

Abstract
The APOBEC3 cytidine deaminases are implicated as the cause of a prevalent somatic mutation pattern found in cancer genomes. The APOBEC3 enzymes act as viral restriction factors by mutating viral genomes. Mutation of the cellular genome is presumed to be an off‐target activity of the enzymes, although the regulatory measures for APOBEC3 expression and activity remain undefined. It is therefore difficult to predict circumstances that enable APOBEC3 interaction with cellular DNA that leads to mutagenesis. The APOBEC3A (A3A) enzyme is the most potent deaminase of the family. Using proteomics, we evaluate protein interactors of A3A to identify potential regulators. We find that A3A interacts with the chaperonin‐containing TCP‐1 (CCT) complex, a cellular machine that assists in protein folding and function. Importantly, depletion of CCT results in A3A‐induced DNA damage and cytotoxicity. Evaluation of cancer genomes demonstrates an enrichment of A3A mutational signatures in cancers with silencing mutations in CCT subunit genes. Together, these data suggest that the CCT complex interacts with A3A, and that disruption of CCT function results in increased A3A mutational activity.
Keywords: APOBEC3A, CCT chaperonin, cytidine deaminase, protein interaction, mutational signatures
Subject Categories: Cancer; DNA Replication, Repair & Recombination; Post-translational Modifications, Proteolysis & Proteomics
The CCT chaperonin complex interacts with the cytidine deaminase APOBEC3A that causes genotoxicity when dysregulated. Depletion of CCT leads to APOBEC3A‐induced DNA damage and cytotoxicity, implicating the CCT complex as a regulator of APOBEC3A activity.
Introduction
The human genome encodes seven APOBEC3 cytidine deaminase enzymes that catalyze the conversion of cytidine bases to uracil on single‐stranded DNA substrates (Jarmuz et al, 2002; Yu et al, 2004; Byeon et al, 2013; Mitra et al, 2014). Subsequent processing of uracil leads to mutations and DNA breaks. Several of the human APOBEC3 enzymes (named A3A through A3H) have the capacity to act as anti‐viral effectors through deamination of viral genomes, resulting in widespread mutations, genome degradation, and restriction of infectivity (Chen et al, 2006; Vartanian et al, 2008; Malim & Bieniasz, 2012; Ooms et al, 2012; Harris & Dudley, 2015). However, in what is presumed to be off‐target activity, APOBEC3 enzymes can also access and deaminate the cellular genome (Landry et al, 2011; Suspene et al, 2011; Burns et al, 2013a; Green et al, 2016). Two of the APOBEC3 family members, A3A and A3B, have been implicated as sources of mutagenesis in human cancers (Nik‐Zainal et al, 2012; Alexandrov et al, 2013; Burns et al, 2013a; Alexandrov et al, 2016; Petljak et al, 2019). Elevated expression of APOBEC3 enzymes, along with mutational patterns consistent with cytidine deaminase activity, have been identified in tumors (Burns et al, 2013a; Roberts et al, 2013; Petljak & Alexandrov, 2016; Cancer Genome Atlas Research Network et al, 2017; Green et al, 2017; Cortez et al, 2019; Jalili et al, 2020). These observations suggest that aberrant activity of APOBEC3 enzymes may contribute to oncogenesis, clonal diversity, tumor evolution, and chemotherapy resistance (Burns et al, 2013a; Roberts et al, 2013; Faltas et al, 2016; Roper et al, 2019).
The concept of off‐target APOBEC3 activity is ill‐defined, since the regulation of APOBEC3 expression and activity under non‐pathogenic conditions remains enigmatic. Basal expression levels of APOBEC3 enzymes are low in most healthy tissues (Koning et al, 2009; Refsland et al, 2010). Expression of two APOBEC3 family members, A3A and A3G, is upregulated by type I interferon stimulation of hematopoietic cells and keratinocytes (Chen et al, 2006; Koning et al, 2009; Wang et al, 2014), likely reflecting an innate immune response to viral pathogens (Peng et al, 2006; Mohanram et al, 2013). However, regulatory measures that ensure the host genome is protected from APOBEC3 activity have not been elucidated. Given the proposed role of APOBEC3 enzymes in cancer mutagenesis, we sought to identify APOBEC3 protein binding partners in order to determine potential regulators of host genome deamination. Here, we show that A3A interacts with and is regulated by the chaperonin‐containing TCP‐1, CCT (also called TRiC) complex.
The CCT complex is a molecular chaperonin that enables folding of newly translated proteins to promote physiologic localization and activity (Yam et al, 2008; Joachimiak et al, 2014; Lopez et al, 2015). CCT is a double‐ring oligomer comprised of eight subunits which encompass client proteins in an ATP‐dependent manner (Booth et al, 2008; Cong et al, 2012; Reissmann et al, 2012). The canonical roles of this complex include folding of nascent proteins and prevention of protein aggregation (Thulasiraman et al, 1999; Kitamura et al, 2006; Tam et al, 2006; Yam et al, 2008). The CCT complex interacts with ˜ 15% of newly synthesized proteins (Yam et al, 2008), and although the fraction that require CCT for folding and proper function is likely smaller, CCT plays an important role in essential cellular processes through protein homeostasis (Tyedmers et al, 2010; Hartl et al, 2011; Lopez et al, 2015). Notable clients of the CCT complex include cytoskeletal components actin and tubulin, oncoproteins TP53, von Hippel–Lindau, and STAT3, and checkpoint complex proteins PLK1 and CDC20 (Gao et al, 1992; Gao et al, 1993; Rommelaere et al, 1993; Feldman et al, 1999; Camasses et al, 2003; Liu et al, 2005; Trinidad et al, 2013; Kasembeli et al, 2014; Kaisari et al, 2017). Experimental deficiency of the CCT complex has resulted in phenotypes ranging from cancer to neurologic disorders, highlighting its broad role in cellular function (Jin et al, 2019).
In this study, we investigated protein interactors of A3A using affinity purification and mass spectrometry (MS). Surprisingly, we identified all eight subunits of the CCT complex by MS upon immunoprecipitation of A3A. Our MS findings were validated across a panel of cell lines by co‐precipitation of A3A and the CCT complex. Furthermore, we demonstrated that depletion of the CCT complex results in increased DNA damage and cytotoxicity caused by A3A. Through in silico evaluation of cancer genome sequences, we found that A3A mutational signatures are enriched in tumors with CCT gene mutations. Together, our data point to the CCT chaperonin as a regulator of A3A that mitigates off‐target deamination of the genome.
Results and Discussion
APOBEC3A interacts with the CCT molecular chaperonin complex
To identify protein interactors of A3A, we developed a U2OS‐HiLo cell line with doxycycline‐inducible HA‐tagged APOBEC3A. The HiLo system enables recombination‐mediated genomic integration of a single transgene per cell, leading to uniform expression levels of inducible genes in each cell (Khandelia et al, 2011). Inducible A3A expression in U2OS‐HiLo cells was evaluated based on dose and duration of doxycycline treatment; a dose of 1 μg/ml for 72 h was selected for immunoprecipitation experiments (Fig EV1A). There is significant variability in subcellular localization among APOBEC3 family members: three are expressed in the cytoplasm (A3F, A3G, A3H) and one is nuclear (A3B) (Lackey et al, 2012; Lackey et al, 2013). Unlike other APOBEC3 enzymes, A3A exhibits pan‐cellular localization (Landry et al, 2011; Lackey et al, 2013). Consistent with previous evaluations, A3A was detected in both nucleus and cytoplasm when induced in U2OS‐HiLo cells (Fig EV1B). Anti‐HA antibody was used to immunoprecipitate A3A‐HA from U2OS‐A3A cell lysates (Figs 1A and EV1C). Doxycycline‐treated parental U2OS‐HiLo cell lysates were used as a control. In order to identify co‐precipitated proteins using a proteomics approach, HA immunoprecipitations were performed in triplicate, verified by immunoblot (Fig 1A), run on SDS–PAGE gels (Fig EV1C), and analyzed by mass spectrometry (Fig EV1D). To identify proteins that interact specifically with A3A, we utilized the Proteome Discoverer proteomics analysis platform. Enriched proteins were compared between HiLo cells induced to express A3A and parental HiLo cells (Fig 1A, Dataset EV1). We found substantial concordance among three replicates; 74% of proteins identified were enriched in A3A‐expressing cells compared to parental HiLo cells in each replicate (Fig 1B). Using a stringent P‐value cut‐off of 0.05 (by one‐tailed t‐test) to define significance, we identified 63 proteins that were significantly enriched by affinity purification from A3A‐expressing U2OS cells as compared to doxycycline‐treated parental cells (Dataset EV1). We used the STRING database (Snel et al, 2000; Szklarczyk et al, 2019) to further investigate functional interactions among A3A co‐precipitated proteins. Gene Ontology analysis of the proteins that were significantly enriched in the A3A dataset when compared to the parental dataset revealed all eight members of the CCT molecular chaperonin complex (CCT1‐8) (Fig 1C and D). In addition, A3A immunoprecipitated with immune response proteins and ribonucleotide‐binding proteins (Fig 1D). Given the striking enrichment of each CCT subunit among proteins that co‐immunoprecipitated with A3A, we chose to further study the role of the CCT complex interaction with A3A (Fig 1C and D).
Figure EV1. Approach for discovery of APOBEC3A protein interactors.
