

Abstract
Introduction
Metastatic prostate cancer is a heterogeneous disease characterized by clinical and genomic heterogeneity. Many prostate cancers harbor mutations causing DNA repair deficiency, specifically homologous recombination deficiency, sensitizing to drugs that inhibit poly ADP-ribose polymerase (PARP). PARP is an enzyme that is involved in single-stranded DNA repair and is the target of newly approved treatments for metastatic prostate cancer.
Areas Covered
Here, the authors’ review the clinical trials leading to the recent approvals of two PARP inhibitors (PARPi), olaparib and rucaparib, specifically TOPARP-A, TOPARP-B, PROfound and TRITON-2. They also compare the different FDA approvals for both of these medications and outline the safety of this class of drugs in prostate cancer.
Expert opinion
Because PARPi are particularly effective in men with somatic or germline alterations in BRCA1 and BRCA2, we recommend that all men be tested for DNA alterations with next-generation sequencing in tumor cells obtained from either tissue or blood. We also recommend that olaparib or rucaparib be considered relatively early in the treatment sequence in metastatic castration-resistant prostate cancer patients with BRCA1 or BRCA2 mutations. Other DNA alterations might also sensitize to PARPi though the response rates are lower, so other standard therapies should be prioritized first.
Keywords: Olaparib, rucaparib, metastatic castration-resistant prostate cancer, PARP inhibitor
1. Introduction
Metastatic castration-resistant prostate cancer (mCRPC) is still a significant health concern for many men today despite recent advances in treatment, and remains an incurable disease with a relatively poor prognosis. Based on recent phase 3 clinical trials, the median overall survival (OS) for men with mCRPC is estimated at only 12 to 33 months depending on the specific biology of the cancer, the treatment sequence used and the number of prior therapies used to treat their disease.[1–4] mCRPC appears to be nearly universally fatal for these men and new therapies are needed to improve the prognosis in this disease. Many therapies have been approved in the recent decades including novel hormone therapies targeting the androgen receptor, chemotherapies, cellular immune therapy, and systemic radiopharmaceuticals with radium-223 being the last FDA approval in 2013. However, despite having eight approved therapies for castration-resistant disease, drug resistance is shared between many of these therapies and limits the clinical benefit.[5,6] The recent approval of rucaparib and olaparib, both in May 2020, represent much needed progress in the treatment of mCRPC in part because these therapies represent the introduction of a novel therapeutic class targeting a specific molecular subtype of prostate cancer. This article will review the trials related to the approval of both of these medications in treating DNA repair-deficient prostate cancer.
2. Targeting DNA repair
DNA damage repair involves multiple proteins with specific functions including: recognition of DNA damage, excision of altered nucleotides, re-insertion of normal nucleotides, control/arresting cell cycle until the DNA is repaired, and annealing strand breaks among other repair functions.[7] DNA damage include single-strand breaks, double strand breaks, crosslinks, DNA adduct formation and mispaired nucleotides. Each DNA damage repair gene has a specific function in one of these repair processes such as base-excision repair, nucleotide-excision repair, non-homologous end joining, mismatch repair and homologous recombination. For example, ATM is involved in arresting cell-cycle progression to allow for the DNA repair process to complete prior to DNA replication. Other genes, such as BRCA1, and BRCA2 are involved in the error-free DNA double-strand repair pathway, homologous recombination repair (HRR). When this repair mechanism is inactivated, for instance with homozygous deletion of BRCA2, then alternate compensatory pathways are involved in the repair of DNA that can lead to introduction of errors in the DNA at a higher rate than HRR. Separate mechanisms exist to repair single-strand DNA breaks. If single-strand breaks are not repaired, these errors in the DNA can progress to double-strand breaks causing catastrophic accumulation of DNA damage leading to cell death. Poly ADP-ribose polymerase (PARP) is an enzyme involved in the repair of DNA single-strand strand breaks. Pre-clinical models with inactivated double-strand repair pathways have shown that PARP inhibitors (PARPi) lead to durable cancer control when the HRR pathway is crippled.[8,9] PARPi are selective for HRR-deficient cancers so are not active in patients with DNA alterations in other DNA repair pathways such as mismatch repair (although these patients may be more sensitive to immunotherapy approaches such as PD-1 inhibition).
3. Recent Clinical Trial Results
3.1. Olaparib
Mateo and colleagues published the first clinical trial of a PARPi in prostate cancer in 2015 with the TOPARP-A study.[10] This was an open-label, single-arm study with a non-standard definition of objective response rate (ORR) as the primary endpoint. The ORR was defined as a composite of either RECIST defined objective response, reduction in PSA level of at least 50% or conversion of circulating tumor cells (CTC) from 5 or more per 7.5mL blood to less than 5/7.5mL blood. Patients were treated with olaparib 400mg twice daily. Patients were eligible if they had progressed after at least one chemotherapy regimen for mCRPC, and no prior PARPi or platinum exposure. According to the design of this study, it would either lead to a second phase (TOPARP-B) with a biomarker-unselected population or a biomarker-selected population.
