

SUMMARY
Nuclear chromosomes transcribe far more RNA than required to encode protein. Here we investigate whether non-coding RNA broadly contributes to cytological-scale chromosome territory architecture. We develop a procedure that depletes soluble proteins, chromatin and most nuclear RNA from the nucleus, but does not delocalize XIST, a known architectural RNA, from an insoluble chromosome “scaffold.” RNA-seq analysis reveals most RNA in the nuclear scaffold is repeat-rich, non-coding, and predominantly derived from introns of nascent transcripts. Insoluble, repeat-rich (C0T-1) RNA co-distributes with known scaffold proteins including scaffold attachment factor A (SAF-A) and distribution of these components inversely correlates with chromatin compaction in normal and experimentally manipulated nuclei. We further show that RNA is required for SAF-A to interact with chromatin and for enrichment of structurally embedded “scaffold-attachment regions” prevalent in euchromatin. Collectively, results indicate that long nascent transcripts contribute a dynamic structural role which promotes the open architecture of active chromosome territories.
eTOC blurb
Creamer et al. report that non-coding, repeat-rich sequences in long nascent transcripts play a dynamic structural role in interphase chromosome architecture. Combining nuclear fractionation, RNA-sequencing and molecular cytology, results indicate insoluble nuclear RNAs platform a non-chromatin scaffold that is integral to open architecture of active chromosome territories.
Graphical Abstract
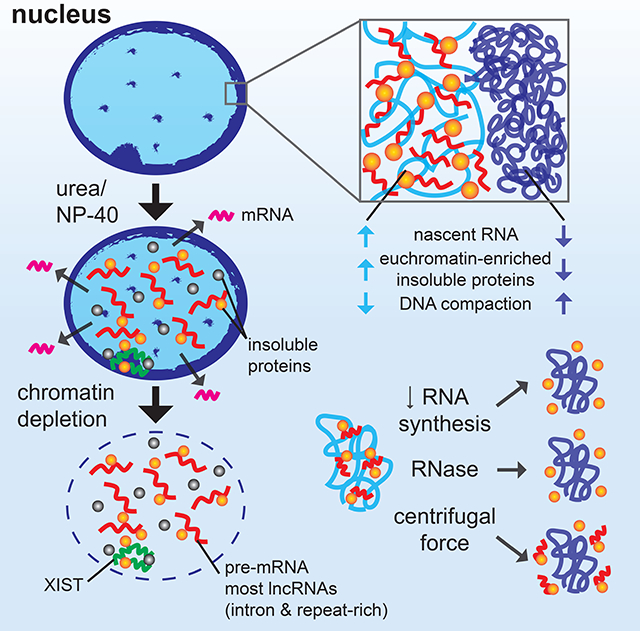
INTRODUCTION
Work in recent decades has led to understanding that the mammalian nucleus is highly compartmentalized (Bickmore and Pombo, 2019), with large regions of visibly condensed inactive heterochromatin in the nuclear periphery and more active and decondensed euchromatin through much of the interior. Only a minor fraction of genomic DNA codes for protein and is expressed in a given cell, yet broad differences in regional chromatin condensation are cytologically apparent, for reasons that are not well understood. It is generally thought that the “open” physical structure of euchromatin is permissive to transcription whereas the condensed state of facultative heterochromatin is refractory to gene expression.
Here we investigate the underlying basis for these cytological-scale differences in regional chromatin architecture with a focus on the role of RNA, particularly the potential for RNA to contribute to physical chromosome structure. Unlike condensed mitotic chromosomes, interphase chromosome territories contain abundant repeat-rich hnRNA (Hall et al., 2014; Pagoulatos and Darnell, 1970). Thousands of low-level long non-coding RNAs (lncRNAs) have been described; the functional significance for most is unknown, but many are cell-type specific and associate with chromatin (Werner et al., 2017). XIST lncRNA established the precedent that a long RNA can directly function in chromosome structure by “coating” the female inactive X-chromosome territory to form the heterochromatic Barr body. XIST RNA is known to induce numerous histone modifications, however how this RNA transforms an open chromosome territory into a dense Barr Body is not clear (Creamer and Lawrence, 2017; Galupa and Heard, 2018). Interestingly, the Barr body is notably depleted of the repeat-rich (C0T-1) heterogeneous RNAs abundant on active chromosomes, suggesting a potential dichotomy between the presence or absence of certain RNAs and cytological-scale chromatin compaction (Hall and Lawrence, 2016).
NEAT1 RNA demonstrates the architectural potential of long RNAs to scaffold protein interactions, forming nuclear bodies known as paraspeckles (Clemson et al., 2009; Sasaki et al., 2009). Different parts of each NEAT1 transcript, as they are transcribed, bind many distinct proteins which align to form paraspeckle substructure; this is facilitated by the propensity of low-complexity domains of RNA binding proteins to form macro-assemblies, such as by phase separation (Yamazaki et al., 2018). Since numerous RNA binding proteins interact with each active genomic region (Xiao et al., 2019), long nascent and chromatin-associated transcripts may also form a network of interactions that influence chromatin architecture.
Whether chromosome territories are shaped by interaction of RNA with architectural proteins is a question of fundamental importance, but has remained unresolved (Fritz et al., 2019). Earlier literature reported that major components of mammalian nuclei remain insoluble after biochemical extraction of proteins and DNA, suggesting a structural framework to the nuclear interior that persists despite removal of most chromatin. This so-called nuclear matrix or scaffold consists largely of lamins, matrins, NUMA, RNA-binding proteins such as scaffold attachment factor A (SAF-A), and was reported to contain most nuclear RNA (Berezney and Coffey, 1974; Capco et al., 1982; Nickerson, 2001). Subsequent studies have affirmed involvement of several of these proteins in nuclear structure, however the specificity and significance of the abundant RNA in this nuclear fraction is not known.
RNase treatment has been shown to disrupt normal nuclear structure and DNA distribution (Caudron-Herger et al., 2011; Ma et al., 1999; Nickerson et al., 1989). To understand the potential significance of this, here we use multiple approaches to investigate RNA’s relationship to chromosome condensation and the hypothesis that RNA physically contributes to nuclear chromosome structure. While many studies focus on potential roles of individual lncRNAs in specific gene regulation, here we investigate a distinct and broader question: whether the great mass of diverse nuclear RNAs that genomes produce serves a more global, structural role integral to large-scale architecture of chromosome territories. RNAs will be associated with chromatin as they are produced, however even nascent pre-mRNA, lncRNAs, or enhancer RNAs could dynamically influence local chromatin architecture (Li and Fu, 2019; Michieletto and Gilbert, 2019).
Our results indicate that abundant, long nascent RNAs, rather than being only mRNA precursors, contribute a structural role with pervasive influence on regional chromatin compaction. Results show that long chromatin-associated RNAs, including pre-mRNAs comprised largely of non-coding, repeat rich sequences, participate in an insoluble RNA-protein network that retains structural integrity independent of canonical chromatin. This dynamic RNA-protein “scaffold” or “meshwork” suggests potential functionality for the unexplained length, abundance and repetitive nature of the “non-coding” nuclear transcriptome.
RESULTS
Repeat-rich hnRNA localization inversely correlates with chromatin compaction
As discussed above, Figure 1A illustrates that nuclear RNA depletion using RNase A disrupts nuclear morphology and causes rapid “collapse” of chromatin into compact regions. The causal basis, and whether this is a direct or indirect effect of RNase on chromatin, is not known; however, the effect is suggestive of a physical role of RNA in chromosome architecture.
Figure 1. C0T-1 RNA is depleted from heterochromatin and inversely correlated with chromatin compaction.

(A) DNA staining and intensity heatmap (below) of permeabilized nuclei treated with RNase A for 10 minutes prior to fixation. Percentages indicate proportion of cells with obvious visible changes in DAPI signal as represented in the image (n=100). (B) RNA FISH on TIG-1 human fibroblasts demonstrating repeat-rich C0T-1 RNA is depleted from peripheral heterochromatin and the Barr body, which is coated by XIST. A line-scan intensity profile of the indicated region is displayed below. Scale bars 10 μm. (C) RNA/DNA FISH using human C0T-1 DNA as a probe on G11687 mouse human hybrid cells containing a single human chromosome 4. C0T-1 RNA and DNA signals overlap marking the human chromosome territory. Cells were either mock or DRB treated for 6 hours. Arrows point to G1 daughter cells that divided during transcription inhibition. (D) RNA FISH analysis of high passage (passage 35) fibroblasts. The cell on the right displays DAPI dense foci characteristic of SAHFs, indicated by arrows in the higher magnification inset. Low passage (passage 18) infrequently had SAHFs (4%, n=100) relative to high passage cells (32%, n=100). C0T-1 RNA was visibly depleted as shown from 96% of individual SAHFs scored (n=100). Scale bars 5 μm. (E) Pixel intensity scatter plots for nuclei shown on left. Linear regression model is indicated by black lines. Pearson correlation coefficients (PCC) between signal intensities were calculated for multiple nuclei and represent the mean with 95% confidence intervals +/− 0.03 (left, n=4) and +/− 0.09 (right, n=4). (F) DAPI staining of control and treated TIG-1 nuclei. Scale bars 10 μm. Examples of cells with medium (*) and high (**) levels of chromatin compaction changes as scored on the right for indicated treatments and times (n=100). (G) RNA FISH of C0T-1 RNA in control and cells treated with transcriptional inhibitors for 3 or 6 hours. A “no probe” (n.p) negative control was also performed. Scale bars 200 μm. On right, C0T-1 RNA signals were quantified per cell, normalized to DAPI intensity and then to the mean intensity of control cells (n=300 for each condition).
Interphase chromosomes are associated with copious RNA which potentially could contribute to structure. As shown in Figure 1B, RNA hybridization using human C0T-1 (highly repetitive) DNA as a probe illuminates abundant repeat-rich hnRNA localized to euchromatin regions (Hall et al., 2014), mostly in the nuclear interior (outside nucleoli) but excluded from peripheral heterochromatin and the inactive X-chromosome, which instead is coated by XIST RNA. C0T-1 hnRNA “coats” and localizes to each actively transcribed chromosome territory, as visualized in mouse-human hybrid cells (Figure 1C) and is “stripped” from chromosomes during mitosis and resynthesized as new G1 daughter nuclei decondense. It was shown that if RNA resynthesis is blocked by transcriptional inhibition with DRB, G1 daughter cells cannot reestablish normal decondensed chromatin distribution (Hall et al., 2014). However, the nature of C0T-1 RNA remained unclear, particularly given the finding that in interphase cells the bright, localized C0T-1 RNA territories remained after long periods of transcriptional arrest (Hall et al., 2014), a phenomenon that will be further examined below.
Cell senescence provides a natural system in which euchromatin and heterochromatin undergo dramatic rearrangement (Yang and Sen, 2018), with large condensed bodies, termed senescence-associated heterochromatin foci (SAHFs), that form within enlarged nuclei (Swanson et al., 2013; Zhang et al., 2007). Hence, we examined whether this marked chromatin compaction coincides with differences in C0T-1 RNA. Indeed, SAHFs are markedly depleted of C0T-1 hnRNA (Figure 1D), similar to the Barr body (Figure 1B). In contrast, C0T-1 RNA signal is robust throughout the highly decondensed chromatin of the enlarged nucleus. Quantitative analysis of normal and SAHF-containing nuclei confirmed that regions of high DNA compaction (DAPI intensity) were depleted of C0T-1 RNA and shows an inverse correlation between C0T-1 RNA and DAPI intensity overall (Figure 1E).
