

Abstract
Objective
Tau hyperphosphorylation at threonine 217 (pT217) in cerebrospinal fluid (CSF) has recently been linked to early amyloidosis and could serve as a highly sensitive biomarker for Alzheimer’s disease (AD). However, it remains unclear whether other tauopathies induce pT217 modifications. To determine if pT217 modification is specific to AD, CSF pT217 was measured in AD and other tauopathies.
Methods
Using immunoprecipitation and mass spectrometry methods, we compared CSF T217 phosphorylation occupancy (pT217/T217) and amyloid‐beta (Aβ) 42/40 ratio in cognitively normal individuals and those with symptomatic AD, progressive supranuclear palsy, corticobasal syndrome, and sporadic and familial frontotemporal dementia.
Results
Individuals with AD had high CSF pT217/T217 and low Aβ42/40. In contrast, cognitively normal individuals and the majority of those with 4R tauopathies had low CSF pT217/T217 and normal Aβ 42/40. We identified a subgroup of individuals with increased CSF pT217/T217 and normal Aβ 42/40 ratio, most of whom were MAPT R406W mutation carriers. Diagnostic accuracies of CSF Aβ 42/40 and CSF pT217/T217, alone and in combination were compared. We show that CSF pT217/T217 × CSF Aβ 42/40 is a sensitive composite biomarker that can separate MAPT R406W carriers from cognitively normal individuals and those with other tauopathies.
Interpretation
MAPT R406W is a tau mutation that leads to 3R+4R tauopathy similar to AD, but without amyloid neuropathology. These findings suggest that change in CSF pT217/T217 ratio is not specific to AD and might reflect common downstream tau pathophysiology common to 3R+4R tauopathies.
Keywords: Alzheimer's Disease, Tauopathies, tau phosphorylation, mass spectrometry, cerebrospinal fluid, biomarker
Introduction
Alzheimer’s disease (AD) is characterized by the plaque deposition of amyloid‐beta 42 peptide (Aβ 42) and aggregation of hyperphosphorylated tau in neurofibrillary tangles, neuritic plaques, and neuropil threads in the brain. Concomitant decrease in Aβ 42/40 ratio and increase of phosphorylated tau (ptau) species in the cerebrospinal fluid (CSF) have been used as biomarkers for AD amyloidosis and as surrogates of AD tau neuropathology, respectively.1, 2 Increasing evidence suggests that either CSF tau phosphorylation at threonine 217 measured as absolute concentration (pT217) or as phosphorylation occupancy (pT217/T217) is a specific and more sensitive biomarker for AD, outperforming the well‐established measure of CSF ptau level at threonine 181 (pT181).3, 4, 5, 6, 7, 8 CSF pT181 level increase is strongly associated with the increase in total CSF tau concentration and is assumed to reflect tau neuropathology. However, CSF pT217 and pT217/T217 more strongly correlate with amyloid neuropathology measured by amyloid Pittsburgh compound B (PiB)‐positron emission tomography (PET) imaging than total CSF tau.3, 9, 10 Moreover, pT217 can predict the beginning of AD clinical symptoms in families with AD mutations better than pT181.3 Thus, it remains unclear if hyperphosphorylation at T217 is an early and direct consequence of amyloidosis or whether it is a downstream marker of tau pathology. In this context, it is also unclear if CSF pT217/T217 changes in primary tauopathies in the absence of amyloid pathology.
Other neurodegenerative dementing illnesses associated with tau neuropathology, including progressive supranuclear palsy (PSP), corticobasal syndrome (CBS), and behavioral variant frontotemporal dementia (bvFTD), currently have no CSF or imaging biomarkers. Diagnosis primarily depends on clinical assessment, which may be confirmed after brain autopsy. The lack of reliable in vivo biomarkers challenges accurate clinical diagnoses, with implications for the design and implementation of clinical trials.11, 12, 13 Previous studies, mostly measuring absolute CSF pT181 concentrations using immunoassays, suggested that increases in pT181 are specific to AD.14, 15, 16, 17, 18 However, global change in CSF tau isoforms concentration may contribute to pT181 absolute concentration without relative change in pT181 phosphorylation.19, 20 pT181 phosphorylation occupancy measured as pT181/T181 ratio is essential to fully interpret the changes in CSF pT181. Furthermore, some recent studies using immunoassays and mass spectrometry (MS) showed that an increase in CSF pT217 concentration is specific to AD and not observed in other neurodegenerative diseases.3, 4, 7, 8 However, previous studies often do not take into account amyloid co‐neuropathology that frequently increases with age and in many neurodegenerative diseases.
In order to evaluate the effect of tau phosphorylation in non‐AD tauopathies, including PSP, CBS, and bvFTD, we measured CSF ptau and CSF Aβ using sequential immunoprecipitation (IP) and MS. CSF pT217/T217 and pT181/T181 ratios were calculated to differentiate tau phosphorylation changes from CSF total tau variation. CSF Aβ 42/40 ratio was calculated within the same participant and used as a surrogate for amyloid neuropathology. A cohort of cognitively normal age‐matched controls (AMC) and individuals with symptomatic AD, PSP, CBS, and sporadic and familial FTD were analyzed. We assessed the correlation between CSF ptau and CSF Aβ 42/40 ratios in each disease group and evaluated diagnostic relevance of CSF ptau alone and in combination with CSF Aβ 42/40 to assess their ability to discriminate individuals with symptomatic AD from those with other neurodegenerative dementing illnesses.
Materials and Methods
Human studies
All studies were approved by the Institutional Review Board at Washington University in St. Louis, MO, USA and the Ethics Committee of the Montpellier University Hospital (CSF‐NeuroBANK #DC‐2008‐417 at the certified NFS 96‐900 CHU resource center BB‐0033‐00031 [http://www.biobanques.eu]). All participants or their delegates consented to the collection and sharing of biofluid samples. Exclusion criteria included contradiction to lumbar punctures (LPs) or lumbar catheters including a bleeding disorder, active anticoagulation, and active infection. Authorization to handle personal data was granted by the French Data Protection Authority (CNIL) under the number 1709743 v0.
AMC and individuals with mild AD were recruited at the Knight Alzheimer Disease Research Center (ADRC) at Washington University School of Medicine as part of Stable Isotope Labeling Kinetics (SILK) studies. AMC are volunteers who were enrolled for research purposes and are cognitively normal. This included two distinct cohorts of symptomatic participants (WashU‐A, and WashU‐B). Individuals from the WashU‐A cohort participated in 36hr lumbar catheter studies.21 Individuals from the WashU‐B cohort participated in the SILK study that involved five LPs over 4 months.22 Individuals were diagnosed by clinical assessment and classified according to the Clinical Dementia Rating (CDR®).23 In addition to interviews of patient and collateral source, brain PET imaging data and diagnostic CSF results were reviewed if available. This cohort includes clinical AD patients who did not have biomarkers consistent with AD, who were classified as non‐AD dementia. WashU‐A cohort was further classified with Amyloid PET positivity based on PiB imaging. Young normal controls (YNC) between the ages of 18–64 without currently diagnosed neurological disorders, were referred from Volunteers for Health at Washington University. Brain tumor patients were referred from Barnes Jewish Hospital. Patients with PSP, CBS, and sporadic bvFTD were referred from affiliated Memory Diagnostic Center and Movement Disorders Clinics. MAPT P301L and R406W mutation carriers were clinically assessed locally at Washington University and referred from the Longitudinal Evaluation of Familial Frontotemporal Dementia Study (LEFFTDS; https://www.allftd.org/artfl‐lefftds; Site PI NG). Eight participants (1 PSP, 3 MAPT R406W, 2 CBS, and 2 AMC) had repeated LPs and CSF collection.
