

Abstract
Epstein-Barr virus (EBV) is a human gammaherpesvirus associated with the development of hematopoietic cancers of B-lymphocyte origin, including AIDS-related non-Hodgkin lymphoma (AIDS-NHL). Primary infection of B-cells with EBV results in their polyclonal activation and immortalization. The transferrin receptor 1 (TfR1), also known as CD71, is important for iron uptake and regulation of cellular proliferation. TfR1 is highly expressed in proliferating cells, including activated lymphocytes and malignant cells. We developed a mouse/human chimeric antibody targeting TfR1 (ch128.1/IgG1) that has previously shown significant anti-tumor activity in immunosuppressed mouse models bearing human malignant B-cells, including multiple myeloma (MM) and AIDS-NHL cells. Here, we examined the effect of targeting TfR1 to inhibit EBV-driven activation and growth of human B-cells in vivo using an immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mouse model. Mice were implanted with T-cell depleted, human peripheral blood mononuclear cells (PBMCs), either without EBV (EBV−), or exposed to EBV in vitro (EBV+) intravenously via the tail vein. Mice implanted with EBV+ cells and treated with an IgG1 control antibody (400 μg/mouse) developed lymphoma-like growths of human B-cell origin that were EBV+, while mice implanted with EBV+ cells and treated with ch128.1/IgG1 (400 μg/mouse) showed increased survival and significantly reduced inflammation and B-cell activation. These results indicate that ch128.1/IgG1 is effective at preventing the growth of EBV+ human B-cell tumors in vivo, thus, indicating that there is significant potential for agents targeting TfR1 as therapeutic strategies to prevent the development of EBV-associated B-cell malignancies.
Keywords: Transferrin receptor 1, Epstein-Barr virus, human B-lymphocytes, antibody cancer therapy, lymphomagenesis
Introduction
Epstein-Barr virus (EBV) is a ubiquitous B-cell-tropic gammaherpesvirus that infects approximately 95% of adults worldwide (1,2). Primary infection with EBV is mostly asymptomatic among healthy, immunocompetent individuals, and in some cases, can result in infectious mononucleosis (3). EBV establishes persistent and lifelong infection of B-cells, a natural reservoir of EBV latency, thus requiring individuals to establish vigorous, life-long humoral and cellular immune responses (2,3). EBV also has significant oncogenic potential. EBV-associated cancers are of particular concern among immunodeficient individuals, such as those infected with human immunodeficiency virus (HIV), or organ-transplant recipients, who are immunosuppressed to avoid transplant rejection, as these persons are unable to effectively control the growth of EBV-infected cells (1,2). EBV infection is associated with the development of approximately 1.5% of all cancers worldwide, and has been implicated in the pathogenesis of several B-cell-derived hematopoietic cancers, such as AIDS-related non-Hodgkin lymphoma (NHL) and Hodgkin lymphoma (HL), as well as endemic (African) Burkitt lymphoma, primary effusion lymphoma (PEL), and post-transplant lymphoproliferative disease (PTLD) (1,2). EBV has also been found to be associated with non-hematopoietic cancers of epithelial cell origin, including nasopharyngeal carcinoma and gastric cancer (4). Initial infection of B-cells with EBV results in polyclonal B-cell activation, driving cellular proliferation, and immunoglobulin (Ig) production (5–7). Days following initial infection, EBV-infected B-cells undergo transformation and immortalization, thus leading to EBV+ clones that grow indefinitely in the absence of effective immune system control (3,5–7). After 6–8 weeks post-EBV infection in vitro, the emergence of transformed, immortalized B-cell lymphoblastoid cell lines (B-LCLs) is seen, characterized by the outgrowth of one or more dominant clones, with a marked increase in cell number and cellular proliferation (2,8,9). EBV-associated hematopoietic cancers arise from ineffective immune surveillance of EBV-infected B-cells, thus, allowing these cells to undergo activation and transformation, and then to evolve into lymphoma-like growths (2,3). Therefore, a therapeutic intervention that can effectively prevent EBV-driven B-cell activation and/or transformation can potentially prevent the development of PTLD or B-cell hematopoietic cancers, such as AIDS-NHL.
The transferrin receptor 1 (TfR1), also known as CD71, is type II transmembrane homodimeric protein involved in iron uptake and regulation of cellular proliferation (10–12). Iron is a cofactor of intracellular enzymes including the ribonucleotide reductase that is coupled to DNA synthesis and, thus, required for cellular proliferation (10–12). TfR1 is aberrantly expressed at high levels on malignant cells and is associated with tumorigenesis and cancer progression, and the poor prognosis of different cancer types including hematopoietic malignancies (10–12). Interestingly, the presence of intracellular iron confers protection to malignant cells from natural killer (NK) cells (13) and the heavy chain of ferritin (iron-storing protein) inhibits apoptosis induced by the tumor necrosis factor alpha (TNFα) due to the suppression of reactive oxygen species (ROS) (14). TfR1 also mediates NF-κB signaling in cancer cells through the interaction with the inhibitor of the NF-κB kinase (IKK) complex, increasing cancer cell survival (15). In addition, NF-κB induces TfR1 expression via regulation of Hypoxia inducible factor-1α (HIF-1α) levels (16–18), creating another connection between NF-κB and TfR1. Moreover, TfR1 can contribute to malignant cell growth and survival modulating mitochondrial respiration and ROS generation (19). Collectively, the TfR1 overexpression on cancer cells and its relevant role in malignant cell pathology makes this receptor an attractive target for antibody therapy.
We have previously developed mouse/human chimeric IgG3/kappa and IgG1/kappa antibodies targeting human TfR1 as a possible cancer therapy (20–26). These chimeric antibodies (ch128.1/IgG3 and ch128.1/IgG1) contain the variable regions of the murine monoclonal antibody 128.1. They do not inhibit the binding of Tf (meaning they are non-neutralizing antibodies) or the hemochromatosis protein (HFE) to TfR1 (24,25), and they do not cross-react with murine TfR1 (27). We have also found that ch128.1/IgG3 and ch128.1/IgG1 exhibit significant anti-tumor activity in immunosuppressed mouse models bearing human malignant B-cells, including multiple myeloma (MM) and AIDS-NHL (20–23,26).
The goal of this study was to determine, for the first time, the effectiveness of the anti-TfR1 antibody ch128.1/IgG1 in its ability to inhibit the activation, growth, and immortalization of EBV-infected B-cells in vivo. To accomplish this, we utilized an immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mouse model in which mice were implanted with EBV-exposed human primary B-cells, or B-cells not exposed to EBV, followed by two treatments of either ch128.1/IgG1 or isotype control IgG1 monoclonal antibody. We measured survival of mice, characterized any lymphoma-like growths that developed, and quantified plasma levels of human inflammatory cytokines, chemokines, and soluble receptors of B-cell activation and proliferation, and measured levels of human antibodies in plasma.
Materials and Methods
Ethics statement
All experimental protocols were approved by the University of California, Los Angeles Animal Research Committee and all local, and national guidelines on the care of animals were strictly adhered to.
Isolation of human B-cell-enriched preparations
Peripheral blood mononuclear cells (PBMCs) isolated from healthy anonymous blood donors were obtained from the UCLA AIDS Institute Virology Core laboratory. B-cell-enriched preparations were obtained by incubating PBMCs with superparamagnetic beads coupled with anti-human CD3 antibody to enable the depletion of human CD3+ T-cells (Dynabeads® CD3, Dynal Biotech, Oslo, Norway, catalog no. 11151D).
Infection of human B-cell enriched preparations by EBV
Human primary B-cells were infected with EBV by exposing T-cell-depleted PBMC preparations to supernatants from B95–8 EBV-infected marmoset cells, which contain a high concentration of infectious EBV, as previously described (5). T-cell depleted PBMCs were exposed to EBV supernatants or media alone for two hours in vitro and then washed withphosphate buffered saline (PBS) (Gibco, Life Technologies, catalog no. 10-019-049). EBV-exposed cells were then cultured in vitro for seven days. After the seven day culture, the EBV+ B-cell enriched preparations were washed twice with PBS, and subsequently administered to mice by intravenous (i.v.) injection via the tail vein.
Preparation of antibodies
For proof-of-principle, we selected the anti-TfR1 antibody ch128.1/IgG1 that was recently developed (21). A mouse/human chimeric IgG1 antibody specific for the hapten dansyl (5-dimethylamino naphthalene-1-sulfonyl chloride) (28) was used as an isotype control antibody (IgG1 control). Both antibodies were expressed in murine myeloma cells, which were grown in roller bottles, and antibodies were purified using affinity chromatography, as previously described (20–25).
