

Abstract
Herpes simplex virus (HSV) establishes latent infection in long‐lived neurons. During initial infection, neurons are exposed to multiple inflammatory cytokines but the effects of immune signaling on the nature of HSV latency are unknown. We show that initial infection of primary murine neurons in the presence of type I interferon (IFN) results in a form of latency that is restricted for reactivation. We also find that the subnuclear condensates, promyelocytic leukemia nuclear bodies (PML‐NBs), are absent from primary sympathetic and sensory neurons but form with type I IFN treatment and persist even when IFN signaling resolves. HSV‐1 genomes colocalize with PML‐NBs throughout a latent infection of neurons only when type I IFN is present during initial infection. Depletion of PML prior to or following infection does not impact the establishment latency; however, it does rescue the ability of HSV to reactivate from IFN‐treated neurons. This study demonstrates that viral genomes possess a memory of the IFN response during de novo infection, which results in differential subnuclear positioning and ultimately restricts the ability of genomes to reactivate.
Keywords: HSV, interferon, latency, neuron, PML‐NB
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Neuroscience
Latent HSV‐1 genomes in peripheral neurons possess a long‐term memory of the IFN response in primary infection. This memory is characterized by association of latent genomes with IFN‐induced PML‐NBs and ultimately restriction of viral reactivation.
Introduction
Herpes simplex virus‐1 (HSV‐1) is a ubiquitous pathogen that persists in the form of a lifelong latent infection in the human host. HSV‐1 can undergo a productive lytic infection in a variety of cell types; however, latency is restricted to post‐mitotic neurons, most commonly in sensory, sympathetic, and parasympathetic ganglia of the peripheral nervous system (Baringer & Swoveland, 1973; Warren et al, 1978; Baringer & Pisani, 1994; Richter et al, 2009). During latent infection, the viral genome exists as an episome in the neuronal nucleus, and there is considerable evidence that on the population level viral lytic gene promoters assemble into repressive heterochromatin (Wang et al, 2005; Cliffe & Knipe, 2008; Cliffe et al, 2009; Kwiatkowski et al, 2009). The only region of the HSV genome that undergoes active transcription, at least in a fraction of latently infected cells, is the latency‐associated transcript (LAT) locus (Stevens et al, 1987; Kramer & Coen, 1995). Successful establishment of a latent gene expression program requires a number of molecular events, likely influenced by both cellular and viral factors, and is not uniform (Efstathiou & Preston, 2005). Significant heterogeneity exists in expression patterns of both lytic and latent transcripts in latently infected neurons, as well as in the ability of latent genomes to reactivate in response to different stimuli (Sawtell, 1997; Proenca et al, 2008; Catez et al, 2012; Ma et al, 2014; Maroui et al, 2016; Nicoll et al, 2016). This heterogeneity could arise from viral genome copy number, exposure to different inflammatory environments, or intrinsic differences in the neurons themselves. Furthermore, there is growing evidence that heterogeneity in latency may ultimately be reflected in part by the association of viral genomes with different nuclear domains or cellular proteins (Catez et al, 2012; Maroui et al, 2016). However, what determines the subnuclear distribution of latent viral genomes is not known. In addition, it is currently unclear whether viral genome association with certain nuclear domains or cellular proteins results in an increased or decreased ability of the virus to undergo reactivation. The aim of this study was to determine whether the presence of interferon during initial HSV‐1 infection can intersect with the latent viral genome to regulate the type of gene silencing and ultimately the ability to undergo reactivation. Because the fate of viral genomes and their ability to undergo reactivation can be readily tracked, latent HSV‐1 infection of neurons also serves as an excellent system to explore how exposure to innate immune cytokines can have a lasting impact on peripheral neurons.
