

Abstract
Dasatinib is an inhibitor of Src that has anti‐tumour effects on many haematological and solid cancers. However, the anti‐tumour effects of dasatinib on human oral cancers remain unclear. In this study, we investigated the effects of dasatinib on different types of human oral cancer cells: the non‐tumorigenic YD‐8 and YD‐38 and the tumorigenic YD‐10B and HSC‐3 cells. Strikingly, dasatinib at 10 µM strongly suppressed the growth and induced apoptosis of YD‐38 cells and inhibited the phosphorylation of Src, EGFR, STAT‐3, STAT‐5, PKB and ERK‐1/2. In contrast, knockdown of Src blocked the phosphorylation of EGFR, STAT‐5, PKB and ERK‐1/2, but not STAT‐3, in YD‐38 cells. Dasatinib induced activation of the intrinsic caspase pathway, which was inhibited by z‐VAD‐fmk, a pan‐caspase inhibitor. Dasatinib also decreased Mcl‐1 expression and S6 phosphorylation while increased GRP78 expression and eIF‐2α phosphorylation in YD‐38 cells. In addition, to its direct effects on YD‐38 cells, dasatinib also exhibited anti‐angiogenic properties. Dasatinib‐treated YD‐38 or HUVEC showed reduced HIF‐1α expression and stability. Dasatinib alone or conditioned media from dasatinib‐treated YD‐38 cells inhibited HUVEC tube formation on Matrigel without affecting HUVEC viability. Importantly, dasatinib's anti‐growth, anti‐angiogenic and pro‐apoptotic effects were additionally seen in tumorigenic HSC‐3 cells. Together, these results demonstrate that dasatinib has strong anti‐growth, anti‐angiogenic and pro‐apoptotic effects on human oral cancer cells, which are mediated through the regulation of multiple targets, including Src, EGFR, STAT‐3, STAT‐5, PKB, ERK‐1/2, S6, eIF‐2α, GRP78, caspase‐9/3, Mcl‐1 and HIF‐1α.
Keywords: apoptosis, dasatinib, HIF‐1α, HSC‐3, Src, YD‐38
1. INTRODUCTION
Oral squamous cell carcinoma (OSCC) accounts for 24% of all head and neck cancers.1 OSCC is caused predominantly through exposure to areca quid chewing, cigarette smoking and alcohol consumption.2, 3 OSCC not only causes significant mortality but is associated with functional alterations in the oromaxillo‐facial region and disfigurement.4 The main treatments for OSCC are surgery, radiotherapy alone, or in combination with chemotherapy.5, 6, 7 However, despite extensive research on treatments for oral cancers, overall survival for patients with OSCC has not improved.8 Therefore, there is a need for both new therapies and biomarkers for OSCC progression.
Src is a non‐receptor protein tyrosine kinase,9, 10 whose overexpression and hyperactivity are associated with tumour mass, metastasis, recurrence, angiogenesis and survival of patients with cancer.14, 15, 16 Of further note, studies have previously demonstrated that Src is overexpressed in OSCC, including tongue cancer, and there is a significant association of Src overexpression with progression, recurrence and prognosis of OSCC.11, 17, 18 These findings strongly suggest Src inhibition as a targeted therapy for solid cancers, including tongue cancer.
Dasatinib (BMS‐354825) is an inhibitor of Bcr‐Abl and Src kinases that has been shown to be an effective treatment of chronic myeloid leukaemia (CML) and Philadelphia chromosome–positive acute lymphoblastic leukaemia.19, 20 Moreover, dasatinib has anti‐proliferative, anti‐invasive, anti‐angiogenic and pro‐apoptotic properties in solid tumours.21, 22, 23 A wealth of information strongly supports that dasatinib is a multi‐targeted tyrosine kinase inhibitor. Dasatinib inhibits the activity of Bcr‐Abl and Src family kinases along with additional receptor/non‐receptor tyrosine kinases and signalling protein kinases, including c‐KIT, platelet‐derived growth factor receptor (PDGFR), epidermal growth factor receptor (EGFR), the ephrin‐A receptor, TEC family kinases and ERK‐1/2.24, 25, 26 These results strongly indicate that dasatinib's anti‐tumour effects on haematological malignancies and solid cancers are largely associated with its multi‐targeted tyrosine kinase inhibition.
At present, the anti‐cancer effect and mechanism of action of dasatinib in human OSCC are not known. In this study, we investigated the inhibitory effects of dasatinib on the growth of four different human oral cancer cell lines (YD‐8, YD‐10B, YD‐38 and HSC‐3) that originated from the tongue (YD‐8, YD‐10B, HSC‐3) or lower gingiva (YD‐38).27, 28 YD‐8 and YD‐38 cells are non‐tumorigenic, whereas YD‐10B and HSC‐3 cells are highly tumorigenic.28, 29 Here, we demonstrate that dasatinib has strong anti‐growth, anti‐angiogenic and pro‐apoptotic effects on YD‐38 and HSC‐3 cells, and these effects are mediated through modulation of the expression and phosphorylation of multiple targets, including Src, EGFR, STAT‐3, STAT‐5, PKB, ERK‐1/2, S6, eIF‐2α, GRP78, caspase‐9/3, Mcl‐1 and HIF‐1α.
2. MATERIAL AND METHODS
2.1. Chemicals and antibodies
Dasatinib was purchased from LC Laboratories. Roswell Park Memorial Institute (RPMI)‐1640 medium, Dulbecco's modified Eagle's medium (DMEM), foetal bovine serum (FBS), penicillin‐streptomycin and phosphate‐buffered saline (PBS) were bought from WelGENE. 3‐(4,5‐dimethylthiazol‐2‐y)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium (MTS) reagent was from Promega. Bradford reagent was from Bio‐Rad. Protease inhibitor cocktail (PIC, 100×) and z‐VAD‐fmk (627610) were purchased from Calbiochem. Control siRNA (sc‐37007) and Src (sc‐29228) were obtained from Santa Cruz Biotechnology. Enzyme‐linked chemiluminescence (ECL) Western detection reagents were bought from Thermo Scientific. PCR primers were purchased from Bioneer. LY294002 and PD98059 were from Biomol Research Lab. Cell culture plastic wares were purchased from SPL Life Sciences. A detailed list of antibodies used in this study is included in Supplementary Table S1.
2.2. Cell culture
Different OSCC (YD‐8, YD‐38 and YD‐10B) cells were procured from the Korean Cell Line Bank. HSC‐3, another human OSCC cell, was bought from American Type Culture Collection. These cells were grown in RPMI‐1640 (YD‐8, YD‐38, and YD‐10B) or DMEM (HSC‐3) supplemented with 10% heat‐inactivated FBS, 100 U/mL penicillin and 100 µg/mL streptomycin at 37ºC in a humidified atmosphere with 5% CO2.
2.3. Cell viability and survival assay
For cell viability assay, cells (1 × 104 cells/mL/100 μL/well) were seeded in a 96‐well plate. After overnight incubation, cells were treated with dasatinib (0, 0.1, 1, and 10 µM) for 24 hours. Cells were then washed twice with PBS, and viability was determined using MTS reagent according to the manufacturer's protocol. Briefly, eight µl of the culture media (RMPI‐1640 or DMEM) and 20 µL of MTS solution was added to each well, and the plate was incubated at 37℃ for 1 hour. The absorbance of each well was measured at 490 nm using a microplate reader (SPECTRA max 340PC; Molecular Devices, LLC). For cell survival, cells (2 × 105 cells/ml/500 μL/well) were seeded in a 24‐well plate overnight. Cells were treated with dasatinib (0, 0.1, 1, and 10 µM) for 24 hours. Cells were then washed twice with PBS. The number of survived cells, which cannot be stained with trypan blue dye, was counted using a phase‐contrast microscope. The cell count assay was performed in triplicate. Data are mean ± standard error (SE) of three independent experiments. Survival is expressed as a percentage of control.
2.4. DNA fragmentation assay
DNA fragmentation was performed as mentioned in our previous study.30 Genomic DNA was extracted and analysed via electrophoresis at 100 V on a 1.7% agarose gel for 20 minutes. The DNA was visualized and photographed under UV illumination after staining with ethidium bromide (0.1 µg/mL) using a gel documentation system (Gel Doc‐XR, Bio‐Rad Laboratories, Inc.).
2.5. Western blot analysis
After treatments, cells were lysed in a modified radioimmunoprecipitation assay buffer (Sigma‐Aldrich; Merck) containing PIC (1×). Proteins (50 µg) were separated by sodium dodecyl sulphate‐polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes (Millipore). The membranes were blocked with 5% (w/v) skim milk in Tris‐buffered saline (TBS) containing 0.1% Tween 20 (TBST) for 2 hours. The membranes were incubated with specific antibodies listed in Supplementary Table 1 at 4 ºC overnight. The membranes were then washed twice with TBST and exposed to secondary antibodies conjugated to horseradish peroxidase (HRP) for 2 hours at room temperature, and immunoreactivity was detected by ECL reagents. β‐actin was used as an equal protein loading control.
