

Abstract
Loss of the retinoblastoma (RB) tumor suppressor protein is a critical step in reprogramming biological networks that drive cancer progression, although mechanistic insight has been largely limited to the impact of RB loss on cell cycle regulation. Here, isogenic modeling of RB loss identified disease stage-specific rewiring of E2F1 function, providing the first-in-field mapping of the E2F1 cistrome and transcriptome after RB loss across disease progression. Biochemical and functional assessment using both in vitro and in vivo models identified an unexpected, prominent role for E2F1 in regulation of redox metabolism after RB loss, driving an increase in the synthesis of the antioxidant, glutathione, specific to advanced disease. These E2F1-dependent events resulted in protection from reactive oxygen species (ROS) in response to therapeutic intervention. On balance, these findings reveal novel pathways through which RB loss promotes cancer progression and highlight potentially new nodes of intervention for treating RB-deficient cancers.
Keywords: prostate cancer, tumor suppressor, RB, E2F1, transcriptional regulation, metabolism, ChIP-seq, RNA-seq, metabolomics
Introduction
The retinoblastoma (RB) tumor suppressor protein serves as a transcriptional corepressor that is frequently altered in cancer and has been linked to both prevention of tumor development and progression. Canonical RB function in cell cycle control is tightly regulated by cyclin dependent kinase (CDK) and cyclin phosphorylation as well as upstream CDK inhibitor (CDKI) proteins. In response to mitogenic stimuli, CDK/cyclin complexes hyperphosphorylate RB promoting a conformational change and allowing for de-repression of activator E2F transcription factors (E2F1, E2F2, and E2F3) (1,2). Loss of function (LOF) of RB alters gene networks, including but not limited to those associated with E2Fs, that have been shown to drive tumorigenesis and disease progression. Common alterations driving RB LOF in human malignancy include RB1 gene mutation, promoter methylation, and deep deletion, in addition to upstream pathway modulation such as upregulation of cyclins and CDKs or loss of CDKI function (3,4). These alterations have been identified across cancers and proven to play a significant role in driving tumor phenotypes. Despite current understanding of RB function in preventing cellular transformation, significant gaps in understanding exist in discerning the role of RB in tumor progression.
Elucidating the role of RB loss in disease progression is critical, as RB LOF has been attributed to poor outcome in a number of tumor types. Specifically, RB loss has been associated with shorter overall survival (OS) in multiple myeloma (MM), bladder cancer, and early stage non-small cell lung cancer (NSCLC) (5–8). Additionally, RB loss has been associated with higher tumor grade and stage in bladder cancer and shorter event-free survival (EFS) in osteosarcoma (9,10). Beyond the loss of RB protein, RB loss gene signatures have been examined in breast and prostate cancers. Elevated expression of the RB loss gene signature has been associated with shorter OS and relapse-free survival (RFS) in breast cancers and shorter progression-free survival (PFS), disease-specific survival (DSS) and OS in prostate cancer (PCa) (11,12). Other studies have linked RB loss to therapeutic relapse. In hormone receptor-positive breast cancers (BrCa) and PCa, RB loss has been associated with tumor relapse and hormone therapy resistance, highlighting the clinical importance of RB loss on disease progression (13–15). Thus, while loss of RB function clearly can promote tumor initiation, the mechanisms by which RB functions to induce the aggressive tumor phenotypes that have been observed in human malignancies remains largely unstudied.
While the tumor suppressive functions of RB in cancer impact cell cycle control, clinical data indicate that functions beyond mitotic regulation likely occur in human disease. Strikingly, in cancers wherein RB loss is strongly associated with poor outcome, such as lung and prostate cancers, there is no correlation between RB loss and hyperproliferative activity (16,17). Studies examining RB loss and Ki67 positivity in lung adenocarcinoma and castration-resistant prostate cancer (CRPC) found no correlation between RB protein negativity and Ki67 positivity indicating that RB regulatory control expands beyond the cell cycle (16,17). Based on these findings and the importance of RB in tumor progression, it is critical to determine the cellular and molecular mechanisms by which RB loss promotes disease progression in advanced cancers.
To address this important gap in understanding, studies herein have discovered unrealized functions of RB in controlling tumor metabolism. Isogenic models of RB loss in early stage and advanced disease were developed, and subsequent molecular assessment identified distinct, stage-specific activities of the RB tumor suppressor protein. Key findings revealed that RB loss in advanced disease redirected E2F1 to serve as a critical regulator of glutathione synthesis, as evident through cistrome, transcriptome, and metabolome profiling. Further investigation revealed that these activities significantly increase glutathione both in vitro and in vivo, and conferred a resulting protection from reactive oxygen species (ROS) generation in response to cytotoxic chemotherapy. These studies shift paradigms in thinking with regard to metabolic control and reveal entirely new activities through which RB and E2F1 function in tumor suppression.
Results
Transcriptional networks governed by RB depletion are disease stage-dependent.
The biological significance of RB1 loss in driving progression to CRPC is well established (15,18). RB loss is infrequent in localized disease, yet is found in 19-35% of CRPC and is associated with poor patient outcome (18–22). Importantly, the molecular consequence of RB1 loss on disease progression extends beyond cell cycle control, highlighting a critical role for RB in regulating a myriad of pro-tumorigenic pathways (16). Analyses of currently available PCa patient cohorts revealed that 72% of RB1 gene alterations are deletions (Fig 1A top), and examination across hormone-therapy sensitive prostate cancer (HSPC) and CRPC cohorts revealed the frequency of this alteration is increased to an average of 10.9% in CRPC compared to 2.4% in HSPC, indicating a distinct function for RB1 depletion in CRPC (Fig 1A bottom). While RB loss induces the CRPC transition, castration resistance can be activated through multiple mechanisms including alternative AR splicing, post-translational modifications, cofactor perturbation, mutations, and intracrine androgen synthesis (23). Here, isogenic modeling was used to assess the relative impact of RB loss in HSPC versus existing CRPC from the identical lineage (Fig 1B top). Thus, HSPC models mimic the “RB1 depletion-induced CRPC” stage of disease (RBD-induced CRPC) as previously described (15,16) while CRPC isogenic models represent the “RB1 depletion-post CRPC” transition” (RBD-post CRPC), a more advanced stage of disease.
Figure 1. Transcriptional networks governed by RB are stage-dependent.
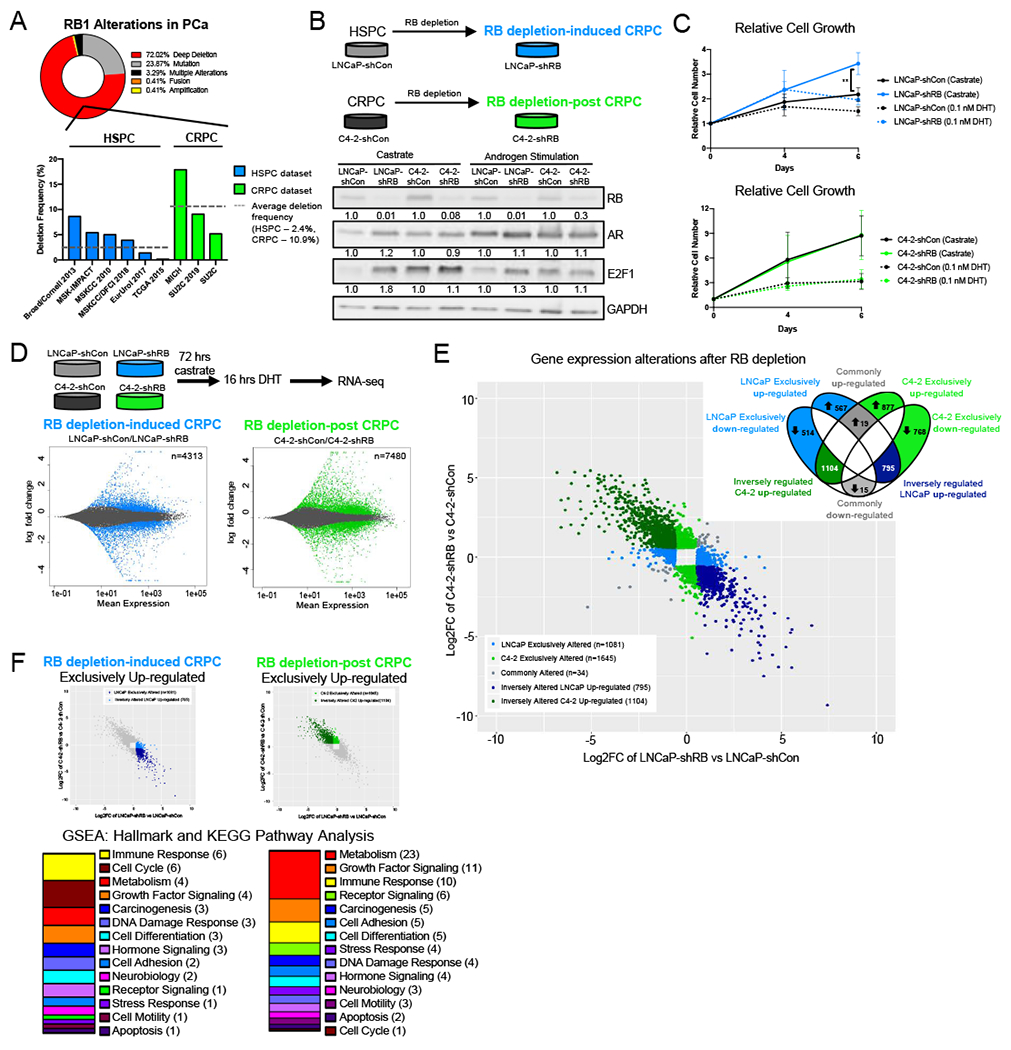
(A) RB1 alterations across prostate adenocarcinoma and CRPC datasets from CBioPortal. Frequency of RB1 gene deep across HSPC and CRPC cohorts. (B) RB1 was depleted via shRNA in LNCaP and C4-2 cell lines to generate RB1 depletion-induced CRPC (RBD-induced CRPC) and RB1 depletion-post CRPC models (RBD-post CRPC), respectively. Relative protein expression of RB, AR, and E2F1 in castrate and androgen stimulated (10 nM DHT) conditions are shown by western blot. (C) Relative growth of LNCaP-shCon, LNCaP-shRB, C4-2-shCon and C4-2-shRB models in castrate and androgen stimulated conditions (0.1 nM DHT). (D) RNA sequencing was performed in LNCaP and C4-2 isogenic models after 72 hours androgen depletion followed by 16 hrs of DHT stimulation. MA plot representing transcript alterations after RB knockdown from RNA-seq in RB depletion induced CRPC and RB depletion post CRPC models. Genes highlighted in blue (LNCaP) and green (C4-2) represent statistically significant differential expression (adjusted p-value < 0.05) (E) Transcripts altered in both models with a fold change >1.5 were plotted on the same graph. Transcripts were divided into those exclusively altered in either model after RB depletion (LNCaP 1081-blue, C4-2 1645-green) and those inversely regulated, upregulated in LNCaP and down regulated in C4-2 after RB depletion (795-dark blue) and downregulated in LNCaP and upregulated in C4-2 (1104-dark green). Transcripts commonly altered in both models are shown in grey (34). (F) GSEA Hallmark and KEGG pathway analysis from the molecular signatures database (MSigDB) of transcripts upregulated in the ONLY in the LNCaP-shRB model and pathway analysis of transcripts upregulated in the ONLY in the C4-2-shRB model. Pathways were categorized into more broad descriptions for comparison.
As RB function is frequently disrupted in cancer, the specific CDK4/6 inhibitor, palbociclib was used to validate functional RB status in parental models utilized herein (24–28). Flow cytometry was performed to measure BrdU incorporation to rigorously assess the number of cells in S-phase in the absence or presence of palbociclib and revealed that both HSPC and CRPC control models showed a significant and similar decrease in S-phase incorporation following CDK4/6 inhibition (Fig S1A). By contrast, RB1 deficient variants proved resistant to palbociclib, confirming loss of RB function. Further, western blot analyses of total and phosphorylated RB protein revealed similar levels of phosphorylated RB present across both models and are both reduced following CDK4/6 inhibition (Fig S1B), thus indicating similar RB functional status.
As AR and E2F1 have both been shown to be sensitive to RB modulation in previous studies (15,16), the effects of RB1 depletion on AR and E2F1 protein expression were examined across all models under castrate and androgen stimulated conditions. In castrate conditions, RB1 depletion promoted a greater than 1.2-fold increase in both AR and E2F1 protein levels in RBD-induced CRPC models consistent with previous publications (15,16). Conversely, no significant changes in AR and E2F1 protein expression were found in RBD-post CRPC models (Fig 1B bottom). Similar changes in AR and E2F1 expression were found in androgen stimulated conditions showing a slight increase in AR and E2F1 protein expression in RBD-induced CRPC models and no change in RBD-post CRPC models (Fig 1B bottom). These data highlight a distinct consequence of RB1 depletion in the transition from HSPC to CRPC; however, they do not clarify the significance of RB1 depletion in CRPC, particularly the impact of RB loss on the pro-tumorigenic functions of E2F1 and AR where no significant change in protein expression was observed. As expected, RBD-induced CRPC models exhibited a growth advantage in castrate conditions (15) (Fig 1C top). However, there was no change in growth in the RBD-post CRPC models (Fig 1C bottom), further underscoring a distinct role for RB loss pre- and post-CRPC transition.
