

Abstract
Although most primary estrogen receptor (ER)–positive breast cancers respond well to endocrine therapies, many relapse later as metastatic disease due to endocrine therapy resistance. Over one third of these are associated with mutations in the ligand-binding domain (LBD) that activate the receptor independent of ligand. We have used an array of advanced computational techniques rooted in molecular dynamics simulations, in concert with and validated by experiments, to characterize the molecular mechanisms by which specific acquired somatic point mutations give rise to ER constitutive activation. By comparing structural and energetic features of constitutively active mutants and ligand-bound forms of ER-LBD with unliganded wild-type (WT) ER, we characterize a spring force originating from strain in the Helix 11–12 loop of WT-ER, opposing folding of Helix 12 into the active conformation and keeping WT-ER off and disordered, with the ligand-binding pocket open for rapid ligand binding. We quantify ways in which this spring force is abrogated by activating mutations that latch (Y537S) or relax (D538G) the folded form of the loop, enabling formation of the active conformation without ligand binding. We also identify a new ligand-mediated hydrogen-bonding network that stabilizes the active, ligand-bound conformation of WT-ER LBD, and similarly stabilizes the active conformation of the ER mutants in the hormone-free state.
Implications:
Our investigations provide deep insight into the energetic basis for the structural mechanisms of receptor activation through mutation, exemplified here with ER in endocrine-resistant metastatic breast cancers, with potential application to other dysregulated receptor signaling due to driver mutations.
Introduction
The increasing number of mutated proteins found from deep DNA sequencing of primary and metastatic tumors (1–3) has presented the cancer field with a number of distinct challenges. Although bioinformatics methods can often distinguish obvious driver mutations from variants of uncertain significance, deciphering the underlying molecular mechanisms whereby mutations alter function requires computationally enabled structural, dynamic, and energetic analyses of the affected proteins (4–7). Here, we have applied molecular dynamics (MD) simulations to define the energetic basis for ligand-independent activation of estrogen receptor alpha (ERα) through specific mutations frequently found in recurrent, metastatic breast cancers.
Approximately 70% of breast cancers are classified as ER+, and many of these respond to endocrine therapies that block the biosynthesis of endogenous estrogens (i.e., aromatase inhibitors, AIs), or directly/competitively antagonize ER function (i.e., selective ER modulators, SERMs, or downregulators/degraders, SERDs; ref. 8). After several years, however, therapy resistance often presents as progressive metastatic disease that no longer responds to these therapies. DNA sequencing of such endocrine therapy-resistant tumors has identified point mutations in the ERα ligand-binding domain (LBD) that convey ligand-independent activity and reduced responsiveness to ER antagonists (9). Y537S and D538G, the most prominent mutations, appear in >30% of endocrine therapy-resistant tumor samples (10, 11), and the mean survival time for patients bearing either mutant is less than those having tumors with wild-type (WT) ERα (10).
ERα Y537S and D538G mutants have pronounced constitutive activity, resulting from structural changes that stabilize the active conformation of the receptor even in the absence of a bound agonist (9, 12, 13). The clustering of several mutations within the loop connecting Helix 11 (H11) to Helix 12 (H12) in the LBD suggested that this loop might provide an energetic barrier controlling the H12 conformational switch that regulates coactivator binding to the receptor and initiates transcriptional cascades. These early studies proposed that differential interactions introduced by specific point mutations were somehow stabilizing the H11-H12 loop or allowing four consecutive hydrophobic residues (533–536, Val-Val-Pro-Leu) to repack against the LBD (9, 12). What has remained unclear, however, were the molecular mechanisms and in particular the underlying energetic perturbations by which such sequence changes give rise to robust transcriptional activity in a ligand-independent manner (14).
Herein, we report extensive use of long-timescale (μs-scale) MD simulations and sophisticated free energy methods to identify and quantitatively characterize specific interactions in a conformational switch by which the Y537S and D538G mutations give rise to ligand-independent receptor activity. Using these data, we developed a “spring-loading” model to account for critical attributes underlying receptor activation, both in the presence of ligand or alternatively in the absence of ligand but the presence of either of these two prevalent mutations. Our computationally generated model is supported experimentally using a set of single and double mutations to study receptor activity in cells and ligand-binding/unbinding kinetics. The simulation-based methods we apply here to understand critical interactions that differentiate WT from mutant forms of the key transcription factor ERα are potentially useful for analyzing other regulatory proteins that are mutated to an active state in cancer.
Materials and Methods
Molecular modeling and system preparation
To characterize the effect of mutation on receptor dynamics, atomic-detailed molecular models were constructed from a high-resolution (2.8 Å) x-ray crystal structure of ERα bound to estradiol (E2; PDB code 1GWR; ref. 15) as previously described (12). Using this protocol, molecular systems were setup for apo-ERα-Y537S and apo-ERα-D538G by removing the E2 ligand and introducing the specified mutation in silico. To facilitate comparisons with WT protein structures, a positive control system, ERα-WT bound to E2 (WT-E2), and a negative control system, apo-ERα (apo-WT), were also prepared. Free energy calculations using the bias-exchange umbrella sampling (BEUS) method (16, 17) require simulating each system across multiple replicas (windows), and are therefore computationally expensive for moderate to large system sizes. To reduce computational costs, a second set of systems were constructed in the same manner as described above, but for a single monomer of the ER complex, reducing system sizes from approximately 100k (dimer) to approximately 35k (monomer) after adding explicit water and ions. Any perturbations to the dimer interface occur far from the ligand-binding pocket and the H11–12 loop, and therefore, should not alter the dynamics directly associated with the free energy pathway under investigation.
Equilibrium MD simulations
Simulations on the timescales of 200 ns were performed using the NAMD2 simulation package (18, 19). Dynamics of the protein, solvent, and ions were described using the CHARMM36 (20–22) molecular force field, which included CMAP backbone corrections and updated NBFIX terms for protein–ion interactions. The TIP3P water model (23) was used for explicit solvent molecules. Ligand parameters describing the ligand (E2) were taken by analogy from the CHARMM General Force Field (24) as assigned by the ParamChem web server (25, 26). Further attempts to refine the ligand parameters using the Force Field Toolkit (27) did not yield significant improvement when comparing molecular mechanics-computed quantities and the quantum mechanical target data.
The detailed simulation setup was applied as follows: Equilibrium simulations were performed using an NPT ensemble at 1.0 atm and 310 K. Temperature and pressure were controlled by a Nosé-Hoover thermostat (28) and a Langevin piston (ref. 29; period = 100 fs, decay = 50 fs, damping coefficient = 0.5 ps–1), respectively. A Verlet integrator was used to compute atomic positions with a 2-fs timestep, and positions were recorded every 500 steps (1 ps). Periodic boundary conditions were used where non-bonded interactions were truncated using a switching function from 10.0 to 12.0 Å, and long-range electrostatics were computed via the particle mesh Ewald (PME) method (30). Bonded and non-bonded forces were computed at every timestep whereas PME forces were computed at every other timestep.
Pre-production simulations were first performed to equilibrate portions of the molecular system that were modeled during structure preparation, for example, missing loops ±2 residues, added solvent molecules and ions, and mutated residues ±2 residues, where applicable. During this phase, all other heavy atoms were restrained using a harmonic potential (k = 1.0 kcal/mol/Å2). An energy minimization routine (10,000 steps, steepest descent algorithm) was used to resolve any high-energy contacts, followed by 1 ns of MD. The resulting coordinates and velocities were used as input for subsequent production simulations (200 ns) performed in triplicate.