2.6. Reverse transcription‐polymerase chain reaction (RT‐PCR)
After treatments, total cellular RNA was isolated using TRIzol reagent (Life Technologies) according to the manufacturer's protocol. Complementary DNA was then prepared using M‐MLV reverse transcriptase (Gibco‐BRL) according to the manufacturer's protocol. The primer sequences used in this study are listed in Table S2. The PCR conditions applied herein were as follows: Mcl‐1, 25 cycles of denaturation at 95℃ for 45 seconds, annealing at 56℃ for 45 seconds and extension at 72℃ for 45 seconds; β‐actin, HIF‐1α and HIF‐1β, 25 cycles of denaturation at 95℃ for 30 seconds, annealing at 56℃ for 30 seconds and extension at 72℃ for 30 seconds. The amplified products were separated by electrophoresis on a 1.2% agarose gel and detected using a gel documentation system (Gel Doc‐XR, Bio‐Rad Laboratories, Inc.).
2.7. siRNA transfection
Small interfering RNA (siRNA) of Src (sc‐29228) and control siRNA (sc‐37007) were obtained from Santa Cruz Biotechnology. YD‐38 cells were transfected with 100 pM of control or Src siRNA using Lipofectamine RNAiMAX (Invitrogen) for 48 hours. The cells were then lysed, and protein expressions were analysed by Western blotting.
2.8. Measurement of HIF‐1α protein stability
YD‐38 cells were initially grown for 8 hours in culture media (without dasatinib) to induce high expression levels of HIF‐1α protein. YD‐38 cells were then treated without or with dasatinib (10 μM), PP1 (25 μM), LY294002 (25 μM) or PD98059 (50 μM) for 5, 10, 15 and 30 minutes in the absence or presence of cycloheximide (CHX), a translation inhibitor. At each time point, whole‐cell lysates were prepared and analysed by Western blotting to measure the amounts of HIF‐1α and HIF‐1β proteins remained in the cells.
2.9. Measurement of HUVEC tube formation
YD‐38 cells were treated without or with dasatinib or phorbol 12‐myristate 13‐acetate (PMA) at the indicated doses for 24 hours. The conditioned media (CM) was then collected and applied to human umbilical vein endothelial cells (HUVEC), which were kindly provided by professor Ho‐Jung Kwon at the Department of Biotechnology in Yonsei University, cultured in Matrigel‐coated plates for an additional 16 hours. Changes in cell morphology (HUVEC tube formation) were captured using an inverted microscope (Nikon Eclipse TS200; Nikon Corp.).
2.10. Statistical analyses
Results are expressed as mean ± standard error (SE) of three independent experiments. The significance of the difference between sample means was determined by one‐way ANOVA, followed by Dunnett's post hoc test, using SPSS 11.5 software (SPSS, Inc.). The difference was considered statistically significant for the value of P < .05.
3. RESULTS
3.1. Dasatinib inhibits growth and induces apoptosis of YD‐38 human oral cancer cells
We initially investigated the effect of 0.1, 1 or 10 µM of dasatinib (Figure 1A) for 24 hours on the viability, survival and apoptosis of YD‐8, YD‐10B and YD‐38 cell lines. Dasatinib induced a concentration‐dependent decrease in the viability of all cell lines tested by MTS assay, with YD‐38 cells exhibiting the highest degree of sensitivity followed by YD‐10B and YD‐8 cells (Figure 1B). By contrast, using trypan blue dye–based cell count analysis, YD‐8 cells were the most sensitive to dasatinib, followed by YD‐38 and YD‐10B cells (Figure 1C). Strikingly, dasatinib only induced a concentration‐dependent accumulation of fragmented DNA in YD‐38 cells (Figure 1D). There was little or no DNA fragmentation in YD‐8 or YD‐10B cells treated with 10 µM of dasatinib for up to 8 hours.
FIGURE 1.
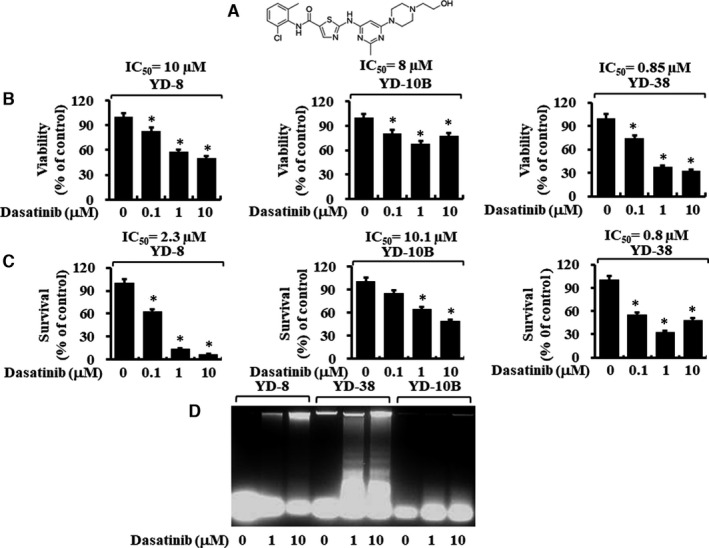
Effects of dasatinib on cell viability, cell survival and apoptosis (DNA fragmentation) on three different human oral cancer cells. A, The chemical structure of dasatinib. B‐C, YD‐8, YD‐38 and YD‐10B cells were treated with dasatinib (0, 0.1, 1 and 10 μM) for 24 h, followed by measurement of cell viability by MTS assay (B) and cell survival by cell count analysis (C). Data are mean ±SE of three independent experiments. *P < .05 compared with the values of control (no dasatinib). D, YD‐8, YD‐38 and YD‐10B cells were treated with dasatinib (0, 1 and 10 μM) for 8 h. Extra‐nuclear fragmented DNA was extracted and analysed on a 1.7% agarose gel
3.2. Dasatinib treatment at 10 µM inhibits the phosphorylation of Src, EGFR, STAT‐3, STAT‐5, PKB and ERK‐1/2 in YD‐38 human oral cancer cells
Given that dasatinib is a multi‐targeted protein kinase inhibitor, we next investigated the treatment effects of dasatinib at 10 µM on the expression and phosphorylation levels of its target Src and its related receptor kinases, signalling kinases and transcription. There were high phosphorylation levels of Src, EGFR, PKB, ERK‐1/2, STAT‐3 and STAT‐5 in YD‐38 cells in the absence of dasatinib at all time points tested (Figure 2A). Dasatinib treatment by 2 hours led to almost complete loss of the phosphorylated Src in YD‐38 cells, supporting the drug's efficacy. Furthermore, dasatinib treatment (10 µM) strongly reduced levels of the phosphorylated EGFR, STAT‐3, STAT‐5, PKB and ERK‐1/2 in YD‐38 cells at all time points tested. Expression levels of total Src, EGFR, STAT‐3, STAT‐5, PKB and ERK‐1/2 remained unchanged under these experimental conditions. We next sought to explore the role of EGFR, STAT‐3/5, PKB and ERK‐1/2 in the growth of YD‐38 cells using each of the selective pharmacological inhibitors: erlotinib (an EGFR inhibitor), AG490 (a JAK/STAT inhibitor), LY294002 (a PI3K/PKB inhibitor) and PD98059 (a MEK 2 and ERK‐1/2 inhibitor). As shown in Figure 2B‐E, individual treatments of erlotinib, AG490, LY294002 and PD98059 all led to a substantial decrease in YD‐38 cell survival.
FIGURE 2.