RB is a well-described transcriptional regulator, as such RNA sequencing (RNA-Seq) was performed in both RBD-induced CRPC and RBD-post CRPC isogenic models after androgen stimulation, so as to understand stage-specific differences. Consistency of biological replicates was confirmed by principal component analysis (PCA) (Fig S1C). As shown, while RBD-induced CRPC exhibited significant changes in 4313 transcripts (Fig 1D – left), RBD-post CRPC resulted in 7480 significantly altered transcripts (Fig 1D – right) (adjusted p-value < 0.05), suggesting a distinct role for RB loss in transcriptional regulation at this later stage of disease. To identify distinct transcriptional alterations regulated by RB between stages of disease, transcriptomic overlay was performed (Fig 1E). Specifically, differentially expressed transcripts were identified compared to stage-specific controls in each model (FC > 1.5, adjusted p-value < 0.05). Expression changes were stratified as described in Figure 1E: exclusively altered in RBD-induced CRPC (1081 transcripts, light blue), exclusively altered in RBD-post CRPC (1645 transcripts, light green), commonly altered in both stages (34 transcripts, gray), and two inversely regulated categories: RBD-induced CRPC upregulated, RBD-post CRPC downregulated (795 transcripts, dark blue) and RBD-post CRPC upregulated, RBD-induced CRPC downregulated (1104 transcripts, dark green). Significantly, the clear differences in transcriptional regulation, including the notable number of inversely regulated transcripts, imply both distinct as well as potentially opposing functions of RB across stages of disease, which has not been previously described. Finally, to identify the putative transcriptional networks driven by these distinct gene expression changes across stages of disease, gene set enrichment analysis (GSEA) was performed, revealing that pathways significantly impacted by RBD-induced CRPC included immune response and cell cycle control related pathways, while those driven by RBD-post CRPC were primarily related to metabolic and growth factor signaling (Fig 1F). Together these data highlight, for the first time, both the divergent transcriptional networks driven by RB1 depletion between stages of disease and the biologically-relevant pathways associated with these changes.
RB depletion-induced E2F1 rewiring is disease stage-specific.
It has been previously reported that RB loss in HSPC results in rewiring of the E2F1 cistrome, and the resulting transcriptional output is associated with poor outcome (16). To discern the relative impact of RB loss in distinct disease states, E2F1 function was assessed via chromatin immunoprecipitation followed by DNA sequencing (ChIP-Seq) across disease progression (Fig 2A top). While 32,235 E2F1 binding sites were identified in the stage specific control, RBD-induced CRPC cells exhibited a reduction in total E2F1 binding sites with 23,950 sites identified after RB1 depletion, with 3597 of those sites exclusive to the RB1 depleted condition (Fig 2A middle). Conversely, RBD-post CPRC induced a marked gain of total E2F1 binding compared to the stage specific control, increasing the number of E2F1 sites from 13,594 to 31,647, with 18,624 of these sites being exclusive to the RB1-depleted condition (Fig 2A bottom). This increase in E2F1 binding suggests a putative gain in E2F1 function when RB is depleted after the CRPC transition, likely contributing to the transcriptional changes seen in this stage as reported in Figure 1.
Figure 2. RB1 depletion induced E2F1 rewiring is disease stage specific.
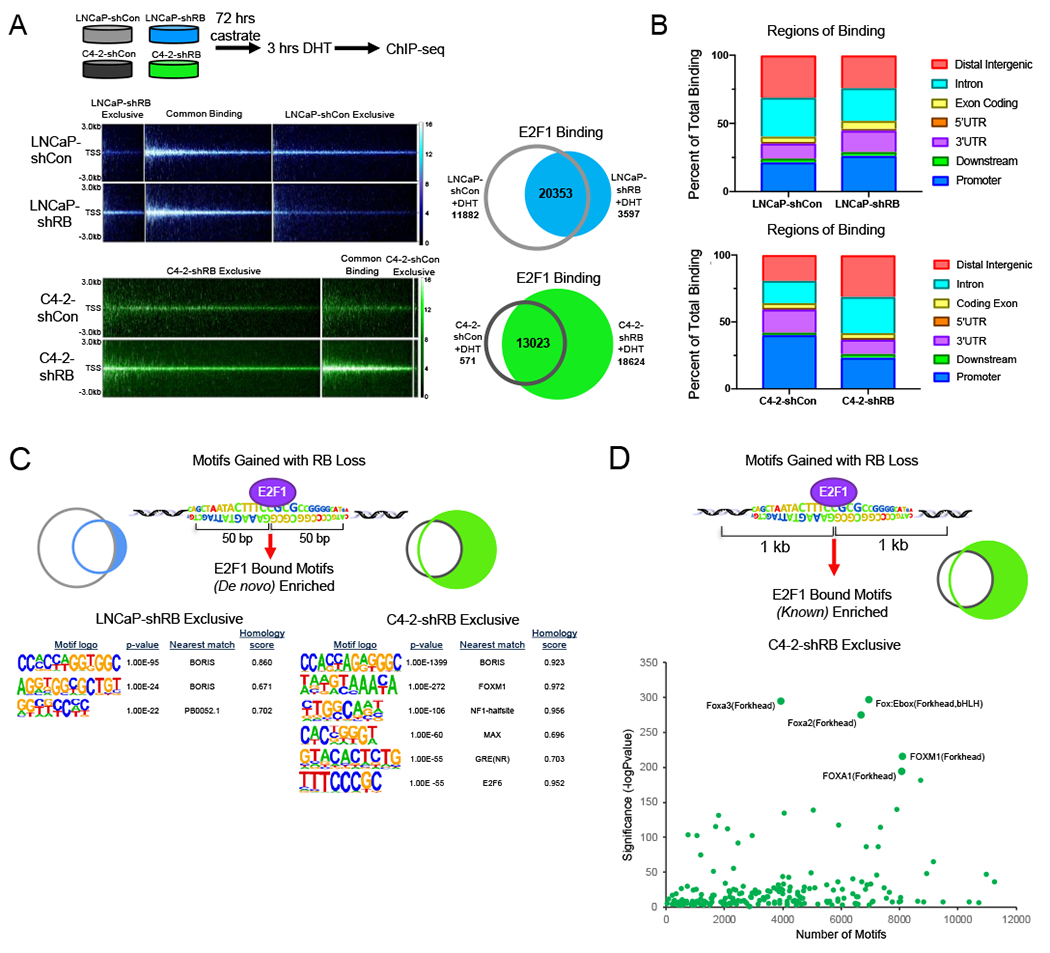
(A) E2F1 ChIP-sequencing was performed in LNCaP-shCon, LNcaP-shRB and C4-2-shCon, C4-2-shRB models after 72 hrs of androgen depletion and 3 hrs of DHT stimulation. Binding intensity plots are shown (left) and number of E2F1 binding sites for each condition is shown via Venn Diagram (right). (B) Regions of binding for each condition in shown for LNCaP (top) and C4-2 (bottom) derived models determined using cis-regulatory element annotation system (CEAS) package. Binding regions are represented as percent of total binding. (C) De novo motif analysis using a 50-bp window from the center of E2F1 binding exclusively after RB1 depletion in LNCaP-shRB and C4-2-shRB models. P-value and homology scores are shown. (D) Known motif analysis using a 1 kb window from the center of E2F1 binding exclusively after RB1 depletion in the C4-2-shRB model. Top motifs enriched are labeled.
As RB loss has previously been shown to alter E2F1 preference for binding at canonically described promoter regions, genomic annotation of E2F1 binding sites was performed for each disease stage. As shown in Figure 2B, RBD-induced CRPC resulted in a slight increase in the percent of E2F1 binding at promoter regions compared to stage specific controls (Fig 2B top). Interestingly, RBD-post CRPC resulted in a significant shift in binding at promoter regions, decreasing from 40% to 23% of total binding, and at intronic and distal intergenic regions, increasing from a combined 40% to 59% of total binding (Fig 2B bottom), suggesting that E2F1 binding shifts towards intronic and distal intergenic regions in CRPC following RB loss. These differential shifts in E2F1 binding further underscore the disparate effects of RB loss pre- and post-transition to CRPC. Given the shift in the genomic elements associated with E2F1 binding after RB depletion, de novo motif analysis was performed to gain insight into the potential mechanism governing these changes. Interestingly, the most significant and highly enriched motif closely resembled BORIS, a known transcription factor that binds at promoters of genes that are frequently elevated in cancer (29) (Fig 2C left). Further, RB1 depleted exclusive E2F1 binding in CRPC was also enriched for motifs resembling BORIS in addition to the pioneer factor, FOXM1 and the NF-1-halfsite motif (Fig 2C, Fig S2) suggesting this gained E2F1 binding may be associated with the function of these additional DNA binding proteins. To further identify potential novel co-factors associated with E2F1, specifically those gained exclusively after RB1 depletion in CRPC (compared to stage-specific controls), known motif analysis was performed. Analysis within a 1 kb window from the center of gained E2F1 binding revealed the most significantly enriched motifs included Foxa3, Foxa2, and Fox:Ebox forkhead motifs (Fig 2D), supporting the potential for E2F1 association with forkhead proteins at these novel binding sites. These data provide insight into the mechanism and putative cofactors driving E2F1 to novel DNA binding sites after RB1 depletion in CRPC.
Given that RB depletion in existing CRPC resulted in distinct expansion of the E2F1 cistrome and concomitant transcriptional rewiring, the RBD-post CRPC associated E2F1 cistrome was mapped to putative E2F1 target genes. Genes with transcriptional start sites (TSS) within 30 kb of the center of E2F1 binding after RB1 depletion were identified (17,393 genes). To prioritize genes for downstream investigation, these binding-associated genes were intersected with those exhibiting an increase in gene expression, specifically those seen to be exclusively or inversely regulated (1981 genes) compared to RBD-induced CRPC. This overlay identified 1599 genes that had E2F1 binding within 30 kb of the TSS in addition to increased expression after RB1 depletion distinct from RBD-induced CRPC. GSEA revealed that 27% of all pathways enriched were related to cell metabolism, with the most significantly enriched pathways involved in sphingolipid and amino acid metabolism (Fig 3A). These data indicate that E2F1 binding in RBD-post CRPC likely drives a specific increase of E2F1-driven expression of genes involved in metabolic pathways and implicates RB as a regulator of cellular metabolism in late stage disease.
Figure 3. E2F1 regulates metabolic gene expression after RB depletion in advanced disease.
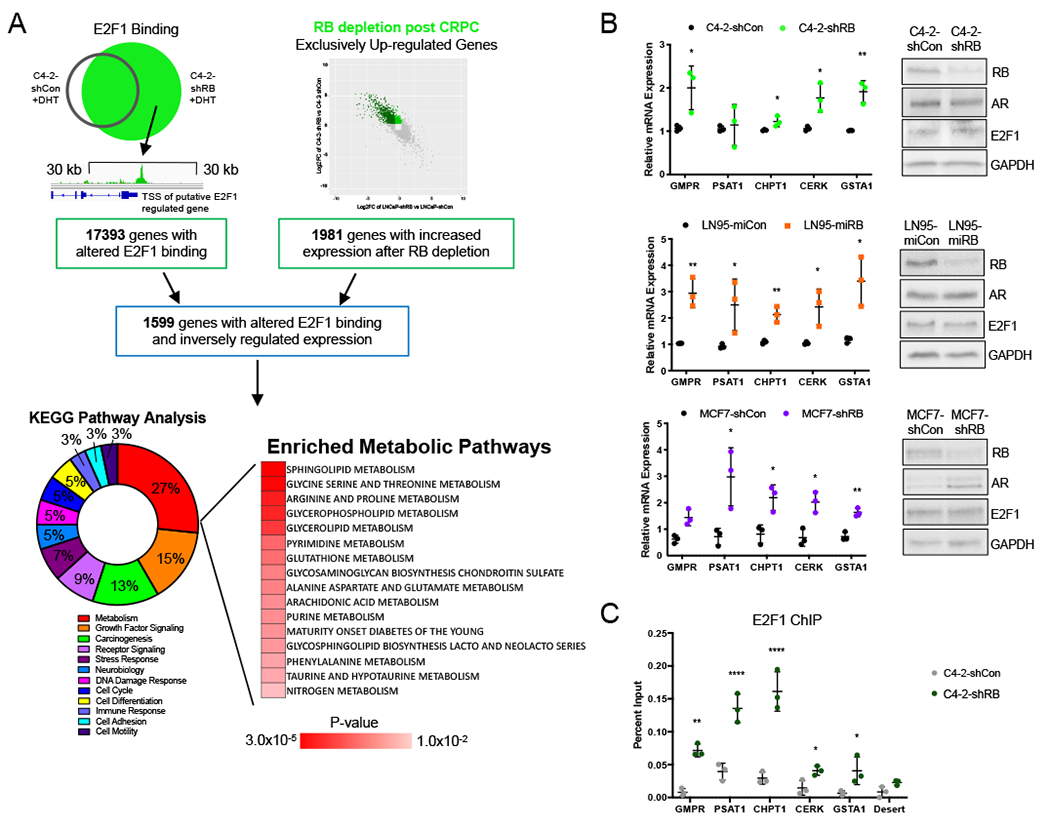
(A) Genes with a TSS within 30 kb of an E2F1 binding site in the C4-2-shRB model were identified. These putative E2F1 targets were compared to transcripts upregulated in CRPC after RB1 depletion to identify genes with altered E2F1 binding and inversely regulated gene expression after RB knockdown in the C4-2 model. GSEA KEGG pathway analysis (MSigDB) of these genes identified 27% being of pathways were metabolic. (B) Validation of increased mRNA expression of select metabolic genes after RB knockdown in C4-2 and LN95 (CRPC models) after 72 hours of androgen deprivation and 16 hours of 10 nM DHT treatment and MCF7 (invasive breast cancer model) in FBS. (C) Validation of E2F1 binding at select metabolic gene promoters after 72 hours of androgen deprivation and 3 hours of 10 nM DHT treatment. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
RB loss in late stage disease results in E2F1-dependent metabolic reprogramming
As E2F1 was identified as a potential, major metabolic regulator in RBD-post CRPC, the assortment of putative metabolic pathways regulated were investigated further. Target genes across pathways including GMPR (purine metabolism), PSAT1 (glycine serine and threonine metabolism), CHPT1 (glycerphospholipid metabolism), CERK (sphingolipid metabolism), and GSTA1 (glutathione metabolism) were confirmed to have a change in mRNA expression and E2F1 binding after RB1 depletion. The mRNA expression of GMPR, CHPT1, CERK and GSTA1 were increased 1.2 to 2.0-fold after RB depletion (Fig 3B, top). When validated in an additional RBD-post CRPC model in which RB1 was downregulated via microRNA, all selected target genes displayed 2.0 to 3.5-fold increases in expression (Fig 3B, middle). Further, expanding beyond PCa, mRNA expression of these targets was confirmed in an invasive BrCa model (Fig 3B, bottom), showing a 1.5 to 3.0-fold increase in mRNA expression after RB1 depletion. Validation of RB, AR and E2F1 protein levels within these additional models is also shown (Fig 3B, right). These mRNA expression data highlight a significant role for RB in regulating the expression of a diverse range of metabolic genes functioning in numerous metabolic pathways across cancer types. To further confirm that these expression changes are likely to be regulated by E2F1 after RB1 depletion, the E2F1 binding sites identified within 30 kb of these gene TSS’s were confirmed with a second antibody recognizing a distinct epitope of E2F1 (30). E2F1 binding was validated across targets, showing a greater than 2-fold increase in binding after RB1 depletion compared to controls (Fig 3C). These data define a distinct role for RB and E2F1 in regulating a collection of metabolic pathways in advanced disease.