Several studies have tested whether ongoing transcription is required to maintain normal chromatin structure using transcription inhibitors under various conditions, with results ranging from little to no change to large-scale compaction (Haaf and Ward, 1996; Hall et al., 2014; Kim et al., 2019; Nickerson et al., 1989; Nozawa et al., 2017; Visvanathan et al., 2013). As shown in Figure 1F, here we found Actinomycin D (ActD) treatment of normal fibroblasts for 6 hours causes progressive chromatin collapse into large DAPI-dense bodies over time.
An interesting contrast is seen in cells treated with 5,6-Dichloro-1-β-d-ribofuranosylbenzimidazole (DRB), which do not show this impact on DNA distribution even after 6 hours. We therefore examined the potential relationship between chromatin condensation and C0T-1 RNA depletion following transcription inhibition and found C0T-1 RNA was greatly reduced in cells with DAPI dense bodies (Act D for 6 hours) but only slightly diminished in DRB-treated cells (Figure 1G, discussed below). This collection of natural and experimental observations consistently suggests that the presence of abundant C0T-1 RNAs may physically antagonize a propensity for chromatin to compact. Therefore, we next undertook a series of experiments to investigate whether some RNAs interact with insoluble elements of the chromosome territory substructure.
XIST and C0T-1 RNA remain with nuclear structure resistant to urea, salt extraction and chromatin removal
We then worked to develop a procedure that removes weakly bound RNAs and proteins as well as chromatin, and selectively enriches for insoluble RNAs that may play a structural role, using XIST RNA as a benchmark. As shown in in Figure 2A, using earlier nuclear matrix protocols that deplete soluble nuclear proteins and chromatin we previously showed that XIST RNA remains tightly localized with residual nuclear material and that C0T-1 RNA similarly remains insoluble (Clemson et al., 2006; Hall et al., 2014). Although several versions of matrix/scaffold isolation protocols exist (Wilson et al., 2016), such procedures generally rely on ionic extraction prior to DNase I digestion (leaving ~5–10% of DNA). Up to 70% of nuclear RNA was found to cofractionates with the nuclear matrix (Fey et al., 1986); however, questions were raised as to whether high salt extraction precipitates weakly bound proteins and RNAs, hence the specificity of the relationship between nuclear RNA and matrix proteins remains unclear.
Figure 2. Detection of chromatin-independent nuclear insoluble RNAs.

(A-C) RNA FISH for XIST and C0T-1 RNA signals after extraction of nuclei for 15 minutes as indicated (AS, ammonium sulfate). Scale bars 5 μm. (D,E) Immunofluorescence and RNA FISH of fixed cells grown on coverslips (top panels) or nuclei after serial extraction as indicated. Scale bars 10 μm.
Recently, extraction with urea and NP-40 containing buffers has become the standard for nuclear fractionation by separating loosely bound proteins and RNA from insoluble material, often referred to as the “chromatin” fraction. Here, to better test the “scaffold/matrix” concept, we developed a progressive nuclear extraction procedure that begins by using urea/NP40 to first remove weakly bound RNAs and proteins (prior to further extraction and chromatin removal). We first tested whether XIST and C0T-1 RNA resist this chaotropic and non-ionic extraction. Bright XIST and C0T-1 RNA signals remained in nuclei incubated in buffers containing 1M urea and 1% NP-40 (Figure 2C); hence, these RNAs are highly resistant to extraction under conditions widely accepted to segregate soluble and insoluble nuclear factors independent of exposure to high salt.
Insolubility of XIST and C0T-1 RNA could reflect only their association with chromatin, hence a fundamentally important distinction is whether XIST, and potentially some other nuclear RNAs, are embedded with insoluble elements of the chromosome territory even after removal of canonical chromatin (genomic DNA and histones). To test this, nuclei were first extracted with urea and NP-40, thereby mitigating potential for artefactual precipitation in downstream steps. Nuclei were further extracted with 0.25M ammonium sulfate (AS), which solubilizes additional proteins by a different non-denaturing mechanism than urea and is not expected to cause precipitation at these concentrations (Duong-Ly and Gabelli, 2014; Slomiany et al., 2000; Wingfield, 2001). Genomic DNA was then thoroughly digested with DNase I for 90 minutes and nuclei washed again in 2M NaCl buffer to extract additional proteins. This procedure dramatically depletes DNA (DAPI signal) and histones (H3) to essentially undetectable levels, as indicated in Figure 2D and further evidenced below.
Even after this thorough extraction a framework nuclear structure persists, with modest changes, indicated by creases in the nuclear lamina (LMNB1). Despite chromatin removal and altered morphology, XIST RNA remains brightly localized to a defined territory (Figure 2E). XIST RNA is a mature transcript which coats and regulates the X-chromosome, yet this RNA fully remains with a chromosome territory “scaffold” that maintains structural integrity independent of chromatin. Perhaps surprisingly, C0T-1 hnRNA is also unperturbed by this extensive nuclear extraction. As will be demonstrated below, the “insoluble” nature of these RNAs that resists extensive extractions, before and after chromatin removal, is not a general property of nuclear RNAs, but a subset of transcripts more embedded with non-chromatin structures.
LncRNAs are preferentially enriched with insoluble, non-chromatin nuclear substructure
We next used RNA sequencing to determine if above findings reflect a general propensity for RNAs to be insoluble under these conditions, or whether we were successful in identifying a subset of RNAs that specifically associate with insoluble structure. Hence, we isolated RNA cofractionating with XIST after serial extractions mirroring the cytological observations above (Figure 3A). Nuclear RNA fractions were analyzed transcriptome-wide by total ribosome-depleted RNA-sequencing analysis.
Figure 3. Nuclear ncRNAs and intronic sequences are enriched with insoluble substructure independent of chromatin.

(A) Schematic of subnuclear fractionation protocol. RNA (red) and associated proteins are solubilized by serial biochemical extraction procedures from TIG-1 nuclei and a pie chart of RNA (average by mass, n=2) extracted for each fraction. (B-D) Relative abundance of XIST, NEAT1, and GAPDH RNAs in transcripts per million (TPM) from RNA-seq. Error bars represent SEM. (E) Enrichment of GENCODE lncRNAs in the CDNI relative to NS fraction. Blue dots indicate significant enrichment (p < 0.05). (F) Boxplot of RNA enrichment grouped by subtype in the CDNI and NS fractions. RNAs binned by mean abundance. (G) SYBR Safe stained 1.5% agarose gel of DNA isolated from CDNI material after DNase I digestion. (H) Relative abundance of lncRNAs in CDNI and CDNS fractions. (I) Stacked bar graph of RNA-seq mapping to Repeatmasker repeats. (J) Bar graph of RNA-seq mapping to indicated repeats relative to their abundance in the genome (LC, low complexity; SR, simple repeats; amb, ambiguous). (K) Percent of CDNI and NS RNA-seq mapping to genomic features. (L) Distribution of total read normalized intron coverage in NS (red) and CDNI (blue) fractions. (M) Distribution of exon-intron splice ratios derived from analysis of spliced and unspliced intron-exon junction read depth (spliced/spliced+unspliced depth) in NS (red) or CDNI (blue) fractions.
Early studies using classic matrix protocols reported most (~70%) nuclear RNA is retained in the matrix (Fey et al., 1986). Here we show addition of more stringent initial urea/NP-40 extraction itself released most RNA (85%) from nuclei (Figure 3A, bottom), affirming the efficacy of this modified protocol to solubilize loosely bound RNAs that might otherwise remain unextracted during high salt extraction or digestion of chromatin.
We first assessed whether known structural lncRNAs co-fractionate with the final chromatin-depleted nuclear insoluble (CDNI) material and, indeed, both XIST and NEAT1 are highly enriched in the CDNI fraction relative to the nuclear soluble (NS) fraction (Figure 3B and 3C). In contrast, GAPDH mRNA was primarily in the NS fraction (Figure 3D), as are other mRNAs (below). Therefore CDNI RNA is not a non-specific residue of nuclear RNA.
Interestingly, most GENCODE lncRNAs were strongly enriched in the CDNI fraction (Figure 3E). Of transcripts detected at >10 transcripts per million (TPM), 84.8 % (581/685) of lncRNAs were enriched in the CDNI fraction compared to just 10% (801/8028) protein-coding. LncRNAs thought to have structural roles or act on chromatin including XIST, MALAT1, NEAT1, GAS5, and MEG3 were enriched in the CDNI fraction. Among the few lncRNAs enriched in the NS fraction (13/685), NORAD and RMRP are thought to have cytoplasmic and mitochondrial functions respectively, suggesting CDNI versus NS enrichment relates to lncRNA function.
An appreciable amount of CDNI RNA (6%) mapped to intergenic regions outside the GENCODE gene annotation. Detection of low-level intergenic RNA is enabled by CDNI fractionation, and such chromatin-associated RNAs may contribute to gene regulation in cis (Werner and Ruthenburg, 2015; Werner et al., 2017). We identified 1209 additional low-level, intergenic transcripts that are not annotated in GENCODE, and have little coding potential or strand preference relative to the nearest protein-coding gene (Figures S1A and S1B). Figure 3F shows that both GENCODE and ‘novel’ lncRNAs were highly enriched in the CDNI fraction compared to protein-coding transcripts of similar abundance, and more so than snoRNA or snRNA (p < 0.01). Thus, this work demonstrates that intergenic non-coding transcripts are bound with the insoluble scaffold with few exceptions.
Previous studies identified intergenic non-coding RNAs that co-fractionate with chromatin (Mayer and Churchman, 2017; Werner and Ruthenburg, 2015), but an important distinction here is that our fractionation procedure uses DNase in large excess and long incubation times to thoroughly digest DNA to undetectable levels (Figure 3G). Remarkably, <10% of nuclear insoluble RNA was eluted during extraction and chromatin removal, indicative of the tenacity of RNA interactions with insoluble proteins. The relative abundance of lncRNAs solubilized during chromatin depletion was nearly identical to the CDNI fraction, indicating a lack of specific RNAs eluted during this step (Figures 3H and Figure S1C). We conclude that with few exceptions, lncRNAs are generally bound with insoluble non-chromatin scaffold factors.
CDNI scaffold RNA is enriched with nascent, intron-containing and repeat-rich transcripts
Human chromosomes are largely non-coding and are interspersed with thousands of transposon-derived repeats. Given that only ~15% of nuclear RNA remains in the scaffold, it is striking that repeat-rich C0T-1 RNA remains undiminished in the residual nuclear structure (Figure 2E). Consistent with this, RNA-seq shows 36.6% of CDNI “scaffold” RNA maps to repeats in contrast to just 9.7% in the NS fraction (Figure 3I). Much CDNI RNA maps to LINE-1 and Alu sequences (10.3% and 8.1% respectively), similar to their genomic abundance (Figure 3J). Hence, the nuclear insoluble transcriptome is not only non-coding but markedly repetitive in nature, comprising “junk” sequences with little known function.
Like intergenic and lncRNAs, pre-mRNA introns are also replete with such sequences, thus we examined their contribution to scaffold RNA. Although lncRNAs are preferentially insoluble (Figure 3E), by mass just 2.8% of CDNI RNA maps to lncRNA “exons” (Figure 3K), ~3 fold less than coding exons. Most striking, 82% of scaffold RNA was derived from introns (Figure 3K), with 71.2% from protein-coding gene introns. We found it remarkable that so much scaffold RNA was intronic, particularly since most introns are thought removed co-transcriptionally (Drexler et al., 2020). To further investigate this, we assessed intron coverage and splicing status of pre-mRNA introns genome-wide using IRFinder (Middleton et al., 2017), which detected 20,368 introns in the CDNI scaffold (Figure 3L). In contrast, only 29 introns, all short, were enriched in NS RNA (Figures S2A and S2B). NS RNA was nearly entirely spliced (Figure 3M), demonstrating that mRNPs in nuclei become soluble upon intron removal, indicating minimal structural attachment clearly distinct from more insoluble scaffold RNAs.