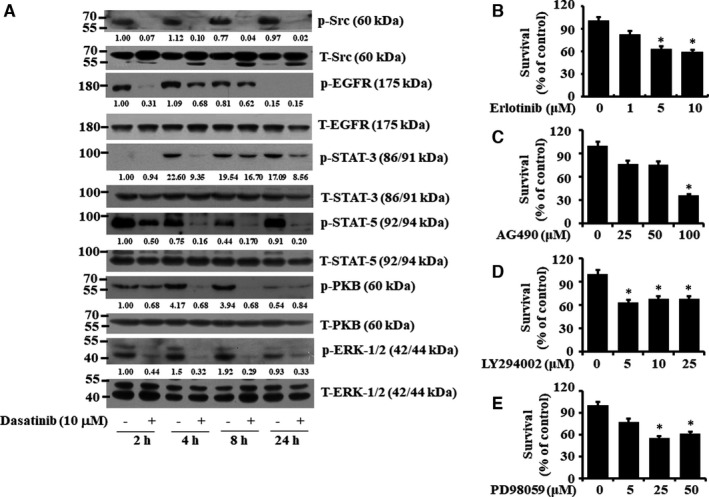
Effects of dasatinib, erlotinib, AG490, LY294002 or PD98059 on expression and phosphorylation of Src, EGFR, STAT‐3, STAT‐5, PKB and ERK‐1/2 in YD‐38 cells and cell survival. A, YD‐38 cells were treated with dasatinib (10 μM) for the indicated times. At each time point, whole‐cell lysates were prepared and analysed by Western blotting. Each picture is representative of three independent experiments. B‐E, YD‐38 cells were treated for 24 h with the indicated doses of erlotinib (B), AG490 (C), LY294002 (D) and PD98059 (E), followed by measurement of cell survival by cell count analysis. Data are mean ±SE of three independent experiments. *P < .05 compared with the values of control (no drug)
3.3. Knockdown of Src causes a strong inhibition of the phosphorylation of EGFR, STAT‐5, PKB and ERK‐1/2 in YD‐38 human oral cancer cells
We hypothesized that dasatinib‐mediated decline of the phosphorylation levels of EGFR, STAT‐3, STAT‐5, PKB and ERK‐1/2 in YD‐38 cells might be mediated through its inhibition of Src. To test this, we transfected YD‐38 cells with control or Src siRNA for 48 hours and analysed the expression and phosphorylation levels of EGFR, STAT‐3, STAT‐5, PKB and ERK‐1/2 in control or Src siRNA–transfected cells by Western blotting. High expression and phosphorylation levels of Src, EGFR, STAT‐3, STAT‐5, PKB and ERK‐1/2 were observed in control siRNA‐transfected YD‐38 cells (Figure 3). By contrast, in YD‐38 cells transfected with Src siRNA, there was little or no expression of total or phosphorylated, supporting a high Src siRNA knockdown efficiency. Strikingly, compared with control siRNA–transfected cells, there was also a marked reduction of the phosphorylation levels of EGFR, PKB, ERK‐1/2 and STAT‐5, but not STAT‐3, in these Src siRNA–transfected YD‐38 cells. Total expression levels of EGFR, STAT‐3, STAT‐5, PKB and ERK‐1/2 remained unchanged under these experimental conditions.
FIGURE 3.
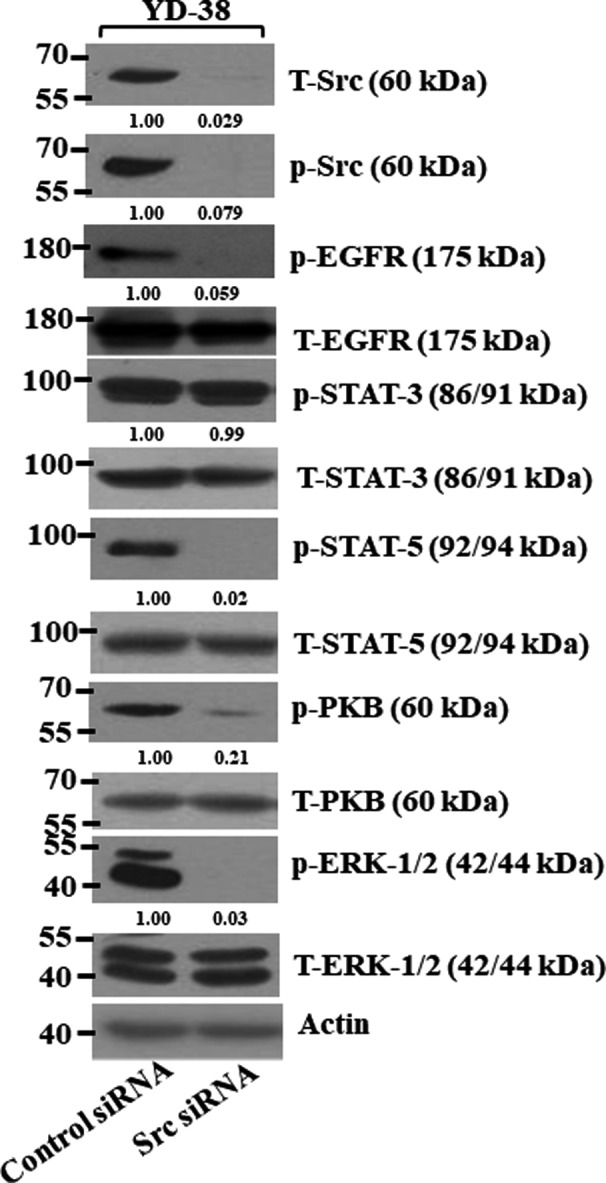
Effects of Src knockdown on expression and phosphorylation of Src, EGFR, STAT‐3, STAT‐5, PKB and ERK‐1/2 in YD‐38 cells. YD‐38 cells were transfected with 100 pM of control or Src siRNA for 48 h. Whole‐cell lysates were prepared and analysed by Western blotting. Each picture is representative of three independent experiments
3.4. Dasatinib induces apoptosis via activation of the intrinsic caspase pathway in YD‐38 human oral cancer cells
Activation of caspases is a principal feature for the execution of cell death by various apoptotic stimuli. Dasatinib treatment led to a time‐dependent increase in cleavage of caspase‐9 and its target PARP in YD‐38 cells (Figure 4A). To see whether dasatinib treatment modulates the extrinsic apoptotic pathway, we next measured the expression levels of death receptor‐5 (DR‐5). Dasatinib treatment did not increase the expression levels of DR‐5 in the cells; rather, a slightly decreased protein expression was observed. In further support of dasatinib actions via the intrinsic pathway, z‐VAD‐fmk, a pan‐caspase inhibitor, greatly attenuated the DNA fragmentation induced by dasatinib in YD‐38 cells (Figure 4B).
FIGURE 4.
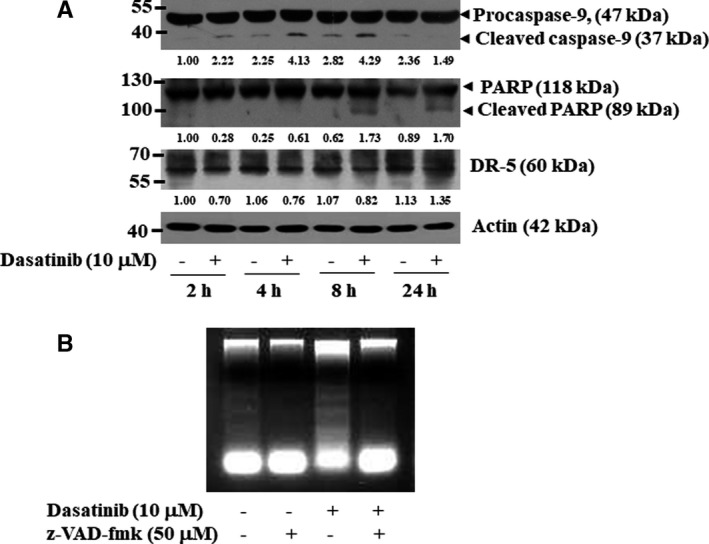
Effects of dasatinib and/or z‐VAD‐fmk on the expression of procaspase‐9, DR‐5 and PARP and DNA fragmentation in YD‐38 cells. A, YD‐38 cells were treated without or with dasatinib (10 μM) for the indicated times. At each time point, whole‐cell lysates were prepared and analysed by Western blotting. B, YD‐38 cells were pre‐treated for 1 h with a pan‐caspase inhibitor z‐VAD‐fmk (50 μM) and then treated with or without dasatinib (10 μM) in the absence or presence of z‐VAD‐fmk for an additional 8 h. The genomic DNA was extracted and analysed on 1.7% agarose gels for DNA fragmentation. Each picture is representative of three independent experiments
3.5. Dasatinib alters expression and phosphorylation levels of Mcl‐2, GRP78, eIF‐2α and S6 in YD‐38 human oral cancer cells
Given that cancer cell growth or apoptosis is influenced by the expression levels of anti‐apoptotic proteins, such as the family of Bcl‐2 and IAPs,31, 32 we next investigated whether dasatinib affects the expression levels of Bcl‐2, Mcl‐1 and XIAP in YD‐38 cells. High expression levels of Bcl‐2, Mcl‐1 and XIAP proteins were observed in untreated YD‐38 cells (Figure 5A). Dasatinib treatment led to a time‐dependent reduction of Mcl‐1 expression but did not influence the expression of Bcl‐2 and XIAP in YD‐38 cells for the times applied. RT‐PCR analysis further revealed that dasatinib reduced mRNA expression levels of Mcl‐1 (Figure 5B), suggesting Mcl‐1 transcriptional down‐regulation by dasatinib. ER stress also mediated cancer cell growth or death.33 GRP78 and eIF‐2α are known as ER stress markers.34 There was a large increase in levels of GRP78 in YD‐38 cells treated with dasatinib for 2, 4 and 8 hours. Furthermore, a time‐dependent increase in the phosphorylation levels of eIF‐2α occurred in YD‐38 cells treated with dasatinib. A ribosomal protein S6 is a component of the 40S ribosomal small subunit and is involved in translation.35 Notably, as further shown in Figure 5A, while dasatinib treatment for 2 hours led to reduced phosphorylation but not total levels of S6, the drug treatment for 4 to 24 hours resulted in reduced phosphorylation and total levels of S6 in YD‐38 cells.