Since these data demonstrate a novel role for RB in regulating cell metabolism in CRPC, to assess biological outcome, steady-state metabolomic analysis was performed in isogenic RBD-post CRPC models. Metabolomics identified 89 significantly altered metabolites after RB1 depletion compared to stage specific controls. KEGG pathway analysis of significantly altered metabolites identified the top pathways modulated after RB1 depletion as: lipid metabolism (49%), amino acid metabolism (28%), and peptide metabolism (12%) (Fig 4A, Fig S3A). These data support a role for RB in regulating distinct lipid and amino acid metabolic pathways. Analysis of significantly altered metabolites within their metabolic pathways (Fig 4B) showed a range of up- and down-regulated lipid metabolites and pathways after RB depletion (Fig S3B). By contrast, there was a distinct upregulation of amino acid metabolites, with the majority exhibiting a 1.5-fold or greater increase in abundance (increased metabolites, red; decreased metabolites, blue; Fig 4C). These data highlight a clear role for RB in regulation of metabolic function in RBD-post CRPC, with a particular effect in amino acid and lipid metabolism.
Figure 4. RB depletion drives increased amino acid metabolism in CRPC.
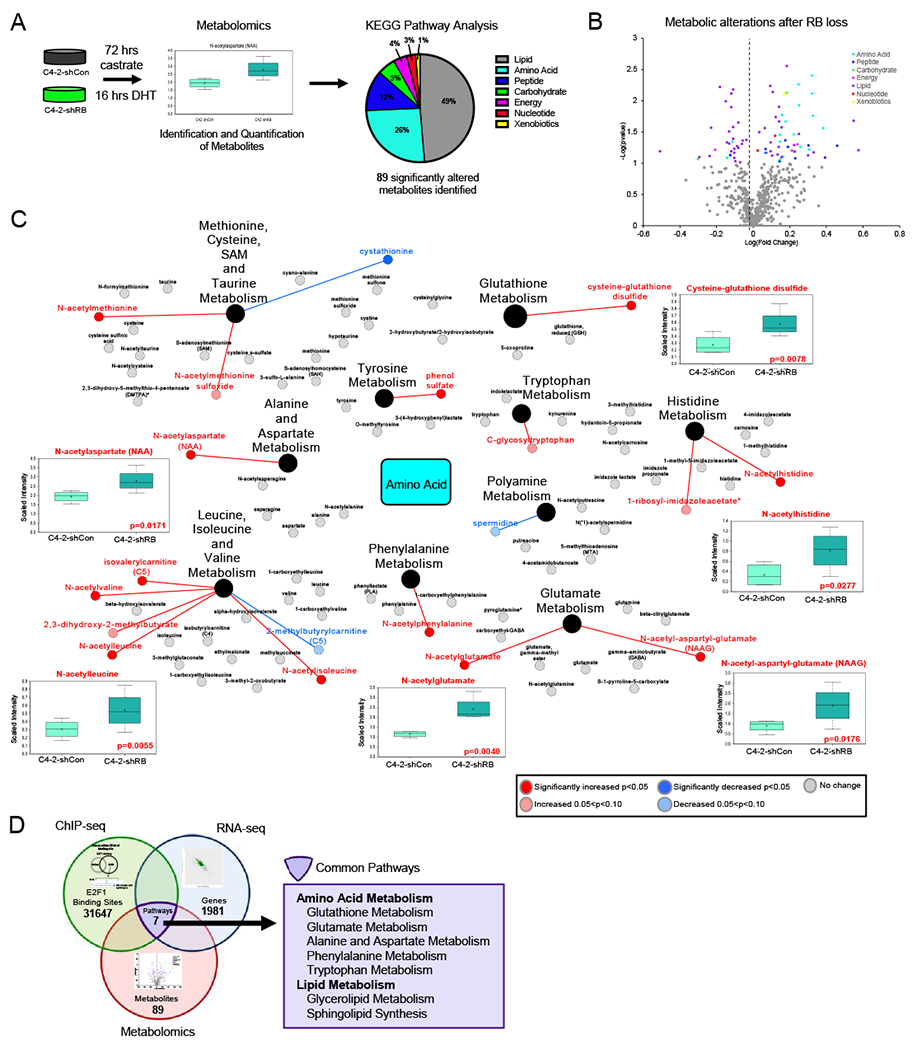
(A) Metabolomics was performed as described in C4-2-shCon and C4-2-shRB models followed by KEGG analysis (B) Metabolites displayed based on pathways. Significant metabolites defined by p<0.05. Gray data points represent metabolites not significantly changed. (C) Map of amino acid metabolism metabolites altered. Increased metabolites are shown in red, decreased metabolites are shown in blue. (D) Pathways that were identified to be altered after RB1 depletion in ChIP-seq, RNA-seq and Metabolomics combined. Overlapping pathways are shown in purple.
To prioritize specific metabolic pathways most likely to be directly regulated by E2F1 after RB1 depletion, pathway analysis from ChIP-seq, RNA-seq, and metabolomics were integrated as shown in Figure 4D. This comparison included the pathways enriched from E2F1 binding-associated genes (31647 binding sites, green circle), the pathways identified from the transcriptional alterations either exclusively or inversely regulated compared to RBD-induced CRPC models (1981 genes, blue circle), and the pathways identified via metabolomic analyses after RB1 depletion in CRPC (89 metabolites, red circle). Integration of these complex datasets revealed seven metabolic pathways commonly altered after RB loss in late stage disease as indicated by novel E2F1 regulation, transcriptional changes, and altered metabolites (Fig 4D). These pathways included five amino acid synthesis pathways including glutamate metabolism and glutathione metabolism, both of which are of strong cancer significance (31). Together, these data identify an unexpected consequence of RB1 depletion on cancer cell metabolic control, reprogramming lipid and amino acid metabolism.
RB1 depletion in late stage disease protects against ROS through increased glutathione synthesis.
Data presented herein indicate E2F1 gain of function after RB1 depletion post-CRPC significantly alters metabolic control, resulting in increased amino acid and glutathione synthesis. Glutathione, an antioxidant that functions by neutralizing intracellular reactive oxygen species (ROS), has been shown to protect cells from DNA damaging agents and promote therapy resistance (32); thus, this pathway was prioritized for further investigation. The synthesis of the tripeptide glutathione requires the amino acids glutamate, cysteine and glycine, which can be either transported into the cell by membrane transporters or metabolized from other sources such as amino acids. Analysis of three common cancer-associated amino acid transporters (SLC1A5/ASCT2, alanine-serine-cysteine transporter 2; LAT1/SLC7A5, L-type amino acid transporter 1; and xCT/SLC7A11 cystine/glutamate transporter), showed a significant increase in expression after RBD-post CRPC (Fig 5A, left, red). ASCT2 imports glutamine which is rapidly converted to glutamate by intracellular glutaminase for both glutamate supply and as a glutamate amine donor in the serine/glycine pathway. xCT exchanges glutamate to facilitate import of cystine, which can subsequently be reduced to cysteine within the cell. LAT1 transports methionine into the cell which can ultimately be converted to cysteine through the trans-sulfuration pathway (33–36). Validation of the mRNA expression of SLC1A5/ASCT2 confirmed a greater than 1.6-fold increase in CRPC, BrCa, non-small cell lung cancer (NSCLC), and bladder cancer models after RB1 depletion. Further, the mRNA of multiple crucial enzymes within the pathway were also found to be increased following RB1 depletion in CRPC. Cystathionase (CTH), the enzyme that converts cystathionine, the output of the transsulfuration pathway, to cysteine was confirmed to have a 1.6-fold mRNA increase in CRPC and NSCLC, and a 1.2-fold increase in bladder cancer models. Lastly, gamma-glutamylcysteine synthetase (GCLC), the rate-limiting enzyme required for the first step of glutathione synthesis, was confirmed to have an average 4.1-fold increase in CRPC and a 2-fold increase in BrCa, NSCLC, and bladder cancer models. The protein expression for each of these validated pathway components was confirmed with a 1.3-fold increase in GCLC and ASCT2 and a 2.2-fold increase in CTH following RB1 depletion in CRPC (Fig 5B). Further, metabolites within the pathway were also changed following RB1 depletion. Cystathionine, the precursor for the reaction catalyzed by CTH was decreased, while glutamine and gamma-glutamylcysteine were increased (Fig 5A, left, blue), highlighting a change in enzymatic activity within the pathway. To further examine the impact of RB loss on glutathione metabolism, the endpoint of the pathway, total glutathione was measured in CRPC, BrCa, NSCLC and bladder cancer disease models. RB1 depletion resulted in an average of a 1.5-fold increase in total glutathione across all models (Fig 5C) further supporting an increase in glutathione synthesis following RB1 depletion in advanced disease and expanding the impact of these findings. These data implicate a significant role for RB in regulating the glutathione synthesis pathway at several key enzymatic points and across multiple advanced disease types.
Figure 5. RB loss protects against ROS through increased glutathione synthesis.
