

Abstract
We describe the isolation and characterization of a novel insect-specific flavivirus (ISFV), tentatively named Aripo virus (ARPV), that was isolated from Psorophora albipes mosquitoes collected in Trinidad. The ARPV genome was determined and phylogenetic analyses showed that it is a dual host associated ISFV, and clusters with the main mosquito-borne flaviviruses. ARPV antigen was significantly cross-reactive with Japanese encephalitis virus serogroup antisera, with significant cross-reactivity to Ilheus and West Nile virus (WNV). Results suggest that ARPV replication is limited to mosquitoes, as it did not replicate in the sandfly, culicoides or vertebrate cell lines tested. We also demonstrated that ARPV is endocytosed into vertebrate cells and is highly immunomodulatory, producing a robust innate immune response despite its inability to replicate in vertebrate systems. We show that prior infection or coinfection with ARPV limits WNV-induced disease in mouse models, likely the result of a robust ARPV-induced type I interferon response.
Keywords: insect-specific flavivirus, Aripo virus, West Nile virus, flavivirus infection, flavivirus pathogenesis, super-infection exclusion
Introduction
The genus Flavivirus (family Flaviviridae) includes medically important pathogens such as dengue (DENV), yellow fever (YFV), West Nile (WNV), St. Louis encephalitis (SLEV), Japanese encephalitis (JEV), and tick-borne encephalitis (TBEV) viruses. Although the classification of many flaviviruses emphasizes nucleotide and deduced amino acid sequences, these viruses can also be classified based on their antigenic characteristics, vector association, host association, and ecological characteristics (Gould and Solomon, 2008; Kuno, 2004).
Flaviviruses have monopartite, positive (+) sense, single-stranded RNA (ssRNA) genomes of approximately 10 – 11,000 nt in length (Simmonds P, 2011), including a single uninterrupted open reading frame (ORF) that encodes three structural proteins and seven non-structural proteins. The single ORF is flanked by two non-coding regions, the 5’ untranslated region (UTR), which possesses a methylated cap that allows translation, and the 3’ UTR, which forms a complex stem-loop secondary structure that facilitates translation and replication (Simmonds P, 2011).
An area of interest to arbovirologists is the discovery and characterization of “insect-specific” viruses, and their application as platforms for a variety of translational products such as vaccines and diagnostics (Erasmus et al., 2017; Erasmus et al., 2015; Erasmus et al., 2018; Hazlewood et al., 2020; Hobson-Peters et al., 2019; Vet et al., 2020). Unlike medically important mosquito- and tick-borne flaviviruses that alternately infect vertebrate and invertebrate hosts, these mosquito-specific flaviviruses appear to replicate only in mosquitoes and mosquito cells. To date, ISFVs have been isolated from and/or detected in mosquitoes collected in Guatemala (Morales-Betoulle et al., 2008), Mexico (Farfan-Ale et al., 2009; Espinoza-Gomez et al., 2011), Finland (Huhtamo et al., 2012; Huhtamo et al., 2009), Cote d’Ivoire (Junglen et al., 2009), Uganda (Cook et al., 2009), Puerto Rico (Cook et al., 2006), Spain (Vazquez et al., 2012), Portugal (Ferreira et al., 2013; Parreira et al., 2012), Japan (Hoshino et al., 2009; Hoshino et al., 2007), China (An et al., 2012; Lee et al., 2013), Kenya (Sang et al., 2003), Peru (Evangelista et al., 2013), the USA (Bolling et al., 2012; Crabtree et al., 2003; Haddow et al., 2013; Tyler et al., 2011; Kim et al., 2009), Thailand (Yamanaka et al., 2013), Brazil (Kenney et al., 2014), Australia (Hobson-Peters et al., 2013; McLean et al., 2015), and Trinidad (Kim et al., 2009). These viruses cluster into two distinct monophyletic groups within the flavivirus phylogeny (Cook et al., 2012). One ISFV clade clusters together with mosquito-borne flaviviruses, and its members are termed dual host associated ISFVs (dhISFVs). The second and larger ISFV clade lies basal in the phylogeny to the vertebrate-infectious flaviviruses (VIFs) and forms two clades: one consisting of the Aedes and Mansonia-associated and the other Culex-associated viruses (Cook et al., 2012). This second clade is collectively termed the classical ISFVs (clISFVs).
Previous studies have shown experimentally (Bolling et al., 2012; Lutomiah et al., 2007) and indirectly through virus isolation from male mosquitoes and immature stages that ISFVs are vertically transmitted in nature (Bolling et al., 2012; Cook et al., 2006; Haddow et al., 2013; Hoshino et al., 2009; Hoshino et al., 2007; Saiyasombat et al., 2011; Sang et al., 2003). Other laboratory studies have demonstrated both horizontal and venereal transmission of Culex flavivirus (CxFV) in Culex pipiens mosquitoes (Bolling et al., 2012; Saiyasombat et al., 2011), indicating that ISFVs may use alternative modes of infection and transmission for natural maintenance, eliminating (or never developing) the need for a vertebrate host.
Although ISFVs are abundant in nature, little is known about their ecological niche, their position in the evolutionary history of flaviviruses, genetic determinants that render them unable to replicate in vertebrate cells, or to what extent they can affect the transmission of VIFs in nature. Previous studies show ISFVs and insect-specific alphaviruses are host-restricted in vertebrate systems at multiple levels, including entry and replication (Colmant et al., 2017; Junglen et al., 2017; Nasar et al., 2015; Piyasena et al., 2017; Tangudu, 2021).
Herein, we report the detection and characterization of a novel ISFV (tentatively named Aripo virus; ARPV) from a pool of Psorophora albipes mosquitoes collected in Trinidad in 2008. We describe its genetic and serological characteristics and show experimentally that ARPV is vertically transmitted in Aedes aegypti mosquitoes. Finally, we explore ARPV’s host restriction and show that ARPV undergoes clathrin-mediated endocytosis into vertebrate cells, and is immunomodulatory, inducing a robust innate immune response in vertebrate systems, even in the absence of replication.
Methods
Mosquito collection and virus isolation
Between 2007 and 2009, more than 185,000 mosquitoes representing 46 species were collected at three locations in Trinidad (Auguste et al., 2010; Auguste et al., 2009). Mosquitoes were sorted into pools, ranging in size from 1 to 50, by species, sex, date and collection site, and frozen at −80°C for subsequent testing for arboviruses by culture in Vero cells, as described previously (Auguste et al., 2009). For the current study, a random sample of 300 unscreened mosquito pools, and pools that failed to produce cytopathic effects (CPE) on Vero cells, were also inoculated onto the Aedes albopictus cell line C6/36 (Igarashi, 1978) for detection of insect-specific viruses.
Briefly, 100 μL of each mosquito pool homogenate were inoculated onto C6/36 cell monolayers in a 24-well plate format, incubated at 28°C for one hour and then 1 mL of overlay medium [5% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml), 1% tryptose phosphate broth solution and amphotericin B (5.6 μg/ml)] was added. Cells were then observed daily for CPE for up to 10 days. If CPE were observed, a sample of the original homogenate was then inoculated onto a monolayer of C6/36 cells in a 12.5 cm2 plastic culture flask for confirmation.
Hemagglutination inhibition assays
The ability of ARPV to agglutinate goose erythrocytes was tested on supernatants obtained from infected and sham-infected control C6/36 cells that were subsequently acetone extracted. Once agglutination was confirmed, the optimal pH was determined to be 6.2, where the C6/36 cell Aripo antigen agglutinated goose erythrocytes up to a 1:512 dilution. This antigen was then subjected to hemagglutination inhibition (HI) tests at pH 6.2 according to Beaty et al., (1989) (Beaty B.J., 1989) with selected mouse immune ascitic fluids representative of various flavivirus serogroups. All flavivirus immune ascitic fluids were obtained from the World Reference Center for Emerging Viruses and Arboviruses (WRCEVA) at the University of Texas Medical Branch (UTMB).
Transmission electron microscopy
Infected C6/36 monolayers were fixed in 2.5% formaldehyde/0.1% glutaraldehyde in 0.05 M cacodylate buffer pH 7.3 containing 0.03% trinitrophenol and 0.03% CaCl2 for at least 1 hour at room temperature. Monolayers were then washed in 0.1 M cacodylate buffer, scraped and pelleted by centrifugation. Pellets were post-fixed in 1% OsO4, 0.1 M cacodylate buffer, pH 7.3, washed with distilled water, en bloc stained with 2% aqueous uranyl acetate for 20 minutes at 60°C, then dehydrated in graded series of ethanol and embedded in Poly/Bed 812 (Polysciences, Warrington, PA). Ultrathin sections were cut on Leica EM UC7 ultramicrotome (Leica Microsystems, Buffalo Grove, IL), stained with lead citrate, and examined in a Philips (Eindhoven, Netherlands) 201 electron microscope at 60 KV.
To visualize ARPV entry into vertebrate cells, Vero cells were infected (MOI 10 genome copies/cell) with ARPV and incubated on ice for 1 hour. Cells were removed from the ice and incubated at 37°C for 15 seconds, 1 minute, or 5 minutes before being fixed in 2.5% formaldehyde/0.1% glutaraldehyde in 0.05 M cacodylate buffer pH 7.3 containing 0.03% trinitrophenol and 0.03% CaCl2 for 1 hour at room temperature. Monolayers were processed for microscopy similarly to C6/36 monolayers as above.
Virus purification and nucleic acid extraction
Virus from the original mosquito homogenate was inoculated onto 80% confluent C6/36 cell monolayers, and culture medium collected six days later and centrifuged at 1,860 × g to pellet cell debris. Polyethylene glycol was added at 7% w/v and held at 4°C for 48 hours prior to centrifugation at 3,100 × g for 40 min as previously described (Auguste et al., 2015). The pellet was resuspended in 250 μl of TEN buffer [10mM Tris-HCl, pH 7.5; 1mM EDTA, pH8.0; and 0.1M NaCl] by repeated pipetting. RNA was then extracted using Trizol LS (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions.
Sequencing
Viral RNA was fragmented by incubation at 94°C for eight (8) minutes in 19.5 μl of fragmentation buffer (Illumina Inc., San Diego, CA). First and second strand synthesis, adapter ligation and amplification of the library were performed using the Illumina TruSeq RNA Sample Preparation kit v2 under conditions prescribed by the manufacturer (Illumina Inc., San Diego, CA).
Cluster formation of the library DNA templates was performed using the TruSeq PE Cluster Kit v3 (Illumina Inc., San Diego, CA) and the Illumina cBot workstation using conditions recommended by the manufacturer. Paired-end 50 base sequencing by synthesis was performed using TruSeq SBS kit v3 (Illumina Inc., San Diego, CA) on an Illumina HiSeq 1000 using protocols defined by the manufacturer. Cluster density per lane was 820–940 k/mm2, and post-filter reads ranged from 148–218 million per lane. Base call conversion to sequence reads was performed using CASAVA-1.8.2. Reads were filtered for quality and adapter sequences were removed, then viral contigs were assembled de novo using AbySS software (Simpson et al., 2009). Assembled contigs were checked using bowtie2 to align reads to the contigs (Langmead and Salzberg, 2012), followed by visualization using the integrative genomics viewer (Robinson et al., 2011).