Montpellier participants were referred from the Memory Resources and Research Center of Montpellier. They were categorized into AD, CBS, PSP, bvFTD, and CBS‐PSP continuum based on clinical, neuropsychological, brain imaging, and follow‐up assessments. CSF biomarkers of Aβ42, tau, pT181 concentrations were also measured with Enzyme‐Linked immunosorbent Assay (ELISA) and Aβ42/40 ratio was calculated.24 AD was diagnosed according to accepted criteria25, 26 and based on the ATN classification27; all AD participants had at least two abnormal CSF biomarkers. This includes AD focal phenotype which refers to predominant language, behavioral, visuospatial, apraxia phenotype with CSF biomarkers of AD. Some PPA endophenotype AD cases were included in the AD focal phenotype (n = 5). CBS and PSP were diagnosed according to international criteria.28 bvFTD may be attributed to frontotemporal lobar degeneration (FTLD)‐tau, FTLD‐TDP, and FTLD‐FUS.29, 30 Some language endophenotype FTLD were included in bvFTD (n = 3). The CBS PSP continuum included patients with CBS clinical phenotypes that evolved into PSP during follow‐up.31
CSF collection
CSF from individuals with AD and AMC in the WashU‐A cohort were collected via a catheter as previously described.21 CSF from AMC and individuals with symptomatic AD, PSP, CBS, and bvFTD in the WashU‐B cohort were obtained via LP with gravity collection and centrifugation as previously described.22 CSF from MAPT mutation families was collected according to the standardized protocol at the Biomarker Core at the Washington University School of Medicine.32 CSF from individuals with brain tumors was obtained via lumbar drain using a catheter before or after surgery.
CSF from the Montpellier cohort was collected using the standardized protocol for the collection, centrifugation, and storage at Memory Resources and Research Center of Montpellier.33, 34, 35 Briefly, the atraumatic needle was used for LP, with CSF collected into 10 mL polypropylene tube (ref 62.610.201, Sarstedt, Germany) and protein low binding Eppendorf® tubes (LoBind microtubes Eppendorf, ref 022431064, Hamburg, Germany). CSF was not centrifuged before aliquoting and storage at −80°C. CSF tau and pT181 concentrations were measured using the standardized commercially available INNOTEST sandwich ELISA X‐MAP following Fujirebio instructions. CSF Aβ 42 and Aβ 40 were measured using INNOTEST sandwich ELISA from Fujirebio.
Sequential IP and MS methods for CSF Aβ and Tau
CSF Aβ was analyzed as previously described36 with the following modifications. Master mix containing detergent and chaotropic reagents (final 1% NP‐40, 5 mmol/L guanidine, protease inhibitor cocktail) and internal standards for tau (15N labeled 2N4R recombinant tau) and Aβ (15N labeled synthetic Aβ 40, and 42) were prepared in low‐binding Axygen tubes (Fisher Scientific, Pittsburgh, PA, USA, MCT‐175). 500–1000 µL of CSF was added and immunoprecipitated with the HJ5.1 mid‐domain Aβ antibody. After washing, samples were digested with LysN protease, desalted, and analyzed by Xevo TQ‐S mass spectrometer (Waters Corporation, Milford, MA, USA).
CSF tau and ptau were analyzed as previously described9, 22 with the following modifications. Post‐HJ5.1 immunoprecipitated CSF samples containing tau internal standards were sequentially immunoprecipitated with Tau1 mid‐domain and HJ8.5 N‐terminus tau antibodies. After washing, samples were digested with trypsin, desalted, and analyzed on Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). MS method measuring pT217 and pT181 was described previously.3
Amyloid and Tau PET imaging
Amyloid PiB‐PET, AV45‐PET, and Tau AV1451‐PET imaging measurements were performed in a subset of AD and AMC participants from the Knight ADRC at Washington University School of Medicine. Data were collected and processed as previously described.22
Statistics
Receiver operating characteristic (ROC) and one‐way ANOVA analyses were performed using GraphPad Prism software (GraphPad, San Diego, CA, USA, v8.3.0.). After confirming the normal distribution of the data, one‐way ANOVA followed by post hoc analyses (Tukey test) were performed. Data were represented as mean ± SEM.
Results
Participants and study workflow
Participants’ demographics and clinical characteristics are summarized in Table 1. For the purpose of the analyses, study cohorts were divided into several groups. The “AD” group (n = 80) included patients with amnestic‐predominant clinically “typical” AD (n = 66) and those with focal variants (n = 14). bvFTD MAPT R406W mutation carriers (n = 5) were grouped as “R406W.” All neurodegenerative diseases other than AD, and MAPT R406W mutation carriers were grouped as “4R tauopathy” (n = 74). This group included individuals with PSP (n = 16), CBS (n = 15), CBS‐PSP continuum (n = 7), sporadic bvFTD (n = 28), and bvFTD MAPT P301L mutation carriers (n = 3), which were primarily 4R tauopathies with 4R tau as primary isoform in the brain aggregates. Note that sporadic bvFTD was listed under “4R tauopathy” group; however, they were pathologically unconfirmed and might contain FTLD‐tau, FTLD‐TDP43, FTLD‐FUS, and small number of 3R tauopathy such as Pick’s disease. One of these participants was later found to have C9orf72 mutation. Cognitively normal controls was named “Control” (n = 98) and included AMC (n = 64), YNC (n = 26), and brain tumor patients (n = 8) who were cognitively normal.
Table 1.
Participants’ demographics and clinical characteristics.
| Groups | AD (n = 80) | 4R tauopathy1 (n = 74) | R406W (n = 5) | Control (N = 98) | All (n = 252) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subcategories | AD (n = 66) | AD focal2 (n = 14) | PSP (n = 16) | CBS (n = 15) | CBS PSP continuum (n = 7) | bvFTD (N = 36) | AMC (n = 64) | YNC (n = 26) | Brain Tumor (n = 8) | Total | ||
| Sporadic bvFTD3 (n = 28) | P301L (n = 3) | R406W (n = 5) | ||||||||||
| Age ± SEM | 74.5 ± 1.9 | 66.7 ± 2.2 | 71.0 ± 2.6 | 68.6 ± 2.6 | 70.7 ± 1.4 | 62.1 ± 1.3 | 37.2 ± 3.6 | 54.8 ± 6.5 | 73.3 ± 0.8 | 42.4 ± 2.4 | 50.2 ± 2.6 | 66.8 ± 0.8 |
| Average age | 73.3 ± 0.9 | 64.5 ± 1.3 | 63.4 ± 1.6 | |||||||||
| Sex (M/F) | 34/32 | 6/8 | 9/7 | 6/9 | 4/3 | 16/12 | 3/0 | 2/3 | 32/32 | 7/19 | 4/4 | 123/129 |
| WashU‐A | 41 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 44 | 0 | 0 | 85 |
| WashU‐B | 11 | 0 | 6 | 3 | 0 | 6 | 3 | 5 | 20 | 26 | 8 | 88 |
| Montpellier | 14 | 14 | 10 | 12 | 7 | 22 | 0 | 0 | 0 | 0 | 0 | 79 |
AD, Alzheimer’s disease; PSP, progressive supranuclear palsy; CBS, corticobasal syndrome; bvFTD, behavioral variant frontotemporal dementia; AMC, age‐matched controls; YNC, Young normal controls; CSF, cerebrospinal fluid.
For the purpose of analyses, all neurodegenerative diseases other than AD including PSP, CBS, CBS PSP continuum, sporadic bvFTD, and FTD MAPT P301L mutation carriers, which are primarily 4R tauopathies are grouped as “4R tauopathies.”
AD focal is defined as individuals with predominant language, behavioral, visuospatial, apraxia phenotype with CSF biomarkers of AD. It is categorized under “AD.”
Sporadic bvFTD is listed under “4R tauopathy” group; however, may contain undiagnosed FTD‐TPD43, FTD‐FUS, and 3R tauopathy cases. N = 1 was later found to have C9orf72 mutation.