Overall they enrolled 49 patients. Median age was 67.5 years (range 40.8–79.3). 23% of men had metastatic disease at diagnosis. Median PSA at study entry was 349.5ng/mL (interquartile range 153 – 806ng/mL). 80% of patient had received at least 4 prior lines of therapy so this was a heavily pre-treated population. Paired samples from tumor biopsies performed prior to initiation of olaparib and on-treatment samples were available for HRR gene testing. Patients were defined as biomarker-positive for DNA repair defects if there was homozygous deletion or a deleterious mutation present to at least one of the following genes reported to be involved with DNA damage repair or known sensitivity to PARP inhibition: BRCA1, BRCA2, ATM, FANCA, CHEK2, PALB2, HDAC2, RAD51, MLH3, ERCC3, MRE11 or NBN. Sixteen patients had alterations in DNA repair genes with BRCA2 being the most frequently observed, found in seven patients. Other DNA repair gene alterations included ATM (5 patients), FANCA (3 patients), CHEK2 (2 patients), BRCA1 (1 patient), PALB2 (1 patient), MRE11 (1 patient) and NBN (1 patient).
The median OS was 10.1 months with a median follow up of 14.4 months. Sixteen patients had an objective response. 14 of 16 biomarker-positive patients (ORR 87.5%) and 2 of 33 biomarker-negative patients (ORR 6%). In total, 11 of 49 patients had at least a 50% reduction in PSA. No complete responses (CR) were seen, but six partial responses (PR) were observed. Median radiographic progression free survival (rPFS) was superior for the biomarker positive group, 9.8 months vs 2.7 months (P < 0.001), as was OS, 13.8 months vs 7.5 months (P = 0.05). The most frequently observed grade-3 or −4 adverse events were anemia (20%), fatigue (12%), thrombocytopenia (6%) and neutropenia (4%). Thirteen patients (20%) required a dose reduction.
Because the efficacy of olaparib in the overall unselected population was not sufficient, the TOPARP-B trial was conducted in biomarker-positive patients using a two-dose pick-the-winner design where each dose cohort was independently assessed for the primary endpoint.[11] This was an open-label randomized phase 2 clinical trial. Eligible patients with mCRPC must have had a known pathogenic mutation or homozygous deletion in a HRR gene (BRCA1, BRCA2, ATM, CDK12, PALB2, ARID1A, ATRX, CHEK1, CHEK2, FANCA, FANCF, FANCG, FANCI, FANCM, NBN, RAD50 and WRN). Progression on at least one chemotherapy regimen was required. Patients could not have previously been exposed to platinum chemotherapy, PARPi, cyclophosphamide or mitoxantrone. Patients were randomized 1:1 to olaparib 300mg or 400mg twice daily. The primary endpoint was ORR, as defined identically in the TOPARP-A trial. The primary purpose of this trial was to validate the predictive gene panel for PARPi. If both cohorts were determined to be successful as assessed by the primary endpoint then the DNA damage gene panel would be considered validated in predicting response to PARPi.
In total 98 patients were randomized, with 46 evaluable in each arm. The baseline characteristics were similar between the two dose cohorts including median age at study entry (67.3 years 300mg cohort; 67.6 years 400mg cohort), metastatic disease at diagnosis (49% 300mg dose; 51% 400mg dose), presence of liver metastasis (22% 300mg cohort; 24% 400mg cohort), median PSA at entry (151.5ng/mL 300mg cohort; 158.0ng/mL 400mg cohort) and CTC count ≥5/7.5mL blood (63% 300mg cohort; 65% 400mg cohort).
Composite overall response was observed in 18 patients (39.1%) and 25 patients (54.3%) in the 300mg and 400mg cohorts, respectively. RECIST-defined ORR was observed in 16.2% and 24.2% of patients, respectively. Overall this suggested a dose-response relation with olaparib in mCRPC with the 400mg cohort meeting the primary endpoint but not the 300mg cohort. One explanation for this difference in efficacy might be the lower dose of olaparib. Alternatively an imbalance between different DNA repair genes could have impacted the results. There were differences in response rates observed in patients between each of the specific DNA repair alterations. For instance, patients with BRCA1 or BRCA2 alterations had the highest response rate of 83.3% (25/30) compared to patients with an ATM alteration having a response rate of only 36.8% (7/19). The adverse event rate and frequency of drug modifications (dose decrease and/or discontinuation) was higher in the 400mg cohort compared with the 300mg cohort.
PROfound was a phase 3 clinical trial confirming the clinical activity of olaparib in mCRPC.[2] In this clinical trial, patients with mCRPC and pathogenic DNA alterations in select HRR genes who had progressed after treatment with either enzalutamide or abiraterone were randomized to olaparib 300mg twice daily or physicians choice of either enzalutamide (160mg daily) or abiraterone (1000mg daily). At the time that PROfound was designed, using a control arm consisting of the alternative AR-targeting agent was deemed appropriate, as this trial was designed before the publication of the CARD trial suggesting that sequential AR-directed therapy may not be the optimal sequencing approach in mCRPC. Two prespecified cohorts were analyzed for each treatment arm: cohort A included patients with DNA alterations in BRCA1, BRCA2 and ATM. Cohort B included alterations in BRIP1, BARD1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D and RAD54L. Both germline and somatic alterations were allowed. The primary endpoint was rPFS in cohort A. Key secondary endpoints included rPFS in the overall population, ORR and OS. DNA alterations were identified based on either fresh or archival tissue biopsy. DNA alterations were detected in 778 patients out of 2792 patients screened (28%), although this might represent an overestimate of the true prevalence. In cohort A, 162 and 83 patients were randomized to olaparib and control respectively. In cohort B, 256 and 131 patients were randomized to olaparib and control respectively. BRCA2 was the most common DNA-repair alteration detected in both cohorts (255/632; 40%), followed by ATM (170/632; 27%). 65% of patients had progressed on at least one cycle of taxane-based chemotherapy (412/632), although this was not required for enrolment.