Sequence analysis
Predicted viral UTR folding patterns were inferred with RNAfold (Gruber et al., 2008) (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) and Mfold (Zuker, 2003) (http://mfold.rna.albany.edu/?q=mfold) programs at both 29°C and 37°C. The NetNGlyc 1.0 server was used to determine potential N-glycosylation sites within the ARPV ORF using a threshold value of 0.5 (http://www.cbs.dtu.dk/services/NetNGlyc/). Polyprotein cleavage sites were identified by visual inspection under comparison with 20 additional flaviviruses. Sequence identities were calculated using GENEIOUS v5.6 (Auckland, New Zealand). Codon usage patterns were estimated for the complete polyprotein sequences for ARPV (MZ358890), WNV (MT968030), CxFV (KU726615) and Modoc virus (MODV; AJ242984) using the codon usage analyzer available at the sequence manipulation suite (Stothard, 2000). Because vertebrates underutilize CG dinucleotides, and flaviviruses that persist in a single host cycle are known to have codon usage patterns similar to their hosts rather than related flaviviruses (Lobo et al., 2009), we estimated codon usage frequencies for threonine, proline, and arginine (i.e. CG dinucleotide containing codons).
Phylogenetic analysis
Representative flavivirus NS5 nucleotide sequences were downloaded from GenBank, aligned with the ARPV NS5 gene sequence using the translation align in the software package GENEIOUS v5.6 (Auckland, New Zealand), and then manually adjusted. A maximum likelihood (ML) phylogenetic tree was then constructed using the general time reversible (GTR + Γ4 + I) model that was identified among the best-fit models of nucleotide substitution using Modeltest (Posada, 2008). Bootstrapping was used to assess the robustness of tree topologies using 1000 replicate neighbor-joining (NJ) trees under the ML substitution model. All analyses were performed with PAUP* version 4.0b (Sinauer Associates, Inc., Sunderland, MA, USA).
In vitro culture of Aripo virus
A variety of vertebrate and insect cell lines derived from monkey (Vero), hamster (BHK), human (MRC5), mosquitoes (Aedes albopictus, Culex tarsalis, Anopheles stephensi, and Toxorhynchites amboinensis), sandflies (Lutzomyia longipalpis), and midges (Culicoides variipennis), obtained from the WRCEVA, was inoculated with ARPV at an MOI of 1 genome copy/cell using cell culture supernatant from infected C6/36 cells as the inoculum. Cultures were observed for CPE for seven days.
In each of the mammalian cell lines, virus was additionally serially blind-passaged three times, using 100ul of supernatant from the previous passage as the inoculum. Cells were observed for CPE and the third passage supernatant screened for ARPV infection by reverse transcriptase PCRs, using primers based on the ARPV envelope (Env-978F – ATGGAACCCAATGCTTAGACG and Env-1981R – TGTGTACGCCACGACCAGAA) and the NS5 (NS5–8754F – GCGTGTGTATGACGGATACCA and NS5–9610R – TTCAGCTTGCCTTATCAGCT) regions. One-step RT-PCRs were performed with the Titan one-step RT-PCR kit (Roche Diagnostics, Indianapolis, Indiana) and cycling conditions were: 45°C for 50 min, 95 °C for 2 min followed by 40 cycles of 95°C × 10 sec, 50°C × 40 sec, and 68°C × 1 min 15 sec, and a final extension of 68°C for 5 min was used.
To confirm whether ARPV could replicate in the Anopheles stephensi or mammalian cell lines (in which it does not cause observable cytopathic effects), a real-time Taqman assay was developed to assess ARPV’s replication kinetics. Taqman primers (7562F – CGGTGTTCATTGAGGATGAC; 7714R - TGATACGTCCAGGTTCGGTA) and a probe (7680F-P - CGCTGCCTCATGGCAATTCG) were designed based on the NS5 gene. Subsequently, a PCR amplicon was generated, purified using the QiaQuick Gel extraction kit (Qiagen, Inc., Valencia, CA), and cloned into a vector using the TOPO TA cloning kit (Invitrogen, Life Technologies, Carlsbad, CA). Purified plasmid DNA was quantified and serially diluted from 108 genome copies/ml to 1 genome copy/ml, and a standard curve was generated. Aedes albopictus (C6/36), A. stephensi, BHK, and Vero cells were infected at an MOI of 1 genome copy/cell, and replication kinetics were assessed at 24 hr time points for seven days. qRT-PCRs were performed with the TaqMan® RNA-to-CT ™ 1-Step Kit (Applied Biosystems, Life Technologies, Carlsbad, CA) in a 96-well plate using the ABI 7900 HT sequence detection system (Applied Biosystems, Life Technologies, Carlsbad, CA) under the following conditions: 48°C × 30 min, 95°C × 10 min, followed by 40 cycles of 95°C × 15 sec and 60°C × 1 min. Results were collected after the elongation step.
Electroporation of Aripo virus and West Nile virus
To determine if the ARPV host restriction occurs at entry, we evaluated ARPV replication kinetics after electroporating ARPV RNA directly into cells. WNV (strain NY99) and ARPV RNA were concentrated and purified as described above, and 6 μg of total RNA were electroporated into Vero cells as previously described (Gorchakov et al., 2012). C7/10 cells were prepared using the same method, but the following electroporation conditions were used: 5 pulses, 99 μsec in length, at 680V with 200 msec periods between pulses. Aliquots were taken to assess replication kinetics at 24 hr time points for seven days. Virus titers were estimated by qRT-PCR as described above.
Intracellular and Extracellular Viral Replication Kinetics
Vero cells were cultured in plates and inoculated (MOI 0.1 genome copy / cell) in triplicate with either ARPV, ZIKV (strain DakAr D 41524), or culture media as a negative control. After infection, cells were washed three-five times with phosphate buffered saline (PBS) before adding maintenance media (culture media with 2% fetal bovine serum). For extraction and quantification of extracellular viral RNA, aliquots of culture supernatant were removed and replaced with an equivalent volume of fresh media at 0, 3, 6, 12, 24, 48, 72, 96, 120, 144, and 168 hours post-infection. To extract intracellular RNAs, cell monolayers were washed five times with PBS, then lysis buffer was applied directly to the cells and incubated for 10 minutes with continuous rocking before continuing with RNA extraction as described above. Viral titers were quantified by RT-qPCR as described above.
Three-dimensional protein modeling
The initial search and template selection was achieved by submitting the ARPV E protein sequence to the universal protein database in an attempt to identify the most similar 3-D protein structure. The template selected was the JEV E crystal structure (PDB ID#: 3p54), which we then used to predict the structure of the ARPV E protein) with the SWISS-MODEL software package (Arnold et al., 2006). Visualization, manipulation, and further comparisons of the final 3-D structure were made using Swiss-PDB Viewer v4.1 (Guex and Peitsch, 1997).
Vertical transmission in A. aegypti
To determine if ARPV is transmitted vertically in mosquitoes, experimental infections were performed using a laboratory colony of A. aegypti established from eggs collected in 2004 from Mae Sot Province, Thailand (Higgs et al., 2006). Mosquitoes were reared at 28°C, ~80% relative humidity, with a photoperiod of 16h:8h (light:dark). One hundred adult females, 3–5 days post-emergence, were cold-anesthetized and intrathoracically inoculated with 4 log10 genome copies of ARPV in 0.3μl of inoculum. Inoculated mosquitoes were transferred to an environmental chamber at 28°C, and at various time points post-inoculation, mosquitoes were removed to test for virus infection by RT-PCR. At seven days post-inoculation, mosquitoes were offered an artificial blood meal, using a membrane feeding system (Hemotek Ltd, Accrington, United Kingdom), in order to stimulate egg production. Eggs were collected five days later, allowed seven days to embryonate, then hatched in plastic pans. The F1 and F2 progeny were reared to adulthood, and freshly emerged adults were tested for ARPV infection by qRT-PCR. The colony was prescreened using ARPV primers and probes to confirm the absence of potentially cross-reactive ISFVS.
Macrophage Infection and Differentially Expressed Gene Studies:
Bone marrow cells were isolated from naïve C57BL/6 mice (Jackson Laboratory) and differentiated into macrophages as previously described (Adam et al., 2021). A total of 5 × 105 cells was then seeded into 96-well plates. Cells were infected in triplicate with one of the following agents: ARPV, UV-inactivated ARPV, ZIKV (strain PRVABC59), UV-inactivated ZIKV (strain PRVABC59), or mock-infected to serve as negative controls. Six and twenty-four hours post-infection, supernatants were collected for analysis of cytokine production using Bioplex (Erasmus et al., 2017) and ELISA (Wang et al., 2004) assays as previously described.
Six hours post-infection, cells were also harvested and RNA was extracted using the miRNeasy Mini kit (QIAGEN) according to the manufacturer’s instructions. RNA was sequenced on an Illumina Novaseq 6000 instrument using a 200 cycle 100bp paired-end read run. After sequencing, adaptors were trimmed, and the data collated. The mouse reference genome and gene sequences and annotation (mm10) were downloaded from UCSC (genome.ucsc.edu). Raw reads were quality controlled and filtered with FastqMcf (Aronesty, 2013). The clean reads were mapped to the gene reference using BWA (Li and Durbin, 2009) with default parameters. The differential expression of genes was calculated using the edgeR package (Robinson et al., 2010) in R software (http://www.r-project.org/), with Benjamini–Hochberg adjusted P-values of 0.05 considered to be significant. Ingenuity Pathways Analysis (IPA) software was then used to analyze the array data.
Animal experiments
All experiments and procedures involving animals were performed according to Virginia Tech approved IACUC protocols. ARPV, YFV 17D, and WNV (strain NY99) inocula used in animal experiments were prepared from infected C6/36 cell cultures. ARPV inocula were concentrated from a 150 cm2 flask by PEG precipitation as previously described (Erasmus et al., 2017). Inocula were diluted with PBS to achieve the desired dosage, and titers were assessed after inoculation by plaque assay on VERO 76 cells for WNV or RT-qPCR for ARPV as described above.
Intracranial inoculation of CD-1 suckling mice
To confirm ARPV’s host restriction and further explore its vertebrate pathogenicity, we intracranially inoculated outbred suckling mice. Gestational age E17 pregnant dams were purchased from Charles River Laboratories (Wilmington, MA, USA). Dams were divided into three groups, including ARPV, WNV, and PBS (unchallenged healthy controls), and allowed to acclimatize and birth pups. Two days after birth, pups were intracranially inoculated with 7 log10 genome copies of ARPV, 3 log10 plaque-forming units (PFU) of WNV, and PBS diluent. Mice were monitored daily for weight loss and signs of disease for 14 days. Sick or moribund animals were euthanized immediately.
CD-1 mouse protection study
Given the immunogenic nature of ARPV in vitro, we sought to explore the effects of ARPV on WNV pathogenicity in murine models using pre- and co-infection studies. Six-week-old CD-1 mice from Charles River labs were randomly divided into nine (9) groups, each with six animals (n=6). Mice were subcutaneously inoculated seven days before or one day before the WNV challenge with ARPV (7 Log10 genome copies), YFV 17D (7 Log10 genome copies), PBS, or C6/36 culture media. One group was also co-inoculated with WNV and ARPV at the time of infection. Mice were challenged subcutaneously with a lethal dose (103 PFU) of WNV. Mice were monitored daily for weight loss and signs of disease for 21 days post-challenge. Mice were immediately euthanized when moribund or lost 20% or more of their original day 0 bodyweight. Six-week-old CD-1 mice (n=5 per group) were also inoculated with ARPV (7 Log10 genome copies) and C6/36 culture media as a control. Four weeks post-inoculation, mice were euthanized and bled by cardiac puncture. Sera were collected and screened for the presence of WNV-neutralizing antibodies against strain NY99 by plaque reduction neutralization tests (PRNTs) on Vero cells as previously described (Webb et al., 2019).