Once spliced most introns are thought rapidly degraded. To assess if introns accumulate in CDNI substructure beyond what could be explained by their pre-mRNA content, we compared mean read depth across introns versus corresponding exon-intron junctions with a per-intron mean ratio of 1.08 indicating most scaffold introns are within pre-mRNAs (Figures S2C and S2D). We then performed meta-analysis of RNA-seq coverage across long (10–50kb) and very long (>50k) introns, which together account for half (50.0%) of CDNI RNA reads. Decline in reads from 5’ to 3’ across introns is suggestive of transcripts being transcribed (Figure S2E). However, the decrease is modest and substantial RNA-seq coverage at the 3’ end of most introns suggests the scaffold contains both introns being transcribed as well as fully transcribed introns not yet excised. Related to this, there is recent evidence that many introns are removed post-transcriptionally and that cleaved, intron-containing transcripts are readily detected in mammalian nuclei (Coté et al., 2020). Hence, the term “nascent” here is used to describe non-coding or coding RNAs in the process of being transcribed or under-processed primary transcripts that remain insoluble after cleavage.
These RNA-seq results indicate that C0T-1 RNA observed cytologically is detecting primarily nascent pre-mRNAs and lncRNAs, raising an intriguing paradox with observations using DRB. As shown above, C0T-1 RNA is lost in cells treated 6 hours with Act D, concomitant with chromatin collapse. However, C0T-1 RNA remains localized to the chromosome territory after 6 hours of DRB and chromatin appears normal, confirming prior results (Figure 1C, 1F, and 1G) (Hall et al., 2014). Consistent with inhibited transcription, DRB depleted coding transcripts from the nucleus and prevented resynthesis of C0T-1 RNA in cells that divided in the presence of DRB (Figures 1C and S3A, see below) (Hall et al., 2014). Thus in contrast to experiments using ActD, examination of DRB-treated nuclei appeared to indicate that the C0T-1 RNA is surprisingly stable in interphase nuclei.
To address these puzzling results, we also sequenced CDNI scaffold RNA from DRB-treated fibroblasts. The total mass of CDNI RNA in DRB-treated cells was maintained with only modest reduction (22%, n=2), and RNA-seq affirmed it remained highly repeat-rich (Figures 4A). However, further analysis revealed that the specific sequence content of CNDI RNA was dramatically altered. Intron-derived RNA was greatly reduced as pre-mRNAs were almost entirely spliced after DRB treatment (Figures 4B–D). Surprisingly, nearly half (48.4%) of CDNI RNA mapped to intergenic regions in DRB-treated cells, in contrast to just 6.0% in control.
Figure 4. DRB can induce insoluble intergenic transcripts.

(A) Percent of CDNI RNA-seq from mock or DRB-treated (4 hours) cells mapping to Repeatmasker repeats. (B) Percent of CDNI RNA-seq from mock or DRB-treated cells mapping to genomic features. Pie chart colors and calculations match those in Figure 3. (C) Distribution of total read normalized intron coverage for introns in the CDNI fraction with (red) and without (blue) DRB treatment. (D) Box plot of intron splice ratios found in RNA-seq fractions (see Figure 3M). (E) CDNI RNA-seq coverage. RefSeq genes, downstream of gene (DOG) regions, and primers used in this study are indicated. (F) Meta-gene heatmap of RNA-seq coverage, log2(1+ RPKM) transformed, in the sense direction only for protein-coding genes on the forward strand. (G) Percent of uniquely mapping reads mapping to DOG regions in CDNI RNA-seq from untreated or DRB-treated cells. (H) RT-PCR analysis of DOG regions from control or DRB-treated cells in indicated fractions. RT-PCR was performed on a per cell equivalent amount of RNA for each fraction. COL1A1 mRNA primers are exon-spanning.
As shown in Figures 4E and 4F, following DRB treatment we observed CDNI RNA-seq reads often mapped to regions downstream of previously expressed genes that was visually consistent with readthrough transcription. Nearly half of CDNI RNA sequenced from cells treated with DRB mapped to these large downstream of gene (DOG) regions (see methods and Figures 4G, S3C and S3D). RT-PCR analysis after fractionation from equal cell numbers confirmed that these transcripts are only detectable with DRB and exclusively in nuclear insoluble fractions (Figure 4H). Quantitative RT-PCR confirmed robust detection of transcripts downstream of the LMO7 and KLF12 genes after DRB, but not in control cells, ActD-treated cells, or negative RT-PCR controls (Figure S3E, see legend for discussion of DRB-induced readthrough). Taken together these results suggest that rather than stabilize transcripts, DRB induced intergenic readthrough transcription that largely maintained insoluble RNA mass and repeat content.
Intergenic transcripts identified here could simply reflect a technical response to DRB treatment, however widespread transcription downstream of genes has also recently identified in the context of osmotic or heat stress (Vilborg et al., 2015). Interestingly, transcription inhibition is known to induce a cellular stress response (Klibanov et al., 2001). We therefore tested whether candidate DRB-induced DOG transcripts identified here could be detected in osmotically stressed cells, and, indeed, they were (Figure S4A). Vilbourg et al. used BAPTA-AM, an intracellular calcium chelator, to show that DOG transcription requires calcium signaling. We no longer detected candidate DOG transcripts and observed markedly reduced C0T-1 RNA signal in cells co-treated with DRB and BAPTA-AM (Figures S4A and S4B). Importantly, reduction in C0T-1 RNA was accompanied by increased chromatin compaction. Collectively, these results reveal continued production and retention of insoluble nascent transcripts in cells treated DRB and indicate that this has the effect of maintaining normal chromatin architecture. In contrast, thorough blockage of nascent transcript production and subsequent depletion in cells leads to increased chromatin compaction.
Specific nuclear scaffold protein distribution inversely correlates with chromatin compaction
The consistent theme shown above supports that presence of long, repeat-rich insoluble RNAs promotes normal chromatin distribution. Removal or redistribution of that RNA leads to rapid euchromatin condensation. Since C0T-1 RNA cofractionates with insoluble non-chromatin elements, we investigated the distribution of several scaffold factors relative to C0T-1 RNA and chromatin by immunofluorescence.
MATR3, NUMA, and SAF-A were identified as abundant scaffold/matrix proteins and have been implicated in multiple nuclear processes (Merdes and Cleveland, 1998; Nozawa et al., 2017; Zeitz et al., 2009). All three have potential to interact with chromatin and RNA via separate DNA and RNA-binding domains. Although they may influence splicing, NUMA, SAF-A, and MATR3 distribute primarily with chromatin, not splicing factor speckles (Figures 5A and S5A). Despite broad distributions, close scrutiny revealed a similar pattern of clear differences relative to condensed DNA. Figure 5A illustrates lack of staining for each of these proteins over the inactive X chromosome, as well as peripheral and nucleolar heterochromatin. We quantified staining for each protein relative to chromatin compaction by segmenting nuclei based on DAPI intensity (Figure 5B) (Cremer et al., 2017). NUMA, MATR3, and SAF-A were depleted from both numerous individual DAPI-dense foci in a single nucleus and DAPI-dense regions in aggregate data from several cells (Figures 5C and 5D).
Figure 5. Specific nuclear scaffold proteins are depleted from heterochromatin.

(A) Immunofluorescence of NUMA, MATR3, and SAF-A in TIG-1 fibroblasts. Higher magnification below. Line-scan of signal intensities for the indicated region shown at right. (B) DAPI intensity segmentation of the above nucleus. Numbers indicate high DAPI intensity bodies (yellow). (C) Log2 ratio of the mean signal intensity within each indicated high intensity body compared to entire nucleus. (D) Log2 ratio of the mean signal intensity within DAPI intensity segmentation classes compared to the entire nucleus. Error bars represent SEM of the log2 ratio determined for multiple nuclei (n=4). (E) IF of NUMA, MATR3, and SAF-A in high passage (35) fibroblasts. Proteins were visibly depleted from high DAPI intensity bodies (SAHFs, arrows) in all cells with greater than 5 SAHFs (n=21). Higher magnification shown below and line-scan for the indicated region on right. Scale bars 5 μm
Intensity correlation analysis across whole nuclei affirmed a negative correlation between SAF-A, MATR3, or NUMA and DAPI DNA signal while pairwise comparisons between these proteins were positively correlated (Figure S5B). An important contrast was found in Lamin A, which is more uniformly distributed through the nucleus and weakly correlated with DAPI (Figures S5C and S5D). Nuclear lamins concentrate in the lamina which tethers peripheral heterochromatin, however lower levels of lamins may also contribute to euchromatin architecture (Gesson et al., 2016). While lamins are also abundant, insoluble scaffold factors, they are not known to bind RNA and are more uniformly distributed, potentially providing a foundational structure for overall nuclear organization distinct from the other three factors with specific euchromatin staining patterns.
The co-distribution of NUMA, SAF-A, and MATR3 staining and the pattern of depletion from condensed chromatin is further evidenced in the condensed SAHFs of senescent fibroblasts (Figure 5E). We also examined SAF-A and NUMA relative to DAPI-dense bodies formed after inhibition with BAPTA-AM/DRB or ActD and observed similar depletion of these factors (Figures 6A and S6).
Figure 6. Euchromatin-associated scaffold proteins are depleted from experimentally condensed chromatin.

(A) IF analysis of cells after treatment with transcriptional inhibitors as indicated. Higher magnification of the indicated region on right. Arrows indicate high DAPI intensity bodies depleted of SAF-A and NUMA. Scale bars 10 μm. (B,C) FISH and IF analysis of cells after high-speed cytocentrifugation. All cells analyzed (n = 200) had many visibly condensed chromatin bodies following high-speed cytospin attachment to coverslips (2000 RPM, 10 minutes) compared to 6% (n = 200) of cells attached to coverslips using a standard cytospin procedure (800 RPM, 3 minutes). High DAPI intensity bodies are indicated by arrows at higher magnification.
SAF-A is an interesting case because it is known to bind diverse nuclear coding and non-coding RNAs, including XIST (McHugh et al., 2015; Minajigi et al., 2015; Puvvula et al., 2014). Yet when bound with XIST RNA on Xi, the endogenous epitope of SAF-A is masked, such that it is only detected there using a GFP tag or if the epitope becomes exposed by antigen retrieval (Helbig and Fackelmayer, 2003). We note a prior observation shows GFP-tagged (and native) MATR3 depleted from the Xi (Zeitz et al., 2009). Hence, results support that MATR3 and NUMA are depleted from heterochromatin, whereas SAF-A is present but packaged differently where it interacts with XIST.
These collective results support that long, repeat-rich RNAs may interact with many RNPs in an insoluble network that bridges with chromatin and physically counters cytological-scale condensation. To further test this concept, we sought a means to physically disrupt the relationship of the proposed RNP network and chromatin. We therefore tested sharply increasing the centrifugal force normally applied for attaching cells to coverslips (prior to fixation) as a potential means to physically disrupt the relationship of the RNP scaffold to chromatin. Remarkably, we found high-speed centrifugation disrupted the distribution of both C0T-1 RNA and scaffold proteins relative to DNA, and simultaneously caused formation of numerous aberrant foci of condensed chromatin (Figures 6B and 6C). These bodies were essentially devoid of C0T-1 RNA, NUMA, MATR3, and SAF-A. Importantly, all four components consistently redistributed together, co-segregating into regions of lower DNA density between them.