Individuals in YNC (42.3 ± 2.4), Brain tumor (50.2 ± 2.6), and MAPT P301L (37.2 ± 3.6) groups were younger than AMC (73.0 ± 0.8) and participants with neurodegenerative diseases including AD (73.3 ± 0.9), CBS (68.6 ± 2.6), CBS PSP continuum (70.7 ± 1.4), PSP (71.0 ± 2.6), and sporadic bvFTD (62.1 ± 1.3) (Table 1, Fig. S1).
All 252 CSF baseline samples and 8 CSF follow‐up samples were measured with sequential IP/MS methods for CSF Aβ 42, Aβ 40, pT217, T217, pT181, and T181 concentrations. CSF Aβ 42/40, pT217/T217, pT181/T181 ratios were calculated. The workflow used to categorize the different clinical groups is described in Figure S2.
Determining cutoffs for IP/MS CSF Aβ 42/40 and CSF pT217/T217
To define amyloid positivity cutoff for CSF Aβ 42/40 measured by IP/MS, we used amyloid PiB‐PET results from 48 participants in the WashU‐A cohort (cutoff 0.18; Fig. S2A, Table 1).21 CSF Aβ 42/40 was significantly decreased in the amyloid‐PiB+ cohort (Fig. S3A). A ROC curve analysis was performed and a Youden’s index value of 0.086 was selected as a cutoff for CSF Aβ 42/40 to maximize discrimination between cohorts (area under the curve [AUC] = 0.921, p < 0.0001; Fig. S3B).
To determine ptau abnormality cutoff for CSF pT217/T217, we used CSF Aβ 42/40 values in amyloid‐PiB+ patients from the WashU‐A cohort (Fig. S2A). We added another subset of 37 participants with only MS CSF Aβ 42/40 measurements. CSF pT217/T217 was significantly increased in amyloid+ individuals based on PiB‐PET and CSF Aβ 42/40 measurements (Fig. S3C). A ROC curve analysis was performed and a Youden’s index value of 4.76 was calculated as a cutoff for CSF pT217/T217 (AUC = 0.983, p < 0.0001; Fig. S3D).
The same ROC analyses were performed for concentrations of CSF pT217, pT181, total tau, and phosphorylation occupancy at T181 (pT181/T181. Fig. S3E–L). AUC for each of these biomarkers was 0.949 (pT217), 0.816 (pT181), 0.698 (total tau), and 0.934 (pT181/T181), respectively, supporting the previous finding that CSF pT217/T217 is a superior discriminative AD biomarker.3, 9
Association between IP/MS CSF Aβ 42/40 and CSF pT217/T217
To evaluate the relationship between IP/MS CSF Aβ 42/40 and pT217/T217, we plotted both ratios for each of the 255 participants. Based on the calculated 0.086 and 4.8 cutoffs for CSF Aβ 42/40 and CSF pT217/T217, respectively, we defined quadrants as follows: I (amyloid−, ptau+), II (amyloid+, ptau+), III (amyloid+, ptau−), and IV (amyloid−, ptau−) (Fig. 1A–E, Fig. S3B).
Figure 1.
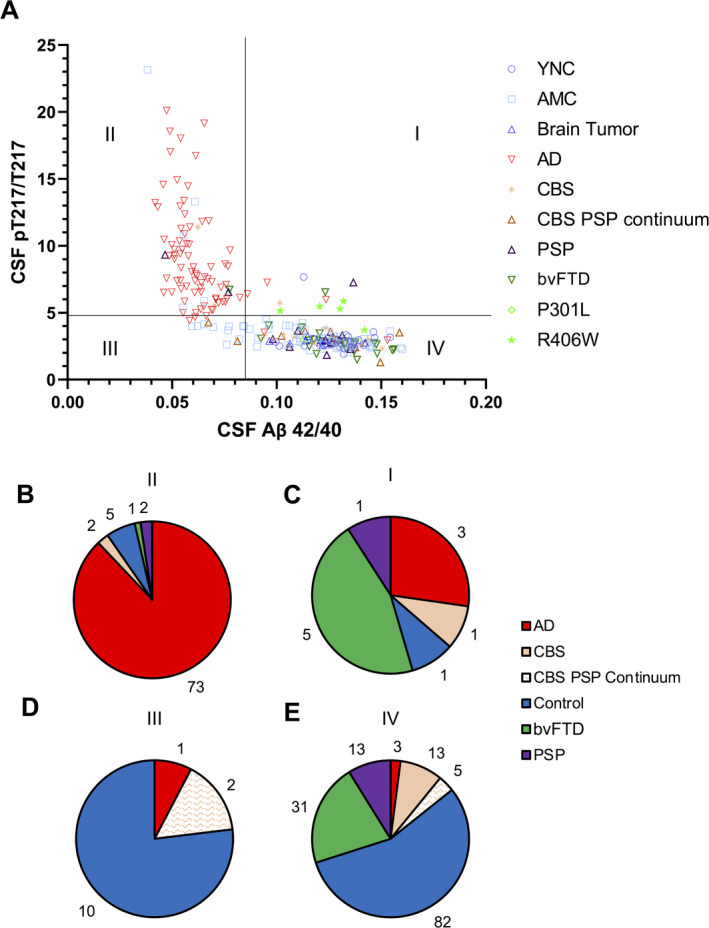
MAPT R406W carriers have increased CSF pT217/T217 without Amyloid pathology. (A) All samples (n = 252) were plotted to quadrant analyses using the cutoffs calculated in the WashU‐A cohort (CSF Aβ 42/40 cutoff = 0.086, CSF pT217/T217 cutoff = 4.76). (B–E) Pie charts showing the number of participants in each clinically classified group for each quadrant: II (B), I (C), III (D), and IV (E). CSF, cerebrospinal fluid.
In quadrant II (amyloid+, ptau+), 88% (73/83) of individuals were clinically identified as AD (Fig. 1B). Overall, 91% (73/80) of individuals with AD were plotted in II (Fig. S4A). Seven participants clinically identified as AD were divided into quadrant I (n = 3), III (n = 1), and IV (n = 3, Fig. 1B–D). A subset of Controls (AMC [n = 5], CBS [n = 2], PSP [n = 2] and bvFTD [n = 1]) were also assigned to quadrant II (Fig. 1A and B).
Eighty four percent (82/98) of controls were plotted in IV and 55% (82/150) of individuals in IV were controls (Fig. 1E, Fig. S4B). Eighty percent (12/15) of CBS and 81% (13/16) of PSP were also in IV (Fig. S4C and D) as well as 71% (5/7) of the CBS PSP continuum.
CSF Aβ 42/40 and CSF pT217/T217 were negatively associated and displayed an L‐shaped curve (Fig. 1). To better understand the dynamic association between CSF Aβ 42/40 and CSF pT217/T217 profile in the context of AD continuum, we assessed amyloid burden measured with PiB‐PET and AV45‐PET imaging and tau aggregation by AV1451‐PET imaging in a subset of participants (Fig. S5). Aβ aggregation measured by both PET tracers gradually and significantly increased from quadrant IV, III to II (Fig. S5A and B). All the Controls in quadrant III were AMC (Fig. 1A) and none belonged to the YNC (age <64). However, within AMC, there was no significant difference in age in quadrant III (74.8 ± 2.4) and quadrant IV (72.4 ± 0.6). These suggest that individuals in quadrant III with amyloid positivity may be defined as presymptomatic AD without abnormal tau phosphorylation.27 In contrast, tau aggregation measured by AV1451‐PET only increased in quadrant II (Fig. S5C), supporting CSF Aβ 42/40 and amyloid PET change before CSF pT217/T217 and tau PET. Importantly, a significant negative correlation between CSF Aβ 42/40 and CSF pT217/T217 was observed in quadrants III and IV (ptau‐) including most controls, PSP, CBS, and bvFTD (Fig. S6).
