (Figure 4H): Lgr5+ cells differentiate in situ in SCZs when organoids inflate; upon collapse, the Lgr5− epithelium buckles, and Lgr5+ cells form nascent buds.
Organoid inflation causes SCZ fission
Does luminal inflation cause SCZ fission? We tested for causality by blocking or triggering inflation using small-molecule inhibitors and agonists of epithelial ion channels (Jaffe et al., 2008) and comparing the frequency of inflation and SCZ fission in the presence or absence of a drug (Figure 5A). Two drugs, CFTRinh-172 (Ma et al., 2002) and forskolin, modulate the activity of the cystic fibrosis transmembrane regulator (CFTR), a key controller of intestinal fluid secretion, and a third, ouabain, blocks organoid swelling by disrupting Na+/K+-ATPase.
Figure 5. Organoid inflation causes SCZ fission.
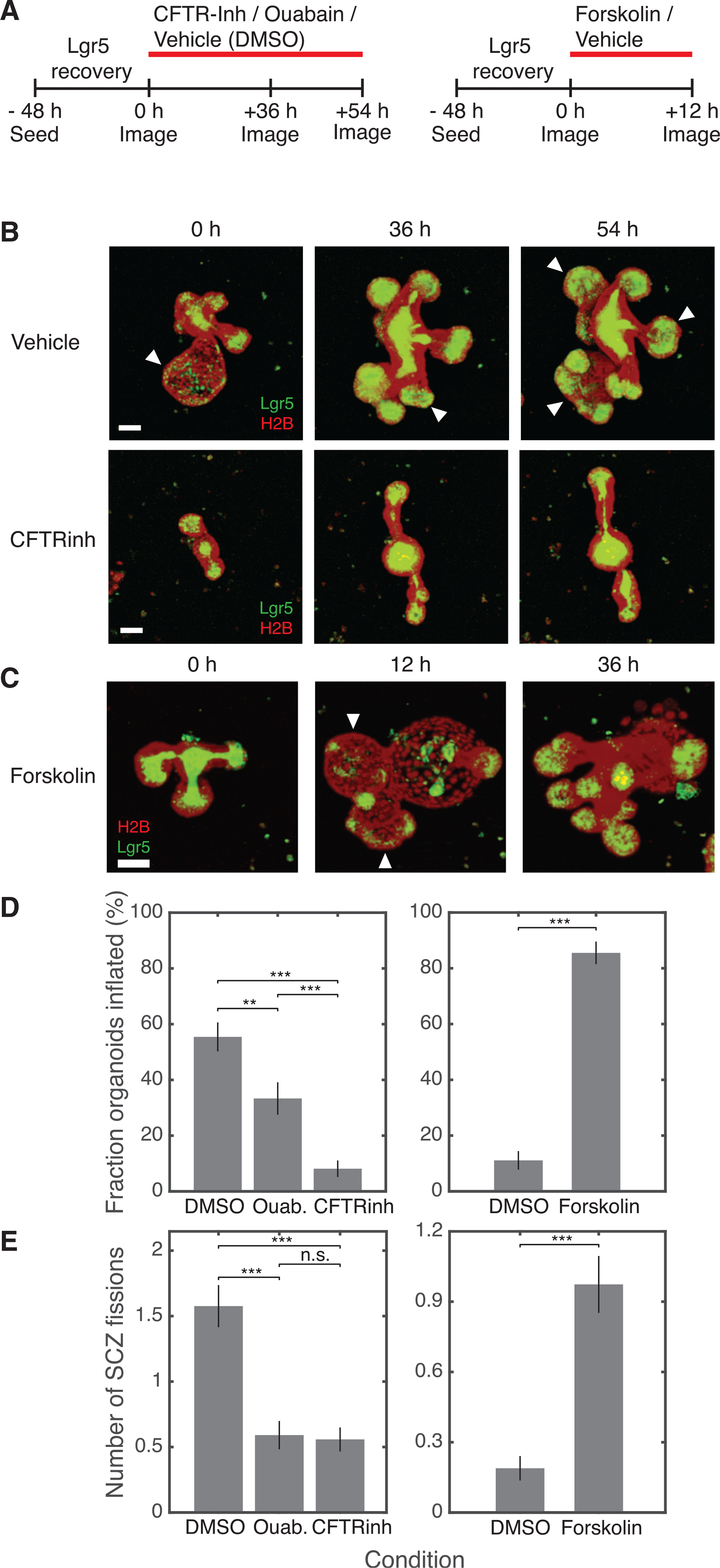
(A) Schematic experimental designs to test the effect of sustained inhibition (left) or acute stimulation (right) of ion channels on SCZ growth and fission.
(B and C) Representative organoids treated with vehicle (DMSO), CFTRinh-172, or forskolin. Images show maximum-intensity projections; white arrows indicate inflated buds. Scale bars, 50 μm.
(D) The fraction of organoids with at least one visibly inflated bud reduces after 36 h and 54 h of ion channel inhibition (left; vehicle, n = 92; ouabain, n = 66; CFTRinh, n = 86; 2 biological replicates per condition) and increases after acute 12-h treatment with forskolin (right; vehicle, n = 90; forskolin, n = 76; 2 biological replicates per condition).
(E) The number of fission events per organoid between 36 h and 54 h of ion channel inhibition reduces compared with vehicle control (left), and increases within 12 h of treatment with forskolin (right). Number fission events, mean increase in SCZ number in time interval.
Error bars indicate SEM. One-sided t test: *p < 0.05, **p < 0.01, ***p < 0.001.
We seeded organoids in triple-decker sandwiches and then treated them with drug or vehicle after 48 h (treatment conditions were assigned randomly to different wells). The number of buds in a randomly selected subset of organoids was compared 36 and 54 h following channel inhibition to allow drugs to equilibrate and after 12 h for the acute response to the agonist (Figure 5A). We followed organoids across 6 replicates after channel inhibition (vehicle, N = 92; ouabain, N = 66; CFTRinh, N = 86) and after channel activation (vehicle, N = 90; forskolin, N = 76).
These tests showed that blocking ion channels reduced SCZ fission, whereas inducing inflation increased fission. Drug effects were apparent by eye (Figures 5B and 5C); vehicle-treated (DMSO) organoids were frequently distended (Figure 5B,white arrows) and had large and complex shapes, whereas organoids treated with CFTRinh-172 rarely inflated and often appeared to be elongated. Activating CFTR with forskolin induced organoids to inflate (Dekkers et al., 2013; Jaffe et al., 2008) and visibly increased SCZ fission (Figure 5C). Some forskolin-treated organoids that were inflated 12 h after drug treatment subsequently ruptured; these collapsed organoids contained multiple new Lgr5+ buds.
We quantified the effects of inducing and inhibiting inflation. Organoids treated with ouabain or CFTRinh-172 showed a significant reduction in the instantaneously inflated fraction (Figure 5D) (40% reduction in ouabain [t test, p = 0.003], 85% reduction in CFTRinh-172 [t test, p = 10−13]) and a 3-fold reduction in SCZ fission under both conditions (Figure 5E; t test p < 10−6). Organoids in forskolin were inflated far more than organoids in vehicle (vehicle, 11% ± 3%; forskolin: 86% ± 4%, p < 10−30) (Figure 5D), and they exhibited five times as many SCZ fission events as organoids in vehicle during the same interval (Figure 5E; t test, p < 10−7). Thus, we established that organoid inflation triggers SCZ fission.
Identifying a stretch-responsive cell state
To investigate how inflation triggers SCZ fission, we analyzed organoids by scRNA-seq after prolonged (36-h) treatment with CFTRinh-172 or acute (12-h) treatment with forskolin alongside time-matched DMSO controls. We co-embedded these data and clustered, classified, and visualized them (uniform manifold approximation and projection [UMAP] plots; Figures 6A and S4A). These analyses revealed all expected cell types and allowed us to quantify changes in abundance (Figures 6B and S4B) and gene expression in response to perturbation.
Figure 6. Dissecting epithelial stretch response by scRNA-seq.
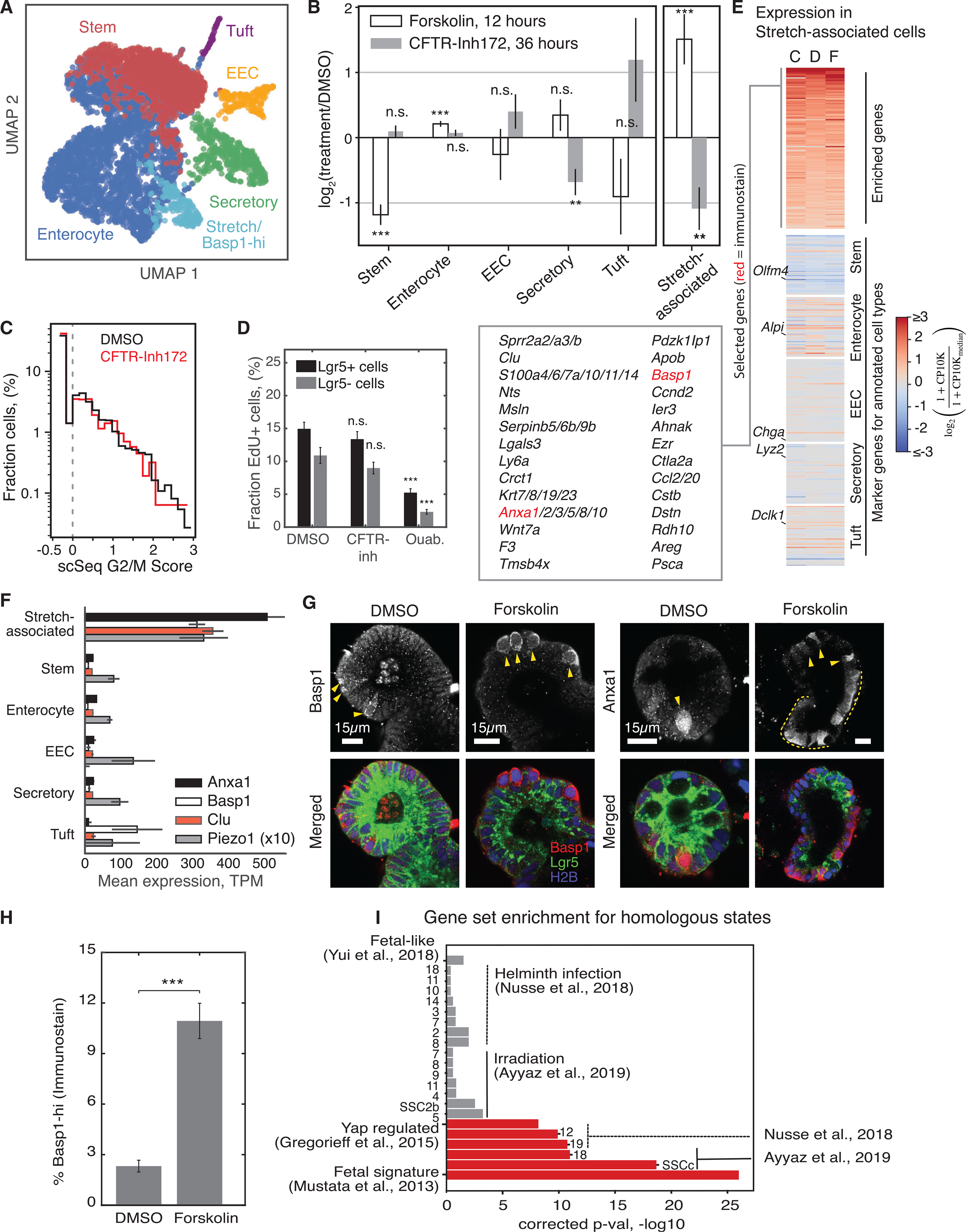
(A) UMAP representation of scRNA-seq of organoids treated with forskolin, CFTRinh-172, or DMSO (5,241 cells × 29,603 genes), colored by annotated cell type.
(B) Fractional change in the abundance of annotated cell types after treatment. Fisher’s exact test after Bonferroni correction: **p < 0.01, ***p < 0.001.
(C) Histogram of G2/M scores calculated from cell transcriptomes shows no change in proliferation with CFTRinh-172.
(D) The fraction of cells in S phase, assessed by 2-h incubation with EdU, does not change significantly after 36 h of treatment with CFTRinh-172. Ouabain reduces the S phase fraction of Lgr5+ and Lgr5− cells. DMSO, n = 66 organoids; CFTRinh, n = 63 organoids; 3 biological replicates per condition; ouabain, n = 35 organoids, 2 biological replicates).
(E) Heatmaps of gene expression in the cluster of stretch-associated cells across different experiments, showing enrichment of multiple genes specific to the state (top) and reduced expression of genes marking canonical cell types of the epithelium (remaining maps). Experiments: CFTRinh-172-treated (C), DMSO-treated (D), and forskolin-treated (F).
(F) Select mean transcript abundances. Error bars indicate SEM.
(G) Representative images of fixed organoids stained for Anxa1 and Basp1.
(H) Fraction of cells scored as Basp1 high in fixed and stained organoids after 12-h treatment with DMSO (n = 1,811 Basp1-low cells; n = 43 Basp1-high cells) or forskolin (n = 790 Basp1-low cells; n = 97 Basp1-high cells). Error bar indicates SEM.
(I) False discovery rate (FDR)-corrected p values from gene set enrichment analysis, comparing stretch-associated marker genes with gene sets enriched in cell states observed in other studies.
After inducing inflation, we expected to observe acute differentiation of ISCs, consistent with live imaging. scRNA-seq revealed a significant reduction in ISC markers after forskolin treatment (Figure S4C) and a reduction in the fraction of cells classified as ISC compared with DMSO-treated controls (Figure 6B). Concurrently, the fraction of cell transcriptomes classified as enterocytes increased proportionately (Figure 6B), as did the expression of marker genes of mature enterocytes (Figure S4C). The fraction of secretory, tuft, and enteroendocrine cells did not change (Figure 6B). These results further support our finding that inflation acutely promotes loss of ISCs and suggest that ISCs commit to an enterocyte fate.
Next we used the scRNA-seq data to find out whether blocking inflation reduces cell proliferation. After 36 h of CFTRinh treatment, the fraction of cell transcriptomes expressing G2/M phase-specific genes showed little change (Figure 6C), suggesting that luminal inflation does not regulate cell division. To confirm this, we fixed organoids after a pulse of EdU to label cells in S phase. Consistent with scRNA-seq, treatment with the CFTR inhibitor did not significantly reduce the percentage of EdU+ cells (Figures 6D and S4D). Ouabain did, however, reproducibly inhibit cell division in organoids, possibly through an inflation-independent mechanism.
scRNA-seq has the potential to reveal previously unannotated cell states that might emerge in response to perturbation. We identified a strong cellular phenotype defining a cluster whose abundance increased when inflation was induced (2.5-fold increase in forskolin-treated cells compared with a matched control cells) and decreased when it was inhibited (2-fold reduction in CFTRinh-172-treated cells) (Figure 6B). These cells were enriched for expression of tens of genes (Figure 6E), including Wnt7a/b, and several genes implicated in calcium signal transduction: Clusterin, Annexins, Basp1, and S100 family transcripts and the mechanosensitive calcium channel Piezo1 (Figures 6E and 6F). These cells had high total transcript counts compared with other cells in the organoid (Figure S4E); they were transcriptionally distinct from stem cells and showed higher expression of enterocyte marker genes than other cell types (Figure 6E).
To identify these putative stretch-dependent cells in situ, we fixed and stained organoids using antibodies for Anxa1 and Basp1 (Figure 6G). Basp1-hi cells were scattered sporadically within the Lgr5+ epithelium of DMSO-treated organoids. Their abundance increased 5-fold after 12 h of forskolin treatment (Figure 6H), consistent with scRNA-seq. These cells had larger nuclei than adjacent Basp1-low cells (Figure S4F), consistent with higher total transcript counts in scRNA-seq data. Like Basp1, Anxa1 stained cells sporadically in the Lgr5+ epithelium of DMSO-treated organoids. scRNA-seq indicated a widespread increase in Anxa1 in ISCs and enterocytes after forskolin treatment. Accordingly, Anxa1 protein expression was detected in broad regions of the epithelium after forskolin treatment, with sporadic cells showing high levels of Anxa1. These experiments support the presence of a sporadic stretch-responsive cell state in organoids.
