

Abstract
Immune checkpoint blockade (ICB) therapy has substantially improved the outcomes of patients with many types of cancers, including renal cell carcinoma (RCC). Initially studied as monotherapy, immunotherapy-based combination regimens have improved the clinical benefit achieved by ICB monotherapy and have revolutionized RCC treatment. While biomarkers like PD-L1 and tumor mutation burden are FDA-approved as biomarkers for ICB monotherapy, there are no known biomarkers for combination immunotherapies. Here, we describe the clinical outcomes and genomic determinants of response from a phase 1b/2 clinical trial on advanced RCC patients evaluating the efficacy of lenvatinib, a multi-kinase inhibitor mainly targeting vascular endothelial growth factor receptor (VEGFR) and fibroblast growth factor receptor (FGFR) plus pembrolizumab, an anti-PD1 immunotherapy. Concurrent treatment with lenvatinib and pembrolizumab resulted in an objective response rate of 79% (19/24) and tumor shrinkage in 96% (23/24) of patients. While tumor mutational burden (TMB) did not predict for clinical benefit, germline HLA-I diversity strongly impacted treatment efficacy. Specifically, HLA-I evolutionary divergence (HED), which measures the breadth of a patient’s immunopeptidome, was associated with both improved clinical benefit and durability of response. Our results identify lenvatinib plus pembrolizumab as a highly active treatment strategy in RCC and reveal HLA-I diversity as a critical determinant of efficacy for this combination. HED also predicted better survival in a separate cohort of RCC patients following therapy with anti-PD-1-based combination therapy.
Keywords: immunotherapy, renal cell carcinoma, cancer, genomics, Major Histocompatibility Complex
INTRODUCTION
Immune checkpoint blockade therapy has improved the clinical outcomes of cancer patients with various types of malignancies including melanoma, lung cancer, lymphoma, colorectal cancer, renal cell carcinoma (RCC), and others. Blockade of the Programmed Cell Death-1 (PD-1) axis with anti-PD-1 or anti-PD-L1 (Programmed Cell Death Ligand 1) antibodies such as pembrolizumab, durvalumab, avelumab, or nivolumab can reinvigorate T cells and promote anti-tumor immunity. Cytotoxic T-cell recognition and targeting of neoantigens presented by major histocompatibility complex (MHC) molecules facilitate the elimination of cancer cells that present them[1, 2]. Anti-PD1/PD-L1 monotherapy has now been approved by the Food and Drug Administration (FDA) for use in a variety of cancers; however, most patients do not benefit, and better immunotherapy-based treatments are needed. Features such as PD-L1 positivity, tumor mutational burden, microsatellite instability, and T cell infiltration have various abilities to identify patients that respond to ICB treatment[3].
In patients with advanced RCC, anti-PD-1 monotherapy improved overall survival in a randomized phase III clinical trial of nivolumab versus everolimus (CheckMate 025)[4]. However, the response rate was only 25%, and most patients did not benefit from either treatment. To further improve outcomes, intense effort has been put into developing immunotherapy-based combination regimens built upon an anti-PD-1 backbone. One promising strategy is to combine multi-tyrosine kinase inhibitors (TKI) with ICB. For example, it has been shown that VEGF can directly inhibit T cell activity, affect the maturation and differentiation of lymphocytes and antigen presenting cells (APC), regulate angiogenesis, and modulate lymphocyte trafficking[5, 6]. The combination of the multi-kinase inhibitor axitinib and anti-PD1/PD-L1 therapy has demonstrated improved progression-free survival compared to TKI monotherapy in randomized phase III clinical trials in comparison to sunitinib[7, 8]. However, the potential of this strategy is far from realized, and there are no effective biomarkers for most immunotherapy-based combination regimens. In fact, PD-L1 quantitation appears to lose predictive ability with combination immunotherapy regimens[9].
Lenvatinib is a multiple receptor TKI targeting VEGFR1-3, FGFR1-4, PDGFRα, Ret, and Kit[10]. The combination of lenvatinib and everolimus is approved for the treatment of advanced RCC after progression on one VEGF-targeted treatment[11]; lenvatinib is also being studied in combination with pembrolizumab in multiple malignancies. Combination regimens that block multiple tyrosine kinases as well as PD-1 are beginning to show exceptional promise[7, 8, 12]. We describe the high clinical efficacy and its genomic correlates in patients with RCC treated with lenvatinib plus pembrolizumab.
MATERIALS AND METHOS
Study design, setting and participants of the phase Ib/II clinical trial
The individuals included in this study consisted of patients who participated in a phase Ib/II, multicenter, open-label clinical trial (NCT02501096)[12] designed to evaluate the efficacy and safety of lenvatinib/pembrolizumab for the treatment of several advanced-stage tumors, including renal cell carcinoma. The trial included individuals with pathologic diagnosis of melanoma, non-small cell lung carcinoma (NSCLC), head and neck squamous-cell carcinoma (HNSCC), renal cell carcinoma (RCC), urothelial carcinoma and endometrial carcinoma. Individuals were treated with pembrolizumab (200 mg intravenous every 3 weeks for up to 35 doses [approximately 2 years]) and lenvatinib (oral). Lenvatinib dosages varied depending on the phase of the clinical trial, and consisted of a 3+3 dose de-escalation strategy starting at 24 mg daily (phase Ib) or 20 mg daily (phase II). Radiographic tumor assessments were performed using computed tomography (CT) scans. Assessments were initially done every 6 weeks (first 24 weeks) and every 9 weeks thereafter. Tumor responses were evaluated by a specialized radiologist using the Immune-related Response Evaluation Criteria in Solid Tumors (irRECIST).
Outcomes and predictors
Baseline characteristics of individuals were assessed at the time of treatment start. Progression-free survival (PFS) was calculated from the time of treatment start to the time of progression or death. For patients who developed objective responses (i.e. a partial [PR] or complete response [CR]), duration of response (DOR) was also calculated. DOR was defined as the time from initial response to progression or death. Individuals who did not die or progress, were considered censored at the time of last follow-up visit. The clinical data included in this study was reviewed by investigators and a database cut-off performed on March 29th, 2019.
Ethics
The protocol for this trial was pre-approved by our institutional review board and ethics committee and the study conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. Written informed consent was obtained prior to any study-related activities. Individuals consented to their clinical records being reviewed and their blood and tumor tissue being profiled as part of the correlative research conducted in the context of the trial. Participants were allowed to withdraw their consent throughout the study period.
Tissue availability and sample procurement
Of the patients who were enrolled in the clinical trial (n=30), only a subset had sufficient tissue for biomarker analysis. Individuals who did not have tumor and germline (i.e., blood-derived) DNA of sufficient quality available were excluded from the analysis (n=6). The final cohort of 24 individuals in this study included only 1 individual treated with the phase Ib dosing scheme. Blood samples were obtained as part of the research protocol during one of the follow-up visits and stored in EDTA-containing tubes frozen at −80°C. Formalin-fixed paraffin-embedded (FFPE) tumor samples were obtained from the pathology facilities where patients were initially diagnosed. Slides stained with hematoxylin and eosin (H&E) were procured and reviewed by a specialized genitourinary pathologist. Based on histologic review of 1–2 reference slides stained with H&E, 5–20 unstained slides per sample were macro-dissected by scalpel (or scraped by razor) to collect tumor material.
PD-L1 staining
Immunohistochemistry (IHC) staining of PD-L1 molecules was performed in unstained slides obtained from the specimen using the PD-L1 IHC 22C3 pharmDx kit (Agilent). Semi-quantitative H-scores were obtained by an experienced pathologist and a cutoff of 1% was used to determine positivity.
Initial sample processing and DNA extraction
Paraffin was removed using heat treatment (90°C for 10’ in 480 μL PBS and 20 μL 10% Tween 20) and ice chill, followed by an incubation overnight in 400 μl of 1M NaSCN for rehydration and impurity removal. Tissues were subsequently digested with 20 μl of Proteinase K (600 mAU/ml) in 360 μl of ATL buffer at 56°C. DNA from whole blood was extracted using the QIAamp DNA Blood Maxi Kit (Qiagen catalog #51194), while DNA from FFPE material was isolated with the DNeasy Blood & Tissue Kit (QIAGEN catalog #69504) according to the manufacturer’s protocol. DNA was eluted in nuclease-free water.