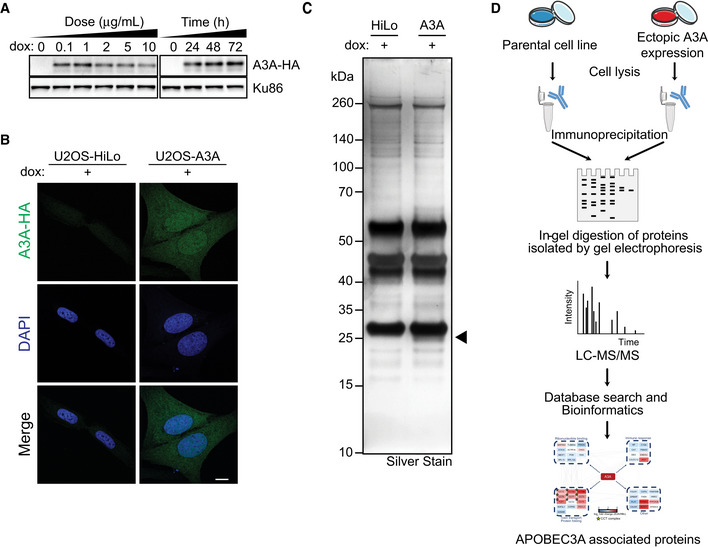
- Ectopically expressed HA‐tagged A3A was induced by treatment with dox at varying concentrations and duration in U2OS‐HiLo cells. Lysates were analyzed by immunoblot using an HA antibody. Ku86 was used as a loading control. Based on these results, proteomics experiments were performed on both the U2OS‐A3A and the parental U2OS‐HiLo cells treated with 1 μg/ml dox for 72 h.
- A3A expression is pan‐cellular in U2OS cells. Parental U2OS‐HiLo cells and U2OS‐A3A cells were treated with dox, stained with HA antibody, and analyzed by confocal microscopy. Nuclei were stained with DAPI. Scale bar is 10 μm.
- Silver stain detection of A3A‐HA immunoprecipitation in U2OS‐HiLo cells. U2OS‐HiLo cells and U2OS‐A3A cells were immunoprecipitated with HA antibody after treatment with 1 μg/ml dox for 72 h. Lysates were separated by PAGE and analyzed by silver stain. APOBEC3A appears under the immunoglobulin light chain at ˜ 24 kDa (indicated by arrow).
- A quantitative proteomics approach was employed to identify protein interactors of A3A. Schematic of the approach for discovery of protein interactors of A3A in a U2OS cell line by immunoprecipitation followed by quantitative proteomics.
Source data are available online for this figure.
Figure 1. APOBEC3A interacts with all subunits of the CCT chaperonin complex.
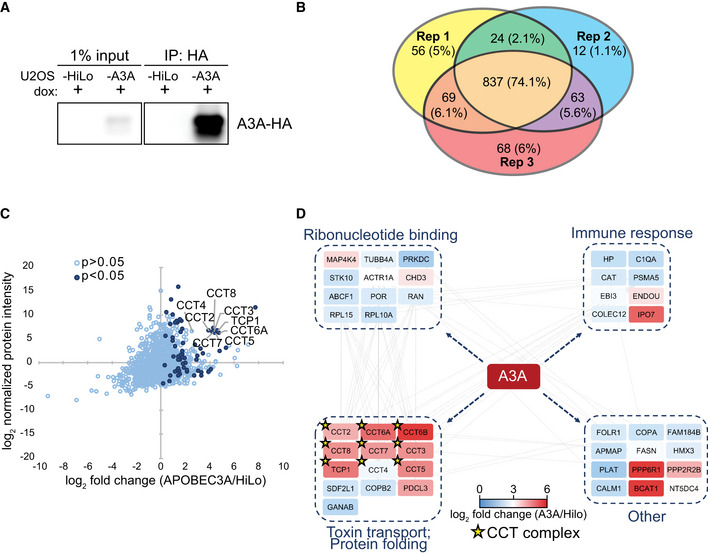
- Immunoprecipitation of A3A. U2OS‐HiLo cells with doxycycline‐inducible, haemagglutinin (HA)‐tagged A3A (U2OS‐A3A) and parental cells (U2OS‐HiLo without transgene) were treated with dox for 72 h. Immunoblot detection of A3A‐HA after immunoprecipitation (IP) of cell lysates with anti‐HA antibody is shown.
- Comparison of mass spectrometry results from three (n = 3) replicates. IP followed by mass spectrometry was performed in triplicate for parental U2OS‐HiLo and U2OS‐A3A cell lines. Venn diagram shows the overlap of proteins identified in three biological replicates of HA immunoprecipitation from U2OS‐A3A cells. Number of proteins identified in each replicate is displayed along with the percentage of protein overlap identified between replicates.
- All eight subunits of the CCT chaperonin complex were enriched in U2OS‐A3A samples. Proteins detected by MS are displayed by fold enrichment (log2) from U2OS‐A3A cells compared to parental cells (x‐axis) and by protein abundance within the U2OS‐A3A samples (y‐axis). Significantly enriched proteins (P < 0.05 by one‐tailed t‐test, n = 3) are denoted in dark blue. Components of the CCT complex are highlighted and labeled.
- Network of specific interactors of APOBEC3A. Only proteins significantly enriched in A3A samples over HiLo were considered for network analysis (log2 fold change > 0; one‐tailed t‐test P < 0.0.5), with the exception of CCT6B and CCT4 (t‐test P > 0.05). Node colors denote enrichment (indicated by heatmap legend) of interacting protein in A3A samples compared to HiLo. Nodes are grouped into boxes according to gene ontology enriched biological processes for interacting proteins (STRING database, FDR < 0.01). Gray edge lines indicate observed interactions between proteins. CCT complex members are denoted by a star.
Source data are available online for this figure.
The CCT complex interacts specifically with A3A
To confirm the interaction between the CCT complex and A3A, we used a lentiviral expression system to engineer inducible A3A expression in two human cell lines (osteosarcoma U2OS cells and hepatic HepaRG cells) and evaluated co‐immunoprecipitation with CCT subunits. First, we immunoprecipitated A3A‐HA and used antibodies to CCT subunits to demonstrate CCT co‐precipitation (Fig 2A). Next, in reciprocal experiments we immunoprecipitated the CCT complex using antibodies to endogenous CCT subunits and found that A3A was pulled down (Fig 2B). To address the interaction in physiologically relevant tissues, we engineered additional cell lines to express inducible A3A including hematopoietic cells (CEM, Ramos, K562) and breast cancer cells (MDA‐MB‐231). Expression and mutational activity of A3A has been previously demonstrated in both hematopoietic cancers and breast tumors (Nik‐Zainal et al, 2012; Burns et al, 2013a; Green et al, 2017; Cortez et al, 2019; Jalili et al, 2020). We immunoprecipitated A3A from several leukemia cell lines and again found that the CCT complex was isolated (Fig 2C). In breast cancer cells, we performed reciprocal co‐immunoprecipitations to demonstrate the A3A‐CCT interaction (Fig 2D). These data validate the interaction between A3A and the CCT complex that we identified by proteomic analysis, including in tissues in which A3A has been shown to impact cancer genomes.
Figure 2. The CCT complex interacts with APOBEC3A in multiple cell types.
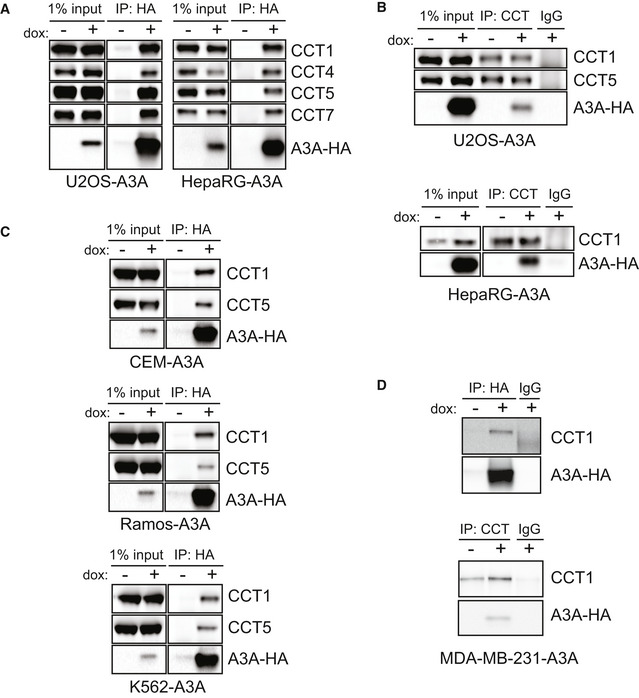
- A3A co‐precipitates with CCT complex subunits. U2OS and HepaRG cells with dox‐inducible A3A‐HA transgenes were treated with dox prior to immunoprecipitation of HA. HA immunoprecipitates were analyzed by immunoblot with antibodies for HA and subunits of the CCT complex.
- Immunoprecipitation of CCT. IP of the CCT complex using antibodies to endogenous CCT subunits was performed in lysates from U2OS‐A3A (top) and HepaRG‐A3A (bottom) cells after dox treatment. Immunoblot analysis with probes for A3A‐HA and indicated subunits of CCT with IgG as a control is shown.
- A3A interacts with CCT in leukemia cells. HA immunoprecipitation in hematopoietic cell lines with inducible A3A‐HA was analyzed by immunoblot. HA, CCT1, and CCT5 antibodies were used to evaluate co‐precipitation of CCT subunits with A3A.
- The CCT‐A3A interaction occurs in breast cancer cells. HA (top) and CCT (bottom) immunoprecipitation in the MDA‐MB‐231 cell line with inducible A3A‐HA expression was analyzed by immunoblot. Anti‐HA and anti‐CCT1 antibodies were used to evaluate reciprocal co‐IPs.
Source data are available online for this figure.