These dramatic changes occur in a timescale (10 min) inconsistent with changes in histone modifications or transcription programming, but indicate a rapid physical impact coincident with stripping RNPs from euchromatin. Results support the hypothesis that physical bridging of an hnRNA/RNP network with DNA counters chromatin compaction.
RNA is required to retain SAF-A with chromatin and influences DNA-scaffold attachment
While the above proteins co-distribute with C0T-1 RNA, distinct interactions in an RNP-chromatin network may be reflected in differences in reliance on RNA to remain bound to structure. We tested this idea using RNase to remove RNA from nuclei. No scaffold protein we tested freely diffuse from control permeabilized nuclei, and NUMA, LMNA and LMNB1 still remained after RNase treatment (Figure S7A). In contrast, we found SAF-A diffused from RNase-treated nuclei (Figure 7A and 7B), consistent with a prior observation (Nozawa et al., 2017). We further show this release of SAF-A is specific and occurs with an immediacy (in just one minute) supporting a requirement for RNA in determining SAF-A localization and solubility.
Figure 7. RNA is required for SAF-A to interact with chromatin and influences nuclear scaffold attachment.

(A) IF or RNA FISH analysis on permeabilized nuclei mock or RNase-treated for 10 minutes at 37°C prior to fixation. Images representative of >95% of nuclei. Scale bars 10 μm. (B) Western blot of protein solubility following gentle centrifugation of mock or RNase-treated nuclei. (C) Schematic of scaffold attachment region isolation and sequencing (SAR-seq) protocol. Soluble proteins and fragmented chromatin are extracted from isolated nuclei. Genomic DNA protected from digestion that remained insoluble after ionic extraction was subjected to paired-end sequencing without fixation or additional fragmentation. (D) Coverage map of CDNI RNA-seq (red) or SAR-seq (blue) at an 18 Mbp region of chr21. Refseq protein-coding genes annotated below. (E) Heatmap and average coverage profiles of CDNI RNA-seq (RPKM), SAR-seq (normalized coverage), highly DNase I digested naked (deproteinized) DNA-seq (normalized coverage), and SAR-seq from nuclei treated with RNase A for 10 minutes. Regions were binned by mean transcript abundance in CDNI and NS fractions. (F) Model summarizing results consistent with a dynamic ribonucleoprotein (RNP) meshwork, or scaffold, platformed on nascent transcripts that antagonizes cytologically observable chromatin compaction. RNP scaffold components (eg. SAF-A, MATR3, and NUMA) co-distribute with nascent RNA. In contrast, nascent RNA and RNP scaffold factors are depleted from regions of heterochromatin and experimentally compacted chromatin. Regions of chromatin near active transcription preferentially interact with the insoluble nuclear scaffold. Results here indicate RNA-dependent interactions between chromatin and the RNP-scaffold may serve to maintain decondensed chromatin structure.
Given SAF-A’s reliance on RNA and evidence SAF-A directly impacts chromatin structure (Fan et al., 2018; Nozawa et al., 2017), we tested one hypothesis for how this relates to the nuclear scaffold concept. Scaffold-attachment factor A (SAF-A) was so named because it was identified as one of several proteins enriched in matrix/scaffold attachment regions (S/MARs), genomic sequences most resistant to digestion and extraction and thus thought to interact with insoluble architectural components (Dobson et al., 2017; Gohring and Fackelmayer, 1997; Heng et al., 2004; Keaton et al., 2011; Pathak et al., 2014; Wilson et al., 2016). Many studies reported S/MARs in specific genes, however genomic-scale analyses are limited. Most importantly, the question of whether RNA is required for DNA “attachment sites” with insoluble scaffold structure has not been tested. Given SAF-A is removed from nuclei by RNase, it was compelling to ask if S/MAR regions in DNA would be compromised by RNA removal.
Since no standard protocol exists (Dobson et al., 2017; Kallappagoudar et al., 2010; Wilson et al., 2016), we modified existing protocols to develop SAR-seq (Figure 7C and see Methods), a genome-wide assay for sites in nuclear chromosomes most resistant to digestion and extraction. Peak calling software identified 293,025 “SARs” comprising ~4% of the genome. As suggested in a prior microarray study (Keaton et al., 2011), we find SARs enriched in protein-coding genes with over half (54.1%) in introns (Figure S7C). SAR-seq signal was visibly highest in regions of high transcriptional activity, both at low-resolution and at the highly expressed COL1A1 gene (Figures 7D and S7D).
Meta-gene analysis showed SAR-seq signal was relatively enriched at gene bodies (Figure 7E, second panel). While enrichment was highest downstream of transcription start and end sites, SAR-seq signal was regionally elevated greater than 100kb away from high and moderately expressed genes. In contrast, SAR-seq coverage was diminished or absent at lowly or unexpressed genes. Cleavage bias is known to complicate genomic assays that use DNase I (Koohy et al., 2013; Lazarovici et al., 2013). Aside from modest enrichment at TSSs irrespective of transcriptional activity, analysis of a published data set of highly digested DNase I-seq performed on deproteinized DNA from Lazarovici et al. did not exhibit these patterns, indicating SAR-seq signals are primarily driven by chromatin solubility and protection rather than intrinsic sequence bias.
Having characterized regions of the genome that remain structurally bound with scaffold material, we addressed the central question as to whether this structural interaction depends on RNA by performing SAR-seq after depleting RNA with RNase A. Remarkably, SARs were largely ablated, not only at gene bodies but at SARs genome-wide (Figure 7E and S7E). Hence, these results support that the above identified classes of nuclear RNAs are not only bound to an insoluble nuclear scaffold, but key to maintaining certain elements of that infrastructure. While the molecular and sequence determinants of chromatin interaction with non-chromatin substructure are not well understood, these results are consistent with a broad and causal relationship between the presence of insoluble hnRNA scaffolds and the cytological appearance of chromatin.
DISCUSSION
It is a long-standing question fundamental to nuclear and genome biology as to the existence and nature of non-chromatin structural elements of chromosome territories. Our results advance this question and at the same time support a new concept whereby the unexplained bulk of long, repeat-rich RNAs the genome produces serves a broad structural role as a platform for RNA binding proteins and associated factors. Despite the dynamic nature of these interactions in living cells, RNP networks can maintain structural integrity even after removal of chromatin. Under a variety of normal and experimental conditions tested we find insoluble repeat-rich hnRNA and specific nuclear scaffold proteins co-distribute within regions of euchromatin but are consistently depleted from compact chromatin. By manipulating insoluble RNA pools, our results indicate nascent RNAs bridge chromatin and insoluble RNP structures to physically antagonize cytological-scale chromatin condensation.
While abundant RNAs associated with chromatin could influence structure in other ways, our results point to the propensity of long RNAs to network with numerous RNA-binding and other proteins to create structures that physically contribute to chromosome territory architecture. Our experiments and models substantially extend earlier work which first provided evidence of an insoluble non-chromatin substructure, termed the nuclear matrix or scaffold. Although some facets of the nuclear matrix were the source of long unresolved controversy (Martelli et al., 2002; Nickerson, 2001; Pederson, 2000; Shopland and Lawrence, 2000), considerable evidence has been presented for the importance of insoluble nuclear substructure, elements of which change during development or become mis-regulated during oncogenic transformation (Munkley et al., 2011; Wilson and Coverley, 2017; Zink et al., 2004). Hallmark proteins earlier identified in the nuclear matrix, most notably lamins and SAF-A, have since been found to regulate chromatin architecture and influence transcription (Fan et al., 2018; Karoutas et al., 2019; Nozawa et al., 2017; Zheng et al., 2018). Hence, while the importance of previously described matrix proteins in nuclear structure is clear, the broad involvement of RNA and potential mechanisms involved have remained an important question.
Criticism of earlier nuclear matrix studies stemmed from lack of in vivo evidence for stable filamentous structures seen by electron microscopy and the potential for proteins to artefactually aggregate during extraction with buffers of high ionic strength. The large amount of RNA found in the nuclear matrix could then potentially include RNAs that co-precipitate with proteins that “salt out” during preparation. For this reason, we developed a progressive and stringent protocol to mitigate these concerns, using initial nonionic and low-ionic strength extractions that are not expected to cause protein precipitation. Remarkably, extensive extraction to remove DNA, most nuclear RNA, and proteins still selectively maintains XIST RNA and C0T-1 hnRNA that localize to chromosome territory substructure. The specific tenacity of XIST RNA to remain in its chromosome territory structure after thorough removal of DNA, in our view, clearly demonstrates existence of a non-chromatin “scaffold”.
RNA-sequencing and transcriptome-wide analysis of the resulting insoluble material provides the most robust demonstration to date that specific RNAs and types of RNA are embedded with insoluble proteins even in the absence of chromatin. Using this more selective fractionation procedure we find most intergenic long non-coding RNAs preferentially cofractionate with chromatin-depleted nuclear insoluble substructure. Although specific lncRNAs (XIST, NEAT1, MALAT1) known to have structural roles in the nucleus were enriched in nuclear insoluble material, more comprehensive analysis of this fraction shows scaffold RNA is overwhelmingly non-coding, with most derived from the introns of nascent precursor transcripts. In contrast, introns were largely absent from protein-coding transcripts in the nucleoplasm.
The vast majority (92%) of human genes contain introns, thousands of which are remarkably long with over 3000 greater than 50 kb in length and replete with interspersed repetitive sequences. Introns are known to contribute toward protein diversity through alternative splicing, however, the excessive length of introns remains largely unexplained given the large energy cost to the cell to transcribe such long pre-mRNA (Shepard et al., 2009). For instance, modern estimates of mammalian RNA Pol II elongation rates suggest it takes approximately 15 minutes to synthesize a single 50 kb intron (Jonkers et al., 2014).
The preponderance of intronic RNA in the nuclear scaffold would not only explain the surprising abundance of repetitive RNA sequences in the nucleus observed by cytology, but potentially give insight into the nature of the nuclear scaffold itself (Hall et al., 2014). While original studies of the nuclear matrix implied the existence of pervasive static structures in the nucleus, recent models have proposed nuclear structure as a dynamic meshwork build on RNA scaffolds (Creamer and Lawrence, 2017; Nozawa and Gilbert, 2019). Though most excised introns are rapidly degraded after excision from pre-cursor transcripts, the extended duration of synthesizing long introns may provide ample opportunity for the assembly of such scaffolds. Here we show nascent RNAs influence chromatin compaction and that intron-containing pre-mRNA account for the bulk of insoluble RNA by mass. However, the interesting possibility remains that lower abundance lncRNAs or intergenic transcripts have a disproportionate influence on chromatin structure. Future studies are needed to definitively address the relative contribution of various transcripts to regulating chromatin compaction.