To characterize the identity of the stretch-responsive cells, we examined the overlap of genes enriched in these cells with those of cell states identified by scRNA-seq or bulk analysis in prior studies (Mustata et al., 2013; Gregorieff et al., 2015; Figure 6I; Table S1). Gene set enrichment analysis showed that the stretch-associated cells are not homologous to cells in homeostatic crypts, but they show significant overlap in gene expression with a rare set of Clusterin-hi/Basp1-hi cells observed to emerge after intestinal injury (Ayyaz et al., 2019). They also reflected genes enriched in organoids derived from fetal ISCs (Mustata et al., 2013; Yui et al., 2018) whose expression is lost upon YAP knockout (Gregorieff et al., 2015).
SCZ fission depends on the activity of mechanosensory Piezo-family ion channels
The presence of a Piezo1-enriched cell that is sensitive to stimulation or inhibition of inflation (Figures 6B and 6F) suggested that inflation-mediated SCZ fission may act via the mechanosensitive ion channel Piezo1. Using the calcium indicator dye Cal-630AM, we quantified calcium influx after blocking or triggering inflation and inhibiting Piezo1 using the calcium channel blocker GdCl3 (Figures 7A–7D). Without forskolin treatment, the intracellular calcium concentration was higher in spontaneously inflated buds than in collapsed ones (Figure 7B). The total intracellular calcium concentration increased after stimulating inflation, decreased after blocking inflation, and decreased further in the presence of GdCl3, even after inflation was induced (Figures 7C and 7D). Thus, calcium signaling in organoids is stretch dependent and acts via a GdCl3-sensitive channel.
Figure 7. Piezo channel activity is necessary for inflation-mediated fission.
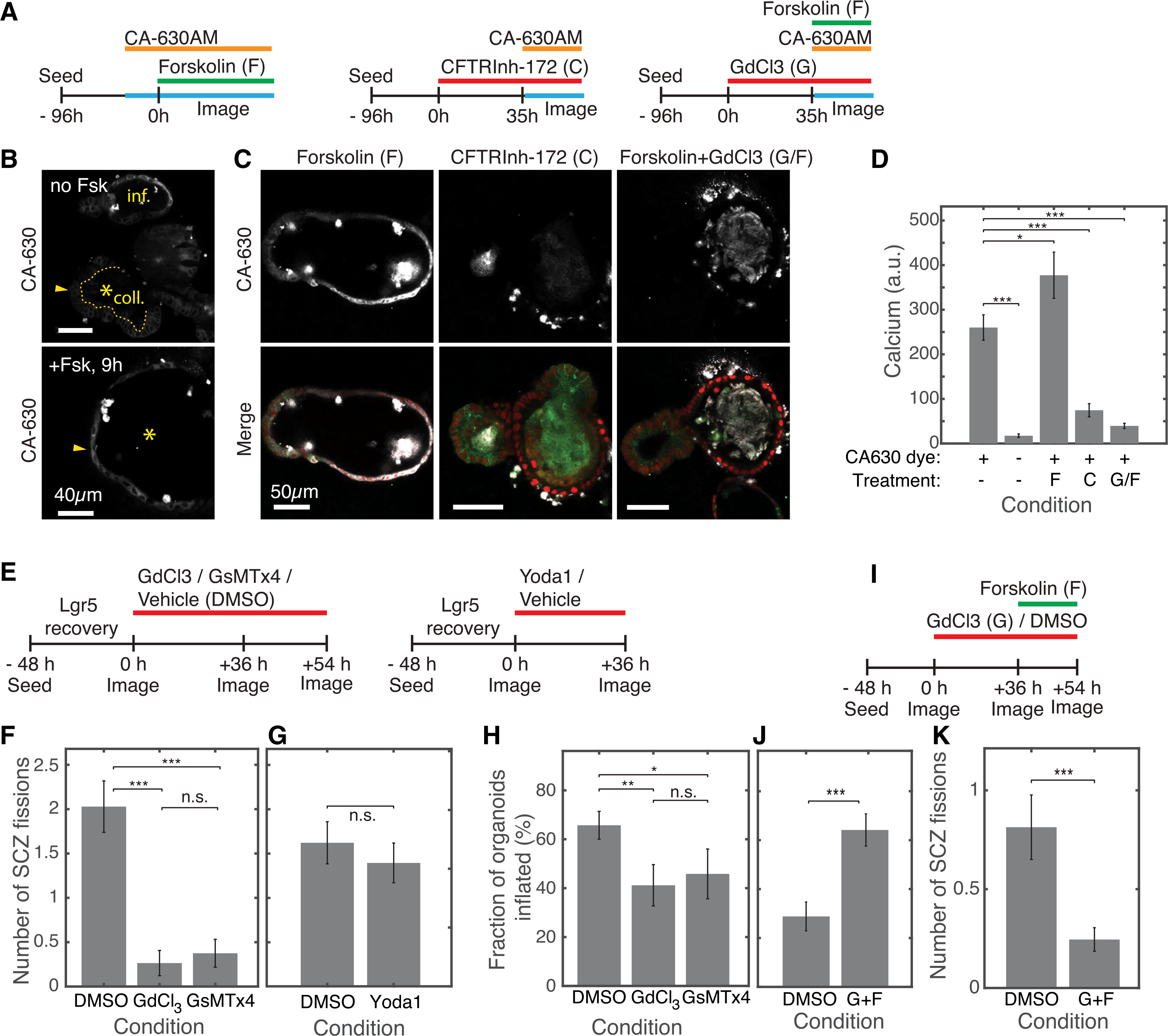
(A) Experimental design for imaging intracellular calcium dynamics after stimulating or inhibiting luminal inflation or Piezo channel activity.
(B) Top: the calcium dye CA-630 shows increased intensity in the epithelium surrounding an inflated (inf.) bud compared with a collapsed (coll.) bud. Bottom: after forskolin treatment, the previously coll. bud (marked by an asterisk) shows increased CA-630 intensity.
(C) Representative images of calcium imaging under perturbation.
(D) Quantification of mean CA-630 dye intensity under different conditions. F, C, G/F as defined in (C) (n = 29 no-drug controls, n = 64 no-dye controls, n = 29 forskolin-treated organoids, n = 31 CFTRinh-172-treated organoids, n = 33 GdCl3 + forskolin-treated organoids; 2 biological replicates per condition). Error bars indicate SEM. One-sided t test: *p < 0.05, **p < 0.01, ***p < 0.001.
(E) Experimental designs to quantify SCZ fission after sustained inhibition or acute activation of Piezo ion channels.
(F) Channel inhibition significantly reduces the number of fission events per organoid compared with vehicle control (n = 70 organoids in DMSO across 6 replicates, n = 34 in GdCl3 across 3 replicates, n = 24 in GsMTx4 across 3 replicates).
(G) Treatment with the Piezo agonist Yoda1 does not significantly alter fission frequencies (n = 61 in DMSO across 6 replicates, n = 38 in Yoda1 across 6 replicates).
(H) Piezo channel inhibition weakly suppresses inflation. Data are from same organoids as in (F).
(I) Experimental design to test for epistasis between inflation and Piezo channel activity on SCZ fission rates.
(J) and K) Dual treatment with the Piezo channel inhibitor GdCl3 and the CFTR agonist forskolin (G+F) increases organoid inflation, but the SCZ fission rate is reduced, consistent with Piezo channel activity acting downstream of inflation to mediate SCZ fission. Experiments were carried out separately from those in (E)–(H), using smaller organoid fragments that exhibited lower inflation and fission rates (n = 59 organoids in DMSO, n = 53 in GdCl3 + forskolin, 6 replicates each).
In light of these results, we wanted to find out whether Piezo channels specifically might underlie inflation-driven SCZ fission. We inhibited these channels using GdCl3 and a peptide (GsMTx4) that blocks Piezo-family channel activity (Bagriantsev et al., 2014) and activated them using the small-molecule Piezo channel agonist Yoda1 (Syeda et al., 2015; Figures 7E–7H). Compared with vehicle controls, Piezo-inhibited organoids exhibited significantly less SCZ fission (vehicle, 2.0 ± 0.3 fissions in 18 h; GdCl3, 0.26 ± 0.14 fissions [p = 1.9 × 10−7]; GsMTx4, 0.38 ± 0.16 fissions, p = 1.3 × 10−6) (Figure 7F). This change in fission rate was as large as or larger than the effect of suppressing inflation directly (5- to 8-fold decrease in fission rates after Piezo channel inhibition compared with a 3-fold decrease after ouabain or CFTR inhibition). However, activating Piezo channels with the agonist Yoda1 did not lead to an acute increase in SCZ fission rate (vehicle, 1.6 ± 0.2 fissions in 36 h; Yoda1, 1.4 ± 0.2 fissions; not significant by t test) (Figure 7G). These results suggest that Piezo channel activity is necessary but not sufficient to trigger SCZ fission.
In principle, Piezo inhibition may reduce SCZ fission by reducing organoid inflation. We did observe a reduction in the frequency of organoid inflation after Piezo1/2 inhibition (30% and 38% reduction in organoid inflation after treatment with GdCl3 and GsMTx4, respectively; Figure 7H), but this effect was noticeably weaker than the reduction in SCZ fission observed in treated organoids.
Piezo activity may act downstream of organoid inflation or collapse to enable fission, or it may influence fission through an independent mechanism. We tested whether Piezo activity is necessary for inflation-mediated fission by stimulating organoids to inflate with forskolin after pre-incubating them with the Piezo inhibitor GdCl3 (Figure 7I). Under these conditions, forskolin did not accelerate SCZ fission even though it induced inflation (Figure 7J); these organoids underwent fission at a significantly lower rate than untreated controls (0.81 ± 0.2 fission events compared with 0.25 ± 0.1 fission events, p = 8 × 10−4; Figure 7K). We conclude that a GdCl3-sensitive channel is required for fission subsequent to inflation.
DISCUSSION
In this study, we improved spatial uniformity, viability, and image quality of intestinal organoids by embedding them in triple-decker hydrogel sandwiches. We then studied how epithelial SCZs obtain their size and clarified cellular events that occur during SCZ fission. Despite stereotyped depictions of organoid SCZs having physiological size (Sato et al., 2009), upon quantification we found them to be significantly more variable in size than in vivo. They show a simple exponential size distribution that can be understood to arise spontaneously from net exponential growth and stochastic fission, which were observed by live imaging. These results quantitatively explained how a typical length scale for SCZs emerges in culture, but they also highlighted a way in which organoid cultures fail to recapitulate in vivo behaviors. It is possible that small changes to organoid culture could be sufficient to recover more uniform behavior. As a case in point, inhibiting luminal inflation reduced fission rates, leading to budding structures more reminiscent of physiological crypts.
The frequent fission of SCZs in organoids, even at small sizes, led us to more closely examine the events that occur as SCZs fragment. We found that fission frequently followed within hours of organoid inflation without prior invagination (Langlands et al., 2016) or progressive growth (Pin et al., 2015), as proposed previously. Suppressing or inducing inflation strongly suppressed or stimulated fission, respectively. Fission occurred when patches of stem cells lost Lgr5 expression during inflation rather than by cell rearrangement. Buds formed only subsequently during luminal collapse, even in organoids grown in Wnt3A, which lack morphological buds but nonetheless show SCZ-like patches of Lgr5+ cells.
Using scRNA-seq and subsequent immunofluorescence, we identified a cell state that is suppressed or stimulated upon suppressing or inducing inflation, respectively. This state was enriched for multiple genes implicated in stretch response and calcium signaling, including Basp1, Anxa1, and the mechanosensory channel Piezo1. Finally, we showed that inflation drives intracellular calcium concentration via a GdCl3-sensitive ion channel and that GdCl3 and a specific inhibitor of Piezo1/2 strongly suppressed fission. These results established strong links between epithelial stretch, calcium dynamics, and the patterning of SCZs.
Volume fluctuations in organoids resemble similar volumetric oscillations reported in other tissues, including the mouse blastocyst (Chan et al., 2019) and the zebrafish inner ear (Mosaliganti et al., 2019). Inflation-collapse cycles have been proposed as an active mechanism for tuning the size of these multicellular structures (Ruiz-Herrero et al., 2017). In intestinal organoids that lack extrinsic information provided by the in vivo niche, inflation and collapse lead to growth and branching rather than stable size control.
Crypt inflation and subsequent fission might not occur during intestinal homeostasis. scRNA-seq of in vivo crypt cells indeed does not reveal the same Anxa1+/Basp1+/Clu+ cell state reported here (Haber et al., 2017). However, after injury, Ayyaz et al. (2019) identified a “revival stem cell” in the mouse intestine that was enriched for most of the marker genes of stretch-responsive cells found here, including Annexins, Basp1, and Clu. Comparable states appear after infection (Nusse et al., 2018) and during development (Mustata et al., 2013). These conditions may induce epithelial stretching. Further, under some conditions, crypt inflation may occur in the intestine. Intestinal distension leads to crypt enlargement within 1 h (Johnson, 1913), and the crypt base also broadens in response to food, meconium, or applied fluid pressure (Harvey, 1908). Thus, crypts may inflate in response to local or global fluid accumulation. Intestinal crypts secrete large volumes of fluid (Kiela and Ghishan, 2009), making it likely that blockages would drive inflation, as observed in organoid buds (e.g., Video S5). Crypt lumens indeed enlarge within 1 h of cell extrusion into the lumen (Choi et al., 2018). Still, we cannot link inflation to crypt fission in health and disease; doing so will likely require intra-vital imaging.
Our observations suggest that if crypt dilation does occur in vivo, then it would be associated with increased crypt fission. Some in vivo evidence supports this notion; the intestine dilates and crypt fission accelerates after surgical resection (Dekaney et al., 2007; Bianchi and Morabito, 2009), intestinal obstruction leads to crypt distension and hyperplasia (Collins et al., 1996), and crypt swelling and branching occur when ion channels are ablated genetically (Schweinfest et al., 1993, 2006). In addition, serrated adenomas show dilatation of the crypt base, luminal blockage by excess mucus, as well as hyperbranching crypts (Snover et al., 2005), and crypts with mutations in the adenomatous polyposis coli tumor suppressor (Apc) appear cyst like, with an expanded lumen (Quyn et al., 2010), prior to adenomatous crypt proliferation.
In summary, we find that the intestinal epithelium in organoids exhibits spontaneous cycles of inflation and collapse that modulate its own patterning, triggering a strong transcriptional response and activating mechanosensitive calcium channels. Our results may point toward approaches to manipulate epithelial stem cell behavior and to more general mechanisms of pressure-driven tissue morphogenesis in development and disease.
Limitations of study
We have discussed in detail that type II fission events might be limited to culture; we do not yet know the role of these phenomena, or if they even occur, in vivo. Organoids in culture differ in specific ways from crypts in vivo. In organoids, secretory cell types, like Paneth cells and goblet cells, are difficult to distinguish transcriptomically, and organoid lumens are small compared with the intestinal lumen. Two further limitations of the study are worth emphasizing. First, the perturbations carried out in this study are pharmacological, and we cannot rule out the possibility of off-target effects. CFTR and Piezo perturbations should ultimately be carried out by genetic means, and the effects of inflation and stretching should be tested further using orthogonal mechanical perturbations. Second, because we were working with double-transgenic animals, our organoids were derived from a single genetic background, albeit with different organoid lines derived from this background. We do not know whether the same events are recapitulated in other animals or in humans. Finally, our study reveals that inflation drives SCZ fission and gives rise to a stretch-associated cell state, but we did not determine whether SCZ fission and the stretch-associated cell state are related.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead contact
All requests for materials and/or reagents used in this study should be directed to the lead contact, Allon M. Klein (allon_klein@hms.harvard.edu).