DNA quality control, exome capture and sequencing
Sample quantity was measured using the Quant-iT PicoGreen dsDNA Assay (ThermoFisher catalog #P11496), while quality was assessed with TapeStation D1000 ScreenTape (Agilent catalog #5067–5582). After PicoGreen quantification and quality control with Agilent 2100 Bioanalyzer Instrument®, 144–500 ng of DNA were used to prepare libraries using the KAPA Hyper Prep Kit (Kapa Biosystems KK8504) with 8–12 cycles of PCR. After sample barcoding, 95–500 ng of library were captured by hybridization using the SureSelectXT Human All Exon V4 (Agilent catalog #5190-4632) according to the manufacturer’s protocol. PCR amplification of the post-capture libraries was carried out for 8 cycles. Samples were then run on a HiSeq 4000 or HiSeq 2500 in Rapid Mode in a 100 bp/100 bp or 125 bp/125 bp paired-end run, using the HiSeq 3000/4000 SBS Kit or HiSeq Rapid SBS Kit v2 (Illumina®). Normal and tumor samples were covered to an average of 118X and 176X, respectively.
Initial data processing and tumor mutational analysis pipeline
Raw sequencing data in FASTQ format was aligned to the human reference genome (b37) using the Burrows–Wheeler aligner (BWA v.0.7.10). Local realignment was done using the Genome Analysis Toolkit (GATK v4.1.4.1)[13] and duplicate reads removed using Picard v.2.13. For mutation calling, we used our integrated pipeline which includes four different variant calling tools: MuTect2 (part of GATKv4.1.4.1)[13], Strelka2 v2.9.10[14], Varscan v2.4.3[15] and Platypus[16]. Only mutations detected by at least two callers were included in the analysis. Additional filters were applied in order to obtain high-accuracy calls. These included a base coverage of at least 10 reads in the tumor, with at least 5 reads supporting the variant and allelic frequency below 2% in the matched-blood sample. Additionally, single-nucleotide variants (SNVs) identified at a frequency higher than 1% in dbSNP[17] or 1000Genomes project[18] were excluded. Dubious mutation calls were manually reviewed by investigators using the Integrative Genome Viewer software v2.4.10 for additional accuracy[19, 20]. (Supplemental Data 1)
Copy-number analysis
Allele-specific copy number analysis (ASCN) was performed using FACETS v0.5.6[21]. This tool provides integer CN values for each tumor allele in addition to conventional outputs. Genomic segments were defined as altered if they had a CN status different than 2:1 (i.e. two total copies, one per allele). Finally, the fraction of non-diploid genome was calculated by normalizing the length of all the CN-altered segments by the total genome length.
HLA-I genotyping and calculation of patient mean HED
As previously described, HLA-I genotyping of blood-derived DNA was performed using WES data. Polysolver, a previously-validated tool, was used with default parameters settings to identify the individuals’ HLA class-I alleles[21–23]. Patients with two different alleles at a specific locus (i.e. HLA-A, HLA-B and HLA-C) were considered heterozygous at that locus, and individuals who were found to have six different HLA class-I alleles were considered fully heterozygous at HLA-I.
HLA evolutionary divergence (HED) was calculated as described by Pierini and Lenz[24]. This approach involves sequence extraction of exons 2 and 3 of each HLA class I allele (i.e. where the peptide-binding domains are located, obtained from the Ensembl database[25]) and retrieval of the corresponding protein sequences from the ImMunoGeneTics/HLA database[26]. After alignment of the two HLA alleles, the Grantham distance metric[27] is calculated for each pair of corresponding amino-acids. The value represents a pairwise distance based on their physio-chemical properties (i.e. composition, polarity and volume). The sum of amino-acidic distances is then normalized by the length of the sequence. The resulting value (i.e. HED) represents a measure of functional divergence between the two HLA alleles. Finally, the values for each of the three HLA-I loci are averaged and reported as ‘Mean HED’. (Supplemental Data 2)
Validation of results - MSK-IMPACT® clear cell RCC cohort
Data from the MSK-IMPACT cohort was obtained for validation purposes[28, 29]. After IRB approval, the medical records of patients who received a PD-1/PD-L1 inhibitor-containing regimen between 2015 and 2018 were reviewed. Only individuals who had histologic evidence of clear cell components in their RCC tumor specimens were included. Patients who were exclusively treated with nivolumab monotherapy were not included in the analysis. Data on therapy administration, life status and previous treatments was analyzed. Overall survival (OS) in this cohort was calculated from the time of ICB therapy initiation to the time of death or last follow-up. MSK-IMPACT is a targeted next-generation sequencing assay performed at a CLIA-certified laboratory and approved by the FDA for the study of solid tumors. It involves the profiling of 468 cancer-associated genes as well as a SNP-assay for copy-number analysis and computational assessment of microsatellite instability[30]. As previously-described[31], we obtained the base sequences corresponding to the HLA class I loci from the BAM files derived from germline DNA sequencing. Only individuals whose normal DNA samples had sufficient coverage at the HLA-class I loci were included in the analysis.
Statistical analyses
For survival analysis, the Kaplan-Meier (KM) method was used to calculate estimates and log-rank tests were used to assess between-group differences. Multivariable Cox proportional hazards regression models were used to assess the relationship between multiple covariates of interest and time-to-event outcomes. Results were reported as point estimates along with its 95% confidence intervals and p-values. To assess potential dose-response relationships between mean HED and time-to-event outcomes, we tested all possible mean HED cut-offs and the hazard ratio estimates were reported. Furthermore, to evaluate the effect on outcomes along the mean HED spectrum, we constructed subpopulation treatment effect pattern plots (STEPP)[32, 33]. An R implementation of this non-parametric approach (‘stepp’ package[34]) was used to estimate the 2-year event-free probabilities across the range of mean HED values observed in both cohorts (parameters r2=20, r1=10, total n=65). Shapiro-Wilk tests were used to assess the mean HED distributions in both cohorts, and Spearman rank correlation test was used to assess the relationship between mean HED and maximum tumor shrinkage. All analyses were performed in the R platform v.3.5.0[35].
RESULTS
Our study was performed in the context of a phase Ib/II, single-arm, multi-institutional clinical trial where patients with advanced stage RCC received combination therapy with lenvatinib and pembrolizumab. The baseline characteristics and a summary of the clinical outcomes for patients with tissue available for genomic analysis (N=24) are shown in Table 1. The median age of the cohort was 60 years (IQR 56–66). When stratifying patients according to the International Metastatic Database Consortium (IMDC) criteria, 54% of individuals were found to have favorable risk and 46% intermediate risk[36]. Twenty-three patients were treated at the recommended phase 2 dose (lenvatinib 20 mg daily + pembrolizumab 200 mg every 3 weeks), and only one individual received treatment at the initial phase Ib starting dose (lenvatinib 24 mg daily + pembrolizumab 200 mg every 3 weeks). Notably, in this group of patients, at a median follow-up of 32.3 months, the objective response rate (ORR) was 79.2% with a disease-control rate of 96% and a median progression-free survival (PFS) of 21.9 months (95%CI 11.7–26.1) (Fig. 1a,b). Notably, the median time to first response was 1.4 months (IQR 1.3–3.4). Furthermore, a high proportion of patients achieved significant tumor shrinkage (Fig. 1b). Representative radiographic responses (Fig. 1c), as well as the timing of responses and clinical course of patients are shown (Fig. 1d). These results highlight the clinical promise of the lenvatinib plus pembrolizumab combination. To our knowledge, the response rate we observe with this treatment is among the highest seen in any therapy for RCC[7, 8, 37]. Taken together, our data from 24 trial subjects suggest that HLA-I diversity is associated with prolonged progression-free intervals as well as treatment durability following therapy with combined multi-kinase and PD1 blockade.
Table 1.
Baseline characteristics and clinical outcomes of the patients included in the study (n=24).
| N=24 | ||
|---|---|---|
| Age (years) (median [IQR]) | 60.0 [56.0 – 66.3] | |
| Female | 4 (16.7) | |
| Other | 5 (20.8) | |
| Third or later | 5 (20.8) | |
| ECOG 1 | 9 (37.5) | |
| Intermediate Risk | 11 (45.8) | |
| Objective Response Rate (%) | 79.2% | |
| Progressive Disease | 1 (4.2) |
Fig. 1. Clinical outcomes of the lenvatinib/pembrolizumab cohort.