We speculate that RNA and DNA binding proteins like SAF-A form local RNA-dependent structures that bridge between a dynamic nuclear meshwork built on nascent transcripts and chromatin (Figure 7F), potentially facilitated by recognition of common repetitive sequences found in RNA and DNA. Our SAR-seq experiments demonstrate that the pattern of structurally embedded sites in genomic DNA is dependent on RNA. Actively transcribed regions preferentially associate with nuclear insoluble substructure, and this relationship depends largely on the physical presence of RNA. Both SAF-A and LMNA have been physically and functionally linked to RNA Polymerase II, and SAF-A, NUMA, and LMNA have all been shown to interact with chromatin in S/MARs (Gohring and Fackelmayer, 1997; Luderus et al., 1994; Spann et al., 2002; Vizlin-Hodzic et al., 2011). Interestingly, a recent study indicates many RNA binding proteins interact with chromatin at actively transcribed regions (Xiao et al., 2019). We suggest that the specific sensitivity of SAF-A and genome-wide SARs to RNase likely reflects different roles and interactions between complex “layers” of insoluble RNA-dependent and independent structures with chromatin and/or transcriptional machinery itself.
Recently it has been proposed that nuclear organization is influenced by phase separation of multivalent factors and chromatin into structured compartments with liquid-like properties (Gibson et al., 2019; McSwiggen et al., 2019; Strom and Brangwynne, 2019). RNA has been shown to be a primary determinant of the nucleation and composition of phase separated bodies, in some cases mediated by repetitive sequences (Garcia-Jove Navarro et al., 2019; Ule and Blencowe, 2019; Yamazaki et al., 2018). RNA Pol II and associated transcriptional machinery have been demonstrated to compartmentalize with co-activators and splicing factors in liquid-like condensates (Cho et al., 2018; Guo et al., 2019). The scaffold protein SAF-A also has low complexity domains, was shown to oligomerize, and has itself been speculated to phase separate (March et al., 2016; Michieletto and Gilbert, 2019; Nozawa et al., 2017; Vizlin-Hodzic et al., 2011).
It’s possible that the presence of large phase-transitioned complexes of transcriptional machinery in euchromatin may be directly responsible for the insolubility of some transcripts, though we note XIST RNA remains localized and insoluble in the absence of chromatin long after it is transcribed. Transcriptional machinery may also contribute to SAR-seq enrichment at gene bodies, an idea consistent with past observations that activated (phosphorylated) RNA Pol II is a component of the nuclear matrix (Mortillaro et al., 1996). Future studies are needed to determine the relative contribution of various factors and precise mechanisms influencing cytological-scale chromatin compaction. Rapid degron-mediated depletion of candidate proteins (e.g. RNA Pol II, SAF-A, or other matrix proteins) may provide insight into the relative contribution of these proteins to RNA localization and chromatin compaction, although we speculate a complex meshwork of RNPs may not be reliant on any single factor (or RNA).
Lastly, our results suggest that chromatin has an inherent propensity to compact at the cytological scale and that euchromatin is regionally “opened” by scaffolds built on nascent transcripts. Thus, the classic decondensed appearance of euchromatin could largely be a downstream consequence of pre-mRNA production rather than a determinant of a chromatin landscape permissive for gene expression. Rather than a purely chicken-or-egg problem, we suggest that gene expression and cytological chromatin compaction are at least partially interdependent, linked by the product of transcription which in turn promotes transcription. Nascent intergenic RNAs have been shown to stimulate nearby gene expression and can recruit transcription factors and chromatin modifying enzymes to chromatin. Based on results here, we envision that nascent transcripts, both intergenic non-coding RNA and precursor mRNAs, collectively contribute to the formation of dynamic RNP matrix microenvironments that not only retain transcription and splicing factors at individual genes, but promote an accessible chromatin environment for efficient gene expression on a regional scale that is reflected in the cytological decondensation of chromatin.
LIMITATIONS
This study restricted most analyses to a line of normal human fibroblasts (TIG-1). As discussed, we anticipate other cell types, particularly transformed cell lines, may not respond to prolonged DRB treatment the same as TIG-1 cells and that this response may be concentration or context dependent. Apart from any influence of DNase I cleavage bias, the proteins and mechanisms responsible for SAR-seq signal (DNA regions resistant to extraction) are not fully understood and warrant future investigation. We broadly attribute to long, nascent transcripts a role in maintaining euchromatin structure, however, targeted studies would be needed to test whether properties of individual transcripts (e.g. repeat content, length, coding potential, recruitment of sequence-specific binding factors, splicing rate) allow for certain transcripts to have a greater contribution to chromosome structure than others. Finally, our results demonstrate that repeat-rich RNAs associated with chromatin maintain open euchromatin structure, and we posit this involves interactions with structural proteins such as SAF-A. However RNA may also affect chromatin via electrostatic forces or molecular crowding (or other unanticipated means) which also may contribute to the cytological de-condensation of chromatin.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeanne Lawrence (Jeanne.Lawrence@umassmed.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
RNA-seq and SAR-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-hnRNP U (SAF-A) | Abcam | Cat# ab10297; RRID: AB_297037 |
| Rabbit polyclonal anti-hnRNP U (SAF-A) | Abcam | Cat# ab20666; RRID: AB_732983 |
| Goat polyclonal anti-MATR3 | Santa Cruz | Cat# sc-55723; RRID: AB_2281755 |
| Goat polyclonal anti-LMNB1 | Santa Cruz | Cat# sc-6217; RRID: AB_648158 |
| Mouse monoclonal anti-RNA Polymerase II (H5) | Covance | Cat# MMS-129R-200; RRID: AB_10143905 |
| Rabbit polyclonal anti-LMNA | Cell Signaling | Cat# 2032; RRID: AB_2136278 |
| Rabbit polyclonal anti-Histone H3 (pan antibody) | Millipore | Cat# 07-690; RRID: AB_417398 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Actinomycin D | Sigma | A1410 |
| 5,6-Dichlorobenzimidazole 1-β-D-ribofuranoside | Sigma | D1916 |
| BAPTA-AM | Abcam | ab120503 |
| C0T-1 DNA (human) | Roche | 11581074001 |
| DNase I recombinant, RNase free | Roche | 04716728001 |
| Turbo™ DNase | Invitrogen | AM2238 |
| RNase A | Thermo Scientific | EN0531 |
| Critical Commercial Assays | ||
| KAPA strand-specific RNA-seq preparation with ribosome depletion | KAPA | KK8483 |
| NEBNext® multiplex oligos for Illumina | NEB | E7335S |
| KAPA Hyper DNA library preparation kits | KAPA | KK8501 |
| iTaq Universal SYBR Green Supermix | BIO-RAD | 1725120 |
| Deposited Data | ||
| RNA-seq and SAR-seq experiments | This paper | GEO: GSE124979 |
| RefSeq Select protein coding transcripts | NCBI | https://genome-preview.soe.ucsc.edu/cgi-bin/hgTables? |
| Ensembl reference transcripts (build 82) | Ensembl | https://grch37.ensembl.org/ |
| DNase I digested deproteinized genomic DNA-seq | Lazarovici et al., 2013 | GEO: SRA068503 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Experimental Models: Cell Lines | ||
| TIG-1 untransformed female human fetal lung fibroblasts | Coriell | AG06173 |
| mouse-human (chr4) somatic cell hybrids | Coriell | GM11684 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| See Table S1 for oligonucleotides used in this study. | ||
| Recombinant DNA | ||
| XIST genomic DNA fragment G1A plasmid | Addgene | Plasmid #24690 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Tophat2 | Kim et al., 2103 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks | Trapnell et al., 2012 | http://cole-trapnell-lab.github.io/cufflinks/ |
| RSEM | Li and Dewey, 2011 | http://deweylab.biostat.wisc.edu/html |
| IRFinder | Middleton et al., 2017 | https://github.com/williamritchie/IRFinder |
| deepTools | Ramirez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| Genomic Association Tester | Heger et al., 2013 | https://gat.readthedocs.io/en/latest/ |
| Fastx Toolkit | Hannon Lab | http://hannonlab.cshl.edu/fastx_toolkit/ |
| R | R Core Team | https://www.r-project.org/ |
| ggplot2 | H. Wickham, 2009 | http://ggplot2.org |
| Other | ||
EXPERIMENTAL MODEL AND SUBJECT DETAILS
TIG-1 female human fetal lung fibroblasts were cultured at 37°C in Minimal Essential Media (MEM) supplemented with 15% FBS. GM11684 mouse-human hybrid cells were cultured at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS. Cells were cultured on 10cm dishes or directly on uncoated coverslips for microscopy. Transcription inhibitors were used by supplementing the above media with at the following concentrations: 6-dichloro-1-β-D-ribofuranosylbenzimidazole (DRB, 50 μg/ml), Actinomycin D (ActD, 5 μg/ml), or BAPTA-AM (50 mM). Osmotic stress was induced with KCl (120 mM).
METHOD DETAILS
RNA isolation and library preparation
Biochemical fractionation was performed on 108 TIG-1 fibroblasts harvested with trypsin and thoroughly washed with ice cold PBS. Buffers at all steps contained protease inhibitors (Roche) and RNase inhibitor where appropriate (RNasin, Promega). First, nuclei were isolated by thoroughly resuspension in 1 ml Isotonic Lysis Buffer (10 mM Tris-HCl pH 7.5, 100 mM NaCl, 3 mM MgCl2, 10% glycerol, 0.5% NP-40, 0.5 mM PMSF) and incubating for 15 min on ice with gentle vortexing every 5 minutes. Nuclei were pelleted by centrifugation at 4°C for 3 min at 1000 g. The cytosolic supernatant was removed and permeabilized nuclei were washed twice in Isotonic Lysis Buffer lacking NP-40 with 5 min incubations on ice and gentle vortexing between and recovered by centrifugation as above. Loosely bound nucleoplasmic proteins and RNAs were then solubilized by suspension in 1 ml ice cold modified urea/NP-40 Buffer (1 M urea, 1% NP-40, 0.5 mM PMSF). Nuclei were incubated for 15 minutes with vigorous vortexing every 5 minutes. The insoluble nuclear material was then pelleted by centrifugation at 4°C for 10 min at 10,000 g. The supernatant was collected as the nuclear soluble (NS) fraction. The insoluble material was resuspended in urea/NP-40 buffer and washed after another incubation of 15 minutes on ice with additional vortexing. The insoluble material was then extracted with 0.5 ml Ammonium Sulfate Extraction Buffer (10 mM Tris-HCl pH 7.5, 250 mM ammonium sulfate, 300 mM sucrose, 3 mM MgCl2, 0.5% Triton X-100, 0.5 mM PMSF) for 10 minutes on ice with vigorous vortexing to disrupt the insoluble pellet. Extracted insoluble material was then pelleted at 4°C for 10 min at 10,000 g. The supernatant was collected and the insoluble pellet was again washed as above in Ammonium Sulfate Extraction Buffer. The insoluble pellet was then washed with 0.5 ml DNase Digestion Buffer (40 mM Trsi-HCl pH 7.9, 10 mM NaCl, 6 mM MgCl2, 1 mM CaCl2, 0.05% NP-40, 0.5 mM PMSF). Insoluble material was again suspended in 0.5 ml DNase Digestion Buffer before addition of 10 μl DNase I (Roche). DNA was digested for 60 minutes at 37°C. Samples were extensively vortexed before an additional 10 μl DNase I (Turbo DNase, Ambion) was added and samples were again incubated for 30 minutes at 37°C.
The insoluble nuclear material was then pelleted by centrifugation at 4°C for 10 min at 10,000 g. Supernatants from this step and the ammonium sulfate extraction were combined as the chromatin-dependent fractions. Insoluble material was subjected to an additional high salt wash by being thoroughly resuspended in 0.5ml High Salt Wash Buffer (10 mM Tris-HCl pH 7.5, 10% glycerol, 2 M NaCl, 3 mM MgCl2, 0.05% NP-40, 0.5 mM PMSF). Samples were vortexed and incubated 5 min on ice. The final insoluble scaffold material was then pelleted by centrifugation at 4°C for 10 min at 10,000 g. RNA was recovered by the addition of Trizol (Invitrogen) and EDTA (10 mM final), following ethanol precipitation where necessary. Samples were heated to 65°C for 15 minutes with intermittent vortexing before RNA isolation according to manufacturer recommendations.