Implantation of human B-cell enriched preparations into NSG mice
Immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were used for these studies (The Jackson Laboratory, Bar Harbor, Maine). A group of mice was implanted with EBV− (no EBV) cells on day 0 (6 × 106 cells in 0.25 ml of PBS per mouse) via i.v. injection. A week later, a second group of mice were implanted with EBV-exposed B-cell enriched preparations (EBV+) (6 × 106 cells in 0.25 ml of PBS per mouse) via i.v. injection.
ch128.1/IgG1 and IgG1 control antibody treatments
Mice were treated by i.v. injections via the tail vein with 400 μg of ch128.1/IgG1 or IgG1 control antibody at 2 days post-cell implantation with EBV− or EBV+ B-cells, respectively. A second antibody treatment (i.v.) was performed 28 days after the first antibody treatment. Animals were monitored for tumor development and euthanized when moribund or when they showed overt signs of illness or physical distress. Survival was recorded as the number of days from cell implantation to euthanasia. At necropsy of mice that did and did not develop tumors, blood was collected via cardiac puncture in anticoagulant-treated (ethylenediamine tetraacetic acid, EDTA) 1.5-ml microcentrifuge tubes and spleen, lymph node, and liver tissues were collected for all mice in PBS in 15 ml conical tubes. Whole blood was centrifuged at 400 × g for 30 min at room temperature; plasma was then collected and stored at −800C. Once all mice were euthanized, stored plasma was used for analysis of human cytokines, B-cell activation molecules, and immunoglobulins (Ig), as described below. Tissues from mice that developed tumor-like growths were also obtained at necropsy. The remaining, surviving mice that did not show signs of malaise or tumor development were euthanized at or after 150 days post-cell implantation with EBV− or EBV+ B-cells.
Flow cytometry
Multicolor flow cytometry was carried out to determine the presence of human immune cells in mice, as previously described (29). Briefly, cells were isolated from tissues by mechanical dissociation using a rubber syringe tip to grind up tissue and liberate cells. Cells were then suspended in red blood cell (RBC) lysis buffer, passed through 70 μm cell strainers (Corning Inc., Corning, New York, catalog no. 352350), and incubated for 5 min at room temperature to lyse RBCs. Cells were then washed once with flow cytometry staining buffer (FACS buffer) (0.5% bovine serum albumin (BSA) in PBS) and then 5 μl of Super Bright Complete Staining Buffer, eBioscience™ (Thermo Fisher Scientific, catalog no. SB-4401–42) was added to each sample prior to introducing the antibody cocktail mix, which is used when more than one Super Bright polymer-dye conjugated antibody is added to the same sample to prevent non-specific polymer interactions. An antibody cocktail was then added consisting of: anti-human monoclonal anotibodies: CD3 (OKT3), Super Bright 600, eBioscience™ (Thermo Fisher Scientific, catalog no. 63-0037-42); CD4 (SK3), PE-Cyanine 7, eBioscience™ (Thermo Fisher Scientific, catalog no. 25-0047-42); CD8a (RPA-T8), APC-eFluor 780, eBioscience™ (Thermo Fisher Scientific, catalog no. 47-0088-42); CD45 (HI30), eFluor 450, eBioscience™ (Thermo Fisher Scientific, catalog no. 48-0459-42); CD19 (SJ25C1), PerCP-Cyanine5.5, eBioscience™ (Thermo Fisher Scientific, catalog no. 45-0198-42); CD274 (PD-L1, B7-H1) (MIH1), APC, eBioscience™ (Thermo Fisher Scientific, catalog no. 17-5983-42); and rat anti-mouse CD45 monoclonal antibody (30-F11), Super Bright 645, eBioscience™ (Thermo Fisher Scientific, catalog no. 64-0451-82). Cells were then incubated with the antibody cocktail for 20 minutes at 4oC, washed once in FACS buffer, and centrifuged at 1,500 rpm for 5 min. After the centrifugation, the solution was discarded and the cell pellet was resuspended in approximately 0.25 ml of cold 1% paraformaldehyde (PFA) solution in PBS. Cells were analyzed by flow cytometry using a BD LSRFortessa™ X-20 Cell Analyzer (BD Biosciences, San Jose, California), and data were analyzed with the FCS Express software program (v7.0, De Novo Software, Pasadena, California).
Immunohistochemistry (IHC) studies
Tissues from mice that developed tumor-like growths were obtained at necropsy, fixed in 10% neutral buffered formalin for 7 days, and then placed in 70% ethanol for 1–3 days. Tissues were then submitted to the Translational Pathology Core Laboratory (TPCL) in the UCLA Department of Pathology and Laboratory Medicine for paraffin embedding, tissue sectioning, and IHC. IHC was performed using the following antibodies: polyclonal rabbit anti-human Kappa (κ) Light Chain (Agilent, catalog no. A019102–2); polyclonal rabbit anti-human Lambda (λ) Light Chain (Agilent, catalog no. A019302–2); monoclonal mouse anti-Epstein-Barr Virus, LMP Clone CS.1–4 (Agilent, catalog no. M0897); polyclonal rabbit anti-human CD3 (Agilent, catalog no. GA50361–2); and monoclonal mouse anti-human CD19 (Clone LE-CD19) (Agilent, catalog no. M729629–2). Tissues were then examined for features characteristic of lymphoma-like tumors, including the presence of B-cells (CD19 positivity), evidence of EBV-infection (LMP1 staining), and evidence for monoclonality such as staining for either human Ig κ or λ light chain (LC). An anti-rabbit HRP (horseradish peroxidase)-labeled polymer conjugated secondary antibody (EnVision+/HRP polymer, rabbit) (Agilent, catalog no. K400311–2) was used to recognize rabbit anti-human primary antibodies. An anti-mouse, HRP-labeled polymer conjugated secondary antibody (EnVision+/HRP polymer, mouse) (Agilent, catalog no. K400111–2) was used to recognize mouse anti-human primary antibodies. For detection of rabbit or mouse specific antibodies, a DAB (3,3’-Diaminobenzidine) chromogen and DAB substrate buffer kit was used (DAB Chromogen Kit, catalog no. DB801L) (Biocare Medical, Pacheco, California).
ELISA and multiplex immunometric assays
Human Ig κ and λ free light chain (FLC) was quantified in mouse plasma samples by ELISA (BioVendor, Brno, Czech Republic, catalog no. RD194088100R) according to manufacturer’s instructions. Multiplexed-immunometric assays were carried out using a custom-made panel (R&D Systems, Minneapolis, Minnesota, catalog no. LHSCM and LXSAHM) consisting of key human pro-inflammatory (IL-1β, IL-2, IL-6, IL-8, IFN-γ, TNF-α, soluble CD163) and anti-inflammatory (IL-10) molecules, chemokines (CXCL10, CXCL13), vascular endothelial growth factor (VEGF), tissue inhibitor matrix metalloproteinase 1 (TIMP-1)), and molecules associated with B-cell activation and/or survival (IL-4, tumor necrosis factor receptor II (TNF-RII), B-cell activating factor (BAFF), soluble CD14 (sCD14), soluble CD25 (sCD25), and soluble CD27 (sCD27)), as previously described (30). A separate multiplex panel was used for the simultaneous quantification of human immunoglobulins (IgA, IgG1, IgG2, IgG3, IgG4, and IgM) (Bio-Rad, Hercules, California, catalog no. 171A3100M). Plasma levels of these molecules were determined using the Luminex (Austin, Texas) multiplex assay platform with the custom-made panels produced by R&D Systems. Briefly, Luminex microparticles precoated with analyte-specific antibodies were incubated with diluted plasma samples, followed by a biotin antibody and by a streptavidin-phycoerythrin conjugate. The fluorescence intensity of each analyte’s microparticles was quantified using a BioPlex 200 (Luminex) System Analyzer (Bio-Rad, Hercules, California), and the data were analyzed using BioPlex Manager (v 4.1.1) software (Bio-Rad, Hercules, California). The lower limit of detection (LLD) for each biomarker was set either as the lowest value that the BioPlex Manager software could calculate using the standard curve or as the lowest value of the standard curve, whichever was smaller. For quality control, samples were equally distributed across reaction plates, and replicates were included across the reaction plates to calculate coefficients of variation. Human plasma samples from healthy blood donors (UCLA Virology Core) were used as positive control/lab quality control samples (Lab QC). Laboratory personnel were blinded to the status of samples.
Statistical analysis
Survival plots, graphical representations, and statistical analysis (log-rank test) was performed using GraphPad Prism Version 9.0 (GraphPad Software, Inc., La Jolla, California). For Kaplan-Meier survival curves and analysis, Mantel-Cox log-rank tests were performed. For data acquired from ELISA and multiplex immunometric assays, nonparametric, unpaired Mann-Whitney tests were performed, where p-values that were ≤ 0.05 were considered statistically significant, and p-values > 0.05 were not significant.
Results
Mice implanted with EBV+ human B-cells succumbed to disease in the absence of treatment with ch128.1/IgG1
To examine the ability of the ch128.1/IgG1 anti-TfR1 antibody to inhibit the outgrowth of EBV-transformed human B-cells, mice were implanted with human T-cell depleted PBMCs exposed to EBV, after which animals were treated with ch128.1/IgG1 or the isotype control IgG1 at 2 and 30 days post-implantation of cells (Fig. 1A). We found that treatment with ch128.1/IgG1 significantly enhanced survival of mice that were implanted with T-cell depleted PBMCs exposed to EBV when compared to mice treated with the IgG1 control monoclonal antibody (Fig. 1B). The difference in survival between ch128.1/IgG1-treated mice and the IgG1 control group was statistically significant (p < 0.0001, log-rank Mantel-Cox test). Moreover, mice treated with ch128.1/IgG1 and implanted with EBV+ B-cells had similar survival trends to a control group of mice implanted with EBV− B-cells (Fig. 1B).