Ifnar1−/− mouse protection study
To further explore the role of innate immunity, and particularly type I interferons in the protection observed in the CD-1 mouse studies, we performed a similarly designed study in Ifnar1−/− mice (i.e. interferon alpha and beta receptor knockout). Six-week-old Ifnar1−/− mice from Jackson Labs (Bar Harbor, ME, USA) were randomly divided into four (4) groups, each consisting of six mice. Mice were subcutaneously inoculated one day before the WNV challenge with ARPV (7 Log10 genome copies) or PBS diluent. Other groups included ARPV (i.e. to evaluate any ARPV pathological effects in these immune-compromised mice) and PBS controls (i.e. healthy controls) with no WNV challenge. As above, mice were challenged subcutaneously with a lethal dose (103 PFU) of WNV and monitored daily for weight loss and signs of disease for seven days post-challenge and euthanized.
Results
Virus isolation on C6/36 cells
A novel ISFV tentatively named ARPV was isolated from a single pool of Psorophora albipes collected on Dec 3rd, 2008. In the initial screen, ARPV-associated CPE was observed on the fifth day after inoculation. On subsequent passages, the virus caused CPE (syncytia formation followed by significant cell lysis and detachment from the plastic surface) within three days. Despite screening five other P. albipes pools (119 mosquitoes) from the same trap from which ARPV was derived, and 17 P. albipes pools from 6 other traps set on the same day (Dec 3rd, 2008) along the same transect, ARPV was not detected in these or other pools from Trinidad.
Sequence analyses
The ARPV genome was determined to be 10,823 nt in length, with the ORF being 10,347 nt (longest reported for a flavivirus); the 5′UTR was 106 nt, and the 3′UTR was 369 nt long. Table S1 shows the ARPV genome organization and the lengths (nucleotide and amino acid) of the 3′ UTR, 5′ UTR, and the ten genes within the ORF as well as putative glycosylation sites in the predicted proteins. Nucleotide and amino acid sequence identities were compared between ARPV and ten genetically diverse flaviviruses (Table S2). These results show the greatest nucleotide similarity with Chaoyang and Donggang viruses (mean identity of 50% for the structural genes and 60% for nonstructural genes), followed by culex-borne flavivirus vertebrate pathogens such as WNV and SLEV, and then the aedes-borne flavivirus pathogens. ARPV shared the least sequence identity with the no known vector, tick-borne and basal insect-specific Culex flavivirus groups, in that order. NS1, NS3, and NS5 consistently had the highest sequence identities.
As expected, given the estimated sequence identities, a maximum likelihood phylogeny grouped ARPV with dhISFVs in the mosquito-borne flavivirus clade (100% bootstrap support) (Fig. 1A). The node supporting the ARPV branch was well supported (100% bootstrap), which, together with the high sequence divergence between ARPV and its closest relatives, suggests that ARPV likely represents a distinct species in the genus.
Figure 1:
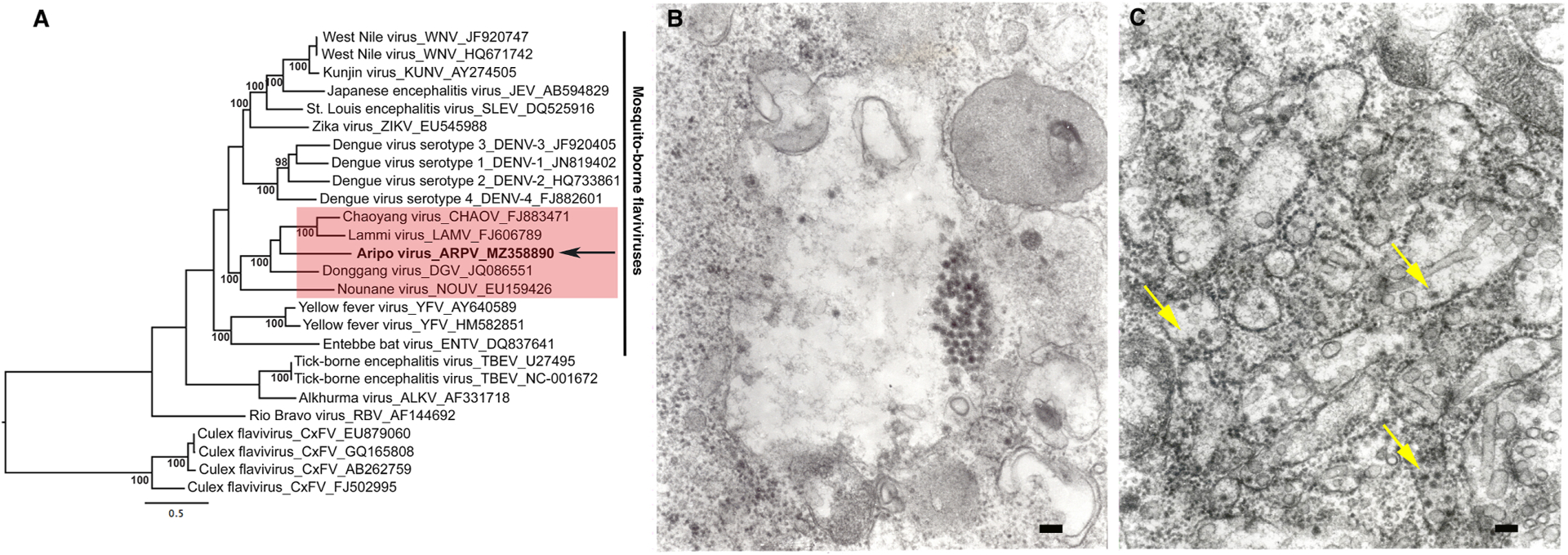
A maximum likelihood (ML) tree was constructed using NS5 gene sequences of representative members of the flavivirus genus (a). The ML phylogeny shows that ARPV clusters together with dual host ISFVs which show a close evolutionary relationship with vertebrate-infectious flaviviruses within the mosquito-borne flavivirus clade. Values at nodes indicate percent bootstrap values. The arrow indicates ARPV and the vertical line indicates vertebrate-infectious flaviviruses, and the red shaded box shows representative dual host associated ISFVs. Taxon labels include virus species name, virus species abbreviation and accession number. The scale bar represents percent nucleotide divergence. Transmission electron micrographs of ARPV in infected C6/36 cells showing (b) a cluster of virus particles inside an expanded vacuole; (c) virus particles are indicated with arrows and smooth membrane structures in the granular endoplasmic reticulum cisterns. Scale bars = 100 nm.
Transmission electron microscopy of ARPV in C6/36 cells
Electron micrographs of ultrathin sections of ARPV-infected C6/36 cells demonstrated spherical virions of ~ 40 nm in diameter with typical flavivirus morphology (Fig. 1B). Virions were mainly distributed inside the cisternae of the granular endoplasmic reticulum and alongside tubular and vesicular smooth membrane structures (Fig. 1C).
Hemagglutination inhibition assays
ARPV antigen agglutinated goose erythrocytes optimally at pH 6.2 to a dilution of 1:512. ARPV antigen cross-reacted with the Japanese encephalitis serogroup antisera (ILHV, WNV, JEV, ROCV, and SLEV) and to a lesser extent with the dengue, Spondweni, and yellow fever serogroup antisera (Table 1). ARPV showed the most cross-reactivity with ILHV and WNV. Additionally, ARPV reacted very strongly with the polyvalent flavivirus antisera used, agglutinating erythrocytes to a dilution of 1:2560.
Table 1:
Hemagglutination inhibition antibody titers showing Aripo virus cross-reactivity with several flavivirus antisera.
| Serogroup | Virus | Antigen | TR9096 Antibody Ht/Ho* |
|---|---|---|---|
| Dengue | DENV-1 | 7.24.70 | 20/640 (32) |
| DENV-2 | 4.5.95 | 80/640 (8) | |
| DENV-3 | 8.2.67 | 80/640 (8) | |
| DENV-4 | 5.75 | 20/640 (32) | |
| Japanese encephalitis | ILHV | PE-163615 | 160/160 (1) |
| WNV | T-34875 | 2560/5120 (2) | |
| JEV | T-36921 | 320/5120 (16) | |
| ROCV | T-36783 | 160/640 (4) | |
| SLEV | T-36388 | 320/5120 (16) | |
| Spondweni | ZIKV | 8.31.65 | 20/640 (32) |
| Yellow fever | WESV | 8.12.65 | 20/640 (32) |
| YFV | T-30906 | 20/640 (32) | |
| Polyvalent Flavivirus | T-37219 | 2560 |
Heterologous divided by homologous titer. Fold Difference is shown in parentheses.
ARPV host range studies
Table 2 summarizes the replication characteristics of ARPV in insect and mammalian cell lines. Figure 2 shows the typical CPE as observed in A. albopictus, C. tarsalis, and T. amboinensis cells. CPE were apparent by day-3 post-infection in all of the mosquito cell lines except A. stephensi, where there was very limited replication detected by qPCR (Fig. 3A). Despite the significant ARPV-induced cytopathic effects observed in C6/36 and C7/10 cell cultures, all attempts to quantify ARPV using conventional plaque assays on these cells were unsuccessful. ARPV did not produce CPE and was not detected by RT-PCR in the Lutzyomyia or Culicoides cells tested. Likewise, there was no evidence of ARPV replication in any of the mammalian cells tested (Table 2, Fig. 3A,S1). Detailed studies on ARPV replication in Vero cell-infected extra- and intra-cellular fractions showed that ARPV successfully attached to Vero cells, and could be detected within the intra-cellular fraction for up to seven days post-infection (Fig. S1). This study further supports an ARPV replication defect, as no evidence of replication was detected in either fraction (Fig. S1). Although ARPV was blind-passaged three times in Vero and BHK cells to allow an opportunity for adaptation, no CPE were observed, and no viral RNA was present after the third passage. Furthermore, none of the intracranially-inoculated suckling mice showed signs of illness or disease over the 14 days that they were monitored.
Table 2:
Replication characteristics of Aripo virus in various cell lines.
| Type | Cell line | CPE (day +) | RT-PCR positive by day 7 |
|---|---|---|---|
| Invertebrate | Aedes. albopictus | Yes (3+) | Yes |
| C6/36 and C7/10 | |||
| Culex tarsalis | Yes (3+) | Yes | |
| Culex | Yes (3+) | Yes | |
| quinquefasciatus | |||
| Toxorynchites | Yes (3+) | Yes | |
| ambionensis | |||
| Anopheles stephensi | No | Yes | |
| Lutzyomyia sp. | No | No | |
| Culicoides sp. | No | No | |
| Vertebrate | Vero-E6*1 | No | No |
| BHK-S*2 | No | No | |
| MRC5*3 | No | No |
Tested after three blind serial passages.
African green monkey kidney cells;
Baby hamster kidney cells;
Human lung fibroblasts
Figure 2:

Phase-contrast micrographs showing the cytopathic effects of ARPV in representative cell lines; (a) negative control for C7/10, (b) C7/10 cells infected with ARPV, (c) negative control for Cx. tarsalis, (d) Cx. tarsalis cells infected with ARPV; (e) negative control for T. ambionensis, and (f) T. ambionensis cells infected with ARPV. Images were taken three days post-infection. Scale bars = 200 μm.