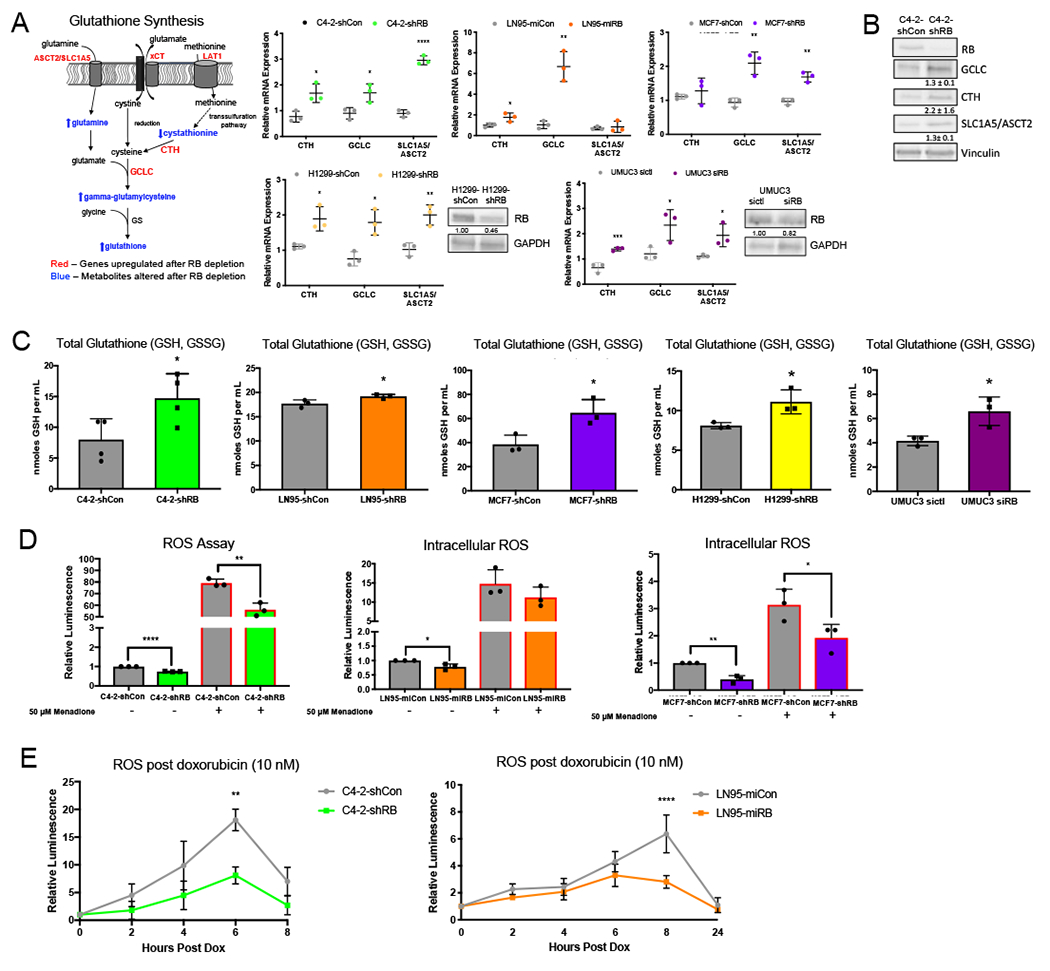
(A) Glutathione synthesis pathway highlighting increased genes after RB knockdown (red) and altered metabolites after RB knockdown (blue). Validation of increased relative mRNA expression of genes required for glutathione metabolism in C4-2, LN95, MCF7, and H1299 derived isogenic models after RB1 depletion and after transient knockdown of RB in UMUC3 models. (B) Validation of protein increase in C4-2-shCon C4-2-shRB. (C) Total glutathione in C4-2, LN95, MCF7, and H1299 derived isogenic models after RB1 depletion and after transient knockdown of RB in UMUC3 models. (D) ROS assay in C4-2, LN95, and MCF7 derived isogenic models. Menadione was used at 50 μM as a positive control for inducing ROS. (E) ROS assay after multiple timepoints of doxorubicin treatment in C4-2 and LN95 derived isogenic models. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
Given that RB1 depletion results in an increase in the antioxidant glutathione, the effect of RB1 depletion on intracellular ROS was examined. Basal levels of intracellular ROS were decreased by 30% after RB1 depletion in CRPC models and 60% in BrCa. Further, when treated with 50 μM menadione, a known inducer of ROS, intracellular ROS continued to display a reduction in the RB1 depleted condition (Fig 5D), suggesting that the increase in glutathione after RB1 depletion lowers intracellular ROS, potentially acting to protect cells from full ROS induction and downstream effects. To examine the response to clinically relevant therapeutics, intracellular ROS was measured in a time-dependent manner following treatment with the cytotoxic agent, doxorubicin (dox). In response to dox, intracellular ROS peaked between 6 and 8 hours after treatment, while this gradual increase and ROS induction was significantly reduced in the absence of RB (Fig 5E), further indicative of a protective role for RB1 LOF through regulation of glutathione production. In sum, these data indicate that RB1 depletion in CRPC and invasive BrCa drives altered metabolic pathways, thereby promoting an increase in glutathione synthesis. This increase in glutathione significantly reduced basal intracellular ROS and protected from ROS induction following treatment with ROS inducers, including doxorubicin, highlighting a protective mechanism in cancer cells against cytotoxic agents currently used to target advanced disease.
Glutathione synthesis is directly regulated by RB loss-induced E2F1 function and is a candidate for therapeutic intervention.
To validate that these biological changes are a direct consequence of RB1 loss, PSM.7-LP, an established, constitutively activate RB was utilized to rescue RB function in RB1 depleted models (37). Use of such a strategy is required, as it is well appreciated the ectopic expression of wild-type RB is rapidly inactivated by endogenous CDKs and does not rescue RB loss(38,39). As such, shRNA-resistant PSM.7-LP variants were generated and subsequently transfected into the RB1 depleted models. Expression of endogenous RB and PSM.7-LP was verified by immunoblot showing depleted full-length RB and the presence of PSM.7-LP (Fig 6A, left). Expression of E2F1 was decreased by 1.4-fold in RB1 depletion models following transfection with PSM.7-LP. This anticipated decrease is a consequence of the presence of constitutively active RB, further validating a functional protein (Fig 6A, left). To examine the effects of rescuing active RB on glutathione synthesis, mRNA expression of E2F1 and glutathione synthesis genes was measured following transfection of PSM.7-LP. Supporting the changes observed at the protein level, expression of PSM.7-LP led to a 2.1-fold reduction in E2F1 mRNA. Further, mRNA expression of CTH, GCLC, and SLC1A5/ASCT2 was also significantly reduced by 1.5 to 2.5-fold following rescue of active RB (Fig 6A, right). These data validate that glutathione synthesis genes are regulated directly by RB and indicate a positive correlation between E2F1 and the expression of CTH, GCLC, and SLC1A5/ASCT2. To further confirm that RB directly regulates glutathione synthesis, total glutathione was also measured following the expression of PSM.7-LP (Fig 6B). Supporting the transcript data, total glutathione was significantly reduced by 1.5-fold with the rescue of active RB, confirming that RB is directly regulating glutathione synthesis in CRPC.
Figure 6. Glutathione synthesis is directly regulated by RB loss-induced E2F1 function and is a candidate for therapeutic intervention.
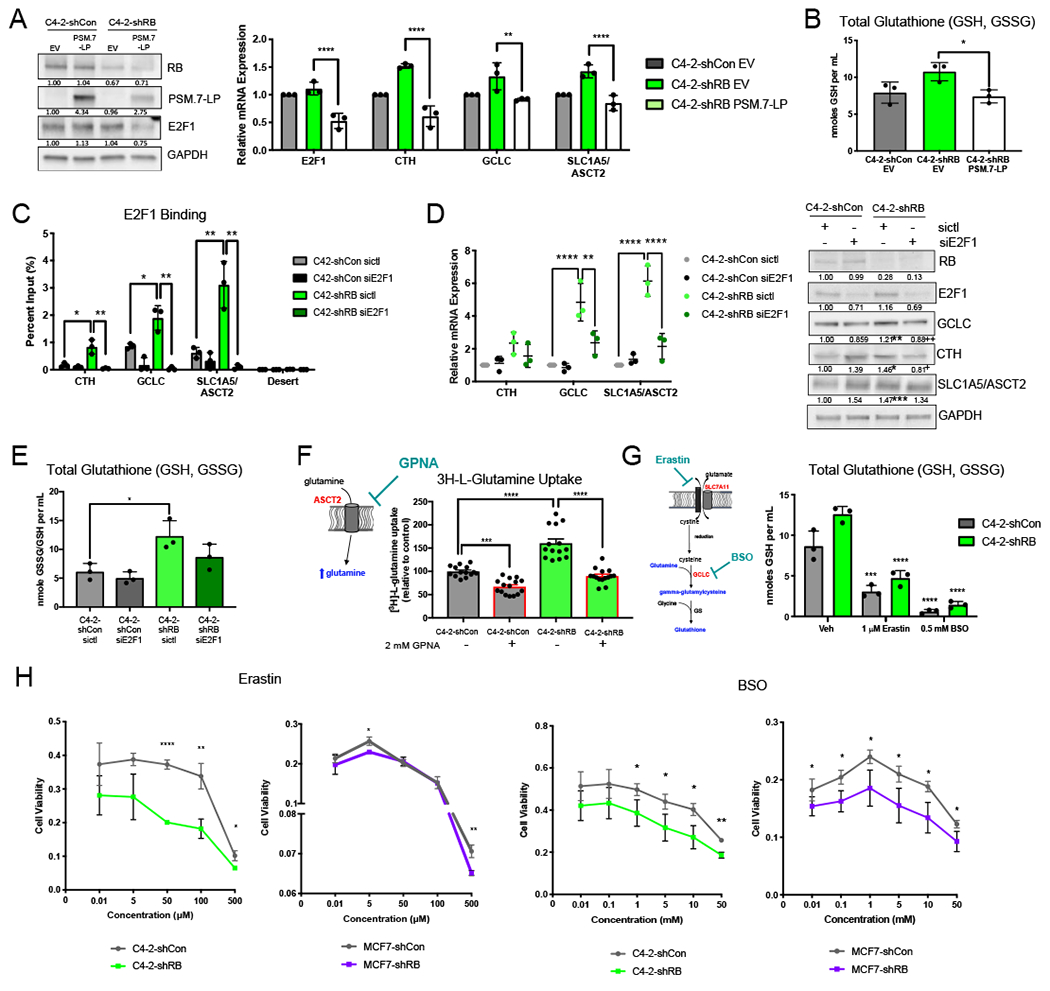
(A) Protein expression of RB, PSM.7-LP, and E2F1 after transfection of 1 μg of PSM.7-LP (left). mRNA expression of E2F1 and glutathione synthesis genes after RB loss and rescue with PSM.7-LP (right). (B) Total glutathione after RB knockdown and rescue with PSM.7-LP. (C) Validation of E2F1 binding at glutathione synthesis gene promoters after RB knockdown. E2F1 binding is lost following transient knockdown of E2F1. (D) mRNA expression of targets after RB knockdown and treatment with siControl or siE2F1 (left). Validation of protein expression of glutathione metabolism enzymes after RB1 depletion with treatment of siControl or siE2F1 (right). Blot quantification is shown below bands. (E) Total glutathione with and without RB knockdown after treatment with siControl or siE2F1. (F) Glutamine uptake assay showing increased glutamine uptake after RB knockdown. C4-2-shCon and C4-2-shRB were treated with 2 mM of the SLC1A5/ASCT2 transporter inhibitor, L-γ-Glutamyl-p-nitroanilide (GPNA). (G) Total glutathione following treatment with either SLC7A11 inhibition via Erastin or GCLC inhibition via BSO, in control and RB knockdown models. (H) Cytotoxicity of varying concentrations of Erastin and BSO in control and RB knockdown CRPC and BrCa models *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
To delineate the underlying mechanism by which RB1 depletion drives increased glutathione synthesis and thus protection from ROS-inducing therapeutics, the potential for E2F1 regulation of glutathione synthesis was examined. ChIP-seq analyses indicated that E2F1 binding was enhanced after RB1 depletion at genes encoding for glutathione synthesis pathway components (Fig S4A). This trend was validated via ChIP-qPCR at the promoters of CTH, GCLC and SLC1A5/ASCT2 where a 5-fold, 2.2-fold, and 5-fold increase in E2F1 binding was observed after RB1 depletion, respectively. This increase in E2F1 binding at glutathione synthesis-related gene promoters was abolished following the knockdown of E2F1 (Fig 6C). Enhanced E2F1 binding was accompanied by an increase in relative mRNA (>2-fold change) and protein expression (>1.2-fold change) of these targets (Fig 6D, left and right, respectively). Importantly, these alterations were abrogated by knockdown of E2F1 (Fig 6D, Fig S4B), suggesting a direct role for E2F1 regulation of glutathione synthesis pathway components by E2F1 after RB1 depletion in advanced CRPC. Further supporting this concept, the marked 2-fold increase in total glutathione following RB1 depletion was largely abrogated after knockdown of E2F1 (Fig 6E). Overall, these data provide a novel link between E2F1 and glutathione synthesis in advanced disease. These data reveal that RB1 loss confers an E2F1-dependent metabolic advantage in CRPC, with potential implications for therapeutic efficacy.
To further investigate the impact of RB regulation on glutathione metabolism, biological function was assessed throughout the pathway. To assess the initial step of glutamine transport, [3H]-L-glutamine uptake was measured before and after RB1 depletion. RB1 depletion alone resulted in a significant 1.6-fold increase in glutamine uptake, which could be inhibited using the ASCT2 inhibitor, L-γ-glutamyl-p-nitroanilide (GPNA) (Fig 6F). Levels of leucine uptake, which is not an ASCT2 substrate, were unchanged after RB1 depletion (Fig S5) indicating that there was a specific increase in ASCT2-mediated glutamine uptake after RB1 depletion and that this increase can be therapeutically targeted. To target this pathway further, total glutathione and cytotoxicity of each isogenic model was assessed in response to xCT (SLC7A11) inhibitor, Erastin and GCLC inhibitor, buthionine sulfoximine (BSO) (Fig 6G, left). Erastin and BSO significantly reduced total glutathione in both RB1 depleted and control models by an average of 11-fold and 2.7-fold respectively, verifying that both inhibitors directly target glutathione synthesis in the models utilized (Fig 6G, right). Critically, inhibition of xCT (SLC7A11) or GCLC resulted in a significant decrease in cell viability at varying concentrations in RB1 depleted models of CRPC and BrCa when compared to the RB1 intact controls (Fig 6H). These data indicate that RB1 depletion leads to increased sensitivity to inhibition of glutathione synthesis suggesting that RB1 depleted tumors are more reliant on this pathway for survival. Additionally, these data reveal novel avenues to clinically target RB1 deficient tumors.