The median rPFS for cohort A strongly favored olaparib over control, 7.4 months vs 3.6 months (HR 0.34, 95% CI 0.25–0.47; P<0.001).[2] rPFS was also improved with olaparib over control in the overall population (cohort A + cohort B), though the magnitude of benefit was somewhat attenuated, 5.8mo vs 3.5mo (HR 0.49, 95% CI 0.38–0.63; P<0.001). A subsequent publication with longer follow-up reported on mature OS data.[12] The OS significantly improved with olaparib compared to control in cohort A, 19.1mo vs 14.7 mo (HR 0.69, 95% CI 0.50–0.97; P=0.02).[9] The protocol allowed for cross-over at the time of disease progression in patents enrolled on the control arm. 67% (56/83) of patients crossed over to olaparib. The OS difference remain significant when adjusting for this effect (HR 0.42, 95% CI 0.19–0.91). The subgroup analysis suggest that all patients benefited with olaparib over control regardless of clinical features with the potential exception of patients with selected DNA alterations (e.g. outcomes appeared worse for men with PPP2R2A mutations). The OS analysis also specifically evaluated cohort B separately (i.e. not combined with cohort A). There was no improvement in OS in this cohort between olaparib and control with a median OS of 14.1mo vs 11.5mo (HR 0.96, 95% CI 0.63–1.49). In cohort B, 30 of 48 patients crossed over to olaparib. When adjusting for cross-over, the difference was even less significant (HR 0.83, 95% CI 0.11–5.98). This suggests that the OS benefit from olaparib is driven by patients harboring BRCA1/2 and ATM mutations, and not all HRR gene mutations. Further, in an exploratory gene-by-gene analysis of OS, the potential survival advantage conferred in those with ATM mutations was less clear than in those with BRCA1/2 mutations.
On the basis of the PROfound study, the FDA approved olaparib for the treatment of HRR-mutated mCRPC patients (germline or somatic mutations in BRCA1/2, ATM, BRIP1, BARD1, CDK12, CHEK1, CHEK2, FANCL, PALB2, RAD51B, RAD51C, RAD51D or RAD54L) who previously received treatment with a novel AR-directed agent with or without prior taxane treatment.
3.2. Rucaparib
Rucaparib received accelerated approval from the FDA based on the TRITON2 clinical trial.[13] This was an open-label, single-arm phase 2 clinical trial. Patients with mCRPC and alterations in HRR genes who had progressed on a next-generation androgen receptor therapy (e.g. abiraterone, enzalutamide) and taxane-based chemotherapy were eligible to enroll. Patients were treated with rucaparib 600mg twice daily. Somatic or germline alterations were allowed. Patients must have pathogenic DNA alterations in either BRCA1, BRCA2, ATM, BARD1, BRIP1, CDK12, CHEK2, FANCA, NBN, PALB2, RAD51, RAD51B, RAD51C, RAD51D, or RAD54L. The primary endpoint was ORR in the RECIST-measurable patients and PSA response rate (50% or greater reduction) in the overall population including patients with RECIST non-measurable disease.
In total 115 patients were enrolled that had BRCA1 or BRCA2 alterations. 62 patients had measurable disease as defined by central radiology review. 43% of patients had a confirmed objective response, 11% complete response (7/62) and 32% partial response (28/62). Clinical benefit rate was observed in 89% of patients. In the overall population, PSA response rates were observed in 55% of patients (63/115). No difference in response rates were observed between BRCA1 and BRCA2 patients (33% vs 45%), but PSA responses were statistically worse in BRCA1 versus BRCA2 patients (15% vs 60%). The median rPFS for the entire BRCA1/2 population was 9.0mo (95% CI 8.3–13.5). The response rate appears to be equivalent regardless of the number of prior lines of therapy and presence or absence of hepatic metastasis or other clinical factors. A subsequent meta-analysis of all published PARP inhibitor trials in prostate cancer suggested diminished efficacy of PARPi in BRCA1- versus BRCA2-mutated mCRPC patients,[14] although these results require validation.
Based on the TRITON2 data, the FDA granted accelerated approval to rucaparib for BRCA1/2-mutated mCRPC, in patients who have previously received an AR-directed therapy and a taxane chemotherapy. Full FDA approval of rucaparib in mCRPC is contingent upon a positive phase 3 trial, TRITON3, which is a randomized study of rucaparib vs. physician’s choice of systemic therapy (abiraterone, enzalutamide or docetaxel) in patients with BRCA1/2 or ATM mutations.