FIGURE 5.
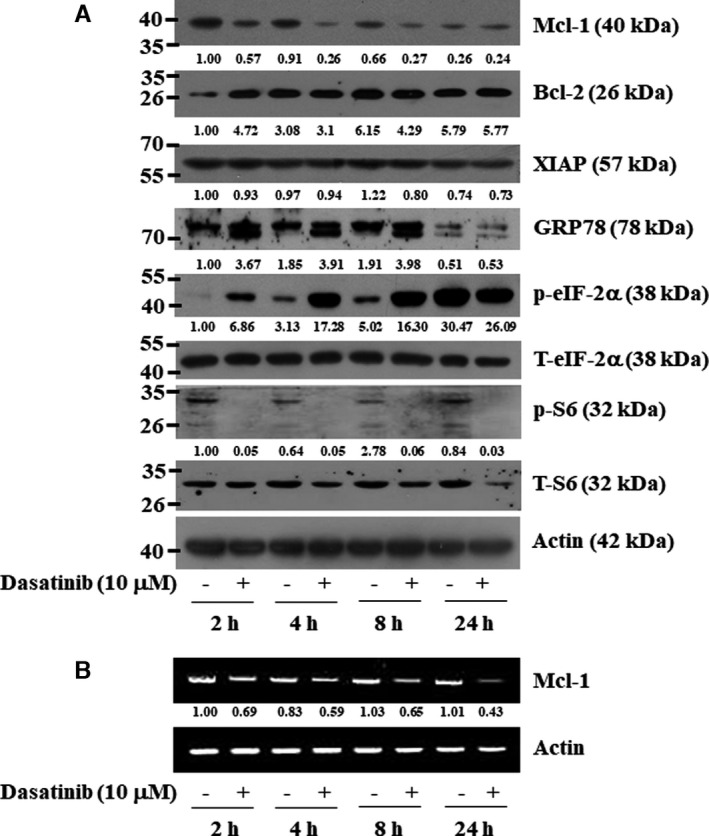
Effects of dasatinib on expression and phosphorylation of Mcl‐1, Bcl‐2, XIAP, GRP78, eIF‐2α and S6 in YD‐38 cells. A, YD‐38 cells were treated with dasatinib (10 μM) for the indicated times. At each time point, whole‐cell lysates were prepared and analysed by Western blotting. Each picture is representative of three independent experiments. B, YD‐38 cells were treated with dasatinib (10 μM) for the indicated times. At each time point, total cellular RNA was prepared and analysed by RT‐PCR analysis. Each picture is representative of three independent experiments
3.6. Dasatinib reduces HIF‐1α at the protein levels in YD‐38 human oral cancer cells
HIF‐1α is an angiogenic transcription factor.36 The HIF‐1α expression is linked to the tumour promotion in human oral cancer37 and correlates with the growth and adhesion in human OSCC cells.38, 39 In this study, we further determined whether HIF‐1α protein is expressed in YD‐38 cells and whether dasatinib modulates it. Of interest, HIF‐1α was highly expressed in YD‐38 and YD‐10B cells, but there was a low expression of HIF‐1α in YD‐8 cells (Figure 6A). Notably, dasatinib treatment at 1 or 10 µM resulted in almost complete down‐regulation of HIF‐1α protein in YD‐38 and YD‐8 cells but had no effect on the expression of HIF‐1α protein in YD‐10B cells. Furthermore, dasatinib treatment at 10 μM greatly down‐regulated HIF‐1α at the protein, but not mRNA, levels in YD‐38 cells at times tested, suggesting post‐transcriptional regulation of HIF‐1α by dasatinib (Figure 6B,C).
FIGURE 6.
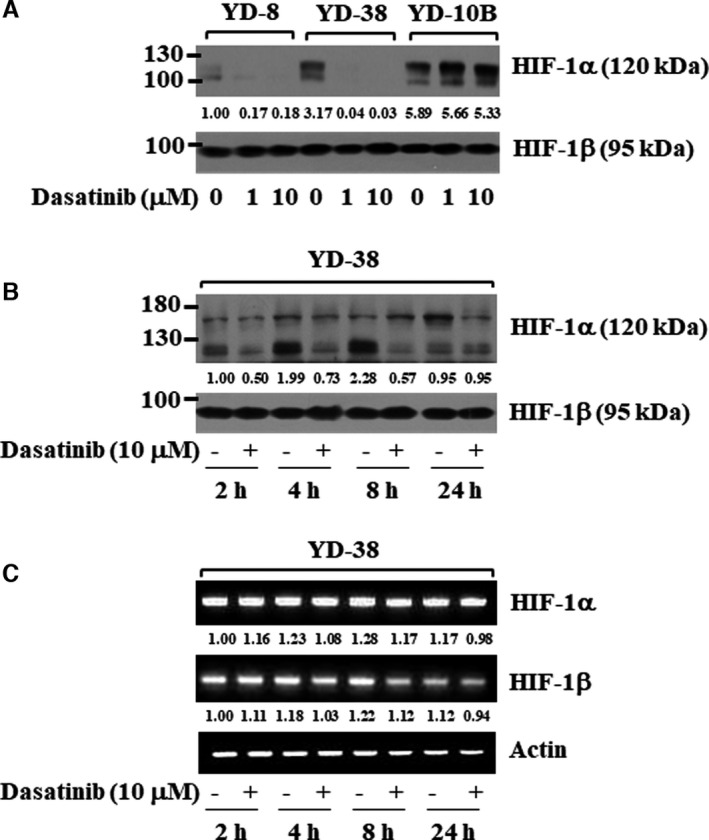
Effects of dasatinib on the expression of HIF‐1α in YD‐8, YD‐10B and YD‐38 cells. A, YD‐8, YD‐38 or YD‐10B cells were treated with dasatinib at the indicated concentrations for 8 h. Whole‐cell lysates were prepared and analysed by Western blotting. B‐C, YD‐38 cells were treated with dasatinib (10 μM) for the indicated times. At each time point, whole‐cell lysates and total cellular RNA were prepared and analysed by Western blotting (B) and RT‐PCR analysis (C), respectively
3.7. Dasatinib induces Src‐, PI3K/PKB‐ and ERK‐1/2‐dependent HIF‐1α destabilization in YD‐38 human oral cancer cells
We next determined whether dasatinib affects the stability of HIF‐1α protein in YD‐38 cells using a CHX‐based protein stability assay. There was a gradual time‐dependent decrease in the amounts of HIF‐1α protein in YD‐38 cells grown in the presence of CHX at times tested (as shown in Figure 7A), suggesting that if the translation is blocked, HIF‐1α protein is intrinsically unstable in these cells. Dasatinib treatment caused a rapid destabilization (or degradation) of HIF‐1α protein in YD‐38 cells at all the time points tested. Given that Src, PI3K/PKB and ERK‐1/2 regulate HIF‐1α protein stability (stabilization),40, 41 and dasatinib inhibits Src, PKB and ERK‐1/2 in YD‐38 cells, we investigated the effects of PP1 (a Src inhibitor), LY294002 (a PI3K/PKB inhibitor) and PD98059 (an ERK‐1/2 inhibitor) on HIF‐1α protein stability in YD‐38 cells. Treatment with PP1 (Figure 7B), LY294002 (Figure 7C) and PD98059 (Figure 7D) decreased HIF‐1α protein in YD‐38 cells. Expression levels of control HIF‐1β and total Src, PKB and ERK‐1/2 proteins remained constant under these experimental conditions.
FIGURE 7.