Figure 1. ch128.1/IgG1 treatment of NSG mice implanted with EBV-exposed human B-cells leads to significantly reduced mortality and inhibits tumor development.
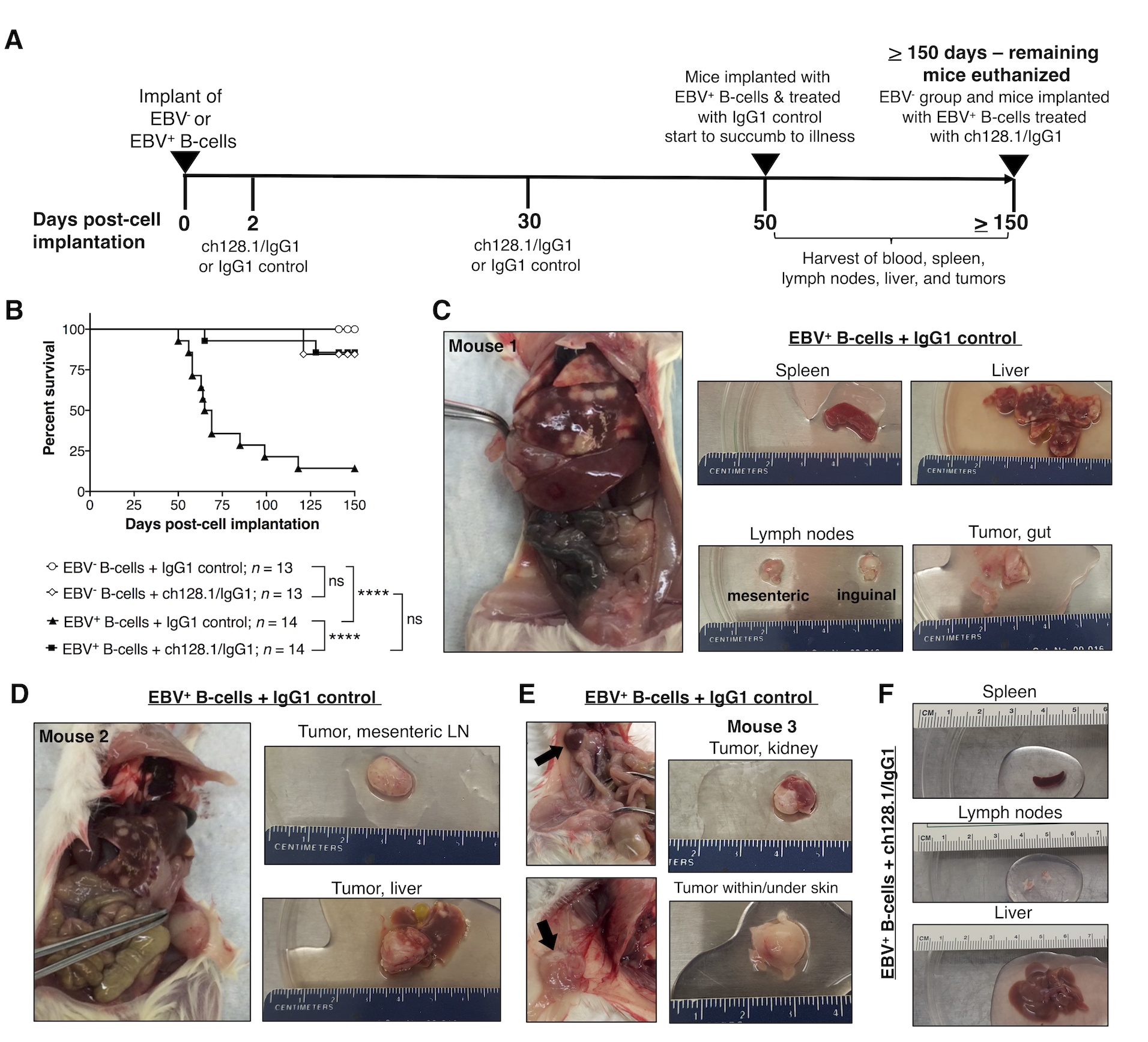
A, Experimental timeline of NSG mice implanted i.v. with 6 × 106 EBV− or EBV+ B-cell enriched preparations (T-cell depleted PBMCs). Two groups of mice received EBV− cells and two groups received EBV+ (cultured in vitro for 7 days with EBV prior to implantation). Mice were then treated with ch128.1/IgG1 (400 μg/mouse) or IgG1 control (400 μg/mouse) antibody at 2 days and 30 days post-cell implantation. Mice implanted with EBV+ cells and treated with IgG1 control started showing signs of disease and/or died after 50 days post-cell implantation, at which point, blood collected from cardiac puncture, spleen, lymph nodes, liver, and tissues with tumors were harvested. All surviving mice were euthanized at or after 150 days post-cell implantation. B, Kaplan-Meier survival curve for mice implanted with EBV− or EBV+ B-cells and treated with ch128.1/IgG1 or IgG1 control. Combined results are from 2 independent experiments. Statistical comparisons were conducted using the Mantel-Cox log-rank test, where **** indicates statistically significant difference (p < 0.0001) and ns, not significant. C, D, E, Necropsy and gross tissue images of mice that developed tumors [C, D, Mouse #1 and #2, respectively, at 58 days post-cell implantation; and E, Mouse #3, 65 days post-cell implantation]. F, Normal gross tissue images from a mouse implanted with EBV-exposed B-cells and treated with ch128.1/IgG1 antibody at 155 days post-cell implantation.
Treatment with ch128.1/IgG1 inhibits EBV-driven lymphoproliferative growth in mice bearing EBV+ human B-cells
Necropsy of mice euthanized due to illness/malaise had signs of lymphoproliferative disorder, typically represented by white spotting in splenic and liver tissues. Fourteen mice implanted with EBV+ B-cells and treated with IgG1 control showed macroscopical signs of disseminated disease with overt tumors in the liver, lymph nodes, kidneys, and in some cases, tumors near the gastrointestinal tract or under the skin (Fig. 1C–E). These mice were also examined pathologically and all showed typical histology of disseminated disease. In contrast, most of the 14 mice treated with ch128.1/IgG1 showed normal tissues without lymphoproliferative growths (Fig. 1F). Only 2 of the 14 mice treated with ch128.1/IgG1 showed disseminated disease and developed tumors in the liver (data not shown).
We examined the tissues collected from mice (blood, spleen, lymph node, liver, and tissues with any tumor growths) as single cell suspensions. Flow cytometry was used to assess the presence of human CD45+ (hCD45+) cells to determine whether the cells present in the tumor-like growths were of human origin. Fig. 2A shows representative flow cytometry plots of splenic single cell suspensions of mice implanted with EBV− or EBV+ B-cells and treated with ch128.1/IgG1 or IgG1 control. Single cell suspensions analyzed from various tissues (peripheral blood, spleen, lymph node, and liver) contained little (< 1.0%) to no detectable human CD3+ T-cells in the spleen (Fig. 2) and other tissues (data not shown). Flow cytometry analysis demonstrated a significant increase in the percentage of hCD45+ leukocytes in mice that were implanted with EBV+ B-cells and treated with the IgG1 control, compared to mice treated with ch128.1/IgG1. Most mice implanted with EBV+ B-cells and treated with ch128.1/IgG1 did not have hCD45+ cells in blood, lymph node, spleen, and liver tissues (Fig. 2B). Only two mice, out of the total of 14 mice treated with ch128.1/IgG1, had detectable hCD45+ cells. The cells detected that were of human origin (hCD45+) were also predominantly CD19+ B-cells (Fig. 2A, C).
Figure 2. Mice implanted with EBV+ B-cell enriched preparations and treated with IgG1 control antibody had human CD45+ cells in blood circulation and in different tissues.