Latent HSV‐1 genomes have been shown to associate with promyelocytic leukemia nuclear bodies (PML‐NBs) in mouse models of infection, as well as in human autopsy material (Catez et al, 2012; Maroui et al, 2016). PML‐NBs are heterogeneous, phase‐separated nuclear condensates that have been associated with the transcriptional activation of cellular genes (Wang et al, 2004; Bernardi & Pandolfi, 2007; Lallemand‐Breitenbach & de The, 2010; Kim & Ahn, 2015; McFarlane et al, 2019), but also can recruit repressor proteins, including ATRX, Daxx, and Sp100, that promote transcriptional repression and inhibition of both DNA and RNA virus replication (Zhong et al, 2000b; Garrick et al, 2004; Bishop et al, 2006; Everett & Chelbi‐Alix, 2007; Xu & Roizman, 2017). In the context of lytic infection of non‐neuronal cells, PML‐NBs have been shown to closely associate with HSV‐1 genomes (Maul et al, 1996; Maul, 1998), and the HSV‐1 viral regulatory protein ICP0 is known to disrupt the integrity of these structures by targeting PML and other PML‐NB‐associated proteins for degradation (Everett & Maul, 1994; Chelbi‐Alix & de The, 1999; Boutell et al, 2002). PML‐NBs entrapment of HSV‐1 genomes during lytic infection of fibroblasts (Alandijany et al, 2018) is hypothesized to create a transcriptionally repressive environment for viral gene expression, as PML directly contributes to the cellular repression of ICP0‐null mutant viruses (Everett et al, 2006). In the context of latency, neurons containing PML‐encased latent genomes exhibit decreased expression levels of the LAT (Catez et al, 2012), suggesting that they are more transcriptionally silent than latent genomes localized to other nuclear domains and raising the question as to whether PML‐NB‐associated genomes are capable of undergoing reactivation. Studies have shown that replication‐defective HSV genomes associated with PML‐NBs are capable of derepressing following induced expression of ICP0 in fibroblasts (Everett et al, 2007; Cohen et al, 2018) and following addition of the histone deacetylase inhibitor trichostatin A (TSA) in cultured adult TG neurons (Maroui et al, 2016). However, it is not known if replication‐competent viral genomes associated with PML‐NBs are capable of undergoing reactivation triggered by activation of cellular signaling pathways in the absence of viral protein.
PML‐NBs can undergo significant changes in number, size, and localization depending on cell type, differentiation stage, and cell–cycle phase, as well as in response to cellular stress and soluble factors (Bernardi & Pandolfi, 2007; Lallemand‐Breitenbach & de The, 2010). Interferon (IFN) treatment directly induces the transcription of PML, Daxx, Sp100, and other PML‐NB constituents, which leads to elevated protein synthesis and a robust increase in both size and number of PML‐NBs (Chelbi‐Alix et al, 1995; Stadler et al, 1995; Grotzinger et al, 1996; Greger et al, 2005; Shalginskikh et al, 2013). During HSV‐1 infection, type I IFNs are among the first immune effectors produced, and they have been shown to restrict HSV viral replication and spread both in vitro and in vivo through multiple pathways (Hendricks et al, 1991; Mikloska et al, 1998; Mikloska & Cunningham, 2001; Sainz & Halford, 2002; Jones et al, 2003). Type I IFNs are elevated within peripheral ganglia during HSV‐1 infection (Carr et al, 1998) and have been linked with control of lytic HSV‐1 replication. In an in vitro model of latency, exogenous type I IFNs also have been shown to induce neuron‐specific antiviral responses that control reactivation (Linderman et al, 2017), but whether type I IFN exposure during initial infection modulates entry into latency is not known. Importantly, exposure to IFN and other cytokines has also been shown to generate innate immune memory or “trained immunity” in fibroblasts and immune cells (Kamada et al, 2018; Moorlag et al, 2018), and PML‐NBs themselves are potentially important in the host innate immune response. A previous study found that the histone chaperone HIRA is re‐localized to PML‐NBs in response to the innate immune defenses induced by HSV‐1 infection, and in this context, PML was required for the recruitment of HIRA to ISG promoters for efficient transcription (McFarlane et al, 2019). Prior exposure to type I interferons has also been shown to promote a transcriptional memory response in fibroblasts and macrophages (Kamada et al, 2018). This interferon memory leads to faster and more robust transcription of ISGs following restimulation and coincided with acquisition of certain chromatin marks and accelerated recruitment of transcription and chromatin factors (Kamada et al, 2018). Thus far, long‐term memory of cytokine exposure has only been investigated in non‐neuronal cells, but it is conceivable that neurons, being non‐mitotic and long‐lived cells, also possess unique long‐term responses to prior cytokine exposure.