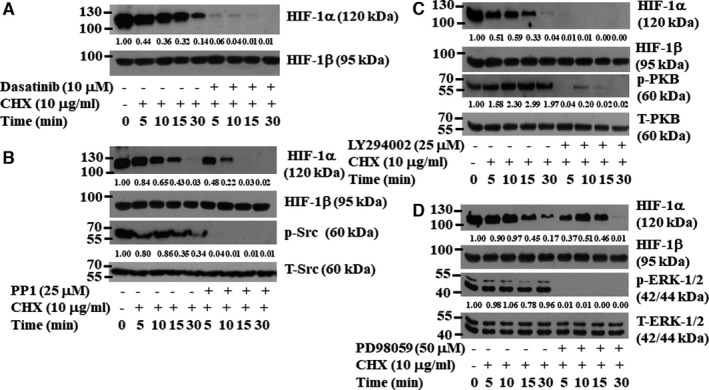
Effects of dasatinib, PP1, LY29402, or PD98059 on HIF‐1α protein stability in YD‐38 cells. A, YD‐38 cells were initially grown for 8 h in culture media to induce high HIF‐1α protein. YD‐38 cells were then treated without or with dasatinib (10 μM) for 5, 10, 15, and 30 min in the absence or presence of CHX, a translation inhibitor. At each time point, whole‐cell lysates were prepared and analysed by Western blotting. (B‐D) YD‐38 cells were initially grown for 8 h in culture media to induce high levels of HIF‐1α protein. YD‐38 cells were then treated without or with PP1 (25 μM) (B), LY294002 (25 μM) (C), or PD98059 (50 μM) (D) for 5, 10, 15, and 30 min in the absence or presence of CHX. At each time point, whole‐cell lysates were prepared and analysed by Western blotting
3.8. Dasatinib blocks tube formation of human umbilical vein endothelial cells
We next investigated the effect of dasatinib on angiogenesis in vitro by measuring the tube formation of HUVEC exposed to the conditioned medium (CM) of YD‐38 cells treated without or with dasatinib. The tube formation of HUVEC was reduced in a dasatinib concentration‐dependent manner when treated with CM from dasatinib‐treated YD‐38 cells compared with control CM (Figure 8A). Moreover, there were substantial levels of HIF‐1α protein in HUVEC cultured with the CM from control YD‐38 cells, but there was no HIF‐1α protein in HUVEC cultured with the CM from dasatinib (10 µM)‐treated YD‐38 cells (Figure 8B). Expression levels of HIF‐1β and actin remained unchanged under these experimental conditions. We next determined whether dasatinib‐mediated inhibition of the tube formation in HUVEC is due to the drug's cytotoxicity. There was no significant change in the viability and survival of HUVEC treated for 24 hours without or with dasatinib at 10 µM, ruling out the possibility of the drug's cytotoxicity (Figure 8C,D). PMA is a known inducer of HUVEC tube formation.42 Treatment with PMA at 10 nM induced tube formation of HUVEC, which was also greatly attenuated by dasatinib treatment (Figure 8E).
FIGURE 8.
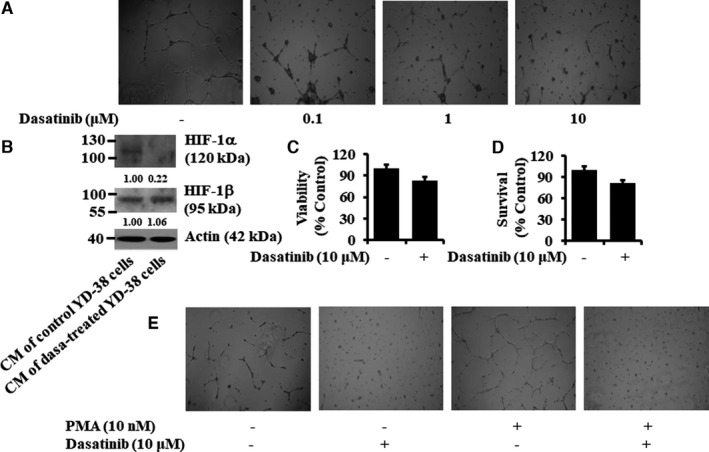
Effects of conditioned media (CM) from control‐ or dasatinib‐treated YD‐38 cells on tube formation, viability, survival, and HIF‐1α protein expression of HUVEC. (A‐B) YD‐38 cells were treated without or with dasatinib at the indicated doses for 24 h. The conditioned media (CM) was then collected and applied to HUVEC cultured in Matrigel‐coated plates for an additional 16 h. Changes in cell morphology (HUVEC tube formation) were captured using an inverted microscope (A). Whole‐cell lysates were prepared and analysed by Western blotting (B). (C‐D) HUVECs were treated without or with dasatinib (10 μM) for 16 h, followed by measurement of cell viability and cell survival by MTS (C) and cell count (D) assays, respectively. Data are the means ±SE of three independent experiments. (E) YD‐38 cells were treated without or with PMA (10 nM), a known inducer of HUVEC tube formation, in the absence or presence of dasatinib (10 µM) for 8 h. The conditioned media (CM) was then collected and applied to HUVEC cultured in Matrigel‐coated plates for an additional 16 h. Changes in cell morphology (HUVEC tube formation) were captured using an inverted microscope
3.9. Dasatinib also has anti‐proliferative, anti‐survival and pro‐apoptotic effects on tumorigenic HSC‐3 cells
To see if dasatinib's anti‐proliferative, anti‐survival and pro‐apoptotic effects are limited to non‐tumorigenic YD‐38 cells, the effect of dasatinib on proliferation, survival and apoptosis of tumorigenic HSC‐3 cells was further investigated. Like YD‐38 cells, dasatinib treatment for 24 hours led to a concentration‐dependent reduction of HSC‐3 cell proliferation (Figure 9A) and survival (Figure 9B). Dasatinib treatment for 8 hours also led to a concentration‐dependent progressive accumulation of fragmented DNA in HSC‐3 cells (Figure 9C). Moreover, dasatinib treatment for 24 hours resulted in a dose‐dependent reduction of the phosphorylation levels of Src, PKB and ERK‐1/2 (Figure 9D). In addition, there was a concentration‐dependent decrease in levels of procaspase‐3, PARP and HIF‐1α in HSC‐3 cells treated with dasatinib for 8 hours. Results of time‐course experiments further revealed the ability of dasatinib (10 μM) to greatly reduce the phosphorylation levels of Src, EGFR, JAK2, STAT‐5, ERK‐1/2 and HIF‐1α in HSC‐3 cells for the time points tested (Figure 9E). Expression of control actin or total Src, EGFR, STAT‐5 and ERK‐2 proteins was not largely changed under these experimental conditions.
FIGURE 9.
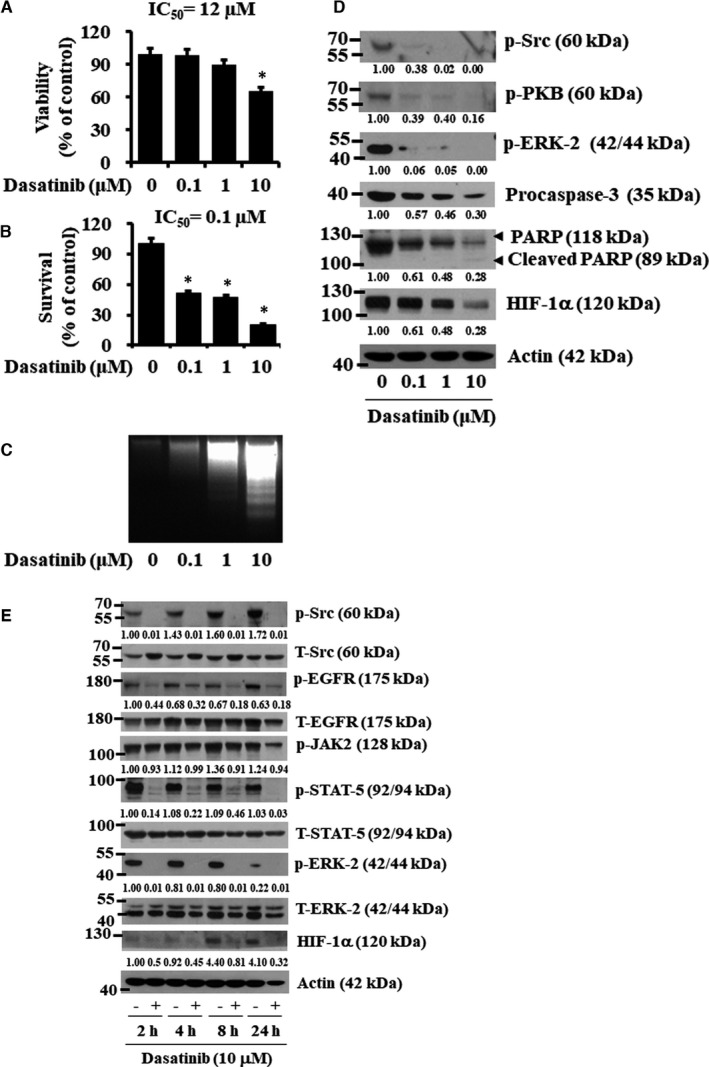
Effects of dasatinib on viability, survival, apoptosis, and expression and phosphorylation of cellular proteins in HSC‐3 cells. (A‐B) HSC‐3 cells were treated for 24 h with the indicated concentration of dasatinib, followed by measurement of cell viability and cell survival by MTS (A) and cell count (B) assays, respectively. Data are mean ±SE of three independent experiments. *P < .05 compared with the values of control (no drug). (C) HSC‐3 cells were treated with dasatinib at the indicated doses for 8 h. Extra‐nuclear fragmented DNA was extracted, and analysed on a 1.7% agarose gel. (D‐E) HSC‐3 cells were treated with dasatinib at the designated doses for 24 h (D) or at the indicated times (E). At the designated time point, whole‐cell lysates were prepared and analysed by Western blotting. Each picture is representative of three independent experiments
4. DISCUSSION
The anti‐cancer effect and the mode of action of dasatinib in human oral cancer are poorly understood. Here, we report that dasatinib has anti‐growth, anti‐angiogenic and pro‐apoptotic effects on both YD‐38 (non‐tumorigenic) and HSC‐3 (tumorigenic) human oral cancer cells, and these effects are mediated through control of the expression and phosphorylation of multiple targets including Src, EGFR, STAT‐3, STAT‐5, PKB, ERK‐1/2, S6, eIF‐2α, GRP78, caspase‐9/3, Mcl‐1 and HIF‐1α.