The consequences of RBD-post CRPC include a rearrangement of transcriptional networks, rewiring of E2F1 function, and reprogramming of cellular metabolism. More specifically, RB1 depletion has been shown to promote a gain in E2F1 binding at the promoters of genes that encode principal components of the glutathione synthesis pathway, with expression and protein changes in these genes dependent on E2F1. Moreover, increased expression of the glutamine transporter, SLC1A5/ASCT2, was demonstrated to drive an increase in glutamine uptake in RBD-post CRPC cells. Further downstream in the pathway, gained E2F1 function after RB1 depletion induced an increase in total glutathione within cancer cells and inhibition at various points within the pathway showed increased sensitivity in the RB1 depleted models. To validate these observations in vivo, tumor xenografts models were utilized. Specifically, isogenic RBD-post CRPC cell models were subcutaneously injected into athymic nude mice. When tumors reached 500 mm3, tumors were harvested and assessed for glutathione, mRNA, and protein. RB1 depletion within the tumors was validated via immunohistochemistry (IHC) (Fig 7A). In agreement with in vitro studies, there was no change in tumor doubling time nor time to tumor take after RB1 depletion in CRPC in vivo (Fig S6). However, total glutathione in the RB1-depleted tumors was significantly increased (2.1-fold) compared to control (Fig 7B). Further validating in vitro findings, mRNA expression of GCLC and SLC1A5/ASCT2 was significantly increased (>1.2 FC) (Fig 7C), while protein expression examined via IHC was also increased following RB1 depletion in vivo (Fig 7D, Fig S7). Thus, these findings further validate the role of RB LOF in glutathione metabolic control in CRPC.
Figure 7. RB1 depletion drives glutathione metabolism in advanced disease.
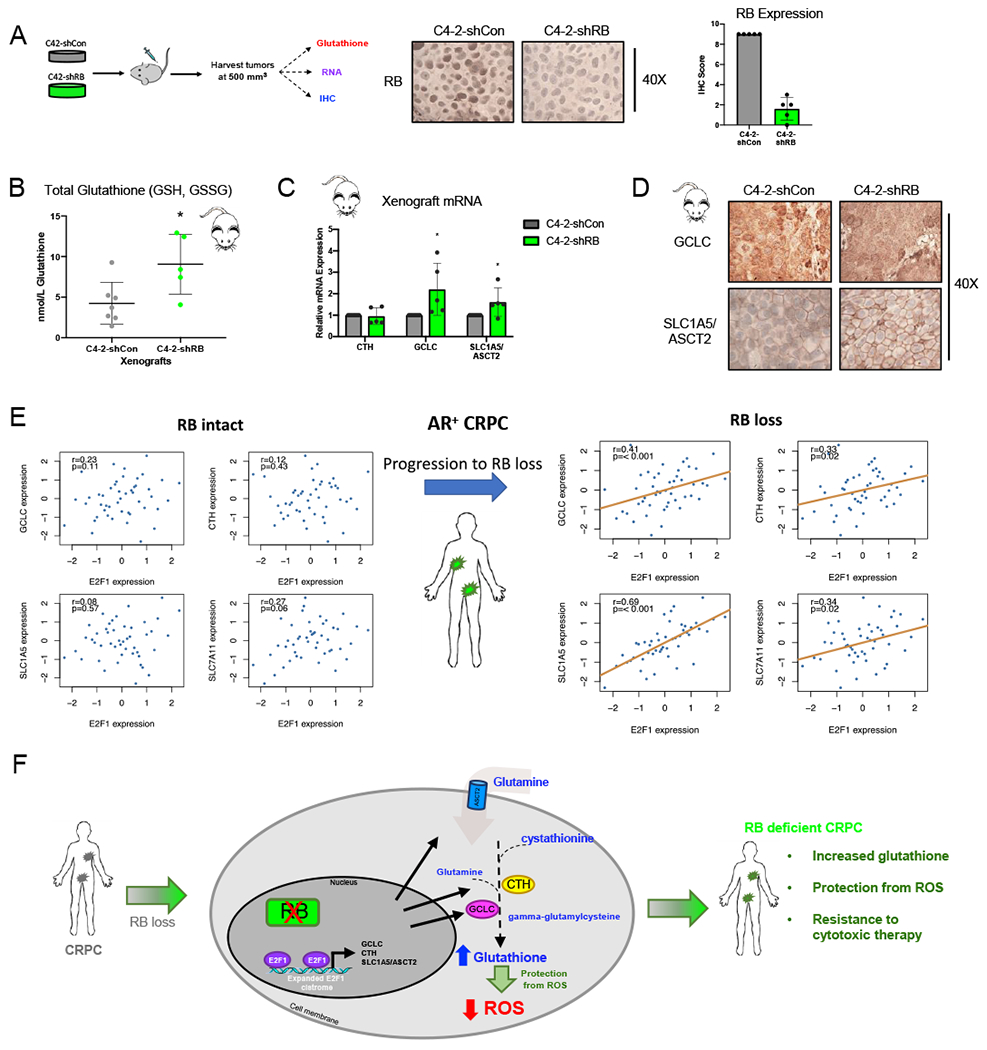
(A) C4-2-shCon or C4-2-shRB cells were subcutaneously injected into both flanks of athymic nude mice. Tumors were harvested at 500 mm3 and sectioned for glutathione, mRNA, and protein analysis. IHC imagine and quantification for RB. (B) Relative glutathione levels in C4-2-shCon and C4-2-shRB tumor xenografts is shown. (C) mRNA expression of E2F1 target genes. (D) Immunohistochemistry staining of target genes from tumor xenografts. Tumor images shown at 20X. (E) RNA-sequencing from fresh tumor biopsies was performed on patient samples from a novel metastatic CRPC patient cohort from Royal Marsden Hospital (RMH) in London, UK (n=98). The cohort was divided into tumors with intact RB and RB depletion. The expression of GCLC, CTH, SLC1A5, and SLC7A11 was examined and compared to expression of E2F1. Of those tumors in the cohort with intact RB, no correlation was observed between E2F1 expression and expression of any of glutathione synthesis genes examined (left) When assessed across tumors with RB depletion, expression of all glutathione synthesis genes examined showed a positive correlation when compared to expression of E2F1 (GCLC, p<0.001; CTH p=0.02; SLC1A5, p<0.001 (right). (F) RB1 depletion results in an expansion of the E2F1 cistrome to drive transcription of genes required for glutathione metabolism thus, promoting protection from intracellular ROS in CRPC. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001.
Importantly, the impact of RB on genes controlling glutathione metabolism was assessed in a novel and unselected metastatic CRPC tumor cohort of 98 patient samples from the Royal Marsden Hospital. RNA-sequencing was performed to identify AR positive tumors with and without intact RB. The expression of GCLC, CTH, SLC1A5, and SLC7A11 was examined across the cohort and compared to E2F1. There was no correlation between the expression of E2F1 and any of the glutathione genes investigated (GCLC, CTH, SLC1A5, and SLC7A11) in tumors with intact RB (Fig 7E, left). However, significant positive correlations were observed between E2F1 and expression of all four glutathione genes in the RB loss tumors; GCLC (Spearman correlation p = <0.001), CTH (p = 0.02), SLC1A5 (p = <0.001), and SLC7A11 (p = 0.02) (Fig 7E, right). These correlations validate our findings that RB loss drives an increase in glutathione synthesis through E2F1 in late stage CRPC tumors.
Together these data are the first to reveal distinct biological and molecular consequences of RB1 depletion in HSPC and CRPC. This study identifies a novel function for E2F1 in rewiring tumor metabolism, driving a redox advantage and protection from cytotoxic therapeutics (Fig 7F); overall implicating RB1 as a biomarker for therapy response in advanced disease.
Discussion
While significant clinical evidence links RB tumor suppressor loss to poor outcome, the underlying mechanisms remain loosely defined. For the first time, the studies herein reveal the disease stage-specific, pro-tumorigenic impact of RB loss on disease progression, supported by the following key findings: (a) RB loss drives distinct biological networks contingent on stage of disease; (b) rewiring of the E2F1 cistrome and transcriptome in response to RB loss is stage-dependent; (c) the RB/E2F1 axis shifts regulation of metabolic pathways exclusively in advanced disease, including lipid and amino acid synthesis; (d) E2F1-mediated networks after RB loss drive increased glutathione synthesis as observed using multiple in vitro and in vivo models of disease, and results in altered ROS production; (e) the relationship between RB and glutathione was confirmed in clinical specimens; and (f) RB-loss induced ROS reduction facilitates bypass of cytotoxic therapy. Taken together, the present study identified the RB/E2F1 pathway as harboring stage-specific functions that regulate critical metabolic pathways in advanced cancers, which impacts therapeutic efficacy.
The data herein reveal striking differences in the biological and molecular outputs governed by RB loss across PCa progression. This evolution of RB regulation was illustrated by transcriptome and E2F1 cistrome analysis, where distinct gene regulation and E2F1 function was observed following RB loss in isogenic models of HSPC compared to models of CRPC derived from the same cell lineage (Figs 1D–F, 2A–B). The study herein is the first to examine the consequence of RB depletion under physiologically-relevant, AR active conditions across stages of disease progression providing the only comparison of the E2F1 cistrome in HSPC and CRPC after RB loss. RB depletion in CRPC resulted in 18,624 gained E2F1 binding sites compared to 3,597 gained sites after RB depletion in HSPC confirming stage-specific control of E2F1. This expansive shift in E2F1 binding after RB depletion in CRPC led to over 30,000 E2F1 binding sites, largely distinct from those identified in early stage disease. This expansion of E2F1 binding after RB loss is supported by previous studies in other PCa models (16), implicating RB loss as a direct modulator of E2F1 function. De novo motif analysis revealed that the expanded E2F1 binding is enriched for additional cancer-associated motifs including BORIS, FOXM1, and NF-1 motifs (40–42) (Fig 2C). BORIS and FOXM1 have been shown to be upregulated in PCa; BORIS positively correlating with Gleason score and AR protein levels, and FOXM1 shown to play a role in increasing migratory, invasive, and proliferative abilities (43,44). Further, NF1 is amplified in neuroendocrine PCa and suggested to regulate AR expression through activation of several kinases including mTOR (45,46). In BrCa, alternative splicing of BORIS has been shown to contribute to the Warburg effect, altering cell metabolism and driving tumorigenesis (47), while overexpression of FOXM1 has been shown to play significant roles in regulating cell cycle progression, invasion, metastasis, and drug resistance (48). This shift in E2F1 binding after RB depletion suggests that E2F1 is rewired to novel sites to drive an expanded set of cancer-related biological networks. Additionally, via known motif analysis, E2F1 is shown to bind in close proximity to a number of forkhead motifs, which are known co-regulators of nuclear receptors, suggesting that these forkhead proteins may also contribute to the recruitment of E2F1 to these novel sites (Fig 2D). Together, these data are the first to reveal a change in RB function across disease progression driven by altered control of E2F1 and provide molecular insight into the basis of E2F1-associated poor outcomes.
As RB loss is associated with poor outcome, these clinically significant changes in RB and E2F1 function emphasize the importance of understanding the timing of RB loss across disease progression. Previous studies have identified multiple RB loss gene signatures that are associated with poor outcome including shorter OS, PFS, and DSS (12,49). Further, it was shown that these signatures can be used to predict RB loss in clinical samples, revealing the clinical significance of RB loss-induced gene expression changes (16). The data herein revealed 1599 genes with increased expression and E2F1 binding within 30 kb of the gene promoter after RB depletion. When compared to previously published RB loss gene signatures, this 1599 gene list included 23% of genes identified in BrCa and 25% of genes identified in PCa signatures associated with shorter OS (12,49). Further, when examined beyond breast and prostate cancers, more than 35% of genes identified in multiple non-small cell lung cancer (NSCLC) gene signatures that predicted for poor therapeutic response and shorter OS were also present in this 1599 gene list (50,51), indicating that a significant number of novel RB/E2F1 regulated genes in CRPC are likely to have clinical impact. After identifying differential functions of RB and E2F1 across disease progression, the differences from the previously developed gene signatures is unsurprising; however, it highlights the importance of examining RB loss in advanced disease. The RB/E2F1 axis drives networks involved in metabolic control specifically in CPRC which is not seen in HSPC (Fig 3A). The shift to metabolic control without the advantage of increased cell growth (Fig 1C) indicate an evolution away from tumor growth and metastasis towards tumor survival and resistance to therapeutics. As the studies herein reveal that RB loss in late stage disease render tumors less responsive to therapy, the potential differences in the early and late stage signatures are of clinical significance.
Although RB loss in advanced disease induced widespread unique gene networks, a critical consequence of RB loss is the observed metabolic shift in lipid and amino acid synthesis. Analysis of the 1599 genes with increase expression and E2F1 binding after RB loss, revealed 16 enriched metabolic pathways suggesting a role for E2F1 and RB in regulating metabolism in advanced disease (Fig 3A). Metabolomic analysis further highlighted a significant change in metabolites involved in lipid and amino acid metabolism (Fig 4). Beyond this, previous studies have linked RB depletion to altered mitochondrial function. RB loss induces a decrease in mitochondrial protein expression in lung and colon cancers (52). Interestingly, the change in protein expression did not correlate with a corresponding change in mRNA level suggesting that mRNA expression of mitochondrial genes does not necessarily correlate with protein expression. While mRNA expression of these mitochondria proteins was also not altered in models herein, further assessment revealed a reduction in oxygen consumption rate (OCR), a measurement of mitochondrial function, in C4-2-shRB cells compared to C4-2-shCon (Fig S8). These data support this previous study revealing first that mitochondrial function can be altered without changes in mitochondrial gene expression at the mRNA level, and second that RB loss does induce a shift in mitochondrial function in CRPC in addition to lung and colon cancer. Overall, these studies support a role for RB in regulating a multitude of metabolic pathways through mechanisms beyond transcriptional control.