Trizol isolated RNA was further cleaned up using RNAEasy columns (Qiagen). Libraries were prepared using a stand specific kit RNA-seq kit with ribosome depletion (KAPA Biosystems) and NEBNext Adapters and Multiplex Oligos for Illumina sequencing. Paired-end 100 bp sequencing was performed on the Illumina HiSeq 2000 platform (UMMS Core Facilities).
RNA-seq analysis
Reads were pre-processed and clipped using the Fastx Toolkit and those mapping to ribosomal RNA were removed prior to subsequent mapping with Bowtie. Non-ribosomal reads were then mapped to the human genome (hg19) using TopHat2 software with default settings. Quantification of transcripts was performed using RSEM on a custom reference prepared from Ensembl build 82 gene annotations including additional intergenic transcripts identified using Cufflinks and the --overlap-radius 1000 parameter on RNA-seq from CDNI, NS, and CDNS fractions from untreated cells only as previously described (Li and Dewey, 2011; Trapnell et al., 2012; Werner and Ruthenburg, 2015). Differential expression by DESeq was performed using counts generated from RSEM. Intron and splicing analysis was performed using IRFinder software and Refseq select protein-coding transcript annotations (Middleton et al., 2017). RNA-seq coverage maps were generated using uniquely mapped reads filtered by the “NH:i:1” flag in TopHat-generated BAM files using Integrated Genome Browser or DeepTools plotHeatmap and bamCoverage software using -bs 10 –normalizeUsing RPKM --ignoreForNormalization chrX chrM parameters (Ramirez et al., 2016).
For genomic distribution calculations either uniquely mapping alignments (relative to genic features) filtered as above or primary alignments (relative to repetitive elements) were used. First, alignments were split using the BEDTools bamtobed tool. Coverage in RefSeq or Repbase-annotated features was then calculated for each alignment using the BEDTools coverage tool. Cumulative coverage was then calculated for all reads for each feature in indicated samples.
Annotation of downstream transcripts
Genome coverage was computed from uniquely mapped reads with TopHat using BEDTools genomecov in CDNI fractions from control and DRB treated cells without using the –split option. Regions with coverage greater than 0.1 reads per million were merged allowing for gaps up to 10kb. Regions were intersected between samples using BEDTools intersect and downstream extensions greater than 10kb were identified in DRB vs control samples. Overlapping extensions were merged and subtracted from the control sample regions using BEDTools merge and subtract tools. Remaining downstream transcripts (regions identified with appreciable coverage in DRB-treated samples but not control) were then selected to be greater than 10kb. Regions lying entirely within Ensembl protein-coding genes were removed as genic or likely artifacts.
RT-PCR
RNA was isolated as described above. cDNA was synthesized using Superscript III Reverse Transcriptase (Thermo Fisher) according to manufacturer instructions. Semi-quantitative PCR was performed using OneTaq polymerase (NEB) using a 58°C annealing temperature. Products were visualized using SYBR Safe (Invitrogen) after agarose gel (1.2%) electrophoresis. Quantitative PCR was performed using iTaq Universal SYBR Green (Bio-Rad) and gene specific primers.
Immunofluorescence and FISH
Immunofluorescence and fluorescence in situ hybridization (FISH) were performed as previously described using C0T-1 DNA (Roche) as a probe or to block non-specific hybridization (Hall et al., 2014). Briefly, cells were grown on coverslips and permeabilized for three minutes in cytoskeleton buffer (CSK) with 0.5% Triton X-100 added on ice. Cells were then fixed in 4% paraformaldehyde in PBS for 10 minutes at room temperature. For antibody staining coverslips were incubated with indicated primary antibodies diluted in 1% BSA in PBS buffer. DNA probes for FISH were nick-translated using biotin-11-dUTP or digoxigenin-16-dUTP (Roche) and hybridization was carried out in buffer containing 50% formamide. Signals were detected using secondary antibodies or streptavidin conjugated with Alexa 488 or Alexa 594 fluorophores (Invitrogen). Cytospin-induced condensation was performed by centrifugation of trypsinized fibroblasts at 2,000 rpm for 10 minutes onto coverslips prior to fixation and analysis. This was compared with typical cytopin attachment (800 rpm, 3 minutes).
In situ nuclear fractionation
To fractionate nuclei in situ, cells grown on coverslips were permeabilized for three minutes in cytoskeleton buffer (CSK) with 0.5% Triton X-100 added on ice. Permeabilized nuclei were then either immediately fixed in 4% paraformaldehyde in PBS for 10 minutes at room temperature (untreated) or extracted as indicated. For 0.25M AS/DNase I extraction, nuclei were extracted on coverslips for 15 minutes in CSK buffer supplemented with 0.25M ammonium sulfate on ice in coplin jars. Coverslips were then washed once with DNase Digestion Buffer (40 mM Tris-HCl pH 7.9, 10 mM NaCl, 6 mM MgCl2, 1mM CaCl2, 300 mM sucrose, 0.05% NP-40, 0.5mM PMSF). Coverslips were then twice incubated in 0.1 ml DNase Digestion Buffer supplemented with 1 μl DNase I (Roche) for 45 minutes at 37°C for a total of 90 minutes, face down on parafilm. Coverslips were then briefly rinsed with CSK prior to fixation as above. Extraction with 1M urea / 1% NP-40 was performed in CSK supplemented with 1M urea and 1% NP-40 for 15 minutes on ice prior to fixation.
For combined urea/scaffold fractionation, cells (~107) were harvested with trypsin and thoroughly washed with ice cold PBS. Cells were then permeabilized for three minutes in 0.5 ml cytoskeleton buffer (CSK) with 0.5% Triton X-100 added on ice and pelleted by centrifugation at 4°C for 3 min at 1000 g. Nuclei were then suspended in 0.1 ml CSK which was then brought to 0.2 ml with CSK supplemented with 2M urea and 2% NP-40. After a 10 minute incubation on ice, approximately 1000 cells were added to 0.15 ml PBS on ice before attachment to coverslips by cytospin centrifugation. Nuclei were then extracted with 0.25M AS and DNase I digested as above. Nuclei were additionally extracted for 10 minutes in coplin jars containing CSK supplemented with 2M NaCl prior to fixation.
Microscopy and image analysis
Nuclei were visualized by Axiovert 200 microscope using a 100X PlanApo objective (NA 1.4) and Chroma 83000 multibandpass dichroic and emission filter sets (Bratteboro). All signals were easily seen by eye and experiments performed at least in duplicate. Images were captured using a cooled charge-coupled device (CCD) camera (200 series, Photometrics). Images were minimally corrected for brightness and contrast using standard practices to best represent signals observed by eye using Axiovision (Zeiss) software.
Image quantification and analysis was performed using ImageJ (Schneider et al., 2012). For intensity calculations background was subtracted using the rolling pin algorithm. Line scans were performed in ImageJ, normalizing signal intensity relative to the maximum intensity found in the line of interest for each channel. Individual nuclei were selected by processing the image as binary and filling holes using the DAPI channel. For intensity scatter plots, images were exported as text files allowing channel intensity and person correlation coefficients between channel intensities to be calculated and graphed using R.
For C0T-1 RNA signal intensity quantification in low magnification images, nuclei were analyzed individually in ImageJ using the Analyze Particles function. C0T-1 RNA was normalized to DAPI intensity within each nucleus. For each condition three separate images were analyzed (100 nuclei each for a total of 300) to minimize potential variability between hybridizations. Intensities were then normalized to the mean normalized C0T-1 RNA intensity in control cells.
Chromatin compaction segmentation analysis was performed as previously described (Cremer et al., 2017). Briefly, z-stacks of individual nuclei were imported and processed in R using the bioimagetools and nucim packages. After masking, nuclei were segmented into class by Gaussian filtering and automatic threshold determination into 7 classes using the classes function (beta=0.1, z=x/z). Classes were merged for simplicity (1 and 2 into segmentation class 1, 3 and 4 into class 2, 5 and 6 into class 3) and the resulting segmentation was then exported to ImageJ for subsequent region of interest area intensity calculations.
RNase treatments
For microscopy analysis, fibroblasts were grown on cover slips and permeabilzed in cytoskeleton buffer (CSK) with 0.5% Triton X-100 for 3 minutes. Cells were washed once with cytoskeleton buffer and then incubated for 5 minutes at 37°C with either RNase A (ThermoFisher, 0.1 mg/ml) or RNase inhibitor (RNasein, Promega) in CSK buffer. Treated nuclei were immediately fixed in 4% paraformaldehyde in PBS for 10 minutes at room temperature and analyzed as described above.
Analysis of protein diffusion from nuclear pellets was performed on 5 million human fibroblasts harvested and washed in PBS buffer. Nuclei were permeabilzed in 0.5 ml cytoskeleton buffer (CSK) with 0.5% Triton X-100 for 3 minutes and pelleted by centrifugation at 4°C for 5 min at 800 g. The supernatant was recovered as the cytoplasmic fractions. Nuclei were washed once in CSK lacking detergent resuspended in CSK. A portion was saved as the nuclear fraction. Nuclei were then incubated for 5 minutes at 37°C with either RNase A (ThermoFisher, 0.1 mg/ml) or RNase inhibitor (RNasein, Promega) in 0.5 ml CSK buffer and again pelleted by centrifugation at 4°C for 5 min at 800 g. The supernatant and pellets were recovered for analysis by western blot.
Scaffold attached DNA (SAR-seq) isolation, sequencing and analysis
Scaffold attached DNA was isolated from 108 TIG-1 fibroblasts harvested with trypsin and thoroughly washed with ice cold PBS. Buffers at all steps contained protease inhibitors (Roche) and RNase inhibitor where appropriate (RNasin, Promega). First, nuclei were isolated by thoroughly resuspending in 1 ml Isotonic Lysis Buffer (10 mM Tris-HCl pH 7.5, 100 mM NaCl, 3 mM MgCl2, 10% glycerol, 0.5% NP-40, 0.5 mM PMSF) and incubating for 5 min. Nuclei were pelleted by centrifugation at 4°C for 3 min at 800 g. The cytosolic supernatant was removed and permeabilized nuclei were washed once in Isotonic Lysis Buffer lacking NP-40. Nuclei were then incubated for 5 minutes at 37°C in 0.5 ml Isotonic Lysis Buffer lacking NP-40 with the addition of RNase inhibitor or RNase A (Thermo Fisher, 0.1 mg/ml) before pelleting as before.
The insoluble material was then extracted with 0.5 ml Ammonium Sulfate Extraction Buffer (10 mM Tris-HCl pH 7.5, 250 mM ammonium sulfate, 300 mM sucrose, 3 mM MgCl2, 0.5% Triton X-100, 0.5 mM PMSF) for 5 minutes on ice with vortexing to disrupt the insoluble pellet. The high salt extracted insoluble material was then pelleted at 4°C for 10 min at 5,000 g. The insoluble pellet was then washed with 0.5 ml DNase Digestion Buffer (40 mM Tris-HCl pH 7.9, 10 mM NaCl, 6 mM MgCl2, 1mM CaCl2, 300 mM sucrose, 0.05% NP-40, 0.5mM PMSF). Insoluble material was again suspended in 0.5 ml DNase Digestion Buffer before addition of 5 μl DNase I (Roche). Nuclei were incubated 30 minutes at 37°C before the addition of an equal volume 4 M High Salt Stop Buffer (10 mM Tis-HCl pH 7.5, 300 mM sucrose, 4M NaCl, 20 mM EDTA). The highly digested, insoluble chromatin was pelleted by centrifugation at 4°C for 10 min at 10,000 g. Time of digestion was titrated to yield majority subnucleosomal DNA fragments. The insoluble pellet was washed once more in in Isotonic Lysis Buffer lacking NP-40 and again harvested by centrifugation.