MAPT R406W carriers have increased pT217/T217 ratio without amyloid pathology
Quadrant I (amyloid−, ptau+) was populated by individuals with bvFTD, PSP, AD, or CBS (Fig. 1C). Interestingly, 45% (5/11) were bvFTD, and all but one (80%, 4/5) were MAPT R406W mutation carriers (Fig. S4F, Table 2). All (5/5) MAPT R406W mutation carriers analyzed in this study were amyloid negative (quadrant I and IV), supporting the absence of amyloid neuropathology in MAPT R406W carriers (Table 2). Only one out of five MAPT R406W mutation carriers who were in their 40s and asymptomatic throughout the study was CSF pT217/T217 negative (quadrant IV) at both baseline and follow‐up visit; all other MAPT R406W mutation carriers were CSF pT217/T217 positive (quadrant I) regardless of their pre/symptomatic status. One participant (#5) who was asymptomatic at baseline and developed dementia in follow‐up visit 4 years later had CSF pT217/T217 just below the threshold at baseline (quadrant IV), but CSF pT217/T217 increased in the follow‐up (quadrant I), suggesting that the longitudinal changes in this biomarker could reflect disease progression. In comparison, five other participants with follow‐up visits within 1 year (1 PSP in quadrant I, 2 CBS, and 2 AMC in quadrant IV) remained in the same quadrant between baseline and follow‐up (Table S1). These results suggest that increasing age and emergence of symptoms associate with increases in CSF pT217/T217 in MAPT R406W mutation carriers.
Table 2.
Demographics and summary of biomarker values for MAPT R406W mutation carriers.
| Participant | Quadrant | Symptomatic/Asymptomatic | Age | Sex |
CSF Aβ 42/40 (cutoff = 0.086) |
CSF pT217/T217 (cutoff = 4.76) |
CSF pT217/T217 × CSF Aβ 42/40 (cutoff for R406W versus control = 0.50) |
CSF pT217/T217 div CSF Aβ 42/40 (cutoff for R406W versus control = 39.9) |
|---|---|---|---|---|---|---|---|---|
| #1 | IV | No dementia | 40 | M | 0.142 | 3.69 | 0.524 | 26.0 |
| IV | No dementia | 41 | 0.152 | 3.75 | 0.572 | 24.6 | ||
| #2 | I | No dementia | 40 | M | 0.130 | 5.28 | 0.688 | 40.6 |
| #3 | I | No dementia | 52 | F | 0.127 | 5.30 | 0.671 | 41.9 |
| I | No dementia | 55 | 0.132 | 5.86 | 0.774 | 44.4 | ||
| #4 | I | Symptomatic | 64 | F | 0.121 | 5.47 | 0.659 | 45.4 |
| #5 | IV | Asymptomatic, other primary | 69 | F | 0.108 | 4.73 | 0.513 | 43.7 |
| I | Symptomatic | 73 | 0.102 | 5.14 | 0.522 | 50.5 | ||
| Average (age <50) | 1 I, 2 IV | No dementia | 40.3 ± 0.40 | ‐ | 0.142 ± 0.0064 | 4.2 ± 0.52 | 0.595 ± 0.049 | 30.4 ± 5.1 |
| Average (age >50) | 4 I, 1 IV | No dementia, Symptomatic, other primary | 63.0 ± 4.1 | ‐ | 0.118 ± 0.0056 | 5.3 ± 0.19 | 0.628 ± 0.049 | 45.2 ± 1.5 |
| Average all | – | – | 54.5 ± 4.8 | ‐ | 0.127 ± 0.0059 | 4.9 ± 0.28 | 0.615 ± 0.049 | 39.6 ± 3.3 |
CSF, cerebrospinal fluid.
Diagnostic values of IP/MS CSF Aβ 42/40, pT217/T217, and composite biomarkers in AD and MAPT R406W mutation carriers
Next, we compared the diagnostic performance of IP/MS CSF Aβ 42/40 and CSF pT217/T217, with composite biomarkers consisting of pT217/T217 multiplied by CSF Aβ 42/40 and CSF pT217/T217 divided by Aβ 42/40 (Fig. 2, Table 3, Fig. S7). For this analysis, the four clinical groups previously defined ("AD,” “R406W,” “4R tauopathy,” and “Control”) were compared. CSF Aβ 42/40 ratio and CSF pT217/T217 used alone only separate AD from the other three groups (Fig. 2A and B). However, the R406W group had significantly increased CSF Aβ 42/40 × pT217/T217 composite biomarker compared to what was observed in the Control and 4R tauopathy groups (Fig. 2C). This composite biomarker demonstrated excellent ability to separate the R406W group from the 4R tauopathy (AUC = 0.948) and Control groups (AUC = 0.961). When this composite biomarker was used, 100% of MAPT R406W mutation carriers were above the cutoff of 0.50 for R406W versus control (Table 3). CSF pT217/T217 divided by CSF Aβ 42/40 performed similarly to CSF Aβ 42/40 and CSF pT2117/T217 alone, and could not distinguish R406W from other groups (Fig. 2D, Table 3).
Figure 2.
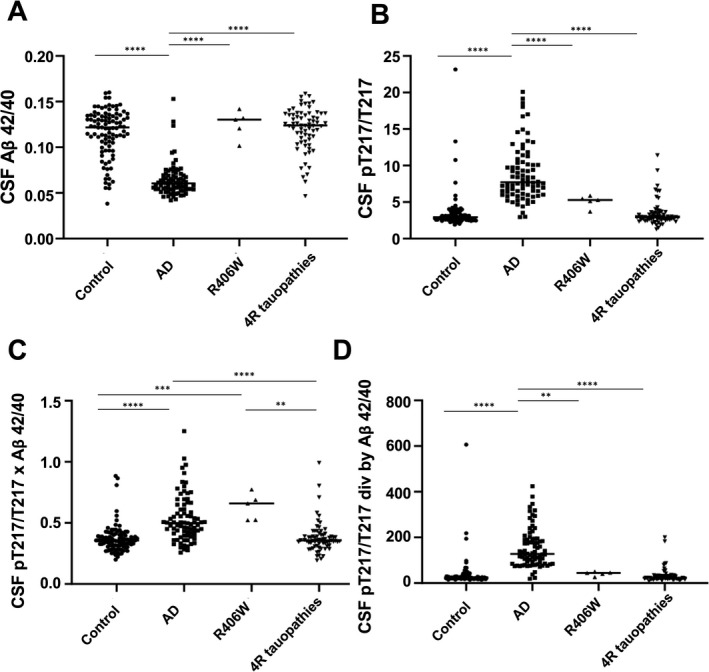
CSF Aβ 42/40, CSF pT217/T217, and composite biomarkers for diagnosis of AD, and MAPT R406W mutation carriers. (A) CSF Aβ 42/40 is significantly decreased in AD (ANOVA, p < 0.0001). (B) CSF pT217/T217 is significantly increased in AD (ANOVA, p < 0.0001). (C) CSF pT217/T217 × CSF Aβ 42/40 is significantly increased in AD compared to Control and 4R tauopathy group consisting mostly of 4R tauopathy (PSP, CBS, FTD‐MAPT P301L) and a subset of sporadic bvFTD (may contain FTD‐TDP, FTD‐FUS and 3R tauopathy. ANOVA, p < 0.0001). This is also significantly increased in MAPT R406W carriers compared to control (ANOVA, p < 0.0001) and 4R tauopathy group (ANOVA, p = 0.0001). (D) CSF pT217/T217 divided by CSF Aβ 42/40, is significantly increased in AD (ANOVA, p < 0.0001). CSF, cerebrospinal fluid; AD, Alzheimer’s disease; PSP, progressive supranuclear palsy; CBS, corticobasal syndrome; bvFTD, behavioral variant frontotemporal dementia.
Table 3.