We then asked whether the CCT complex interacts with other APOBEC3 family members. The APOBEC3 proteins are highly similar; the degree of homology between A3A and A3B or A3G is higher than 50% (Jarmuz et al, 2002; Narvaiza et al, 2009; Bulliard et al, 2011). Thus, we sought to determine whether homologous APOBEC3 enzymes also interact with CCT. Ectopic expression of HA‐tagged A3A, A3B, and A3G was achieved by transfection of expression vectors into 293T cells. Immunoprecipitation of the CCT complex demonstrated co‐precipitation with only A3A, and not A3B or A3G (Fig 3A). The converse experiment, in which the APOBEC3 proteins were immunoprecipitated, revealed that only A3A pulldown resulted in co‐precipitation of CCT complex subunits (Fig 3B). To ensure that this result was generalizable across cellular contexts, we immunoprecipitated doxycycline‐inducible HA‐tagged APOBEC3 proteins expressed in U2OS cell lines. We evaluated A3A and A3B and found that A3A, but not A3B, co‐precipitated with the CCT complex (Fig 3C). To determine whether catalytic activity of A3A is required for CCT binding, we performed immunoprecipitation of the catalytically inactive A3A‐C106S mutant (Bogerd et al, 2006; Chen et al, 2006; Narvaiza et al, 2009). We found that the mutant co‐precipitated with CCT subunits as efficiently as wild‐type A3A (Fig 3C). Thus, a functional deaminase domain of A3A is not essential for CCT interaction. Our findings demonstrate that the APOBEC3‐CCT interaction occurs specifically with A3A.
Figure 3. The APOBEC3‐CCT interaction is specific to A3A.
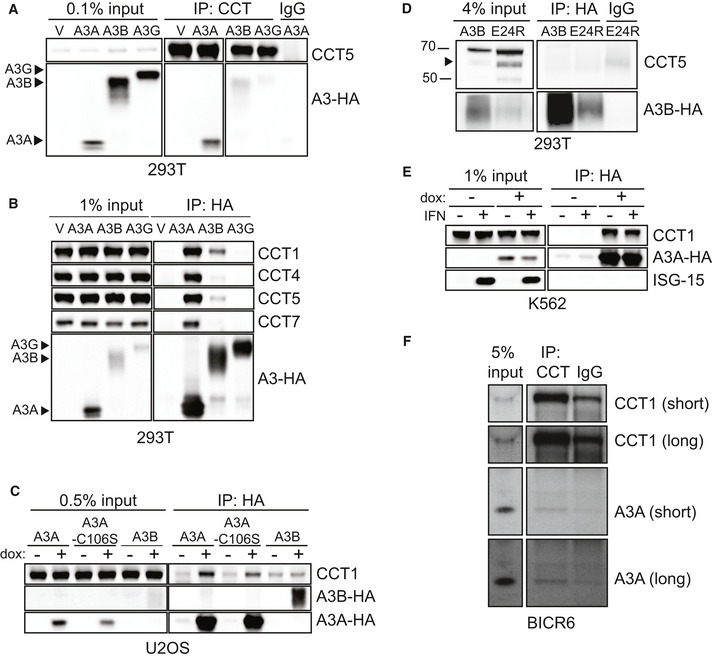
- CCT co‐precipitates with A3A but not A3B or A3G. 293T cells were transfected with empty vector (V), A3A‐HA, A3B‐HA, or A3G‐HA. CCT IP was performed from transfected cells and evaluated by immunoblot analysis using antibodies for CCT5 and HA.
- A3A, but not other APOBEC3 enzymes, interacts with CCT complex subunits. After transfection of 293T cells with HA‐tagged APOBEC3 expression plasmids, IP was performed with anti‐HA antibody and analyzed by immunoblot using antibodies to HA and CCT subunits. Arrows mark location of each APOBEC3 protein on HA immunoblot.
- HA immunoprecipitation was performed from U2OS cells with dox‐inducible A3A, A3A‐C106S (catalytically inactive mutant), and A3B transgenes. IP lysates were evaluated by immunoblot using antibodies to HA and CCT1.
- Immunoprecipitation of HA‐tagged A3B constructs transfected into 293T cells. IP of A3B‐HA and A3B‐E24R‐HA (cytoplasmic localization) was evaluated by immunoblot analysis using antibodies to HA and CCT1. Arrow indicates CCT5 band.
- CCT interacts with A3A both with and without interferon. Immunoprecipitation of A3A‐HA in K562 cells treated with and without type I interferon (IFN). Anti‐ISG15 antibody serves as a control for IFN induction.
- CCT co‐precipitates with endogenous A3A. Immunoprecipitation of the CCT complex in BICR6 cells was analyzed by immunoblot using antibodies to endogenous A3A and CCT1. Chemiluminescent exposure times described as short and long are indicated beside the blot. IgG antibody was used as a control throughout.
Source data are available online for this figure.
Since the subcellular localization of A3A differs from that of A3B, we asked whether cytosolic CCT required cytoplasmic localization of client APOBEC3 proteins for interaction. The A3B E24R mutant re‐localizes nuclear A3B to the cytoplasm (Salamango et al, 2018). We immunoprecipitated wild‐type A3B and A3B‐E24R mutant and found that, although the A3B‐E24R mutant appears to be less stable than wild‐type A3B, neither co‐precipitated with CCT complex subunits (Fig 3D). These data suggest that specificity of CCT interaction with A3A is not simply due to subcellular localization and may not be influenced by APOBEC3 amino acid sequence given the significant homology between A3A and other APOBEC3 proteins.
The A3A gene is interferon‐stimulated and expression is increased significantly upon treatment with type I interferon (Chen et al, 2006; Koning et al, 2009). Although no prior reports have addressed the effect of interferon on CCT interaction with client proteins, we evaluated the impact on the A3A‐CCT interaction. To accomplish this, we treated an interferon‐responsive hematopoietic cell line, K562, with type I interferon. ISG‐15, a canonical interferon‐stimulated gene, was used as a control for interferon treatment. A3A was immunoprecipitated from cells treated with and without interferon, and there was no difference in CCT co‐precipitation (Fig 3E). Thus, interferon does not appear to influence the interaction of A3A and CCT.
We addressed whether A3A and/or A3B, when endogenously expressed, interact with CCT. To do so, we utilized the BICR6 squamous cell carcinoma line in which A3A is expressed. Upon immunoprecipitation of the CCT complex, we found co‐precipitation of A3A (Fig 3F). These data demonstrate that the A3A‐CCT interaction occurs in the context of endogenous A3A expression.
Cytotoxicity from A3A is increased upon CCT depletion
Based on the specific and conserved interaction between A3A and CCT, we hypothesized that the CCT complex serves as a regulatory measure of the A3A protein. Since A3A can deaminate ssDNA at replication forks of cycling cells (Green et al, 2016; Hoopes et al, 2016; Seplyarskiy et al, 2016), we hypothesized that the CCT complex may prevent A3A from deaminating and damaging cellular DNA. To evaluate the impact of the A3A‐CCT interaction on the genome, we investigated the outcome of disrupting the interaction by depleting CCT complex proteins. The CCT complex consists of stoichiometrically equal amounts of each protein, and free CCT complex subunits are rapidly degraded (Kunisawa & Shastri, 2003; Kasembeli et al, 2014). Consistent with previous reports (Kunisawa & Shastri, 2003; Kasembeli et al, 2014), we found that short interfering RNA (siRNA)‐mediated knockdown of one CCT complex protein resulted in depletion of the remaining CCT complex members (Fig 4A), leading to effective suppression of CCT complex activities. We used siRNA to knock down CCT4 and CCT7 in U2OS cells, which resulted in significant protein depletion (Fig 4B immunoblots). We evaluated the impact of A3A expression with CCT depletion in inducible cell lines and found that the combination of A3A expression and CCT depletion resulted in significantly decreased cell viability by WST‐8 assay (Fig 4B bar graphs). We also evaluated cell death by live‐dead staining in CCT‐depleted cells and found a significant increase upon expression of A3A, greater than CCT depletion alone (Fig 4C). We hypothesized that the catalytic activity of A3A on the genome was responsible for increased genome instability resulting in cell death upon CCT depletion; thus, we evaluated the effect of a catalytically inactive A3A mutant (A3A‐C106S) on cytotoxicity. Regardless of CCT depletion, we found no increase in cell death when the inactive A3A mutant was expressed (Fig 4D). Since A3A activity has been shown to cause DNA damage and elicit DNA damage response signaling including phosphorylation of histone variant H2AX (γH2AX), a marker of DNA breaks (Landry et al, 2011; Burns et al, 2013a; Mussil et al, 2013; Green et al, 2016), we addressed whether A3A‐mediated DNA damage was altered upon CCT depletion. To do so, CCT7 was depleted by siRNA targeting in K562 cells with ectopic inducible A3A expression. The combination of CCT7 depletion and A3A expression resulted in significantly increased γH2AX as compared to controls (Fig 4E). Together, these data suggest that the interaction between CCT and A3A mitigates genome deamination, DNA damage, and genotoxicity.
Figure 4. CCT complex depletion leads to decreased cell viability when A3A is expressed.
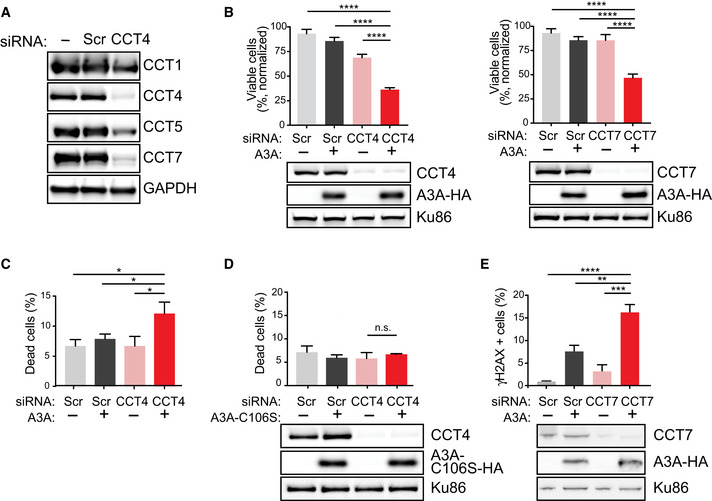
- Knockdown of one CCT complex member results in depletion of other CCT subunits. Following siCCT4 or siCCT7 transfection, cell lysates were evaluated by immunoblot for expression of additional CCT subunits by endogenous antibodies for CCT1, CCT4, CCT5, and CCT7. GAPDH was used as a loading control.