The TRITON2 authors separately reported on the clinical outcomes for those patients with non-BRCA alterations (Table 1).[15] 78 patients were enrolled including those with RECIST-measurable and non-measurable disease. 49 patients total had ATM alterations (62%), 15 had CDK12 alterations (19%), 12 had CHEK2 (15%) and 14 had other alterations (18%). The responses varied by the specific DNA alteration but were consistently lower in non-BRCA mutations compared to BRCA1/2 alterations. (Table 1). The diminished efficacy of PARP inhibitors in mCRPC patients with ATM and CDK12 mutations has also been suggested by other retrospective series.[16,17]
Table 1:
Response Assessment in Non-BRCA alterations in TRITON2
| Cohort | Measurable Disease, N | Total, N | ORR (%) | Confirmed PSA Response Rate (%) | 12 Month Clinical Benefit Rate, All patients % (N) |
|---|---|---|---|---|---|
| ATM | 19 | 49 | 2 (10.5%) | 2 (4.1%) | 16.7% (3/18) |
| CDK12 | 10 | 15 | 0 (0%) | 1 (6.7%) | 7.1% (1/14) |
| CHEK2 | 9 | 12 | 1 (11.1%) | 2 (16.7%) | 0% |
| Other genes | 14 | 14 | 4 (28.6%) | 5 (35.7%) | Not reported |
4. Safety of PARP inhibitors
The toxicities reported for olaparib and rucaparib are generally consistent regarding the type and severity of adverse events. (Table 2) The most common toxicities include: asthenia/fatigue, nausea, anorexia, diarrhea and cytopenias. Most patients generally appear to tolerate PARPi therapy regardless of the number of prior lines of therapy, although myelosuppression may be more pronounced in those who have previously received chemotherapy.
Table 2:
Selected Adverse Events for Olaparib and Rucaparib
| Olaparib (N = 256)[2, 9] | Rucaparib (N = 115)[10] | |||
|---|---|---|---|---|
| Adverse Event | All Grades (%) | Grades ≥ 3 (%) | All Grades (%) | Grades ≥ 3 (%) |
| Anemia | 199 (46) | 55 (21) | 50 (43.5) | 29 (25.2) |
| Anorexia | 77 (30) | 3 (1) | 32 (27.8) | 2 (1.7) |
| Asthenia/Fatigue | 104 (41) | 7 (3) | 71 (61.7) | 10 (8.7) |
| Creatinine elevation[8] | 9 (18%) | 0 (0) | 18 (15.7) | 1 (0.9) |
| Diarrhea | 54 (21) | 2 (<1) | 23 (20.0) | 0 |
| Nausea | 106 (41) | 3 (1) | 60 (52.2) | 3 (2.6) |
| Neutropenia | NR | 10 (3.9) | 12 (10.4) | 8 (7.0) |
NR Not reported
Adverse effects with PARPi are frequent and can significantly impact either quality of life or safety. 46% of patients required a dose interruption of olaparib due to adverse events. However, most patients tolerate PARPi therapy with dose modifications including dose reductions and/or dose delays. As reported in the initial publication of PROfound, 18% of patients (46/256) discontinued treatment due to adverse effects. As reported in the second publication of PROfound, 20% (51/256) of patients discontinued treatment due to adverse effects demonstrating most of these toxicities are not cumulative over time allowing most patients to tolerate long-term drug exposure. However, about 10% of patients receiving these drugs may require red blood cell transfusions for treatment-related anemia. Additionally, there appeared to be a signal of excess thromboembolic events (DVT, PE) as well as pulmonary effects (cough, dyspnea and pneumonitis) in the olaparib arm of the PROfound study, a phenomenon that should be carefully monitored moving forward.
With respect to rucaparib, 7.8% of patients (9/115) discontinued this drug due to adverse events. 63.5% of patients (73/115) required at least one dose modification (dose delay or dose decrease) due to an adverse event. In the TRITON2 clinical trial grade 1 or 2 AST/ALT elevated occurred in 33% of patients but resolved over time without dose modifications. Elevation in creatinine was similarly observed.
Finally, a class effect of PARP inhibitors is the potential to induce or accelerate the development of cytopenias with the theoretical risk of bone marrow failure syndromes such as myelodysplastic disease or acute myeloid leukemia. Thus, careful hematological monitoring of patients receiving PARP inhibitors, especially those on long-term treatment, is required. As reported in the PROfound trial, the frequency of these events is low, significantly less than 3%. Due to the rarity of bone marrow failure syndromes, defining the exact risk to patients is difficult and longer follow up might further clarify this risk. It is theoretically possible that a longer duration of treatment with PARPi might increase this risk. MDS and/or AML have not yet been reported in any patient in any of the previously described prostate cancer trials. However these medications are used in other cancers where these adverse events have been reported.[18]
5. Expert Opinion
PARPi are a novel therapeutic class for prostate cancer treatment and represent a new standard of care for selected patients with HRR alterations. Even though the concept of DNA damage response and HRR deficiency are becoming increasingly understood, neither of these terms has an accepted definition in clinical practice. Different gene panels have been used to define HRR populations in prior clinical trials. The subgroup analysis from these studies consistently demonstrates differential responses between the various specific genomic alterations, as evidenced by the responses with BRCA1/2 compared with ATM, CDK12 or CHEK2 alterations.