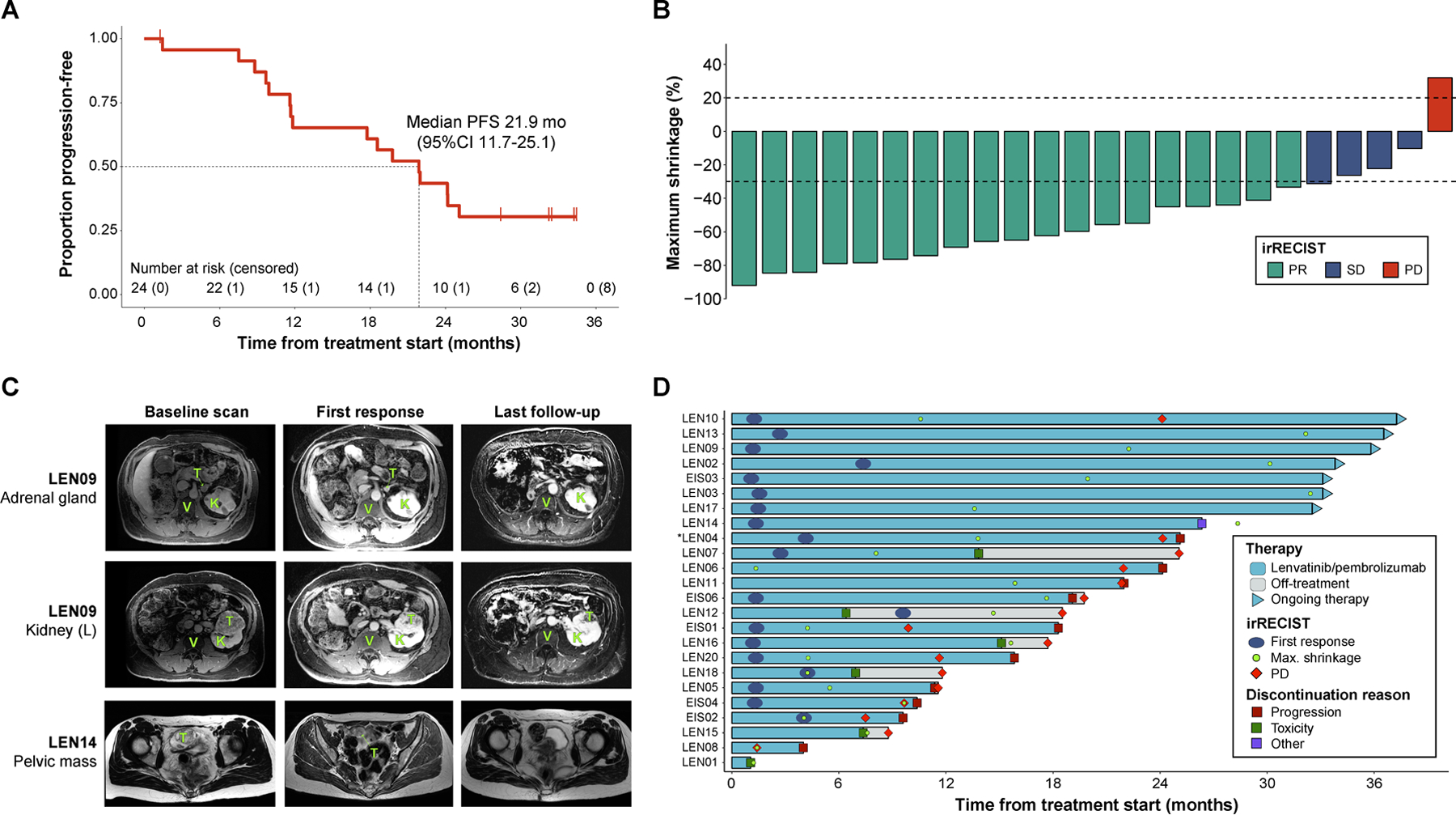
(a) Kaplan-Meier curve showing the progression-free survival estimates of the 24 patients included in the study. Median survival along with its 95% confidence interval are reported. (b) Maximum tumor shrinkage (in percentage) in the target lesions measured as part of patient assessment. Tumor responses were evaluated using irRECIST. (c) Magnetic resonance imaging (MRI) studies performed during patient management to assess therapy response. The representative images are from patients that showed exceptional responses to the combination regimen. Legend: T: tumor, K: normal kidney, V: vertebral body. (d) Swimmer plot showing the sequence of therapy-related events during patient management. Bars represent the timeline of each individual from therapy start to disease progression, treatment discontinuation or last-follow-up, whichever occurred later. Patients still on therapy are represented by the triangles at the end of the bars.
To assess potential genomic correlates of treatment response and durability, we performed whole exome sequencing on tumor and matched normal DNA (n=24, see Methods). Tumor from each individual was obtained before therapy was initiated. We also determined PD-L1 staining status of the tumors (Fig. 2 and Methods). Somatic mutations, tumor mutational burden (TMB), arm-level copy-number alterations, the fraction of genome bearing copy number aberrations (FCNA), HLA-I zygosity, and other metrics were determined using well-validated computational pipelines (Fig. 2)[13, 23, 31, 38]. The overall spectrum of somatic mutations was consistent with previously-reported results from The Cancer Genome Atlas Consortium (TCGA) and other landmark genomic studies on RCC[39–41]. As expected from the histologic subtype of these tumors (23/24 clear cell RCC), there was a high-prevalence of specific driver mutations in VHL (75%), PBRM1 (42%) and BAP1 (29%) (Fig. 2). The number of exonic, non-synonymous somatic mutations identified was also consistent with previous reports, with a median mutation count of 48.5 (IQR 40–67) and median TMB of 1.31 mutations per DNA megabase (mut/Mb). As expected in this tumor type, we also observed a relatively high insertion and deletion (indel) occurrence rate (median 13.7%), which is consistent with results from the TCGA and others where a relatively low TMB and higher indel proportion (around 9–12%) have been reported for RCC when compared to other cancer types[42]. Nearly all tumors displayed genomic losses involving cytoband 3p21 (92%). Other common events identified in these tumors were 14q loss (58%), 5q gain (54%) and 9p loss (42%). These results are in line with previous studies in clear cell RCC that have demonstrated a near-ubiquitous presence of biallelic VHL inactivation through a combination of mutations and 3p21 losses[39–41], recurrent 5q gains[39, 41], and an enrichment of 14q/9p losses in metastases (usually involving the CDKN2A/2B loci)[43]. We also calculated the fraction of the genome bearing CN alterations (FCNA) in each tumor sample[44]. The median FCNA was found to be 0.17 (IQR 0.24–0.35), this result is also consistent with previous reports[39, 45, 46]. In summary, the mutational and copy-number landscape of the RCC tumors obtained from our trial patients have genomic characteristics that are typical of this tumor type.
Fig. 2. Genomic characterization of the study cohort.