Materials availability
Mice crossed to generate the organoids used in this study, and the organoids used in this study are available upon request. The plasmid generated in this study is also available upon request.
Data and code availability
Single cell RNA-sequencing data generated during this study are publicly available at the Gene Expression Omnibus (GEO: GSE164638). Jupyter notebooks and inDrops.py pipeline scripts used for single cell RNA-seq analysis are publicly available on Github: https://github.com/AllonKleinLab/paper-data/tree/master/Tallapragada_Cambra_2021. SPRING plots of datasets as processed and annotated in this study are available; see Key resources table for publicly accessible links to each SPRING plot. Imaging data and MATLAB analysis scripts are available from the corresponding author upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Annexin A1 | Abcam | Cat# ab214486 |
| anti-BASP1 | Abcam | Cat# ab214322 |
| anti-Chromogranin A | Abcam | Cat# ab15160; RRID: AB_301704 |
| anti-CLCA1 | Abcam | Cat# ab180851; RRID: AB_2722611 |
| anti-Lysozyme | Agilent | Cat# F037201–1; RRID: AB_578661 |
| anti-Rabbit IgG (secondary, 555) | Thermo Fisher Scientific | Cat# A21428; RRID: AB_2535849 |
| anti-Rabbit IgG (secondary, 647) | Life Technologies | Cat# A21245; RRID: AB_2535813 |
| anti-Villin | Santa Cruz Biotechnology | Cat# sc-58897-AF647; RRID: AB_2304475 |
| Biological Samples | ||
| Intestinal organoids from Mouse: Lgr5-DTR-EGFP | This paper | N/A |
| Intestinal organoids from Mouse: Lgr5-DTR-EGFP/Villin-H2B-mCherry | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| B27 | Thermo Fisher Scientific | Cat# 17504–044 |
| Cell Recovery Solution | Corning | Cat# 354253 |
| CFTRinh-172 | Selleck Chemicals | Cat# S7139 |
| Complete crypt culture medium containing EGF, Noggin, and R-Spondin-1 (ENR) | R&D Systems Protocols | https://resources.rndsystems.com/images/site/dw_r-spondinmediumprotocol_34749-web.pdf?v=1 |
| Complete crypt culture medium containing Wnt3a, EGF, Noggin, and R-Spondin-1 (WENR) | Miyoshi and Stappenbeck, 2013 | N/A |
| Diethylaminoethyl (DEAE)-Dextran | Millipore Sigma | SKU- D9885 |
| Forskolin | Selleck Chemicals | Cat# S2449 |
| GdCl3 | Millipore Sigma | SKU- G7532 |
| Glutamax | Thermo Fisher Scientific | Cat# 35050–079 |
| GsMTx4 | Abcam | Cat# ab141871 |
| Hanks’ Buffer with 20 mM Hepes | AAT Bioquest | Cat# 20011 |
| Matrigel, growth factor reduced, phenol red-free | Corning | Cat# 356231 |
| N-acetyl cysteine 500 mM stock solution | Millipore Sigma | SKU- A7250 |
| N2 | Thermo Fisher Scientific | Cat# 17502–048 |
| Noggin | Millipore Sigma | SKU- SRP3227 |
| Ouabain | Tocris | Cat# 1076 |
| Pluronic® F-127 | AAT Bioquest | Cat# 20050 |
| Poly(2-hydroxyethyl methacrylate) | Millipore Sigma | SKU- P3932 |
| Primocin | Invivogen | Cat# ant-pm-1 |
| Probenecid | AAT Bioquest | Cat# 20060 |
| Puromycin dihydrochloride | Thermo Fisher Scientific | Cat# A1113803 |
| Recombinant mouse EGF | Thermo Fisher Scientific | Cat# PMG8041 |
| Yoda1 | Tocris | Cat# 5586 |
| Critical Commercial Assays | ||
| Calcium indicator kit | AAT Bioquest | Cat# 20530 |
| EdU staining kit (AlexaFluor647) | Thermo Fisher Scientific | Cat# C10640 |
| NEBNext Ultra II FS DNA Library Prep kit | NEB | Cat# E7805S |
| Deposited Data | ||
| In vivo reference data for Figure 1D | Haber et al., 2017 | GEO: GSE92332 |
| Mouse genome build 38 (GRCm38) | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/ |
| Raw and analyzed single cell RNA-sequencing data | This paper | GEO: GSE164368 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| Mouse: 293T-HA-RspoI-Fc | Trevigen | Cat# 3710–001-01; RRID: CVCL_RU08 |
| Mouse: L-WRN | ATCC | Cat# CRL-3276; RRID: CVCL_DA06 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6;129S-Gt(ROSA)26Sortm1.1Ksvo/J | Jackson Laboratories | Cat# 023139; RRID:IMSR_JAX:023139 |
| Mouse: B6.Cg-Tg(Vil1-cre)997Gum/J | Jackson Laboratories | Cat# 004586; RRID:IMSR_JAX:004586 |
| Mouse: Lgr5-DTR-EGFP | Tian et al., 2011 | N/A |
| Mouse: Lgr5-DTR-EGFP/Villin-H2B-mCherry | This paper | N/A |
| Oligonucleotides | ||
| RT primers on hydrogel beads: 5’CGATTGATCAACGTAATACGACTCACTATAGGGTGTCGGGTGCAG[bc1,8nt]GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG[bc2,8nt]NNNNNNTTTTTTTTTTTTTTTTTTTV- 3’ | This paper; HMS Single Cell Core | N/A |
| TSO primer: 5’-AAGCAGTGGTATCAACGCAGAGTACATrGrGrG-3’ | Datlinger et al., 2019 | N/A |
| Fwd cDNA primer: 5’- CACTATAGGGTGTCGGGTGCAG-3’ | This paper; HMS Single Cell Core | N/A |
| Rev cDNA primer: 5’-AAGCAGTGGTATCAACGCAGAGT-3’ | This paper; HMS Single Cell Core | N/A |
| Ligation Fwd primer: 5’-/5Phos/GATCGGAAGAGCACACGTCTGAACTCCAGTCAC/3ddC | This paper; HMS Single Cell Core | N/A |
| Ligation Rev primer: 5’- /5AmMC6/GCTCTTCCGATCT -3’ | This paper; HMS Single Cell Core | N/A |
| Indexing primer PCR_p7_r2: 5’-CAAGCAGAAGACGGCATACGAGATGGGTGTCGGGTGCAG-3’ | This paper, HMS Single Cell Core | N/A |
| Indexing primer PCR_p5_r1_ixx: 5’-AATGATACGGCGACCACCGAGATCTACAC[i5]TCGTCGGCAGCGTC-3’ | This paper; HMS Single Cell Core | N/A |
| Recombinant DNA | ||
| Plasmid: pLVX-H2B-iRFP670 | This paper; Kyogo Kawaguchi | N/A |
| Software and Algorithms | ||
| 3D Distance Transform algorithm | Mishchenko, 2015 | https://link.springer.com/article/10.1007%2Fs11760-012-0419-9 |
| Exact unbiased inverse of the Anscombe variance-stabilizing transformation – MATLAB implementation | Makitalo and Foi, 2011 | https://ieeexplore.ieee.org/document/5721817 |
| Image Denoising by Sparse 3-D Transform-Domain Collaborative Filtering – MATLAB implementation | Dabov et al., 2007 | https://ieeexplore.ieee.org/document/4271520 |
| Fiji | Schindelin, J., Arganda-Carreras, I. & Frise, E. et al., 2012 | https://imagej.net/Fiji |
| Imaris 8.4 | Oxford Instruments | https://imaris.oxinst.com/support/imaris-release-notes/8-4-0 |
| inDrops pipeline package | Klein et al., 2015; Github | https://github.com/indrops/indrops |
| Scanpy – Single Cell Analysis in Python | Wolf et al., 2018 | https://scanpy.readthedocs.io/en/stable/ |
| Scrublet – Single Cell Removal of Doublets | Wolock et al., 2019 | https://github.com/AllonKleinLab/scrublet |
| Other | ||
| Interactive visualization of scRNA-seq data with SPRING - Sandwich and dome data (without stretch signature annotation) | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/SPRING_private/mouse_intestinal_organoids_21/murine_intestinal_organoid_sandwich_dome_culture_merged_no_stretch_signature/all_cells |
| Interactive visualization of scRNA-seq data with SPRING - Sandwich and dome data (with stretch signature annotation) | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/SPRING_private/mouse_intestinal_organoids_21/murine_intestinal_organoid_sandwich_dome_culture_merged/all_cells |
| Interactive visualization of scRNA-seq data with SPRING - Perturbation data | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/SPRING_private/mouse_intestinal_organoids_21/murine_intestinal_organoid_inflation_perturbation_expt/all_cells |
| Jupyter notebooks for scRNA-seq data analysis | This paper | https://github.com/AllonKleinLab/paper-data/tree/master/Tallapragada_Cambra_2021 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All mouse work was approved by Institutional Animal Care and Use Committees at UCSF and Harvard Medical School. With the exception of organoids imaged to generate Figures 1J, 1K, S1H, and 2G, all organoids in this study were derived from Lgr5-DTR-EGFP/Villin-H2B-mCherry mice whose intestines were stably labeled for both nuclei and stem cells. In order to generate this strain, Villin-Cre (B6.Cg-Tg(Vil1-cre)997Gum/J; JAX 004586) and R26 LSL H2B mCherry (B6;129S-Gt(ROSA)26Sortm1.1Ksvo/J; JAX 023139) mice purchased from The Jackson Laboratory were crossed to generate progeny with fluorescently-labeled nuclei in the intestines. These offspring were then crossed with Lgr5-DTR-EGFP mice (Tian et al., 2011) to produce the desired strain.
METHOD DETAILS
Organoid WENR and ENR media
Cell lines for producing conditioned media were tested for the presence of mycoplasma (Lonza MycoAlert PLUS kit, LT07–701). Media containing Wnt3A, EGF, Noggin and R-spondin1 (WENR media) was generated by harvesting conditioned media from cultured L-WRN cells (ATCC CRL-3276) at 2X dilution in DMEM and supplemented with 50 ng/mL recombinant mouse EGF (ThermoFisher; PMG8041) as previously described (Miyoshi and Stappenbeck, 2013). Media containing EGF, Noggin and R-spondin1 but not Wnt3A (ENR media) was prepared by first harvesting conditioned media from cultured 293T-HA-RspoI-Fc cells (Trevigen Cat# 3710–001-01) as described (R&D Systems Protocol; https://resources.rndsystems.com/images/site/dw_r-spondinmediumprotocol_34749-web.pdf?v=1). 10 mL of conditioned media was then added to 86 mL DMEM/F12 (ThermoFisher, Cat# 11320–033), 1 mL Glutamax (ThermoFisher, Cat# 35050–079), 1 mL HEPES (ThermoFisher, Cat# 15630–080), 200 μL Primocin (Invivogen, ant-pm-1), 1 mL B27 (Thermo, Cat# 17504–044), 0.5 mL N2 (ThermoFisher, Cat# 17502–048), 250 μl N-acetyl cysteine 500 mM stock solution (Sigma, SKU-A7250–10G), with EGF at final concentration (50 ng/mL), and Noggin (MilliporeSigma; SKU-SRP3227–20UG) at final concentration 100 ng/mL. Media were filter-sterilized and stored for up to 6 months at −20°C. 50 mL aliquots of media were thawed and stored at 4°C as necessary to feed organoids.
Establishing organoids from primary tissue
Organoid cultures were established from primary tissue as previously described (Sato and Clevers, 2013; Sato et al., 2009). Briefly, 6 to 12-week old Lgr5-DTR-EGFP mice, or Lgr5-DTR-EGFP/Villin-H2B-mCherry mice, were sacrificed and dissected to harvest the proximal small intestine. This tissue was chemically and mechanically dissociated to separate epithelium from mesenchyme and crypts from villi as previously described (Sato et al., 2009). Isolated crypts were resuspended in Matrigel and deposited on cell culture plates to initiate organoid cultures (defining passage P0). In order to generate enough material for frozen stocks, organoids were expanded for several passages in WENR media. Organoids were frozen in STEM-CELLBANKER (ZENOAQ/AMSBIO, Cat# 11890) within 4 weeks of initial setup, primarily at passage P3. Lgr5-DTR-EGFP/Villin-H2B-mCherry organoids were used for all experiments except where noted otherwise below (see “Lentiviral infection for nuclear labeling”), where Lgr5-DTR-EGFP organoids were used. Organoids were tested for the presence of mycoplasma (Lonza MycoAlert PLUS kit, Cat# LT07–701).
Maintaining organoid cultures
Organoid cultures were fed and passaged as previously described (Sato and Clevers, 2013; Sato et al., 2009), with a few modifications to ensure that low-passage organoids were consistently available for experiments. Organoids were maintained in WENR media to enrich for stem cells and progenitors that could form organoids on the next passage. Each week a subset of these organoids were split into new wells and cultured in ENR media without Wnt3A. Organoids recovered budding morphologies in ENR media, after Wnt3A was withdrawn. These organoids were allowed to grow for 5–7 days in ENR media before they were used to set up new plates for imaging experiments. Ongoing cultures in WENR media were discarded after 3 months (after P16) and restarted from the P4 frozen stock.
Lentiviral infection for nuclear labeling
The organoids used to generate Figures 1J, 1K, S1H, and 2G; Videos S1 and S3; and example 3 in Video S8 were derived from Lgr5-DTR-EGFP mice without a fluorescent nuclear label. To label cell nuclei in these organoids, we transduced them with a custom-made lentivirus (pLVX-H2B-iRFP670), with the pLVX backbone carrying a gene for the far-red fluorescent protein iRFP670 (Shcherbakova and Verkhusha, 2013) fused to the histone H2B protein and puromycin resistance (generated by Dr. Kyogo Kawaguchi). We followed a published transduction protocol (Maru et al., 2016) with one modification – using cationic Diethylaminoethyl (DEAE)-Dextran (MilliporeSigma, Cat# D9885) in PBS at final concentration 0.1 mg/mL, instead of TransDux, to facilitate cell entry. Viral supernatant for transduction was produced in HEK293T cells lipofected with pLVX-H2B-iRFP670 as well as 3rd generation lentiviral proteins. We selected for transduced cells by treating organoids with 2 μg/mL puromycin dihydrochloride (ThermoFisher, Cat# A1113803) at this step and during the next passage. H2B-iRFP transgene expression was verified by far-red wavelength (640 nm) confocal imaging, which revealed distinct nuclei in transduced organoids and minimal mosaicism in expression.
Triple-decker hydrogel sandwich cultures
To prepare triple-decker hydrogel sandwich cultures, 20 mg/mL Poly(2-hydroxyethyl methacrylate) (PolyHEMA) (MilliporeSigma; SKU- P3932) solution was prepared by dissolving PolyHEMA in 95% ethanol at 65°C on a hot plate for 3 hours with a magnetic stir bar. The solution was covered with Parafilm M (Bemis) during this time to prevent evaporation. Once cooled, the solution was filtered through a cellulose acetate filter with 0.22 μm pore size (Corning Cat# 430513) and then degassed for 30 minutes in a vacuum desiccator. 187.5 μL of the PolyHEMA solution was pipetted onto coverslips in 12-well glass bottom dishes (Mattek, Cat# P12G-1.5–14-F) and allowed to dry overnight in a dessicator (Nalgene/VWR, Cat# 24987–004) under vacuum in a tissue culture hood. This step was repeated twice, followed by at least one day of drying at room temperature and pressure in a tissue culture hood. Tissue culture-grade 1X PBS (Corning, Cat# 21–040-CV) was added to every PolyHEMA-coated well before placing the multi-well plate in a 37°C incubator for at least 3 hours. This procedure simultaneously washed wells (to remove precipitated PolyHEMA, debris, etc.) and brought to the surface any gas bubbles still trapped in the PolyHEMA film that could compromise the long-term durability of the hydrogel sandwich. PolyHEMA-coated plates were prepared up to one month in advance of further use, with no apparent downstream effects on organoid growth and viability, and possibly could be prepared even earlier.