- Decreased cell viability upon CCT depletion and A3A expression. CCT4 (left) and CCT7 (right) subunits were depleted by siRNA transfection in U2OS cells with dox‐inducible A3A transgenes. Immunoblot analysis of cell lysates showed decreased expression of targeted CCT subunit, but no alteration in A3A expression. Scr indicates non‐targeted siRNA, used as control. Ku86 is a loading control. Following siRNA transfection to deplete CCT subunits, U2OS‐A3A cells were treated with dox. Viability was determined by colorimetric change after addition of a water‐soluble tetrazolium salt. Percent viability was normalized to untreated controls. Statistical analysis was performed using a paired two‐tailed t‐test, n = 3 biological replicates; error bars, SEM.
- Increased cell death resulting from A3A expression along with depletion of CCT. U2OS‐A3A cells were depleted of CCT4 by siRNA, treated with dox, or combination. Cell death was measured by staining cells with fluorescent‐labeled calcein AM (live) and DNA (dead) stains. Bar chart shows quantitation of FACS results averaged over three biological replicates. Statistical analysis was performed using a paired two‐tailed t‐test, n = 3; error bars, SEM.
- Viability of cells with catalytically inactive A3A. U2OS‐A3A‐C106S cells were induced with dox to express the catalytically inactive A3A mutant (C106S), depleted of CCT4 by siRNA, or combination. Cells were subsequently evaluated by live‐dead staining assessed by FACS. Bar chart shows quantitation of FACS results averaged over three biological replicates. Statistical analysis was performed using a paired two‐tailed t‐test, n = 3; error bars, SEM. Immunoblot analysis of cell lysates showed decreased expression of CCT4 after siRNA targeting. Ku86 is a loading control.
- Increased DNA damage signaling upon CCT depletion and A3A expression. Depletion of CCT7 in K562‐A3A cells was achieved by siRNA targeting. Expression of A3A was induced by dox treatment. Intracellular staining with anti‐γH2AX antibody was analyzed by flow cytometry. Bar chart shows quantification of FACS results obtained over three biological replicates. Statistical analysis was performed using a two‐tailed t‐test, n = 3; error bars, SEM. Immunoblot analysis of cell lysates showed decreased expression of CCT7 after siRNA targeting and A3A expression upon dox treatment. Ku86 is a loading control. **P‐value < 0.01, ***P‐value < 0.01, ****P‐value < 0.0001, and n.s. non‐significant.
Source data are available online for this figure.
A3A mutational signatures are enriched in cancers with deleterious CCT mutations
We next evaluated human tumors from The Cancer Genome Atlas (TCGA) to determine the effect of CCT depletion on A3A mutagenesis. APOBEC3 activity has been extrapolated from mutational patterns identified in cancer genome sequences, and the mutational hallmarks of A3A activity are thought to include a high burden of mutations with predominantly C→T transitions (Nik‐Zainal et al, 2012; Alexandrov et al, 2013). We classified tumors with mutations within CCT genes that are predicted to cause negative effects on protein function, such as frameshift or non‐sense mutations or indels, as having deleterious CCT gene mutations (Choi et al, 2012; McLaren et al, 2016). Tumors with deleterious mutations in one or more CCT complex proteins were compared to tumors with intact CCT genes (Fig 5). In total, 326 tumors had a deleterious mutation in one or more CCT genes as compared to 9,876 genomes with intact CCT complex genes. Mutations of individual CCT complex member genes were relatively evenly distributed in this data set, ranging from 8 to 20% of the 326 tumors identified with CCT complex mutations. We found that tumors with CCT gene mutations had a higher overall burden of mutations, and in particular C→T transitions, than those with intact CCT genes (Fig 5A). Mutational signatures, comprised of patterns of single base substitutions (SBS) identified within a trinucleotide context, have been used to determine mutagenic processes active in cancers (Alexandrov et al, 2020). Within the COSMIC database, mutational signatures attributed to APOBEC3 activity are denoted SBS2 and SBS13 (Alexandrov et al, 2020). Both signatures consist of predominantly cytidine mutations within a TC dinucleotide context, which is consistent with the context preference in which A3A deaminates cytidine bases (Chen et al, 2006). We evaluated the frequency of SBS2 and SBS13 in genomes from tumors with deleterious CCT gene mutations and found a significant increase in both SBS2 and SBS13 as compared to tumors with intact CCT genes (Fig 5B). As a control, we performed a parallel evaluation of tumors with deleterious mutations in a randomly chosen set of nine genes which were of similar size to the CCT complex genes and were mutated with similar frequency to the CCT complex genes (Fig EV2). Although the random gene set‐mutated tumors harbored mutational burdens similar to CCT‐mutated tumors (Fig EV2A), we found that the increase in SBS2 and SBS13 occurred more so in the CCT‐mutated tumors (though not significant for SBS2, Fig EV2B). Thus, it appears that the mutational pattern of APOBEC3 activity is enriched more so in cancer genomes from tumors with dysfunctional CCT complexes.
Figure 5. A3A mutational signatures are enriched in cancers with CCT mutations.
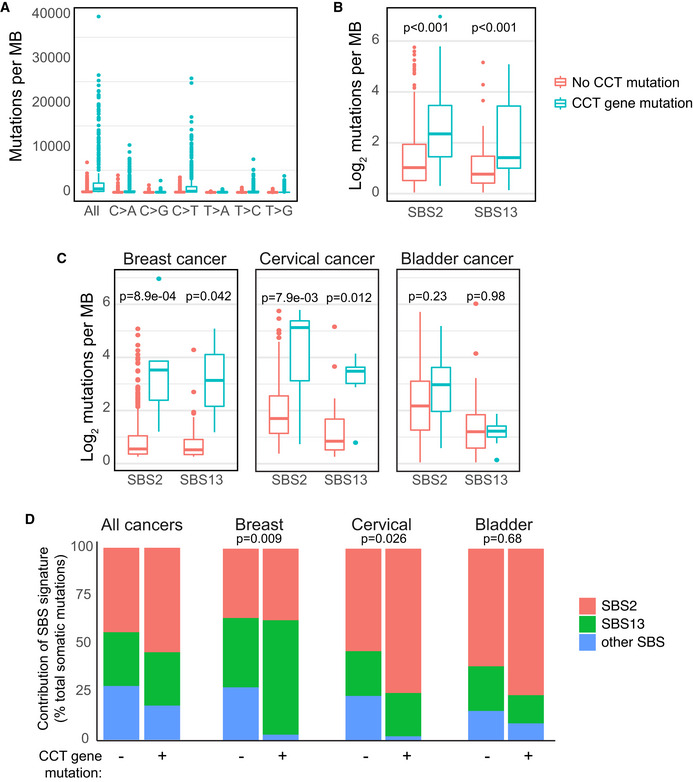
- Tumors with CCT gene mutations have an increased mutational burden. All cancer genome sequences in TCGA were evaluated for mutation burden and specific base substitutions. Cancers with CCT gene mutations predicted to cause a negative impact on protein function were classified as deleterious and were compared to those with no mutations in any CCT gene. Wilcoxon rank sum test applied to all paired comparisons between tumors with and without CCT gene mutations yielded P < 0.001.
- The APOBEC3 mutational signature is enriched in tumors with CCT gene mutations. Single base substitution (SBS) patterns, as defined by COSMIC Mutational Signatures v2, were evaluated in CCT‐mutated and CCT non‐mutated cancer genome sequences. Depicted are the mutational signatures attributable to APOBEC3 activity (SBS2 and SBS13). Denoted P‐values determined by Wilcoxon rank sum test.
- Quantification of APOBEC3 mutational signatures among three tumor types that have previously been associated with high APOBEC3 activity. Breast, bladder, and cervical cancer samples from the TCGA database were divided into CCT‐mutated and non‐mutated genomes, as above. The number of mutations contributing to SBS2 and SBS13 in each tumor type was quantified and displayed as an average. Comparison by Wilcoxon rank sum test yielded P‐values denoted.
- The contribution of APOBEC3 mutational signatures (SBS2 and SBS13) and all other SBS signatures defined by COSMIC v2 was evaluated in all tumors and independently in breast, bladder, and cervical cancer genomes within the TCGA database. A comparison of mutational signature contributions between CCT‐mutated and non‐mutated tumors is shown. A z‐test of proportions was used to compare whether the percentage of “other” signatures is significantly different between CCT‐mutated and non‐mutated tumors, and P‐values show statistical significance in breast and cervical cancers but not in bladder cancer.
Data information: Box and whisker plots depict median and interquartile range (25–75%).
Figure EV2. Comparison of APOBEC mutational signature burden in CCT‐deficient and control tumors.