Dasatinib exerts anti‐tumour effects on blood malignancies and solid cancers by inhibiting cell proliferation/survival and inducing cell apoptosis.19, 20, 21, 22, 43, 44 It had been previously shown that dasatinib at 10 µM inhibits the viability and induces the apoptosis of HSC‐3 cells.45 Extending on this, we demonstrated that dasatinib at 10 µM markedly inhibits the proliferation and survival of YD‐8, YD‐10B, HSC‐3 and YD‐38 cells; the most sensitive being YD‐38 cells. Dasatinib's growth‐suppressive and apoptosis‐inducing effects are therefore not limited to HSC‐3 cells.
Dasatinib is an inhibitor of the non‐receptor tyrosine kinase Src.19, 20, 46 Src is highly expressed and phosphorylated (activated) in many solid tumours.11, 12, 13 High levels of phosphorylated Src are detected in most tongue cancer biopsies of human tongue cancer patients, and the outcome in patients with tongue cancer inversely correlates with Src hyperphosphorylation,17, 18 highlighting the prognostic role of Src overexpression/hyperactivation in tongue cancer. In this study, YD‐38 (and HSC‐3) cells express high levels of phosphorylated Src, but dasatinib at 10 µM almost completely down‐regulates them in these cells, supporting the drug's efficacy. Given that Src overexpression/hyperactivation contributes to cancer cell proliferation and survival,14, 15, 16 we further sought to explore the role of Src hyperphosphorylation in YD‐38 cells. Pharmacological inhibition of Src by PP1 (a selective inhibitor of Src) causes a substantial decrease in YD‐38 cell survival. These results confirm that Src is a survival factor in YD‐38 cells, and thus, dasatinib's anti‐growth effect on YD‐38 (and HSC‐3) is in part mediated through Src inhibition.
In agreement with previous studies,45 we found herein that dasatinib at 10 µM largely inhibits the phosphorylation of EGFR, PKB and ERK‐1/2 in both YD‐38 and HSC‐3 cells, further advocating dasatinib's multi‐targeted tyrosine kinase inhibition in both non‐tumorigenic and tumorigenic human oral cancer cells. Moreover, we demonstrated that respective pharmacological inhibition of EGFR, PI3K/PKB and ERK‐1/2 causes a substantial reduction of YD‐38 cell viability and survival. These results collectively suggest that EGFR, PI3K/PKB and ERK‐1/2 also act as survival factors in YD‐38 cells, and thus, dasatinib's anti‐growth effect on YD‐38 (and HSC‐3) cells is associated with inhibition of EGFR, PI3K/PKB and ERK‐1/2 signalling pathways.
STAT proteins including STAT‐3 and STAT‐5 are transcription factors involved in cancer cell proliferation and survival by regulating the transcription of cell cycle–related, survival‐related and apoptosis‐related genes.47 Notably, dasatinib inhibits STAT‐5 signalling associated with apoptosis in K562 CML cells,48 and it inhibits Src and STAT‐5 phosphorylation along with apoptosis induction in acute lymphoblastic leukaemia cells.49 These findings address that dasatinib‐mediated Src and STAT‐5 inhibition is crucial for the induction of apoptosis in these leukaemia cells. These findings address that dasatinib‐mediated Src and STAT‐5 inhibition is crucial for the induction of apoptosis in these leukaemia cells. The ability of dasatinib to regulate STAT proteins in human oral cancer cells is not known. Of interest, in the current study, dasatinib rapidly inhibits the phosphorylation of STAT‐3 and STAT‐5 without influencing their total protein expression in YD‐38 cells. Given that pharmacological inhibition of STAT‐3 and STAT‐5 by AG490 (a JAK/STAT inhibitor) results in a substantial reduction of YD‐38 cell survival, it is likely that STAT‐3 and STAT‐5 also facilitate the survival of YD‐38 cells, and thus, inhibition of STAT‐3 and STAT‐5 further contributes to dasatinib's growth‐suppressive and/or pro‐apoptotic effects on YD‐38 cells.
Src participates in much of the crosstalk between the receptor tyrosine kinases (RTKs) and cytoplasmic protein tyrosine kinases. Truly, a wealth of information illustrates that Src regulates EGFR, PKB and ERK‐1/2 in multiple cancers.50, 51, 52, 53 At present, the crosstalk between Src and the RTKs or cytoplasmic protein tyrosine kinases in human oral cancer cells remains elusive. Profoundly, we observed that knockdown of Src causes strong and selective inhibition of the phosphorylation of EGFR, STAT‐5, PKB and ERK‐1/2 in YD‐38 cells, but it does not induce cell apoptosis (data not shown). These results suggest that Src lies upstream of EGFR, STAT‐5, PKB and ERK‐1/2 in YD‐38 cells, and thus, dasatinib's anti‐growth effect on these cells is likely to be further mediated through Src inhibition–mediated suppression of EGFR, STAT‐5, PKB and ERK‐1/2.
The induction of cell apoptosis is mostly mediated via the intrinsic (mitochondrial)‐ and extrinsic (DR)‐mediated pathways.54 It is documented that members of the Bcl‐2 family, including Bcl‐2 and Mcl‐1, regulate apoptosis and caspase activation by regulating the mitochondrial membrane integrity,55 and the decline of the expression levels of Bcl‐2 members contributes to apoptosis induced by some anti‐cancer agents.56, 57 Human IAPs, including XIAP, additionally regulate apoptosis by inhibiting caspase‐9/3.31, 58, 59 Here, dasatinib induced the apoptosis of YD‐38 cells via the intrinsic apoptotic pathways but not an extrinsic pathway, as evidenced by nuclear DNA fragmentation, PARP cleavage and activation of caspase‐9 without affecting the expression of DR‐5, Bcl‐2 and XIAP and reducing the expression of Mcl‐1. Given that pharmacological blockade of caspase activation by z‐VAD‐fmk (a pan‐caspase inhibitor) greatly abolished the ability of dasatinib to trigger the apoptosis of YD‐38 cells, it is evident that the intrinsic activation of the caspase pathway is important for the induction of apoptosis in these cells. It is also likely that this Mcl‐1 down‐regulation may further contribute to caspase activation and induction of apoptosis in YD‐38 cells in response to dasatinib.
Induction of ER stress is common to many anti‐cancer drugs and/or agents.60, 61 ER stress occurs when cells have dysfunction of protein synthesis and excessive accumulation of misfolded proteins in the ER62 and high phosphorylation levels of eIF‐2α, a translation regulatory protein.63 The phosphorylation of eIF‐2α is indicative of its inactivation and global translational inhibition.64 It is known that the phosphorylation of S6, a ribosomal protein involved in translation, is closely associated with global translational activation.35 It is worth mentioning previous reports that the regulation of ER stress is identified as a mechanism for the anti‐cancer effects of dasatinib in head and neck cancers.65, 66 Since dasatinib increased the expression of GRP78 and the phosphorylation of eIF‐2α while decreasing the phosphorylation of S6 in YD‐38 cells, induction of ER stress and global translational inhibition in YD‐38 cells may therefore contribute to the drug's growth‐suppressive and pro‐apoptotic effects.
The transcription factor HIF‐1α is involved in tumour angiogenesis by regulating the transcriptional induction of angiogenesis‐related genes, such as VEGF, that contain HRE cis‐acting elements within their promoters.34, 67HIF‐1α is currently regarded as a prime target for anti‐cancer therapies.68, 69 At present, the mechanisms underlying high HIF‐1α protein expression and dasatinib regulation of HIF‐1α expression in oral cancer cells are poorly understood. In this study, we found high HIF‐1α protein expression in all four human oral cancer cell lines. Distinctly, the present study demonstrated that dasatinib greatly down‐regulated HIF‐1α protein in YD‐8, YD‐38 and HSC‐3, but not YD‐10B cells. These results indicate that dasatinib differentially regulates HIF‐1α expression in a cell type–dependent manner. A further striking finding herein is that dasatinib down‐regulates HIF‐1α at the protein, but not mRNA level, which is due to an increase in HIF‐1α protein turnover/destabilization through inhibition of Src, PI3K/PKB and ERK‐1/2. It is thus likely that dasatinib‐induced HIF‐1α protein down‐regulation and turnover in YD‐38 cells are associated with the drug's ability to inhibit these multi‐kinases.