Diagnostic accuracy of combinations of CSF Aβ 42/40 and CSF pT217T217 biomarkers for AD and FTD‐MAPT R406W.
| Test | Diagnostic groups | n per group | AUC | 95% CI | p value | Sensitivity% | Specificity% | Cutoff |
|---|---|---|---|---|---|---|---|---|
| CSF Aβ 42/40 | AD versus R406W | 80 versus 5 | 0.978 | 0.947–1.000 | 0.0004 | 100.0 | 96.3 | >0.0985 |
| AD versus 4R tauopathy | 80 versus 69 | 0.946 | 0.905–0.988 | <0.0001 | 94.2 | 90.0 | >0.0768 | |
| Control versus AD | 98 versus 80 | 0.925 | 0.882–0.969 | <0.0001 | 91.3 | 87.8 | <0.0798 | |
| Control versus R406W | 98 versus 5 | 0.588 | 0.384–0.791 | 0.5094 | 60.0 | 66.3 | >0.1302 | |
| 4R tauopathy versus R406W | 69 versus 5 | 0.548 | 0.329–0.767 | 0.7223 | 55.4 | 62.5 | <0.1298 | |
| Control versus 4R tauopathy | 98 versus 69 | 0.543 | 0.454–0.631 | 0.3459 | 56.5 | 55.1 | >0.1231 | |
| CSF pT217/T217 | Control versus AD | 98 versus 80 | 0.951 | 0.916–0.987 | <0.0001 | 96.3 | 92.9 | >4.325 |
| AD versus 4R tauopathy | 80 versus 69 | 0.945 | 0.907–0.983 | <0.0001 | 88.4 | 96.3 | <4.338 | |
| Control versus R406W | 98 versus 5 | 0.908 | 0.829–0.987 | 0.0021 | 100.0 | 76.5 | >3.678 | |
| AD versus R406W | 80 versus 5 | 0.878 | 0.799–0.956 | 0.0048 | 100.0 | 80.0 | <5.917 | |
| 4R tauopathy versus R406W | 69 versus 5 | 0.875 | 0.797–0.954 | 0.0053 | 81.2 | 100.0 | <3.675 | |
| Control versus 4R tauopathy | 98 versus 69 | 0.525 | 0.435–0.615 | 0.5895 | 65.2 | 45.9 | <3.037 | |
| CSF pT217/T217 × CSF Aβ 42/40 | Control versus R406W | 98 versus 5 | 0.961 | 0.924–0.998 | <0.0001 | 100.0 | 94.9 | >0.522 |
| 4R tauopathy versus R406W | 69 versus 5 | 0.948 | 0.897–0.999 | 0.0009 | 92.8 | 100.0 | <0.522 | |
| Control versus AD | 98 versus 80 | 0.814 | 0.748–0.880 | <0.0001 | 66.3 | 90.8 | >0.448 | |
| AD versus 4R tauopathy | 80 versus 69 | 0.793 | 0.719–0.868 | <0.0001 | 85.5 | 68.8 | <0.436 | |
| AD versus R406W | 80 versus 5 | 0.733 | 0.602–0.863 | 0.0824 | 100.0 | 60.0 | >0.515 | |
| Control versus 4R tauopathy | 98 versus 69 | 0.521 | 0.432–0.611 | 0.6375 | 73.9 | 36.7 | >0.332 | |
| CSF pT217/T217 divided by CSFAβ 42/40 | AD versus R406W | 80 versus 5 | 0.963 | 0.922–1.000 | 0.0005 | 100.0 | 95.0 | <61.1 |
| AD versus 4R tauopathy | 80 versus 69 | 0.953 | 0.915–0.990 | <0.0001 | 92.8 | 95.0 | <67.6 | |
| Control versus AD | 98 versus 80 | 0.950 | 0.912–0.987 | <0.0001 | 95.0 | 94.9 | >69.8 | |
| 4R tauopathy versus R406W | 69 versus 5 | 0.817 | 0.701–0.934 | 0.0184 | 85.5 | 80.0 | <38.0 | |
| Control versus R406W | 98 versus 5 | 0.804 | 0.694–0.914 | 0.0222 | 80.0 | 82.7 | >39.9 | |
| Control versus 4R tauopathy | 98 versus 69 | 0.533 | 0.443–0.623 | 0.4746 | 14.5 | 94.9 | <17.0 |
CSF, cerebrospinal fluid; AD, Alzheimer’s disease; AUC, area under the curve; FTD, frontotemporal dementia.
IP/MS CSF total tau and ptau concentrations are not efficient biomarkers for MAPT R406W carriers
Neither CSF pT217, pT181, total tau, nor phosphorylation occupancy at T181 (pT181/T181) were as efficient as the composite biomarker CSF pT217/T217 × CSF Aβ 42/40 at separating MAPT R406W mutation carriers from individuals with other neurodegenerative dementing illnesses (Figs. S8, S9). CSF pT181/T181 was significantly decreased in sporadic bvFTD (including FTLD‐tau, FTLD‐TDP43, and FTLD‐FUS) compared to AD, AMC, and MAPT R406W mutation carriers (Figs. S8D, S9D). When regrouped, CSF pT181/T181 was able to separate sporadic bvFTD group from AD (AUC = 0.959), Control (AUC = 0.752), and other tauopathies including CBS, PSP, and FTD‐MAPT including R406W and P301L (AUC = 0.707. Fig. S10). However, the specificity and sensitivity of CSF pT181/T181 to separate sporadic bvFTD from other tauopathies and control were not as high as that of CSF pT217/T217 in identifying MAPT R406W mutation carriers.
Discussion
MAPT R406W mutation carriers have increased pT217/T217 without amyloid pathology
CSF pT217/T217 strongly correlates with amyloid pathology measured by amyloid PET in AD,9 but it was unproven whether pT217/T217 was a readout for CSF amyloid pathology or tau pathology. In this study, we first showed a specific correlation between CSF Aβ 42/40 and CSF pT217/T217 in individuals with symptomatic AD. Even in presymptomatic AD, CSF Aβ 42/40 and CSF pT217/T217 correlate when slight changes in phosphorylation are observed, consistent with previous reports showing a correlation between PiB‐PET and CSF pT217/T217.3, 9 Neither CSF Aβ 42/40 nor CSF pT217/T217 were altered in other tauopathies, including PSP, CBS, and most sporadic and familial FTD. However, we found that MAPT R406W mutation carriers have increased CSF pT217/T217 independent of amyloid pathology, demonstrating that increased CSF pT217/T217 is, indeed, a biomarker of pathological tau modification common to AD and MAPT R406W associated dementia and that amyloid pathology is not a prerequisite to this modification. This suggests that there is a common tau pathology downstream of AD and MAPT R406W mutation carriers, which results in specific tau phosphorylation changes in the brain, leading to an increase in CSF pT217/T217. Alternatively, two distinct upstream mechanisms, one involving amyloid deposition and the second involving MAPT R406W mutation, could lead to the activation of a similar pathway, ultimately leading to tau hyperphosphorylation and aggregation.
MAPT R406W mutation’s similarity to AD
MAPT R406W mutation‐related pathology shares multiple clinical and neuropathological similarities with AD. Unlike other MAPT mutation carriers, MAPT R406W mutation carriers have later ages‐at‐symptomatic onset, with clinical symptoms including memory loss emerging, on average, in the mid‐50s with slow progression.37 Most pathological MAPT mutations such as P301L are located in or around exon 10 and typically lead to 4R tau isoform aggregation, resulting in 4R tauopathies. In contrast, the MAPT mutations such as R406W and V337M are located in the C‐terminus of the tau protein in a domain common to both 3R and 4R tau isoforms, resulting in 3R+4R mixed brain pathologies.38, 39 The MAPT R406W mutation, like AD, can thus be categorized as a 3R+4R tauopathy and is differentiated from other 4R (CBS, PSP, bvFTD related to MAPT mutations located on exon 10) or 3R (Pick’s disease) tauopathies.