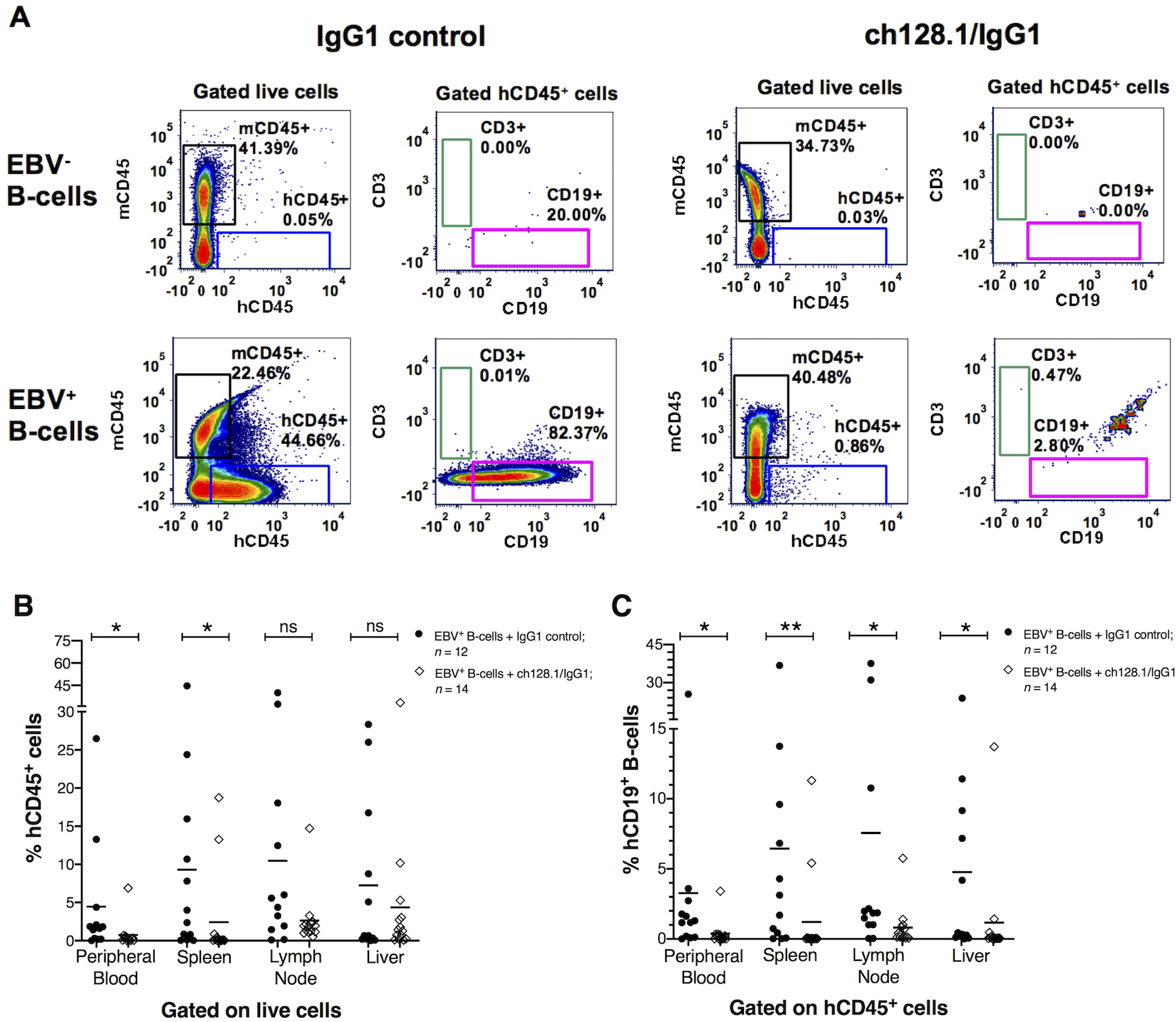
A, Representative flow cytometry plots of cells isolated from spleens of mice implanted with EBV− or EBV+ B-cell enriched preparations and treated with ch128.1/IgG1 or IgG1 control monoclonal antibody. Plots compare the percentage of murine CD45+ (mCD45+) and human CD45+ (hCD45+) cell populations (gated from live cells). Human CD3+ T-cell and CD19+ B-cell populations were gated from hCD45+ cells. B, Percentage of hCD45+ cells in peripheral blood, spleen, lymph node, and liver tissues for each EBV+ group treated with ch128.1/IgG1 or IgG1 control. The bars represent mean values of %hCD45+ B-cells; EBV+ group treated with ch128.1/IgG1: peripheral blood mean = 0.7%, spleen mean = 2.4%, lymph node mean = 2.6%, liver mean = 4.4%; EBV+ group treated with IgG1 isotype control: peripheral blood mean = 4.5%, spleen mean = 9.3%, lymph node mean = 10.5%, liver mean = 7.3%. C, Percentage of hCD19+ B-cells in peripheral blood, spleen, lymph node, and liver tissues. Percentages of hCD19+ B-cells were calculated from the total percentage of hCD45+ cells for each sample [(percentage of hCD19+) × (percentage of hCD45+ / 100%)]. The bars represent mean values of %hCD19+ B-cells; EBV+ group treated with ch128.1/IgG1: peripheral blood mean = 0.4%, spleen mean = 1.2%, lymph node mean = 0.8%, liver mean = 1.2%; EBV+ group treated with IgG1 isotype control: peripheral blood mean = 3.3%, spleen mean = 6.4%, lymph node mean = 7.6%, liver mean = 4.8%. Combined results shown in B and C are from 2 independent experiments. The results shown here for the EBV+ group treated with IgG1 control are from 12 of the 14 mice presented in Fig. 1B as two mice died before acquiring tissue at necropsy. Statistical comparisons were made for ch128.1/IgG1 versus IgG1 control treatments using nonparametric, unpaired Mann-Whitney tests, where *p = 0.01–0.05, **p = 0.001–0.01, and ns, not significant.
We then measured the expression of programmed death ligand 1 (PD-L1) on CD19+ B-cells by flow cytometry. PD-L1 is highly expressed on tumor cells of several human malignancies and various hematological malignancies, including aggressive EBV+ B-cell lymphomas (31,32). Fig. S1 shows representative flow cytometry histograms of PD-L1 expression in human CD19+ B-cell suspensions isolated from tumor tissues of mice implanted with EBV-exposed cells and treated with IgG1 control. PD-L1+ B-cells were observed in cell suspensions acquired from different tumor tissues and in different mice, including tumor cells of human B-cell origin in liver, inguinal, and mesenteric lymph nodes, kidney, tumor near the gastrointestinal tract, and a subcutaneous tumor mass in the back of the mouse (Supplementary Fig. S1). In contrast, human CD19+ B-cells did not express PD-L1 in cells obtained from peripheral blood, splenic, lymph node, or liver tissues of mice in the EBV− control groups, which did not develop any tumors, or mice in the experimental group implanted with EBV+ B-cells and treated with ch128.1/IgG1 (data not shown).
We then performed IHC staining to determine if the tumor growths observed in mice were indeed EBV-infected and of human B-cell lymphoma origin. Human tonsillar tissue sections were used as positive controls to assess the presence of human CD3+ T-cells and CD19+ B-cells in serial sections of mouse tissue (Fig. 3, left panels). We stained for EBV LMP1 to show EBV infection in tumor tissues. LMP1 is an oncogenic protein that has been shown to be essential for the in vitro transformation of B-cells (33). We found that mice implanted with EBV-exposed cells and treated with IgG1 control monoclonal antibody developed lymphoma-like growths that were of human B-cell origin (CD19+) and EBV LMP1+ (Fig. 3 and Table 1). Only in a few cases did we observe the presence of human CD3+ T-cells in spleen and liver tumor tissues (Table 1), which were mainly composed of human CD19+ B-cells (data not shown). In order to assess the clonality of tumors, we determined the expression of human Ig κ or Ig λ light chain (LC). Although most tumor tissues showed both positive staining for human Ig κ and Ig λ, some had exclusively κ or λ LC positivity, suggesting that these were monoclonal B-cell proliferations (Fig. 3; right panel Mouse #9 and Table 1).
Figure 3. NSG mice implanted with EBV+ B-cells and treated with IgG1 control antibody developed lymphoproliferative growths that were of human CD19+ B-cell origin and EBV-positive.