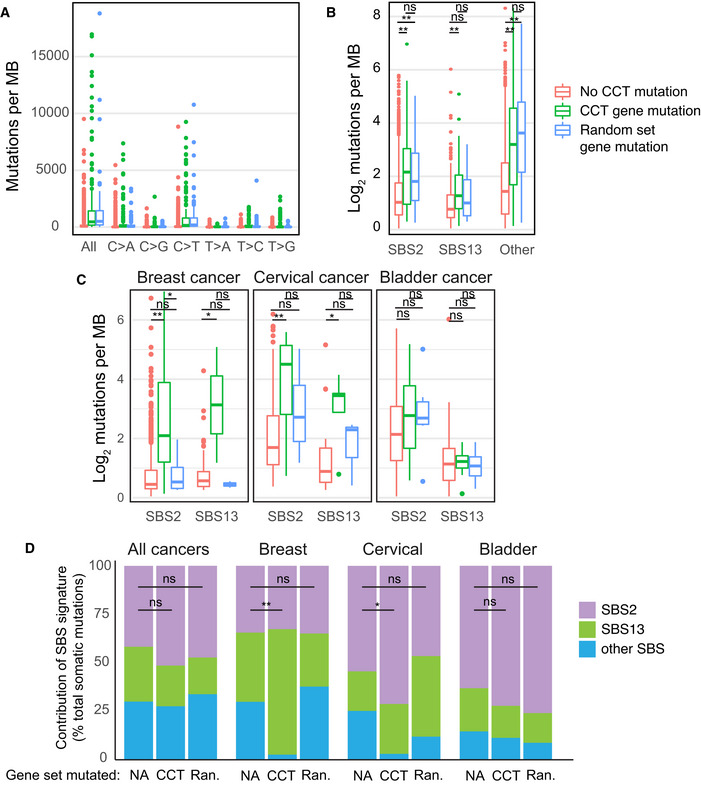
- All cancer genome sequences in TCGA were evaluated for mutation burden and specific base substitutions. Cancers with deleterious mutations in one or more CCT complex genes were compared to a control group of primary cancers that harbor deleterious mutations in a random gene set. The random gene set was constructed from 9 genes of similar size to those that comprise CCT members, and with similarly frequent mutational rate among the 9 genes relative to mutation rate among CCT complex genes. Wilcoxon rank sum test was used for statistical comparison. For each column on the x‐axis, a comparison between tumors without CCT mutations and those with CCT mutations is significant (P < 0.01). Similarly, for each x‐axis column a comparison between tumors without CCT mutations and those with random gene mutations is significant (P < 0.01). For each x‐axis column, a comparison between tumors with CCT gene mutations and tumors with random gene mutations is not statistically significant.
- The burden of mutations attributed to APOBEC3 activity (SBS2 and SBS13) was evaluated in CCT‐mutated, random gene set‐mutated, and all other cancer genome sequences. Depicted is the quantity of mutations within mutational signatures attributable to APOBEC3 activity (SBS2 and SBS13) in comparison with all other SBS mutational patterns (other). Wilcoxon rank sum test was used for statistical comparison. **P < 0.01 and ns non‐significant.
- Quantification of APOBEC3 mutational signatures among breast, bladder, and cervical cancer samples from the TCGA database was divided into CCT‐mutated, random gene set‐mutated, and non‐mutated genomes, as above. The number of mutations contributing to SBS2 and SBS13 in each tumor type is quantified and displayed as an average. Wilcoxon rank sum test was used for statistical comparison. **P < 0.01, *P < 0.05, and ns non‐significant.
- The contribution of SBS2, SBS13, and all other SBS signatures was evaluated in all tumors and independently in breast, bladder, and cervical cancer genomes within the TCGA database. A comparison of mutational signature contributions between CCT‐mutated, random gene set‐mutated (Ran.), and non‐mutated tumors is shown. A z‐test of proportions was used to compare whether the percentage of “other” signatures is significantly different between CCT‐mutated and non‐mutated tumors. **P < 0.01, *P < 0.05, and ns non‐significant.
Next, we addressed mutational signatures in a subset of tumors in which APOBEC3 activity or expression has been specifically reported including breast, cervical, and bladder cancer (Nik‐Zainal et al, 2012; Burns et al, 2013a; Faltas et al, 2016; Robertson et al, 2017). APOBEC3 mutational signatures SBS2 and SBS13 were quantified in the aforementioned tumors from TCGA (Fig 5C). In all tissue types, the APOBEC3 signatures were increased in tumors with CCT gene mutations compared to those with intact CCT genes. Finally, the contribution of all COSMIC SBS signatures to total mutation burden was assessed in TCGA samples. The contribution of SBS2 and SBS13 was higher in tumors which had CCT gene mutations (Fig 5D). Of note, the combined contribution of all other SBS signatures was lower in tumors with CCT gene mutations (Fig 5D), which suggests that APOBEC3 deamination is a predominant mutagenic process. We independently evaluated breast, cervical, and bladder tumors with and without CCT gene mutations and found an increased contribution of APOBEC3‐associated SBS2 and SBS13 in tumors with CCT gene mutations as compared to tissue‐matched controls with intact CCT genes (Fig 5D). When SBS2 and SBS13 were evaluated in control tumors with mutations in a random gene set, we found no increase in breast cancer genomes and a modest increase in cervical cancer genomes which was lower than the SBS2/13 mutations in CCT‐mutated tumors (Fig EV2C and D). In bladder cancers, the contribution of SBS2 and SBS13 was similar in control tumors and those with CCT gene mutations (Fig EV2D). Taken together, these findings suggest overall that CCT dysfunction in human tumors is associated with increased APOBEC3 activity on the genome.
The role of APOBEC3 enzymes in cancer mutagenesis remains enigmatic, in part because our mechanistic understanding of these endogenous DNA mutators in relation to the host genome is incomplete. The two main bodies of evidence that link APOBEC3 enzymes to cancer indicate that (i) APOBEC3 enzymes are expressed at abnormally high levels in a subset of human cancers and (ii) a mutational pattern consistent with APOBEC3 deaminase activity is widespread in cancer genome sequences. However, correlation between APOBEC3 expression and activity has been weak at best, both experimentally and in primary tumor samples (Burns et al, 2013b; Chan et al, 2015; Cortez et al, 2019). In other words, the majority of cancer genome sequences which display APOBEC3 mutational hallmarks do not have elevated APOBEC3 expression levels (Petljak et al, 2019). This discord has been explained by hypothetical transient expression of APOBEC3 proteins which, upon genotoxicity, become downregulated (Roberts & Gordenin, 2014). However, this hypothesis does not explain the converse discordance, i.e., when high APOBEC3 expression is found in a tumor, the APOBEC3 mutational signature is often not enriched in the corresponding genome (Roberts et al, 2013; Petljak et al, 2019). This observation suggests that regulatory measures within a cell could mitigate APOBEC3 activity on the genome despite high expression.
In this study, we sought to identify protein interactors that influence the activity of A3A on the cellular genome. Immunoprecipitation of A3A followed by mass spectrometry unexpectedly demonstrated an interaction with all subunits of the CCT chaperonin complex. The CCT chaperonin interacts with up to 15% of all cytosolic proteins, including an array of highly varied clients (Yam et al, 2008). The canonical client proteins of CCT are large and complex and include beta‐rich strands and/or WD repeats (Thulasiraman et al, 1999; Kubota et al, 2006; Yam et al, 2008; Freund et al, 2014; Miyata et al, 2014). While A3A is not large, it has the classical structure of a DNA cytidine deaminase consisting of a fold composed of 5 beta‐strands, 6 alpha‐helices, and a zinc‐coordinated domain (Byeon et al, 2013; Bohn et al, 2015). Thus, A3A shares some features with typical CCT client proteins.
The A3A‐CCT interaction was reproducible across many cellular contexts and appears to be specific to A3A since no interaction between CCT subunits and A3B or A3G was observed. This specificity is curious given the high degree of homology between A3A and the C‐terminal domains of A3B (93%) and A3G (66%). CCT is primarily found in the cytoplasmic compartment and A3A is expressed in both nucleus and cytoplasm, suggesting the interaction likely occurs in the cytoplasm similar to most CCT client proteins (Thulasiraman et al, 1999). In contrast to A3A, the A3B protein is located exclusively in the nucleus, and therefore, the interaction with CCT may have been limited by disparate subcellular localization. However, we also could not detect interaction with CCT for cytoplasmic A3B. The A3B protein has a similar DNA cytidine deaminase fold to that of A3A (Byeon et al, 2016); thus, we suspect that factors exogenous to the protein structure confer the specificity of the A3A‐CCT interaction, presenting an area for future study.
Few protein interactors of A3A have been identified to date. The TRIB3 pseudokinase was previously reported to interact with A3A (Aynaud et al, 2012), and the interaction was suggested to inhibit A3A deamination of nuclear DNA (Aynaud et al, 2012). Similarly, we found that depletion of CCT resulted in increased deaminase‐dependent DNA damage and cytotoxicity induced by A3A, suggesting that the CCT complex serves as a negative regulator of A3A activity on the genome. It is possible that this occurs indirectly through, for example, CCT interaction with a protein client that mediates repair of A3A‐induced DNA damage. However, given our finding of a physical interaction between A3A and the CCT complex, we favor the likelihood of a direct impact of CCT on the A3A protein. Most substrates are released from the CCT complex upon folding, but in some cases, CCT can act as a holdase. For example, the von Hippel–Lindau (vHL) protein is retained by CCT until the VHL binding partner, elongin‐BC, interacts with VHL and enables release from CCT (Feldman et al, 1999). Similarly, a recent study demonstrated that CCT functions as a holdase for the reovirus capsid protein σ3 to prevent aggregation of the protein prior to assembly into functional oligomeric complexes (Knowlton et al, 2018). It is possible that the CCT complex similarly functions as a holdase for A3A, thereby preventing access to the genome to minimize deamination and mutation. Further studies are required to determine the mechanism by which interaction with CCT mitigates A3A‐induced mutagenesis. If the CCT‐A3A interaction functions to sequester A3A, the signal for release may be related to the anti‐viral properties of A3A. A possible model for the CCT‐A3A interaction is that, upon viral infection, A3A is released from the CCT holdase and is therefore allowed to act on viral genomes leading to deamination, genome degradation, and viral restriction. In this model, disruption of the A3A‐CCT interaction would allow for A3A mutagenesis of the cellular genome. Although our study did not find that treatment with type I IFN changed the A3A‐CCT interaction, we did not directly assess whether viral infection disrupted the protein interaction.
The APOBEC3 mutational signature is among the most prevalent pattern of somatic mutagenesis in all sequenced cancer genomes (Petljak & Alexandrov, 2016). Here, we demonstrate that the hallmarks of APOBEC3 mutagenesis are increased in tumors with silencing mutations in CCT subunit genes. This provides one of the first examples of a cellular context that alters APOBEC3‐mediated genotoxicity. It was recently suggested that targeting CCT may present a therapeutic anti‐viral opportunity, since CCT is required for folding and assembly of viral capsid proteins (Knowlton et al, 2018). Inhibition of CCT may also present an opportunity to enable mutagenesis and genotoxicity selectively in cancer cells with high levels of A3A expression.