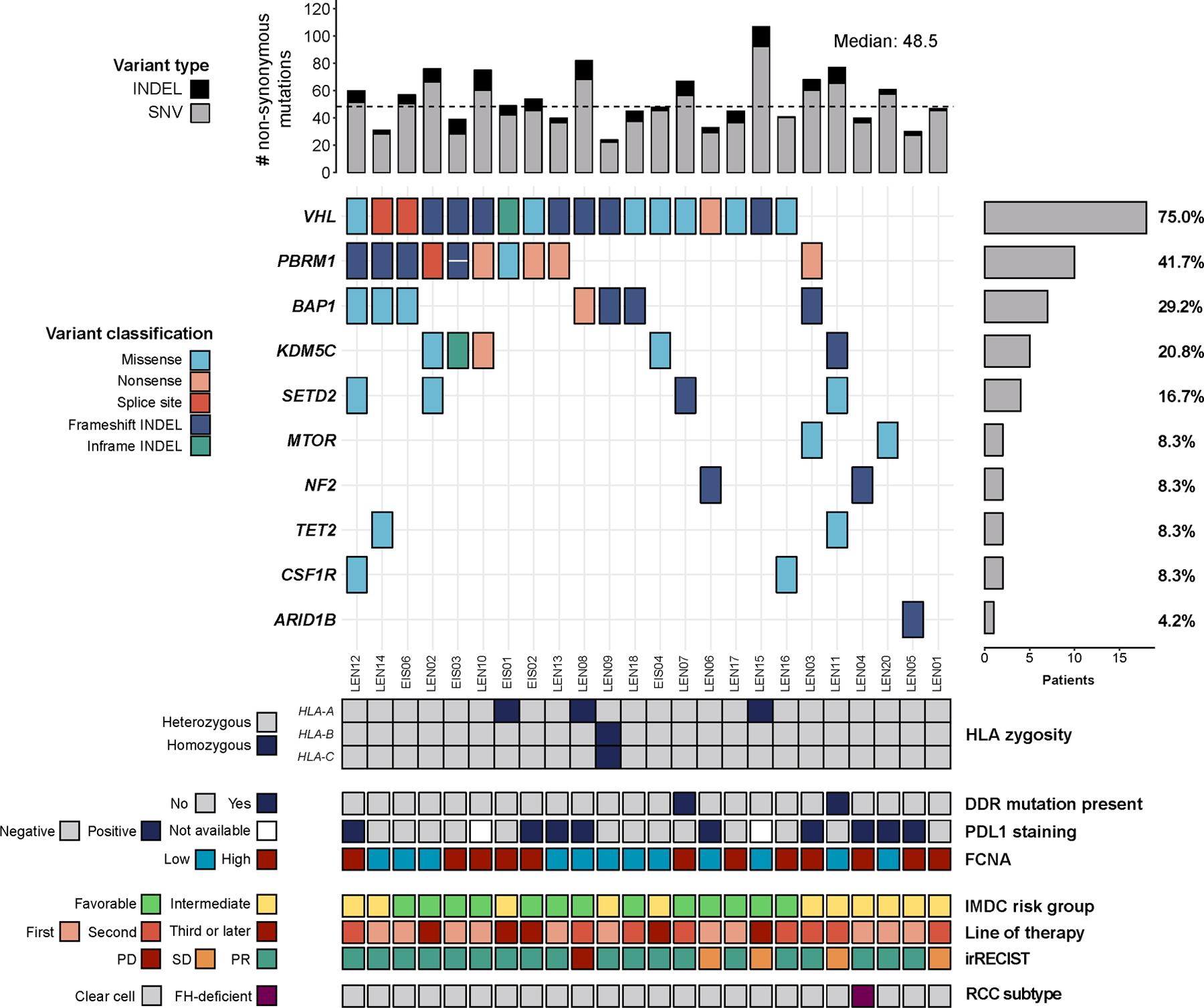
The oncoprint shows the most common nonsynonymous somatic mutations in cancer-associated genes identified by exome-sequencing. The top bars represent the number of variants identified in each sample, while the side bars represent the number (and percent) of individuals displaying mutations in each gene. The bottom tiles contain additional genomic and clinical information evaluated as part of this study.
There is no actionable biomarker that is currently approved for use in RCC patients treated with immune checkpoint therapy or ICB-based combinations. We, therefore, examined the associations between clinical outcomes and different molecular correlates. Several groups of investigators have observed that HLA genotype can affect ICB response[31, 47]. Reduced MHC function can lead to reduced antigen presentation, resulting in insufficient T-cell dependent tumor-cell killing[48–52]. Four patients in the study cohort were found to be homozygous at any HLA-I gene and these individuals showed worse outcomes (Fig. 2). Although a trend was observed in survival analysis when comparing homozygous to fully-heterozygous individuals (median 22 vs 9.4 mo, log-rank, p=0.2, HR 0.46 [0.13, 1.64]), our power to detect any meaningful associations was limited. Strikingly, however, HLA-class I evolutionary diversity (HED) was associated with longer progression-free intervals and greater durability of clinical response (Fig. 3). Our group has recently demonstrated the value of quantifying the overall evolutionary divergence at the HLA class-I alleles[24] (i.e. mean HED) in patients treated with ICB therapy[53]. HED is a measure that represents the molecular diversity of a patient’s HLA-I alleles. Greater HLA diversity is associated with the ability to present a wider antigen repertoire[24], higher T-cell clonal activation status following ICB exposure[2], and improved response to ICIs[31, 47, 53]. In our cohort, the distribution of mean HED was found to be non-normal (Shapiro-Wilk, p=0.01) with a median mean HED of 6.9 (range 0.69, 9.0).
Fig. 3. Association between HLA-I evolutionary divergence (HED) and outcomes.
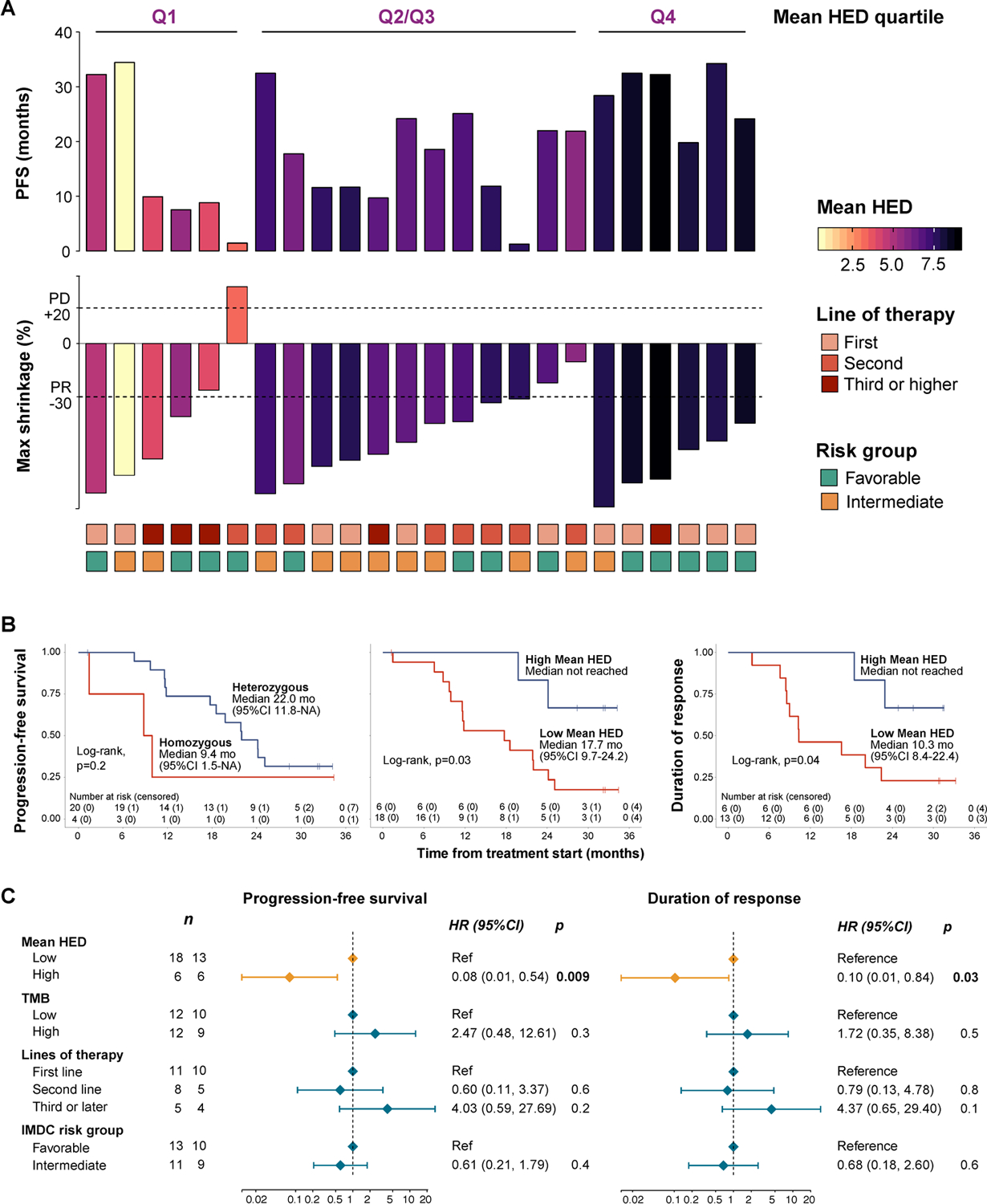
(a) Clinical outcomes of the cohort in relation to the mean HED values calculated for each patient. Each column represents an individual. The outcomes are represented by the top bars which are colored by the mean HED value. The bottom tiles represent relevant prognostic factors for RCC that were used for adjustment in multivariable analysis. (b) Kaplan-Meier curves showing the relationship between HLA diversity and clinical outcomes. Zygosity at HLA-class I loci was not significantly associated with progression-free survival (PFS), while high mean HED was found to be predictive of longer progression-free survival, as well as longer durability of response (DOR). (c) Forest plots showing results from the multivariable Cox regression analysis. Mean HED was found to be significantly associated with improved PFS and DOR even after adjusting for known prognostic factors.