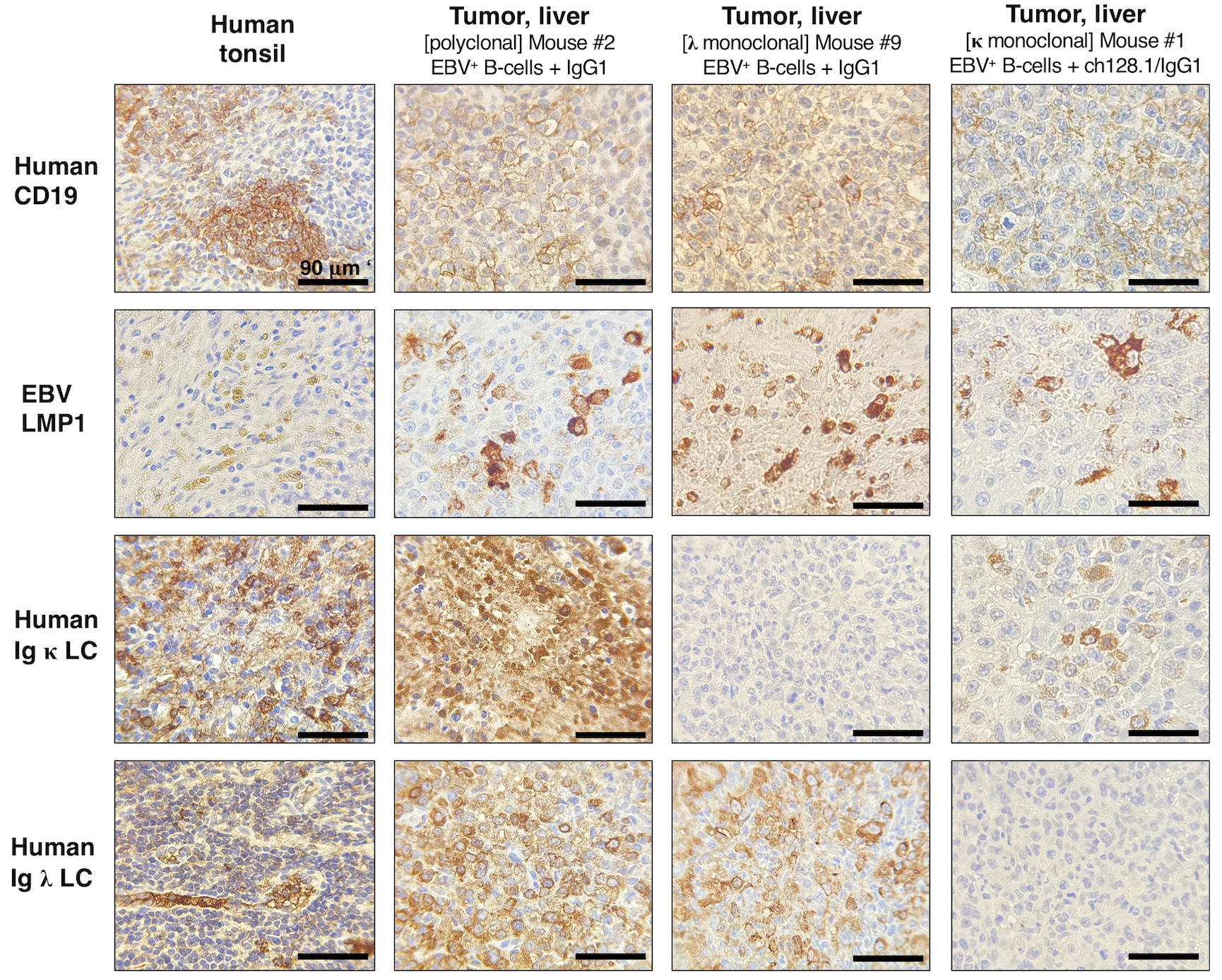
Left panels, Human tonsil tissue showing positive staining for human CD19+ B-cells, human Ig κ or Ig λ LC, and EBV infection (EBV LMP1+). Middle panels, Representative IHC staining in liver tissue sections from mice implanted with EBV+ B-cell enriched preparations and treated with IgG1 control monoclonal antibody. This mouse had human CD19+ and EBV+ polyclonal tumor growths (derived from different EBV+ B-cell clones) in liver tissue (middle panels), and mesenteric lymph nodes (images of Mouse #2 are shown in Fig. 1D and IHC results are summarized in Table 1). Right panels, IHC staining in liver tissue sections from a mouse implanted with EBV+ B-cells and treated with IgG1 control. This mouse had tumor growths in liver (shown) and splenic tissue sections (not shown) that were of human CD19+ B-cell origin and EBV LMP1+, as summarized in Table 1. Liver cells were human Ig κ LC negative and Ig λ LC positive, suggesting that these were monoclonal lymphoproliferative growths.
Table 1.
Summary of IHC staining results for mice implanted with EBV+ human B-cells and treated with IgG1 control monoclonal antibody that developed lymphoma-like growths.
| Immunohistochemistry | Circulating plasma levels | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mouse # | Euthanized (Days post-cell implantation) | Tissue section | Human CD3 | Human CD19 | EBV LMP1 | Human Ig κ LC | Human Ig λ LC | Human Ig κ FLC (μg/l) | Human Ig λ FLC (μg/l) |
| 1 | Day 58 | 402 | 18,544 | ||||||
| Tumor near the gastrointestinal tract | + | + | + | + | + | ||||
| 2 | Day 58 | 623 | 3,120 | ||||||
| Tumor, liver | Negative | + | + | + | + | ||||
| 3 | Day 65 | 3,854 | 8,363 | ||||||
| Tumor, liver | + | + | + | + | + | ||||
| 4 | Day 99 | 111,110 | 1,649 | ||||||
| Liver (white spots) | Negative | + | + | + | Negative | ||||
| 5 | Day 50 | Subcutaneous tumor mass in the back of the neck | Negative | + | + | + | + | 305 | 3,855 |
| 6 | Day 64 | Tumor, kidney | Negative | + | + | + | + | 8,956 | 1,010 |
| 7 | Day 69 | 1,214 | 2,589 | ||||||
| Tumor, inguinal lymph node | Negative | + | + | + | + | ||||
| 8 | Day 69 | Tumor, kidney | Negative | + | + | + | + | 6,255 | 963 |
| 9 | Day 85 | 778 | 150,319 | ||||||
| Liver (white spots) | + | + | + | Negative | + | ||||
| 10 | Day 118 | 12,982 | 1,574 | ||||||
| Liver (white spots) | Negative | + | + | + | Negative | ||||
EBV infection drives B-cell proliferation and clonal expansion of activated B-cells (2,3). Clonality of these EBV+ cellular expansions can be assessed by measurement of cell surface Ig LC, as this expression is clonally restricted; therefore, EBV+ monoclonal growths can be either Ig κ or λ positive, but not positive for both Ig light chains. Mature B-cells produce Ig heavy and light chains, and elevated levels of unbound Ig light chains can be released into peripheral blood circulation as Ig κ and λ free-light chain (FLC). We therefore isolated plasma from blood collected at time of necropsy and subsequently measured human Ig κ and λ FLC by ELISA. We found that human κ and λ FLC concentrations were significantly more elevated in mice implanted with EBV+ B-cells and treated with IgG1 control (Ig κ FLC geometric mean = 2,059.7 μg/l; Ig λ FLC geometric mean = 3,397.1 μg/l), compared to animals in the EBV− control group, EBV− B-cell implanted mice treated with IgG1 control (Ig κ FLC geometric mean = 129.6 μg/l; Ig λ FLC geometric mean = 1,076.2 μg/l), indicating increased B-cell activation in mice implanted with EBV+ B-cells (Fig. 4). Furthermore, mice implanted with EBV+ cells treated with ch128.1/IgG1 showed significantly reduced levels of Ig κ FLC (geometric mean = 391.6 μg/l) compared to EBV-infected mice in the IgG1 control group (Fig. 4A), but no statistically significant differences in Ig λ FLC levels were observed between the two groups (EBV+ group treated with ch128.1/IgG1 geometric mean = 1,724.4 μg/l versus EBV+ group treated with IgG1 control, geometric mean = 3,397.1 μg/l) (Fig. 4B).
Figure 4. Mice implanted with EBV+ human B-cells show significantly increased plasma levels of human Ig κ and λ FLC.
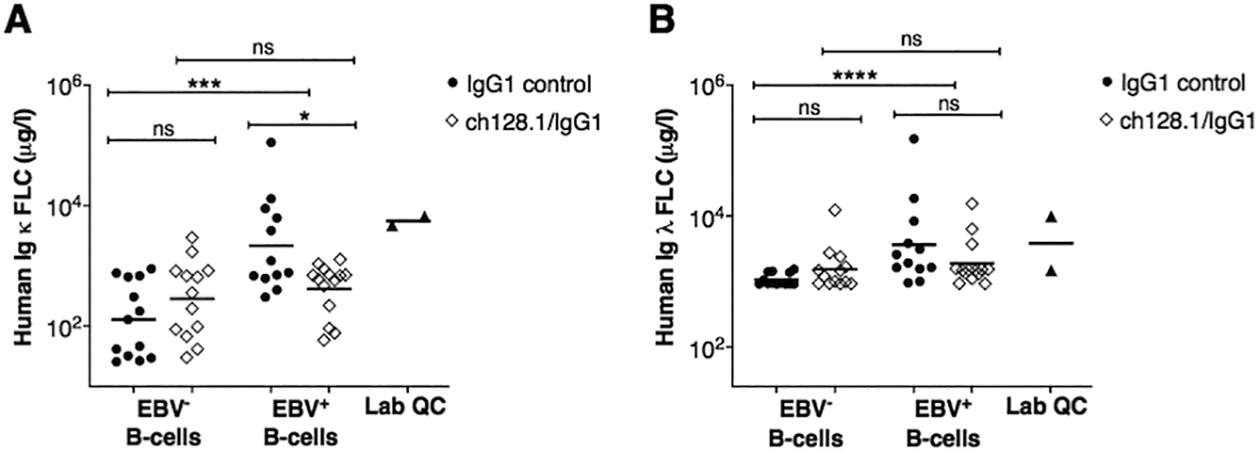
A, Plasma levels of human Ig κ and B, Ig λ FLC, as measured by ELISA. Results are provided for each experimental group and included are the combined results of two-independent experiments for mice implanted EBV− and EBV+ B-cells, and treated with ch128.1/IgG1 or IgG1 control. Bars are geometric mean values. Also shown are mean values of human Ig κ and λ FLC in healthy human plasma samples (Lab QC). Nonparametric, unpaired Mann-Whitney tests were conducted, where *p = 0.01–0.05, ***p = 0.0001–0.001, ****p < 0.0001, and ns, not significant.