Materials and Methods
Cell lines
All cell lines were purchased from ATCC and tested for mycoplasma contamination every 6 months during experimentation. 293T, MDA‐MB‐231, and U2OS cells were maintained in DMEM supplemented with penicillin and streptomycin (100 U/ml) and 10% tetracycline‐free fetal bovine serum (TF‐FBS). BICR6 cells were maintained in DMEM supplemented with penicillin and streptomycin (100 U/ml), 10% fetal bovine serum (FBS), and 0.4 mg/ml hydrocortisone. HepaRG cells were maintained in William’s Medium E, supplemented with penicillin and streptomycin (100 U/ml), 10% TF‐FBS, L‐glutamine (2 mM), hydrocortisone (0.5 μM), and insulin (5 μg/ml). CEM, Ramos, and K562 cells were maintained in RPMI media supplemented with 10% TF‐FBS, penicillin and streptomycin (100 U/ml), and L‐glutamine (2 mM).
Inducible U2OS‐A3A, U2OS‐A3B, U2OS‐A3AC106S, MDA‐MB‐231‐A3A, HepaRG‐A3A, CEM‐A3A, Ramos‐A3A, and K562‐A3A cells were generated by lentiviral transduction using the pSLIK‐A3A lentivector with neomycin resistance, as previously described (Green et al, 2017). Inducible U2OS HiLo A3A cells were constructed by transfection of parental U2OS HiLo acceptor cells with inducible plasmid cassette containing HA‐tagged A3A along with plasmid expressing Cre recombinase, as previously described (Khandelia et al, 2011; Avgousti et al, 2016). All cells were induced by culture in the presence of doxycycline (0.1–5 μg/ml). All cells were grown at 37°C under 5% atmospheric CO2.
Antibodies
Commercially available antibodies used for immunoblotting, immunofluorescence, and intracellular staining were obtained from Abnova (CCT1, CCT5, CCT7), Santa Cruz Biotechnology (CCT4, ISG‐15, Ku86, Tubulin), GeneTex (GAPDH), BD Biosciences (γH2AX‐488), BioLegend (HA), Cell Signaling (HA), and the NIH AIDS Reagent Program (APOBEC3A/G). Secondary antibodies for immunoblotting included goat anti‐rabbit IgG and goat anti‐mouse IgG (Jackson ImmunoResearch). Secondary antibodies for immunofluorescence included anti‐Rabbit IgG (H + L), Alexa Fluor 488 (Invitrogen). For CCT immunoprecipitation, 1ug of each antibody was conjugated to protein G beads (Bio‐Rad): CCT1 clone 2B2‐D6 (Abnova), CCT4 clone H‐1 (Santa Cruz), CCT5 clone 4E5‐4B1 (Abnova), and CCT7 clone 1D6 (Abnova).
Plasmids, siRNA, and transfection
Expression vectors containing wild‐type and mutant APOBEC3 cDNA with C‐terminal HA epitopes have been previously described (Chen et al, 2006; Landry et al, 2011). Transfection of expression vectors was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Short interfering RNA (siRNA) targeting the CCT complex subunits was purchased from the Mission siRNA library (Sigma). Transfection of siRNA was achieved using the RNAiMAX transfection reagent (Invitrogen) according to the manufacturer’s protocol.
Immunoblotting, immunofluorescence, and intracellular staining
For immunoblotting, lysates were harvested by boiling for 15 min at 95°C in 10% LDS sample buffer (Invitrogen) and 5% B‐Mercaptoethanol (Sigma). Samples were run on Bis‐Tris gels and transferred to nitrocellulose membrane (Amersham). For immunofluorescence, cells were fixed with 4% paraformaldehyde (15 min) and permeabilized with 0.5% Triton‐X (10 min). Nuclei were visualized by 4,6‐diamidino‐2‐phenylindole (DAPI, Sigma). Images were acquired by confocal microscopy. For intracellular antibody staining of γH2AX, cells were harvested, fixed, and stained using the BD Cytofix/Cytoperm Kit according to the manufacturer’s protocol. Events were acquired using an LSRFortessa flow cytometer (BD Biosciences) and analyzed by FlowJo (version 10.7).
HA immunoprecipitation
Cells were harvested in RIPA buffer, lysed on ice for 10 min, sonicated for 5 min at 4°C in Bioruptor (Diagenode UCD‐200), and centrifuged to clear debris (25,000 g, 10 min, 4°C). Normalized lysates (1–3 mg total protein per 1 ml) were incubated with protein G beads (Invitrogen or Bio‐Rad) on rotator (30 min, 4°C) and then incubated with Pierce™ Anti‐HA Magnetic Beads (Thermo Scientific) on rotator (1‐4 h, 4°C). Beads were washed three times in RIPA buffer and eluted with LDS sample buffer (Invitrogen) by boiling (15 min, 95°C). For immunoblot analysis, 20% of eluate was run on NuPAGE™ 4‐12% Bis‐Tris Protein Gel (Invitrogen). For silver stain gel, 80% of eluate was run on NuPAGE™ 10% Bis‐Tris Protein Gel (Invitrogen) and stained using the Silver Stain kit (Pierce) according to the manufacturer’s protocol.
CCT complex immunoprecipitation
Cells were fixed with 1% paraformaldehyde (20 min) and quenched with 1.25 M glycine. Fixed cells were lysed as above. Normalized lysates (1.0 mg total protein in 1 ml) were incubated with protein G beads (Invitrogen or Bio‐Rad) on rotator (30 min, 4°C). Antibody‐conjugated protein G beads were incubated with normalized cell lysate on rotator (4 h, 4°C) and then washed and eluted as above. For experiments performed in BICR6 cells, fixation was not performed prior to immunoprecipitation. Lysates were prepared, normalized, cleared as above, and then incubated with antibody overnight (4°C) prior to 1‐h incubation with protein G beads. Beads were then washed and protein was eluted as above.
Cell proliferation and viability assays
U2OS cells were transfected with siRNA for 72 h prior to evaluation. For proliferation assays, cells were seeded in 96‐well plates, and water‐soluble CCK‐8 reagent (Dojindo) was added 2–4 h prior to analysis. Data were collected using the Infinity M1000 Pro plate reader (Tecan). To evaluate viability, cells were stained using the Live/Dead kit (Invitrogen) according to the manufacturer’s instructions. Data were collected using an Accuri C6 Flow Cytometer (BD Biosciences).
Sample preparation for proteomic analysis
All chemicals used for preparation of nanoflow liquid chromatography–tandem mass spectrometry (nLC‐MS/MS) samples were sequencing grade (Sigma‐Aldrich), unless otherwise stated. The immunoprecipitation eluate (80‐100%) was separated by SDS–PAGE using NuPAGE 1DE System (NuPAGE Novex 10% Bis‐Tris gels, Thermo Fisher Scientific). Visualization of separated proteins was performed by overnight staining with Coomassie blue G‐250 solution (Thermo Fisher Scientific). The in‐gel tryptic digestion followed by peptide extraction from the gel bands was performed according to previously published protocols (Shevchenko et al, 1996). The extracted peptides were desalted using Poros Oligo R3 RP (PerSeptive Biosystems) P200 columns with C18 3M plug (3M Bioanalytical Technologies) prior to nLC–MS/MS analysis.
Nanoflow liquid chromatography–tandem mass spectrometry (nLC–MS/MS)
The peptide mixture was resuspended in buffer containing 0.1% formic acid and loaded onto an Easy‐nLC system (Thermo Fisher Scientific), coupled online with an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific). Peptides were loaded into a picofrit 18 cm long fused silica capillary column (75 μm i.d., 360 μm o.d.) packed in‐house with reversed‐phase ReproSil‐Pur C18‐AQ 3 μm resin. The gradient length was 75 min. The gradient was from 2–26% buffer containing 100% ACN/0.1% formic acid at a flow rate of 300 nl/min. The MS method was set up in a data‐dependent acquisition (DDA) mode. For full MS scan, the mass range of 350–1200 m/z was analyzed in the Orbitrap at 120,000 FWHM (200 m/z) resolution and 5 × 10e5 AGC target value. HCD collision energy was set to 27, AGC target to 10e4, and maximum injection time to 120 ms. Detection of MS/MS fragment ions was performed in the ion trap in the rapid mode using the TopSpeed mode (3 s).
Protein identification and quantification
The raw mass spectrometer files were processed for protein identification using the Proteome Discoverer (v2.3, Thermo Scientific) and the Sequest HT algorithm with a peptide mass tolerance of 10 ppm, fragment m/z tolerance of 0.25 Da, and a false discovery rate (FDR) of 1% for proteins and peptides. All peak lists were searched against the UniProtKB/Swiss‐Prot database of Human (March 2019, 20417 entries) sequences using the parameters as follows: enzyme, trypsin; maximum missed cleavages, 2; fixed modification, carbamidomethylation (C); variable modifications, oxidation (M), protein N‐terminus acetylation. Protein quantifications were log2‐transformed and normalized using the median of the distribution for each sample. Every MS analysis was performed with three biological replicates to provide enough statistical power to apply parametric tests (either homoscedastic or heteroscedastic one‐tailed t‐test, depending on the statistical value of the F‐test; heteroscedastic if F‐test P‐value < 0.05). The t‐test was considered as a valuable statistical test because binary comparisons were performed, and the number of replicates was limited. No samples were excluded as outliers. Proteins with a t‐test P‐value < 0.05 were considered as significantly altered between the two tested conditions. Data distribution was assumed to be normal, but this was not formally tested. Gene Ontology (GO) biological process and protein–protein interaction information were obtained from STRING database (Snel et al, 2000; Szklarczyk et al, 2019) with false discovery rate (FDR) < 0.01.