Filament structures in tau aggregates have been recently resolved by cryo‐electron microscopy for different tauopathies such as AD and chronic traumatic encephalopathy (CTE) (3R+4R), CBS (4R), and Pick’s disease (3R).40, 41, 42, 43, 44 Consistent with neuropathological findings, tau domains shared by 3R and 4R isoforms are involved in AD and CTE tau aggregates, while 4R and 3R specific domains are, respectively, involved in corticobasal degeneration and Pick’s disease aggregates. Though no such structural data is available for MAPT R406W, AD, MAPT R406W, and V337M have paired helical filament structures,37, 45 and AD Tau PET tracers such as AV1451 bind to some extent in presymptomatic MAPT R406W and V337M mutation carriers but not in other tauopathies,45, 46, 47, 48, 49 suggesting that tau aggregates in these 3R+4R tauopathies have similar characteristics. However, how hyperphosphorylation at T217 contributes to or associates with paired helical filament formation remains to be addressed. Previous studies suggest that CSF T217 is hyperphosphorylated in the early presymptomatic stages of AD, and is detectable more than 20 years before the emergence of clinical symptoms, while tau aggregates detected by PET imaging increase near symptom onset.9 We speculate that in 3R+4R tauopathies including MAPT R406W mutation carriers, (1) CSF pT217/T217 becomes abnormal prior to symptom onset when tau paired helical filament formation begins but it is below the detection limit by tau PET imaging followed by evident changes in Tau PET imaging; or, (2) CSF T217 hyperphosphorylation is not directly associated with the formation of neurofibrillary tangles but reflects an abnormal cellular metabolism affecting tau and leading ultimately to tau aggregation.
Composite biomarker of CSF pT217/T217 × CSF Aβ 42/40 serves as a sensitive biomarker for MAPT R406W mutation carriers
We evaluated diagnostic values of CSF pT217/T217 and CSF Aβ 42/40 alone and in combination. CSF pT217/T217 levels were increased in MAPT R406W mutation carriers. However, the degree of increase was much smaller compared to that of AD and we could not separate MAPT R406W mutation carriers from the Control or 4R tauopathy groups including PSP, CBS, sporadic bvFTD, and FTD‐MAPT P301L by CSF pT217/T217 alone (Fig. 2A, 2B, Fig. S6). Previous studies have indicated an increase of CSF and plasma pT181 concentrations in some cases of MAPT R406W mutation carriers, but the increase was mild.50, 51, 52 These are consistent with the insufficient sensitivity obtained from this study using only CSF concentrations of pT181, pT217, or phosphorylation occupancies at T181 and T217 (pT181/T181 and pT217/T217) as R406W biomarkers. Through quadrant analysis, we demonstrated that both CSF pT217/T217 and CSF Aβ 42/40 are necessary to distinguish MAPT R406W mutation carriers with high accuracy. We also demonstrated that a composite biomarker, CSF pT217/T217 × CSF Aβ 42/40, contained sufficient sensitivity and specificity to distinguish MAPT R406W mutation carriers from the controls and 4R tauopathies (Fig. 2C, Fig. S7C. AUC = 0.934, 0.960, respectively). This is comparable to the high specificity and sensitivity of CSF Aβ 42/40 and CSF pT217/T217 to distinguish AD from the controls (AUC = 0.926, 0.952, respectively). A combination of CSF Aβ 42/40 and pT217/T217 ratios could be used in future trials to select for presymptomatic MAPT R406W mutation carriers and possibly other 3R+4R tauopathies such as V337M. Moreover, longitudinal measures of CSF pT217/T217 could reflect disease progression, suggesting that CSF Aβ 42/40, CSF pT217/T217, and composite biomarkers may serve as new sensitive readouts in drug clinical trials against tauopathies that can assess target engagement in MAPT R406W mutation carriers.
CSF pT181/T181 may decrease in sporadic bvFTD
Previous studies using immunoassays showed mixed results in bvFTD, PSP, and CBS patients showing no or mild changes in CSF total tau or pT181.16, 17, 18, 53, 54, 55, 56, 57 Consistent with multiple reports, our study did not show significant differences in CSF total tau or CSF pT181 concentrations alone between bvFTD, PSP, CBS, and Control groups. However, by calculating the phosphorylation occupancies within the same participant, we showed that CSF pT181/T181 significantly decreases in sporadic bvFTD. This may be achieved by normalizing the changes in pT181 by T181, accounting for any physiological increase in pT181 as total tau increases, and individual variabilities such as age, sex, and genotype. Specificity and sensitivity of CSF pT181/T181 biomarker in identifying sporadic bvFTD from Controls or other tauopathies (AUC <0.8) were not as high as a composite biomarker, CSF pT217/T217 × CSF Aβ 42/40, in identifying MAPT mutation carriers (AUC >0.9). This may be due to the heterogeneity of the sporadic bvFTD cohort including FTLD‐tau, FTLD‐TDP, and FTLD‐FUS. Previous studies showed that FTLD‐TDP has lower CSF pT181/T181,58, 59 which could be consistent with our results if the sporadic bvFTD cohort included FTLD‐TDP.
Limitations of this study and future directions
The relatively small number of participants in some subgroups and the inclusion of pathologically unconfirmed tauopathy and sporadic bvFTD cases may have decreased diagnostic accuracies. MAPT V337M mutation carriers may also be interesting to evaluate in the context of 3R+4R tauopathy. Future studies utilizing pathologically confirmed cases, longitudinal samples with clinical and PET assessments, and a larger cohort may facilitate additional analyses in ptau or tau that may be specific to MAPT R406W mutation carriers, sporadic bvFTD, or other subgroups of tauopathies.
Author Contributions
CS, NRB, and AG conceived the project. NRB, CS, and NM developed tau and Aβ sequential IP/MS methods. CS, NRB, and NM designed and performed IP/MS experiments. CS and NRB analyzed and interpreted the data. CS developed IRB protocols and collected the majority of non‐AD tauopathy CSF in the WashU‐B cohort. NG, BAW, GSD, AAD referred bvFTD and tauopathy patients at Washington University. CS and GSD recruited YNCs. AHK and GJZ referred brain tumor patients. AG collected CSF at University of Montpellier. RJB provided CSF from the entire WashU‐A cohort and part of the WashU‐B cohort, mass spectrometry resources, and mentorship. CS, NRB, AG, NG, RJB wrote the initial draft of the paper; all authors made substantial contributions to the subsequent version of the manuscript and approved the final version for submission.
Conflict of Interests
Washington University, with CS, NRB, and RJB as co‐inventors, have submitted the U.S. provisional patent application “Methods to detect novel tau species in CSF and use thereof to track tau neuropathology in AD and other tauopathies.” and “CSF phosphorylated tau and Amyloid beta profiles as biomarkers of tauopathies” to Washington University. RJB and NRB as co‐inventors, have submitted the non‐provisional patent application “Methods of Diagnosing and Treating Based on Site‐Specific Tau Phosphorylation.” RJB has received honoraria from AC Immune, Janssen, Pfizer, and Roche as a speaker, from AC Immune, Amgen, Eisai, and Janssen as a consultant, and from Roche as an advisory board member. RJB has an equity ownership interest in C2N Diagnostics and receives royalty income based on technology licensed by Washington University to C2N Diagnostics. RJB receives income from C2N Diagnostics for serving on the scientific advisory board. NG has participated or is currently participating in clinical trials of anti‐dementia drugs sponsored by the following companies: Bristol Myers Squibb, Eli Lilly/Avid Radiopharmaceuticals, Janssen Immunotherapy, Novartis, Pfizer, Wyeth, SNIFF (The Study of Nasal Insulin to Fight Forgetfulness) study, and A4 (The Anti‐Amyloid Treatment in Asymptomatic Alzheimer’s Disease) trial. She receives research support from NIH, Tau Consortium, and the Association for Frontotemporal Dementia. BAW participates in research sponsored by Acadia, Biogen, Global Kinetics, Neurocrine, Roche, Vaccinex, and Wave Biosciences. GSD is supported by grants from NIH/NIA (K23AG064029); personal fees from Parabon NanoLabs, Inc, personal fees from DynaMed (EBSCO Health), outside the submitted work; and is the clinical director for the Anti‐NMDA Receptor Encephalitis Foundation (uncompensated). AHK is a consultant for Monteris Medical and has received research funding from Monteris Medical, Stryker, and Collagen Matrix.