EBV transforms B-cells in vitro, generating immortalized B-LCLs that produce and secrete Ig (2,8,9). EBV-transformed B-cells isolated from peripheral blood or tissue of human adults show heterogeneity in Ig class and isotype (IgM, IgG, or IgA), according to their tissue origin (34). We found that EBV infection significantly induced B-cell secretion of all classes of Ig molecules in mice treated with ch128.1/IgG1 or IgG1 control antibody compared to mice implanted with EBV− B-cells (Supplementary Fig. S2). Moreover, no significant differences in plasma levels of human IgA, IgG1 - IgG4 were observed for mice implanted with EBV+ B-cells and treated with ch128.1/IgG1 versus those treated with IgG1 control antibodies (Supplementary Fig. S2A–E). However, IgM levels were significantly lower in mice implanted with EBV+ B-cells and treated with ch128.1/IgG1 compared to mice treated with IgG1 control (Supplementary Fig. S2F).
Treatment with ch128.1/IgG1 significantly reduced plasma levels of markers of inflammation in mice implanted with EBV+ B-cells
A characteristic feature of many EBV-associated cancers is the involvement of long-term (chronic) inflammation. Therefore, we measured inflammation-associated and immune cell markers of B-cell proliferation and activation in plasma of mice using a custom panel designed for multiplexed Luminex-based immunometric assay. This panel consisted of key pro-inflammatory (IL-1β, IL-2, IL-6, IL-8, IFN-γ, TNF-α, and soluble CD163) and anti-inflammatory (IL-10) molecules, chemokines (CXCL10 and CXCL13), growth factors (VEGF and TIMP-1), and molecules associated with B-cell activation and/or survival (IL-4, TNF-RII, BAFF, sCD14, sCD25 and sCD27). As expected, infection with EBV increased the levels of inflammatory cytokines and this increase was diminished by treatment with ch128.1/IgG1 (Fig. 5A–H). We found that treatment with ch128.1/IgG1 resulted in significantly reduced plasma levels of human pro-inflammatory molecules IL-6, IL-8, IFN-γ, TNF-α, and CXCL10 (Fig. 5A–E). In addition, reduced levels of IL-10, sCD25, and sCD27 were observed (Fig. 5F–H). No statistically significant differences were seen in plasma levels of several other human molecules (IL-2, IL-4, IL-1β, VEGF, sCD163, sCD14, CXCL13, TIMP-1, BAFF, sTNF-RII) between the ch128.1/IgG1 and isotype control antibody treatment groups implanted with EBV+ B-cells (data not shown).
Figure 5. NSG mice implanted with EBV+ human B-cells and treated with ch128.1/IgG1 showed significantly reduced plasma levels of human inflammatory cytokines, immunoregulatory molecules, and soluble factors.
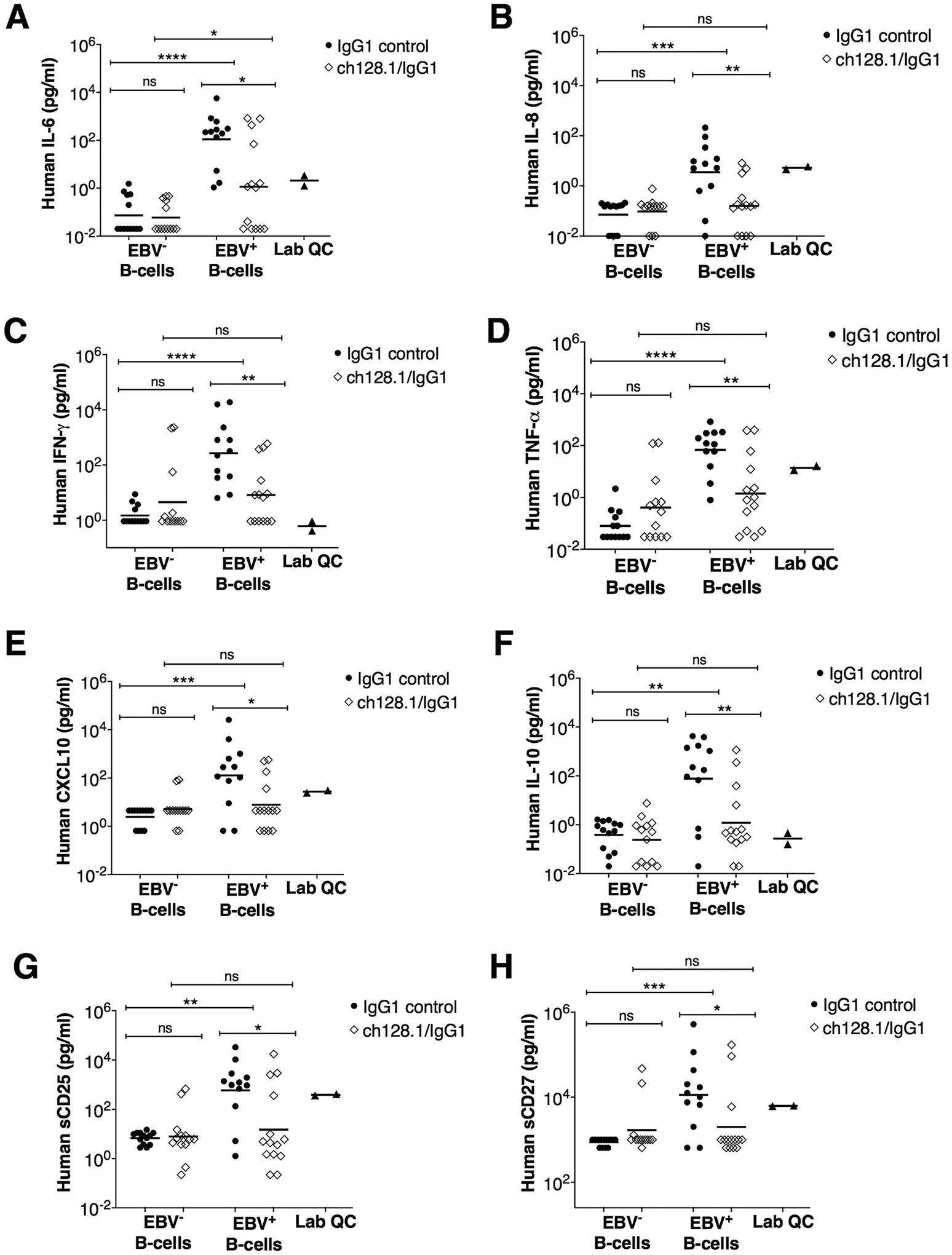
Plasma levels of human IL-6 (A), human IL-18 (B), human IFN-γ (C), human TNF-α (D), human CXCL10 (E), human IL-10 (F), human sCD25 (G), and sCD27 (H) were measured by multiplex immunometric assays. Results are provided for each experimental group and included are the combined results of 2-independent experiments for mice implanted with EBV− or EBV+ B-cells and treated with ch128.1/IgG1 or IgG1 control. Bars indicate geometric mean values. Also shown are mean values for each molecule in healthy human plasma samples (Lab QC). Nonparametric, unpaired Mann-Whitney tests were conducted, where *p = 0.01–0.05, **p = 0.00–0.01, ***p = 0.0001–0.001, ****p <0.0001, and ns, not significant.
Collectively, these results suggest that ch128.1/IgG1 treatment effectively inhibits EBV-driven lymphomagenesis by preventing the growth of EBV+ B-cells, thus blocking tumor development. Treatment with ch128.1/IgG1 also appeared to decrease systemic inflammation in NSG mice implanted with EBV+ B-cells.
Discussion
Our studies show that in an NSG mouse model implanted with EBV+ B-cells, ch128.1/IgG1 inhibits the proliferation of B-cells into malignant, lymphoma-like cells. Mice implanted with EBV+ B-cells and treated with IgG1 control antibody developed lymphoma-like cancers, causing mortality in mice beginning at 50 days post-cell implantation. However mice implanted with EBV+ B-cells and treated with ch128.1/IgG1 did not develop disease and survived for more than 150 days post-cell implantation, after which they were euthanized. These results demonstrate that ch128.1/IgG1 protects mice from developing lymphoma-like disease and ultimately death. These lymphoma-like growths were EBV+ and of human CD19+ B-cell origin, and in some cases, with individual tumor foci often being monoclonal. Moreover, treatment with ch128.1/IgG1 resulted in decreased plasma levels of human cytokines and chemokines associated with inflammation (IL-6, IL-8, IFN-γ, TNF-α, and CXCL10), and molecules associated with B-cell activation and proliferation (IL-10, sCD25, sCD27) and that have, in some cases, been shown to be signature molecules of EBV-transformed B-cell lymphoblastoid cell lines (35–40). IL-6 is a B-cell-stimulatory cytokine that is associated with the development of lymphoma and other lymphopoietic tumors (35,41–45). IL-10 is a pleiotropic cytokine that regulates the survival, proliferation, and differentiation of T-cells, B-cells, and monocytes/macrophages. Malignant cells overproduce IL-10, which can serve as an autocrine growth factor, and thus contribute to the pathogenesis of B-cell lymphomas, including NHL (37,45,46). Moreover, EBV co-opts host B-cell microRNA machinery to promote IL-10 production in B-cell lymphomas (47). Both IL-6 and IL-10 have been proposed as diagnostic or prognostic makers of EBV-associated B-cell malignancies (35,37,38). In addition, high levels of sCD27 have been reported in serum or cerebrospinal fluid of patients with B-cell malignancies (48) or AIDS-associated lymphoma (49).