RNA was removed from fragmented, insoluble genomic DNA by the addition of RNase A (ThermoFisher, 0.1 mg/ml) in TE buffer and incubation for 30 minutes at 37°C. Remaining protein was then digested by the addition of SDS to 0.5 % and Proteinase K to 1 mg/ml and incubating for 90 minutes at 37°C. Samples were the subjected to phenol:chloroform:isoamyl alcohol extraction and ethanol precipitated.
Libraries were prepared from isolated DNA using the Hyper Prep Kit from Kapa Biosystems using NEB TruSeq adapters and indexed primers for amplification. Paired-end read libraries were mapped to the human genome (hg19) using Bowtie2 with –no-mixed –nodiscordant –very-sensitive parameters (Langmead and Salzberg, 2012). Genome coverage was computed and normalized using uniquely mapped reads (filtered using Samtools view -F 260 -q 10) and DeepTools bamCoverage using -bs –maxFragmentLength 120 –normalizeTo1x – ignoreFoorNormalization chrX chrM parameters (Li et al., 2009).
SAR-seq peaks were called using MACS2 software and IDR analysis (Zhang et al., 2008). Briefly, peaks were called using macs2 callpeak BAMPE -p 0.01 for each SAR-seq experiment from control cells and then analyzed using the batch-consistency-analysis.R script. Peaks not found in ENCODE blacklist regions and satisfying a 5% FDR were annotated as SARs. Peak intersection analysis was performed using Genomic Association Tester --num-samples 1000 excluding ENCODE blacklist regions (Heger et al., 2013).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance for RNA-seq transcript enrichment was determined using p-values calculated using DESeq2 (n=2 for each condition or fraction). Student t-tests comparing means of groups were calculated in R software using the t.test() function testing the alternative hypothesis. Error bars represent standard error of the mean (SEM) with indicated sample sizes. Pearson correlation coefficients were calculated in R. Linear regression lines were calculated and drawn using the ggplot2 package in R (Wickham, 2009). Statistical significance for genomic association tests were determined using Genomic Association Tester software.
KEY RESOURCES TABLE
Supplementary Material
HIGHLIGHTS.
Repeat-rich RNA is depleted from heterochromatin and experimentally compacted DNA
Selective nuclear fractionation identifies RNAs in non-chromatin scaffold substructures
Scaffold RNA is rich in repetitive non-coding sequences of pre-mRNAs and lncRNAs
Long transcripts in euchromatin platform scaffolds that counter DNA compaction
Acknowledgements
The authors would like to acknowledge other members of the Lawrence lab for feedback and support with the preparation of this manuscript, particularly Lisa L. Hall. This work was supported by the NIH R35 GM122597.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Berezney R, and Coffey DS (1974). Identification of a nuclear protein matrix. Biochem Biophys Res Commun 60, 1410–1417. [DOI] [PubMed] [Google Scholar]
- Capco DG, Wan KM, and Penman S (1982). The nuclear matrix: three-dimensional architecture and protein composition. Cell 29, 847–858. [DOI] [PubMed] [Google Scholar]
- Caudron-Herger M, Muller-Ott K, Mallm JP, Marth C, Schmidt U, Fejes-Toth K, and Rippe K (2011). Coding RNAs with a non-coding function: maintenance of open chromatin structure. Nucleus 2, 410–424. [DOI] [PubMed] [Google Scholar]
- Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, and Cisse II (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM, Hall LL, Byron M, McNeil J, and Lawrence JB (2006). The X chromosome is organized into a gene-rich outer rim and an internal core containing silenced nongenic sequences. Proc Natl Acad Sci U S A 103, 7688–7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A, and Lawrence JB (2009). An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell 33, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coté A, Coté C, Bayatpour S, Drexler HL, Alexander KA, Chen F, Wassie AT, Boyden ES, Berger S, Churchman LS, et al. (2020). The spatial distributions of pre-mRNAs suggest post-transcriptional splicing of specific introns within endogenous genes. bioRxiv, 2020.2004.2006.028092. [Google Scholar]
- Creamer KM, and Lawrence JB (2017). XIST RNA: a window into the broader role of RNA in nuclear chromosome architecture. Philos Trans R Soc Lond B Biol Sci 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer M, Schmid VJ, Kraus F, Markaki Y, Hellmann I, Maiser A, Leonhardt H, John S, Stamatoyannopoulos J, and Cremer T (2017). Initial high-resolution microscopic mapping of active and inactive regulatory sequences proves non-random 3D arrangements in chromatin domain clusters. Epigenetics Chromatin 10, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson JR, Hong D, Barutcu AR, Wu H, Imbalzano AN, Lian JB, Stein JL, van Wijnen AJ, Nickerson JA, and Stein GS (2017). Identifying Nuclear Matrix-Attached DNA Across the Genome. J Cell Physiol 232, 1295–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler HL, Choquet K, and Churchman LS (2020). Splicing Kinetics and Coordination Revealed by Direct Nascent RNA Sequencing through Nanopores. Mol Cell 77, 985–998 e988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong-Ly KC, and Gabelli SB (2014). Salting out of proteins using ammonium sulfate precipitation. Methods Enzymol 541, 85–94. [DOI] [PubMed] [Google Scholar]
- Fan H, Lv P, Huo X, Wu J, Wang Q, Cheng L, Liu Y, Tang QQ, Zhang L, Zhang F, et al. (2018). The nuclear matrix protein HNRNPU maintains 3D genome architecture globally in mouse hepatocytes. Genome Res 28, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fey EG, Ornelles DA, and Penman S (1986). Association of RNA with the cytoskeleton and the nuclear matrix. J Cell Sci Suppl 5, 99–119. [DOI] [PubMed] [Google Scholar]
- Fritz AJ, Sehgal N, Pliss A, Xu J, and Berezney R (2019). Chromosome territories and the global regulation of the genome. Genes Chromosomes Cancer 58, 407–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galupa R, and Heard E (2018). X-Chromosome Inactivation: A Crossroads Between Chromosome Architecture and Gene Regulation. Annu Rev Genet 52, 535–566. [DOI] [PubMed] [Google Scholar]
- Garcia-Jove Navarro M, Kashida S, Chouaib R, Souquere S, Pierron G, Weil D, and Gueroui Z (2019). RNA is a critical element for the sizing and the composition of phase-separated RNA-protein condensates. Nat Commun 10, 3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesson K, Rescheneder P, Skoruppa MP, von Haeseler A, Dechat T, and Foisner R (2016). A-type lamins bind both hetero- and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res 26, 462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson BA, Doolittle LK, Schneider MWG, Jensen LE, Gamarra N, Henry L, Gerlich DW, Redding S, and Rosen MK (2019). Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484 e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohring F, and Fackelmayer FO (1997). The scaffold/matrix attachment region binding protein hnRNP-U (SAF-A) is directly bound to chromosomal DNA in vivo: a chemical cross-linking study. Biochemistry 36, 8276–8283. [DOI] [PubMed] [Google Scholar]
- Guo YE, Manteiga JC, Henninger JE, Sabari BR, Dall’Agnese A, Hannett NM, Spille JH, Afeyan LK, Zamudio AV, Shrinivas K, et al. (2019). Pol II phosphorylation regulates a switch between transcriptional and splicing condensates. Nature 572, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaf T, and Ward DC (1996). Inhibition of RNA polymerase II transcription causes chromatin decondensation, loss of nucleolar structure, and dispersion of chromosomal domains. Exp Cell Res 224, 163–173. [DOI] [PubMed] [Google Scholar]
- Hall LL, Carone DM, Gomez AV, Kolpa HJ, Byron M, Mehta N, Fackelmayer FO, and Lawrence JB (2014). Stable C0T-1 repeat RNA is abundant and is associated with euchromatic interphase chromosomes. Cell 156, 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall LL, and Lawrence JB (2016). RNA as a fundamental component of interphase chromosomes: could repeats prove key? Curr Opin Genet Dev 37, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heger A, Webber C, Goodson M, Ponting CP, and Lunter G (2013). GAT: a simulation framework for testing the association of genomic intervals. Bioinformatics 29, 2046–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig R, and Fackelmayer FO (2003). Scaffold attachment factor A (SAF-A) is concentrated in inactive X chromosome territories through its RGG domain. Chromosoma 112, 173–182. [DOI] [PubMed] [Google Scholar]
- Heng HH, Goetze S, Ye CJ, Liu G, Stevens JB, Bremer SW, Wykes SM, Bode J, and Krawetz SA (2004). Chromatin loops are selectively anchored using scaffold/matrix-attachment regions. J Cell Sci 117, 999–1008. [DOI] [PubMed] [Google Scholar]
- Jonkers I, Kwak H, and Lis JT (2014). Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife 3, e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallappagoudar S, Varma P, Pathak RU, Senthilkumar R, and Mishra RK (2010). Nuclear matrix proteome analysis of Drosophila melanogaster. Mol Cell Proteomics 9, 2005–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoutas A, Szymanski W, Rausch T, Guhathakurta S, Rog-Zielinska EA, Peyronnet R, Seyfferth J, Chen HR, de Leeuw R, Herquel B, et al. (2019). The NSL complex maintains nuclear architecture stability via lamin A/C acetylation. Nat Cell Biol 21, 1248–1260. [DOI] [PubMed] [Google Scholar]
- Keaton MA, Taylor CM, Layer RM, and Dutta A (2011). Nuclear scaffold attachment sites within ENCODE regions associate with actively transcribed genes. PLoS One 6, e17912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Han KY, Khanna N, Ha T, and Belmont AS (2019). Nuclear speckle fusion via long-range directional motion regulates speckle morphology after transcriptional inhibition. J Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klibanov SA, O’Hagan HM, and Ljungman M (2001). Accumulation of soluble and nucleolar-associated p53 proteins following cellular stress. J Cell Sci 114, 1867–1873. [DOI] [PubMed] [Google Scholar]
- Koohy H, Down TA, and Hubbard TJ (2013). Chromatin accessibility data sets show bias due to sequence specificity of the DNase I enzyme. PLoS One 8, e69853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarovici A, Zhou T, Shafer A, Dantas Machado AC, Riley TR, Sandstrom R, Sabo PJ, Lu Y, Rohs R, Stamatoyannopoulos JA, et al. (2013). Probing DNA shape and methylation state on a genomic scale with DNase I. Proc Natl Acad Sci U S A 110, 6376–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, and Fu XD (2019). Chromatin-associated RNAs as facilitators of functional genomic interactions. Nat Rev Genet 20, 503–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luderus ME, den Blaauwen JL, de Smit OJ, Compton DA, and van Driel R (1994). Binding of matrix attachment regions to lamin polymers involves single-stranded regions and the minor groove. Mol Cell Biol 14, 6297–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Siegel AJ, and Berezney R (1999). Association of chromosome territories with the nuclear matrix. Disruption of human chromosome territories correlates with the release of a subset of nuclear matrix proteins. J Cell Biol 146, 531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March ZM, King OD, and Shorter J (2016). Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res 1647, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli AM, Falcieri E, Zweyer M, Bortul R, Tabellini G, Cappellini A, Cocco L, and Manzoli L (2002). The controversial nuclear matrix: a balanced point of view. Histol Histopathol 17, 1193–1205. [DOI] [PubMed] [Google Scholar]