Figure 3:
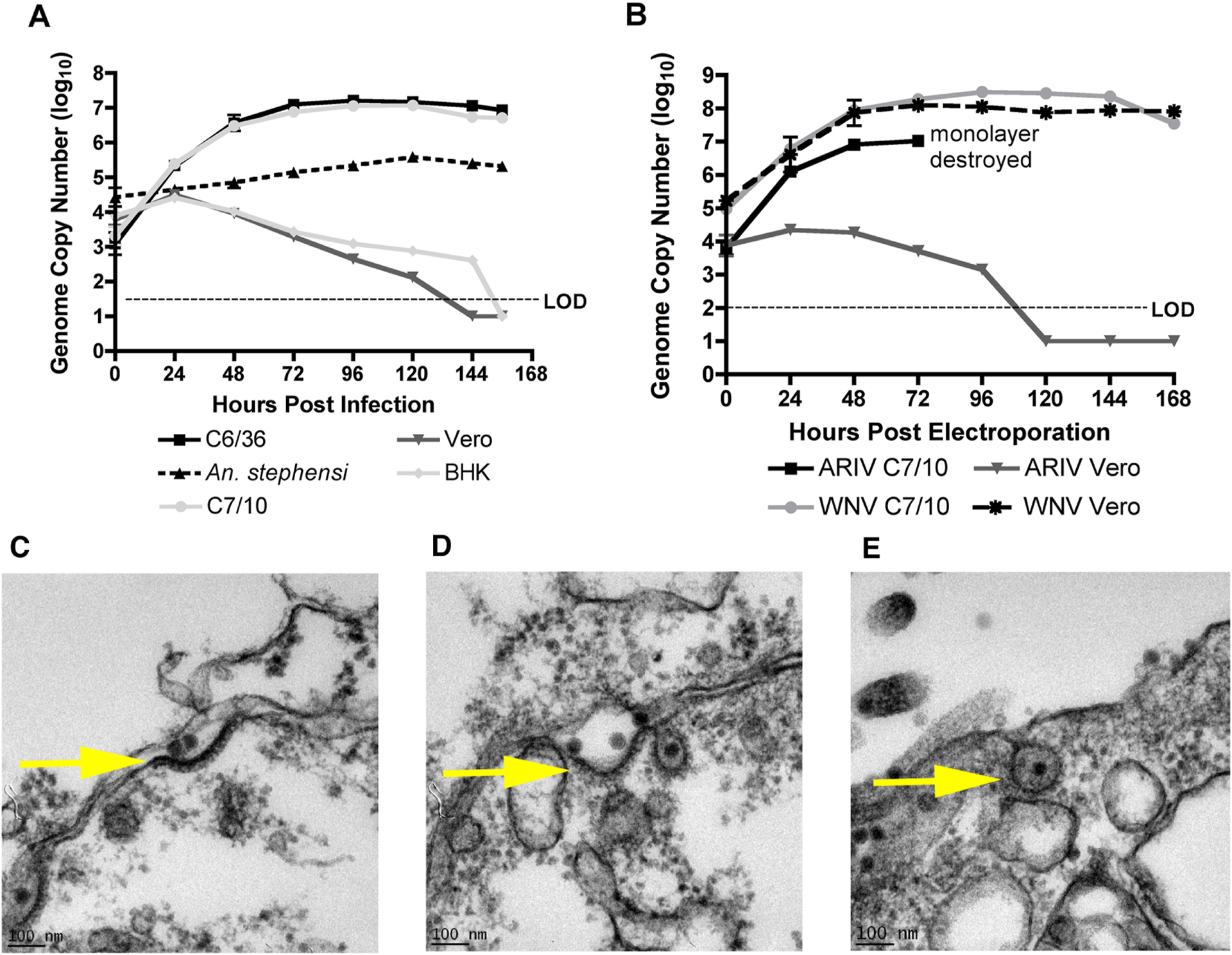
Replication kinetics of ARPV in representative mosquito and vertebrate cell lines post-infection (A) and post-electroporation (B). Monolayers were inoculated with an MOI of ~0.1. Data points represent the mean number of genome copies/ml for duplicate infections titrated in triplicate using qPCR tests in (3A) and triplicate infections titrated in triplicate in (3B). Error bars indicate the standard deviation of the mean. Transmission electron micrographs of ARPV-infected Vero cells show that ARPV enters by clathrin-mediated endocytosis by 15 sec (c), 1 min (d), and 5 mins (e) post-infection. An ARPV virion is localized at the surface of the cellular plasma membrane (c). A clathrin-coated pit is formed, and endocytosis is initialized after viral protein-host cell receptor binding (d) prior to ARPV entry via endocytosis (e). Scale bar (100 nm) is shown in the bottom left of each micrograph.
WNV and ARPV replication kinetics post-electroporation
To determine whether the block to ARPV replication in vertebrate cells is at the level of entry or post-entry, we electroporated WNV and ARPV RNA extracted from purified virus into Vero and C7/10 cells and assessed their replication kinetics by qPCR. WNV readily replicated to high titers (108 genome copies/ml) in both cell lines (Fig. 3B) and ARPV replicated efficiently in C7/10 cells achieving titers of 107 genome copies/ml but showed no replication in Vero cells (Fig. 3B).
Transmission electron microscopy of ARPV in Vero cells
ARPV demonstrated a unique phenotype unlike other insect-specific viruses (Colmant et al., 2017; Junglen et al., 2017; Nasar et al., 2015; Tangudu, 2021); it was capable of vertebrate cell entry via receptor mediated endocytosis as observed in Vero cells using transmission electron microscopy (Fig. 3c–e). Results showed that ARPV is associated with the cell membrane upon infection and enters via clathrin-mediated endocytosis within minutes of infection.
Vertical transmission of ARPV in A. aegypti
ARPV was detected by RT-PCR in 100% (n=12) of intrathoracically inoculated female A. aegypti (Thailand) mosquitoes tested on days 2, 5 or 15 post-inoculation. Infection rates in the F1 and F2 progeny (reared to adulthood) were 100% and 80% respectively, indicating efficient vertical transmission.
ARPV sequence characteristics
Figure S2 shows the inferred folding pattern for the 5′ and 3′ ARPV UTRs. These patterns were similar to those inferred by Parreira et al., (2012) (Parreira et al., 2012) for C. theileri flavivirus. However, that study did not determine the complete sequence of both UTRs, resulting in fewer stem-loop (SL) structures than in our predicted ARPV structures. The 5′ SL structure (Fig. S2) also included the viral AUG translation initiation start codon, supporting Parreira’s suggestion that this UTR may play a role in initiating translation (Parreira et al., 2012).
Table S3 shows the inferred polyprotein cleavage sites, which were similar to those of other flaviviruses. The predicted cleavage sites for ARPV’s serine protease included virC/CTHD, NS2A/NS2B, NS2B/NS3, NS3/NS4A, NS4A/2K, and NS4B/NS5 junctions. They typically occurred after C-terminal residues such as KR, RR, or QR and immediately before short-chain amino acids such as G or S. Sites cleaved by host signalase include VirC-CTHD/preM, Pr/M, M/E, E/NS1, NS1/NS2A, 2K/NS4B.
Eleven potential glycosylation sites were detected, including one each in the PrM and E protein, five in the NS1 protein, and one each in the NS2A, NS3, NS4B, and NS5 proteins. ARPV has the most potential glycosylation sites (5) identified to date in NS1; other flaviviruses usually have 2/3 glycosylation sites in this protein, with the exception of CFAV (Crabtree et al., 2003) and LAMV (Huhtamo et al., 2009), which have four sites. It is unlikely that the cytoplasmic proteins are glycosylated.
Codon usage analysis of CG-containing dinucleotide codons for proline, arginine and threonine from ARPV, WNV, CxFV and MODV showed a significantly reduced bias against encoded CG dinucleotide codons for ARPV, WNV and CxFV in comparison to MODV. As expected, MODV showed significant bias against CG dinucleotide codons (CCG, CGT, CGC, CGA, CGG, and ACG), ranging between 5 and 9 percent usage frequencies. ARPV showed similar CG codon usage frequencies to WNV and CxFV, and greater similarity to WNV. Overall codon usage frequencies ranged between 25–65% for ARPV, 11–45% for WNV, and 30–80% for CxFV among the proline, arginine and threonine residues evaluated.
3-D protein modeling
The 3-D structure of the ARPV E protein was predicted based on the previously published JEV envelope structure (PDB # 3p54A) (Luca et al., 2012). The inferred structure had a mean Z-score of −3.79. Figure S3 shows the predicted 3-D ARPV and JEV envelope protein structures and highlights the differences observed among the major cellular receptor binding sites. Table S4 shows the results of the amino acid comparisons between the heparin sulfate-binding region and antigenic sites among six envelope proteins including JEV (PDB # 3p54A), SLEV (PDB # 4FG0) (Luca et al., 2013), DENV (PDB # 3J27) (Zhang et al., 2013), WNV (PDB # 2I69) (Kanai et al., 2006) and the predicted ARPV and Lammi virus (LAMV) structures. A total of 8 amino acid changes were observed between ARPV and the VIFs tested, including three antigenic sites and five sites within the heparin-binding region.
Mouse monocyte-derived macrophage infections with ARPV
Bone marrow-derived macrophages from C57BL/6 mice infected with media (mock), ARPV, ZIKV, and UV-inactivated virus controls were analyzed for differential gene expression, and corresponding pathways analyzed using Ingenuity Pathway Analysis (IPA). Figure 4A shows volcano plots of the differentially expressed genes among representative comparisons of the various groups studied. Correlation among replicates was >0.97 in all cases showing strong consistency. Our data show that ARPV infection induces an antiviral response, including a significant (z score > 4) interferon response (Fig. 4B). ARPV infection significantly upregulated cell recruitment pathways and pattern recognition receptors (PRR) cascades, the latter of which correlated with robust induction of MYD88 and DDX58 pathways. Upregulated downstream cascades included STAT1, STAT2, IRF3, IRF7, and IRF9. These cascades are associated with increased cytokine and interferon signaling, such as IL-17, IL-1ß, IFN-γ, IFN-α, and IFN-ß.
Figure 4:
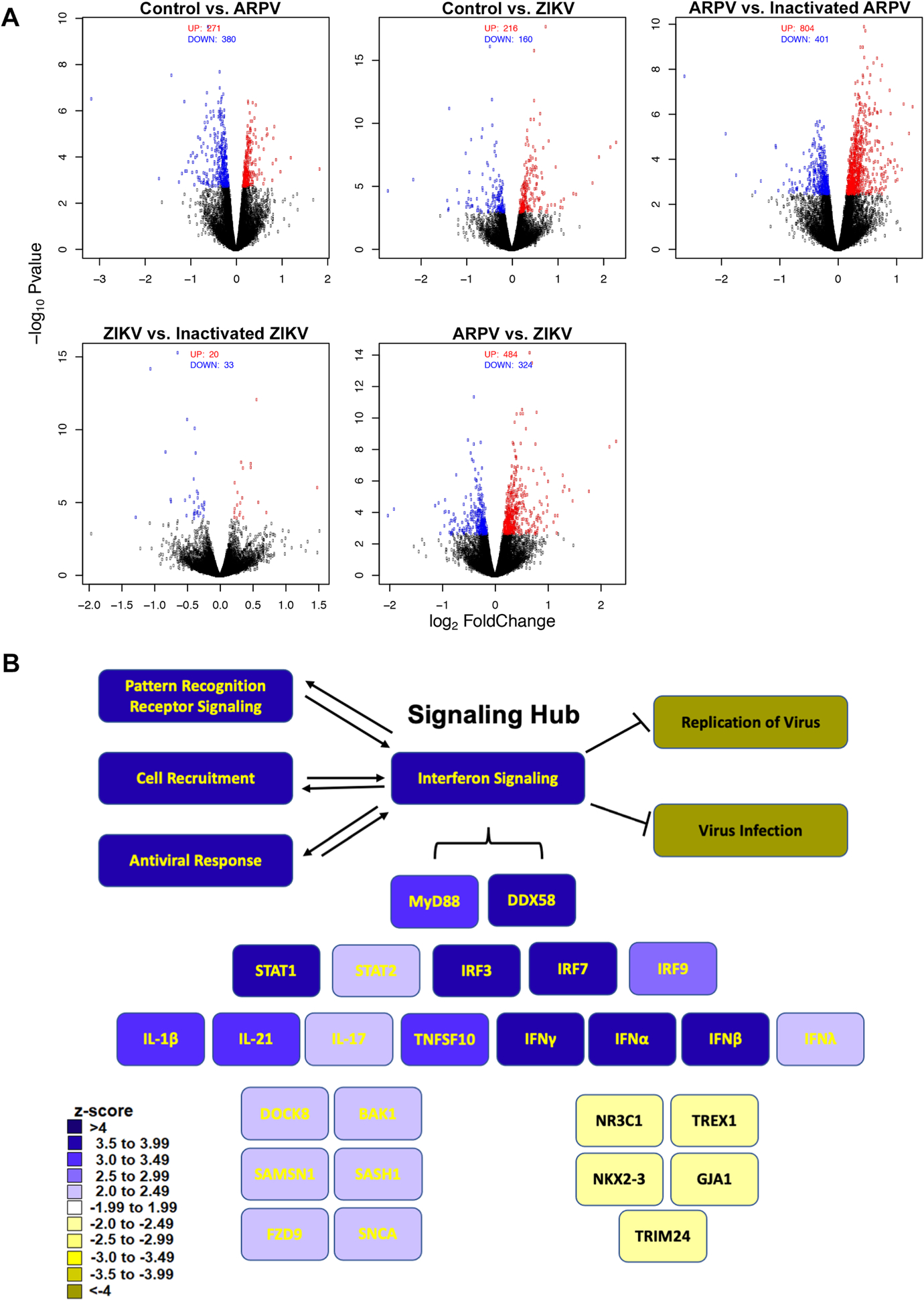
ARPV infection of macrophages results in robust expression of interferon-associated gene responses. Bone-marrow-derived macrophages from naïve C57BL/6 mice were inoculated with ARPV, UV-inactivated ARPV, ZIKV, or culture media as a negative control. RNA was harvested six hours post-infection for DEG analysis. Gene expression data from each of the experimental groups were analyzed using edgeR to generate (a) volcano plots of differential gene expression and using Ingenuity Pathway Analysis (IPA) to generate (b) a heatmap schematic to illustrate signaling pathways identified as significantly up- or down-regulated following ARPV infection relative to controls. Color intensity represents the respective z-score range provided by IPA. Green squares represent up-regulated, and red squares represent down-regulated pathways. Significant pathways identified were grouped and displayed as either upstream regulators, canonical pathways activated or predicted biological functions impacted by ARPV infection.