To reach microsecond simulation timescales, a single snapshot was taken for each system at t = 100 ns from one of the production simulations described above (replicate 1 for WT-E2, apo-Y537S, apo-D538G, and replicate 2 for apo-WT) and converted to run using the DESMOND simulation program (31) on the special purpose Anton supercomputer (32). Because of the burden of storing a high trajectory frame rate for microsecond simulations, the coordinates were saved less frequently: Every 12 ps for 1 μs of simulation.
Trajectory analysis
The conformation of residue 537 was visualized by tracking the position of the side-chain hydroxyl as either the phenol of tyrosine (WT) or alcohol of serine (Y537S mutant) at 200-ps intervals over the replicate 200-ns simulation trajectories (3,000 positions) and projected onto protein structure observed in the final frame of the last replicate simulation. The specific hydrogen-bonding contacts of residue 537 were further quantified by measuring the distance between the alcohol oxygen of Y/S537 and the side-chain heteroatoms of known interacting residues, Asn348 and Asp351, reporting the shortest of the two measured distances for each interaction (2 monomers × 3 replicates × 20,000 frames/simulation = 120,000 data points). A histogram of the observed distances was then constructed by sorting data points into bins of 0.5 Å width, reporting the aggregated frequency of each bin observed for both monomers in all three 200-ns simulation trajectories.
The conformational dynamics of residue Leu536, and the interplay with preceding hydrophobic residues 533–535 (Val-Val-Pro), were characterized by computing the solvent accessible surface area (SASA) in VMD (ref. 33; “measure sasa”) of the side-chain atoms for all replicates of the 200-ns and the long-timescale (microsecond) simulation trajectories. The data were smoothed using a Gaussian-weighted running average (σ = 5) and visualized as time series.
Visual inspection of the long-timescale (microsecond) simulation for the WT-E2 system revealed a ligand-mediated hydrogen-bonding network initiated by interaction of the D-ring hydroxyl of the E2 ligand with His524:Nδ. Because of all other systems lacked a bound ligand, only the subsequent interactions of the network were characterized. The interaction of His524:Nε with the carbonyl of Glu419 was quantified by measuring the heteroatom–heteroatom distance over the course of the simulation, followed by smoothing using a Gaussian-weighted running average (σ = 5). The terminal interaction of the network—a salt bridge formed between the side chains of Glu419 and Lys531—was assessed using the Hydrogen Bonds plugin of VMD with a 3.0 Å donor–acceptor distance threshold and 30° angle cutoff (Donor–Acceptor-H, equivalent to a Donor-H–Acceptor angle of 150°) to digitize the presence or absence of this interaction while taking both distance and geometric constraints into account.
BEUS-free energy calculation
Free energy calculations using the BEUS method (16, 17, 34) were performed to quantitate the energetics associated with rotating residue Leu536, located within the H11–12 loop region, from solvent exposed to a buried position. Simulations were performed in NAMD2 (18, 19) using a combination of the Colvars (35) and Replica Exchange modules. The reaction coordinate was defined using the Coordination Number collective variable (colvar, ξ) for which the Leu536 side-chain heavy atoms formed one group, whereas all other heavy atoms in the protein formed the second group. Appropriate coverage of the reaction coordinate space was determined by first reanalyzing the 1-μs equilibrium simulations, which captured the Leu536 conformational states visited under equilibrium, providing a range of the relevant colvar values (16 < ξ < 42). Short pilot simulations applying harmonic biases to the colvar were then performed to probe the ends of this range to ensure that the energy profile extends beyond the local minima representing the “exposed” and “buried” side-chain conformational states, yielding a broadened colvar range of 6 < ξ <53.
Starting from the structures obtained after pre-production simulations, 1-ns biased simulations were performed using a moving harmonic potential applied to the ξ colvar (k = 0.5 kcal/mol), driving the Leu536 side chain through the entirety of the extended colvar range. Conformations from these short, driven simulations were binned on the basis of ξ, from which snapshots were randomly selected to seed the umbrella sampling windows (23 windows; width = 2; centers evenly distributed from ξ = 7 to 51). Driven simulations for the WT-E2 system did not capture any conformations for the lowest bin (i.e., 6 < ξ <8); accordingly, the range was shifted by one window width for this system (i.e., centers evenly distributed from ξ = 9 to 53), whereas all other parameters remained consistent across all four systems. The snapshot assigned to each window was then equilibrated for 4 ns while applying a harmonic restraint (k = 0.5 kcal/mol) to keep ξ near the window center. Short simulations were performed to tune the force constant associated with the harmonic potential applied to each window until the exchange probability between adjacent windows reached 20%–40%. For all windows and all systems, it was determined that a force constant of k = 0.09 kcal/mol was sufficient to achieve the desired exchange probability. Production simulations were performed in 20-ns blocks until the resulting free energy profile no longer changed with additional sampling (total of 100 ns/window). The last 60 ns of simulation were used to compute the free energy profiles by sorting the simulation trajectories and applying the Generalized Weighted Histogram Analysis Method (GWHAM; refs. 36–38) with Bayesian bootstrapping (ref. 39; 4 blocks, 100 iterations) to yield an average relative free energy and standard deviation for each window.
Binding and unbinding kinetics of single and double mutants
The tetrahydrochrysene (THC) was synthesized in our laboratory as previously reported (40). It binds to ERα with an affinity of 68% ± 4% compared with E2 set to 100%. This is equivalent to a Kd of 0.29 nmol/L. The His-6–tagged ERα-LBDs, amino acids 304–554, were expressed from pET-15b vectors in BL21(DE3)pLysS E. coli and purified as described previously (41). Mutations were introduced by site-directed mutagenesis. The kinetic experiments were with 3 nmol/L ER and 30 nmol/L THC. The data were collected using a constant wavelength analysis with excitation at 370 nm and emission at 570 nm, with 5-mm slits. The association kinetics were followed in real time, taking time points every 15 seconds for 120 seconds and then every 60 seconds for a total of 14 minutes. For the dissociation experiments, 1,500 nmol/L (50×) unlabeled E2 was added (dilution effect 0.2%), and the kinetics were followed with time points every 5 minutes for 60 minutes, in a Spex FluoroMax-3 (Jobin Yvon Horiba) fluorometer. The data were transformed and plotted with GraphPad Prism 4.
Activity of ER single and double mutants in breast cancer cells by luciferase assay
MCF7 cells were seeded into 24-well plates at a density of 0.15 × 106 cells per well in an estrogen-deprived medium a day before transfection. Each well of cells was then transfected with 0.125 μg of either WT or mutant ER, 0.315 μg of 3xERE-Firefly luciferase and 0.06 μg of pRL-TK (Renilla) using Xtremegene HP transfection reagent (Roche) according to the manufacturer's instructions. The luciferase activity of the cells was then determined 48 hours post-transfection using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Luciferase bioluminescence measurements were performed with the Veritas Microplate Luminometer (Promega). All experiments were conducted in triplicate and the Firefly luciferase activity was normalized with the Renilla luciferase activity of each sample.