Supporting information
Figure S1. Age of the participants in each subgroup.
Figure S2. Flowchart of cohort analyzed in this study.
Figure S3. ROC analyses of IP/MS CSF Aβ 42/40, CSF tau, and ptau.
Figure S4. Quadrant analyses by diagnosis.
Figure S5. Amyloid and tau PET Imaging by quadrant.
Figure S6. CSF Aβ 42/40 and CSF pT217/T217 correlate in quadrants III and IV.
Figure S7. Subcategory of diagnosis are shown from Figure 2.
Figure S8. Quadrant analyses using CSF Aβ 42/40 and CSF total tau and ptau.
Figure S9. IP/MS CSF total tau and ptau in subgroups of tauopathies.
Figure S10. Sporadic bvFTD containing FTLD‐tau, FTLD‐TDP, FTLD‐FUS may be separated from Control, AD and other tauopathies with CSF pT181/T181.
Table S1. Demographics and summary of biomarker values for participants with follow‐up visits.
Acknowledgments
We thank the participants and families for their contribution to this study. We thank Megan Arb, Theresa Arb, Melissa Sullivan, Wendy Sigurdson, Tamara Donahue for IRB protocol development and recruitment at Washington University, and Dr. Sylvain Lehmann, Karim Bennys, Cecilia Marelli, Christophe Hirtz at Montpellier. We thank Kathryn Draege for the brain tumor CSF collection. We thank Drs. Robert Swarm, Lesley Rao, Jacob Aubuchon for performing lumbar punctures. We thank members of the Bateman lab for discussion, especially Dr. Kanta Horie for initial feedback and Andrew Espeland for the initial analyses and exploration of the data. We thank the Clinical, Biomarker, and Imaging Cores at the Washington University School of Medicine for participant evaluation, samples, and data collection.
Funding Information
This work was supported by Barnes Jewish Hospital Foundation (BJHF) pilot grant #3945 (CS) and NIH/NIA K01AG062796 (CS). Additionally, samples analyzed in this study were obtained with support from Tau Foundation Plan Alzheimer (AG), BIRCWH K12 HD001459 (NG), NIH/NIA K23 AG064029 (GSD), NIH K08 NS101118 (AAD), NIH/NINDS RF1NS103276 (GJZ), NIH R01NS065667 (RJB), NIH R01NS095773 (RJB), Rainwater Charitable Foundation (RJB), The Association for Frontotemporal Degeneration (RJB), and Tau SILK Consortium (RJB). This work was supported by cores, resources, and effort provided by Washington University Biomedical Mass Spectrometry Research Facility (NIH P41 GM103422) and access to equipment made possible by the Hope Center for Neurological Disorders, and the Departments of Neurology and Psychiatry at the Washington University School of Medicine.
Funding Statement
This work was funded by Tau Consortium ; Association for Frontotemporal Degeneration ; Barnes Jewish Hospital Foundation grant 3945; BIRCWH grant K12 HD001459; NIH grants P41 GM103422, R01NS065667, R01NS095773, RF1NS103276, K08 NS101118, K23 AG064029, and K01 AG062796; Tau Foundation Plan Alzheimer; Washington University Biomedical Mass Spectrometry Research Facility.
Contributor Information
Chihiro Sato, Email: satochihiro@wustl.edu.
Audrey Gabelle, Email: a-gabelle@chu-montpellier.fr.
Nicolas R. Barthélemy, Email: barthelemy.nicolas@wusltl.edu.
References
- 1.Fagan AM, Head D, Shah AR, et al. Decreased cerebrospinal fluid Aβ42 correlates with brain atrophy in cognitively normal elderly. Ann Neurol 2009;65:176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 2010;6:131–144. [DOI] [PubMed] [Google Scholar]
- 3.Barthélemy NR, Bateman RJ, Hirtz C, et al. Cerebrospinal fluid phospho‐tau T217 outperforms T181 as a biomarker for the differential diagnosis of Alzheimer’s disease and PET amyloid‐positive patient identification. Alzheimer's Res Ther 2020;12:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janelidze S, Stomrud E, Smith R, et al. Cerebrospinal fluid p‐tau217 performs better than p‐tau181 as a biomarker of Alzheimer’s disease. Nat Commun 2020;11:1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Triana‐Baltzer G, Van Kolen K, Theunis C, et al. Development and validation of a high sensitivity assay for measuring p217 + tau in cerebrospinal fluid. J Alzheimers Dis 2020;77:1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suárez‐Calvet M, Karikari TK, Ashton NJ, et al. Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer’s continuum when only subtle changes in Aβ pathology are detected. EMBO Mol Med 2020;12:e12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanes J, Kovac A, Kvartsberg H, et al. Evaluation of a novel immunoassay to detect p‐tau Thr217 in the CSF to distinguish Alzheimer's disease from other dementias. Neurology 2020;95:e3026–e3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karikari TK, Emeršič A, Vrillon A, et al. Head‐to‐head comparison of clinical performance of CSF phospho‐tau T181 and T217 biomarkers for Alzheimer’s disease diagnosis. Alzheimers Dement 2021;17:755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barthélemy NR, Li Y, Joseph‐Mathurin N, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med 2020;26:398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mattsson‐Carlgren N, Andersson E, Janelidze S, et al. Aβ deposition is associated with increases in soluble and phosphorylated tau that precede a positive Tau PET in Alzheimer’s disease. Sci Adv 2020;6:eaaz2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Day GS, Gordon BA, Jackson K, et al. Tau‐PET binding distinguishes patients with early‐stage posterior cortical atrophy from amnestic Alzheimer's disease dementia. Alzheimer Dis Assoc Disord 2017;31:87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irwin DJ, Trojanowski JQ, Grossman M. Cerebrospinal fluid biomarkers for differentiation of frontotemporal lobar degeneration from Alzheimer’s disease. Front Aging Neurosci 2013;5:6. 10.3389/fnagi.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meeter LH, Kaat LD, Rohrer JD, van Swieten JC. Imaging and fluid biomarkers in frontotemporal dementia. Nat Rev Neurol 2017;13:406–419. [DOI] [PubMed] [Google Scholar]
- 14.Skillbäck T, Farahmand BY, Rosén C, et al. Cerebrospinal fluid tau and amyloid‐β1‐42 in patients with dementia. Brain 2015;138(Pt 9):2716–2731. [DOI] [PubMed] [Google Scholar]
- 15.van Harten AC, Kester MI, Visser P‐J, et al. Tau and p‐tau as CSF biomarkers in dementia: a meta‐analysis. Clin Chem Lab Med 2011;49:353–366. [DOI] [PubMed] [Google Scholar]
- 16.Hampel H, Teipel SJ. Total and phosphorylated tau proteins: evaluation as core biomarker candidates in frontotemporal dementia. Dement Geriatr Cogn Disord 2004;17:350–354. [DOI] [PubMed] [Google Scholar]
- 17.Wagshal D, Sankaranarayanan S, Guss V, et al. Divergent CSF τ alterations in two common tauopathies: Alzheimer’s disease and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 2015;86:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosso SM, van Herpen E, Pijnenburg YAL, et al. Total tau and phosphorylated tau 181 levels in the cerebrospinal fluid of patients with frontotemporal dementia due to P301L and G272V tau mutations. Arch Neurol 2003;60:1209–1213. [DOI] [PubMed] [Google Scholar]
- 19.Skillbäck T, Rosén C, Asztely F, et al. Diagnostic performance of cerebrospinal fluid total tau and phosphorylated tau in Creutzfeldt‐Jakob disease: results from the Swedish Mortality Registry. JAMA Neurol 2014;71:476–483. [DOI] [PubMed] [Google Scholar]