In addition to the studies herein showing that RB and E2F1 function to regulate metabolic control through transcription, E2F1 has also been shown to regulate nucleotide synthesis through transcriptional regulation of thymidine kinase (TK1), and dihydrofolate reductase (DHFR), and has been shown to regulate glucose oxidation through transcription of pyruvate dehydrogenase (PDK1) (53–55). Interestingly, blocking amino acid pathways via inhibition of ASCT2 or LAT1 has been shown to directly shut down E2F transcriptional activation, suggesting a reciprocal feedback between amino acid metabolism and E2F regulation that should be further examined (56,57). Further, the RB/E2F1 axis has been associated with metabolic control through overexpression of c-Myc. While c-Myc is a known regulator of numerous metabolic transcripts (58), E2F1 occupancy at the c-Myc promoter and mRNA expression of c-Myc is unchanged after RB loss in the models utilized herein. Even with this lack of change in regulation, c-Myc may still have a significant role in metabolism and should be investigated further in the context of RB loss. The RB/E2F1 regulated metabolic genes identified within this study expands on previous RB loss gene signatures. While genes such as TK1 and ribonucleotide reductases regulator subunit M2 (RRM2) have already been recognized as altered after RB loss (49), 87 of the 92 increased metabolic genes identified here, are exclusive to RB loss in advanced disease. Thus, the studies herein reveal a novel RB loss/E2F1 regulated metabolic gene signature for advanced disease. This study is the first to explore the genome-wide contribution of E2F1 to metabolic control and highlights that this metabolic control is specific to RB loss in advanced disease.
Further investigation of the RB loss-induced metabolic shift revealed coordinated alteration in glutathione synthesis and downstream ROS generation. Under normal physiological conditions, cells maintain a redox balance by controlling intracellular ROS with ROS scavengers, such as glutathione. Genes involved in the glutathione synthesis pathway such as GCLC, CTH, and SLC1A5/ASCT2, and the increase in glutathione observed after RB depletion were found to be directly regulated by E2F1 (Figs 5A, C & 6C–E). This study supports previous data in multiple model systems that RB depletion drives an increase in glutathione. In mouse embryonic fibroblasts (MEFs) triple knockout (TKO) of all RB family members (Rb-1, Rbl1 and Rbl2) revealed a change in glutamine uptake and increased glutathione production controlled by E2F3. This increase in glutamine uptake and utilization was required for increase proliferation, and modulation of E2F1 had no effect on glutamine uptake or cell proliferation (59). Though the study herein does not examine the effects of RB loss on E2F3 regulation of metabolism, this may be of interest for future studies. Further, increased glutathione was seen in a Drosophila model system with mutant RBF1 through a flux of glutamine, suggesting that this mechanism is conserved across species (60). The elevated glutathione observed herein coincided with lower intracellular basal ROS levels and a lower ROS induction following treatment with cytotoxic agents (Figs 5D, E). As cancer cells display elevated basal levels of ROS due to the increased proliferation and altered metabolism, this shift in redox homeostasis is often exploited therapeutically through the application of ROS inducing agents (61,62). These cytotoxic agents, such as cisplatin and doxorubicin, generate high levels of ROS to induce oxidative stress that leads to damage of protein, lipids, and DNA, promoting apoptosis, necrosis, and autophagic cell death (63). Thus, alterations within this redox control can have significant clinical implications. These studies are the first to confirm that the glutathione increase after RB depletion is directly controlled by E2F1 function and that this increase coincides with protection from ROS induction following treatment with clinically approved cytotoxic therapy.
Taken together, the studies herein are the first to demonstrate stage-specific consequences of RB loss in advanced disease. Through transcriptome, cistrome, and metabolome analysis, RB depletion rewired E2F1 to drive glutathione synthesis exclusively in advanced disease. This increase in glutathione synthesis drives an acquired protection from ROS generation in response to cytotoxic chemotherapy. These studies reveal a novel role for RB/E2F1 in regulating cell metabolism and implicate RB loss as a stage-specific driver of clinical response to treatment.
Material and Methods
Cell lines, cell culture and reagents.
LNCaP, C4-2, MCF7, H1299, and UMUC3 cells were obtained from ATCC. LnCaP95 cells were a kind gift from Dr. Jun Luo at John Hopkins University. C4-2, MCF7, and LNCaP derived cell lines were maintained in Improved Minimum Essential Medium (IMEM) (Thermo Fisher Scientific, 10024CV) supplemented with 5% fetal bovine serum (FBS), heat inactivated, 1% L-glutamine (2 mmol/l), and 1% penicillin-streptomycin (100 units/ml). H1299 and UMUC3 cells were maintained in IMEM supplemented with 10% heat inactivated FBS, 1% L-glutatmine (2 mmol/l), and 1% penicillin-streptomycin (100 units/ml). LnCaP95 derived cell lines were maintained in Modified IMEM (Thermo Fisher Scientific, A1048801) supplemented with 10% CDT and 1% penicillin-streptomycin. All cells were maintained at 37°C with 5% CO2. LNCaP, C4-2, MCF7, H1299, UMUC3 cell lines were authenticated by ATCC. All cell lines were checked for mycoplasma upon thawing.
RB1 knockdown cell lines.
LNCaP, C4-2, H1299, and MCF7 isogenic models were previously developed as described (64,65). Briefly, cells were transfected with either a shRNA plasmid targeting RB (sequence: 5′-CGCATACTCCGGTTAGGACTGTTATGAA-3′) or a control plasmid (MSCV donor) using FuGENE Transfection Reagent. Stable clones were isolated after 3-4 days of selection with 2.5 μg/mL of puromycin (13). LNCaP95 isogenic models were developed as previously described (65). Cells were transfected with either a plasmid targeting RB (sequence: 5′-GCAGTTCGATATCTACTGAAA-3′) or a non-specific control (sequence: 5′-GCTGAGGTGATAAACAGTTACA-3′). Newly generated LnCaP95 RB depleted (miCon and miRB) cell lines underwent at least three rounds of antibiotic selection with 2.5 μg/ml puromycin. These models were labeled LN95-miCon for the control and LN95-miRB for the RB depleted line.
In vitro RNA-sequencing.
RNA was extracted and purified utilizing TRIzol reagent and RNeasy Mini Kit (Qiagen) following manufacturer’s instructions. RNA-seq libraries were constructed using TruSeq RNA Sample Prep Kit v2 (Illumina, RS-122-2001) and sequenced on the NExtSeq 500 at the Dana-Farber Cancer Institute Molecular Biology Core Facility. RNA-seq was aligned against the hg19 human genome using STAR v2.5.2a (66). Differential gene expression was generated using DESeq2 v1.12.4 (67). Gene set enrichment analysis was performed through GSEA using gene sets from the Molecular Signature Database (MSigDB) (68). RNA-seq data has been deposited in GEO under accession number GSE154190.
Proliferation assays.
LNCaP-shCon, LNCaP-shRB, C4-2-shCon, and C4-2-shRB cells were plated at equal densities in 5% charcoal dextran-treated (CDT) media supplemented with 1% penicillin and 1% L-glutamine, to measure relative cell growth in castrate conditions. To measure cell growth in response to androgen stimulation, cells were treated with 0.1 nM DHT after 72 hours which was replenished every 48 hours. Day 0 timepoints represent cell number 24 hours after plating or 24 hours after DHT treatment. Cell number was quantified at day 4 and day 6 utilizing the Quanti-IT Pico Green dsDNA assay kit (Thermo Fisher).
RB Rescue.
PSM.7-LP, an established, constitutively activated RB was utilized as previously described (37). C4-2 isogenic cells were seeded at equal densities in 5% CDT and transfected using Liofectamine 2000 (Thermo Fisher Scientific 11668019) with 1 μg of either empty vector (EV) or PSM.7-LP DNA for 24 hours. Cells were treated with 10 nM DHT or either 16 hours (mRNA) or 24 hours (protein or glutathione) before harvest. Use of such a strategy is required, as it is well appreciated the ectopic expression of wild-type RB is rapidly inactivated by endogenous CDKs and does not rescue RB loss (38,39). As such, shRNA-resistant PSM.7-LP variants were generated and subsequently transfected into the RB knockdown models.
Chromatin Immunoprecipitation (ChIP)-Sequencing.
E2F1 ChIP-seq were performed as previously described (16). Briefly, cells were cross-linked with 1% fresh formaldehyde for 10 minutes at room temperature. Chromatin was sheared to approximately 200 bp using a Diaganode Ultrasonicator for 30 cycles (30 seconds on, 30 seconds off). E2F1 antibody utilized for ChIP-sequencing was purchased from Santa Cruz Biotechnology (sc-193). Validation ChIP-qPCR was performed with E2F1 antibody purchased from Bethyl Laboratories (A300-766A), recognizing epitopes distinct from the antibody purchased form Santa Cruz Biotechnology to avoid any accessibility concerns. E2F1 ChIP-Seq libraries were constructed using ThruPLEX-FD Prep Kit (Rubicon Genomics). Sequencing was performed on NextSeq 500 at the Dana-Farber Cancer Institute Molecular Biology Core Facility. E2F1 ChIP-seq deposited in GEO repository under accession number GSE154191. ChIP-Seq was analyzed as previously described(16). Cis-regulatory element analysis was performed using CEAS v1.0.2. Motif analyses were performed through Homer v4.8.3 (69).
Gene Expression.
Cells were plated at equal densities in hormone-deficient media for 72 hours followed by 16 hours of 10 nM DHT treatment. RNA was isolated using TRIzol (Invitrogen). RT-PCR was performed using SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific). ABI StepOne Real-Time PCR using PowerSybr (Fisher Scientific 43-676-59) was utilized to perform quantitative PCR (qPCR) analyses. Primer sequences: GMPR (Fw: 5-’CTCAAGCTCTTCTACGGGA-3’; Rev: 5’-CTCAGAGGCTCTGTACTCAG-3’); PSAT1 (Fw: 5’-AAATGAGTCACAGGTCATCAG-3’; Rev: 5’-CTGGAACAGCTAGCAATTCC-3’); CHPT (Fw: 5’-AGAAGAACAAACTCTTGTTCCC-3’; Rev: 5’-CACTGCCATAAATACTGTGGA-3’); CERK (Fw: 5’-CTCATCCGGAAATGCTCCA-3’; Rev: 5’-AGTGAAGTCAAACTGGTCCT-3’); GSTA1 (Fw: 5’-AAGCTCCACTACTTCAATGC-3’; Rev: 5’-CTCTTCAAACTCTACTCCAGC-3’); CTH (Fw: 5’-ATAGCCGTTCTGGAAATCC-3’; Rev: 5’-AGGCCAAACAGTACTTAGC-3’); GCLC (Fw: 5’-AACCCAAACCATCCTACCC-3’; Rev: 5’-ACATTGTTCCTCCGTAGGG-3’); SLC1A5 (Fw: 5’-TCCTGGATCATGTGGTACG-3’; Rev: 5’-TAAACCCACATCCTCCATCTC-3’); 18S (Fw: 5’-CGGCGACGACCCATTCGAAC-3’; Rev: 5’-GAATCGAACCCTGATTCCCCGTC-3’).
Immunoblotting.
LNCaP, and C4-2 derived cells were plated in equal densities 5% CDT media for 72 hours followed by 24 hours of 10 nM DHT. LN95 derived cells were plated in equal densities in 10% CDT media for 24 hours and treated with 10 nM DHT for 24 hours. MCF7 derived cells were plated in equal densities in 5% FBS media and harvested after 48 hours. UMUC3 cells were transfected with siCtl or siRB as described below in 10% FBS media. Cells were harvested after 72 hours. Cell lysates were generated as previously described(70). Lysate was resolved by SDS-PAGE and transferred to polyvinylidene (PVDF) membrane. Proteins were analyzed using the following antibodies at 1:1000 dilution – RB (BD Pharmingen 554136), phospho-RB (Cell Signaling 9307S), AR (N-20, directed against amino acids 1-20 by Bethyl Laboratories), E2F1 (Cell Signaling Technologies 3742S), GAPDH (Santa Cruz Biotechnology sc-25778), CTH (Santa Cruz Biotechnology sc-365382), GCLC (Abcam ab53179), SLC1A5/ASCT2 (Cell Signaling Technologies 8057S), and Vinculin (Sigma-Aldrich V9264).
Flow Cytometry.
LNCaP and C4-2 isogenic cell models were plated in triplicate and treated for 24 hours with 0.5 μM of CDK4/6 inhibitor, Palbociclib (Selleck Chemicals S1116). Adherent and non-adherent cells were harvested together following 2 hours of 1:1000 BrdU labeling (Life Technologies 00-0103) and fixed with 100% ethanol. After additional propidium iodide (PI) staining, flow cytometry was performed using a Guava easyCyte flow cytometer. At least 10,000 events were analyzed per sample. Analysis was performed using the Guava InCyte software.