To confirm the differentially expressed gene (DEG) data, a separate study was performed to assess cytokine production after infection of murine bone marrow-derived macrophages. We observed robust inflammatory and anti-inflammatory cytokine generation at 6 and/or 24 hours post-infection with significant production of IFN-γ, TNF-α, IL-6, IL-12, IL-10, and IL-17 (Fig. 5). ARPV-infected macrophages also resulted in robust type I IFN (IFN-α, IFN-ß) production at 24 and 96 hours post-infection (Fig. S4)
Figure 5:
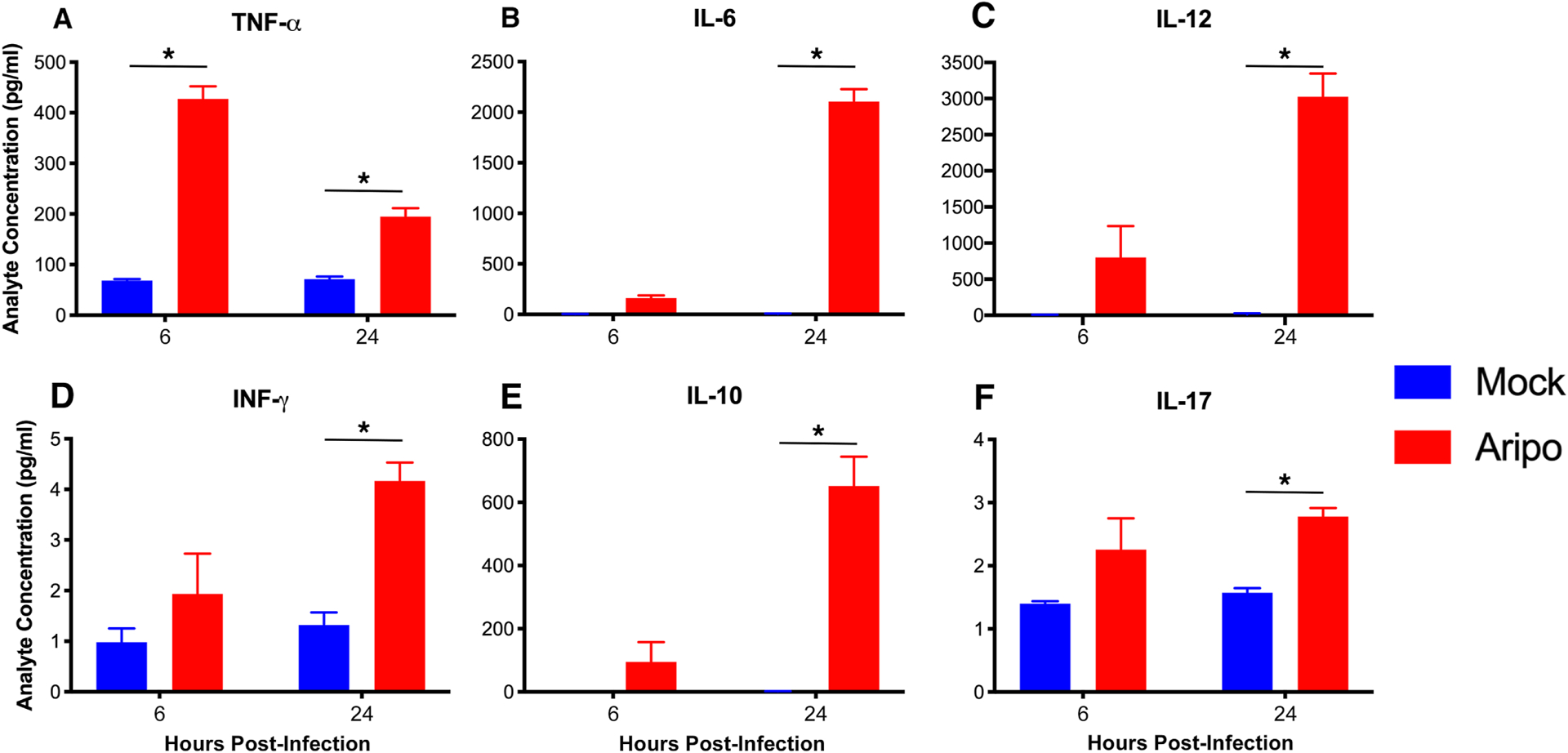
ARPV infection of macrophages results in robust cytokine production. Bone-marrow-derived macrophages from naïve C57BL/6 mice were inoculated with ARPV or culture media as a negative control (mock). Cytokine levels in cell culture supernatant were determined by bead-based Bioplex immunoassay. Infected macrophages produced significant levels of cytokines (a) TNF-α, (b) IL-6 and (c) IL-12, (d) IFN-γ, (e) IL-10, and (f) IL-17 at both 6- and 24-hours post-infection. Data points represent mean values, and error bars indicate standard deviation of the mean for triplicate infections. Significance was determined by two-way ANOVA with ad hoc Tukey’s test. Statistically significant differences are denoted by * p<0.05.
ARPV exposure limits WNV-induced disease in murine models
Given our observation that ARPV induces a potent anti-viral state, we explored the potential for ARPV to limit WNV disease in pre-infection and co-infection scenarios using immune-competent mice. Our data show that ARPV administered one day before challenge completely protected mice from WNV disease and mortality (Fig. 6A,6B). When ARPV was administered simultaneously at the time of challenge, co-infected mice showed reduced mortality and morbidity in contrast to WNV-only infected controls. YFV 17D pre-infected controls did not yield any detectable protective effects against WNV-induced disease or death (Fig. 6A,6B).
Figure 6:
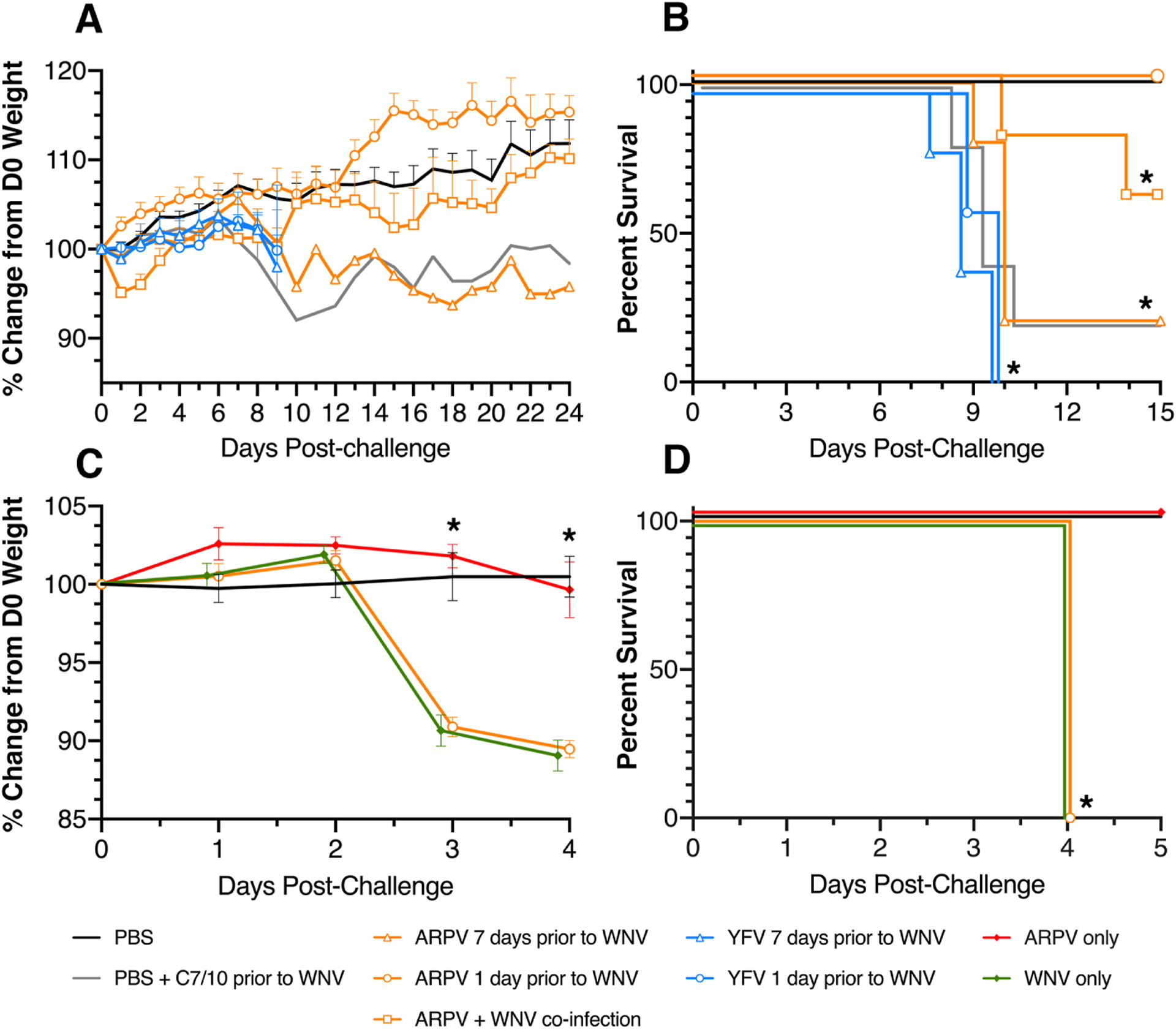
ARPV limits WNV disease in mice due to a robust innate immune response. Six-week-old CD-1 mice were divided into nine (9) groups of n=6 and subcutaneously inoculated seven days before or one day before the WNV challenge with ARPV, YFV 17D, PBS, or C6/36 culture media. One group was also simultaneously co-inoculated with WNV and ARPV. Mice were then challenged with 103 PFU of WNV and were monitored daily for weight loss (a) and survival (b) for 21 days post-challenge. Six-week-old Ifnar1−/− mice were also divided into groups of n=6 and subcutaneously inoculated one day before the WNV challenge with ARPV or PBS. Mice were then challenged with 103 PFU of WNV and monitored daily for weight loss (c) and survival (d) for five days post-challenge. Data points indicate mean values, and error bars indicate standard deviation of the mean. Significance was determined by two-way ANOVA (a,c) and by log-rank (Mantel-Cox) test (b,d). Statistically significant values are denoted by * p<0.05.