We observed a statistically significant association between high mean HED and better outcomes in patients treated with lenvatinib plus pembrolizumab. More specifically, when using the top quartile to stratify patients, progression-free survival (PFS) was found to be longer in patients with high mean HED (median not reached vs 17.7 mo, log-rank, p=0.03, HR 0.23 [0.05, 1]). Importantly, mean HED was also associated with greater duration of treatment response (median not reached vs 10.3 mo, log-rank p=0.045, HR: 0.23 [0.05, 1.09]) (Fig. 3b). In a multivariable Cox proportional hazards model, the association between mean HED and improved outcomes was found to be significant (HR 0.08 [0.01–0.54], p=0.009) even after adjusting for potential confounders such as the number of previous therapy lines, IMDC risk group and TMB. Similar results were observed with regards to response durability (Fig. 3c). Notably, there was a trend between the degree of HED diversity and PFS; with higher mean HED linked to longer PFS and tumor shrinkage in a “dose-dependent fashion” (Fig. 4a, b). When assessing different HED thresholds, we observed that increasing HED was associated with longer PFS (Fig. 4c, d). This relationship has been previously described for HED[53] as well as TMB[38, 47], and different optimal thresholds may be needed to predict response to ICB therapy in different disease and therapeutic contexts. Despite the fact that immunotherapy combinations are quickly becoming standard of care, no biomarkers currently exist for immunotherapy-based combination regimens. Our results, from this biomarker correlative analysis of clinical trial data, demonstrate that HLA-I evolutionary divergence is associated with clinical benefit in patients with RCC treated with combined ICB/multi-kinase inhibitor therapy.
Fig. 4. Dose-response relationship between mean HED and clinical outcomes.
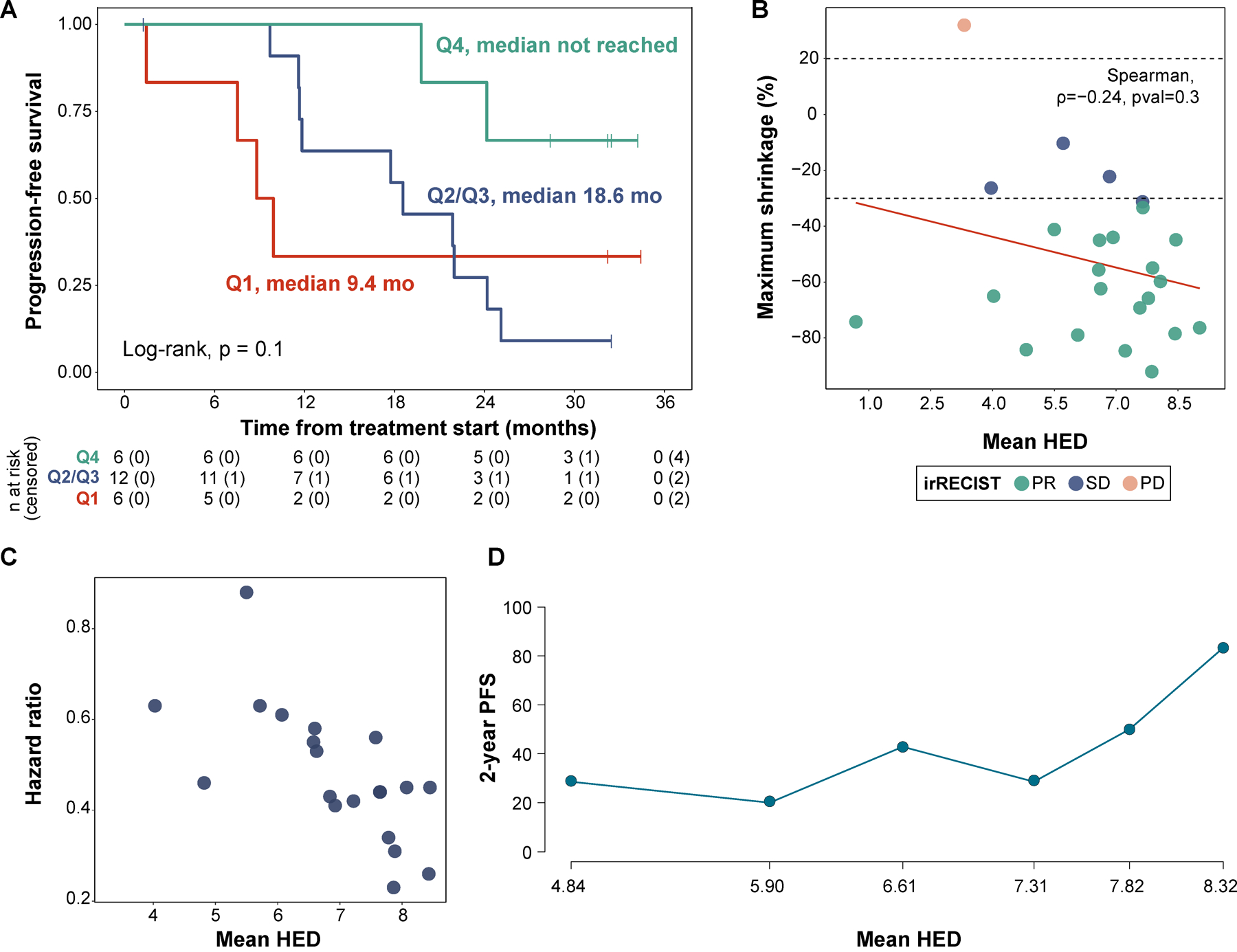
(a) Kaplan-Meier curve showing progression-free survival (PFS) stratifying patients by mean HED quartile (Q1 vs Q2/Q3 vs Q4). (b) Association between mean HED and maximum tumor shrinkage achieved in target lesions. The Spearman rank correlation test showed a negative correlation (rho: −0.24), but results were not statistically-significant. (c) Effect of mean HLA-I evolutionary divergence on hazard ratio for PFS across all possible cutpoints. The plot shows a negative relationship between mean HED and hazard ratio, indicating improved PFS as mean HED increases. (d) Subpopulation treatment effect pattern plot (STEPP) showing the 2-year progression-free probability across the mean HED range, higher HLA-I divergence was found to be associated with improved outcomes.
We sought to validate these findings in a separate cohort of RCC patients treated with ICB. We identified 41 metastatic ccRCC patients that were treated with ICB combination therapy and had their tumors analyzed using MSK-IMPACT[28–30]. This is a targeted next-generation sequencing assay approved by the FDA for the study of solid tumors (see Methods). We explored the relationship between HLA diversity and clinical benefit in this set of patients and observed that individuals with high mean HED (stratified by the median) who were treated with ICB therapy showed improved overall survival when compared to the rest (median not reached vs 35.4 months, log-rank, p=0.03) (Fig. 5a,b). The distribution of HED in this cohort did not reach the threshold for non-normality on statistical testing (Shapiro-Wilk, p=0.5) (Fig. 5c). To confirm that the dose-response association followed the same trend in both groups of patients, we used the STEPP approach to assess the 2-year event-free probability along the mean HED range. In both cohorts, higher mean HED values were found to be associated with improved outcomes. However, the threshold at which these benefits were observed differed between the two sets of patients (Fig. 4d, 2d). Taken together, these data suggest that the overall degree of HLA diversity, which shapes the size and diversity of an individual’s immunopeptidome, is associated with the depth and persistence of clinical benefit to ICB therapy in renal cell carcinoma.
Fig. 5. Validation of results in the MSK-IMPACT cohort.