For plating organoids, a thin layer of growth factor-reduced, phenol red-free Matrigel (Corning, Cat# 356231) was deposited on top of each PolyHEMA coating by adding 1.2 mL of 5% Matrigel in DMEM/F12 (ThermoFisher, Cat# 11320033) to each well and incubating the plate at 37°C for 45 minutes. This Matrigel coating facilitated the subsequent deposition of cells. Simultaneously, organoids were dissociated by incubation in Cell Recovery Solution (Corning, Cat# 354253) for 45 minutes, followed by centrifugation and pelleting, re-suspension in ENR media, and vigorous pipetting. Once dissociation was complete, organoid fragments in media were passed through a 70 μm cell strainer (pluriSelect Cat# 43–50070-5) to remove large debris before deposition on each Matrigel-coated well. After allowing cells to settle and attach to the Matrigel basecoat for at least 30 minutes, media was aspirated, and each well was top-coated with 70 μL of ice-cold 100% Matrigel deposited slowly directly onto the base Matrigel layer. After the addition of this topcoat, cells were incubated at 37°C for 45 minutes before being fed 1.5 mL ENR media. Any empty wells were filled with tissue culture-grade 1X PBS.
Plate set-up for long-term imaging
For Videos S1, S2, S3, S4, S5, S6, S7, S8, and S9, organoids were imaged continuously for > 12 hours on the microscope. Long-term imaging required the following additional considerations in cell culture and sample preparation. Organoids were plated in triple-decker hydrogel sandwich cultures. Only the 2 central wells of a 12-well plate were used, to minimize variation in temperature and humidity between wells during imaging (Ewald, 2013). To counteract evaporation, organoids in these wells were fed extra media (2 mL instead of 1.5 mL media, every two days). Additionally, 2mL PBS was added to surrounding empty wells. Organoids were maintained at 37°C and 5% CO2 during imaging using a stage-top incubator (Tokai Hit).
Microscopy
Images and time-lapse movies were acquired using an inverted Nikon A1R point-scanning confocal microscope with solid-state lasers in red (555 nm) for H2B-mCherry, far-red (640 nm) for H2B-iRFP and SiR-Actin, and green (488 nm) for Lgr5-DTR-EGFP. SiR-Actin (Cytoskeleton, Cat# CY-SC001) was used only for Figure 2G. For Video S4 and Figures 2H, 3C, and 3E, a 10X magnification dry objective was used to capture whole-well images and tiled movies of large fields of view. Whole-well images were stitched automatically during image capture using NIS-Elements software, as described below (“Quantification and statistical analysis: Distribution and viability of organoids”). For Video S7, all other analyses in Figure 2, all drug treatment analyses in Figures 5, and 7F–7K, a 20X dry objective (NA 0.75) was used to capture images of tens of organoids at intermediate resolution for segmenting SCZ sizes. All remaining movies and images were acquired using a 40X water immersion objective (NA 1.15) with Immersol W 2010 (Zeiss, Cat# 444969–0000-000) as the immersion fluid for high-resolution time-lapse. 3D volumetric images and movies were rendered in Imaris 8.4 (Oxford Instruments).
Immunostaining
Organoids for immunostaining were grown in dome cultures (for Figure S1) or sandwich cultures (for Figures S1 and 6) in 12-well glass bottom plates (MatTek, Cat# P12G-1.5–14-F), and liberated from Matrigel using a protocol adapted from MilliporeSigma [https://www.sigmaaldrich.com/technical-documents/protocols/biology/organoid-antibody-staining.html]. All pipette tips and plasticware used in the liberation procedure (dishes, falcon tubes) were pre-washed with 10% w/v BSA to prevent organoids from sticking to surfaces, except for the final 96-well glass bottom plate (MatTek, Cat# P96G-1.5–5-F) that the organoids were deposited into for blocking and staining steps.
Dome cultures
Media was aspirated and organoids were washed twice with 1X PBS. Cultures were subject to incubation in Cell Recovery Solution (Corning, Cat# 354253) for 30 minutes and the dome fragments were then transferred using a cut 1mL pipette to 10 mm Petri dishes. Organoids were treated with 4% paraformaldehyde in 1X PBS (PFA) for 30 minutes on a rotator at room temperature. A subset of the fixed organoids are liberated; these were transferred to a collection tube (50 mL falcon tube) with a cut 1-mL pipette tip. The Petri dishes containing remaining organoids were washed three times at room temperature, with gentle rocking, for 15 minutes each in Wash Solution (0.75% Glycine, 5% w/v BSA in 1X PBS). After each wash, organoids that become liberated were transferred to the collection tube and allowed to settle by gravity for 15 minutes.
Wash Solution was aspirated, and organoids were resuspended in 1 mL of Wash Solution and transferred in 100 uL aliquots (containing 1 to 10 organoids) into 10–12 glass bottom 96 well plates (MatTek, Cat# P96G-1.5–5-F) with a cut 1-mL pipette tip. In each well, the wash solution was aspirated and 200 uL of Permeabilization Solution (0.5% v/v Triton X-100 in 1X PBS) was added, followed by incubation for 45 minutes with gentle rocking at room temperature. The Permeabilization Solution was then removed and replaced with 200 μL of Blocking Buffer (10% v/v goat serum, 5% w/v BSA, 0.3 M glycine, and 0.5% v/v Triton X-100 in 1X PBS). Organoids were incubated overnight at 4°C with gentle rocking. After blocking overnight (12–16 hours), Blocking Buffer was replaced with fresh Blocking Buffer containing primary antibodies at the concentrations specified (see table below in this section), and as indicated for each image shown in Figures S1 and 6. Organoids were then incubated for one day (16–28 hours) at 4°C with gentle rocking. The Blocking Buffer containing primary antibodies was then removed and the wells were washed three times with 250 μL Antibody Wash Buffer (0.2% v/v Triton X-100, 0.04% v/v Tween-20 in 1X PBS) for 20 minutes at room temperature with gentle rocking. For primary antibodies that were directly conjugated to a fluorophore, the wells were wrapped in foil to protect them from light prior to the overnight incubation, and after each of the above wash steps. After the third wash, 200 μL of fresh Blocking Buffer containing secondary antibodies was added followed by incubation overnight (up to one day) at 4°C with gentle rocking. The wells were wrapped in foil to protect them from light prior to incubation, and after each of the following wash steps. Organoids were washed twice with Antibody Wash Buffer for 20 minutes at room temperature with gentle rocking. Optionally, the second wash step included DAPI at 1:1000 (Sigma, SKU- MBD0015–1ML). Organoids were then washed with 1X PBS for 20 minutes at room temperature with gentle rocking. After washing, 1X PBS was added to samples and the wells were transferred to the microscope for confocal imaging.
Sandwich cultures
Sandwich cultures were prepared for immunostaining following the same protocol as described above for dome cultures, with the following modification to the start of the protocol. Media was aspirated and organoids were washed twice with 1X PBS, as with dome cultures. Cultures were then subject to incubation in Cell Recovery Solution (Corning, Cat# 354253) for 10 minutes on a rotator, at room temperature, followed by incubation in 4% paraformaldehyde in 1X PBS (PFA) in the same wells for 15 minutes on a rotator, at room temperature. A subset of the fixed organoids are liberated; these were transferred to a collection tube (50 mL falcon tube) with a cut 1-mL pipette tip. The original culture wells containing remaining organoids were washed three times at room temperature, with gentle rocking, for 15 minutes each in Wash Solution (0.75% Glycine, 5% w/v BSA in 1X PBS). After each wash, organoids that become liberated were transferred to the collection tube and allowed to settle by gravity for 15 minutes. All subsequent steps are as described for dome cultures. See Key resources table for the full list of antibodies used in this study.
EdU staining
For Figure 4E, organoids were grown in sandwich cultures for 48 hours in ENR media prior to 36 hours treatment with 100 μM CFTRinh, 100 μM Ouabain, or DMSO. Culture media was then replaced fresh ENR media containing 10 μM EdU, and organoids were incubated for a further 2 hours at 37.5°C, then fixed for 15 minutes in 4% PFA at room temperature with gentle rocking, followed by two washes with 5% w/v BSA, 0.75% glycine in 1X PBS for 5 minutes each with gentle rocking at room temperature. The fixed organoids were permeabilized for 20 minutes in 0.5% Triton X-100 at room temperature with gentle rocking, then washed twice more at room temperature with gentle rocking for 5 minutes each with 5% w/v BSA, 0.75% glycine in 1X PBS. Immediately after the final wash, incorporated EdU was conjugated to Alexa Fluor 647 using the Click-IT EdU kit (Thermo Fisher Scientific, Cat# C10640) according to manufacturer instructions. Organoids were incubated in the dark with the kit reaction mix for 30 minutes at room temperature with gentle rocking. Each well was then washed once with 5% w/v BSA, 0.75% glycine in 1X PBS for 5 minutes with gentle rocking at room temperature. After washing, 1X PBS was added to samples. Samples were subjected to confocal imaging in aqueous immersion with Immersol (representative image in Figure S6D). A total of n = 164 organoids were imaged, over 3 replicate experiments carried out in separate weeks for CFTRinh (n = 66) and DMSO (n = 63), and 2 replicate experiments for Ouabain (n = 35).
Diffusion equilibration experiments
Dome and sandwich cultures were prepared as described above, with the following modifications. First, no organoids were embedded in these cultures. Second, dye-free PBS containing 1% w/v BSA was added to these wells instead of ENR media, which was not suitable as it contains Phenol Red. BSA adjusted solution osmolarity so that Matrigel would not swell and deform during the dye diffusion experiment. Wells were imaged on the confocal microscope as described above using a dry 20X objective. We focused on a single focal plane in the Matrigel immediately above the glass coverslip (and PolyHEMA layer, in the case of a sandwich). Images of this single z section in each well were obtained in the red channel (555 nm excitation). We captured images tiling a line along the diameter of the Matrigel such that the central image contained the center of the Matrigel dome or sandwich and the edge images contained the edges of the dome or sandwich. At t = 0, test molecules were added to the top of each well: Texas Red-dextran [average mol. wt. 3 kDa; ThermoFisher #D3329; 1:1000 dilution of 10 mg/mL stock in dH2O], fluorescently-labeled IgG [goat anti-rabbit Alexa Fluor 555; (4 μg/mL; 1:250 dilution from stock) ThermoFisher Cat# A27039], or Phenol Red [i.e., DMEM/F12 containing 8.63 mg/L Phenol Red-Na]. A time series of the well diameter was then collected at 1-hour time intervals. For each time point, the mean image intensity (smoothed over a 1mm window using the MATLAB built-in function movmean) was calculated as a function of distance from the image mid-point, which represented the center of the Matrigel, as shown in Figure 1F. Equilibration time across the base of the Matrigel (Figure S1G) was defined as the earliest time point in the time series at which the mean fluorescent intensity at the Matrigel center reached 80% of the mean value at the Matrigel edge.
Laser ablation
WENR organoids were re-plated in sandwich culture and allowed to grow for 3 days prior to ablation. Laser ablation was conducted by exposing one 40μm × 40μm (512px × 512px) patch of each organoid studied to 405nm laser light at maximum power for 9 minutes, the maximum exposure time allowed by the confocal microscope at the pixel resolution selected. This procedure triggered cell death in the ablated patch within 30 minutes of photoexposure, rupturing the epithelium and driving rapid luminal collapse.
Plate set-up for drug perturbations
Perturbation experiments (Figures 5 and 6) began 48 hours after organoids were seeded in triple-decker sandwiches in order to allow organoids to recover from passaging and to begin to express the stem cell marker Lgr5. To select a subset of organoids for analysis, initially whole wells of organoids were imaged, and the stage position of every organoid was recorded in every well. A subset of stage positions was then selected at random (using the MATLAB datasample function, without replacement) for subsequent imaging. Treatment conditions were randomly assigned to wells. Full z stack images of the organoids were first collected after 48 hours of plating, defining t = 0 hours for perturbation experiments.
Ion channel inhibition and stimulation
Organoids were plated as described above (“Plate set-up for drug perturbations”). At 48 hours post-plating, organoid wells were treated by replacing media with fresh media containing 100 μM ouabain (Tocris, Cat# 1076), 100 μM CFTRinh-172 (Selleckchem, Cat# S7139) or 100 μM forskolin (Selleckchem, Cat# S2449) in tissue culture-grade dimethyl sulfoxide (DMSO) (MilliporeSigma, Cat# D2650). Although the CFTRInh-172 dosage used here exceeds its Ki value (300nM), the combined results of EdU staining and scRNA-Seq (Figure 6) show no evidence of adverse effects on cell proliferation, viability or induction of stress. Control wells were treated with DMSO alone. Media with inhibitors was left in the wells for the rest of the experiment.
Piezo channel perturbation
Organoids were plated as described above (“Plate set-up for drug perturbations”). At 48 hours post-plating, organoid wells were treated by replacing media with fresh media containing 20 μM gadolinium chloride hexahydrate (GdCl3 · 6H2O) (MilliporeSigma, Cat# G7532) or 10 μM GsMTx4 (Abcam, Cat# ab141871) in UltraPure distilled water (ThermoFisher, Cat# 10977015). Control wells were subject to fresh media change alone. To activate Piezo1, organoids were treated with 30 μM Yoda1 (Tocris, Cat# 5586) in DMSO. Organoids used for epistatic analysis (GdCl3 + forskolin) were from a different primary culture, with different passage numbers.
Live imaging intracellular calcium
Organoids were grown for 96 hours in ENR media, then treated and imaged according to the schedules shown in Figure 7A. For CA-630AM addition, media was replaced with 0.5 mL of 1X ENR, and 0.5 mL of 2X Calcium Working Solution [10 uM Cal-630 AM (AAT Bioquest Cat# 20530), 0.08% Pluronic® F-127 (Cat# 20050), 2 mM Probenecid (Cat# 20060), Hanks’ Buffer with 20 mM HEPES (AAT Bioquest Cat# 20011) at 0.9X dilution] to give a final 1X ENR/CW Solution [0.5X ENR, 5 μM Cal-630 AM (AAT Bioquest Cat# 20530), 0.04% Pluronic® F-127 (AAT Bioquest Cat# 20050), 1 mM Probenecid (AAT Bioquest Cat# 20060), Hanks’ Buffer with 20 mM HEPES (AAT Bioquest Cat# 20011) at 0.45X dilution]. As indicated in Figure 7A, prior to or concurrently with addition of the ENR/CW Solution, the organoids were treated with drugs: (1) for forskolin treatment the organoids were incubated for 2 hours in ENR/CW Solution, imaged without forskolin, and then the media was supplemented to achieve a final concentration of 1X ENR/CW with 100 μM forskolin. (2) For CFTRinh-172 treatment, organoids were incubated for 35 hours in 100 μM CFTRinh-172, after which the media was supplemented to achieve a final concentration of of 1X ENR/CW Solution containing 100 μM CFTRinh-172. (3) For GdCl3 and forskolin treatment, organoids were incubated in 20 μM GdCl3 for 35 hours, after which the media was supplemented to achieve a final concentration of 1X ENR/CW Solution containing 20μM GdCl3, and 100 μM forskolin. Representative images shown in Figures 7B and 7C. A total of n = 122 organoids were analyzed across 4 dye-treated conditions: no-drug control (n = 29), forskolin (n = 29), CFTRinh-172 (n = 31), GdCl3 + forskolin (n = 33).These organoids were compared to no-dye controls (n = 64), which were measured by imaging organoids in the CFTRinh-172 and GdCl3 + forskolin conditions before the addition of drugs and ENR/CW Solution. Organoids in all conditions were imaged using the same camera settings, which enabled quantitative comparison of mean calcium intensity across conditions; Figure 7D shows results 2 hours after ENR/CW addition.