Importantly, there appear to be no significant differences in response rates between patients with somatic compared with germline alterations based on the available data thus far. Olaparib and rucaparib both appear to be highly effective in biomarker-selected patients (especially those with BRCA1/2 mutations) and generally appear to be tolerable with chronic use. There are some important differences in the FDA approvals for each agent though. Olaparib is approved after at least one line of novel hormone therapy, does not require failure of a prior taxane agents, and can be used in a broader range of HRR alterations beyond BRCA1 and BRCA2. In contrast, rucaparib is approved after progression on both a novel hormone therapy and a taxane-based chemotherapy, and only in patients with BRCA1 or BRCA2 alterations. Thus, we predict that olaparib will be utilized more frequently than rucaparib for mCRPC patients in the real-world setting.
Due to the high response rate in BRCA1/2 patients, we suggest that PARPi therapy be considered early in the mCRPC treatment course. Clinical judgement is required in optimally sequencing treatment with hormone therapy, chemotherapy and PARPi since there are no prospective trials to guide decision making. For instance, in the subgroup analysis in the PROfound trial, patients with prior taxane chemotherapy benefited from olaparib over alternative hormone therapy. In contrast patients without prior taxane use did not clearly benefit from olaparib over alternative hormone therapy use though the number of patients was small limiting interpretation (HR 1.12, 95% CI 0.69–1.85). We do not recommend PARPi therapy prior to treatment with a NHT though. We also recommend that PARPi may be used in patients with other HRR alterations but it should be used after the patient has exhausted other standard of care options due to the lower response rates in these patients (e.g. ATM or CDK12 mutations). It is also possible that mCRPC patients harboring other non-BRCA mutations may benefit more from combinatorial approaches that enhance synthetic lethality. For example, ATM-altered patients may require the combination of a PARP inhibitor with an ATR inhibitor,[16] and CDK12-altered patients may do best when combining a PARP inhibitor with a PD-1 inhibitor.[19]
Finally, use of PARP inhibitors requires that patients be tested for HRR alterations. This can be done with either tissue or liquid biopsies. We agree with the NCCN Guidelines (version 2.2020) and strongly recommend that all patients be tested for both somatic and germline DNA-repair gene alterations as these results can impact patient care. However, sample selection for HRR mutations remains challenging, and clinicians are faced with many options. In our opinion, we would recommend tumor DNA testing using a hierarchical scale: new metastatic biopsy is best, archival tumor sample is second, circulating tumor DNA is third, and germline-only testing is fourth.[20] Using this testing approach, the majority of mCRPC patients will be amenable to somatic testing, and only the minority will be offered germline-only testing (which will miss about half of all HRR gene mutations). Archival tissue can be used before proceeding with a fresh tissue biopsy since many of these gene alterations are early events.[21] However, there is a higher incidence of qualifying HRD alterations in fresh tissue obtained from patients with mCRPC compared with localized prostate cancer.[12–13,22]
5.1. Future Directions
Further refinement of which patients benefit the most from PARPi treatment is still needed. Theoretically, those with bi-allelic mutations would be expected to benefit more from PARPi treatment than those with mono-allelic mutations. Similarly, homozygous deletions of key HRR genes might be expected to produce longer remissions that smaller sequence alterations (which might be more prone to acquired reversion mutations).[23] Also unknown is the potential impact of other concurrent somatic mutations on PARPi response. For example, BRCA1/2-altered cancers with concurrent TP53 or RB1 mutations might be less sensitive to these drugs. Ultimately, a functional assay of HRR activity would be needed to best predict responsiveness (and primary resistance) to PARPi therapy, although such assays have been challenging to develop.
Many new studies are currently ongoing to further advance the impact of PARPi in prostate cancer. (Tables 3,4) The clinical trials discussed in this article explored PARPi therapy as monotherapy in the later stages of the disease process whereas many newer studies are exploring PARPi earlier in the disease process or in novel combinations. In particular, many of the combination clinical trials are exploring the efficacy in the HRR wild-type population with the hypothesis that combinations will sensitize all prostate cancer patients to PARPi. These combinations are based on strong pre-clinical data that has been reviewed in other articles.[24] Finally, there is active research investigating novel DNA repair targets beyond PARP for those patients with alterations that do not appear to have significant responses to PARPi. For instance ATR inhibitors in patients with ATM alterations.[25–26] We anticipate that many of these studies will lead to further advances in the clinical management for our patients.