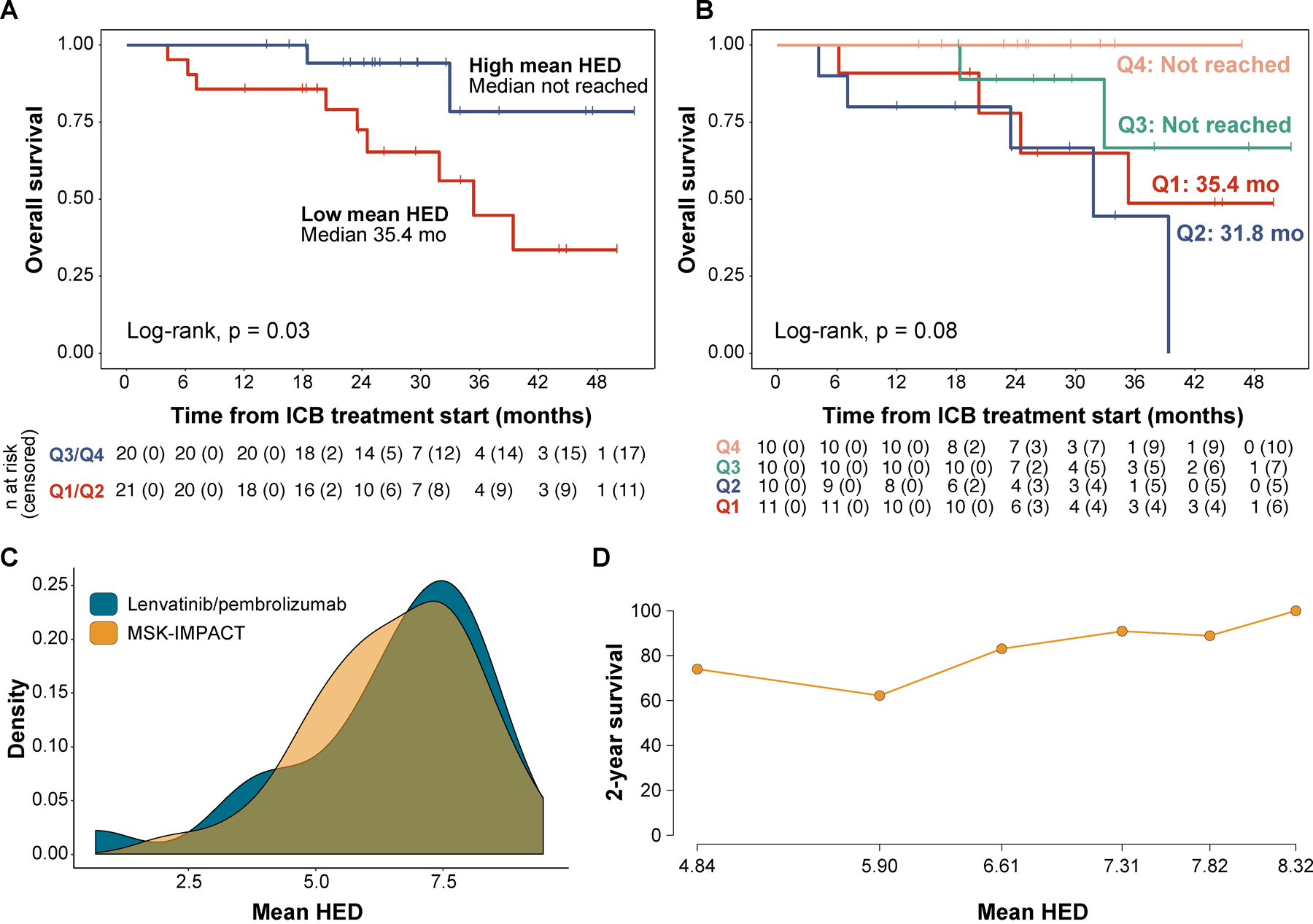
(a) Kaplan-Meier curve showing the overall survival (OS) estimates, stratifying patients by the median value of mean HED. (b) Kaplan-Meier curve showing OS estimates stratifying patients by mean HED quartiles. (c) Density plots showing the distribution of mean HED values in the discovery (lenvatinib/pembrolizumab, n=24) and validation cohorts (MSK-IMPACT, n=41). (d) Subpopulation treatment effect pattern plot (STEPP) showing the 2-year survival probability across the mean HED range, higher divergence was found to be associated with improved outcomes.
Finally, to investigate the independence of the predictive ability of mean HED from other known prognostic factors and previously-reported molecular correlates of response (not yet validated in RCC)[3], we developed a multivariable Cox model including HED as well as clinical and genomic covariates (Fig. 6a–c). Tumor mutational burden (TMB), defined as the number of nonsynonymous exonic mutations per DNA megabase, has been widely validated as a measure that is associated with improved outcomes after treatment with ICIs in many cancer types[38, 47, 54]. We did not observe any association between TMB and outcomes in any of the cohorts analyzed (stratified by the median, Log-rank, p=0.14 for the lenvatinib/pembrolizumab [PFS, Fig. 6a] and p=0.72 for the MSK-IMPACT cohort [OS, not shown]), nor did we find any associations with PD-L1 status and clinical benefit in the lenvatinib/pembrolizumab cohort (Log-rank, p=0.3, not shown). Another genomic measure that has been shown to be associated with immune evasion and worse survival after immunotherapy is the overall burden of copy-number alterations in the genome (FCNA)[45, 55]. We did not observe an association between FCNA (stratified by the median) and response to ICB therapy in these patients (Fig. 6b). Additional features, such as somatic PBRM1 loss-of-function mutations, have shown mixed results in studies evaluating their ability to predict outcomes after ICB therapy[48, 56, 57]. In multivariate analysis, loss-of-function mutations in PBRM1 were not an independent predictor of response (Fig. 6c).
Fig. 6. Other potential biomarkers of response to ICB therapy and their association with progression-free survival in RCC.
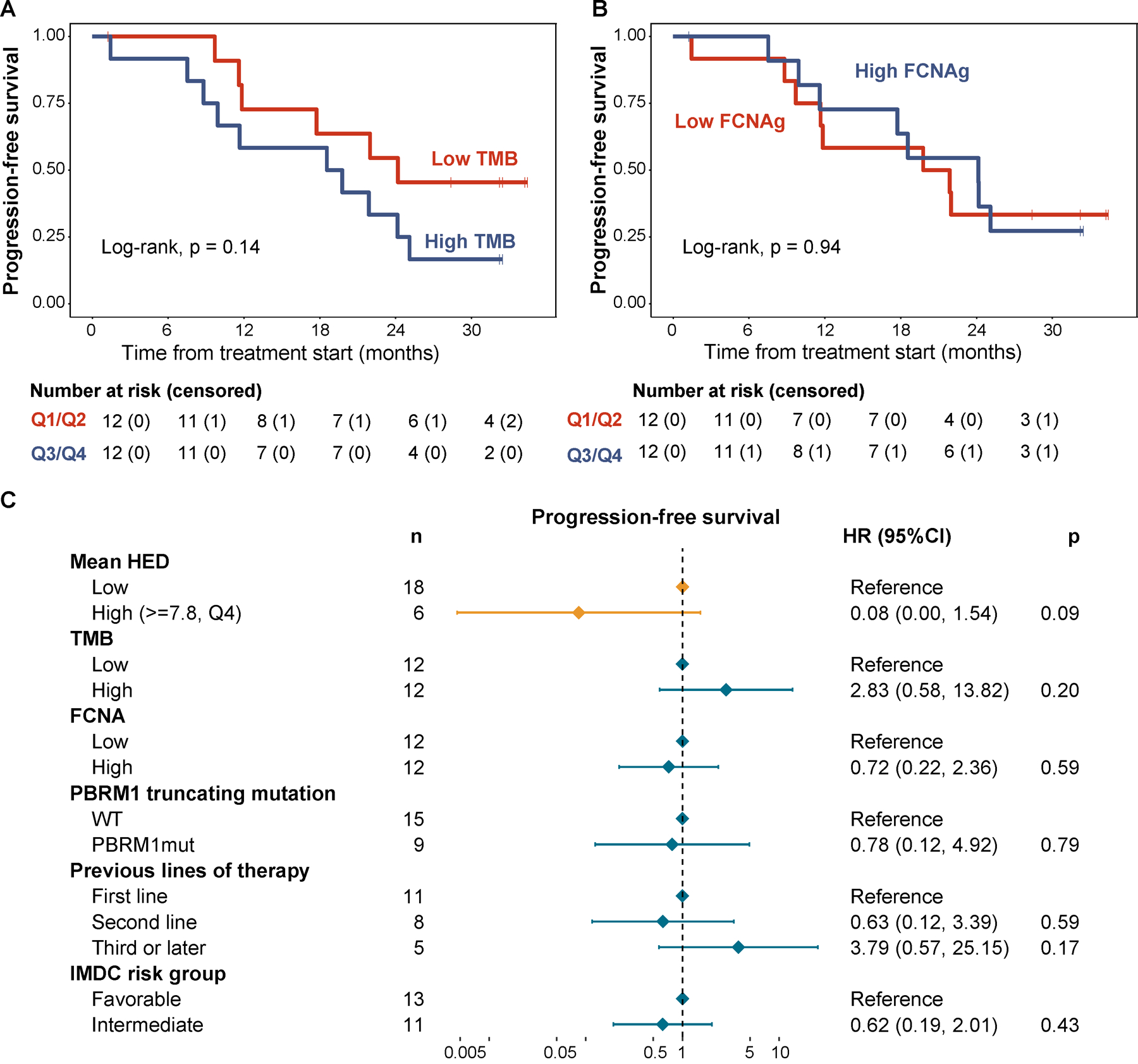
(a) Kaplan-Meier curve showing PFS by tumor mutational burden (stratified by the median). (b) Kaplan-Meier curve showing PFS by the fraction of non-diploid genome (stratified by the median). (c) Forest plot showing the results from a multivariable Cox regression model including genomic biomarkers of response to ICB tested in the study as well as additional relevant prognostic factors.