Since mice are not susceptible to infection with EBV, in vivo murine models of EBV infection have required the implantation of human cells, from some source. Two prior in vivo murine models for the study of therapeutic interventions to prevent or treat EBV-associated lymphoproliferative disease and EBV-associated malignancies are the human peripheral blood leukocytes-SCID model (hu-PBL-SCID), as well as more recent humanized mouse models (50). The severe combined immunodeficiency (SCID) mutation impairs the development of antigen receptors in both B- and T-cells. As SCID mice do not have functional mature B- and T-cells, these animals cannot reject cells of human origin, allowing these mice to be used to create a xenograft model when implanted with human PBL. Hu-PBL-SCID mice often show the outgrowth of EBV+ B-cells and lymphoproliferative disease, which arise from the outgrowth of mature, EBV+ B-cells. While the hu-PBL-SCID model has provided valuable information as a platform to test potential therapies, it had significant drawbacks that limited its widespread use. For instance, the hu-PBL-SCID model, in which PBMCs from EBV+ donors are administered into SCID mice, has shown significant variability in the emergence of lymphoproliferative disease, depending on the donor and their EBV infection status, as well as other factors (50).
More recently, several humanized mouse xenograft models have been developed, allowing the study of human cells in mice (50). Immunocompetent human immune system (HIS) mice are generated by transplantation of immunodeficient mouse strains with human fetal liver and thymus organoids, along with hematopoietic progenitor cells (CD34+) from the fetal liver. While HIS mouse models have significant advantages over the hu-PBL-SCID model, those model systems are hampered by variability in the extent of implantation of human immune cells, by variability in the subsets of human immune cells engrafted in mice, by the number of mice that can be generated from a single donor, by the spontaneous development of tumors, by the technical complexity and expense of generating HIS mice, and by the requirement for the use of human fetal tissues, which can present legislative impediments in some localities. Additionally, while most HIS mouse models develop functional T-cells, they display poor implantation and development of human B-cell populations, as these mice do not develop human secondary lymphoid organs and germinal centers, and therefore, cannot support the development of mature B-cells from hematopoietic stem cells (50).
In the studies described here, we utilized a novel human/murine xenograft system to assess the effect of a therapeutic intervention in preventing EBV-driven activation and growth of B-cells and lymphoproliferative disease. This system, which utilizes PBMC-derived, B-cell-enriched (T-cell-depleted) preparations that are infected with EBV in vitro, prior to administration into NSG mice, leads to mice that consistently develop EBV+ tumor-like growths of human B-cell origin, providing an alternative, novel, tractable model for the assessment of therapeutic interventions.
In previous work, we have described the anti-tumor activity of the anti-TfR1 antibody (ch128.1/IgG1 or ch128.1/IgG3) to target and eliminate malignant cells in xenograft mouse models of human MM and NHL, including AIDS-NHL (20–23,26). The in vivo anti-tumor activity of ch128.1/IgG1 was shown to be dependent on the Fc fragment, similar to its IgG3 counterpart, in which macrophages play a major role in the in vivo anti-tumor activity (21,23). This study describes, for the first time, that ch128.1/IgG1 can be used as a direct anti-cancer agent as a means of passive immunotherapy to inhibit EBV-driven B-cell activation and carcinogenesis. Further studies are needed to determine the mechanisms governing the prevention of lymphomagenesis of EBV+ B-cells by this anti-TfR1 antibody. Since ch128.1/IgG1 does not cross-react with mouse TfR1, further studies are needed to fully define the therapeutic potential and the toxicity profile of ch128.1/IgG1.
Targeting TfR1 as a tool to prevent EBV-carcinogenesis provides new and exciting opportunities for meaningful clinical intervention and/or passive immunotherapy for B-cell associated malignancies (such as AIDS-NHL), PTLD, and/or the treatment of HIV-infected individuals at risk for developing NHL. Currently, there are no established technologies aimed at eliminating EBV-driven activation or EBV-infected pre-malignant B-cells in HIV-infected individuals and immunodeficient persons. Thus, prophylactic treatment with anti-TfR1 monoclonal antibody is a novel approach that has the potential to “reset the B-cell clock” and eliminate pre-malignant cells to prevent the development of NHL. Since chronic polyclonal B-cell activation also contributes to immune deficiency by subverting normal B-cell immune responses, targeting TfR1 with antibodies has the potential to restore normal B-cell function. Additionally, it is possible that this strategy may prevent EBV-driven activation and growth not only of B-cells, but also other cell types known to be infected by EBV and involved in tumor development.
Supplementary Material
Significance:
An anti-TfR1 antibody, ch128.1/IgG1, effectively inhibits the activation, growth, and immortalization of EBV+ human B-cells in vivo, as well as the development of these cells into lymphoma-like tumors in immunodeficient mice.
Acknowledgments
The authors would like to express their gratitude to Valerie Rezek and Nianxin Zhong for their excellent technical assistance with our in vivo work at the UCLA Center for AIDS Research (CFAR) Humanized Mouse Core Laboratory. We also thank Ko Kiehle and Ngan B. Doan for their excellent assistance with tissue handling, processing, and IHC at the Translational Pathology Core Laboratory (TPCL) in the UCLA Department of Pathology and Laboratory Medicine and a UCLA JCCC Shared Facility.
Financial support.
This work was supported in part by NIH grants R01-CA196266, R01-CA196266–01A1:S1, R01-CA196266–03 revised, R01-CA196266–04S1, R21-CA220475, R01-CA228157; UCLA Tumor Immunology T32 Training Grant - Postdoctoral Fellowship to Dr. Martínez (2T32CA009120–41A1 and 5T32CA009120–42), and the UCLA AIDS Institute and UCLA Center for AIDS Research (CFAR) (P30-AI028697). Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and the CFAR Flow Cytometry Core Facility that is supported by NIH awards P30-CA016042 and P30-AI028697, and by the UCLA JCCC, the UCLA AIDS Institute, the David Geffen School of Medicine at UCLA, the UCLA Chancellor’s Office, and the UCLA Vice Chancellor’s Office of Research. This work was also supported by the Pendleton Charitable Trust and the McCarthy Family Foundation.
Footnotes
Conflict of interest disclosure statement. Dr. Manuel Penichet has a financial interest in Stellar Biosciences, Inc. The Regents of the University of California are in discussions with Stellar Biosciences to license a technology invented by Dr. Penichet to this firm. In addition, Dr. Penichet has a financial interest in Klyss Biotech, Inc. All others authors have declared that there are no financial conflicts of interest with regard to this work.