Since ARPV pre-infection only protected mice when administered 1-day prior but not earlier, we sought to determine if this protection is mediated by type-1 interferon signaling. Ifnar1−/− mice pre-inoculated with ARPV 1-day prior to challenge showed no protection from WNV-induced weight loss or mortality (Fig. 6C,6D). ARPV inoculated, and WNV unchallenged mice showed no weight loss or mortality. WNV-only infected mice showed significant weight loss and 100% mortality 4 DPI, similar to the ARPV pre-inoculated mice (Fig. 6C,6D). CD-1 mice inoculated with ARPV showed no detectable WNV-cross-neutralizing antibodies by PRNT or HI assays at 7- or 28-days post-infection.
Discussion
The isolation of ARPV from Psorophora mosquitoes represents the first detection of an ISFV from this important genus that is known to be a bridge vector of several arbovirus pathogens (Laporta et al., 2012; Pitzer et al., 2009; Turell et al., 1999; Turell et al., 2006; Turell et al., 2000; Unlu et al., 2010). In the flavivirus phylogeny, insect-specific viruses typically cluster with other viruses from the same mosquito genus and, as such, are generally described as Aedes- or Culex-flaviviruses. Although this nomenclature suggests a narrow mosquito host range, ARPV is now the third example of an insect-specific flavivirus isolated from non-Aedes and non-Culex species. The other two include Nakiwogo virus from Mansonia sp. mosquitoes (Cook et al., 2009), and Nounane virus from Uranotaenia sp. (Junglen et al., 2009). In addition to demonstrating efficient vertical transmission in A. aegypti mosquitoes, our in vitro results show that ARPV can replicate in Aedes, Culex, Anopheles, and Toxorhynchites cell cultures. However, it failed to replicate in the midge (Culicoides) and sandfly (Lutzyomyia) cell lines, suggesting its invertebrate host range may be limited to mosquitoes only. It remains to be seen whether the broad mosquito host range demonstrated in vitro reflects its transmission potential in nature, but we did not detect ARPV in mosquito pools of any other species despite continued surveillance.
ARPV antigen showed strong cross-reactivity with other flavivirus antibodies by HI tests, with the strongest reaction against JE serogroup viruses. Of these, it reacted most strongly with WNV antibodies (Ht/Ho titer of 2560/5120) and those induced by ILHV (Ht/Ho titer of 160/160), indicating a close antigenic relationship. Most ISFVs identified to date are phylogenetically distinct from the mosquito-borne flavivirus pathogens of vertebrates and form a distinct basal grouping within the genus (i.e., clISFVs). In contrast, ARPV is among a smaller group of dhISFVs that cluster within the main mosquito-borne VIFs clade and thus share a more recent evolutionary history with the medically important flaviviruses. While ARPV falls within the same lineage, it is phylogenetically (mean nucleotide divergence 50%; 100% bootstrap support) and serologically distinct and thus may represent a new species.
With regard to mammalian hosts, ARPV did not cause CPE in vitro, nor did it replicate in either the interferon-deficient or -competent mammalian cells tested. It also did not cause any detectable illness in intracranially inoculated suckling mice. The CPE assays, PCR and inoculation of suckling mice used here and previously (Haddow et al., 2013; Huhtamo et al., 2009; Junglen et al., 2009; Kim et al., 2009) cannot rule out low-level replication of ISFVs. However, given that we used a sensitive real-time Taqman assay (limit of detection = 100 genome copies/ml) and mammalian cells that are among the most permissive for flavivirus replication, our results suggest an inability of ARPV to replicate in mammalian cells at 37°C. The small increase detected in the number of genome copies/ml observed at the 24 hr time point in the vertebrate cells tested may reflect previously bound virus that later detached and was released into the supernatant, suggesting inefficient attachment and entry into vertebrate cells. Furthermore, ARPV was serially blind-passaged to provide an opportunity for adaptation to BHK and Vero cells, and no viral RNA was detected after these passages.
Electroporation of concentrated WNV RNA into both C7/10 and Vero cells resulted in efficient replication. ARPV RNA also replicated after electroporation into C7/10 but not Vero cells indicating a fundamental block in RNA replication and/or genomic RNA translation in vertebrate cells. This replication defect is further supported by the absence of detectable replication in intracellular and extracellular fractions of ARPV-infected Vero cells, despite detection of genomic RNA in both fractions for up to 7 days post-infection (Fig. S1). Insect-specific alphaviruses and flaviviruses have previously been shown to be host restricted at the RNA replication level (Colmant et al., 2017; Junglen et al., 2017; Nasar et al., 2015; Piyasena et al., 2017; Tangudu, 2021). Scrutiny of the ARPV replication machinery may lead to new approaches to attenuate pathogenic flaviviruses. Results also show that ARPV is not temperature-restricted in vertebrate cells, as it did not replicate in Vero or BHK cells maintained at 29°C after infection (data not shown). The recently characterized Rabensburg virus (Aliota et al., 2012) represents an intermediate between mosquito-specific and VIFs, and was shown to be temperature restricted in vertebrate cells (Aliota and Kramer, 2012), but this virus is more similar genetically and phenotypically to WNV than are dhISFVs.
Although previous studies show that ISFVs are incapable of vertebrate cell entry (Colmant et al., 2017; Junglen et al., 2017; Nasar et al., 2015; Piyasena et al., 2017; Tangudu, 2021), ARPV shows evidence of vertebrate attachment, entry, and induces a robust innate immune response to infection. Previous studies revolved around clISFVs, which are exceptionally genetically divergent and likely originate from a different ancestor that has never been associated with vertebrate cell infection, unlike dhISFVs that have likely lost the ability to replicate in these cells. Our DEG studies show a significant interferon response, which was further supported by cytokine expression analysis and mouse infection studies. It is unlikely that this interferon response restricts ARPV replication, as no replication was detected in BHK or Vero (i.e., interferon defective) cells, and no RNAemia was detected in ARPV-inoculated Ifnar1−/− mice. The inability of ARPV to suppress interferon production, a trait common to VIFs is indeed interesting. This may suggest that ARPV lost or never acquired the ability to evade vertebrate responses. Future studies are needed to explore if ARPV employs any vertebrate immune response evasive strategies, which will provide a better understanding of the evolutionary history of dhISFVs. Nonetheless, we hypothesize that mosquito-borne flaviviruses are derived from a common ancestor that was restricted to mosquitoes, and subsequently acquired the ability for amplification in vertebrates. We propose that dhISFVs subsequently lost the ability to replicate in vertebrates, while acquiring an adaptation for a competitive advantage in vector transmission, such as vertical transmission. Our data on CG codon usage frequencies for ARPV, a dhISFV, support this hypothesis, and suggest that ARPV likely lost the ability to replicate in vertebrates given the similarities to WNV. Alternatively, ARPV may utilize a non-arthropod host with altered CG codon usage frequencies from those observed in vertebrates.
Our data indicating that ARPV and WNV co-infection results in reduced WNV disease and death are also interesting. Further studies are needed to explore whether or not co-infected mosquitoes that transmit VIFs to vertebrates show reduced arbovirus vector competence or altered pathogenesis, especially for those VIFs that show increased susceptibility to interferon.
ARPV provided complete protection from a lethal WNV challenge when administered one day prior to infection. Although YFV is a potent agonist of the innate immune response (Hacker et al., 1998; Querec et al., 2006; Sinigaglia et al., 2018), YFV-inoculated control mice were not protected from WNV infection, suggesting that the inclusion of an interferon inducer such as polyI:C is needed to more accurately compare the observed protection. Ifnar1−/− mice pre-inoculated with ARPV 1 day prior to challenge showed no protection from WNV disease. Furthermore, mice inoculated with ARPV 7 days prior to WNV challenge were not protected from WNV-induced morbidity or mortality, further supporting the role of innate immunity, in particular type I interferons in protection. CD-1 mice inoculated with ARPV showed no detectable WNV cross-neutralizing antibodies by PRNT or HI assays at 7- or 28-days post-infection, suggesting protection is unlikely to be antibody-mediated.
Our in vitro binding assays show that ARPV is less efficient at binding and entry into Vero cells than ZIKV (Fig. S1). Comparison of the predicted 3-D structure of the ARPV E-glycoprotein with previously published VIFs revealed a number of important epitope features that could potentially account for ARPV’s less efficient attachment and entry into mammalian cells: (i) a change from lysine to the more hydrophobic alanine at the antigenic site at position 138 resulted in internalization and loss of this binding site. This site has been previously shown in JEV to be important for attachment and penetration into BHK-21 cells (Liu et al., 2004); (ii) an aspartate to glycine change at position 390. A mutation from aspartate to histidine at the equivalent site in dengue virus type 2 has been shown to increase mouse neurovirulence, while aspartate to asparagine resulted in attenuated neurovirulence (Sanchez and Ruiz, 1996); (iii) five (5) changes in the heparin-sulfate binding region that could potentially affect receptor binding. Of these, two resulted in small changes in hydrophobicity, two resulted in a loss of positive charge and one in a loss of negative charge. Site-directed mutagenesis studies are needed to determine if these or other residues in the ARPV E protein reduce the efficacy of this virus’ attachment and entry.
The predicted 5′ and 3′ RNA structures for ARPV are the first to be reported for a dhISFV. The predicted structures (Fig. S2) are similar to those described by Parreira et al. (2012) (Parreira et al., 2012) for Cx. theileri flavivirus, but with additional SL structures. The folding patterns for ARPV were identical at both 29°C and 37°C, suggesting that temperature may not be an important factor in determining ARPV replication, at least pertaining to the UTR’s influence on replication. The conserved pentanucleotide sequence 5′-CACAG-3′ found in tick- and mosquito-borne viruses is also conserved within ARPV. Translation reporter assays employing ARPV UTRs are needed to determine if these sequences are incapable of translation in vertebrate cells.
In summary, the isolation and characterization of ARPV broaden our knowledge and understanding of flavivirus diversity, distribution, host range, transmission dynamics, and genomic characteristics. ARPV’s vertebrate host restriction likely occurs at the level of RNA replication and/or translation, but its ability to enter vertebrate cells results in robust innate immune responses that may influence the transmission and pathogenesis of VIFs.
Supplementary Material
Highlights.
Aripo virus is a novel dual host insect-specific flavivirus isolated from Trinidad.
Aripo virus antigen shows strong cross-reactivity with Japanese encephalitis virus serogroup antisera.
Aripo virus infection generates a robust innate immune response despite an inability to replicate in vertebrate systems.
Prior infection or coinfection with Aripo virus limits West Nile virus-induced disease in mouse models
Acknowledgements
This work was supported by grants from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Numbers K22AI125474 and R01AI153433 to AJA, and R01AI 121452 and R24AI120942 to SCW. We thank Steven Widen, Thomas Wood and Jill Thompson for assistance with sequencing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The Aripo virus complete genome sequence is available in GenBank under accession number MZ358890. Aripo virus is available from the WRCEVA or the authors upon request.