- Mayer A, and Churchman LS (2017). A Detailed Protocol for Subcellular RNA Sequencing (subRNA-seq). Curr Protoc Mol Biol 120, 4 29 21–24 29 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh CA, Chen CK, Chow A, Surka CF, Tran C, McDonel P, Pandya-Jones A, Blanco M, Burghard C, Moradian A, et al. (2015). The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSwiggen DT, Mir M, Darzacq X, and Tjian R (2019). Evaluating phase separation in live cells: diagnosis, caveats, and functional consequences. Genes Dev 33, 1619–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merdes A, and Cleveland DW (1998). The role of NuMA in the interphase nucleus. J Cell Sci 111 ( Pt 1), 71–79. [DOI] [PubMed] [Google Scholar]
- Michieletto D, and Gilbert N (2019). Role of nuclear RNA in regulating chromatin structure and transcription. Curr Opin Cell Biol 58, 120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton R, Gao D, Thomas A, Singh B, Au A, Wong JJ, Bomane A, Cosson B, Eyras E, Rasko JE, et al. (2017). IRFinder: assessing the impact of intron retention on mammalian gene expression. Genome Biol 18, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minajigi A, Froberg JE, Wei C, Sunwoo H, Kesner B, Colognori D, Lessing D, Payer B, Boukhali M, Haas W, et al. (2015). Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortillaro MJ, Blencowe BJ, Wei X, Nakayasu H, Du L, Warren SL, Sharp PA, and Berezney R (1996). A hyperphosphorylated form of the large subunit of RNA polymerase II is associated with splicing complexes and the nuclear matrix. Proc Natl Acad Sci U S A 93, 8253–8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkley J, Copeland NA, Moignard V, Knight JR, Greaves E, Ramsbottom SA, Pownall ME, Southgate J, Ainscough JF, and Coverley D (2011). Cyclin E is recruited to the nuclear matrix during differentiation, but is not recruited in cancer cells. Nucleic Acids Res 39, 2671–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson J (2001). Experimental observations of a nuclear matrix. J Cell Sci 114, 463–474. [DOI] [PubMed] [Google Scholar]
- Nickerson JA, Krochmalnic G, Wan KM, and Penman S (1989). Chromatin architecture and nuclear RNA. Proc Natl Acad Sci U S A 86, 177–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa RS, Boteva L, Soares DC, Naughton C, Dun AR, Buckle A, Ramsahoye B, Bruton PC, Saleeb RS, Arnedo M, et al. (2017). SAF-A Regulates Interphase Chromosome Structure through Oligomerization with Chromatin-Associated RNAs. Cell 169, 1214–1227 e1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa RS, and Gilbert N (2019). RNA: Nuclear Glue for Folding the Genome. Trends Cell Biol 29, 201–211. [DOI] [PubMed] [Google Scholar]
- Pagoulatos GN, and Darnell JE (1970). A comparison of the heterogeneous nuclear RNA of HeLa cells in different periods of the cell growth cycle. J Cell Biol 44, 476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak RU, Srinivasan A, and Mishra RK (2014). Genome-wide mapping of matrix attachment regions in Drosophila melanogaster. BMC Genomics 15, 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson T (2000). Half a century of “the nuclear matrix”. Mol Biol Cell 11, 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puvvula PK, Desetty RD, Pineau P, Marchio A, Moon A, Dejean A, and Bischof O (2014). Long noncoding RNA PANDA and scaffold-attachment-factor SAFA control senescence entry and exit. Nat Commun 5, 5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki YT, Ideue T, Sano M, Mituyama T, and Hirose T (2009). MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc Natl Acad Sci U S A 106, 2525–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard S, McCreary M, and Fedorov A (2009). The peculiarities of large intron splicing in animals. PLoS One 4, e7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shopland LS, and Lawrence JB (2000). Seeking common ground in nuclear complexity. J Cell Biol 150, F1–4. [DOI] [PubMed] [Google Scholar]
- Slomiany BA, Kelly MM, and Kurtz DT (2000). Extraction of nuclear proteins with increased DNA binding activity. Biotechniques 28, 938–942. [DOI] [PubMed] [Google Scholar]
- Spann TP, Goldman AE, Wang C, Huang S, and Goldman RD (2002). Alteration of nuclear lamin organization inhibits RNA polymerase II-dependent transcription. J Cell Biol 156, 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AR, and Brangwynne CP (2019). The liquid nucleome - phase transitions in the nucleus at a glance. J Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson EC, Manning B, Zhang H, and Lawrence JB (2013). Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J Cell Biol 203, 929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, and Blencowe BJ (2019). Alternative Splicing Regulatory Networks: Functions, Mechanisms, and Evolution. Mol Cell 76, 329–345. [DOI] [PubMed] [Google Scholar]
- Vilborg A, Passarelli MC, Yario TA, Tycowski KT, and Steitz JA (2015). Widespread Inducible Transcription Downstream of Human Genes. Mol Cell 59, 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visvanathan A, Ahmed K, Even-Faitelson L, Lleres D, Bazett-Jones DP, and Lamond AI (2013). Modulation of Higher Order Chromatin Conformation in Mammalian Cell Nuclei Can Be Mediated by Polyamines and Divalent Cations. PLoS One 8, e67689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizlin-Hodzic D, Runnberg R, Ryme J, Simonsson S, and Simonsson T (2011). SAF-A forms a complex with BRG1 and both components are required for RNA polymerase II mediated transcription. PLoS One 6, e28049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner MS, and Ruthenburg AJ (2015). Nuclear Fractionation Reveals Thousands of Chromatin-Tethered Noncoding RNAs Adjacent to Active Genes. Cell Rep 12, 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner MS, Sullivan MA, Shah RN, Nadadur RD, Grzybowski AT, Galat V, Moskowitz IP, and Ruthenburg AJ (2017). Chromatin-enriched lncRNAs can act as cell-type specific activators of proximal gene transcription. Nat Struct Mol Biol 24, 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2009). ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York: ISBN 978–3-319–24277-4. [Google Scholar]
- Wilson RH, Hesketh EL, and Coverley D (2016). The Nuclear Matrix: Fractionation Techniques and Analysis. Cold Spring Harb Protoc 2016, pdb top074518. [DOI] [PubMed] [Google Scholar]
- Wilson RHC, and Coverley D (2017). Transformation-induced changes in the DNA-nuclear matrix interface, revealed by high-throughput analysis of DNA halos. Sci Rep 7, 6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingfield P (2001). Protein precipitation using ammonium sulfate. Curr Protoc Protein Sci Appendix 3, Appendix 3F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R, Chen JY, Liang Z, Luo D, Chen G, Lu ZJ, Chen Y, Zhou B, Li H, Du X, et al. (2019). Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription. Cell 178, 107–121 e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Souquere S, Chujo T, Kobelke S, Chong YS, Fox AH, Bond CS, Nakagawa S, Pierron G, and Hirose T (2018). Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation. Mol Cell 70, 1038–1053 e1037. [DOI] [PubMed] [Google Scholar]
- Yang N, and Sen P (2018). The senescent cell epigenome. Aging (Albany NY) 10, 3590–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitz MJ, Malyavantham KS, Seifert B, and Berezney R (2009). Matrin 3: chromosomal distribution and protein interactions. J Cell Biochem 108, 125–133. [DOI] [PubMed] [Google Scholar]
- Zhang R, Chen W, and Adams PD (2007). Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol 27, 2343–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Hu J, Yue S, Kristiani L, Kim M, Sauria M, Taylor J, Kim Y, and Zheng Y (2018). Lamins Organize the Global Three-Dimensional Genome from the Nuclear Periphery. Mol Cell 71, 802–815 e807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink D, Fischer AH, and Nickerson JA (2004). Nuclear structure in cancer cells. Nat Rev Cancer 4, 677–687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and SAR-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-hnRNP U (SAF-A) | Abcam | Cat# ab10297; RRID: AB_297037 |
| Rabbit polyclonal anti-hnRNP U (SAF-A) | Abcam | Cat# ab20666; RRID: AB_732983 |
| Goat polyclonal anti-MATR3 | Santa Cruz | Cat# sc-55723; RRID: AB_2281755 |
| Goat polyclonal anti-LMNB1 | Santa Cruz | Cat# sc-6217; RRID: AB_648158 |
| Mouse monoclonal anti-RNA Polymerase II (H5) | Covance | Cat# MMS-129R-200; RRID: AB_10143905 |
| Rabbit polyclonal anti-LMNA | Cell Signaling | Cat# 2032; RRID: AB_2136278 |
| Rabbit polyclonal anti-Histone H3 (pan antibody) | Millipore | Cat# 07-690; RRID: AB_417398 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Actinomycin D | Sigma | A1410 |
| 5,6-Dichlorobenzimidazole 1-β-D-ribofuranoside | Sigma | D1916 |
| BAPTA-AM | Abcam | ab120503 |
| C0T-1 DNA (human) | Roche | 11581074001 |
| DNase I recombinant, RNase free | Roche | 04716728001 |
| Turbo™ DNase | Invitrogen | AM2238 |
| RNase A | Thermo Scientific | EN0531 |
| Critical Commercial Assays | ||
| KAPA strand-specific RNA-seq preparation with ribosome depletion | KAPA | KK8483 |
| NEBNext® multiplex oligos for Illumina | NEB | E7335S |
| KAPA Hyper DNA library preparation kits | KAPA | KK8501 |
| iTaq Universal SYBR Green Supermix | BIO-RAD | 1725120 |
| Deposited Data | ||
| RNA-seq and SAR-seq experiments | This paper | GEO: GSE124979 |
| RefSeq Select protein coding transcripts | NCBI | https://genome-preview.soe.ucsc.edu/cgi-bin/hgTables? |
| Ensembl reference transcripts (build 82) | Ensembl | https://grch37.ensembl.org/ |
| DNase I digested deproteinized genomic DNA-seq | Lazarovici et al., 2013 | GEO: SRA068503 |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Experimental Models: Cell Lines | ||
| TIG-1 untransformed female human fetal lung fibroblasts | Coriell | AG06173 |
| mouse-human (chr4) somatic cell hybrids | Coriell | GM11684 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| See Table S1 for oligonucleotides used in this study. | ||
| Recombinant DNA | ||
| XIST genomic DNA fragment G1A plasmid | Addgene | Plasmid #24690 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Tophat2 | Kim et al., 2103 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks | Trapnell et al., 2012 | http://cole-trapnell-lab.github.io/cufflinks/ |
| RSEM | Li and Dewey, 2011 | http://deweylab.biostat.wisc.edu/html |
| IRFinder | Middleton et al., 2017 | https://github.com/williamritchie/IRFinder |
| deepTools | Ramirez et al., 2016 | https://deeptools.readthedocs.io/en/develop/ |
| Genomic Association Tester | Heger et al., 2013 | https://gat.readthedocs.io/en/latest/ |
| Fastx Toolkit | Hannon Lab | http://hannonlab.cshl.edu/fastx_toolkit/ |
| R | R Core Team | https://www.r-project.org/ |
| ggplot2 | H. Wickham, 2009 | http://ggplot2.org |
| Other | ||