Bioinformatic analysis
Single base substitutions (SBSs) and mutational signature information for TCGA tumors were obtained from the COSMIC database at https://cancer.sanger.ac.uk/cosmic/signatures (v2). Mutation calls made for the CCT complex genes were downloaded from the Genomic Data Commons Data Portal at https://docs.gdc.cancer.gov/ (v22.0; Grossman et al, 2016). The data from these two databases were merged, and only tumors with one or more deleterious mutations in the CCT complex genes or tumors with intact CCT genes were kept for further analysis. All samples were subdivided by tumor type and mutation status of all CCT complex members. A tumor was considered wild‐type when there were no single nucleotide variant or indels in any of the CCT complex genes, while mutations predicted by Ensembl VEP (McLaren et al, 2016) and SIFT (Choi et al, 2012) to cause a negative effect on protein function including indels, substitutions, frameshifts, and non‐sense mutations were classified as deleterious. Additionally, tumors with non‐deleterious mutations in the CCT complex genes as well as tumors without any somatic substitutions per megabase pairs were excluded for clarity of analysis. A control set of tumors was defined by random selection of nine genes (CPNE1, RAD51B, NMUR2, KRT32, ALO24498.2, CFAP52, RSPH6A, PLSCR3, and SERPINB7) of analogous size to CCT complex genes and with similar mutation frequency (293 tumors with deleterious mutations in the random gene set compared to 9557 tumors without). Analysis of mutations and APOBEC enrichment between samples was computed and visualized using the R libraries ggplot2, dplyr, and tidyr, as well as a two‐sided Mann–Whitney test to assess statistical significance.
Statistical analysis
Statistical details are reported in each figure legend. Each statistical analysis was performed on at least three biological replicates. The t‐test was utilized for binary comparison of limited replicates. Analysis resulting in a P‐value < 0.05 was considered to be significant.
Author contributions
AMG and MDW conceived the project. RAD, DRO, ARH, KK, ASD, JHS, and KEH performed experiments. BAG supervised mass spectrometry and proteomics. AMG, RAD, DRO, KK, KEH, and MDW designed experiments and analyzed the data. AMG and MDW supervised the project, acquired funding, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank all members of the Green and Weitzman laboratories for critical evaluation of experimental data and thoughtful review of the manuscript. We are thankful to our colleagues and collaborators for experimental discussions and manuscript review, in particular Dr. Daphne Avgousti, Dr. Nima Mosammaparast, and Dr. Nathan Singh. The results contained in this manuscript are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. This work was supported by a Young Investigator Award from the Alex’s Lemonade Stand Foundation (AMG), K08 CA212299 (AMG), R01 CA181359 (MDW), R21 CA185799 (MDW), P01 CA196539 (BAG), and R01 AI118891 (BAG) from the National Institutes of Health, and support from the Children’s Discovery Institute and the Washington University School of Medicine (AMG).
EMBO reports (2021) 22: e52145.
Contributor Information
Abby M Green, Email: abby.green@wustl.edu.
Matthew D Weitzman, weitzmanm@email.chop.edu.
Data availability
The datasets produced in this study are available in the following databases: Mass spectrometry proteomics data have been deposited to ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez‐Riverol et al, 2019) with the dataset identifier PXD024191 or at https://www.ebi.ac.uk/pride/archive/projects/PXD024191.
References
- Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik‐Zainal S, Totoki Y, Fujimoto A, Nakagawa H, Shibata Tet al (2016) Mutational signatures associated with tobacco smoking in human cancer. Science 354: 618–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom ENet al (2020) The repertoire of mutational signatures in human cancer. Nature 578: 94–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik‐Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen‐Dale A‐Let al (2013) Signatures of mutational processes in human cancer. Nature 500: 415–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avgousti DC, Herrmann C, Kulej K, Pancholi NJ, Sekulic N, Petrescu J, Molden RC, Blumenthal D, Paris AJ, Reyes EDet al (2016) A core viral protein binds host nucleosomes to sequester immune danger signals. Nature 535: 173–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aynaud MM, Suspene R, Vidalain PO, Mussil B, Guetard D, Tangy F, Wain‐Hobson S, Vartanian JP (2012) Human Tribbles 3 protects nuclear DNA from cytidine deamination by APOBEC3A. J Biol Chem 287: 39182–39192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR (2006) APOBEC3A and APOBEC3B are potent inhibitors of LTR‐retrotransposon function in human cells. Nucleic Acids Res 34: 89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn MF, Shandilya SMD, Silvas TV, Nalivaika EA, Kouno T, Kelch BA, Ryder SP, Kurt‐Yilmaz N, Somasundaran M, Schiffer CA (2015) The ssDNA Mutator APOBEC3A Is Regulated by Cooperative Dimerization. Structure 23: 903–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth CR, Meyer AS, Cong Y, Topf M, Sali A, Ludtke SJ, Chiu W, Frydman J (2008) Mechanism of lid closure in the eukaryotic chaperonin TRiC/CCT. Nat Struct Mol Biol 15: 746–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulliard Y, Narvaiza I, Bertero A, Peddi S, Rohrig UF, Ortiz M, Zoete V, Castro‐Diaz N, Turelli P, Telenti Aet al (2011) Structure‐function analyses point to a polynucleotide‐accommodating groove essential for APOBEC3A restriction activities. J Virol 85: 1765–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JBet al (2013a) APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494: 366–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Temiz NA, Harris RS (2013b) Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet 45: 977–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon IJ, Ahn J, Mitra M, Byeon CH, Hercik K, Hritz J, Charlton LM, Levin JG, Gronenborn AM (2013) NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat Commun 4: 1890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon IJ, Byeon CH, Wu T, Mitra M, Singer D, Levin JG, Gronenborn AM (2016) Nuclear magnetic resonance structure of the APOBEC3B catalytic domain: structural basis for substrate binding and DNA deaminase activity. Biochemistry 55: 2944–2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camasses A, Bogdanova A, Shevchenko A, Zachariae W (2003) The CCT chaperonin promotes activation of the anaphase‐promoting complex through the generation of functional Cdc20. Mol Cell 12: 87–100 [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network , Albert Einstein College of Medicine , Analytical Biological Services , Barretos Cancer Hospital , Baylor College of Medicine , Beckman Research Institute of City of Hope , Buck Institute for Research on Aging , Canada's Michael Smith Genome Sciences Centre , Harvard Medical School , Helen F. Graham Cancer Center & Research Institute at Christiana Care Health Services et al (2017) Integrated genomic and molecular characterization of cervical cancer. Nature 543: 378–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K, Roberts SA, Klimczak LJ, Sterling JF, Saini N, Malc EP, Kim J, Kwiatkowski DJ, Fargo DC, Mieczkowski PAet al (2015) An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat Genet 47: 1067–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD (2006) APOBEC3A is a potent inhibitor of adeno‐associated virus and retrotransposons. Curr Biol 16: 480–485 [DOI] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012) Predicting the functional effect of amino acid substitutions and indels. PLoS One 7: e46688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y, Schröder GF, Meyer AS, Jakana J, Ma B, Dougherty MT, Schmid MF, Reissmann S, Levitt M, Ludtke SLet al (2012) Symmetry‐free cryo‐EM structures of the chaperonin TRiC along its ATPase‐driven conformational cycle. EMBO J 31: 720–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez LM, Brown AL, Dennis MA, Collins CD, Brown AJ, Mitchell D, Mertz TM, Roberts SA (2019) APOBEC3A is a prominent cytidine deaminase in breast cancer. PLoS Genet 15: e1008545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faltas BM, Prandi D, Tagawa ST, Molina AM, Nanus DM, Sternberg C, Rosenberg J, Mosquera JM, Robinson B, Elemento Oet al (2016) Clonal evolution of chemotherapy‐resistant urothelial carcinoma. Nat Genet 48: 1490–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE, Thulasiraman V, Ferreyra RG, Frydman J (1999) Formation of the VHL‐elongin BC tumor suppressor complex is mediated by the chaperonin TRiC. Mol Cell 4: 1051–1061 [DOI] [PubMed] [Google Scholar]
- Freund A, Zhong FL, Venteicher AS, Meng Z, Veenstra TD, Frydman J, Artandi SE (2014) Proteostatic control of telomerase function through TRiC‐mediated folding of TCAB1. Cell 159: 1389–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Thomas JO, Chow RL, Lee GH, Cowan NJ (1992) A cytoplasmic chaperonin that catalyzes beta‐actin folding. Cell 69: 1043–1050 [DOI] [PubMed] [Google Scholar]
- Gao Y, Vainberg IE, Chow RL, Cowan NJ (1993) Two cofactors and cytoplasmic chaperonin are required for the folding of alpha‐ and beta‐tubulin. Mol Cell Biol 13: 2478–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AM, Budagyan K, Hayer KE, Reed MA, Savani MR, Wertheim GB, Weitzman MD (2017) Cytosine deaminase APOBEC3A sensitizes leukemia cells to inhibition of the DNA replication checkpoint. Cancer Res 77: 4579–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AM, Landry S, Budagyan K, Avgousti DC, Shalhout S, Bhagwat AS, Weitzman MD (2016) APOBEC3A damages the cellular genome during DNA replication. Cell Cycle 15: 998–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, Staudt LM (2016) Toward a shared vision for cancer genomic data. N Engl J Med 375: 1109–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Dudley JP (2015) APOBECs and virus restriction. Virology 479–480: 131–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer‐Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475: 324–332 [DOI] [PubMed] [Google Scholar]
- Hoopes JI, Cortez LM, Mertz TM, Malc EP, Mieczkowski PA, Roberts SA (2016) APOBEC3A and APOBEC3B preferentially deaminate the lagging strand template during DNA replication. Cell Rep 14: 1273–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili P, Bowen D, Langenbucher A, Park S, Aguirre K, Corcoran RB, Fleischman AG, Lawrence MS, Zou L, Buisson R (2020) Quantification of ongoing APOBEC3A activity in tumor cells by monitoring RNA editing at hotspots. Nat Commun 11: 2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N (2002) An anthropoid‐specific locus of orphan C to U RNA‐editing enzymes on chromosome 22. Genomics 79: 285–296 [DOI] [PubMed] [Google Scholar]