- 20.Barthélemy NR, Mallipeddi N, Moiseyev P, et al. Tau phosphorylation rates measured by mass spectrometry differ in the intracellular brain vs. extracellular cerebrospinal fluid compartments and are differentially affected by Alzheimer’s disease. Front Aging Neurosci 2019;11:121. 10.3389/fnagi.2019.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patterson BW, Elbert DL, Mawuenyega KG, et al. Age and amyloid effects on human CNS amyloid‐beta kinetics. Ann Neurol 2015;78:439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sato C, Barthélemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron 2018;97:1284–1298.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 24.Dumurgier J, Schraen S, Gabelle A, et al. Cerebrospinal fluid amyloid‐β 42/40 ratio in clinical setting of memory centers: a multicentric study. Alzheimers Res Ther 2015;7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brookmeyer R, Evans DA, Hebert L, et al. National estimates of the prevalence of Alzheimer’s disease in the United States. Alzheimers Dement 2011;7:61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging‐Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jack CR, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boeve BF. The multiple phenotypes of corticobasal syndrome and corticobasal degeneration: implications for further study. J Mol Neurosci 2011;45:350–353. [DOI] [PubMed] [Google Scholar]
- 29.Josephs KA, Hodges JR, Snowden JS, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011;122:137–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ling H. Clinical approach to progressive supranuclear palsy. J Mov Disord 2016;9:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewczuk P, Matzen A, Blennow K, et al. Cerebrospinal fluid Aβ42/40 corresponds better than Aβ42 to amyloid PET in Alzheimer’s disease. J Alzheimers Dis 2016;55:813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.del Campo M, Mollenhauer B, Bertolotto A, et al. Recommendations to standardize preanalytical confounding factors in Alzheimer’s and Parkinson’s disease cerebrospinal fluid biomarkers: an update. Biomark Med 2012;6:419–430. [DOI] [PubMed] [Google Scholar]
- 34.Perret‐Liaudet A, Pelpel M, Tholance Y, et al. Risk of Alzheimer’s disease biological misdiagnosis linked to cerebrospinal collection tubes. J Alzheimers Dis 2012;31:13–20. [DOI] [PubMed] [Google Scholar]
- 35.Dumurgier J, Vercruysse O, Paquet C, et al. Intersite variability of CSF Alzheimer’s disease biomarkers in clinical setting. Alzheimers Dement 2012;9:406–413. [DOI] [PubMed] [Google Scholar]
- 36.Horie K, Barthélemy NR, Mallipeddi N, et al. Regional correlation of biochemical measures of amyloid and tau phosphorylation in the brain. Acta Neuropathol Commun 2020;8:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reed LA, Grabowski TJ, Schmidt ML, et al. Autosomal dominant dementia with widespread neurofibrillary tangles. Ann Neurol 1997;42:564–572. [DOI] [PubMed] [Google Scholar]
- 38.de Silva R, Lashley T, Strand C, et al. An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R‐ and 4R‐specific tau monoclonal antibodies. Acta Neuropathol 2006;111:329–340. [DOI] [PubMed] [Google Scholar]
- 39.Lindquist SG, Holm IE, Schwartz M, et al. Alzheimer's disease‐like clinical phenotype in a family with FTDP‐17 caused by a MAPT R406W mutation. Eur J Neurol 2008;15:377–385. [DOI] [PubMed] [Google Scholar]
- 40.Fitzpatrick AWP, Falcon B, He S, et al. Cryo‐EM structures of tau filaments from Alzheimer’s disease. Nature 2017;547:nature23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Falcon B, Zhang W, Murzin AG, et al. Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 2018;561:137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falcon B, Zivanov J, Zhang W, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 2019;568:420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arakhamia T, Lee CE, Carlomagno Y, et al. Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell 2020;180:633–644.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang W, Tarutani A, Newell KL, et al. Novel tau filament fold in corticobasal degeneration. Nature 2020;580:283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones DT, Knopman DS, Graff‐Radford J, et al. In vivo 18F‐AV‐1451 tau PET signal in MAPT mutation carriers varies by expected tau isoforms. Neurology 2018;90:e947–e954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith R, Puschmann A, Schöll M, et al. 18F‐AV‐1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain 2016;139:2372–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsai RM, Bejanin A, Lesman‐Segev O, et al. 18F‐flortaucipir (AV‐1451) tau PET in frontotemporal dementia syndromes. Alzheimers Res Ther 2019;11:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spina S, Schonhaut DR, Boeve BF, et al. Frontotemporal dementia with the V337M MAPT mutation. Neurology 2017;88:758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolters EE, Papma JM, Verfaillie SCJ, et al. [18F]Flortaucipir PET across various MAPT mutations in presymptomatic and symptomatic carriers. Neurology 2021: 10.1212/WNL.0000000000012448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ygland E, van Westen D, Englund E, et al. Slowly progressive dementia caused by MAPT R406W mutations: longitudinal report on a new kindred and systematic review. Alzheimers Res Ther 2018;10:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tolboom N, van der Flier WM, Boverhoff J, et al. Molecular imaging in the diagnosis of Alzheimer’s disease: visual assessment of [11C]PIB and [18F]FDDNP PET images. J Neurol Neurosurg Psychiatry 2010;81:882–884. [DOI] [PubMed] [Google Scholar]
- 52.Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat Med 2020;26:387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aerts MB, Esselink RAJ, Bloem BR, Verbeek MM. Cerebrospinal fluid tau and phosphorylated tau protein are elevated in corticobasal syndrome. Mov Disord 2011;26:169–173. [DOI] [PubMed] [Google Scholar]
- 54.Green AJE, Harvey RJ, Thompson EJ, Rossor MN. Increased tau in the cerebrospinal fluid of patients with frontotemporal dementia and Alzheimer’s disease. Neurosci Lett 1999;259:133–135. [DOI] [PubMed] [Google Scholar]
- 55.Urakami K, Wada K, Arai H, et al. Diagnostic significance of tau protein in cerebrospinal fluid from patients with corticobasal degeneration or progressive supranuclear palsy. J Neurol Sci 2001;183:95–98. [DOI] [PubMed] [Google Scholar]
- 56.Bibl M, Mollenhauer B, Lewczuk P, et al. Cerebrospinal fluid tau, p‐tau 181 and amyloid‐β38/40/42 in frontotemporal dementias and primary progressive aphasias. Dement Geriatr Cogn Disord 2011;31:37–44. [DOI] [PubMed] [Google Scholar]
- 57.Grossman M, Farmer J, Leight S, et al. Cerebrospinal fluid profile in frontotemporal dementia and Alzheimer’s disease. Ann Neurol 2005;57:721–729. [DOI] [PubMed] [Google Scholar]
- 58.Borroni B, Benussi A, Archetti S, et al. Csf p‐tau181/tau ratio as biomarker for TDP pathology in frontotemporal dementia. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:86–91. [DOI] [PubMed] [Google Scholar]
- 59.Hu WT, Watts K, Grossman M, et al. Reduced CSF p‐TAU181 to Tau ratio is a biomarker for FTLD‐TDP. Neurology 2013;81:1945–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Age of the participants in each subgroup.
Figure S2. Flowchart of cohort analyzed in this study.
Figure S3. ROC analyses of IP/MS CSF Aβ 42/40, CSF tau, and ptau.
Figure S4. Quadrant analyses by diagnosis.
Figure S5. Amyloid and tau PET Imaging by quadrant.
Figure S6. CSF Aβ 42/40 and CSF pT217/T217 correlate in quadrants III and IV.
Figure S7. Subcategory of diagnosis are shown from Figure 2.
Figure S8. Quadrant analyses using CSF Aβ 42/40 and CSF total tau and ptau.
Figure S9. IP/MS CSF total tau and ptau in subgroups of tauopathies.
Figure S10. Sporadic bvFTD containing FTLD‐tau, FTLD‐TDP, FTLD‐FUS may be separated from Control, AD and other tauopathies with CSF pT181/T181.
Table S1. Demographics and summary of biomarker values for participants with follow‐up visits.