Results
Specific point mutations change the dynamics of known hydrogen-bonding contacts for residue 537 in the H11–12 loop of ERα
The availability of multiple high-resolution x-ray crystal structures for ligand-bound WT and both Y537S and D538G receptors has highlighted the consistent formation of hydrogen bonds between Tyr537 and Asn348, or for the case of the Y537S mutation, Ser537 and Asp351 (9, 12). There are no published structures of WT ER in the apo state, and thus, changes in residue interactions upon ligand binding remain unknown. The full ensemble of side-chain conformations, the stability of these interactions, and the important consequences of removing the ligand all remain open questions. To address this, MD simulations were performed in the presence and absence of estradiol (E2) ligand in the WT sequence (WT-E2 or apo-WT) or in the absence of ligand with point mutations (apo-Y537S, apo-D538G). Extensive sampling of receptor conformations was achieved by combining 200-ns simulations, performed in triplicate, and by combining the data measured for each monomer in the dimeric simulation system, yielding 1.2 μs of aggregate sampling for each of the four molecular systems. These extended simulations yielded robust statistics and enabled determination of accurate energy profiles for the conformation of the critical residue at position 537 (vide infra).
The dynamic nature of residue 537, as a tyrosine or serine, is revealed by tracking the position of the side-chain hydroxyl throughout the simulation trajectory both through visual inspection (Fig. 1, top) and by computing the frequency of distances observed between the alcohol oxygen of the side chain and hydrogen-bonding partners, Asn348 and Asp351 (Fig. 1, bottom). The phenolic side chain of Tyr537 in the ligand-bound WT receptor (WT-E2, green) adopts two different conformations: One forming a hydrogen bond to Asn348, and a second directed into bulk solvent. Removal of the ligand (apo-WT, red) appears to loosen the Tyr537–Asn348 interaction as additional clusters of side-chain conformations are observed, which result in new minor populations in the interaction distance histogram. However, the hydrogen-bonded cluster with Asn348 remains the dominant conformation (light red frequency peak near 2.4 Å). Mutating residue 537 to serine (Y537S, blue) changes the hydrogen-bonding partner from Asn348 to strongly favor interaction with Asp351, as also indicated by a reversal in the frequency peaks [351 peak (dark blue) shifts to near 2.4 Å, whereas the 348 peak (light blue) shifts to >4 Å], compared with the patterns observed for the WT sequence both with and without ligand. These data recapitulate the wholesale shift in hydrogen-bonding partner from Asn348 to Asp351 observed in all high-resolution x-ray crystal structures with the Y537S mutation. Finally, mutating residue 538 from aspartic acid to glycine (D538G, purple), a known “helix breaker,” perturbs the structure near the beginning of H12, as observed from the twisted ribbon in the final structure from simulation. Compared with the other molecular systems, the frequency peak heights for hydrogen bonding to Asn348 or Asp351 are significantly reduced, indicating a reduced prominence of these interactions in stabilizing receptor conformation for the D538G mutant, which is consistent with published x-ray crystal structures (12).
Figure 1.

Effect of ligand binding and mutation on hydrogen-bonding contacts. The dynamics of hydrogen-bonding partners Y/S537 (H12) with N348 and D351 (H3) were evaluated from replicate 200-ns simulations of dimeric ERα complexes (1.2 μs aggregate sampling for each system). Top, the position of the Y/S537 side-chain oxygen is shown every 200 ps (3,000 spheres; only monomer A is shown for clarity). Bottom, the shortest distance between side-chain heteroatoms of Y/S537(O) and N348(O/N) or D351(O/O) were binned (width = 0.5 Å) to yield frequency histograms of each interaction.
The positioning of leucine 536 is an indicator of strain in the loop between H11 and H12
The strong dependence of WT ER on ligand binding to adopt an active conformation suggests that an energy barrier is associated with the positioning of H12 over the ligand-binding pocket. We hypothesized that the short H11–12 loop, consisting of consecutive hydrophobic residues (Val-Val-Leu-Pro), was a key feature based on side-chain packing (12); however, the specific role of Leu536 in establishing an energy barrier foundational to the conformational switch eluded our initial studies. In the absence of this insight, one aspect of the x-ray crystal structure of ER (WT) complexed to E2 and the steroid receptor coactivator peptide (SRC2; ref. 15) remained unexplained: Leu536, located in the middle of the H11–12 loop, is buried away from solvent in monomer A but projects into bulk solvent in monomer B (15). The exposure of this hydrophobic side chain to water was not the result of crystal contacts with the neighboring unit cell and thus would result in an energy penalty, an observation that ran counter to the prevailing hypothesis that optimized side-chain packing of the hydrophobic loop was responsible for stabilizing the active conformation of H12.
To clarify the predominance of buried versus exposed positions, and to establish which conformation is most representative, all structures of agonist-conformation ERα deposited in the PDB (n = 185) were binned on the basis of sequence (i.e., WT, Y537S, or D538G) and analyzed in terms of the conformation of key residues (Table 1). The conformation of Leu536 observed in WT structures was mixed, with approximately 18% in the buried position and 82% solvent exposed. In striking contrast, Leu536 was overwhelmingly observed in the buried position for structures containing either Y537S (>98%, n = 170) or D538G (100%, n = 2) point mutations.
Table 1.
Conformations of Leu536 observed in high-resolution x-ray crystal structures of ERα.
| Individual | Position of Leu536 | ||||
|---|---|---|---|---|---|
| Sequence | # Structures | Monomers | Buried | Exposed | Unresolved |
| Wild-type | 13 | 28 | 5 (17.9%) | 23 (82.1%) | 0 (0%) |
| Y537S | 170 | 346 | 340 (98.3%) | 3 (0.9%) | 3 (0.9%) |
| D538G | 2 | 4 | 4 (100%) | 0 (0%) | 0 (0%) |
The strong correlation between the buried positioning of Leu536 in the structures that bear activating mutations, in contrast with the mixed conformational states observed for WT sequences, provides compelling evidence that the activating mutations introduce structural changes that alter the H11–12 loop dynamics. Accordingly, the MD simulation trajectories were analyzed to compute the SASA for each replicate of the four ER variants simulated. Furthermore, the SASA was decomposed to individually assess Leu536 versus the preceding hydrophobic residues (Val-Val-Pro) in each monomer (Fig. 2). The resulting time series show that monomer A, for which Leu536 starts in the buried conformation, remains stable for the complete duration of the simulation with only one exception (apo-WT, replicate 3). The conformations observed in monomer B, for which Leu536 starts in a solvent-exposed conformation, are more dynamic. Only one of the three replicates converged to the buried conformation in the apo-WT system, whereas the other two maintain the solvent-exposed conformation. Including the ligand (WT-E2) or introducing either of the mutations (apo-Y537S or apo-D538G) appears to favor the Leu536-buried conformation as 2/3 of the simulations converged to this conformation.
Figure 2.

Solvent exposure of consecutive hydrophobic residues in the H11–12 loop. The solvent accessible surface area (SASA) was measured for residues 533–535 (Val, Val, Pro, dark coloring) and for 536 (Leu, light coloring), separately, in replicate 200-ns (left 2) and in single 1-μs (right 2) equilibrium simulations.
Concerned that the apparent rigidity of the loop may exhibit conformational transition times that exceeded the 200 ns afforded to each replicate, we greatly extended the simulation timescale by branching one of the replicates for each system (branched at 100 ns) and extending the simulation for an additional 1 μs using the Anton special-purpose supercomputer (32). For all systems, the buried conformation of monomer A remained stable for the duration of the simulation, and monomer B converged to the buried conformation within 0.5 μs in all cases.