Table 3:
Clinical Trials with Rucaparib in Prostate Cancer
| Clinical Trial Name | NCI Study Number | Patient Population | Phase | Primary Endpoint | Intervention | Biomarker Selected Enrollment* | Estimated Number of Patients |
|---|---|---|---|---|---|---|---|
| Metastatic Disease | |||||||
| CASPAR | NCT04455750 | CRPC | 3 | rPFS | E+R vs E | No | 1002 |
| TRITON3 | NCT02975934 | CRPC | 3 | rPFS | R vs Physician Choice | Yes | 400 |
| CheckMate 9KD | NCT03338790 | CRPC | 2 | ORR, PSA-RR | N+R vs N+D vs N+E | No | 330 |
| TRIUMPH | NCT03413995 | HSPC | 2 | PSA-RR | R, no ADT given | Yes | 30 |
| PLATI-PARP | NCT03442556 | CRPC | 2 | rPFS | Carbo+D+R | Yes | 20 |
| {none} | NCT04253262 | CRPC | 1,2 | Safety | C+R | Yes (phase 2); No (phase 1) | 44 |
| {none} | NCT03572478 | CRPC or endometrial cancer | 1,2 | Safety, T-cell infilatration in tumor | N+R | No | 12 |
| {none} | NCT03840200 | CRPC, breast or ovarian cancer | 1,2 | Safety | I+R | No | 51 |
| RAMP | NCT04179396 | CRPC | 1 | PK of R | E+R vs E+A | No | 60 |
| Localized Prostate Cancer or PSA Relapse After Definitive Treatment | |||||||
| ROAR | NCT03533946 | HSPC | 2 | PSA-RR | R, no ADT given | Yes | 32 |
R= rucaparib; E= enzalutamide; D= docetaxel; A = abiraterone; I = Ipatasertib; C = copanlisib; N = nivolumab
PK = pharmacokinetics; PSA-RR = PSA response rate; rPFS = radiographic progression-free survival
Enrollment criteria varies for each biomarker selected trial for the allowed DNA alterations or other molecular criteria
Table 4:
Clinical Trials with Olaparib in Prostate Cancer
| Clinical Trial Name | Study Number | Patient Population | Phase | Primary Endpoint | Intervention | Biomarker Selected Enrollment* | Estimated Number of Patients |
|---|---|---|---|---|---|---|---|
| Immune Therapy Combinations in Metastatic Disease | |||||||
| KEYLYNK-010 | NCT03834519 | CRPC | 3 | rPFS, OS | P+O vs (E or A) | No | 780 |
| KEYNOTE-365 | NCT02861573 | CRPC | 1,2 | Safety, PSA-RR, ORR | P+O vs P+D vs P+E vs P+A | No | 400 |
| Other Combinations in Metastatic Disease | |||||||
| PROpel Study | NCT03732820 | CRPC | 3 | rPFS | A+O vs A+Placebo | No | 720 |
| BRCAAway | NCT03012321 | CRPC | 2 | rPFS | A vs O vs A+O | Yes | 70 |
| {none} | NCT03263650 | CRPC (specifically in AVPC) | 2 | PFS | O vs Placebo maintenace after Cabaz+Carbo | No | 123 |
| IMANOL | NCT03434158 | CRPC | 2 | rPFS | O maintenance after D | Yes | 27 |
| {none} | NCT02893917 | CRPC | 2 | rPFS | O+C vs O | No | 90 |
| TRAP | NCT03787680 | CRPC | 2 | ORR | AZD6738+O** | No | 47 |
| COMRADE | NCT03317392 | CRPC | 1,2 | Safety, rPFS | O + Ra | No | 120 |
| {none} | NCT04556617 | 1,2 | Safety, ORR | PLX2853+O+A*** | No | 110 | |
| LuPARP | NCT03874884 | CRPC | 1 | Safety | Lu+O | Yes (PSMA positive scan) | 52 |
| Localized Prostate Cancer or PSA Relapse After Definitive Treatment | |||||||
| {none} | NCT04336943 | PSA relapse HSPC | 2 | Undetectable PSA | Durv + O | Yes | 30 |
| {none} | NCT03810105 | PSA relapsed HSPC | 2 | Undetectable PSA at 24 months | Durv+O, No ADT given | Yes | 32 |
| {none} | NCT03047135 | PSA relapsed HSPC | 2 | PSA-RR | O, no ADT given | No (Yes for enrichment cohort if added) | 50 |
| BrUOG 337 | NCT03432897 | Localized Prostate Cancer | 2 | PSA-RR | Neoadjuvant O prior to prostatectomy | Yes | 13 |
| {none} | NCT03570476 | Localized Prostate Cancer | 2 | Safety, Pathologic CR | Neoadjuvant O prior to prostatectomy | Yes | 2 |
| CaNCaP03 | NCT02324998 | Localized Prostate Cancer | 1 | Safety, Change in PARP expression | Neoadjuvant O+Degarelix | No | 20 |
O = olaparib; P = pembrolizumab; E = enzalutamide; A = abiraterone; Cabaz = cabazitaxel; Carbo = carboplatin; D = docetaxel; Durv = Durvalumab; Ra = radium-223; ADT = androgen deprivation therapy; Lu = 177Lu-PSMA
OS = overall survival; AVPC = aggressive variant prostate cancer[20]; rPFS = radiographic progression-free survival; ORR = objective response rate; CR = complete response
Enrollment criteria varies for each biomarker selected trial for the allowed DNA alterations or other molecular criteria
AZD6738 is an ATR inhibitor
PLX2853 is a BRD4 inhibitor
Figure 1.