ChIP-qPCR.
qPCR analyses were performed on the ABI StepOne Real-Time PCR using PowerUPsybr (Fisher Scientific, A25742) Primer sequences are listed below. For ChIP-qPCR, C4-2-shCon and C4-2-shRB cells were transfected as described below. Cells were transferred 5% CDT media following transfection for 72 hours and treated with 10 nM DHT for 3 hours. Primer Sequences: GMPR (Fw: 5’-TGTCCCTAACCAGTCTGCCT-3’; Rev: 5’-ACGCCGCGGTCTTAAACA-3’); PSAT1 (Fw: 5’-CACCTTCTTCTGGTTTGGGCT-3’; Rev: 5’-CGAGTGAGCTGGAAACCTGC-3’); CHPT (Fw: 5’-TGTTCCGACCCCTAGGAGAG-3’; Rev: 5’-CGCCAACGGCAACTTGTTAT-3’); CERK (Fw: 5’-CAGCCAGTCTGGAAGAGCAG-3’; Rev: 5’-AGCACTGTGCAAGCCAAATG-3’); GSTA1 (Fw: 5’-CTCAACGGAGTCCCACAGAC-3’; Rev: 5’-TTCCAGGAGAACACCCAAGC-3’); CTH (Fw: 5’-GGCTTACGTCTTTTAGCACCG-3’; Rev: 5’-GGTCCAAGTGGTGTTAGTCCC-3’); GCLC (Fw: 5’- AGGCCGATTCCCAGTAGTGT -3’; Rev: 5’-GGGGGCTGTATCACAACAAC -3’); SLC1A5 (Fw: 5’-CTGCCAGGGATAAGGGTAGG-3’; Rev: 5’-AACCCCTGTGGTTTAAGGGC -3’); Desert (Fw: 5’-CTAGGAGGGTGGAGGTAGGG-3’; Rev: 5’-GCCCCAAACAGGAGTAATGA-3’).
Metabolomics.
Metabolomics were performed by Metabolon, Inc (Durham, NC). C4-2-shCon and C4-2-shRB cells were seeded at 1.5 million cells per plate in 5% CDT media for 72 hours followed by 10 nM DHT treatment for 16 hours in quadruplicate. Cell pellets were collected following Metabolon sample guidelines, flash frozen and stored at −80 °C until shipment. Metabolite identification was performed through ultra-high-performance liquid-phase chromatography and gas-chromatography separation, coupled with mass spectrometry. Compounds were identified by comparison to libraries maintained by Metabolon. Data visualization was performed using Metabolon’s custom-built Cytoscape plugin.
Amino Acid Uptake Assay.
C4-2 derived cells were assessed using a [3H]-labelled amino acid uptake assay, as described previously (71,72). Briefly, cells (1 × 105/well) were incubated at 37°C in 96-well plates with [3H]-L-glutamine (400 nM; Perkin Elmer) or [3H]-L-leucine (200 nM; Perkin Elmer) in glutamine-free RPMI or leucine-free RPMI media (Invitrogen) for 15 min in the presence or absence of 2 mM l-γ-glutamyl-p-nitroanilide (GPNA; MP Biochemicals; diluted in H2O). Cells were collected and transferred to filter paper using a 96-well plate harvester (Wallac, PerkinElmer), dried, exposed to scintillation fluid (PerkinElmer) and [3H] levels analyzed using a liquid scintillation counter (PerkinElmer).
Glutathione Assay.
C4-2, LN95, MCF7, and H1299 derived cells were seeded at equal densities in 5% CDT media for 72 hours followed by 24 hours 10 nM DHT treatment. Relative glutathione was measured using a glutathione assay kit (Sigma-Aldrich, CS0260) following manufacturer’s instructions. UMUC3 cells were transfected with siCtl or siRB as described below and harvested after 72 hours in 10% FBS IMEM. For assays with siE2F1, transfection was performed as described below in 5% CDT media for 72 hours following transfection and treated with 10 nM DHT for 24 hours. For assays with drug treatments, C42 isogenic cells were seeded at equal densities in 5% CDT for 72 hours. Cells were treated with 10 nM DHT for 24 hours before an additional 24-hour treatment with either 0.5 mM L-Buthionine-sulfoximine (BSO) (Sigma-Aldrich B2515) or 1 μM Erastin (Sigma-Aldrich E7781) prior to harvest. RB rescue with PSM.7-LP assays were performed following 72 hours of transfection as described above in 5% CDT and 24 hours of 10 nM DHT treatment. Relative fluorescence was measured on a BioTek Synergy HT microplate reader and analysis using BioTek Gen5 2.09 software.
ROS Assay.
C4-2, LN95, and MCF7 derived cell lines were seeded at equal densities. C4-2 cells were plated in 5% CDT media, LN95 were plated in 10% CDT media, and MCF7 cells were plated in 5% FBS media. C4-2 and LN95 isogenic cells were treated with 10 nM DHT for 24 hours following 72 hours of CDT. Menadione (Sigma-Aldrich, M5625) treatment was utilized as a positive control. Relative ROS levels were measured by fluorescence using the ROS-Glo Assay (Promega, G8821). Fluorescence was measure on a BioTek Synergy HT microplate reader and analysis using BioTek Gen5 2.09 software.
RNA Interference.
C4-2-shCon and C4-2-shRB cell lines were seeded on poly-L-lysine-coated plates in 5% FBS media for 24 hours. Cells were transfected with either scramble control (C4-2-shCon siCtl, C4-2-shRB siCtl) or E2F1 (C4-2-shCon siE2F1, C4-2-shRB siE2F1) siRNA pools (Dharmacon) for 8 hours according to the manufacturer’s protocol. Following transfection, the cells were moved 5% CDT media for 72 hours followed by appropriate 10 nM DHT treatment. UMUC3 cells were seeded on poly-L-lysine-coated plates in 10% FBS media for 24 hours. Cells were transfected with either scramble control (UMUC3 siCtl) or RB (UMUC3 siRB) siRNA pools (Dharmacon) for 8 hours according to manufacturer’s protocol. Cells were harvested after 72 hours.
MTT Cell Viability Assay.
C4-2 and MCF7 isogenic pairs were seeded at equal densities in a 96-well plate. C4-2 derived models were seeded in 5% CDT for 72 hours followed by 24 hours of 10 nM DHT. MCF7 derived models were seeded in 5% FBS for 48 hours. Cells were treated with varying concentrations of Erastin (0.1 μM to 500 μM) or BSO (0.01 mM to 50 mM) for 24 hours MTT assay was performed according to the manufacturer’s instructions (Sigma-Aldrich 11465007001). Absorbance was measured at 625 nm on a BioTek Synergy HT microplate reader and analysis using BioTek Gen5 2.09 software.
Xenografts.
C4-2-shCon and C4-2-shRB cells were resuspended in 100 μL of saline with 50% Matrigel (BD Biosciences 354234). Cell mixture (3x106 cells per injection) was injected subcutaneously into the flank of 5 to 6-week-old athymic nude male mice following protocols approved by IACUC at Thomas Jefferson University. Tumor volume was measured with digital calipers multiple times per week. Mice were sacrificed, and tumors were harvested once tumors reached an approximate size of 500 mm3. Tumors were cut into four pieces for glutathione, mRNA, and IHC. Glutathione analysis was performed following manufacturer’s guidelines using a glutathione assay kit (Sigma-Aldrich, CS0260). For RNA extraction, tumor tissue was homogenized, and RNA was extracted using TRIzol (Invitrogen). Immunohistochemistry (IHC) staining was performed by the Sidney Kimmel Cancer Center Shared Resource Translational Pathology Core. Antibodies utilized included RB (Abcam ab226979), GCLC (Abcam ab 53179), and SLC1A5/ASCT2 (Cell Signaling Technology 8057S).
Royal Marsden Hospital Cohort.
RMH cohort patient samples (n=98) that were collected and analysed with written informed consent under the CCR2472 protocol approved by the Ethics Committee at the Royal Marsden NHS Foundation Trust Hospital (London, UK).
RMH sample RNA-sequencing.
RNA, from fresh tissue biopsy, quality was analyzed using Agilent RNA ScreenTape assay (Agilent). 500 ng of total RNA was used for library preparation using the NEBNext rRNA depletion kit followed by NEBNext Ulta II directional RNA assay kit as per manufacturers protocol (New England Biolabs). Library quality was confirmed using the Agilent High sensitivity D1000 ScreenTape Assay (Agilent). The libraries were quantified and normalized by qPCR using Generead Library Quant Kit (Qiagen). Library clustering and sequencing were performed on the Illumina NovaSeq 6000. The libraries were run across two lanes of an Illumina NovaSeq S2 flowcell using 150 base pair pair-end v1 Kit and eight base pair dual indexes. Base-calling and quality scoring were performed using Real-Time Analysis (version v3.4.4) and FASTQ file generation and de-multiplexing using Illumina bcl2fastq2 (version 2.20). CRPC transcriptomes reads were aligned to the human reference genome (GRCh37/hg19) using TopHat2 (version 2.0.7). Gene expression, fragments per kilobase of transcript per million mapped reads (FPKM), was calculated using Cufflinks. AR activity score was an accumulation measurement of AR pathway activity based on 43 genes regulated by AR in prostate cancer cell lines and metastatic prostate cancer as previously described (73).
Seahorse Assay.
Cellular oxygen consumption rate (OCR) was measured with a Seahorse XF96e bioanalyzer (Seahorse Bioscience, MA) using a Cell Mito Stress Test Kit according to manufacturer’s instructions. Briefly, C4-2-shCon or C4-2-shRB cells (1.5 × 104 cells/well) were seeded in 4 wells (technical replicates) each of a Seahorse XF 96-well assay plate in full growth medium. After overnight attachment, the medium was carefully washed and replaced with pre-warmed running medium (consisting of non-buffered DMEM (Agilent Technologies, CA) supplemented with 1 mM sodium pyruvate, 2 mM glutamine and 10 mM glucose, pH 7.4). Plates were incubated for 60 min in a non-CO2 incubator at 37°C before four basal measurements were undertaken determining oxygen and proton concentration in the medium. The ATP synthase inhibitor oligomycin (1 μM) was immediately added followed by four further measurements; Carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP; 1 μM) was then added with another four further measurements, before addition of the complex I and III inhibitors rotenone/antimycin A (0.5 μM) and a final four further measurements taken.
Statistical analysis.
All experiments were performed in technical triplicate with at least 3 biological replicates per condition. Data are displayed as mean ± standard error of the mean (SEM). Statistical significance (p < 0.05) was determined using Student’s t-test, one-way ANOVA, and two-way ANOVA on GraphPad Prism Software as appropriate.
Supplementary Material
Statement of Significance.
This study identifies stage-specific consequences of RB loss across cancer progression that have a direct impact on tumor response to clinically utilized therapeutics. The study herein is the first to investigate the effect of RB loss on global metabolic regulation and link RB/E2F1 to redox control in multiple advanced diseases.
Acknowledgements
This work was supported by NIH T32 grant to Amy C Mandigo (GM100836); NIH R01 grants to Karen E Knudsen (5R01CA17640105, 5R01CA18256905, 5R01CA21732903); NCI F99 grant to Jennifer McCann (F99CA212225); and the Sidney Kimmel Cancer Center (SKCC) Support Grant (5P30CA056036). Additional support was provided by the SKCC Cancer Genomics, Translational Research/Pathology and Biostatistics core services, specifically Dr. Benjamin Leiby. We would like to thank Dr. Jun Lou (John Hopkins University) for providing the LnCaP95 cells utilized in this study. The sponsors were critical in study design, data collection, analysis, interpretation, and review of the manuscript.
Conflicts of Interest Disclosure: The following disclosures for Karen E Knudsen are unrelated to this work and included: Research Support (Celgene, CellCentric) and Consultant (CellCentric, Sanofi, Atrin, Celgene, Janssen, Genentech). The following disclosures for Jeff Holst: Founder and Chief Scientific Officer of MetabloQ Pharmaceuticals, Research Support and Consultant (MetabloQ Pharmaceuticals). No disclosures for the other authors.