Preparation of single cells for scRNA-Seq
Organoids were grown in either sandwich or dome culture in glass-bottom 12 well plates (Mattek, Cat# P12G-1.5–14-F). Before dissociation, all pipette tips and transfer vessels were coated with 10% w/v BSA to prevent loss of material. Media was aspirated and culture wells were washed twice with 1X PBS before incubating in 1mL TrypLE Express (ThermoFisher, Cat# 12604–021). Upon addition of TrypLE Express, a 1 mL pipette tip was used to mechanically detach organoids embedded in Matrigel from either sandwich or dome cultures, taking care to scrape the bottom of the well with such force so as not to detach the glass bottom coverslip, but to visibly remove the Matrigel from the coverslip. Organoids were incubated at 37°C for between 20–30 minutes, monitoring every 5 minutes to check on the status of dissociation, and to repeat brief mechanical disruption by trituration. Multiple wells (between 2 and 10) per condition were pooled into a 15 mL falcon tube and diluted 1:1 with ice cold DMEM, and strained (40 μm [pluriSelect, Cat# 43–10040-60]) into a 15 mL falcon tube. This cell solution was then centrifuged at 250xg in a spinning bucket centrifuge for 1 minute at 4°C, and the suspension was aspirated. Cells were resuspended in 200 μL of 1% w/v BSA in 1X PBS, kept on ice, and cell count and viability were measured with a hemocytometer immediately prior to single cell encapsulation with the inDrops method (Klein et al., 2015).
Single cell RNA Sequencing
Encapsulation of single cells was performed as previously described using inDrops at the Harvard Medical School Single Cell Core (Zilionis et al., 2017).
We used the following RT primers on hydrogel beads:
5′CGATTGATCAACGTAATACGACTCACTATAGGGTGTCGGGTGCAG[bc1,8nt]GTCTCGTGGGCTCGGAGATGTGTATAAGAGA CAG[bc2,8nt]NNNNNNTTTTTTTTTTTTTTTTTTTV- 3′
Cells were processed as described until step 105 in Zilionis et al. (2017), and then proceeded with the following modifications that implement a template-switching approach followed by PCR, instead of linear amplification originally used in Zilionis et al. 2017). For the sandwich and dome comparison (Figure 1), cells were co-encapsulated with barcoded hydrogel beads in the presence of 1x Maxima Buffer, 1 U/uL of Ribolock RNase inhibitor (Thermo Fisher Scientific, Cat# EO0381), 10 U/uL of Maxima H minus reverse transcriptase (Thermo Fisher Scientific, Cat# EP0751), 0.25 μM TSO primer (5′-AAGCAGTGGTATCAACGCAGAGTACATrGrGrG-3′), 0.5 mM of dNTPs and 0.3% of NP40. Following encapsulation, emulsions were incubated for 60 minutes at 42°C (i.e., Reverse transcription) followed by 5 minutes at 85°C (enzyme inactivation). Droplets were then broken with 20% 1H, 1H, 2H, 2H- perfluorooctanol (PFO) and spun down for 30 s at 300xg. Without pipetting oil, the supernatant containing the emulsion was transferred onto a Costar Spin-X centrifuge tube filter (Sigma-Aldrich, Cat# CLS8160) and centrifuged for 2 minutes at 20,000xg. The fractions were washed using 1.2X volume of SPRI Select (Beckman Coulter, Cat# B23318) to reaction volume. The cDNA was then amplified using Kapa 2X HiFi HotStart PCR mix (Kapa Biosystems, Cat# KK2601) and primers (Fwd cDNA primer- 5′-CACTATAGGGTGTCGGGTGCAG-3′, Rev cDNA primer- 5′-AAGCAGTGGTATCAACGCAGAGT-3′) with the following thermocycler program: 1) 98°C x 3 minutes, 2) 98°C x 15 s, 3) 67°C x 20 s, 4) 72°C x 1 minute [go to 2) x (12–16 cycles depending on the cell type)], 5) 72°C x 1 minute, 6) hold at 4C. Amplified cDNA was size-selected using 1.2X volume of SPRI Select to reaction volume and quantified using Agilent Bioanalyzer system. Half of the cDNA was used for library preparation. Fragmentation, end repair and adaptor ligation (Ligation Fwd primer: 5′-/5Phos/GATCGGAAGAGCACACGTCTGAACTCCAGTCAC/3ddC, Ligation Rev primer: 5′- /5AmMC6/GCTCTTCCGATCT 3′) was carried out using the NEBNext Ultra II FS DNA Library Prep kit (NEB, Cat# E7805S) following manufacturer’s recommendations, followed by purification by 0.8X volume of SPRI Select to reaction volume. Library indexing PCR was carried out using 1X KAPA HiFi ReadyMix and 0.5 uM indexing primers (PCR_p7_r2: 5′-CAAGCAGAAGACGGCATACGAGATGGGTGTCGGGTGCAG-3′, PCR_p5_r1_ixx: 5′-AATGATACGGCGACCACCGAGATCTACAC[i5]TCGTCGGCAGCGTC-3′) and using the following thermocycler program: 1) 98°C x 45 s, 2) 98°C x 20 s, 3) 54°C x 30 s, 4) 72°C x 20 s [go to 2) x (12–16 cycles depending on the PCR input)], 5) 72°C x 1 minute, 6) hold at 4°C. The final library was then purified using double-sided size selection using 0.6X- 0.8X volume of SPRI Select to reaction volume. Final library was then quantified using the Agilent Bioanalyzer system and sequenced using a NextSeq500 Instrument PE 2×100 (Illumina).
The perturbation experiment (Figure 6) was carried out at a later time, with several modifications to the TSO protocol as follows. Cells were co-encapsulated with barcoded hydrogel beads in the presence of 1X Maxima Buffer, 1 U/uL of Ribolock RNase inhibitor (Thermo Fisher Scientific, Cat# EO0381), 10 U/uL of Maxima H minus reverse transcriptase (Thermo Fisher Scientific, Cat# EP0751), 0.5 mM of dNTPs and 0.3% of NP40. Following encapsulation, emulsions were incubated for 60 minutes at 50°C (i.e., Reverse transcription) followed by 5 minutes at 85°C (enzyme inactivation). The fractions were washed using Dynabeads MyOne Silane (Thermo Fisher Scientific, Cat# 37002D) followed by template-switching reaction outside of the droplet. The Template-Switching reaction outside of the droplet was performed by combining the purified hybrid cDNA/RNA with 1X Maxima Buffer, 1 U/uL of Ribolock, 10 U/uL of Maxima RNase H− Reverse Transcriptase, 1 mM of dNTPs, 2.5 uM TSO primer and 4% Ficoll P-400, and incubating for 30 minutes at 25°C, 90 minutes at 42°C with lid set to 50°C. After Template-Switching reaction a 1X volume of SPRI Select cleanup was carried out, and cDNA hybrid was ready for cDNA amplification. The cDNA was then amplified using Kapa 2X HiFi HotStart PCR mix (Kapa Biosystems, Cat# KK2601; PCR_I_fw primer_v2: 5′- CACTATAGGGTGTCGGGTGCAG – 3′, PCR_I_rev primer: 5′- AAG CAGTGGTATCAACGCAGAGTACAT-3′) with the following thermocycler program: 1) 95°C x 3 minutes, 2) 98°C x 20 s, 3) 59°C x 30 s, 4) 72°C x 1 minute [go to 2) x (12–16 cycles depending on the cell type)], 5) 72°C x 1 minute, 6) hold at 4C. Amplified cDNA was size-selected using 0.6X volume of SPRI Select to reaction volume and quantified using Agilent Bioanalyzer system. Half of the cDNA was used for library preparation. Fragmentation, end repair and adaptor ligation (Ligation Fwd primer: /5Phos/CTGTCTCTTATACACATCTGACGCTGCCGACGA,
Ligation Rev primer: AGATGTGTATAAGAGACAG*T) was carried out using the NEBNext Ultra II FS DNA Library Prep kit (NEB, Cat#E7805S) following manufacturer’s recommendations, followed by purification by 0.8X volume of SPRI Select to reaction volume. The final steps (Library indexing PCR onward) were carried out as for sandwich and dome cultures above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Distribution and viability of organoids
The XYZ centroid positions for all organoids in each well were extracted from tiled whole-well z stacks (images acquired at 10X magnification as described above). Whole-well images (Figures 1E and 1F) were automatically stitched together using NIS-Elements AR 4.51 (Nikon), using the Large Image setting in ND Acquisition to capture a scan area of 7 mm x 7 mm (dome) or 14.5 mm x 14.5 mm (triple-decker sandwich) centered at the middle of each well, with stitching enabled (using all channels for stitching) and 1% overlap between separate fields of view. Custom MATLAB code was used to (1) downsample the tiled images; (2) Gaussian blur the images; (3) threshold the H2B-mCherry channel to produce a binary image; (4) call the MATLAB regionprops3 subroutine to segment the organoids and identify their centroids; and (5) exclude small regions that corresponded to debris.
Spatial density profiles (Figures 1E and 1F) were calculated from a list of centroids in each well using the following formula. Let Q be the radius of the hydrogel, and let2 be the XY centroid position of the i-th organoid in a plate with a total of N organoids, and let be the set of points at a distance less than Q-a from the edge of the hydrogel. Then the coarse-grained density at a point
where H(x) = {1 for x > 0; 0 otherwise}. Plots in Figures 1E and 1F show the results of averaging over three independent wells of organoids for each condition after 6 days post-plating, using a = 1.5 mm.
To generate viability plots (Figure 1G), spatial density profiles were first calculated as above for the three plates imaged at 1 day and 6 days post-plating, giving and respectively, with denoting averaging over independent wells. The plots then show the radially-averaged fold-change in density between 1 and 6 days in culture, calculated as the average over points :
with L being the width of the moving window. The plot was generated using a value of L = Q/4.
Image denoising
Microscopy images were subject to computational denoising using MATLAB image processing libraries from (Dabov et al., 2007), with the exception of Figures S1A, S1B, S1H, S1I, and S2 and Videos S5, S6, and S8 which are rendered without denoising. The workflow consists of (1) a variance-stabilizing Anscombe transformation (Mäkitalo and Foi, 2011 ) that changes the image noise profile from Poisson to Gaussian; (2) noise reduction using the block matching and 3D filtering (BM3D) algorithm (Dabov et al., 2007); (3) numerical inversion of the Anscombe transform (Mäkitalo and Foi, 2011); (4) background subtraction to remove constant bias added to the image by the filtering procedure; (5) mean filtering with kernel size of 3 pixels, applied to the H2B-mCherry channel only. In addition, a subset of images (Figures 1J, 1K, 3A, 3B, 4A, 5B, and 5C; Videos S1, S3, and S7, and examples 3–7 of Videos S8), and all images processed for segmentation (see below) were subject to a preliminary step of Intensity Correction that preserved structure in z sections far from the objective that was otherwise lost during denoising. Intensity Correction was carried out by (1) determining foreground pixels in each z section independently using Otsu’s threshold, and (2) setting the intensity of foreground pixels in all z sections to a constant IF. The value IF = 1000A.U. (in 12-bit scale images) was chosen as close to the mode of the foreground pixels in the sections closest to the coverslip.
Quantifying total organoid size
For Figures S1F and 3C organoid volumes were calculated through the following workflow, implemented in MATLAB. (1) Denoised H2B-mCherry z stack images were blurred using a 3D Gaussian filter with a 5 μm radius (i.e., the size of a typical cell). (2) The blurred images were binarized using the lower level of a two-level multi-Otsu threshold calculated using the multithresh function in MATLAB (version R2018b, argument N = 2). (3) Binary masks were flood-filled to close holes; (operation carried out separately for each z-slice). (4) Objects smaller than 250 voxels were removed from the mask using the bwareaopen function in MATLAB. (5) Segmented volumes were evaluated in voxel units using the regionprops3 function in MATLAB, and then converted to μm3. For Figure 2H, organoid surface areas were determined by following steps 1–4 above, then using regionprops3 to measure 2D surface areas of 3D organoid masks in voxel units and converting these quantities to μm2.
Quantifying stem cell zone size
We defined Stem Cell Zones (SCZs) as contiguous patches of the epithelium positive for Lgr5-DTR-EGFP. In order to quantify SCZ size and generate the plots in Figures 2E, 2G, and 2J, we took a two-step approach that allowed us to estimate the number of Lgr5+ stem cells in an SCZ with both high accuracy and high throughput. First, we generated standard curves to convert SCZ tissue volume to total cell number (Figure S2H) and Lgr5+ stem cell number (Figure S2I), which subsequently enabled us to measure SCZ volumes semi-automatically and rapidly estimate cell numbers from these measurements. We prepared standard curves by using ImageJ to manually segment n = 61 SCZs and count the number of cells that each SCZ contained; we determined the number of Lgr5+ cells per SCZ indirectly, by immunostaining organoids for the Paneth cell marker Lysozyme and subtracting the number of Lyz+ Paneth cells from the total number of nuclei in each SCZ.
We segmented SCZs and measured their volumes semi-automatically using a MATLAB script that implemented the following steps. (1) A 3D binary mask of each entire organoid was generated as described in steps 1–3 of the above methods section, “Quantifying total organoid size.” (2) A 3D binary mask of the luminal core of each entire organoid was generated by (2a) calculating the distance of each voxel within the organoid to the surface using the 3D Distance Transform algorithm (Mishchenko, 2015); and (2b) defining the 15% voxels with greatest distance to the surface as the luminal core (these are the deepest voxels). For the following step we denote the luminal core mask as M, where Mi,j,k = 1 if voxel (i,j,k) is in the organoid luminal core, and 0 otherwise. (3) LGR5-DTR-EGFP z stack images of the organoid were filtered to mask out the luminal core. Let the intensity-corrected and denoised LGR5-DTR-EGFP image be X, where Xi,j,k[REMOVED HYPERLINK FIELD] is the recorded fluorescence intensity of voxel (i,j,k). The filtered image is . This step removes voxels that show autofluorescence in the organoid luminal core. (4) The image X’ was binarized using a single-level Otsu threshold applied to the organoid image. (5) The binary masks were converted into editable Surface objects in Imaris 8.4 (Oxford Instruments). (6) The segmented Lgr5+ masks were visually inspected in Imaris software, and regions that were deemed poorly segments were either manually corrected or excluded from further analysis. In this step, SCZs were inspected for (a) under-segmentation: where two or more distinct zones were merged together or where spurious debris can be fused to an SCZ; and (b) incomplete segmentation: where the segmented volume did not account for the majority of the volume of the zone. For case (a), we used the Surface editing tools in Imaris to fix the segmentation. For (b), we discarded these surfaces. (7) SCZ volumes were quantified as the number of voxels in segmented regions, converted to units of μm3.
For Figure 2G only, we measured SCZ size in Lgr5-DTR-EGFP organoids that lacked nuclear labeling. For these organoids we followed the same workflow described above in “Quantifying stem cell zone size,” but replaced steps 1–2 with the following step: (Combined 1–2) the luminal core binary mask M is defined as voxels saturated in the the SiR-Actin channel. All remaining steps were as described. This modification took advantage of the strong accumulation of SiR-Actin dye in the organoid lumen.
Calculating SCZ fission number and rate
SCZs were counted manually by identifying and tabulating the number of disconnected regions of Lgr5 expression after segmentation as described above (steps 1–6 in “Quantifying stem cell zone size”). Using Imaris software, we used Spots objects to mark SCZs over time, and we used Tracks to connect SCZs across time points into lineage trees by connecting parent SCZs at time points before fission with their children at time points after fission. For Figure 2E, we tabulated from these tracks giving the scalar size s, the binary indicator F = {1 if fission occurs, 0 otherwise}, and the imaging time interval Δt for each time point t in the k-th organoid. The size-dependent fission rate was then calculated as
For Figure 4C, to generate the histogram of daughter SCZs per fission event, we tabulated the number of children per branch point in the lineage trees generated as described.