Although in vivo models are incredibly powerful tools to investigate the contribution of the host immune response to HSV infection, they are problematic for investigating how individual components of the host’s immune response specifically regulate neuronal latency. Conversely, in vitro systems provide a simplified model that lacks many aspects of the host immune response. Therefore, to investigate the role of type I IFN on HSV‐1 latency and reactivation, we utilized a model of latency in primary murine sympathetic neurons (Cliffe et al, 2015), which allowed us to manipulate conditions during initial HSV‐1 infection and trigger synchronous robust reactivation. Using this model, we show that primary neurons isolated from mouse peripheral ganglia are largely devoid of detectable PML‐NBs but PML‐NBs form following type I IFN exposure and persist even when ISG gene expression and production of other antiviral proteins have returned to baseline. Neither exogenous type I IFN nor detectable PML‐NBs are required for HSV gene silencing and entry into latency in this model system, but, importantly, the presence of IFNα specifically at the time of initial infection results in the entrapment of viral genomes in PML‐NBs and a more restrictive form of latency that is less able to undergo reactivation. This study therefore demonstrates how the viral latent genome has a long‐term memory of the innate response during de novo HSV infection that results in entrapment of genomes in PML‐NBs and a more repressive form of latency.
Results
Interferon induces the formation of detectable PML‐NBs in primary sympathetic and sensory neurons isolated from postnatal and adult mice
We initially set out to investigate the contribution of PML‐NBs to HSV latency and reactivation using primary sympathetic and sensory neurons that have been well characterized as in vitro models of HSV latency and reactivation (Wilcox & Johnson, 1987; Wilcox et al, 1990; Camarena et al, 2010; Cliffe et al, 2015; Ives & Bertke, 2017; Cuddy et al, 2020). In addition, primary neuronal systems allow for much more experimental control of specific conditions during de novo infection and can be easily manipulated either immediately prior to or following infection. Peripheral neurons were isolated from the superior cervical ganglia (SCG) or trigeminal ganglia (TG) from young (post‐natal day; P1) or adult (> P28) mice and cultured for 6 days prior to staining. PML‐NBs were defined as detectable punctate nuclear structures by staining for PML protein. Strikingly, we observed that both SCG and TG neurons were largely devoid of detectable PML‐NBs (Fig 1A).
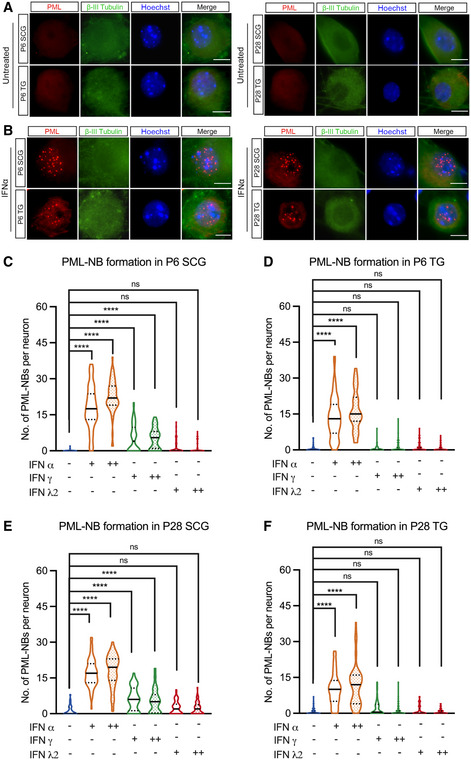
Figure 1. Type I IFN induces the formation of PML‐NBs in primary peripheral neurons.
-
ARepresentative images of primary neurons isolated from superior cervical ganglia (SCG) and sensory trigeminal ganglia (TG) of postnatal (P6) and adult (P28) mice stained for PML and the neuronal marker BIII‐tubulin.
-
BSCG and TG neurons isolated from P6 and P28 mice were treated with interferon (IFN)α (600 IU/ml) for 18 h and stained for PML and BIII‐tubulin.
-
C–FQuantification of detectable PML puncta in P6 and P28 neurons following 18‐h treatment with IFNα (150 IU/ml, 600 IU/ml), IFNγ (150 IU/ml, 500 IU/ml), and IFNλ2 (100 ng/ml, 500 ng/ml). Data information: Data represent the mean ± SEM. n = 60 cells from 3 biological replicates. Statistical comparisons were made using a one‐way ANOVA with Tukey’s multiple comparison (ns not significant, ****P < 0.0001). Scale bar, 20 μm.
Source data are available online for this figure.