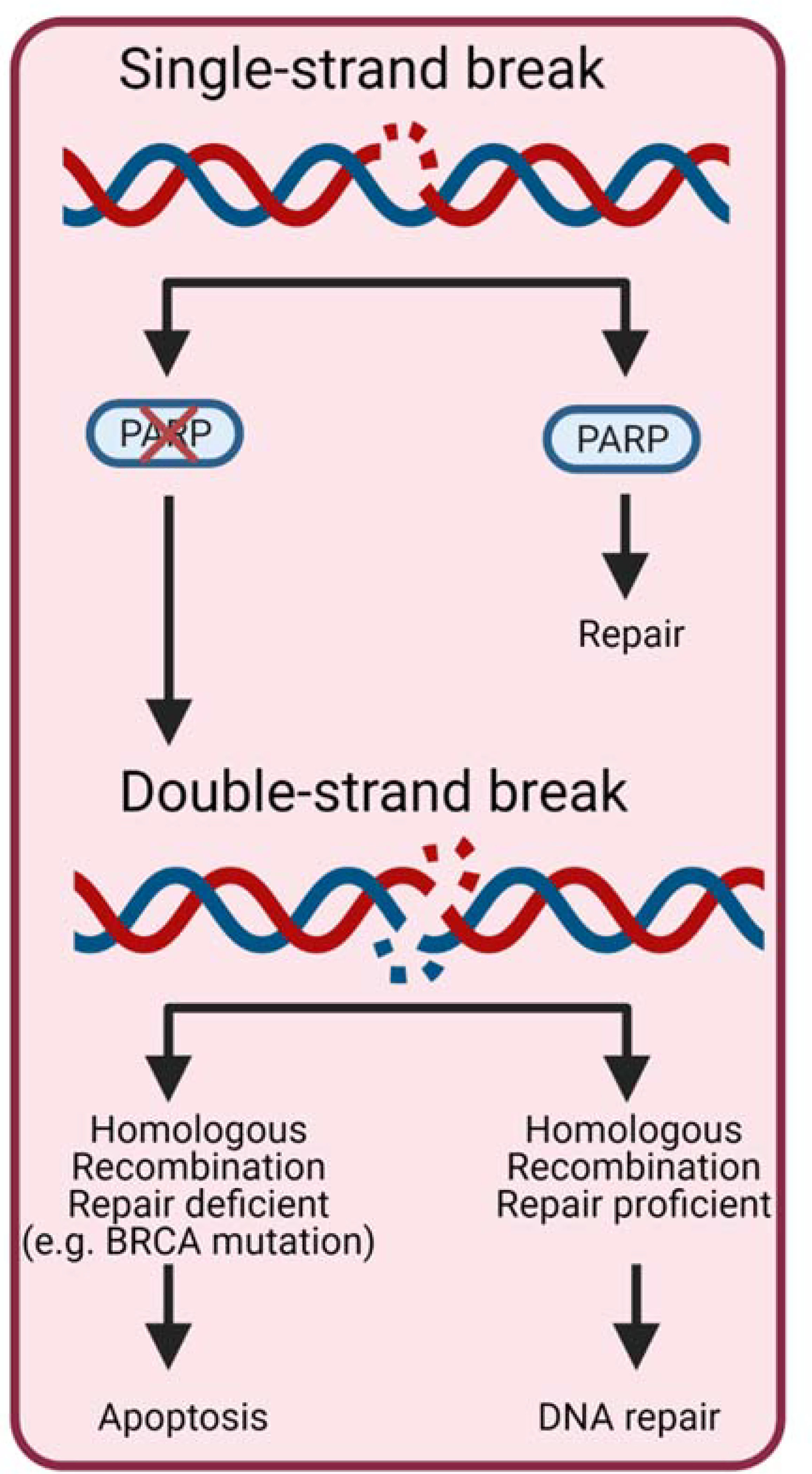
Mechanism of PAPRPi in cancer. PARP: Poly (ADP-ribose) polymerase HRR: Homologous recombination repair. PARP is a key protein in repairing single-strand DNA breaks. If the cell has lost PARP function then the single-strand breaks accumulate leading to double-strand DNA breaks. If the HRR repair pathway is nonfunctional then the cell undergoes apoptosis.
Article Highlights:
Alterations to DNA damage and repair genes, such as homologous recombination repair (HRR), is common in metastatic prostate cancer.
PARP inhibitors are effective in treating patients with HRR alterations.
Efficacy is variable depending on the HRR gene that is mutated.
PARP inhibitors are generally well-tolerated, with common adverse events including fatigue, nausea, and cytopenias with anemia being the most frequent.
Olaparib and rucaparib are both FDA approved for metastatic castration-resistant prostate cancer with HRR mutations and BRCA1/2 mutations, respectively.
Funding:
This work was partially supported by National Institutes of Health Cancer Center Support Grant P30CA006973
Footnotes
Declaration of Interest:
BL Maughan is a paid consultant/advisor to Janssen, Astellas, Bristol-Myers Squibb, Clovis, Tempus, Merck, Exelixis, Bayer Oncology and Peloton Therapeutics. He has also received research funding to his institution from Exelixis, Bavarian-Nordic, Clovis and Bristol-Myers Squibb. ES Antonarakis is a paid consultant/advisor to Janssen, Astellas, Sanofi, Dendreon, Pfizer, Amgen, AstraZeneca, Bristol-Myers Squibb, Clovis, and Merck. He has received research funding to his institution from Janssen, Johnson & Johnson, Sanofi, Dendreon, Genentech, Novartis, Tokai, Bristol- Myers Squibb, AstraZeneca, Clovis, and Merck. He is also the co-inventor of an AR-V7 biomarker technology that has been licensed to Qiagen. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures:
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
- 1.de Wit R, de Bono J, Sternberg CN, et al. Cabazitaxel versus Abiraterone or Enzalutamide in Metastatic Prostate Cancer. N Engl J Med, 2019. 381(26): p. 2506–2518. [DOI] [PubMed] [Google Scholar]
- 2. de Bono J, Mateo J, Fizazi K, et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med, 2020. 382(22): p. 2091–2102. ** The PROfound study demonstrates the clinical benefit of olaparib over standard of care for patients with HRR alterations.