In summary, we demonstrate that dasatinib has strong anti‐growth, anti‐angiogenic and pro‐apoptotic effects on YD‐38 and HSC‐3 cells, and these effects are mediated through the regulation of multiple cellular targets and pathways. This work suggests that dasatinib could be a possible regimen for the treatment of human oral cancer.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Nam‐Sook Park: Investigation (equal); Methodology (equal). Yu‐Kyoung Park: Investigation (equal); Methodology (equal). Anil Kumar Yadav: Data curation (equal); Investigation (equal); Writing‐original draft (equal). Young‐Min Shin: Formal analysis (equal); Validation (equal). David Bishop Bailey: Formal analysis (equal); Validation (equal); Writing‐review & editing (equal). Jong‐Soon Choi: Formal analysis (equal); Validation (equal). Jong Wook Park: Formal analysis (equal); Validation (equal). Byeong‐Churl Jang: Conceptualization (equal); Formal analysis (equal); Supervision (equal); Validation (equal); Visualization (equal); Writing‐original draft (equal).
Supporting information
Table S1‐S2.
ACKNOWLEDGEMENTS
This study was supported by the Korea Basic Science Institute Grant to JS Choi (C180310) and in part the National Research Foundation of Korea (NRF) Grant funded by the Korea Government (MSIP) (No. 2014R1A5A2010008).
Park N‐S, Park Y‐K, Yadav AK, et al. Anti‐growth and pro‐apoptotic effects of dasatinib on human oral cancer cells through multi‐targeted mechanisms. J Cell Mol Med. 2021;25:8300–8311. 10.1111/jcmm.16782
Nam‐Sook Park and Yu‐Kyung Park contributed equally to this work.
Contributor Information
Jong‐Soon Choi, Email: jschoi@kbsi.re.kr.
Jong Wook Park, Email: jangbc123@gw.kmu.ac.kr, Email: j303nih@dsmc.or.kr, Email: jschoi@kbsi.re.kr.
Byeong‐Churl Jang, Email: jangbc123@gw.kmu.ac.kr.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Warnakulasuriya S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009;45:309‐316. [DOI] [PubMed] [Google Scholar]
- 2.Johnson DE, Burtness B, Leemans CR, et al. Head and neck squamous cell carcinoma. Nat Rev Dis Primers. 2020;6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Madani AH, Dikshit M, Bhaduri D, et al. Phase II study of a novel oral formation of 5‐fluorouracil in combination with low‐dose cisplatin as preoperative chemotherapy of oral squamous cell carcinoma. Int J Clin Pharmacol Res. 2005;25:115‐122. [PubMed] [Google Scholar]
- 4.Panarese I, Aquino G, Ronchi A, et al. Oral and Oropharyngeal squamous cell carcinoma: prognostic and predictive parameters in the etiopathogenetic route. Expert Rev Anticancer Ther. 2019;19:105‐119. [DOI] [PubMed] [Google Scholar]
- 5.Nakazawa M, Ohnishi T, Ohmae M, et al. Phase II study of a novel oral formation of 5‐fluorouracil in combination with low‐dose cisplatin as preoperative chemotherapy of oral squamous cell carcinoma. Int J Clin Pharmacol Res. 2005;25:115‐122. [PubMed] [Google Scholar]
- 6.Huang SH. Oral cancer: current role of radiotherapy and chemotherapy. Med Oral Patol Oral Cir Bucal. 2013;18:e233‐e240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bulsara VM, Worthington HV, Glenny AM, et al. Interventions for the treatment of oral and oropharyngeal cancers: surgical treatment. Cochrane Database Syst Rev. 2018;12:CD006205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim UK, Geum DH, Cheon GB, et al. The impact factors on 5‐year survival rate in patients operated with oral cancer. J Korean Assoc Oral Maxillofac Surg. 2013;39:207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636‐5642. [DOI] [PubMed] [Google Scholar]
- 10.Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4:470‐480. [DOI] [PubMed] [Google Scholar]
- 11.Hermida‐Prado F, Granda‐Díaz R, Del‐Río‐Ibisate N, et al. The differential impact of SRC expression on the prognosis of patients with head and neck squamous cell carcinoma. Cancers. 2019;11:1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lutz MP, Esser IB, Flossmann‐Kast BB, et al. Overexpression and activation of the tyrosine kinase Src in human pancreatic carcinoma. Biochem Biophys Res Commun. 1998;243:503‐508. [DOI] [PubMed] [Google Scholar]
- 13.Verbeek BS, Vroom TM, Adriaansen‐Slot SS, et al. c‐Src protein expression is increased in human breast cancer. An immunohistochemical and biochemical analysis. J Pathol. 1996;180:383‐388. [DOI] [PubMed] [Google Scholar]
- 14.Wilson GR, Cramer A, Welman A, et al. Activated c‐SRC in ductal carcinoma in situ correlates with high tumour grade, high proliferation and HER2 positivity. Br J Cancer. 2006;95:1410‐1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Florio A, Capurso G, Milione M, et al. Src family kinase activity regulates adhesion, spreading and migration of pancreatic endocrine tumour cells. Endocr Relat Cancer. 2007;14:111‐124. [DOI] [PubMed] [Google Scholar]
- 16.Leung EL, Tam IY, Tin VP, et al. Src promotes survival and invasion of lung cancers with epidermal growth factor receptor abnormalities and is a potential candidate for molecular‐targeted therapy. Mol Cancer Res. 2009;7:923‐932. [DOI] [PubMed] [Google Scholar]
- 17.Cheng SJ, Kok SH, Lee JJ, et al. Significant association of SRC protein expression with the progression, recurrence, and prognosis of oral squamous cell carcinoma in Taiwan. Head Neck. 2012;34:1340‐1345. [DOI] [PubMed] [Google Scholar]
- 18.Ben‐Izhak O, Cohen‐Kaplan V, Nagler RM. The prognostic role of phospho‐Src family kinase analysis in tongue cancer. J Cancer Res Clin Oncol. 2010;136:27‐34. [DOI] [PubMed] [Google Scholar]
- 19.Foa R, Vitale A, Vignetti M, et al. Dasatinib as first‐line treatment for adult patients with Philadelphia chromosome–positive acute lymphoblastic leukemia. Blood. 2011;118:6521‐6528. [DOI] [PubMed] [Google Scholar]
- 20.Keskin D, Sadri S, Eskazan AE. Dasatinib for the treatment of chronic myeloid leukemia: patient selection and special considerations. Drug Des Devel Ther. 2016;10:3355‐3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farshchian M, Nissinen L, Grénman R, et al. Dasatinib promotes apoptosis of cutaneous squamous carcinoma cells by regulating activation of ERK1/2. Exp Dermatol. 2017;26:89‐92. [DOI] [PubMed] [Google Scholar]
- 22.Shor AC, Keschman EA, Lee FY, et al. Dasatinib inhibits migration and invasion in diverse human sarcoma cell lines and induces apoptosis in bone sarcoma cells dependent on SRC kinase for survival. Cancer Res. 2007;67:2800‐2808. [DOI] [PubMed] [Google Scholar]
- 23.Seo S, Suh W. Antiangiogenic effect of dasatinib in murine models of oxygen‐induced retinopathy and laser‐induced choroidal neovascularization. Mol Vis. 2017;23:823‐831. [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Z, Lee FY, Bhalla KN, et al. Potent inhibition of platelet‐derived growth factor‐induced responses in vascular smooth muscle cells by BMS‐354825 (dasatinib). Mol Pharmacol. 2006;69:1527‐1533. [DOI] [PubMed] [Google Scholar]
- 25.Nautiyal J, Majumder P, Patel BB, et al. Src inhibitor dasatinib inhibits growth of breast cancer cells by modulating EGFR signaling. Cancer Lett. 2009;283:143‐151. [DOI] [PubMed] [Google Scholar]
- 26.Wu J, Liao X, Yu B, et al. Dasatinib inhibits primary melanoma cell proliferation through morphology‐dependent disruption of Src‐ERK signaling. Oncol Lett. 2013;5:527‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park NS, Park YK, Ramalingam M, et al. Meridianin C inhibits the growth of YD‐10B human tongue cancer cells through macropinocytosis and the down‐regulation of Dickkopf‐related protein‐3. J Cell Mol Med. 2018;22:5833‐5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee G, Kim HM. JNK‐associated scattered growth of YD‐10B oral squamous carcinoma cells while maintaining the epithelial phenotype. Biochem Biophys Res Commun. 2017;487:862‐867. [DOI] [PubMed] [Google Scholar]