DISCUSSION
Combination therapy with PD-1 axis/multi-kinase blockade constitutes one of the most promising treatment strategies for RCC and other cancers with the regulatory approval of multiple PD-1 axis/multi-kinase regimens as first line therapy[7, 8]. Previous studies have reported that some of the ligands of these kinases, such as VEGF, can directly inhibit T-lymphocyte maturation and activity, and lead to dedifferentiation of lymphocytes and antigen-presenting cells (APCs)[5, 6]. Furthermore, clear cell RCC tumors have been shown to display a high degree of angiogenesis signaling mediated to a significant extent by increased VEGF pathway activity[58]. Extrapolation of these findings to the systemic therapy setting provides a biological basis for the additional anti-tumor efficacy demonstrated by the lenvatinib/pembrolizumab combination, and a working model to investigate biomarkers of response to these ICB-based therapies.
Genomic analyses were undertaken in our cohort to explore the significance of emerging biomarkers of response to ICB-based therapies in this context, and our findings were largely consistent with previous reports. Consistent with previous studies, our results failed to demonstrate a significant association between TMB and response to ICB-based combination therapy in this cohort. Although TMB has proven to be a useful predictor of response to ICB therapy in many different solid tumors[38, 47, 54], these findings have not been recapitulated in RCC[38, 57, 59] and the biological basis underlying these observations remains to be elucidated.
Antigen recognition by CD8+ T lymphocytes has become a topic of particular interest due to its well-established role in the development of adaptive immune responses[60]. T cell dynamics have not only been directly implicated in response to ICB therapy, but T-cell receptor (TCR) repertoires have been shown to vary by therapeutic context[2, 31, 61–63]. Sequencing platforms now allow for the assessment of antigen specificity and clonal heterogeneity of CD8+ T cells in the tumor microenvironment at useful resolution[60]. Although the utility of TCR repertoire features in predicting response to ICB-based therapies is far from clear [61–70], clonal T cell populations have been shown to develop in ICB responders and their presence has been associated with tumor neoantigen depletion[2]. Notably, our group has previously shown TCR clonality to be directly associated with HLA class I genetic variability and evolutionary divergence, features that were in turn predictive of ICB therapy response in a cohort of patient with advanced melanoma[31, 53]. Although not explored as part of this study due to technical limitations, research efforts aimed at further elucidating these findings should consider assessing the relationship between HED and TCR repertoire composition in different contexts.
The role of loss-of-function mutations in the PBRM1 gene, a recurrent molecular feature in clear cell RCC, was explored. Truncating PBRM1 variants have been reported in association with a distinct non-immunogenic tumor phenotype leading to ICB-resistance[71], as well as with IFN-γ-associated expression programs that promote tumor clearance after ICB therapy[56, 72]. Contradicting evidence also exists on the role of PBRM1 mutations in the immunotherapy context[56, 57, 59], and studies have yet to provide conclusive evidence about their utility in this setting. Similar to previous reports, our results did not support a significant association between PBRM1 loss-of-function mutations and clinical benefit after treatment with lenvatinib/pembrolizumab.
Taken together, our data suggest that HLA-I diversity is associated with prolonged progression-free intervals as well as treatment durability following therapy with combined multi-kinase and PD1 blockade. Questions still remain about the optimal HED thresholds required to predict response to ICB, and it is likely that, much like TMB, optimization to each therapeutic context will be needed in the future.
Supplementary Material
Implications:
These findings have substantial implications for RCC therapy and for understanding immunogenetic mechanisms of efficacy and warrants further investigation.
ACKNOWLEDGEMENTS
We thank the Chan lab members for helpful discussions. We thank the staff and physicians of the MSK Department of Medicine Kidney Program and the Urology Service for helpful suggestions. We acknowledge the use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. This work was in part supported by grants NIH R35 CA232097 and DOD KC180165 (TAC). This work was also supported by the Mellnikoff Fund (RJM and TAC) and the Weiss Family Fund (AAH, RJM). CV acknowledges grant funding from ‘Fundación Alfonso Martín Escudero’. We thank Rodolfo Perini of Merck pharmaceuticals for his support.
FUNDING
Study 111 (NCT02501096) was funded by Eisai Inc., Woodcliff Lake, NJ, USA, and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. Patients treated at Memorial Sloan Kettering Cancer Center were supported in part by a Memorial Sloan Kettering Cancer Center Support Grant/Core Grant (P30 CA008748)
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
T.A.C. is a co-founder of Gritstone Oncology and holds equity. T.A.C. holds equity in An2H. T.A.C. acknowledges grant funding from Bristol-Myers Squibb, AstraZeneca, Illumina, Pfizer, An2H, and Eisai. T.A.C. has served as an advisor for Bristol-Myers, MedImmune, Squibb, Illumina, Eisai, AstraZeneca, and An2H. T.A.C., L.G.T.M, and D.C. hold ownership of intellectual property on using tumor mutation burden to predict immunotherapy response, with pending patent, which has been licensed to PGDx. RJM served in a consultancy or advisory role for Pfizer, Novartis, Merck, Genentech/Roche, Eisai and Exelixis, and received research funding from Bristol-Myers Squibb, Merck, Pfizer, Genentech/Roche, Eisai, Exelixis, and Novartis. C.H.L. served in a consultancy or advisory role to Amgen, Bristol-Myer Squibb, Exelixis, Merck, Pfizer, EMD Serono, and Eisai, and also received research funding from Bristol-Myers Squibb, Calithera, Eisai, Exelixis, Eli Lilly, Merck, and Pfizer.
Footnotes
DATA AVAILABILITY
All whole-exome sequencing data produced in this study are deposited on the Sequence Read Archive (SRA) under the accession number PRJNA610643. All the data from the validation set can be explored at http://cbioportal.org. Any other relevant data is available from the corresponding authors upon reasonable request.