Computation of energy profiles for the buried versus solvent-exposed transition for Leu536: Defining the “spring”-like nature of the H11–12 loop in WT ER and its perturbation by ligand binding or mutation
The results above, highlighted in Table 1 and Fig. 2, provide a qualitative picture for how ligand binding and/or the presence of specific mutations alters H11–12 loop dynamics and illustrate the consequences of these differences through the conformational preferences of Leu536. To directly and quantitatively assess the flexibility of the H11–12 loop, we used the advanced free energy technique of the BEUS method (16, 17, 34) with the GWHAM (36–38) to compute the free energy profiles describing the transition pathway of Leu536 between the buried and solvent-exposed conformations and how the energy profile is impacted by ligand binding or activating mutations.
The free energy profile describing the Leu536 transition was computed for each system (apo-WT, WT-E2, apo-Y537S, apo-D538G) and aligned to a common energy scale based on the global minimum corresponding to the buried state (Fig. 3), highlighting distinct differences in the transition pathways. Notably, the buried conformation of Leu536 is substantially lower in energy than the solvent-exposed conformation by 2.00–2.75 kcal/mol, for the WT-E2, apo-WT, and apo-Y573S systems. When comparing the apo-WT (red) to the ligand-bound receptor (WT-E2, green), ligand binding raises the transition state energy by approximately 1 kcal/mol in addition to raising the energy of the solvent-exposed state by approximately 0.5 kcal/mol, suggesting that ligand binding acts to tighten the receptor by burying Leu536. The profile of the apo-Y537S (blue) system closely resembles the ligand-bound profile, despite lacking bound ligand. The profile for the apo-D538G system (purple), in contrast, is profoundly different: The energy of the transition state is reduced by half (1.5 kcal/mol) compared with the apo-WT system, and the energy of the solvent-exposed state is approximately equivalent to that of the buried state. The clear differences in the effects of mutation, as demonstrated by these free energy profiles, suggest that they stabilize the agonist conformation of the receptor by different molecular mechanisms.
Figure 3.
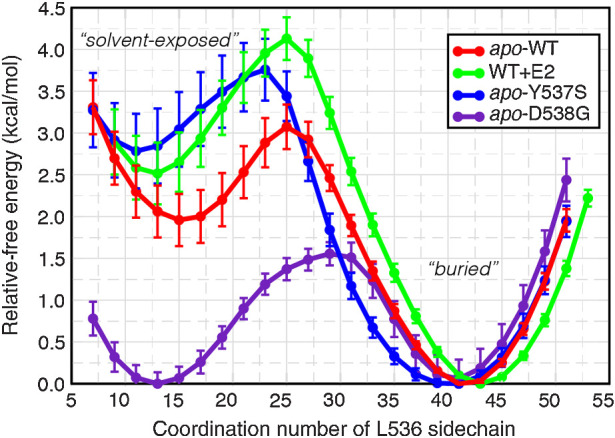
Free energy profile of Leu536 conformational change. The average relative free energy and associated error were computed for conformations of Leu536 rotating from a solvent-exposed conformation (low coordination number) to a buried conformation (high coordination number).
Identifying and characterizing an extended hydrogen-bonding network that serves to overcome the spring-tension of the H11–12 loop and “locks” the active conformation of the receptor
Specific interactions mediating the high affinity of E2 for the ligand-binding pocket are known from structure-activity relationships (42) and more recently, structural biology (43, 44). The extensive sampling accrued during the microsecond-timescale simulations performed here afforded an excellent opportunity to search beyond the obvious ligand-contacting residues for additional ligand-mediated conformational effects, with potential impact on protein–protein interactions that may also be influenced by the activating mutations under investigation. Through careful analysis of the simulation trajectories, tracing out from direct ligand–receptor interactions, we identified an extended hydrogen-bonding network (Fig. 4), which is ligand-initiated and crisscrosses helices critical to establish the active conformation of the receptor. The D-ring hydroxyl group of E2 forms a highly stable interaction with His524, orienting the proton-bearing ε-nitrogen within hydrogen-bonding distance of the Glu419 backbone carbonyl (Fig. 4A), located in a short turn between H7 and H8 that forms a portion of the ligand-binding pocket (along with H3 and H11). Furthermore, the side chain of Glu419 was observed to form a salt bridge with Lys531, located at the C-terminal end of H11, just before the beginning of the H11–12 loop. Because all other systems were simulated in the apo state, only the last two elements of this interaction network were quantified for simulation trajectories. The His524–Glu419 interaction was monitored as the distance between the Nε hydrogen-bond donor and the Glu419 carbonyl oxygen as the acceptor (Fig. 4B). The terminal Glu419–Lys531 salt bridge, however, is a slightly more complicated interaction due to the two oxygen of the glutamic acid for which both the distance and proper geometry required for a strong hydrogen bond need to be accounted for. The presence or absence of this salt bridge was digitized using a hydrogen-bonding distance threshold of 3.0 Å and donor-H–acceptor angle 150° < θ ≤ 180° (Fig. 4C).
Figure 4.

Ligand-mediated hydrogen-bonding network correlates to receptor activity. The stability of key interactions forming a ligand-mediated hydrogen-bond network was quantified from microsecond simulations. A, The network is initiated by the 17β-hydroxyl group of E2 and proceeds through His524:Nε hydrogen bond to the carbonyl of Glu419, terminating with a salt bridge formed between Lys531 and Glu419. B, The His524–Glu419 interaction was monitored by measuring the distance between His524:Nε (donor) and Glu419 carbonyl oxygen (acceptor), whereas (C) the presence of the terminal salt bridge between Glu419 and Lys531 was monitored by digitizing the signal based on geometric constraints.
The presence of the ligand proved critical to stabilizing the His524 conformation required to propagate the interaction network, as the His524–Glu419 interaction was only observed for the WT-E2 system. In the apo-WT system, the terminal salt-bridge interaction between Glu419 and Lys531 was never formed with any prolonged stability in monomer A, and was rapidly disrupted in monomer B (Fig. 4C). It is of note that termination of the initially stable salt bridge in monomer B correlated with the conformational shift of the Val-Val-Pro loop from buried to solvent-exposed (compare loop shift with solvent-exposed for monomer B at t = 200 ns in Fig. 2, Long-Timescale Simulations, with the disruption of the salt bridge in Fig. 4C). In distinct contrast with WT ER, the two mutants were capable of establishing stable Glu419–Lys531 salt-bridge interactions throughout the simulation despite the absence of ligand and without the His524–Glu419 interaction needed to prime the Glu419–Lys531 interaction in WT-E2.
The above observations suggest that the activating mutations change the dynamics of receptor in such a way that Lys531 is capable of adopting a conformation that retains the interaction with Glu419, bypassing the ligand-mediated interactions through His524 that are needed to initiate the activation network in WT receptor. The location of the salt bridge, spanning two helices that form one end of the ligand-binding pocket in a suture-like manner, suggests that it might influence the dynamics of ligand interaction with the LBD by functioning as a “lock” on the closed-pocket conformation, effectively acting as a gate regulating ligand access or egress. Also, because the stability of the Glu419–Lys531 salt bridge requires ligand binding in WT ER but not in the Y537S and D538G mutants, it may be more important in supporting ligand binding and the active state of WT ER than the two activating mutants. On the basis of the hypotheses suggested by this model, we investigated the effects of mutations disrupting the salt bridge experimentally, both as single mutants in WT ER and as double mutants paired with the Y537S or D538G-activating mutations, with the aim of probing the importance of the terminal salt bridge in regulating ligand-binding kinetics and establishing the ligand-independent activity of the two activating ER mutants (vide infra).