Calculating SCZ size distribution with PBE
For Figure 2I, we plotted the quantitative form of SCZ size distributions using numerical solutions to the Population Balance Equation (PBE). The PBE is a non-local partial differential equation of the form,
where D is a diffusion constant reflecting stochasticity in stem cell division and differentiation, v(s) is the size-dependent SCZ growth rate, γ(s) is the size-dependent SCZ fission rate, and
is the rate at which SCZs of size s emerge from fission of larger SCZs, with P(q, s) as the probability that an SCZ of size s gives rise to an SCZ fragment of size qs. This equation was solved numerically using the MATLAB function ode45, numerically calculating θ[N; s; t] at each step of the numerical solver. We restricted our analysis to the generation of two fragments per fission, although Type II fission events lead to the formation of multiple fragments (see Figure 4). We assumed a uniform and size-independent distribution in q, P(q, s) = 1, but we found that a range of distributions over q provided comparable results. The PBE equation excluded terms for SCZ fusion because we observed it to be extremely rare in live movies of growing organoids. It also excluded terms reflecting SCZ emergence de novo, as we did not observe this to occur outside of the first 24–48 hours after passaging consistent with previous reports (Fordham et al., 2013; Mustata et al., 2013; Serra et al., 2019) and we started all experiments after this initial interval. The exponential steady-state solution of the PBE in Figure 2I can also be derived analytically (see Rulands et al., 2018).
Detection of inflation and collapse events
To detect inflation-collapse events (Figures 3C and 3E), time-series of organoid volume were analyzed using a pulse detection algorithm implemented in MATLAB. The algorithm identified local maxima and minima in a time series and generated a distribution of the magnitude of volume changes between each peak (maximal events) and its subsequent trough in volume (minimal events), allowing for any time interval between peak and trough. To enrich for bona fide inflation/collapse events in our correlation analysis (Figure 3E), we selected events that were in the top 10% by volume change.
Cell density during inflation and collapse
To quantify changes in cell density in an organoid bud undergoing inflation and collapse (Figure 4E), we measured a proxy, the mean inter-nuclear distance between cells in Lgr5+ and Lgr5- regions of the tissue. We used the line segment tool in ImageJ to measure inter-nuclear distances manually in 9 buds x 3 xy cross-sections per bud, at 3 time points – pre-inflation (t1), peak inflation (t2), and post-collapse (t3). We partitioned these measurements into two groups: distances between Lgr5+ cells, and distances between Lgr5− cells. For each group, within each bud, we averaged inter-nuclear distances and calculated the fractional change in this average relative to its value pre-inflation. These measurements of tissue strain within individual buds, separated by Lgr5 expression, are shown in gray in Figure 4E. The average across buds, again separated by Lgr5 expression, is shown in black. Red arrows in Figure 4E indicate statistical significance (p < 0.05), as ascertained by one-tailed t tests comparing the mean strain across buds at a given time point with the null hypothesis of zero strain.
Dynamics of Lgr5 expression and tissue deformation during inflation
The following quantities were analyzed in 8 different time series of bud inflation: maximum cross-sectional bud area (Figure S3A); Lgr5 coverage, as a fraction of that area (Figures S3B and S3C); and total Lgr5 intensity in each bud (Figure S3C). Bud area and Lgr5+ area were determined by outlining H2B-mCherry+ and Lgr5-EGFP+ regions in maximum intensity projections of buds along the z axis, using the Freehand ROI tool in ImageJ. Total Lgr5 intensity was calculated by summing pixel values in the EGFP channel in these segmented regions. For each bud, we used the trace of maximum cross-sectional area, and luminal distension visible in a bright-field image of that cross-section, to identify the time of inflation onset. We aligned all time-series data such that inflation onset corresponded to t = 0, and considered dynamics from 4 hours prior to inflation to 12 hours after inflation onset. In all cases we smoothed raw data using a moving average filter (MATLAB built-in function smooth) with a 2-hour window, and we converted raw data to fractional changes relative to values at inflation onset.
To identify the time at which Lgr5 signal loss began (Figure S3B, indicated by red triangles), relative to the onset of inflation, we fit the fold change in Lgr5 area to a piecewise linear model comprising two parts: a plateau before the onset of Lgr5 loss, and a linear decrease thereafter. We defined the time of Lgr5 loss as the junction point between these two regimes that minimized the sum of squared errors of the piecewise fit, subject to the constraint that Lgr5 signal drops for at least 2 hours after this point. This constraint ensured that we detected persistent decreases in Lgr5 signal rather than transient fluctuations.
For comparison of inter-nuclear distances at the onset of inflation and Lgr5 signal loss (Figures S3D and S3E), we measured inter-nuclear distances as above (“Cell density during inflation and collapse”), but with two modifications. (1) We considered two time points: the onset of inflation, and the onset of Lgr5 signal loss. (2) We analyzed only two xy cross-sections per bud – the cross-section closest to the microscope objective, and the “midplane” of the bud with maximum cross-sectional area.
Relationship between Paneth cell positions and Lgr5 loss
For analysis of Paneth cell proximity to cells losing Lgr5 (Figure S3F), we extended the analysis from the previous section (“Dynamics of Lgr5 expression and tissue deformation during inflation”) by marking the positions of Paneth cells in the movies analyzed. We manually identified Paneth cells based on their distinct shape and granular appearance in bright-field z stacks of these buds. We recorded their xyz-positions at two time points – inflation onset, and 12 hours thereafter – using the Point ROI tool in ImageJ; and we scored Paneth cell contact with Lgr5+ cells by calculating the fraction of xy-positions at each time point contained within Lgr5+ and Lgr5− regions of buds, as manually segmented previously.
Laser ablation of budding phenotypes
The effects of collapse on organoid morphology were determined by imaging WENR organoids immediately before ablation and every 30 minutes for 18 hours after photo-exposure. Collapse was deemed to have induced Lgr5+ bud formation in a WENR organoid when the following criteria were met: (1) before ablation, the organoid had at least one Lgr5+ SCZ; (2) within 2 hours of ablation, the organoid epithelium buckled to form at least one new bud with higher outward curvature than the pre-ablation cyst; and (3) at least one of these nascent buds contained Lgr5+ cells at its tip. These criteria were applied uniformly both to organoids that reinflated after wound healing and to organoids that remained collapsed for the duration of the experiment.
Quantifying perturbation phenotypes
The effect of drug treatment on inflation was assessed by measuring the fraction of organoids in a given condition that showed morphological evidence of inflation at the time points indicated for each of the experiments in Figure 5A. Each organoid under perturbation or control conditions was scored as “inflated” or “not inflated” manually, as defined by the presence of one or more of the following morphological criteria: (1) thinning of the epithelium evident in brightfield images; (2) thinning of the epithelium evident in the H2B-mCherry channel as elongated nuclei consistent with squamous cell shape; and (3) luminal dilation visible in brightfield images. For all inhibition experiments, the effect of drug treatment on SCZ fission rate was defined as the change in the manually-counted number of SCZs between 36 and 54 hours in the presence of drug or control at steady state, divided by the number of SCZs observed at 36 hours. For forskolin treatments, the same analysis was carried out, but with time points as indicated in figures.
Processing sequencing reads for scRNA-Seq
Per-cell gene expression counts from raw sequencing reads were generated using the v3 iteration of the publicly available (https://github.com/indrops) data processing pipeline as described (Klein et al., 2015). Parameters used with the indrop.py pipeline are specified in yaml files as provided on GitHub (https://github.com/AllonKleinLab/paper-data/tree/master/Tallapragada_Cambra2021). Briefly, raw reads (fastq) were filtered for sequencing quality and expected structure, sorted based on barcode sequences, and aligned to the 2018 version of mm10 cDNA reference with separately added mitochondrial cDNA sequences and GFP sequence. To quantify gene expression while correcting for amplification biases, we used unique molecular identifiers (UMIs) introduced during RT in drops. The output of low-level processing is a genes x cells expression matrix.
Single cell data filtering and normalization
To remove noise introduced by technical aspects of collection and sequencing of single cell data, we filtered out cells with few counts, high mitochondrial gene fraction, high ribosomal gene fraction, low total number of unique genes expressed, and optionally with non-coding RNA gene fraction. With the sandwich and dome comparison data (Figures 1C, 1D, S1C, and S1D), we retained transcriptomes with a mitochondrial fraction < 25%, and a ribosomal fraction fraction < 25%, and at least 1000 total UMI-filtered mapped reads. A minimum gene count of 200 genes was used to filter cells, and genes expressed in less than 3 cells were excluded from further analysis, using the scanpy functions sc.pp.filter_cells and sc.pp.filter_genes, respectively. With the perturbation experiment (Figure 6), we excluded a small number of cells (24 transcriptomes) expressing the genes (gm26917, gm42418, gm25580, gm24139, gm24146) at an abundance of > 4% of total counts, as these were deemed to reflect dead or stressed cells. For one sample (forskolin-treated), owing to lower library depth the total count threshold was altered to 150 UMI Filtered Mapped Reads and the ribosomal fraction threshold was raised to < 45%. All data was subject to doublet detection using the python package Scrublet (Wolock et al., 2019), but we rejected no cells with a Scrublet score threshold of 0.85. After filtering, single cell transcriptomes were normalized by the total counts per cell to 10,000 counts per cell (CP10K).
Data visualization and cell classification
Following filtering and normalization, scRNA-seq data was log-scaled with a pseudocount of 1 CP10K, then z-scored (scanpy function sc.pp.scale) and subject to dimensionality reduction by PCA using the highly variable genes calculated and selected using the scanpy function sc.pp.highly_variable_genes, with the following thresholding parameters applied to both datasets: min_mean = 0.0125, max_mean = 8, min_disp = 0.5. 15 principle components were used for embedding for the sandwich and dome data, and 11 principle components were chosen for embedding the perturbation data, based on the ‘elbow rule’ after plotting the explained variance ratio for each dataset. Batch correction was performed on the sandwich and dome data using batch balanced KNN, using the scanpy wrapper sc.external.pp.bbknn (Polanski et al., 2019). This KNN graph was then used subsequently for final visualization via UMAP (McInnes et al., 2018). Batch correction was not performed on the perturbation dataset, and instead a KNN graph was constructed using 10 nearest neighbors with final visualization via UMAP.
The sandwich and dome dataset and perturbation dataset were both annotated using marker genes selected from the literature for the major cell types, with the consideration that we grouped all enterocyte subsets together, as well as goblet and secretory cells (resulting in longer gene lists for these sets). We should note that goblet and secretory cells frequently co-localize in murine intestinal organoids, leading to the decision to merge these two cell states into the Secretory state. Specific gene lists used to develop cell type scores are given in Table S1. For the perturbation experiment, we included the additional stretch-responsive state alongside the annotated cell types identified in the literature; we assembled a list of marker genes from this state, also included in Table S1. We assigned each cell transcriptome to a cell type as follows. For observed transcriptome i, we defined a score for each of cell types j = 1,..,n, as zij = zscoreiðsijÞ, where is the normalized expression of the k-th marker gene for cell type j, and mjis the total number marker genes for cell type j given in Table S1. The denserank function is implemented using scipy.stats.rankdata. For each observed transcriptome, the highest scoring cell type (argmaxj(zij)) was chosen as the annotation. To denoise cell type annotations, the cells were then clustered using the leiden algorithm (scanpy function sc.tl.leiden, resolution = 0.12), and the frequency of each cell type in each cluster was calculated. Cells whose annotation appeared at a frequency less than fmin in each cluster were considered classification errors, and re-assigned the majority annotation. We used fmin = 0.3 for the data in Figure 1, and fmin = 0.2 for the perturbation data in Figure 6. To generate the comparison to in vivo data in Figure 1D, the same process of classification was applied to the in vivo reference data from Haber et al., 2017, with 20 PCs, batch correction across each replicate, with resolution = 1 for leiden clustering. Fully processed and annotated single cell RNA-sequencing data for each dataset was exported to SPRING as described in Weinreb et al. (2018) from Jupyter notebooks (https://github.com/AllonKleinLab/paper-data/tree/master/Tallapragada_Cambra_2021). See Key resources table for links to interactive single cell representations of perturbation and sandwich and dome data.
Marker gene identification
‘Marker’ genes are those genes plotted in the heatmap in Figure S1D to enable visual inspection of consistency of gene expression between dome and sandwich culture, and again in Figure 6E to enable visual inspection of cell type-specific gene expression in stretch-associated cells. The genes identified for sandwich and dome data and perturbation data are given in Sheets 4 and 5 of Table S1, respectively. To identify marker genes, for each annotated cell type j we first calculated the mean expression of each gene k as , where the sum i is over cells annotated to cell type j, is the unnormalized sum, and xik is the raw (unnormalized) number of counts for gene j in cell transcriptome i. We define as the ratio of the maximal value of Xjk to the second-maximal value of Xjk for gene k, corrected by a pseudovalue of 1 CP10K. With these definitions, Gene k was defined as a marker gene of state j if it satisfied the following criteria (adapted from Zilionis et al., 2019):
Gene k is maximally expressed in state j (argmaxi(Xik) = j)
The gene is at least 1.3-fold higher in expression in the state than in the next-highest state after pseudo-count correction .
In Figure 6E, marker genes for the stretch signature were calculated as described above. To visualize marker genes for other annotated cell states (Figure 6E, bottom), the same analysis was repeated but first excluding the stretch response state, i.e., including only: stem, enterocyte, enteroendocrine, secretory, and tuft states. Marker genes for the stretch state were then excluded as markers for these cell types. For visualization, the marker genes were ordered by the pseudocount-adjusted max-to-median expression ratio, . All marker genes are indicated in Table S1 (sheets 4,5). The first 10 markers are shown in Figure S1D, and up to 100 are shown in Figure 6E.
Measuring abundance changes in scRNA-seq data
In condition c, let the number of cells classified to each annotated cell state j set be . The fraction of cells in each state was then . Log-abundance Pearson correlation within states between treatment conditions for Figure 1D were calculated as . The log-fold change in abundances between inflation perturbation treatment and controls shown in Figure 6B were calculated as with p = forskolin, c = (DMSO, 12h) for one data series, and p = CFTRinh-172, c = (DMSO, 36h) for the second data series shown. DMSO samples are time-matched respectively to forskolin and CFTRinh-172 treatment times. Error bars shown in Figure 6B show the standard errors for log-abundance ratios, calculated using the approximate error propagation formula for sampling error,
Significance of changes between perturbed and controlled samples (Figures 6B and S3B) was assessed using the Fisher exact test (scipy.stats.fisher_exact), and p values were corrected using Bonferroni’s method.
Measuring stained stretch-responsive cells
We analyzed DMSO- and forskolin-treated organoids (n = 12 organoids, 6 per condition) for immunostained Basp1 (Figure 6H) as described above (see Immunostaining). We manually scored cells as Basp1-hi if they showed membrane staining (as indicated by yellow arrows in Figure 6H, in at least 3 consecutive z-slices, with a 1.5 μm interval between slices), and then calculated the fraction of Basp1-hi cells by dividing by the total number of nuclei in the H2B-mCherry channel in the same sections. Care was taken not to double-count nuclei in consecutive z sections. All analyses were carried out in ImageJ. To measure the average size and intensity of Basp1-hi and Basp1-low cells (Figure S4F), we used the polygonal ROI tool in ImageJ to outline the nuclei of Basp1-high cells and their Basp1-low neighbors, and recorded cross-sectional area and integrated H2B-mCherry fluorescent intensity.
Gene set enrichment analysis
Gene sets were obtained from Tables S3 and S5 in Ayyaz et al. (2019) for clusters 1–19 and SSC2a-c, Table S1 from Mustata et al. (2013) and Table S3 from Yui et al. (2018) for intestinal fetal signature gene expression, enriched genes for each cluster calculated from available single cell data collected from Sca1+/− sorted cells from organoids exposed to helminth infection (GEO:GSE108233; Nusse et al., 2018) with a wilcoxon rank sum test using sc.pp.rank_genes_groups after processing single cell data as described here in, and Table S1 from Gregorieff et al. (2013) for YAP response. For the Gregorieff data, we selected the 500 most downregulated genes after YAP knockout as the YAP regulated gene list. The gene sets are aggregated in Table S1 (Sheet 6) of this paper. Set enrichment was evaluated using Fisher’s exact test, with one-tailed p values, and subject to Benjamini-Hochberg multiple hypothesis correction to control for FDR. For this test, the background gene list was defined as the highly-variable genes in our scRNA-Seq dataset [n = 4,567 genes, see Table S1 (Sheet 6)].