- 29.Kim KR, Park KK, Chung WY, et al. The inhibitory effect of buddlejasaponin IV on the growth of YD‐10B human oral squamous cell carcinoma cells. J Cancer Prev. 2013;18:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yadav AK, Kumar V, Bailey DB, et al. AZD1208, a Pan‐Pim kinase inhibitor, has anti‐growth effect on 93T449 human liposarcoma cells via control of the expression and phosphorylation of Pim‐3, mTOR, 4EBP‐1, S6, STAT‐3 and AMPK. Int J Mol Sci. 2019;20:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang J, Zhao W, Tong P, et al. Comprehensive molecular characterization of inhibitors of apoptosis proteins (IAPs) for therapeutic targeting in cancer. BMC Med Genomics. 2020;13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kale J, Osterlund EJ, Andrews DW. BCL‐2 family proteins: changing partners in the dance towards death. Cell Death Differ. 2018;25:65‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7:1013‐1030. [DOI] [PubMed] [Google Scholar]
- 34.Sano R, Reed JC. ER stress‐induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460‐3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci. 2006;31:342‐348. [DOI] [PubMed] [Google Scholar]
- 36.Hashimoto T, Shibasaki F. Hypoxia‐inducible factor as an angiogenic master switch. Front Pediatr. 2015;3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brennan PA, Mackenzie N, Quintero M. Hypoxia‐inducible factor 1α in oral cancer. Oral Pathol Med. 2005;34:385‐389. [DOI] [PubMed] [Google Scholar]
- 38.Song Y, Wang W, Qu X, et al. Effects of hypoxia inducible factor‐1 alpha (HIF‐1alpha) on the growth and adhesion in tongue squamous cell carcinoma cells. Indian J Med Res. 2009;129:154‐163. [PubMed] [Google Scholar]
- 39.Jo JR, Park YK, Jang BC. Short‐term treatment with glucosamine hydrochloride specifically downregulates hypoxia‐inducible factor‐1α at the protein level in YD‐8 human tongue cancer cells. Int J Oncol. 2014;44:1699‐1706. [DOI] [PubMed] [Google Scholar]
- 40.Jang BC. The fruit juice of Morinda citrifolia (noni) downregulates HIF‐1α protein expression through inhibition of PKB, ERK‐1/2, JNK‐1 and S6 in manganese‐stimulated A549 human lung cancer cells. Int J Mol Med. 2012;29:499‐504. [DOI] [PubMed] [Google Scholar]
- 41.Jiang BH, Agani F, Passaniti A, et al. V‐SRC induces expression of hypoxia‐inducible factor 1 (HIF‐1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF‐1 in tumor progression. Cancer Res. 1997;57:5328‐5335. [PubMed] [Google Scholar]
- 42.Taylor CJ, Motamed K, Lilly B. Protein kinase C and downstream signaling pathways in a three‐dimensional model of phorbol ester‐induced angiogenesis. Angiogenesis. 2006;9:39‐51. [DOI] [PubMed] [Google Scholar]
- 43.Gencer EB, Ural AU, Avcu F, et al. A novel mechanism of dasatinib‐induced apoptosis in chronic myeloid leukemia; ceramide synthase and ceramide clearance genes. Ann Hematol. 2011;90:1265‐1275. [DOI] [PubMed] [Google Scholar]
- 44.Song Y, Sun X, Bai WL, et al. Antitumor effects of Dasatinib on laryngeal squamous cell carcinoma in vivo and in vitro. Eur Arch Otorhinolaryngol. 2013;270:1397‐1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin YC, Wu MH, Wei TT, et al. Degradation of epidermal growth factor receptor mediates dasatinib‐induced apoptosis in head and neck squamous cell carcinoma cells. Neoplasia. 2012;14:463‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lombardo LJ, Lee FY, Chen P, et al. Discovery of N‐(2‐chloro‐6‐methyl‐phenyl)‐2‐(6‐(4‐(2‐hydroxyethyl)‐piperazin‐1‐yl)‐2‐methylpyrimidin‐4‐ylamino) thiazole‐5‐carboxamide (BMS‐354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658‐6661. [DOI] [PubMed] [Google Scholar]
- 47.Loh CY, Arya A, Naema AF, et al. Signal transducer and activator of transcription (STATs) proteins in cancer and inflammation: functions and therapeutic implication. Front Oncol. 2019;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nam S, Williams A, Vultur A, et al. Dasatinib (BMS‐354825) inhibits Stat5 signaling associated with apoptosis in chronic myelogenous leukemia cells. Mol Cancer Ther. 2007;6:1400‐1405. [DOI] [PubMed] [Google Scholar]
- 49.Fei F, Stoddart S, Müschen M, et al. Development of resistance to dasatinib in Bcr/Abl‐positive acute lymphoblastic leukemia. Leukemia. 2010;24:813‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvarez RH, Kantarjian HM, Cortes JE. The role of Src in solid and hematologic malignancies: development of new‐generation Src inhibitors. Cancer. 2006;15(107):1918‐1929. [DOI] [PubMed] [Google Scholar]
- 51.Nautiyal J, Majumder P, Patel BB, et al. Src inhibitor dasatinib inhibits growth of breast cancer cells by modulating EGFR signaling. Cancer Lett. 2009;8:143‐151. [DOI] [PubMed] [Google Scholar]
- 52.Qian XL, Zhang J, Li PZ, et al. Dasatinib inhibits c‐src phosphorylation and prevents the proliferation of Triple‐Negative Breast Cancer (TNBC) cells which overexpress Syndecan‐Binding Protein (SDCBP). PLoS One. 2017;12:e0171169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nijhuis E, Lammers JW, Koenderman L, et al. Src kinases regulate PKB activation and modulate cytokine and chemoattractant‐controlled neutrophil functioning. J Leukoc Biol. 2002;71:115‐124. [PubMed] [Google Scholar]
- 54.Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Festjens N, van Gurp M, van Loo G, et al. Bcl‐2 family members as sentinels of cellular integrity and role of mitochondrial intermembrane space proteins in apoptotic cell death. Acta Haematol. 2004;111:7‐27. [DOI] [PubMed] [Google Scholar]
- 56.Jang BC, Paik JH, Jeong HY, et al. Leptomycin B‐induced apoptosis is mediated through caspase activation and down‐regulation of Mcl‐1 and XIAP expression, but not through the generation of ROS in U937 leukemia cells. Biochem Pharmacol. 2004;68:263‐274. [DOI] [PubMed] [Google Scholar]
- 57.Rao J, Xu DR, Zheng FM, et al. Curcumin reduces expression of Bcl‐2, leading to apoptosis in daunorubicin‐insensitive CD34+ acute myeloid leukemia cell lines and primary sorted CD34+ acute myeloid leukemia cells. J Transl Med. 2011;9:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abbas R, Larisch S. Targeting XIAP for promoting cancer cell death‐the story of ARTS and SMAC. Cells. 2020;9:663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shiozaki EN, Chai J, Rigotti DJ, et al. Mechanism of XIAP‐mediated inhibition of caspase‐9. Mol Cell. 2003;11:519‐527. [DOI] [PubMed] [Google Scholar]
- 60.Han M, Gao H, Xie J, et al. Hispidulin induces ER stress‐mediated apoptosis in human hepatocellular carcinoma cells in vitro and in vivo by activating AMPK signaling pathway. Acta Pharmacol Sin. 2019;40:666‐676. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 61.Su CC, Lee KI, Chen MK, et al. Cantharidin induced oral squamous cell carcinoma cell apoptosis via the JNK‐regulated mitochondria and endoplasmic reticulum stress‐related signaling pathways. PLoS One. 2016;11:e0168095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee AS. Glucose‐regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wek RC. Role of eIF2α kinases in translational control and adaptation to cellular stress. Cold Spring Harb Perspect Biol. 2018;10:a032870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DuRose JB, Scheuner D, Kaufman RJ, et al. Phosphorylation of eukaryotic translation initiation factor 2α coordinates rRNA transcription and translation inhibition during endoplasmic reticulum stress. Mol Cell Biol. 2009;29:4295‐4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lin YC, Wu MH, Wei TT, et al. Metformin sensitizes anticancer effect of dasatinib in head and neck squamous cell carcinoma cells through AMPK‐dependent ER stress. Oncotarget. 2014;5:298‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cole DW, Svider PF, Shenouda KG, et al. Targeting the unfolded protein response in head and neck and oral cavity cancers. Exp Cell Res. 2019;382: 111386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pagès G, Pouysségur J. Transcriptional regulation of the Vascular Endothelial Growth Factor gene–a concert of activating factors. Cardiovasc Res. 2005;65:564‐573. [DOI] [PubMed] [Google Scholar]
- 68.Qin J, Liu Y, Lu Y, et al. Hypoxia‐inducible factor 1 alpha promotes cancer stem cells‐like properties in human ovarian cancer cells by upregulating SIRT1 expression. Sci Rep. 2017;7:1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burroughs SK, Kaluz S, Wang D, et al. Hypoxia inducible factor pathway inhibitors as anticancer therapeutics. Future Med. Chem. 2013;5:553‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S2.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.