References
- 1.Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev. Genes Dev; 2016;30:1492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van den Heuvel S, Dyson NJ. Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol. Nat Rev Mol Cell Biol; 2008;9:713–24. [DOI] [PubMed] [Google Scholar]
- 3.Di Fiore R, D’Anneo A, Tesoriere G, Vento R. RB1 in cancer: Different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J Cell Physiol. 2013;228:1676–87. [DOI] [PubMed] [Google Scholar]
- 4.Knudsen ES, Knudsen KE. Tailoring to RB: Tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8:714–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brambilla E, Moro D, Gazzeri S, Brambilla C. Alterations of expression of rb , p16 ink4a and cyclin d1 in non-small cell lung carcinoma. J Pathol. 1999;188:351–60. [DOI] [PubMed] [Google Scholar]
- 6.Zojer N, Königsberg R, Ackermann J, Fritz E, Dallinger S, Krömer E, et al. Deletion of 13q14 remains an independent adverse prognostic variable in multiple myeloma despite its frequent detection by interphase fluorescence in situ hybridization. Blood. 2000;95:1925–30. [PubMed] [Google Scholar]
- 7.Xu HJ, Cagle PT, Hu SX, Li J, Benedict WF. Altered retinoblastoma and p53 protein status in non-small cell carcinoma of the lung: potential synergistic effects on prognosis. Clin Cancer Res. American Association for Cancer Research; 1996;2:1169–76. [PubMed] [Google Scholar]
- 8.Cordon-Cardo C, Wartinger D, Petrylak D, Dalbagni G, Fair WR, Fuks Z, et al. Altered expression of the retinoblastoma gene product: prognostic indicator in bladder cancer. J Natl Cancer Inst. 1992;84:1251–6. [DOI] [PubMed] [Google Scholar]
- 9.Xu HJ, Cairns P, Hu SX, Knowles MA, Benedict WF. Loss of RB protein expression in primary bladder cancer correlates with loss of heterozygosity at the RB locus and tumor progression. Int J cancer. 1993;53:781–4. [DOI] [PubMed] [Google Scholar]
- 10.Patiño-García A, Piñeiro ES, Díez MZ, Iturriagagoitia LG, Klüssmann FA, Ariznabarreta LS. Genetic and epigenetic alterations of the cell cycle regulators and tumor suppressor genes in pediatric osteosarcomas. J Pediatr Hematol Oncol. 2003;25:362–7. [DOI] [PubMed] [Google Scholar]
- 11.Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. BioMed Central; 2008;10:R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen WS, Alshalalfa M, Zhao SG, Liu Y, Mahal BA, Quigley DA, et al. Novel Rb1-loss transcriptomic signature is associated with poor clinical outcomes across cancer types. Clin Cancer Res. 2019;25:4290–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bosco EE, Wang Y, Xu H, Zilfou JT, Knudsen KE, Aronow BJ, et al. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest. 2007;117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lehn S, Fernö M, Jirström K, Rydén L, Landberg G. A non-functional retinoblastoma tumor suppressor (RB) pathway in premenopausal breast cancer is associated with resistance to tamoxifen. Cell Cycle. 2011;10:956–62. [DOI] [PubMed] [Google Scholar]
- 15.Sharma A, Yeow W-S, Ertel A, Coleman I, Clegg N, Thangavel C, et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J Clin Invest. 2010;120:4478–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McNair C, Xu K, Mandigo AC, Benelli M, Leiby B, Rodrigues D, et al. Differential impact of RB status on E2F1 reprogramming in human cancer. J Clin Invest. 2018;128:341–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cecchini MJ, Ishak Bsc CA, Passos Dvm DT, Warner Msc A, Palma DA, Howlett CJ, et al. Loss of the retinoblastoma tumor suppressor correlates with improved outcome in patients with lung adenocarcinoma treated with surgery and chemotherapy ☆,☆☆. Hum Pathol. 2015;46:1922–34. [DOI] [PubMed] [Google Scholar]
- 18.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2011;18:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson DR, Wu Y, Lonigro RJ, Vats P, Cobain E, Everett J, et al. Integrative Clinical Genomics of Metastatic Cancer. 2018;548:297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cooney KA, Wetzel JC, Merajver SD, Macoska JA, Singleton TP, Wojno KJ. Distinct regions of allelic loss on 13q in prostate cancer. Cancer Res. 1996;56:1142–5. [PubMed] [Google Scholar]
- 21.Ittmann MM, Wieczorek R. Alterations of the retinoblastoma gene in clinically localized, stage B prostate adenocarcinomas. Hum Pathol. 1996;27:28–34. [DOI] [PubMed] [Google Scholar]
- 22.Jarrard DF, Modder J, Fadden P, Fu V, Sebree L, Heisey D, et al. Alterations in the p16/pRb cell cycle checkpoint occur commonly in primary and metastatic human prostate cancer. Cancer Lett. 2002;185:191–9. [DOI] [PubMed] [Google Scholar]
- 23.Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. Trends Endocrinol Metab; 2010;21:315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Comstock CES, Augello MA, Goodwin JF, Leeuw R de, Schiewer MJ, Ostrander WF, et al. Targeting cell cycle and hormone receptor pathways in cancer. Oncogene. Nature Publishing Group; 2013;32:5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Leeuw R, McNair CM, Schiewer MJ, Poudel Neupane N, Brand LJ, Augello MA, et al. MAPK reliance via acquired CDK4/6 inhibitor resistance in cancer HHS Public Access. Clin Cancer Res. 2018;24:4201–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dean JL, McClendon AK, Hickey TE, Butler LM, Tilley WD, Witkiewicz AK, et al. Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle. Taylor and Francis Inc.; 2012;11:2756–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M, et al. Retinoblastoma-protein-dependent cell-cycle inhibition by the tumour suppressor p16. Nature. Nature; 1995;375:503–6. [DOI] [PubMed] [Google Scholar]
- 28.Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, et al. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N Engl J Med. 2015;373:209–19. [DOI] [PubMed] [Google Scholar]
- 29.Pugacheva EM, Rivero-Hinojosa S, Espinoza CA, Méndez-Catalá CF, Kang S, Suzuki T, et al. Comparative analyses of CTCF and BORIS occupancies uncover two distinct classes of CTCF binding genomic regions. Genome Biol. Genome Biol; 2015;16:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Z, Wang Y, Kong L, Yue Z, Ma Y, Chen X. Expression of ADAM12 is regulated by E2F1 in small cell lung cancer. Oncol Rep. Oncol Rep; 2015;34:3231–7. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017;36:1302–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lv H, Zhen C, Liu J, Yang P, Hu L, Shang P. Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxid Med Cell Longev. Hindawi; 2019;2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (London, England). BioMed Central; 2018;38:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh N, Ecker GF. Insights into the structure, function, and ligand discovery of the large neutral amino acid transporter 1, lat1. Int J Mol Sci. 2018;19:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu J, Berisa M, Schwörer S, Qin W, Cross JR, Thompson CB. Transsulfuration Activity Can Support Cell Growth upon Extracellular Cysteine Limitation. Cell Metab. 2019;30:865–876.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freidman N, Chen I, Wu Q, Briot C, Holst J, Font J, et al. Amino Acid Transporters and Exchangers from the SLC1A Family: Structure, Mechanism and Roles in Physiology and Cancer. Neurochem Res. Neurochem Res; 2020;45:1268–86. [DOI] [PubMed] [Google Scholar]
- 37.Knudsen ES, Wang JY. Dual mechanisms for the inhibition of E2F binding to RB by cyclin-dependent kinase-mediated RB phosphorylation. Mol Cell Biol. American Society for Microbiology Journals; 1997;17:5771–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang JYJ, Knudsen ES, Welch PJ. The retinoblastoma tumor suppressor protein. Adv. Cancer Res. Adv Cancer Res; 1994;64:25–85. [DOI] [PubMed] [Google Scholar]
- 39.Zacksenhaus E, Bremner R, Jiang Z, Gill RM, Muncaster M, Sopta M, et al. Unraveling the function of the retinoblastoma gene. Adv Cancer Res. Adv Cancer Res; 1993;61:115–41. [DOI] [PubMed] [Google Scholar]
- 40.Debruyne DN, Dries R, Sengupta S, Seruggia D, Gao Y, Sharma B, et al. BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature. Nature; 2019;572:676–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang C, Qiu Z, Wang L, Peng Z, Jia Z, Logsdon CD, et al. A novel FoxM1-caveolin signaling pathway promotes pancreatic cancer invasion and metastasis. Cancer Res. Cancer Res; 2012;72:655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh J, Murata K, Itahana Y, Desprez P-Y. Constitutive expression of the Id-1 promoter in human metastatic breast cancer cells is linked with the loss of NF-1/Rb/HDAC-1 transcription repressor complex. Oncogene. Oncogene; 2002;21:1812–22. [DOI] [PubMed] [Google Scholar]
- 43.Cheema Z, Hari-Gupta Y, Kita G-X, Farrar D, Seddon I, Corr J, et al. Expression of the cancer-testis antigen BORIS correlates with prostate cancer. Prostate. John Wiley & Sons, Ltd; 2014;74:164–76. [DOI] [PubMed] [Google Scholar]
- 44.Pan H, Zhu Y, Wei W, Shao S, Rui X. Transcription factor FoxM1 is the downstream target of c-Myc and contributes to the development of prostate cancer. World J Surg Oncol. World J Surg Oncol; 2018;16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Philpott C, Tovell H, Frayling IM, Cooper DN, Upadhyaya M. The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics. BioMed Central; 2017;11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weindorf SC, Taylor AS, Kumar-Sinha C, Robinson D, Wu Y-M, Cao X, et al. Metastatic castration resistant prostate cancer with squamous cell, small cell, and sarcomatoid elements—a clinicopathologic and genomic sequencing-based discussion. Med Oncol. Springer; 2019;36:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Singh S, Narayanan SP, Biswas K, Gupta A, Ahuja N, Yadav S, et al. Intragenic DNA methylation and BORIS-mediated cancer-specific splicing contribute to the Warburg effect. Proc Natl Acad Sci U S A. National Academy of Sciences; 2017;114:11440–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song X, Fiati Kenston SS, Zhao J, Yang D, Gu Y. Roles of FoxM1 in cell regulation and breast cancer targeting therapy. Med Oncol. Springer; 2017;34:41. [DOI] [PubMed] [Google Scholar]
- 49.Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, et al. RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle. Taylor & Francis; 2010;9:4153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang H, Xiao G, Behrens C, Schiller J, Allen J, Chow C-W, et al. A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients. Clin Cancer Res. Clin Cancer Res; 2013;19:1577–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen D-T, Hsu Y-L, Fulp WJ, Coppola D, Haura EB, Yeatman TJ, et al. Prognostic and predictive value of a malignancy-risk gene signature in early-stage non-small cell lung cancer. J Natl Cancer Inst. J Natl Cancer Inst; 2011;103:1859–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicolay BN, Danielian PS, Kottakis F, Lapek JD, Sanidas I, Miles WO, et al. Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev. Cold Spring Harbor Laboratory Press; 2015;29:1875–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hsieh MCF, Das D, Sambandam N, Zhang MQ, Nahlé Z. Regulation of the PDK4 Isozyme by the Rb-E2F1 Complex. J Biol Chem. 2008;283:27410–7. [DOI] [PubMed] [Google Scholar]
- 54.Jensen DE, Black AR, Swick AG, Azizkhan JC. Distinct roles for Sp1 and E2F sites in the growth/cell cycle regulation of the DHFR promoter. J Cell Biochem. 1997;67:24–31. [DOI] [PubMed] [Google Scholar]
- 55.Tommasi S, Pfeifer GP. Constitutive protection of E2F recognition sequences in the human thymidine kinase promoter during cell cycle progression. J Biol Chem. J Biol Chem; 1997;272:30483–90. [DOI] [PubMed] [Google Scholar]
- 56.Wang Q, Tiffen J, Bailey CG, Lehman ML, Ritchie W, Fazli L, et al. Targeting amino acid transport in metastatic castration-resistant prostate cancer: Effects on cell cycle, cell growth, and tumor development. J Natl Cancer Inst. J Natl Cancer Inst; 2013;105:1463–73. [DOI] [PubMed] [Google Scholar]
- 57.Wang Q, Hardie RA, Hoy AJ, Van Geldermalsen M, Gao D, Fazli L, et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol. 2015;236:278–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clem BF, Chesney J. Molecular pathways: Regulation of metabolism by RB. Clin Cancer Res. 2012;18:6096–6100. [DOI] [PubMed] [Google Scholar]
- 59.Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, et al. Control of glutamine metabolism by the tumor suppressor Rb. Oncogene. Nature Publishing Group; 2014;33:556–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nicolay BN, Gameiro PA, Tschöp K, Korenjak M, Heilmann AM, Asara JM, et al. Loss of RBF1 changes glutamine catabolism. Genes Dev. 2013;27:182–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J. Biochem J; 2009;418:29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Luo B, Li X, Lu W, Yang J, Hu Y, et al. Inhibition of cancer growth in vitro and in vivo by a novel ROS-modulating agent with ability to eliminate stem-like cancer cells. Cell Death Dis. Cell Death Dis; 2017;8:e2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim SJ, Kim HS, Seo YR. Understanding of ROS-Inducing Strategy in Anticancer Therapy. Oxid Med Cell Longev. 2019;2019:5381692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharma A, Comstock CES, Knudsen ES, Cao KH, Hess-Wilson JK, Morey LM, et al. Retinoblastoma Tumor Suppressor Status Is a Critical Determinant of Therapeutic Response in Prostate Cancer Cells. Cancer Res. 2007;67:6192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thangavel C, Dean JL, Ertel A, Knudsen KE, Aldaz CM, Witkiewicz AK, et al. Therapeutically activating RB: reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr Relat Cancer. Endocr Relat Cancer; 2011;18:333–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. Bioinformatics; 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. Bioinformatics; 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. National Academy of Sciences; 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. Mol Cell; 2010;38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. Cancer Discov; 2013;3:1254–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Q, Beaumont KA, Otte NJ, Font J, Bailey CG, van Geldermalsen M, et al. Targeting glutamine transport to suppress melanoma cell growth. Int J cancer. Int J Cancer; 2014;135:1060–71. [DOI] [PubMed] [Google Scholar]
- 72.Wang Q, Bailey CG, Ng C, Tiffen J, Thoeng A, Minhas V, et al. Androgen receptor and nutrient signaling pathways coordinate the demand for increased amino acid transport during prostate cancer progression. Cancer Res. Cancer Res; 2011;71:7525–36. [DOI] [PubMed] [Google Scholar]
- 73.Welti J, Sharp A, Yuan W, Dolling D, Rodrigues DN, Figueiredo I, et al. Targeting Bromodomain and Extra-Terminal (BET) family proteins in Castration-Resistant Prostate Cancer (CRPC). Clin Cancer Res. Clin Cancer Res; 2018;24:3149–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.