- 3.Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014July31;371(5):424–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013January10;368(2):138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maughan BL, Luber B, Nadal R, et al. Comparing Sequencing of Abiraterone and Enzalutamide in Men With Metastatic Castration-Resistant Prostate Cancer: A Retrospective Study. Prostate, 2017. 77(1): p. 33–40. [DOI] [PubMed] [Google Scholar]
- 6.Khalaf DJ, Annala M, Taavitsainen S, et al. Optimal sequencing of enzalutamide and abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer: a multicentre, randomised, open-label, phase 2, crossover trial. Lancet Oncol, 2019. 20(12): p. 1730–1739. [DOI] [PubMed] [Google Scholar]
- 7.Helleday T, Petermann E, Lundin C, et al. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008March;8(3):193–204. [DOI] [PubMed] [Google Scholar]
- 8.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature, 2005. 434(7035): p. 917–21. [DOI] [PubMed] [Google Scholar]
- 9.Asim M, Tarish F, Zecchini HI, et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun, 2017. 8(1): p. 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mateo J, Carreira S, Sandhu S, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med, 2015. 373(18): p. 1697–708. * The TOPARP-A study was the first prospective clinical trial of PARPi in prostate cancer.
- 11.Mateo J, Porta N, Bianchini D, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol, 2020. 21(1): p. 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hussain M, Mateo J, Fizazi K, et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N Engl J Med, 2020. [DOI] [PubMed] [Google Scholar]
- 13. Abida W, Patnaik A, Campbell D, et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J Clin Oncol, 2020: p. JCO2001035. ** The TRITON2 trial confirmed the efficacy of rucaparib in mCRPC leading to FDA approval.
- 14.Markowski MC, Antonarakis ES. BRCA1 Versus BRCA2 and PARP Inhibitor Sensitivity in Prostate Cancer: More Different Than Alike? J Clin Oncol, 2020: p. JCO2002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abida W, Campbell D, Patnaik A, et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin Cancer Res, 2020. 26(11): p. 2487–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marshall CH, Sokolova AO, McNatty AL, et al. Differential Response to Olaparib Treatment Among Men with Metastatic Castration-resistant Prostate Cancer Harboring BRCA1 or BRCA2 Versus ATM Mutations. Eur Urol, 2019. 76(4): p. 452–458. * The magnitude of clinical benefit from PARPi depends on the specific gene altered.
- 17.Antonarakis ES, Velho PI, Fu W, et al. CDK12-Altered Prostate Cancer: Clinical Features and Therapeutic Outcomes to Standard Systemic Therapies, Poly (ADP-Ribose) Polymerase Inhibitors, and PD-1 Inhibitors. JCO Precis Oncol, 2020. 4: p. 370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poveda A, Floquet A, Ledermann JA, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2021March18;S1470–2045(21)00073–5. [DOI] [PubMed] [Google Scholar]
- 19.Antonarakis ES. Cyclin-Dependent Kinase 12, Immunity, and Prostate Cancer. N Engl J Med, 2018. 379(11): p. 1087–1089. [DOI] [PubMed] [Google Scholar]
- 20.Antonarakis ES, Gomella LG, Petrylak DP. When and How to Use PARP Inhibitors in Prostate Cancer: A Systematic Review of the Literature with an Update on On-Going Trials. Eur Urol Oncol 2020October;3(5):594–611. [DOI] [PubMed] [Google Scholar]
- 21.Marshall CH, Fu W, Wang H, et al. Prevalence of DNA repair gene mutations in localized prostate cancer according to clinical and pathologic features: association of Gleason score and tumor stage. Prostate Cancer Prostatic Dis. 2019March;22(1):59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015November5;163(4):1011–25 ** HRR alterations are common in prostate cancer suggesting that many patients are likely to benefit from this treatment strategy.
- 23.Pettitt SJ, Frankum JR, Punta M, et al. Clinical BRCA1/2 Reversion Analysis Identifies Hotspot Mutations and Predicted Neoantigens Associated with Therapy Resistance. Cancer Discov, 2020. 10(10): p. 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gourley C, Balmana J, Ledermann JA, et al. Moving From Poly (ADP-Ribose) Polymerase Inhibition to Targeting DNA Repair and DNA Damage Response in Cancer Therapy. J Clin Oncol, 2019. 37(25): p. 2257–2269. [DOI] [PubMed] [Google Scholar]
- 25.Rafiei S, Fetzpatrick K, Liu D, et al. ATM Loss Confers Greater Sensitivity to ATR Inhibition Than PARP Inhibition in Prostate Cancer. Cancer Res, 2020. 80(11): p. 2094–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wengner AM, Siemeister G, Lucking U, et al. The Novel ATR Inhibitor BAY 1895344 Is Efficacious as Monotherapy and Combined with DNA Damage-Inducing or Repair-Compromising Therapies in Preclinical Cancer Models. Mol Cancer Ther, 2020. 19(1): p. 26–38. [DOI] [PubMed] [Google Scholar]