REFERENCES:
- 1.Kalaora S, et al. , Combined Analysis of Antigen Presentation and T-cell Recognition Reveals Restricted Immune Responses in Melanoma. Cancer Discov, 2018. 8(11): p. 1366–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riaz N, et al. , Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell, 2017. 171(4): p. 934–949 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Havel JJ, Chowell D, and Chan TA, The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer, 2019. 19(3): p. 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Motzer RJ, et al. , Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med, 2015. 373(19): p. 1803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long J, et al. , Vascular endothelial growth factor (VEGF) impairs the motility and immune function of human mature dendritic cells through the VEGF receptor 2-RhoA-cofilin1 pathway. Cancer Sci, 2019. 110(8): p. 2357–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohm JE, et al. , VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood, 2003. 101(12): p. 4878–86. [DOI] [PubMed] [Google Scholar]
- 7.Motzer RJ, et al. , Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med, 2019. 380(12): p. 1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rini BI, et al. , Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med, 2019. 380(12): p. 1116–1127. [DOI] [PubMed] [Google Scholar]
- 9.Gandhi L and Garassino MC, Pembrolizumab plus Chemotherapy in Lung Cancer. N Engl J Med, 2018. 379(11): p. e18. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto Y, et al. , Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell, 2014. 6: p. 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Motzer RJ, et al. , Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol, 2015. 16(15): p. 1473–1482. [DOI] [PubMed] [Google Scholar]
- 12.Taylor MH, et al. , Phase IB/II Trial of Lenvatinib Plus Pembrolizumab in Patients With Advanced Renal Cell Carcinoma, Endometrial Cancer, and Other Selected Advanced Solid Tumors. J Clin Oncol, 2020. 38(11): p. 1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DePristo MA, et al. , A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet, 2011. 43(5): p. 491–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim S, et al. , Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods, 2018. 15(8): p. 591–594. [DOI] [PubMed] [Google Scholar]
- 15.Koboldt DC, et al. , VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res, 2012. 22(3): p. 568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rimmer A, et al. , Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet, 2014. 46(8): p. 912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherry ST, et al. , dbSNP: the NCBI database of genetic variation. Nucleic Acids Res, 2001. 29(1): p. 308–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Genomes Project C, et al. , A map of human genome variation from population-scale sequencing. Nature, 2010. 467(7319): p. 1061–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnell EK, et al. , Standard operating procedure for somatic variant refinement of sequencing data with paired tumor and normal samples. Genet Med, 2019. 21(4): p. 972–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thorvaldsdottir H, Robinson JT, and Mesirov JP, Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform, 2013. 14(2): p. 178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matey-Hernandez ML, et al. , Benchmarking the HLA typing performance of Polysolver and Optitype in 50 Danish parental trios. BMC Bioinformatics, 2018. 19(1): p. 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiyotani K, Mai TH, and Nakamura Y, Comparison of exome-based HLA class I genotyping tools: identification of platform-specific genotyping errors. J Hum Genet, 2017. 62(3): p. 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shukla SA, et al. , Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol, 2015. 33(11): p. 1152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pierini F and Lenz TL, Divergent Allele Advantage at Human MHC Genes: Signatures of Past and Ongoing Selection. Mol Biol Evol, 2018. 35(9): p. 2145–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunt SE, et al. , Ensembl variation resources. Database (Oxford), 2018. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson J, et al. , The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res, 2015. 43(Database issue): p. D423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grantham R, Amino acid difference formula to help explain protein evolution. Science, 1974. 185(4154): p. 862–4. [DOI] [PubMed] [Google Scholar]
- 28.Cerami E, et al. , The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov, 2012. 2(5): p. 401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao J, et al. , Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal, 2013. 6(269): p. pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng DT, et al. , Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn, 2015. 17(3): p. 251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chowell D, et al. , Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science, 2018. 359(6375): p. 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonetti M and Gelber RD, Patterns of treatment effects in subsets of patients in clinical trials. Biostatistics, 2004. 5(3): p. 465–81. [DOI] [PubMed] [Google Scholar]
- 33.Lazar AA, et al. , Evaluation of treatment-effect heterogeneity using biomarkers measured on a continuous scale: subpopulation treatment effect pattern plot. J Clin Oncol, 2010. 28(29): p. 4539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yip WK, et al. , Subpopulation Treatment Effect Pattern Plot (STEPP) analysis for continuous, binary, and count outcomes. Clin Trials, 2016. 13(4): p. 382–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feigelson ED and Babu GJ, Statistical challenges in modern astronomy V. Lecture notes in statistics,. 2013, New York: Springer. xxiii, 559 p. [Google Scholar]
- 36.Heng DY, et al. , External validation and comparison with other models of the International Metastatic Renal-Cell Carcinoma Database Consortium prognostic model: a population-based study. Lancet Oncol, 2013. 14(2): p. 141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Motzer RJ, et al. , Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med, 2018. 378(14): p. 1277–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Samstein RM, et al. , Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet, 2019. 51(2): p. 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cancer Genome Atlas Research, N., Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature, 2013. 499(7456): p. 43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitchell TJ, et al. , Timing the Landmark Events in the Evolution of Clear Cell Renal Cell Cancer: TRACERx Renal. Cell, 2018. 173(3): p. 611–623 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sato Y, et al. , Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet, 2013. 45(8): p. 860–7. [DOI] [PubMed] [Google Scholar]
- 42.Turajlic S, et al. , Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol, 2017. 18(8): p. 1009–1021. [DOI] [PubMed] [Google Scholar]
- 43.Turajlic S, et al. , Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell, 2018. 173(3): p. 581–594 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bielski CM, et al. , Genome doubling shapes the evolution and prognosis of advanced cancers. Nat Genet, 2018. 50(8): p. 1189–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor AM, et al. , Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell, 2018. 33(4): p. 676–689 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turajlic S, et al. , Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell, 2018. 173(3): p. 595–610 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anagnostou V, et al. , Multimodal genomic features predict outcome of immune checkpoint blockade in non-small-cell lung cancer. Nat Cancer, 2020. 1(1): p. 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McGranahan N, et al. , Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell, 2017. 171(6): p. 1259–1271 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paulson KG, et al. , Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun, 2018. 9(1): p. 3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodig SJ, et al. , MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci Transl Med, 2018. 10(450). [DOI] [PubMed] [Google Scholar]
- 51.Sade-Feldman M, et al. , Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun, 2017. 8(1): p. 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaretsky JM, et al. , Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med, 2016. 375(9): p. 819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chowell D, et al. , Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nat Med, 2019. 25(11): p. 1715–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Snyder A, et al. , Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med, 2014. 371(23): p. 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davoli T, et al. , Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science, 2017. 355(6322). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miao D, et al. , Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science, 2018. 359(6377): p. 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McDermott DF, et al. , Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med, 2018. 24(6): p. 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hakimi AA, et al. , Transcriptomic Profiling of the Tumor Microenvironment Reveals Distinct Subgroups of Clear Cell Renal Cell Cancer: Data from a Randomized Phase III Trial. Cancer Discov, 2019. 9(4): p. 510–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hakimi AA, et al. , A pan-cancer analysis of PBAF complex mutations and their association with immunotherapy response. Nat Commun, 2020. 11(1): p. 4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DiNatale RG, Hakimi AA, and Chan TA, Genomics-based immuno-oncology: bridging the gap between immunology and tumor biology. Hum Mol Genet, 2020. 29(R2): p. R214–R225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reuben A, et al. , Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom Med, 2017. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Snyder A, et al. , Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: An exploratory multi-omic analysis. PLoS Med, 2017. 14(5): p. e1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hopkins AC, et al. , T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight, 2018. 3(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Massa C, et al. , Identification of patient-specific and tumor-shared T cell receptor sequences in renal cell carcinoma patients. Oncotarget, 2017. 8(13): p. 21212–21228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo L, et al. , Characteristics, dynamic changes, and prognostic significance of TCR repertoire profiling in patients with renal cell carcinoma. J Pathol, 2020. 251(1): p. 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerlinger M, et al. , Ultra-deep T cell receptor sequencing reveals the complexity and intratumour heterogeneity of T cell clones in renal cell carcinomas. J Pathol, 2013. 231(4): p. 424–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ross-Macdonald P, et al. , Molecular correlates of response to nivolumab at baseline and on treatment in patients with RCC. J Immunother Cancer, 2021. 9(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Forde PM, et al. , Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N Engl J Med, 2018. 378(21): p. 1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roh W, et al. , Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med, 2017. 9(379). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tumeh PC, et al. , PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature, 2014. 515(7528): p. 568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu XD, et al. , PBRM1 loss defines a nonimmunogenic tumor phenotype associated with checkpoint inhibitor resistance in renal carcinoma. Nat Commun, 2020. 11(1): p. 2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pan D, et al. , A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science, 2018. 359(6377): p. 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.