Quantifying epithelial calcium signal
We quantified the average epithelial calcium signal in different treatment conditions (Figure 7D), using the following workflow implemented in MATLAB to automatically segment and measure signal intensity in the epithelium over time. (1) At each time point we segmented organoids in three dimensions, as described above (“Quantifying total organoid size”). (2) We detected the surface of each organoid by identifying all voxels contained wholly within the organoid segmentation mask (using the imerode function with a 3px x 3px structuring element) and removing them from the mask. (3) We generated an epithelial mask for each organoid by using the imdilate function (with a 15 μm x 15 μm structuring element) to expand the boundary detected in the previous step, and retaining only the voxels within the original segmentation mask. (4) We defined the average calcium signal in the epithelium of each organoid, at each time point, as the average of the denoised calcium signal contained within this epithelial mask. For Figure 7D, images were analyzed at 2 hours after addition of Cal-630AM dye and/or drug as per the schematic in Figure 7A, with the exception of the no-dye control, which reflects a time point prior to dye addition.
Hypothesis testing and reproducibility
All imaging experiments presented in this study were repeated on at least two separate occasions, with the number of wells and number of organoids per well as described in the paper. All single cell RNA-Seq experiments combined cells from two organoid cultures that were passaged independently and combined prior to cell encapsulation into droplets. Gene expression signatures were reproducible between multiple conditions as shown in Figures S1D and 6E, and the specific predictions from this work were validated through imaging experiments (EdU staining, Basp1 and Anxa1 staining, cell size analyses and stretch-mediated differentiation of stem cells). P values report the results of t test comparisons with null models of zero correlation (Figure 3E) or zero difference in mean effect between two experimental conditions (Figures 5, 6, and 7). In Figures 5 and 7, all t tests were one-sided, and statements about significant increases (decreases) in effects were accompanied by p values reporting upper (lower) tail probabilities. * = p < 0.05; ** = p < 0.01; *** = p < 0.001, unless otherwise indicated.
Supplementary Material
Highlights.
Triple-decker sandwich organoid cultures allow long-term live imaging
Ex vivo stem cell zone formation is dominated by CFTR-mediated inflation events
Inflation-mediated fission requires Piezo
Rare stretch-responsive cell abundance changes with inflation
ACKNOWLEDGMENTS
A.M.K. acknowledges support from NIH grant R01HD096755. O.D.K. acknowledges support from NIH grant U01DK103147 from the Intestinal Stem Cell Consortium, a collaborative research project funded by the National Institute of Diabetes and Digestive and Kidney Diseases and the National Institute of Allergy and Infectious Diseases. N.P.T. was supported by an NSF graduate research fellowship. We thank Ysbrand Nusse for support with mouse genetics and generating organoid cultures; Frederic de Sauvage for Lgr5-DTR-EGFP transgenic mice and comments; Kyogo Kawaguchi and James Briggs for feedback on experimental design; Nancy Chen for help with image annotation; and Kalki Kukreja, Tal Scully, and Qiu Wu for feedback on figures. Finally, we thank the Nikon Imaging Center at Harvard Medical School for microscopy guidance and the Single Cell Core at Harvard Medical School for support with scRNA-seq experiments.
Footnotes
DECLARATION OF INTERESTS
A.M.K. is a founder of 1CellBio, Inc.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.stem.2021.04.002.
REFERENCES
- Ayyaz A, Kumar S, Sangiorgi B, Ghoshal B, Gosio J, Ouladan S, Fink M, Barutcu S, Trcka D, Shen J, et al. (2019). Single-cell transcriptomes of the regenerating intestine reveal a revival stem cell. Nature 569, 121–125. [DOI] [PubMed] [Google Scholar]
- Bagriantsev SN, Gracheva EO, and Gallagher PG (2014). Piezo proteins: regulators of mechanosensation and other cellular processes. J. Biol. Chem 289, 31673–31681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker AM, Cereser B, Melton S, Fletcher AG, Rodriguez-Justo M, Tadrous PJ, Humphries A, Elia G, McDonald SA, Wright NA, et al. (2014). Quantification of crypt and stem cell evolution in the normal and neoplastic human colon. Cell Rep. 8, 940–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N (2014). Adult intestinal stem cells: critical drivers of epithelial homeostasis and regeneration. Nat. Rev. Mol. Cell Biol 15, 19–33. [DOI] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, and Clevers H (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003–1007. [DOI] [PubMed] [Google Scholar]
- Batlle E, Henderson JT, Beghtel H, van den Born MM, Sancho E, Huls G, Meeldijk J, Robertson J, van de Wetering M, Pawson T, and Clevers H (2002). Beta-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/ephrinB. Cell 111, 251–263. [DOI] [PubMed] [Google Scholar]
- Berlanga-Acosta J, Playford RJ, Mandir N, and Goodlad RA (2001). Gastrointestinal cell proliferation and crypt fission are separate but complementary means of increasing tissue mass following infusion of epidermal growth factor in rats. Gut 48, 803–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi A, and Morabito A (2009). The dilated bowel: a liability and an asset. Semin. Pediatr. Surg 18, 249–257. [DOI] [PubMed] [Google Scholar]
- Bjerknes M, and Cheng H (1981a). The stem-cell zone of the small intestinal epithelium. I. Evidence from Paneth cells in the adult mouse. Am. J. Anat 160, 51–63. [DOI] [PubMed] [Google Scholar]
- Bjerknes M, and Cheng H (1981b). The stem-cell zone of the small intestinal epithelium. III. Evidence from columnar, enteroendocrine, and mucous cells in the adult mouse. Am. J. Anat 160, 77–91. [DOI] [PubMed] [Google Scholar]
- Bruens L, Ellenbroek SIJ, van Rheenen J, and Snippert HJ (2017). In Vivo Imaging Reveals Existence of Crypt Fission and Fusion in Adult Mouse Intestine. Gastroenterology 153, 674–677.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buske P, Przybilla J, Loeffler M, Sachs N, Sato T, Clevers H, and Galle J (2012). On the biomechanics of stem cell niche formation in the gut–modelling growing organoids. FEBS J. 279, 3475–3487. [DOI] [PubMed] [Google Scholar]
- Chan CJ, Costanzo M, Ruiz-Herrero T, Mönke G, Petrie RJ, Bergert M, Diz-Muñoz A, Mahadevan L, and Hiiragi T (2019). Hydraulic control of mammalian embryo size and cell fate. Nature 571, 112–116. [DOI] [PubMed] [Google Scholar]
- Choi J, Rakhilin N, Gadamsetty P, Joe DJ, Tabrizian T, Lipkin SM, Huffman DM, Shen X, and Nishimura N (2018). Intestinal crypts recover rapidly from focal damage with coordinated motion of stem cells that is impaired by aging. Sci. Rep 8, 10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H (2013). The intestinal crypt, a prototype stem cell compartment. Cell 154, 274–284. [DOI] [PubMed] [Google Scholar]
- Collins J 3rd, Vicente Y, Georgeson K, and Kelly D (1996). Partial intestinal obstruction induces substantial mucosal proliferation in the pig. J. Pediatr. Surg 31, 415–419. [DOI] [PubMed] [Google Scholar]
- Cummins AG, Catto-Smith AG, Cameron DJ, Couper RT, Davidson GP, Day AS, Hammond PD, Moore DJ, and Thompson FM (2008). Crypt fission peaks early during infancy and crypt hyperplasia broadly peaks during infancy and childhood in the small intestine of humans. J. Pediatr. Gastroenterol. Nutr 47, 153–157. [DOI] [PubMed] [Google Scholar]
- Dabov K, Foi A, Katkovnik V, and Egiazarian K (2007). Image denoising by sparse 3-D transform-domain collaborative filtering. IEEE Trans. Image Process. 16, 2080–2095. [DOI] [PubMed] [Google Scholar]
- Datlinger P, Rendeiro AF, Boenke T, Krausgruber T, Barreca D, and Bock C (2019). Ultra-high throughput single-cell RNA sequencing by combinatorial fluidic indexing. bioRxiv. 10.1101/2019.12.17.879304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, and Brugge JS (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268. [DOI] [PubMed] [Google Scholar]
- Dekaney CM, Fong JJ, Rigby RJ, Lund PK, Henning SJ, and Helmrath MA (2007). Expansion of intestinal stem cells associated with long-term adaptation following ileocecal resection in mice. Am. J. Physiol. Gastrointest. Liver Physiol 293, G1013–G1022. [DOI] [PubMed] [Google Scholar]
- Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssens HM, de Winter-de Groot KM, Brandsma AM, de Jong NW, Bijvelds MJ, Scholte BJ, et al. (2013). A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med 19, 939–945. [DOI] [PubMed] [Google Scholar]
- Donati G, and Watt FM (2015). Stem cell heterogeneity and plasticity in epithelia. Cell Stem Cell 16, 465–476. [DOI] [PubMed] [Google Scholar]
- Drasdo D (2000). Buckling instabilities of one-layered growing tissues. Phys. Rev. Lett 84, 4244–4247. [DOI] [PubMed] [Google Scholar]
- Dunn SJ, Appleton PL, Nelson SA, Näthke IS, Gavaghan DJ, and Osborne JM (2012). A two-dimensional model of the colonic crypt accounting for the role of the basement membrane and pericryptal fibroblast sheath. PLoS Comput. Biol 8, e1002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien CB, Morcos PA, and Rosenblatt J (2012). Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 484, 546–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald AJ (2013). Practical considerations for long-term time-lapse imaging of epithelial morphogenesis in three-dimensional organotypic cultures. Cold Spring Harb. Protoc 2013, 100–117. [DOI] [PubMed] [Google Scholar]
- Farin HF, Van Es JH, and Clevers H (2012). Redundant sources of Wnt regulate intestinal stem cells and promote formation of Paneth cells. Gastroenterology 143, 1518–1529.e7. [DOI] [PubMed] [Google Scholar]
- Farin HF, Jordens I, Mosa MH, Basak O, Korving J, Tauriello DV, de Punder K, Angers S, Peters PJ, Maurice MM, and Clevers H (2016). Visualization of a short-range Wnt gradient in the intestinal stem-cell niche. Nature 530, 340–343. [DOI] [PubMed] [Google Scholar]
- Fordham RP, Yui S, Hannan NR, Soendergaard C, Madgwick A, Schweiger PJ, Nielsen OH, Vallier L, Pedersen RA, Nakamura T, et al. (2013). Transplantation of expanded fetal intestinal progenitors contributes to colon regeneration after injury. Cell Stem Cell 13, 734–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewska M, and McNally S (2017). Using 3D Culture of Primary Mammary Epithelial Cells to Define Molecular Entities Required for Acinus Formation: Analyzing MAP Kinase Phosphatases. Methods Mol. Biol 1501, 199–216. [DOI] [PubMed] [Google Scholar]
- Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, and Wrana JL (2015). Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 526, 715–718. [DOI] [PubMed] [Google Scholar]
- Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y, et al. (2017). A single-cell survey of the small intestinal epithelium. Nature 551, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannezo E, Prost J, and Joanny JF (2011). Instabilities of monolayered epithelia: shape and structure of villi and crypts. Phys. Rev. Lett 107, 078104. [DOI] [PubMed] [Google Scholar]
- Harvey RW (1908). Variations in the wall of the large intestine and in the number and staining properties of the goblet cells. Anat. Rec 2, 129–142. [Google Scholar]
- He L, Si G, Huang J, Samuel ADT, and Perrimon N (2018). Mechanical regulation of stem-cell differentiation by the stretch-activated Piezo channel. Nature 555, 103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hild M, and Jaffe AB (2016). Production of 3-D Airway Organoids From Primary Human Airway Basal Cells and Their Use in High-Throughput Screening. Curr. Protoc. Stem Cell Biol 37, IE.9.1–IE.9.15. [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Kaji N, Durgan J, and Hall A (2008). Cdc42 controls spindle orientation to position the apical surface during epithelial morphogenesis. J. Cell Biol 183, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson FP (1913). The effects of distention of the intestine upon the shape of villi and glands. Am. J. Anat 14, 235–250. [Google Scholar]
- Kiela PR, and Ghishan FK (2009). Ion transport in the intestine. Curr. Opin. Gastroenterol 25, 87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivsky PL, Redner S, and Ben-Naim E (2010). A kinetic view of statistical physics (Cambridge University Press; ). [Google Scholar]
- Langlands AJ, Almet AA, Appleton PL, Newton IP, Osborne JM, and Näthke IS (2016). Paneth Cell-Rich Regions Separated by a Cluster of Lgr5+ Cells Initiate Crypt Fission in the Intestinal Stem Cell Niche. PLoS Biol. 14, e1002491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblond CP (1981). The life history of cells in renewing systems. Am. J. Anat 160, 114–158. [DOI] [PubMed] [Google Scholar]
- Lee GY, Kenny PA, Lee EH, and Bissell MJ (2007). Three-dimensional culture models of normal and malignant breast epithelial cells. Nat. Methods 4, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Garcia C, Klein AM, Simons BD, and Winton DJ (2010). Intestinal stem cell replacement follows a pattern of neutral drift. Science 330, 822–825. [DOI] [PubMed] [Google Scholar]
- Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, and Verkman AS (2002). Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest 110, 1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkitalo M, and Foi A (2011). A closed-form approximation of the exact unbiased inverse of the Anscombe variance-stabilizing transformation. IEEE Trans. Image Process 20, 2697–2698. [DOI] [PubMed] [Google Scholar]
- McInnes L, Healy J, Saul N, and Großberger L (2018). UMAP: Uniform Manifold Approximation and Projection. J. Open Source Softw 3, 861. [Google Scholar]
- Meran L, Baulies A, and Li VSW (2017). Intestinal Stem Cell Niche: The Extracellular Matrix and Cellular Components. Stem Cells Int. 2017, 7970385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merker SR, Weitz J, and Stange DE (2016). Gastrointestinal organoids: How they gut it out. Dev. Biol 420, 239–250. [DOI] [PubMed] [Google Scholar]
- Mishchenko Y (2015). A fast algorithm for computation of discrete Euclidean distance transform in three or more dimensions on vector processing architectures. Signal Image Video Process. 9, 19–27. [Google Scholar]
- Miura S, and Suzuki A (2017). Generation of Mouse and Human Organoid-Forming Intestinal Progenitor Cells by Direct Lineage Reprogramming. Cell Stem Cell 21, 456–471.e5. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, and Stappenbeck TS (2013). In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc 8, 2471–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosaliganti KR, Swinburne IA, Chan CU, Obholzer ND, Green AA, Tanksale S, Mahadevan L, and Megason SG (2019). Size control of the inner ear via hydraulic feedback. eLife 8, e39596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroue R, and Bissell MJ (2013). Three-dimensional cultures of mouse mammary epithelial cells. Methods Mol. Biol. 945, 221–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustata RC, Vasile G, Fernandez-Vallone V, Strollo S, Lefort A, Libert F, Monteyne D, Pérez-Morga D, Vassart G, and Garcia MI (2013). Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep. 5, 421–432. [DOI] [PubMed] [Google Scholar]