The transcriptional activity of constitutively active mutant ERs relies on formation of the glu419–lys531 salt bridge
To investigate the importance of the Glu419–Lys531 salt bridge for ER activity in a cellular context, we assayed ER transcriptional activation in hormone-dependent MCF7 breast cancer cells in which we disrupted the salt bridge by mutation (Fig. 5A). As expected, Y537S and D538G mutants alone have estrogen-independent activity 4–5x greater than WT ER (13). Abrogation of the Glu419–Lys531 salt bridge by mutating either residue to alanine in cis markedly reduced the transcriptional signal from the Y537S and D538G mutant ERs, with E419A diminishing their activity to nearly that of WT ER and K531A also having a marked effect. These results are consistent with the Glu419–Lys531 salt bridge providing substantial stabilization of the active form of the ER LBD that underlies the constitutive activity of the unliganded Y537S and D538G mutant ERs, but having essentially no effect on WT ER basal activity in which it does not form.
Figure 5.

Ligand-binding kinetics and cellular activity of the ER. A, MCF-7 cells were transfected with plasmids for control, WT ERα and the eight indicated mutant ERs, as well as an ER-responsive luciferase plasmid, and constitutive transcriptional activity was monitored in the absence of added estrogen. The * indicates a significance of <0.05 and ** a significance <0.01. B, Ligand association and dissociation rates of the LBDs of WT, Y537S, and D538G ERα were monitored under pseudo first-order conditions using the fluorescent ligand, THC-ketone. Rate constants are shown in the presence of the Glu419–Lys531 salt bridge (gray bars) and in its absence due to the additional E419A (stippled bars). The fold increase in ligand association rate from removal of the salt bridge is indicated by the number above the bars for each receptor.
Differential effects of the Glu419–Lys531 salt bridge on ligand association rates highlights an open, disordered conformation of apo-WT ER versus closed, active conformations of apo-Y537S and D538G ERs.
To measure the coupling between the Glu419–Lys531 salt bridge and the kinetics of ligand binding, we used our inherently solvatochromic fluorescent ligand, THC-ketone (THC), with which one can follow ER–ligand interaction spectroscopically in real time (40, 45). The rate constants for THC association with and dissociation from the LBD (amino acids 304–554) of WT, Y537S or D538G ERs are shown in Fig. 5B, both in the presence of the salt bridge (gray bars) or when the salt bridge is obviated by the E419A mutation (stippled bars), the change that had the greatest effect on ER activity (Fig. 5A).
The different mutations have pronounced effects on ligand association rates, with changes being related to the constitutive activity of the ER: Without the E419A mutation (gray bars), Y537S and D538G ERs have 4- to 6-fold slower association rates than WT ER, suggesting that the Glu419–Lys531 lock, which in the apo state is prominent only in the two mutant ERs, serves as a gate blocking ligand access. Elimination of the salt bridge through E419A mutation has a much greater effect on the ligand association of the two mutant forms (23x and 8.6x for Y537S and D538G, respectively) than the WT (2.5x), with changes essentially eliminating the marked differences in ligand association rates between the WT and activated mutant forms (stippled bars). The effect of the salt-bridge lock on ligand dissociation rates of all three forms of ER (WT, Y537S, or D538G) is less pronounced. Because with ligand bound, the lock is present in all three ER variants, eliminating the salt bridge has a small (2–3-fold) and quite uniform effect on all three ERs.
These findings suggest that the Glu419–Lys531 salt-bridge lock stabilizes a closed, active state of the LBD in WT ER only with bound ligand, but in the Y537S and D538G mutants without needing bound ligand. Notably, in WT ER, ligand binding to the more open ligand-binding pocket is rapid and unimpeded, whereas in apo-ER Y537S and D538G the ligand-binding pocket is closed to the active state that is enforced by formation of the Glu419–Lys531 salt bridge; whereas this conformation supports constitutive activity, it also makes it more difficult for the ligand to bind. Eliminating formation of the salt bridge by the E419A mutation abrogates the difference between the activated mutant ERs and WT ER.
Discussion
In this study, we have used an extensive set of MD simulations and free energy calculations to describe a critical network of interactions in the LBD of ERα that constitutes a conformational switch coupling various structural signals to activation of the receptor. This activation of ER requires folding of H12 in the LBD over the ligand-binding pocket, creating the surface to which coactivators subsequently bind. In WT-ER, the H11–12 loop has a spring-like character that resists this bending and requires stabilization from agonist ligand binding to attain the active conformation; this characteristic of being off without ligand and on with ligand is a fundamental necessity for a ligand-regulated protein (9). By contrast, the two activating ERα mutants can reach the active conformation in the absence of ligand binding: They alter the nature of the H11–12 loop through additional stabilizing interactions, such as by an optimal “latching” hydrogen bond in the case of the Y537S mutation or by release of backbone strain in the H11–12 loop due to the conformationally “relaxing” D538G mutation. A newly recognized, suturing hydrogen-bonding Glu419–Lys531 salt-bridge network also serves as a “lock” that stabilizes the active conformation, operating in the absence of agonist ligand with either activating mutation but requiring ligand binding with WT-ER. We have also verified the effect of these mutations and interactions experimentally. Our use of extended MD simulations to reveal key structural and energetic aspects of constitutively active ER mutant forms and also of unliganded WT ER (for which a crystal structure is not available) could also prove useful in studying the functional nature of aberrant receptor signaling due to mutations in other ligand-regulated proteins.
The Y537S- and D538G-activating mutations use distinct mechanisms to overcome the spring-like nature of H11–12 and access the active conformation without bound ligand
Although both mutations give rise to ligand-independent receptor activity, each mutation operates by a different mechanism (Fig. 6). Mutating Tyr537 to serine changes the hydrogen-bonding partner on H3 from Asn348 to Asp351, with the latter interaction adopting an optimized geometry to yield a more stable hydrogen bond. In the spring-loading model, the optimized hydrogen bond acts as a “latch,” fastening H12 in the active conformation. Mutating Asp538 to glycine, by contrast, changes the backbone torsional profiles of the H11–12 loop to allow for optimal packing of four sequential hydrophobic residues (Val-Val-Pro-Leu). Functionally, this lengthens the spring to “relax” loop strain and lowers the energy associated with the conformational switch of H12. Via these two mechanisms, the activating mutations allow the unliganded receptor to adopt and maintain receptor conformations quantitatively similar to liganded WT ER (Supplementary Fig. S1). The energetic basis for both of these activating mutations was quantitated by the substantial change in the free energy profile of Leu536 rotation between buried and exposed positions. The distinct—latching versus relaxing—mechanisms by which the two mutations overcome the strain of the H11–12 loop in WT ER reflect differences found in both the level of constitutive activity and the effectiveness of antiestrogens in suppressing cell and tumor growth driven by Y537S versus D538G ERs (13, 46, 47).