References
- 1.Adam A, Luo H, Osman SR, Wang B, Roundy CM, Auguste AJ, Plante KS, Peng BH, Thangamani S, Frolova EI, Frolov I, Weaver SC, Wang T, 2021. Optimized production and immunogenicity of an insect virus-based chikungunya virus candidate vaccine in cell culture and animal models. Emerg Microbes Infect 10, 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aliota MT, Jones SA, Dupuis AP, 2nd, Ciota AT, Hubalek Z, Kramer LD, 2012. Characterization of Rabensburg virus, a flavivirus closely related to West Nile virus of the Japanese encephalitis antigenic group. PLoS One 7, e39387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aliota MT, Kramer LD, 2012. Replication of West Nile virus, Rabensburg lineage in mammalian cells is restricted by temperature. Parasit Vectors 5, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.An SY, Liu JS, Ren Y, Wang ZS, Han Y, Ding J, Guo JQ, 2012. [Isolation of the Culex flavivirus from mosquitoes in Liaoning Province, China]. Bing Du Xue Bao 28, 511–516. [PubMed] [Google Scholar]
- 5.Arnold K, Bordoli L, Kopp J, Schwede T, 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201. [DOI] [PubMed] [Google Scholar]
- 6.Aronesty E, 2013. Comparison of sequencing utility programs. The open bioinformatics journal 7. [Google Scholar]
- 7.Auguste AJ, Adams AP, Arrigo NC, Martinez R, Travassos da Rosa AP, Adesiyun AA, Chadee DD, Tesh RB, Carrington CV, Weaver SC, 2010. Isolation and characterization of sylvatic mosquito-borne viruses in Trinidad: enzootic transmission and a new potential vector of Mucambo virus. Am J Trop Med Hyg 83, 1262–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Auguste AJ, Kaelber JT, Fokam EB, Guzman H, Carrington CV, Erasmus JH, Kamgang B, Popov VL, Jakana J, Liu X, Wood TG, Widen SG, Vasilakis N, Tesh RB, Chiu W, Weaver SC, 2015. A newly isolated reovirus has the simplest genomic and structural organization of any reovirus. J Virol 89, 676–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Auguste AJ, Volk SM, Arrigo NC, Martinez R, Ramkissoon V, Adams AP, Thompson NN, Adesiyun AA, Chadee DD, Foster JE, Travassos Da Rosa AP, Tesh RB, Weaver SC, Carrington CV, 2009. Isolation and phylogenetic analysis of Mucambo virus (Venezuelan equine encephalitis complex subtype IIIA) in Trinidad. Virology 392, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beaty BJ, C.C.H., Shope RE, 1989. Arboviruses. In: Schmidt NJ, Emmons RW, editors. Diagnostic procedures for viral, rickettsial and chlamydial infections. Washington DC: American Public Washington DC: American Public Health Association;. [Google Scholar]
- 11.Bolling BG, Olea-Popelka FJ, Eisen L, Moore CG, Blair CD, 2012. Transmission dynamics of an insect-specific flavivirus in a naturally infected Culex pipiens laboratory colony and effects of co-infection on vector competence for West Nile virus. Virology 427, 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colmant AMG, Hobson-Peters J, Bielefeldt-Ohmann H, van den Hurk AF, Hall-Mendelin S, Chow WK, Johansen CA, Fros J, Simmonds P, Watterson D, Cazier C, Etebari K, Asgari S, Schulz BL, Beebe N, Vet LJ, Piyasena TBH, Nguyen HD, Barnard RT, Hall RA, 2017. A New Clade of Insect-Specific Flaviviruses from Australian Anopheles Mosquitoes Displays Species-Specific Host Restriction. mSphere 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook S, Bennett SN, Holmes EC, De Chesse R, Moureau G, de Lamballerie X, 2006. Isolation of a new strain of the flavivirus cell fusing agent virus in a natural mosquito population from Puerto Rico. J Gen Virol 87, 735–748. [DOI] [PubMed] [Google Scholar]
- 14.Cook S, Moureau G, Harbach RE, Mukwaya L, Goodger K, Ssenfuka F, Gould E, Holmes EC, de Lamballerie X, 2009. Isolation of a novel species of flavivirus and a new strain of Culex flavivirus (Flaviviridae) from a natural mosquito population in Uganda. J Gen Virol 90, 2669–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cook S, Moureau G, Kitchen A, Gould EA, de Lamballerie X, Holmes EC, Harbach RE, 2012. Molecular evolution of the insect-specific flaviviruses. J Gen Virol 93, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crabtree MB, Sang RC, Stollar V, Dunster LM, Miller BR, 2003. Genetic and phenotypic characterization of the newly described insect flavivirus, Kamiti River virus. Arch Virol 148, 1095–1118. [DOI] [PubMed] [Google Scholar]
- 17.Erasmus JH, Auguste AJ, Kaelber JT, Luo H, Rossi SL, Fenton K, Leal G, Kim DY, Chiu W, Wang T, Frolov I, Nasar F, Weaver SC, 2017. A chikungunya fever vaccine utilizing an insect-specific virus platform. Nat Med 23, 192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erasmus JH, Needham J, Raychaudhuri S, Diamond MS, Beasley DW, Morkowski S, Salje H, Fernandez Salas I, Kim DY, Frolov I, Nasar F, Weaver SC, 2015. Utilization of an Eilat Virus-Based Chimera for Serological Detection of Chikungunya Infection. PLoS Negl Trop Dis 9, e0004119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Erasmus JH, Seymour RL, Kaelber JT, Kim DY, Leal G, Sherman MB, Frolov I, Chiu W, Weaver SC, Nasar F, 2018. Novel Insect-Specific Eilat Virus-Based Chimeric Vaccine Candidates Provide Durable, Mono- and Multivalent, Single-Dose Protection against Lethal Alphavirus Challenge. J Virol 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Espinoza-Gomez F, Lopez-Lemus AU, Rodriguez-Sanchez IP, Martinez-Fierro ML, Newton-Sanchez OA, Chavez-Flores E, Delgado-Enciso I, 2011. Detection of sequences from a potentially novel strain of cell fusing agent virus in Mexican Stegomyia (Aedes) aegypti mosquitoes. Arch Virol 156, 1263–1267. [DOI] [PubMed] [Google Scholar]
- 21.Evangelista J, Cruz C, Guevara C, Astete H, Carey C, Kochel TJ, Morrison AC, Williams M, Halsey ES, Forshey BM, 2013. Characterization of a novel flavivirus isolated from Culex (Melanoconion) ocossa mosquitoes from Iquitos, Peru. J Gen Virol 94, 1266–1272. [DOI] [PubMed] [Google Scholar]
- 22.Farfan-Ale JA, Lorono-Pino MA, Garcia-Rejon JE, Hovav E, Powers AM, Lin M, Dorman KS, Platt KB, Bartholomay LC, Soto V, Beaty BJ, Lanciotti RS, Blitvich BJ, 2009. Detection of RNA from a novel West Nile-like virus and high prevalence of an insect-specific flavivirus in mosquitoes in the Yucatan Peninsula of Mexico. Am J Trop Med Hyg 80, 85–95. [PMC free article] [PubMed] [Google Scholar]
- 23.Ferreira DD, Cook S, Lopes A, de Matos AP, Esteves A, Abecasis A, de Almeida AP, Piedade J, Parreira R, 2013. Characterization of an insect-specific flavivirus (OCFVPT) co-isolated from Ochlerotatus caspius collected in southern Portugal along with a putative new Negev-like virus. Virus Genes 47, 532–545. [DOI] [PubMed] [Google Scholar]
- 24.Gorchakov R, Wang E, Leal G, Forrester NL, Plante K, Rossi SL, Partidos CD, Adams AP, Seymour RL, Weger J, Borland EM, Sherman MB, Powers AM, Osorio JE, Weaver SC, 2012. Attenuation of Chikungunya virus vaccine strain 181/clone 25 is determined by two amino acid substitutions in the E2 envelope glycoprotein. J Virol 86, 6084–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gould EA, Solomon T, 2008. Pathogenic flaviviruses. Lancet 371, 500–509. [DOI] [PubMed] [Google Scholar]
- 26.Gruber AR, Lorenz R, Bernhart SH, Neubock R, Hofacker IL, 2008. The Vienna RNA websuite. Nucleic Acids Res 36, W70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guex N, Peitsch MC, 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- 28.Hacker UT, Jelinek T, Erhardt S, Eigler A, Hartmann G, Nothdurft HD, Endres S, 1998. In vivo synthesis of tumor necrosis factor-alpha in healthy humans after live yellow fever vaccination. J Infect Dis 177, 774–778. [DOI] [PubMed] [Google Scholar]
- 29.Haddow AD, Guzman H, Popov VL, Wood TG, Widen SG, Haddow AD, Tesh RB, Weaver SC, 2013. First isolation of Aedes flavivirus in the Western Hemisphere and evidence of vertical transmission in the mosquito Aedes (Stegomyia) albopictus (Diptera: Culicidae). Virology 440, 134–139. [DOI] [PubMed] [Google Scholar]
- 30.Hazlewood JE, Rawle DJ, Tang B, Yan K, Vet LJ, Nakayama E, Hobson-Peters J, Hall RA, Suhrbier A, 2020. A Zika Vaccine Generated Using the Chimeric Insect-Specific Binjari Virus Platform Protects against Fetal Brain Infection in Pregnant Mice. Vaccines (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higgs S, Vanlandingham DL, Klingler KA, McElroy KL, McGee CE, Harrington L, Lang J, Monath TP, Guirakhoo F, 2006. Growth characteristics of ChimeriVax-Den vaccine viruses in Aedes aegypti and Aedes albopictus from Thailand. Am J Trop Med Hyg 75, 986–993. [PubMed] [Google Scholar]
- 32.Hobson-Peters J, Harrison JJ, Watterson D, Hazlewood JE, Vet LJ, Newton ND, Warrilow D, Colmant AMG, Taylor C, Huang B, Piyasena TBH, Chow WK, Setoh YX, Tang B, Nakayama E, Yan K, Amarilla AA, Wheatley S, Moore PR, Finger M, Kurucz N, Modhiran N, Young PR, Khromykh AA, Bielefeldt-Ohmann H, Suhrbier A, Hall RA, 2019. A recombinant platform for flavivirus vaccines and diagnostics using chimeras of a new insect-specific virus. Sci Transl Med 11. [DOI] [PubMed] [Google Scholar]
- 33.Hobson-Peters J, Yam AW, Lu JW, Setoh YX, May FJ, Kurucz N, Walsh S, Prow NA, Davis SS, Weir R, Melville L, Hunt N, Webb RI, Blitvich BJ, Whelan P, Hall RA, 2013. A new insect-specific flavivirus from northern Australia suppresses replication of West Nile virus and Murray Valley encephalitis virus in co-infected mosquito cells. PLoS One 8, e56534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoshino K, Isawa H, Tsuda Y, Sawabe K, Kobayashi M, 2009. Isolation and characterization of a new insect flavivirus from Aedes albopictus and Aedes flavopictus mosquitoes in Japan. Virology 391, 119–129. [DOI] [PubMed] [Google Scholar]
- 35.Hoshino K, Isawa H, Tsuda Y, Yano K, Sasaki T, Yuda M, Takasaki T, Kobayashi M, Sawabe K, 2007. Genetic characterization of a new insect flavivirus isolated from Culex pipiens mosquito in Japan. Virology 359, 405–414. [DOI] [PubMed] [Google Scholar]
- 36.Huhtamo E, Moureau G, Cook S, Julkunen O, Putkuri N, Kurkela S, Uzcategui NY, Harbach RE, Gould EA, Vapalahti O, de Lamballerie X, 2012. Novel insect-specific flavivirus isolated from northern Europe. Virology 433, 471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huhtamo E, Putkuri N, Kurkela S, Manni T, Vaheri A, Vapalahti O, Uzcategui NY, 2009. Characterization of a novel flavivirus from mosquitoes in northern europe that is related to mosquito-borne flaviviruses of the tropics. J Virol 83, 9532–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Igarashi A, 1978. Isolation of a Singh’s Aedes albopictus cell clone sensitive to Dengue and Chikungunya viruses. J Gen Virol 40, 531–544. [DOI] [PubMed] [Google Scholar]