References
- 1.Farrell PJ. Epstein-Barr Virus and Cancer. Annu Rev Pathol 2019;14:29–53 [DOI] [PubMed] [Google Scholar]
- 2.Young LS, Yap LF, Murray PG. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer 2016;16:789–802 [DOI] [PubMed] [Google Scholar]
- 3.Taylor GS, Long HM, Brooks JM, Rickinson AB, Hislop AD. The immunology of Epstein-Barr virus-induced disease. Annu Rev Immunol 2015;33:787–821 [DOI] [PubMed] [Google Scholar]
- 4.Tsao SW, Tsang CM, To KF, Lo KW. The role of Epstein-Barr virus in epithelial malignancies. J Pathol 2015;235:323–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez-Maza O, Britton S. Frequencies of the separate human B cell subsets activatable to Ig secretion by Epstein-Barr virus and pokeweed mitogen. J Exp Med 1983;157:1808–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosen A, Gergely P, Jondal M, Klein G, Britton S. Polyclonal Ig production after Epstein-Barr virus infection of human lymphocytes in vitro. Nature 1977;267:52–4 [DOI] [PubMed] [Google Scholar]
- 7.Yarchoan R, Tosato G, Blaese RM, Simon RM, Nelson DL. Limiting dilution analysis of Epstein-Barr virus-induced immunoglobulin production by human B cells. J Exp Med 1983;157:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henderson E, Miller G, Robinson J, Heston L. Efficiency of transformation of lymphocytes by Epstein-Barr virus. Virology 1977;76:152–63 [DOI] [PubMed] [Google Scholar]
- 9.Sugden B, Mark W. Clonal transformation of adult human leukocytes by Epstein-Barr virus. J Virol 1977;23:503–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol 2006;121:144–58 [DOI] [PubMed] [Google Scholar]
- 11.Daniels-Wells TR, Penichet ML. Transferrin receptor 1: a target for antibody-mediated cancer therapy. Immunotherapy 2016;8:991–4 [DOI] [PubMed] [Google Scholar]
- 12.Shen Y, Li X, Dong D, Zhang B, Xue Y, Shang P. Transferrin receptor 1 in cancer: a new sight for cancer therapy. Am J Cancer Res 2018;8:916–31 [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang XP, Elliott RL. Decreased Iron in Cancer Cells and Their Microenvironment Improves Cytolysis of Breast Cancer Cells by Natural Killer Cells. Anticancer Res 2017;37:2297–305 [DOI] [PubMed] [Google Scholar]
- 14.Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 2004;119:529–42 [DOI] [PubMed] [Google Scholar]
- 15.Kenneth NS, Mudie S, Naron S, Rocha S. TfR1 interacts with the IKK complex and is involved in IKK-NF-kappaB signalling. Biochem J 2013;449:275–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tacchini L, Gammella E, De Ponti C, Recalcati S, Cairo G. Role of HIF-1 and NF-kappaB transcription factors in the modulation of transferrin receptor by inflammatory and anti-inflammatory signals. J Biol Chem 2008;283:20674–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J 2008;412:477–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Uden P, Kenneth NS, Webster R, Muller HA, Mudie S, Rocha S. Evolutionary conserved regulation of HIF-1beta by NF-kappaB. PLoS Genet 2011;7:e1001285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeong SM, Hwang S, Seong RH. Transferrin receptor regulates pancreatic cancer growth by modulating mitochondrial respiration and ROS generation. Biochem Biophys Res Commun 2016;471:373–9 [DOI] [PubMed] [Google Scholar]
- 20.Daniels TR, Ortiz-Sanchez E, Luria-Perez R, Quintero R, Helguera G, Bonavida B, et al. An antibody-based multifaceted approach targeting the human transferrin receptor for the treatment of B-cell malignancies. J Immunother 2011;34:500–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daniels-Wells TR, Candelaria PV, Leoh LS, Nava M, Martinez-Maza O, Penichet ML. An IgG1 Version of the Anti-transferrin Receptor 1 Antibody ch128.1 Shows Significant Antitumor Activity Against Different Xenograft Models of Multiple Myeloma: A Brief Communication. J Immunother 2020;43:48–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Daniels-Wells TR, Widney DP, Leoh LS, Martinez-Maza O, Penichet ML. Efficacy of an Anti-transferrin Receptor 1 Antibody Against AIDS-related Non-Hodgkin Lymphoma: A Brief Communication. J Immunother 2015;38:307–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leoh LS, Kim YK, Candelaria PV, Martinez-Maza O, Daniels-Wells TR, Penichet ML. Efficacy and Mechanism of Antitumor Activity of an Antibody Targeting Transferrin Receptor 1 in Mouse Models of Human Multiple Myeloma. J Immunol 2018;200:3485–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng PP, Helguera G, Daniels TR, Lomas SZ, Rodriguez JA, Schiller G, et al. Molecular events contributing to cell death in malignant human hematopoietic cells elicited by an IgG3-avidin fusion protein targeting the transferrin receptor. Blood 2006;108:2745–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez JA, Helguera G, Daniels TR, Neacato II, Lopez-Valdes HE, Charles AC, et al. Binding specificity and internalization properties of an antibody-avidin fusion protein targeting the human transferrin receptor. J Control Release 2007;124:35–42 [DOI] [PubMed] [Google Scholar]
- 26.Daniels-Wells TR, Candelaria PV, Kranz E, Wen J, Weng L, Kamata M, Almagro JC, Martinez-Maza O and Penichet ML. Efficacy of antibodies targeting TfR1 in xenograft mouse models of AIDS-related non-Hodgkin lymphoma American Association for Cancer Research (AACR) 111th Annual Meeting (AACR Virtual Meeting II, June 22–24, 2020). Abstract No. 56552020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helguera G, Jemielity S, Abraham J, Cordo SM, Martinez MG, Rodriguez JA, et al. An antibody recognizing the apical domain of human transferrin receptor 1 efficiently inhibits the entry of all new world hemorrhagic Fever arenaviruses. J Virol 2012;86:4024–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tao MH, Canfield SM, Morrison SL. The differential ability of human IgG1 and IgG4 to activate complement is determined by the COOH-terminal sequence of the CH2 domain. J Exp Med 1991;173:1025–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Epeldegui M, Conti DV, Guo Y, Cozen W, Penichet ML, Martinez-Maza O. Elevated numbers of PD-L1 expressing B cells are associated with the development of AIDS-NHL. Sci Rep 2019;9:9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Epeldegui M, Magpantay L, Guo Y, Halec G, Cumberland WG, Yen PK, et al. A prospective study of serum microbial translocation biomarkers and risk of AIDS-related non-Hodgkin lymphoma. AIDS 2018;32:945–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin Cancer Res 2013;19:3462–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xue T, Wang WG, Zhou XY, Li XQ. EBV-positive diffuse large B-cell lymphoma features PD-L1 protein but not mRNA overexpression. Pathology 2018;50:725–9 [DOI] [PubMed] [Google Scholar]
- 33.Kaye KM, Izumi KM, Kieff E. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc Natl Acad Sci U S A 1993;90:9150–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyawaki T, Kubagawa H, Butler JL, Cooper MD. Ig isotypes produced by EBV-transformed B cells as a function of age and tissue distribution. J Immunol 1988;140:3887–92 [PubMed] [Google Scholar]
- 35.Breen EC, van der Meijden M, Cumberland W, Kishimoto T, Detels R, Martinez-Maza O. The development of AIDS-associated Burkitt’s/small noncleaved cell lymphoma is preceded by elevated serum levels of interleukin 6. Clin Immunol 1999;92:293–9 [DOI] [PubMed] [Google Scholar]
- 36.Emilie D, Coumbaras J, Raphael M, Devergne O, Delecluse HJ, Gisselbrecht C, et al. Interleukin-6 production in high-grade B lymphomas: correlation with the presence of malignant immunoblasts in acquired immunodeficiency syndrome and in human immunodeficiency virus-seronegative patients. Blood 1992;80:498–504 [PubMed] [Google Scholar]
- 37.Emilie D, Touitou R, Raphael M, Peuchmaur M, Devergnee O, Rea D, et al. In vivo production of interleukin-10 by malignant cells in AIDS lymphomas. Eur J Immunol 1992;22:2937–42 [DOI] [PubMed] [Google Scholar]
- 38.Pastore C, Gaidano G, Ghia P, Fassone L, Cilia AM, Gloghini A, et al. Patterns of cytokine expression in AIDS-related non-Hodgkin’s lymphoma. Br J Haematol 1998;103:143–9 [DOI] [PubMed] [Google Scholar]
- 39.Setsuda J, Teruya-Feldstein J, Harris NL, Ferry JA, Sorbara L, Gupta G, et al. Interleukin-18, interferon-gamma, IP-10, and Mig expression in Epstein-Barr virus-induced infectious mononucleosis and posttransplant lymphoproliferative disease. Am J Pathol 1999;155:257–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharma V, Zhang L. Interleukin-8 expression in AIDS-associated lymphoma B-cell lines. Biochem Biophys Res Commun 2001;282:369–75 [DOI] [PubMed] [Google Scholar]
- 41.De Roos AJ, Mirick DK, Edlefsen KL, LaCroix AZ, Kopecky KJ, Madeleine MM, et al. Markers of B-cell activation in relation to risk of non-Hodgkin lymphoma. Cancer Res 2012;72:4733–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jenkins BJ, Roberts AW, Greenhill CJ, Najdovska M, Lundgren-May T, Robb L, et al. Pathologic consequences of STAT3 hyperactivation by IL-6 and IL-11 during hematopoiesis and lymphopoiesis. Blood 2007;109:2380–8 [DOI] [PubMed] [Google Scholar]
- 43.Kato H, Kinoshita T, Suzuki S, Nagasaka T, Hatano S, Murate T, et al. Production and effects of interleukin-6 and other cytokines in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma 1998;29:71–9 [DOI] [PubMed] [Google Scholar]
- 44.Kurzrock R, Redman J, Cabanillas F, Jones D, Rothberg J, Talpaz M. Serum interleukin 6 levels are elevated in lymphoma patients and correlate with survival in advanced Hodgkin’s disease and with B symptoms. Cancer Res 1993;53:2118–22 [PubMed] [Google Scholar]
- 45.Levin LI, Breen EC, Birmann BM, Batista JL, Magpantay LI, Li Y, et al. Elevated Serum Levels of sCD30 and IL6 and Detectable IL10 Precede Classical Hodgkin Lymphoma Diagnosis. Cancer Epidemiol Biomarkers Prev 2017;26:1114–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cortes J, Kurzrock R. Interleukin-10 in non-Hodgkin’s lymphoma. Leuk Lymphoma 1997;26:251–9 [DOI] [PubMed] [Google Scholar]
- 47.Harris-Arnold A, Arnold CP, Schaffert S, Hatton O, Krams SM, Esquivel CO, et al. Epstein-Barr virus modulates host cell microRNA-194 to promote IL-10 production and B lymphoma cell survival. Am J Transplant 2015;15:2814–24 [DOI] [PubMed] [Google Scholar]
- 48.Kersten MJ, Evers LM, Dellemijn PL, van den Berg H, Portegies P, Hintzen RQ, et al. Elevation of cerebrospinal fluid soluble CD27 levels in patients with meningeal localization of lymphoid malignancies. Blood 1996;87:1985–9 [PubMed] [Google Scholar]
- 49.Widney D, Gundapp G, Said JW, van der Meijden M, Bonavida B, Demidem A, et al. Aberrant expression of CD27 and soluble CD27 (sCD27) in HIV infection and in AIDS-associated lymphoma. Clin Immunol 1999;93:114–23 [DOI] [PubMed] [Google Scholar]
- 50.Ahmed EH, Baiocchi RA. Murine Models of Epstein-Barr Virus-Associated Lymphomagenesis. ILAR J 2016;57:55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.