Figure 6.

The “Spring-Loading” Model consolidating the effects of ligand binding and the activating mutations in ER. In WT receptor, a ligand-mediated hydrogen-bonding network forms, crisscrossing H7, H8, and H11, and terminating with a salt bridge formed across the base of the ligand-binding pocket. In the absence of ligand, His524 is no longer ordered, and the remainder of the network fails to form. Introduction of either the Y537S or D538G mutations, however, overcomes the strain energy of the H11–12 loop to allow the terminal salt bridge to form in the absence of ligand. Specifically, the Y537S mutation yields an optimal hydrogen bond between H3 and H12, operating as a “latch” holding H12 in the activated conformation. The D538G mutation, by contrast, induces a partial unwinding of H12, which serves to relax the backbone strain energy of the spring-like loop.
Elucidating the specific details of each mutation observed to drive ligand-independent activation of ER within the context of the spring-loading model also provides a framework to understand additional mutations that have arisen since our first investigations (9, 48). At position Tyr537, additional mutations (Asn, Asp, Cys) giving rise to ligand-independent activity have been reported (9), all of which potentially change the hydrogen bonding preferences similar to that observed for the serine mutant (Y537S) described herein. Although not explicitly discussed above, Leu536 occupies the fourth position of the hydrophobic sequence in the H11–12 loop and its presence is critical to providing the loop strain associated with the fidelity of the on–off conformational switching. Point mutations to Arg, His, Asn, or Pro at this position have been observed in metastatic breast cancers and show ligand-independent activity, likely due to reduced loop strain by converting a hydrophobic residue to charged or polar—whereby the side chain adopts a less strained, solvent-exposed position—or by the different backbone preference of the cyclic proline residue.
A hydrogen-bonding network that is ligand-dependent in WT ER but intrinsic in the mutants, locks a fully closed form of the ligand-binding pocket and functions as a gateway for ligand transit
Our extended simulation dataset combined with advanced free energy techniques allowed us to characterize the dynamics of several structural elements that were well correlated with receptor activity. We identified an extended hydrogen-bonding network initialized in WT ER through a ligand interaction with His524, propagating through Glu419, and terminating with Lys531. This newly recognized network crisscrosses between elements of the helix bundle that form the base of the ligand-binding pocket (Helices 7, 8, 11), holding them together like a suture that “locks” the pocket shut. Although the Glu419–Lys531 salt bridge was not observed in WT ER in the absence of ligand, the Y537S or D538G mutations alter receptor dynamics sufficiently to maintain the terminal interaction of the Glu419–Lys531 hydrogen-bonding network without requiring the presence of ligand, thereby bypassing the initiating His524–E419 ligand-binding interactions required for WT ER activation.
The Glu419–Lys531 salt bridge affects both ER activity and ligand-binding kinetics, which we have probed by disrupting it by mutation. Changing either salt-bridge partner to alanine markedly reduced the constitutive activity of the Y537S and D538G mutant ERs, but had little effect on WT ER. We found that the activating mutant ER LBDs have markedly slower rates of ligand association than WT ER, confirming that even in the absence of ligand, the two activating mutations stabilize an active form of the LBD with H12 folded to cover and seal an empty ligand-binding pocket, whereas the pocket is open and more accessible in apo WT ER. Consequently, the E419A mutation, which disrupts the locking salt bridge, greatly accelerates ligand association for the Y537S and D538G mutant ER but has little effect on WT ER.
The H11–12 spring force keeping WT-ER LBD off in the absence of ligand gives it the “fly-casting” characteristics of intrinsically disordered domains and results in a pronounced ligand-dependent on–off character
In our model for receptor activation, the short H11–12 loop acts as a spring that presents an energetic barrier for the H12 conformational switch from an inactive to an active state in the absence of ligand. The LBD of apo-WT ER adopts an inactive conformation that appears to be intrinsically disordered (41, 45). This has been an obstacle to obtaining crystal structures of this unliganded domain, and in cells, apo-WT ER is known to be bound by HSPs (1). Ligand binding drives a conformational change that releases the HSPs, resulting in the recruitment of coactivator proteins and initiation of the transcriptional cascade driving expression of ER-responsive genes (49, 50). The intrinsic disorder of WT-ER LBD is found in many binding proteins (51), and it conveys an open-like character that enables them to search for, recognize, and bind cognate ligands rapidly and efficiently through a so-called “fly-casting” mechanism (52, 53).
The activation of WT ER by ligand binding requires that the opposing spring force of the H11–12 loop be overcome as H12 moves into the active conformation. The energy to overcome this spring force is presumed to come both from new ligand–protein interactions as well as favorable protein–protein contacts in protein–ligand complex, which is now sufficiently ordered such that crystal structures can be obtained. The equilibrium between the disordered off-state dominating the apo-WT ER and more ordered state of liganded WT-ER determines the magnitude of the on–off signaling.
Combining our spring-loading model for ligand signaling with the fly-casting model of ligand-induced receptor folding provides a direct mechanism whereby subtle changes to receptor structure that mimic ligand-binding events can erode the fidelity of the on–off switch in ER and give rise to ligand-independent activation, as observed for the Y537S and D538G mutations found in metastatic breast cancers. They also highlight the Glu419–Lys531 salt bridge as one of the protein–protein stabilizing interactions in the active conformation. Very likely, there are other protein–protein interactions of similar nature that could potentially stabilize the active conformation of the ER LBD and result in ligand-independent activity.
Marshalling the computational and analytic tools needed to characterize the energy landscape of mutated proteins in cancer
The analyses we have undertaken in this report have relied on two critical—often limiting—factors in MD simulations: Long-timescale simulations and strategic replica-based free energy calculations. By using a computer with special facility for MD simulations (Anton; refs. 32, 54), we are able to address a major practical barrier, which is to sample enough in time to capture evidence of the biophysical phenomena under observation. In our case, by sampling up to one microsecond, we were finally able to see a convergence in Leu536 positioning that we suspected from other x-ray crystal structures but eluded us in the shorter, replicate runs. And, by using sophisticated biasing methods with careful selection of reaction coordinates, we are able to address a technical barrier in achieving converged free energy calculations, to rigorously quantify biophysical changes that are directly linked to structural changes driving receptor activation. It is worth noting that, more recently, the traditional limitations in computational resources are being overcome by advances in GPU computing (55–62) and wider access to new academic supercomputers (63, 64). Furthermore, replica-based free energy methods, such as BEUS, while challenging to perform, have become more accessible to non-mathematicians by recent advances in best practices (34, 65, 66) and the availability of several toolsets (35, 67–74). Hence, extended MD simulations and energy landscape exploration should be easier to carry out in the future by experts and non-experts alike.
Finally, by identifying a critical suturing network spanning H11 with the H7/8 turn that serves to lock the ER LBD in an active conformation—without ligand in the ER mutants but requiring ligand in WT ER—we have localized a portion of the LBD that might be targeted through future design of inhibitors tailored more specifically to inhibit these constitutively active ERα mutant forms. Where good crystal structures are available for cancer regulatory proteins in which mutations have important effects on activity, the extended MD simulations enabling detailed energy landscape analyses illustrated here might be applied fruitfully to reveal specific interactions that are responsible for the phenotypic behavior of the mutant protein.