- 39.Junglen S, Kopp A, Kurth A, Pauli G, Ellerbrok H, Leendertz FH, 2009. A new flavivirus and a new vector: characterization of a novel flavivirus isolated from uranotaenia mosquitoes from a tropical rain forest. J Virol 83, 4462–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Junglen S, Korries M, Grasse W, Wieseler J, Kopp A, Hermanns K, Leon-Juarez M, Drosten C, Kummerer BM, 2017. Host Range Restriction of Insect-Specific Flaviviruses Occurs at Several Levels of the Viral Life Cycle. mSphere 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanai R, Kar K, Anthony K, Gould LH, Ledizet M, Fikrig E, Marasco WA, Koski RA, Modis Y, 2006. Crystal structure of west nile virus envelope glycoprotein reveals viral surface epitopes. J Virol 80, 11000–11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kenney JL, Solberg OD, Langevin SA, Brault AC, 2014. Characterization of a novel insect-specific flavivirus from Brazil: potential for inhibition of infection of arthropod cells with medically important flaviviruses. J Gen Virol 95, 2796–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim DY, Guzman H, Bueno R Jr., Dennett JA, Auguste AJ, Carrington CV, Popov VL, Weaver SC, Beasley DW, Tesh RB, 2009. Characterization of Culex Flavivirus (Flaviviridae) strains isolated from mosquitoes in the United States and Trinidad. Virology 386, 154–159. [DOI] [PubMed] [Google Scholar]
- 44.Kuno G, 2004. A survey of the relationships among the viruses not considered arboviruses, vertebrates, and arthropods. Acta Virol 48, 135–143. [PubMed] [Google Scholar]
- 45.Langmead B, Salzberg SL, 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laporta GZ, Ribeiro MC, Ramos DG, Sallum MA, 2012. Spatial distribution of arboviral mosquito vectors (Diptera, Culicidae) in Vale do Ribeira in the South-eastern Brazilian Atlantic Forest. Cad Saude Publica 28, 229–238. [DOI] [PubMed] [Google Scholar]
- 47.Lee JS, Grubaugh ND, Kondig JP, Turell MJ, Kim HC, Klein TA, O’Guinn ML, 2013. Isolation and genomic characterization of Chaoyang virus strain ROK144 from Aedes vexans nipponii from the Republic of Korea. Virology 435, 220–224. [DOI] [PubMed] [Google Scholar]
- 48.Li H, Durbin R, 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu H, Chiou SS, Chen WJ, 2004. Differential binding efficiency between the envelope protein of Japanese encephalitis virus variants and heparan sulfate on the cell surface. J Med Virol 72, 618–624. [DOI] [PubMed] [Google Scholar]
- 50.Lobo FP, Mota BE, Pena SD, Azevedo V, Macedo AM, Tauch A, Machado CR, Franco GR, 2009. Virus-host coevolution: common patterns of nucleotide motif usage in Flaviviridae and their hosts. PLoS One 4, e6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luca VC, AbiMansour J, Nelson CA, Fremont DH, 2012. Crystal structure of the Japanese encephalitis virus envelope protein. J Virol 86, 2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luca VC, Nelson CA, Fremont DH, 2013. Structure of the St. Louis encephalitis virus postfusion envelope trimer. J Virol 87, 818–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lutomiah JJ, Mwandawiro C, Magambo J, Sang RC, 2007. Infection and vertical transmission of Kamiti river virus in laboratory bred Aedes aegypti mosquitoes. J Insect Sci 7, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McLean BJ, Hobson-Peters J, Webb CE, Watterson D, Prow NA, Nguyen HD, Hall-Mendelin S, Warrilow D, Johansen CA, Jansen CC, van den Hurk AF, Beebe NW, Schnettler E, Barnard RT, Hall RA, 2015. A novel insect-specific flavivirus replicates only in Aedes-derived cells and persists at high prevalence in wild Aedes vigilax populations in Sydney, Australia. Virology 486, 272–283. [DOI] [PubMed] [Google Scholar]
- 55.Morales-Betoulle ME, Monzon Pineda ML, Sosa SM, Panella N, Lopez MR, Cordon-Rosales C, Komar N, Powers A, Johnson BW, 2008. Culex flavivirus isolates from mosquitoes in Guatemala. J Med Entomol 45, 1187–1190. [DOI] [PubMed] [Google Scholar]
- 56.Nasar F, Gorchakov RV, Tesh RB, Weaver SC, 2015. Eilat virus host range restriction is present at multiple levels of the virus life cycle. J Virol 89, 1404–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parreira R, Cook S, Lopes A, de Matos AP, de Almeida AP, Piedade J, Esteves A, 2012. Genetic characterization of an insect-specific flavivirus isolated from Culex theileri mosquitoes collected in southern Portugal. Virus Res 167, 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pitzer JB, Byford RL, Vuong HB, Steiner RL, Creamer RJ, Caccamise DF, 2009. Potential vectors of West Nile virus in a semiarid environment: Dona Ana County, New Mexico. J Med Entomol 46, 1474–1482. [DOI] [PubMed] [Google Scholar]
- 59.Piyasena TBH, Setoh YX, Hobson-Peters J, Newton ND, Bielefeldt-Ohmann H, McLean BJ, Vet LJ, Khromykh AA, Hall RA, 2017. Infectious DNAs derived from insect-specific flavivirus genomes enable identification of pre- and post-entry host restrictions in vertebrate cells. Sci Rep 7, 2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Posada D, 2008. jModelTest: phylogenetic model averaging. Mol Biol Evol 25, 1253–1256. [DOI] [PubMed] [Google Scholar]
- 61.Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, Akira S, Ahmed R, Pulendran B, 2006. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med 203, 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP, 2011. Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson MD, McCarthy DJ, Smyth GK, 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Saiyasombat R, Bolling BG, Brault AC, Bartholomay LC, Blitvich BJ, 2011. Evidence of efficient transovarial transmission of Culex flavivirus by Culex pipiens (Diptera: Culicidae). J Med Entomol 48, 1031–1038. [DOI] [PubMed] [Google Scholar]
- 65.Sanchez IJ, Ruiz BH, 1996. A single nucleotide change in the E protein gene of dengue virus 2 Mexican strain affects neurovirulence in mice. J Gen Virol 77 (Pt 10), 2541–2545. [DOI] [PubMed] [Google Scholar]
- 66.Sang RC, Gichogo A, Gachoya J, Dunster MD, Ofula V, Hunt AR, Crabtree MB, Miller BR, Dunster LM, 2003. Isolation of a new flavivirus related to cell fusing agent virus (CFAV) from field-collected flood-water Aedes mosquitoes sampled from a dambo in central Kenya. Arch Virol 148, 1085–1093. [DOI] [PubMed] [Google Scholar]
- 67.Simmonds P, B. P, Collett MS, Gould EA, Heinz FX, Meyers G, Monath T, Pletnev A, Rice CM, Stinsny K, Thiel HJ, Weiner A, Bukh J, 2011. Family Flaviviridae. In: Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses.,9th ed. Elsevier, Academic Press. [Google Scholar]
- 68.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I, 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19, 1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sinigaglia L, Gracias S, Decembre E, Fritz M, Bruni D, Smith N, Herbeuval JP, Martin A, Dreux M, Tangy F, Jouvenet N, 2018. Immature particles and capsid-free viral RNA produced by Yellow fever virus-infected cells stimulate plasmacytoid dendritic cells to secrete interferons. Sci Rep 8, 10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stothard P, 2000. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 28, 1102, 1104. [DOI] [PubMed] [Google Scholar]
- 71.Tangudu CS, C.J.; Nunez-Avellaneda D; Hargett AM; Brault AC; Blitvich BJ, 2021. Chimeric Zika viruses containing structural protein genes of insect-specific flaviviruses cannot replicate in vertebrate cells due to entry and post-translational restrictions. Virology 559, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Turell MJ, Barth J, Coleman RE, 1999. Potential for Central American mosquitoes to transmit epizootic and enzootic strains of Venezuelan equine encephalitis virus. J Am Mosq Control Assoc 15, 295–298. [PubMed] [Google Scholar]
- 73.Turell MJ, Dohm DJ, Fernandez R, Calampa C, O’Guinn ML, 2006. Vector competence of Peruvian mosquitoes (Diptera: Culicidae) for a subtype IIIC virus in the Venezuelan equine encephalomyelitis complex isolated from mosquitoes captured in Peru. J Am Mosq Control Assoc 22, 70–75. [DOI] [PubMed] [Google Scholar]
- 74.Turell MJ, Jones JW, Sardelis MR, Dohm DJ, Coleman RE, Watts DM, Fernandez R, Calampa C, Klein TA, 2000. Vector competence of Peruvian mosquitoes (Diptera: Culicidae) for epizootic and enzootic strains of Venezuelan equine encephalomyelitis virus. J Med Entomol 37, 835–839. [DOI] [PubMed] [Google Scholar]
- 75.Tyler S, Bolling BG, Blair CD, Brault AC, Pabbaraju K, Armijos MV, Clark DC, Calisher CH, Drebot MA, 2011. Distribution and phylogenetic comparisons of a novel mosquito flavivirus sequence present in Culex tarsalis Mosquitoes from western Canada with viruses isolated in California and Colorado. Am J Trop Med Hyg 85, 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Unlu I, Kramer WL, Roy AF, Foil LD, 2010. Detection of West Nile virus RNA in mosquitoes and identification of mosquito blood meals collected at alligator farms in Louisiana. J Med Entomol 47, 625–633. [DOI] [PubMed] [Google Scholar]
- 77.Vazquez A, Sanchez-Seco MP, Palacios G, Molero F, Reyes N, Ruiz S, Aranda C, Marques E, Escosa R, Moreno J, Figuerola J, Tenorio A, 2012. Novel flaviviruses detected in different species of mosquitoes in Spain. Vector Borne Zoonotic Dis 12, 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vet LJ, Setoh YX, Amarilla AA, Habarugira G, Suen WW, Newton ND, Harrison JJ, Hobson-Peters J, Hall RA, Bielefeldt-Ohmann H, 2020. Protective Efficacy of a Chimeric Insect-Specific Flavivirus Vaccine against West Nile Virus. Vaccines (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA, 2004. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 10, 1366–1373. [DOI] [PubMed] [Google Scholar]
- 80.Webb EM, Azar SR, Haller SL, Langsjoen RM, Cuthbert CE, Ramjag AT, Luo H, Plante K, Wang T, Simmons G, Carrington CVF, Weaver SC, Rossi SL, Auguste AJ, 2019. Effects of Chikungunya virus immunity on Mayaro virus disease and epidemic potential. Sci Rep 9, 20399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yamanaka A, Thongrungkiat S, Ramasoota P, Konishi E, 2013. Genetic and evolutionary analysis of cell-fusing agent virus based on Thai strains isolated in 2008 and 2012. Infect Genet Evol 19, 188–194. [DOI] [PubMed] [Google Scholar]
- 82.Zhang X, Ge P, Yu X, Brannan JM, Bi G, Zhang Q, Schein S, Zhou ZH, 2013. Cryo-EM structure of the mature dengue virus at 3.5-A resolution. Nat Struct Mol Biol 20, 105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zuker M, 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Aripo virus complete genome sequence is available in GenBank under accession number MZ358890. Aripo virus is available from the WRCEVA or the authors upon request.