- Jin M, Liu C, Han W, Cong Y (2019) TRiC/CCT chaperonin: structure and function. Subcell Biochem 93: 625–654 [DOI] [PubMed] [Google Scholar]
- Joachimiak LA, Walzthoeni T, Liu CW, Aebersold R, Frydman J (2014) The structural basis of substrate recognition by the eukaryotic chaperonin TRiC/CCT. Cell 159: 1042–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaisari S, Sitry‐Shevah D, Miniowitz‐Shemtov S, Teichner A, Hershko A (2017) Role of CCT chaperonin in the disassembly of mitotic checkpoint complexes. Proc Natl Acad Sci U S A 114: 956–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasembeli M, Lau WC, Roh SH, Eckols TK, Frydman J, Chiu W, Tweardy DJ (2014) Modulation of STAT3 folding and function by TRiC/CCT chaperonin. PLoS Biol 12: e1001844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelia P, Yap K, Makeyev EV (2011) Streamlined platform for short hairpin RNA interference and transgenesis in cultured mammalian cells. Proc Natl Acad Sci U S A 108: 12799–12804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura A, Kubota H, Pack CG, Matsumoto G, Hirayama S, Takahashi Y, Kimura H, Kinjo M, Morimoto RI, Nagata K (2006) Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat Cell Biol 8: 1163–1170 [DOI] [PubMed] [Google Scholar]
- Knowlton JJ, Fernandez de Castro I, Ashbrook AW, Gestaut DR, Zamora PF, Bauer JA, Forrest JC, Frydman J, Risco C, Dermody TS (2018) The TRiC chaperonin controls reovirus replication through outer‐capsid folding. Nat Microbiol 3: 481–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH (2009) Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol 83: 9474–9485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota S, Kubota H, Nagata K (2006) Cytosolic chaperonin protects folding intermediates of Gbeta from aggregation by recognizing hydrophobic beta‐strands. Proc Natl Acad Sci U S A 103: 8360–8365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunisawa J, Shastri N (2003) The group II chaperonin TRiC protects proteolytic intermediates from degradation in the MHC class I antigen processing pathway. Mol Cell 12: 565–576 [DOI] [PubMed] [Google Scholar]
- Lackey L, Demorest ZL, Land AM, Hultquist JF, Brown WL, Harris RS (2012) APOBEC3B and AID have similar nuclear import mechanisms. J Mol Biol 419: 301–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey L, Law EK, Brown WL, Harris RS (2013) Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle 12: 762–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry S, Narvaiza I, Linfesty DC, Weitzman MD (2011) APOBEC3A can activate the DNA damage response and cause cell‐cycle arrest. EMBO Rep 12: 444–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Lin CY, Lei M, Yan S, Zhou T, Erikson RL (2005) CCT chaperonin complex is required for the biogenesis of functional Plk1. Mol Cell Biol 25: 4993–5010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez T, Dalton K, Frydman J (2015) The mechanism and function of group II chaperonins. J Mol Biol 427: 2919–2930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malim MH, Bieniasz PD (2012) HIV restriction factors and mechanisms of evasion. Cold Spring Harb Perspect Med 2: a006940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F (2016) The ensembl variant effect predictor. Genome Biol 17: 122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra M, Hercik K, Byeon I‐J, Ahn J, Hill S, Hinchee‐Rodriguez K, Singer D, Byeon C‐H, Charlton LM, Nam Get al (2014) Structural determinants of human APOBEC3A enzymatic and nucleic acid binding properties. Nucleic Acids Res 42: 1095–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata Y, Shibata T, Aoshima M, Tsubata T, Nishida E (2014) The molecular chaperone TRiC/CCT binds to the Trp‐Asp 40 (WD40) repeat protein WDR68 and promotes its folding, protein kinase DYRK1A binding, and nuclear accumulation. J Biol Chem 289: 33320–33332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanram V, Skold AE, Bachle SM, Pathak SK, Spetz AL (2013) IFN‐alpha induces APOBEC3G, F, and A in immature dendritic cells and limits HIV‐1 spread to CD4+ T cells. J Immunol 190: 3346–3353 [DOI] [PubMed] [Google Scholar]
- Mussil B, Suspene R, Aynaud MM, Gauvrit A, Vartanian JP, Wain‐Hobson S (2013) Human APOBEC3A isoforms translocate to the nucleus and induce DNA double strand breaks leading to cell stress and death. PLoS One 8: e73641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narvaiza I, Linfesty DC, Greener BN, Hakata Y, Pintel DJ, Logue E, Landau NR, Weitzman MD (2009) Deaminase‐independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog 5: e1000439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik‐Zainal S, Alexandrov L, Wedge D, Van Loo P, Greenman C, Raine K, Jones D, Hinton J, Marshall J, Stebbings Let al (2012) Mutational processes molding the genomes of 21 breast cancers. Cell 149: 979–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms M, Krikoni A, Kress AK, Simon V, Munk C (2012) APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T‐lymphotropic virus type 1. J Virol 86: 6097–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng G, Lei KJ, Jin W, Greenwell‐Wild T, Wahl SM (2006) Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon‐induced anti‐HIV‐1 activity. J Exp Med 203: 41–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Csordas A, Bai J, Bernal‐Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher Met al (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47: D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petljak M, Alexandrov LB (2016) Understanding mutagenesis through delineation of mutational signatures in human cancer. Carcinogenesis 37: 531–540 [DOI] [PubMed] [Google Scholar]
- Petljak M, Alexandrov LB, Brammeld JS, Price S, Wedge DC, Grossmann S, Dawson KJ, Ju YS, Iorio F, Tubio JMCet al (2019) Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell 176: 1282–1294.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS (2010) Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV‐1 restriction. Nucleic Acids Res 38: 4274–4284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissmann S, Joachimiak LA, Chen B, Meyer AS, Nguyen A, Frydman J (2012) A gradient of ATP affinities generates an asymmetric power stroke driving the chaperonin TRIC/CCT folding cycle. Cell Rep 2: 866–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Gordenin DA (2014) Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer 14: 786–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena Get al (2013) An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 45: 970–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson AG, Kim J, Al‐Ahmadie H, Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA, Akbani Ret al (2017) Comprehensive molecular characterization of muscle‐invasive bladder cancer. Cell 171: 540–556.e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommelaere H, Van Troys M, Gao Y, Melki R, Cowan NJ, Vandekerckhove J, Ampe C (1993) Eukaryotic cytosolic chaperonin contains t‐complex polypeptide 1 and seven related subunits. Proc Natl Acad Sci U S A 90: 11975–11979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper N, Gao S, Maity TK, Banday AR, Zhang Xu, Venugopalan A, Cultraro CM, Patidar R, Sindiri S, Brown A‐Let al (2019) APOBEC mutagenesis and copy‐number alterations are drivers of proteogenomic tumor evolution and heterogeneity in metastatic thoracic tumors. Cell Rep 26: 2651–2666.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salamango DJ, McCann JL, Demir O, Brown WL, Amaro RE, Harris RS (2018) APOBEC3B nuclear localization requires two distinct N‐terminal domain surfaces. J Mol Biol 430: 2695–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seplyarskiy VB, Soldatov RA, Popadin KY, Antonarakis SE, Bazykin GA, Nikolaev SI (2016) APOBEC‐induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res 26: 174–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins silver‐stained polyacrylamide gels. Anal Chem 68: 850–858 [DOI] [PubMed] [Google Scholar]
- Snel B, Lehmann G, Bork P, Huynen MA (2000) STRING: a web‐server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res 28: 3442–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suspene R, Aynaud MM, Guetard D, Henry M, Eckhoff G, Marchio A, Pineau P, Dejean A, Vartanian JP, Wain‐Hobson S (2011) Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc Natl Acad Sci U S A 108: 4858–4863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta‐Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork Pet al (2019) STRING v11: protein‐protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res 47: D607–D613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam S, Geller R, Spiess C, Frydman J (2006) The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit‐specific interactions. Nat Cell Biol 8: 1155–1162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thulasiraman V, Yang CF, Frydman J (1999) In vivo newly translated polypeptides are sequestered in a protected folding environment. EMBO J 18: 85–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinidad AG, Muller PA, Cuellar J, Klejnot M, Nobis M, Valpuesta JM, Vousden KH (2013) Interaction of p53 with the CCT complex promotes protein folding and wild‐type p53 activity. Mol Cell 50: 805–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J, Mogk A, Bukau B (2010) Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 11: 777–788 [DOI] [PubMed] [Google Scholar]
- Vartanian JP, Guetard D, Henry M, Wain‐Hobson S (2008) Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 320: 230–233 [DOI] [PubMed] [Google Scholar]
- Wang Z, Wakae K, Kitamura K, Aoyama S, Liu G, Koura M, Monjurul AM, Kukimoto I, Muramatsu M (2014) APOBEC3 deaminases induce hypermutation in human papillomavirus 16 DNA upon beta interferon stimulation. J Virol 88: 1308–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam AY, Xia Y, Lin HT, Burlingame A, Gerstein M, Frydman J (2008) Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol 15: 1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR (2004) Single‐strand specificity of APOBEC3G accounts for minus‐strand deamination of the HIV genome. Nat Struct Mol Biol 11: 435–442 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Data Availability Statement
The datasets produced in this study are available in the following databases: Mass spectrometry proteomics data have been deposited to ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez‐Riverol et al, 2019) with the dataset identifier PXD024191 or at https://www.ebi.ac.uk/pride/archive/projects/PXD024191.