Authors’ Disclosures
C.G. Mayne reports other support from Celgene Corporation and Loxo Oncology at Lilly outside the submitted work; as well as patent 20200199073 and 20200199074 pending. K.E. Carlson reports grants from NIH and Breast Cancer Research Foundation during the conduct of the study. G.L. Greene reports grants, personal fees, and non-financial support from Sermonix Pharmaceuticals and grants and non-financial support from Olema Pharmaceuticals outside the submitted work. S. Chandarlapaty reports personal fees from Sanofi, Novartis, Lilly, as well as grants from Daiichi-Sankyo, and grants and personal fees from Paige.ai, as well as personal fees from Sermonix, Context Therapeutics, and Bristol-Myers Squibb outside the submitted work; as well as a patent for ER PROTACs pending. J.A. Katzenellenbogen reports grants from NIH-NCI, NIH-DDK, and Breast Cancer Research Foundation during the conduct of the study. No disclosures were reported by the other authors.
Acknowledgments
The authors acknowledge funding from the National Institutes of Health grants P41-GM104601 (to E. Tajkhorshid), R01-CA204999 and P30-CA008748 (to S. Chandarlapaty) and R01-CA220284 (to J.A. Katzenellenbogen and B.S. Katzenellenbogen), and from the Breast Cancer Research Foundation grant BCRF 18–083 (to J.A. Katzenellenbogen). This work used computational resources provided by XSEDE, supported by the National Science Foundation grant number ACI-1548562 through allocation MCA06N060, and the Pittsburgh Supercomputing Center (PSC), supported by the National Institutes of Health grant number R01-GM116961 through allocation PSCA16082P (both to E. Tajkhorshid).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Authors' Contributions
C.G. Mayne: Conceptualization, data curation, software, formal analysis, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. W. Toy: Data curation, formal analysis, investigation, writing–review and editing. K.E. Carlson: Formal analysis, investigation, visualization, writing–review and editing. T. Bhatt: Investigation. S.W. Fanning: Data curation, formal analysis, writing–review and editing. G.L. Greene: Conceptualization, resources, supervision, funding acquisition, writing–original draft, project administration, writing–review and editing. B.S. Katzenellenbogen: Conceptualization, supervision, writing–original draft, writing–review and editing. S. Chandarlapaty: Conceptualization, resources, supervision, funding acquisition, validation, writing–original draft, project administration, writing–review and editing. J.A. Katzenellenbogen: Conceptualization, resources, supervision, funding acquisition, validation, writing–original draft, project administration, writing–review and editing. E. Tajkhorshid: Conceptualization, resources, supervision, funding acquisition, writing–original draft, project administration, writing–review and editing.
References
- 1. The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 2018;34:427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria JC, et al. Mutational profile of metastatic breast cancers: a retrospective analysis. PLoS Med 2016;13:e1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature 2007;450:964–72. [DOI] [PubMed] [Google Scholar]
- 5. Berendsen H. Collective protein dynamics in relation to function. Curr Opin Struct Biol 2000;10:165–9. [DOI] [PubMed] [Google Scholar]
- 6. Adcock SA, McCammon JA. Molecular dynamics: survey of methods for simulating the activity of proteins. Chem Rev 2006;106:1589–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang LQ, Sang P, Tao Y, Fu YX, Zhang KQ, Xie YH, et al. Protein dynamics and motions in relation to their functions: several case studies and the underlying mechanisms. J Biomol Struct Dyn 2013;32:372–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Puhalla S, Bhattacharya S, Davidson NE. Hormonal therapy in breast cancer: a model disease for the personalization of cancer care. Mol Oncol 2012;6:222–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Katzenellenbogen JA, Mayne C, Katzenellenbogen BS, Greene GL, Chandarlapaty S. Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat Rev Cancer 2018;18:377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D, et al. Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol 2016;2:1310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun 2016;7:11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fanning SW, Mayne C, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. eLife 2016;5:e12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov 2017;7:277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Papaleo E, Saladino G, Lambrughi M, Lindorff-Larsen K, Gervasio FL, Nussinov R. The role of protein loops and linkers in conformational dynamics and allostery. Chem Rev 2016;116:6391–423. [DOI] [PubMed] [Google Scholar]
- 15. Wärnmark A, Treuter E, Gustafsson JÅ, Hubbard RE, Brzozowski AM, Pike ACW. Interaction of transcriptional intermediary factor 2 nuclear receptor box peptides with the coactivator binding site of estrogen receptor alpha. J Biol Chem 2002;277:21862–8. [DOI] [PubMed] [Google Scholar]
- 16. Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett 1999;314:141–51. [Google Scholar]
- 17. Sugita Y, Kitao A, Okamoto Y. Multidimensional replica-exchange method for free-energy calculations. J Chem Phys 2000;113:6042–51. [Google Scholar]
- 18. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem 2005;26:1781–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Phillips JC, Hardy DJ, Maia JDC, Stone JE, Ribeiro JV, Bernardi RC, et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J Chem Phys 2020;153:044130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yin D, MacKerell AD. Combined ab initio/empirical approach for optimization of Lennard–Jones parameters. J Comput Chem 1998;19:334–48. [DOI] [PubMed] [Google Scholar]
- 21. MacKerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 2004;25:1400–15. [DOI] [PubMed] [Google Scholar]
- 22. Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ1 and χ2 dihedral angles. J Chem Theory Comput 2012;8:3257–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983;79:926–35. [Google Scholar]
- 24. Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem 2010;31:671–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vanommeslaeghe K, MacKerell AD. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J Chem Inf Model 2012;52:3144–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vanommeslaeghe K, Raman EP, MacKerell AD. Automation of the CHARMM general force field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model 2012;52:3155–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mayne C, Saam J, Schulten K, Tajkhorshid E, Gumbart JC. Rapid parameterization of small molecules using the force field toolkit. J Comput Chem 2013;34:2757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys 1994;101:4177–89. [Google Scholar]
- 29. Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: the langevin piston method. J Chem Phys 1995;103:4613–21. [Google Scholar]
- 30. Darden T, York D, Pedersen L. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J Chem Phys 1993;98:10089–92. [Google Scholar]
- 31. Bowers KJ, Chow E, Xu H, Dror RO, Eastwood MP, Gregersen BA, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. SC '06 In: Proceedings of the 2006 ACM/IEEE conference on Supercomputing; 2006; New York, NY: ACM; 2006:84. [Google Scholar]
- 32. Shaw DE, Deneroff MM, Dror RO, Larson RH, Salmon JK, Young C, et al. Anton, a special-purpose machine for molecular dynamics simulation. Comput Architect News ACM; 2007;35:1–12. [Google Scholar]
- 33. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics 1996;14:33–8. [DOI] [PubMed] [Google Scholar]