- Nelson MR, King JR, and Jensen OE (2013). Buckling of a growing tissue and the emergence of two-dimensional patterns. Math. Biosci 246, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse YM, Savage AK, Marangoni P, Rosendahl-Huber AKM, Landman TA, de Sauvage FJ, Locksley RM, and Klein OD (2018). Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature 559, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin C, Parker A, Gunning AP, Ohta Y, Johnson IT, Carding SR, and Sato T (2015). An individual based computational model of intestinal crypt fission and its application to predicting unrestrictive growth of the intestinal epithelium. Integr. Biol 7, 213–228. [DOI] [PubMed] [Google Scholar]
- Polański K, Young MD, Miao Z, Meyer KB, Teichmann SA, and Park JE (2020). BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quyn AJ, Appleton PL, Carey FA, Steele RJ, Barker N, Clevers H, Ridgway RA, Sansom OJ, and Näthke IS (2010). Spindle orientation bias in gut epithelial stem cell compartments is lost in precancerous tissue. Cell Stem Cell 6, 175–181. [DOI] [PubMed] [Google Scholar]
- Ross ST, Allen JR, and Davidson MW (2014). Practical considerations of objective lenses for application in cell biology. Methods Cell Biol. 123, 19–34. [DOI] [PubMed] [Google Scholar]
- Ruiz-Herrero T, Alessandri K, Gurchenkov BV, Nassoy P, and Mahadevan L (2017). Organ size control via hydraulically gated oscillations. Development 144, 4422–4427. [DOI] [PubMed] [Google Scholar]
- Rulands S, Lescroart F, Chabab S, Hindley CJ, Prior N, Sznurkowska MK, Huch M, Philpott A, Blanpain C, and Simons BD (2018). Universality of clone dynamics during tissue development. Nat. Phys 14, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarela U, Akram SU, Desgrange A, Rak-Raszewska A, Shan J, Cereghini S, Ronkainen VP, Heikkilä J, Skovorodkin I, and Vainio SJ (2017). Novel fixed z-direction (FiZD) kidney primordia and an organoid culture system for time-lapse confocal imaging. Development 144, 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs N, Papaspyropoulos A, Zomer-Van Ommen DD, Heo I, Böttinger L, Klay D, Weeber F, Huelsz-Prince G, Iakobachvili N, Amatngalim GD, et al. (2019). Long-term expanding human airway organoids for disease modeling 38, e100300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai Y (2013). Cytosystems dynamics in self-organization of tissue architecture. Nature 493, 318–326. [DOI] [PubMed] [Google Scholar]
- Sato T, and Clevers H (2013). Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science 340, 1190–1194. [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, and Clevers H (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265. [DOI] [PubMed] [Google Scholar]
- Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, and Clevers H (2011a). Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 141, 1762–1772. [DOI] [PubMed] [Google Scholar]
- Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, and Clevers H (2011b). Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scadden DT (2006). The stem-cell niche as an entity of action. Nature 441, 1075–1079. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwank G, Andersson-Rolf A, Koo BK, Sasaki N, and Clevers H (2013). Generation of BAC transgenic epithelial organoids. PLoS ONE 8, e76871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweinfest CW, Henderson KW, Suster S, Kondoh N, and Papas TS (1993). Identification of a colon mucosa gene that is down-regulated in colon adenomas and adenocarcinomas. Proc. Natl. Acad. Sci. USA 90, 4166–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweinfest CW, Spyropoulos DD, Henderson KW, Kim JH, Chapman JM, Barone S, Worrell RT, Wang Z, and Soleimani M (2006). slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct up-regulation of ion transporters in the colon. J. Biol. Chem 281, 37962–37971. [DOI] [PubMed] [Google Scholar]
- Serra D, Mayr U, Boni A, Lukonin I, Rempfler M, Challet Meylan L, Stadler MB, Strnad P, Papasaikas P, Vischi D, et al. (2019). Self-organization and symmetry breaking in intestinal organoid development. Nature 569, 66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazi MN, Siggia ED, and Zernicka-Goetz M (2019). Self-organization of stem cells into embryos: A window on early mammalian development. Science 364, 948–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova DM, and Verkhusha VV (2013). Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods 10, 751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyer AE, Huycke TR, Lee C, Mahadevan L, and Tabin CJ (2015). Bending gradients: how the intestinal stem cell gets its home. Cell 161, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons BD, and Clevers H (2011). Strategies for homeostatic stem cell self-renewal in adult tissues. Cell 145, 851–862. [DOI] [PubMed] [Google Scholar]
- Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, Klein AM, van Rheenen J, Simons BD, and Clevers H (2010). Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 143, 134–144. [DOI] [PubMed] [Google Scholar]
- Snover DC, Jass JR, Fenoglio-Preiser C, and Batts KP (2005). Serrated polyps of the large intestine: a morphologic and molecular review of an evolving concept. Am. J. Clin. Pathol 124, 380–391. [DOI] [PubMed] [Google Scholar]
- Spit M, Koo BK, and Maurice MM (2018). Tales from the crypt: intestinal niche signals in tissue renewal, plasticity and cancer. Open Biol. 8, 180120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Nystul T, Lighthouse D, Morris L, Fox D, Cox R, Tootle T, Frederick R, and Skora A (2008). Stem cells and their niches: integrated units that maintain Drosophila tissues. Cold Spring Harb. Symp. Quant. Biol 73, 49–57. [DOI] [PubMed] [Google Scholar]
- Sumigray KD, Terwilliger M, and Lechler T (2018). Morphogenesis and Compartmentalization of the Intestinal Crypt. Dev. Cell 45, 183–197.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syeda R, Xu J, Dubin AE, Coste B, Mathur J, Huynh T, Matzen J, Lao J, Tully DC, Engels IH, et al. (2015). Chemical activation of the mechanotransduction channel Piezo1. eLife 4, e07369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalheim T, Quaas M, Herberg M, Braumann UD, Kerner C, Loeffler M, Aust G, and Galle J (2018). Linking stem cell function and growth pattern of intestinal organoids. Dev. Biol 433, 254–261. [DOI] [PubMed] [Google Scholar]
- Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, and de Sauvage FJ (2011). A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 478, 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirouvanziam R, Makam M, and Péault B (2009). Lung. In Human Adult Stem Cells, Masters JR and Palsson BØ, eds. (Springer; ), pp. 91–112. [Google Scholar]
- Trier JS, and Moxey PC (1979). Morphogenesis of the small intestine during fetal development. Ciba Found. Symp 3–29. [DOI] [PubMed] [Google Scholar]
- Vijftigschild LAW, Berkers G, Dekkers JF, Zomer-Van Ommen DD, Matthes E, Kruisselbrink E, Vonk A, Hensen CE, Heida-Michel S, Geerdink M, et al. (2016). b2-Adrenergic receptor agonists activate CFTR in intestinal organoids and subjects with cystic fibrosis. Eur. Respir. J 48, 768–769. [DOI] [PubMed] [Google Scholar]
- Weinreb C, Wolock S, and Klein AM (2018). SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 34, 1246–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, and Klein AM (2019). Scrublet: Computational identification of cell doublets in single-cell transcriptomic data. Cell Syst. 8, 281–291.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yui S, Azzolin L, Maimets M, Pedersen MT, Fordham RP, Hansen SL, Larsen HL, Guiu J, Alves MRP, Rundsten CF, et al. (2018). YAP/TAZ-Dependent Reprogramming of Colonic Epithelium Links ECM Remodeling to Tissue Regeneration. Cell Stem Cell 22, 35–49.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilionis R, Nainys J, Veres A, Savova V, Zemmour D, Klein AM, and Mazutis L (2017). Single-cell barcoding and sequencing using droplet microfluidics. Nat. Protoc 12, 44–73. [DOI] [PubMed] [Google Scholar]
- Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM, et al. (2019). Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single cell RNA-sequencing data generated during this study are publicly available at the Gene Expression Omnibus (GEO: GSE164638). Jupyter notebooks and inDrops.py pipeline scripts used for single cell RNA-seq analysis are publicly available on Github: https://github.com/AllonKleinLab/paper-data/tree/master/Tallapragada_Cambra_2021. SPRING plots of datasets as processed and annotated in this study are available; see Key resources table for publicly accessible links to each SPRING plot. Imaging data and MATLAB analysis scripts are available from the corresponding author upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-Annexin A1 | Abcam | Cat# ab214486 |
| anti-BASP1 | Abcam | Cat# ab214322 |
| anti-Chromogranin A | Abcam | Cat# ab15160; RRID: AB_301704 |
| anti-CLCA1 | Abcam | Cat# ab180851; RRID: AB_2722611 |
| anti-Lysozyme | Agilent | Cat# F037201–1; RRID: AB_578661 |
| anti-Rabbit IgG (secondary, 555) | Thermo Fisher Scientific | Cat# A21428; RRID: AB_2535849 |
| anti-Rabbit IgG (secondary, 647) | Life Technologies | Cat# A21245; RRID: AB_2535813 |
| anti-Villin | Santa Cruz Biotechnology | Cat# sc-58897-AF647; RRID: AB_2304475 |
| Biological Samples | ||
| Intestinal organoids from Mouse: Lgr5-DTR-EGFP | This paper | N/A |
| Intestinal organoids from Mouse: Lgr5-DTR-EGFP/Villin-H2B-mCherry | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| B27 | Thermo Fisher Scientific | Cat# 17504–044 |
| Cell Recovery Solution | Corning | Cat# 354253 |
| CFTRinh-172 | Selleck Chemicals | Cat# S7139 |
| Complete crypt culture medium containing EGF, Noggin, and R-Spondin-1 (ENR) | R&D Systems Protocols | https://resources.rndsystems.com/images/site/dw_r-spondinmediumprotocol_34749-web.pdf?v=1 |
| Complete crypt culture medium containing Wnt3a, EGF, Noggin, and R-Spondin-1 (WENR) | Miyoshi and Stappenbeck, 2013 | N/A |
| Diethylaminoethyl (DEAE)-Dextran | Millipore Sigma | SKU- D9885 |
| Forskolin | Selleck Chemicals | Cat# S2449 |
| GdCl3 | Millipore Sigma | SKU- G7532 |
| Glutamax | Thermo Fisher Scientific | Cat# 35050–079 |
| GsMTx4 | Abcam | Cat# ab141871 |
| Hanks’ Buffer with 20 mM Hepes | AAT Bioquest | Cat# 20011 |
| Matrigel, growth factor reduced, phenol red-free | Corning | Cat# 356231 |
| N-acetyl cysteine 500 mM stock solution | Millipore Sigma | SKU- A7250 |
| N2 | Thermo Fisher Scientific | Cat# 17502–048 |
| Noggin | Millipore Sigma | SKU- SRP3227 |
| Ouabain | Tocris | Cat# 1076 |
| Pluronic® F-127 | AAT Bioquest | Cat# 20050 |
| Poly(2-hydroxyethyl methacrylate) | Millipore Sigma | SKU- P3932 |
| Primocin | Invivogen | Cat# ant-pm-1 |
| Probenecid | AAT Bioquest | Cat# 20060 |
| Puromycin dihydrochloride | Thermo Fisher Scientific | Cat# A1113803 |
| Recombinant mouse EGF | Thermo Fisher Scientific | Cat# PMG8041 |
| Yoda1 | Tocris | Cat# 5586 |
| Critical Commercial Assays | ||
| Calcium indicator kit | AAT Bioquest | Cat# 20530 |
| EdU staining kit (AlexaFluor647) | Thermo Fisher Scientific | Cat# C10640 |
| NEBNext Ultra II FS DNA Library Prep kit | NEB | Cat# E7805S |
| Deposited Data | ||
| In vivo reference data for Figure 1D | Haber et al., 2017 | GEO: GSE92332 |
| Mouse genome build 38 (GRCm38) | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/ |
| Raw and analyzed single cell RNA-sequencing data | This paper | GEO: GSE164368 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| Mouse: 293T-HA-RspoI-Fc | Trevigen | Cat# 3710–001-01; RRID: CVCL_RU08 |
| Mouse: L-WRN | ATCC | Cat# CRL-3276; RRID: CVCL_DA06 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6;129S-Gt(ROSA)26Sortm1.1Ksvo/J | Jackson Laboratories | Cat# 023139; RRID:IMSR_JAX:023139 |
| Mouse: B6.Cg-Tg(Vil1-cre)997Gum/J | Jackson Laboratories | Cat# 004586; RRID:IMSR_JAX:004586 |
| Mouse: Lgr5-DTR-EGFP | Tian et al., 2011 | N/A |
| Mouse: Lgr5-DTR-EGFP/Villin-H2B-mCherry | This paper | N/A |
| Oligonucleotides | ||
| RT primers on hydrogel beads: 5’CGATTGATCAACGTAATACGACTCACTATAGGGTGTCGGGTGCAG[bc1,8nt]GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG[bc2,8nt]NNNNNNTTTTTTTTTTTTTTTTTTTV- 3’ | This paper; HMS Single Cell Core | N/A |
| TSO primer: 5’-AAGCAGTGGTATCAACGCAGAGTACATrGrGrG-3’ | Datlinger et al., 2019 | N/A |
| Fwd cDNA primer: 5’- CACTATAGGGTGTCGGGTGCAG-3’ | This paper; HMS Single Cell Core | N/A |
| Rev cDNA primer: 5’-AAGCAGTGGTATCAACGCAGAGT-3’ | This paper; HMS Single Cell Core | N/A |
| Ligation Fwd primer: 5’-/5Phos/GATCGGAAGAGCACACGTCTGAACTCCAGTCAC/3ddC | This paper; HMS Single Cell Core | N/A |
| Ligation Rev primer: 5’- /5AmMC6/GCTCTTCCGATCT -3’ | This paper; HMS Single Cell Core | N/A |
| Indexing primer PCR_p7_r2: 5’-CAAGCAGAAGACGGCATACGAGATGGGTGTCGGGTGCAG-3’ | This paper, HMS Single Cell Core | N/A |
| Indexing primer PCR_p5_r1_ixx: 5’-AATGATACGGCGACCACCGAGATCTACAC[i5]TCGTCGGCAGCGTC-3’ | This paper; HMS Single Cell Core | N/A |
| Recombinant DNA | ||
| Plasmid: pLVX-H2B-iRFP670 | This paper; Kyogo Kawaguchi | N/A |
| Software and Algorithms | ||
| 3D Distance Transform algorithm | Mishchenko, 2015 | https://link.springer.com/article/10.1007%2Fs11760-012-0419-9 |
| Exact unbiased inverse of the Anscombe variance-stabilizing transformation – MATLAB implementation | Makitalo and Foi, 2011 | https://ieeexplore.ieee.org/document/5721817 |
| Image Denoising by Sparse 3-D Transform-Domain Collaborative Filtering – MATLAB implementation | Dabov et al., 2007 | https://ieeexplore.ieee.org/document/4271520 |
| Fiji | Schindelin, J., Arganda-Carreras, I. & Frise, E. et al., 2012 | https://imagej.net/Fiji |
| Imaris 8.4 | Oxford Instruments | https://imaris.oxinst.com/support/imaris-release-notes/8-4-0 |
| inDrops pipeline package | Klein et al., 2015; Github | https://github.com/indrops/indrops |
| Scanpy – Single Cell Analysis in Python | Wolf et al., 2018 | https://scanpy.readthedocs.io/en/stable/ |
| Scrublet – Single Cell Removal of Doublets | Wolock et al., 2019 | https://github.com/AllonKleinLab/scrublet |
| Other | ||
| Interactive visualization of scRNA-seq data with SPRING - Sandwich and dome data (without stretch signature annotation) | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/SPRING_private/mouse_intestinal_organoids_21/murine_intestinal_organoid_sandwich_dome_culture_merged_no_stretch_signature/all_cells |
| Interactive visualization of scRNA-seq data with SPRING - Sandwich and dome data (with stretch signature annotation) | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/SPRING_private/mouse_intestinal_organoids_21/murine_intestinal_organoid_sandwich_dome_culture_merged/all_cells |
| Interactive visualization of scRNA-seq data with SPRING - Perturbation data | This paper | https://kleintools.hms.harvard.edu/tools/springViewer_1_6_dev.html?datasets/SPRING_private/mouse_intestinal_organoids_21/murine_intestinal_organoid_inflation_perturbation_expt/all_cells |
| Jupyter notebooks for scRNA-seq data analysis | This paper | https://github.com/AllonKleinLab/paper-data/tree/master/Tallapragada_Cambra_2021 |