- 34. Moradi M, Tajkhorshid E. Computational recipe for efficient description of large-scale conformational changes in biomolecular systems. J Chem Theory Comput 2014;10:2866–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fiorin G, Klein ML, Hénin J. Using collective variables to drive molecular dynamics simulations. Mol Phys 2013;111:3345–62. [Google Scholar]
- 36. Bennett CH. Efficient estimation of free energy differences from Monte Carlo data. J Comput Phys 1976;22:245–68. [Google Scholar]
- 37. Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. The weighted histogram analysis method for free-energy calculations on biomolecules. I. the method. J Comput Chem 1992;13:1011–21. [Google Scholar]
- 38. Bartels C. Analyzing biased Monte Carlo and molecular dynamics simulations. Chem Phys Lett 2000;331:446–54. [Google Scholar]
- 39. Rubin DB. The bayesian bootstrap. Annal Stat 1981;9:130–4. [Google Scholar]
- 40. Hwang KJ, Carlson KE, Anstead GM, Katzenellenbogen JA. Donor-acceptor tetrahydrochrysenes, inherently fluorescent, high-affinity ligands for the estrogen receptor: binding and fluorescence characteristics and fluorometric assay of receptor. Biochemistry 1992;31:11536–45. [DOI] [PubMed] [Google Scholar]
- 41. Carlson KE, Choi I, Gee A, Katzenellenbogen BS, Katzenellenbogen JA. Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry 1997;36:14897–905. [DOI] [PubMed] [Google Scholar]
- 42. Wallace OB, Richardson TI, Dodge JA. Estrogen receptor modulators: relationships of ligand structure, receptor affinity and functional activity. Curr Top Med Chem 2003;3:1663–80. [DOI] [PubMed] [Google Scholar]
- 43. Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engström O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997;389:753–8. [DOI] [PubMed] [Google Scholar]
- 44. Pike ACW, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, et al. Structural insights into the mode of action of a pure antiestrogen. Structure 2001;9:145–53. [DOI] [PubMed] [Google Scholar]
- 45. Gee AC, Carlson KE, Martini PGV, Katzenellenbogen BS, Katzenellenbogen JA. Coactivator peptides have a differential stabilizing effect on the binding of estrogens and antiestrogens with the estrogen receptor. Mol Endocrinol 1999;13:1912–23. [DOI] [PubMed] [Google Scholar]
- 46. Zhao Y, Laws MJ, Guillen VS, Ziegler Y, Min J, Sharma A, et al. Structurally novel antiestrogens elicit differential responses from constitutively active mutant estrogen receptors in breast cancer cells and tumors. Cancer Res 2017;77:5602–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell 2018;33:173–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weis KE, Ekena K, Thomas JA, Lazennec G, Katzenellenbogen BS. Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol Endocrinol 1996;10:1388–98. [DOI] [PubMed] [Google Scholar]
- 49. Smith DF, Toft DO. Minireview: the intersection of steroid receptors with molecular chaperones: observations and questions. Mol Endo 2008;22:2229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med 2003;228:111–33. [DOI] [PubMed] [Google Scholar]
- 51. Staby L, O'Shea C, Willemoës M, Theisen F, Kragelund BB, Skriver K. Eukaryotic transcription factors: paradigms of protein intrinsic disorder. Biochem J 2017;474:2509–32. [DOI] [PubMed] [Google Scholar]
- 52. Jain VP, Tu RS. Coupled folding and specific binding: fishing for amphiphilicity. Int J Mol Sci 2011;12:1431–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Trizac E, Levy Y, Wolynes PG. Capillarity theory for the fly-casting mechanism. Proc Natl Acad Sci USA 2010;107:2746–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shaw DE, Grossman JP, Bank JA, Batson B, Butts JA, Chao JC, et al. Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. In SC'14: Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; 2014; New Orleans, LA;41–52. [Google Scholar]
- 55. Stone JE, Phillips JC, Freddolino PL, Hardy DJ, Trabuco LG, Schulten K. Accelerating molecular modeling applications with graphics processors. J Comput Chem 2007;28:2618–40. [DOI] [PubMed] [Google Scholar]
- 56. Harvey MJ, Giupponi G, Fabritiis GD. ACEMD: accelerating biomolecular dynamics in the microsecond time scale. J Chem Theory Comput 2009;5:1632–9. [DOI] [PubMed] [Google Scholar]
- 57. Eastman P, Pande VS. Efficient nonbonded interactions for molecular dynamics on a graphics processing unit. J Comput Chem 2010;31:1268–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, et al. OpenMM 7: rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol 2017;13:e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J Chem Theory Comput 2013;9:3878–88. [DOI] [PubMed] [Google Scholar]
- 60. Kutzner C, Páll S, Fechner M, Esztermann A, de Groot BL, Grubmüller H. Best bang for your buck: GPU nodes for GROMACS biomolecular simulations. J Comput Chem 2015;36:1990–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stone JE, Hynninen A-P, Phillips JC, Schulten K. Early experiences porting the NAMD and VMD molecular simulation and analysis software to GPU-accelerated OpenPOWER platforms. High Perform Comput 2016;9945:188–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen H, Maia JDC, Radak BK, Hardy DJ, Cai W, Chipot C, et al. Boosting free-energy perturbation calculations with GPU-Accelerated NAMD. J Chem Inf Model 2020;60:5301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A, et al. XSEDE: accelerating scientific discovery. Comput Sci Eng 2014;16:62–74. [Google Scholar]
- 64. Bode B, Butler M, Dunning T, Hoefler T, Kramer W, Gropp W, et al. The blue waters super-system for super-science. Contemporary high performance computing. Chapman and Hall/CRC; Boca Raton, FL, 2013. p. 339–66. [Google Scholar]
- 65. Hansen N, van Gunsteren WF. Practical aspects of free-energy calculations: a review. J Chem Theory Comput 2014;10:2632–47. [DOI] [PubMed] [Google Scholar]
- 66. Pietrucci F. Strategies for the exploration of free energy landscapes: unity in diversity and challenges ahead. Rev Phys 2017;2:32–45. [Google Scholar]
- 67. Roe DR, Cheatham TE. III. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 2013;9:3084–95. [DOI] [PubMed] [Google Scholar]
- 68. Tribello GA, Bonomi M, Branduardi D, Camilloni C, Bussi G. PLUMED 2: new feathers for an old bird. Comput Phys Commun 2014;185:604–13. [Google Scholar]
- 69. Jiang W, Phillips JC, Huang L, Fajer M, Meng Y, Gumbart JC, et al. Generalized scalable multiple copy algorithms for molecular dynamics simulations in NAMD. Comput Phys Commun 2014;185:908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kobayashi C, Jung J, Matsunaga Y, Mori T, Ando T, Tamura K, et al. GENESIS 1.1: a hybrid-parallel molecular dynamics simulator with enhanced sampling algorithms on multiple computational platforms. J Comput Chem 2017;38:2193–206. [DOI] [PubMed] [Google Scholar]
- 71. Ferguson AL. BayesWHAM: a Bayesian approach for free energy estimation, reweighting, and uncertainty quantification in the weighted histogram analysis method. J Comput Chem 2017;38:1583–605. [DOI] [PubMed] [Google Scholar]
- 72. Lervik A, Riccardi E, van Erp TS. PyRETIS: a well-done, medium-sized python library for rare events. J Comput Chem 2017;38:2439–51. [DOI] [PubMed] [Google Scholar]
- 73. Sidky H, Colón YJ, Helfferich J, Sikora BJ, Bezik C, Chu W, et al. SSAGES: software suite for advanced general ensemble simulations. J Chem Phys 2018;148:044104. [DOI] [PubMed] [Google Scholar]
- 74. Treikalis A, Merzky A, Chen H, Lee T, York DM, Jha S. RepEx: a flexible framework for scalable replica exchange molecular dynamics simulations [abstract]. In: Proceedings of the 2016 45th International Conference on Parallel Processing (ICPP): ICPP; 2016628–37. [Google Scholar]