In certain cell types, the transcription of certain PML‐NB‐associated proteins, including PML, can be induced by either type I or type II interferon (IFN) treatment, which is correlated with an increase in PML‐NB size and/or number per cell (Chelbi‐Alix et al, 1995; Stadler et al, 1995). Therefore, we were interested in determining whether exposure of primary sensory or sympathetic neurons to different types of IFN resulted in PML‐NB formation. Type I IFN treatment using IFN‐alpha (IFNα) (Fig 1B, C–F) or IFN‐beta (Fig EV1A) led to a significant induction of detectable PML‐NBs in both sensory and sympathetic neurons isolated from postnatal and adult mice. Representative images of IFNα‐treated neurons are shown (Fig 1B), and the number of detectable PML‐NBs per neurons is quantified (Fig 1C–F). The increase in detectable PML‐NBs was comparable for both 150 IU/ml and 600 IU/ml of IFNα. Type II IFN (IFNγ) led to a more variable response with a small but significant increase in detectable PML‐NBs in a subpopulation of sympathetic neurons. However, IFNγ treatment of sensory neurons did not result in the formation of detectable PML‐NBs. Exposure of neurons to IFN‐lambda 2 (IFN‐λ2), a type III IFN, did not induce the formation of detectable PML‐NBs in either sympathetic or sensory neuron cultures (Figs 1C–F and EV1B and C). Therefore, PML‐NBs are largely undetectable in primary sympathetic and sensory neurons but can form upon exposure to type I IFNs.
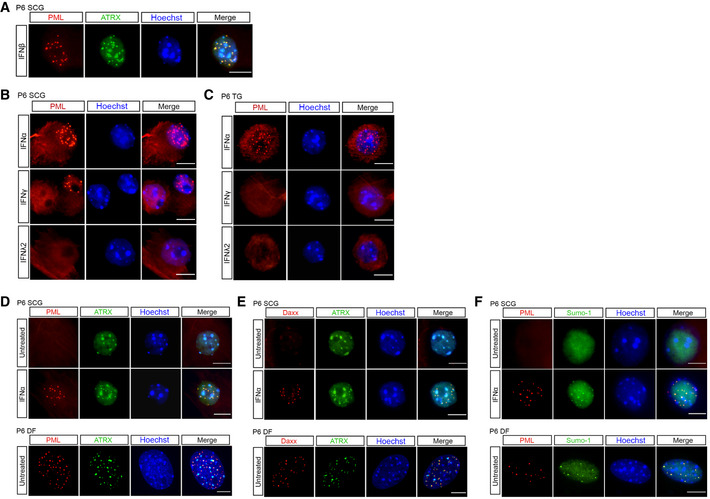
Figure EV1. Type I IFN alters the sub‐cellular localization of ATRX, Daxx, and SUMO‐1 in primary peripheral neurons.
-
ARepresentative images of P6 SCG neurons treated with IFNβ (150 IU/ml) and stained for PML and ATRX.
-
BRepresentative images of P6 SCG treated with IFNα (600 IU/ml), IFNγ (500 IU/ml) and IFNλ2 (500 ng/ml) and stained for PML.
-
CRepresentative images of P6 TG treated with IFNα (600 IU/ml), IFNγ (500 IU/ml), and IFNλ2 (500 ng/ml) and stained for PML.
-
D–FRepresentative images of untreated or IFNα (600 IU/ml)‐treated P6 SCG neurons stained for PML and ATRX (D), Daxx and ATRX (E), and PML and SUMO‐1 (F). P6 dermal fibroblasts (DF) isolated from the same mice were used as a non‐neuronal control (D–F).
Data information: Scale bars, 20 μm.
The absence of detectable PML‐NBs in untreated primary neurons prompted us to investigate other known components of PML‐NBs. We were particularly interested in ATRX and Daxx because like PML they have previously been found to be involved in restricting HSV lytic replication in non‐neuronal cells (Lukashchuk & Everett, 2010; Alandijany et al, 2018; Cabral et al, 2018; McFarlane et al, 2019). Therefore, we investigated the localization of ATRX and Daxx in primary peripheral neurons. ATRX is a multifunctional, heterochromatin‐associated protein that is localized to PML‐NBs in human and mouse mitotic cells and is largely characterized as interacting with the Daxx histone chaperone (Lewis et al, 2010; Clynes et al, 2013). In untreated neurons, we observed abundant ATRX staining throughout the nucleus in regions that also stained strongly with Hoechst (Fig EV1D and E). This potential colocalization of ATRX with regions of dense chromatin is consistent with a previous study demonstrating that in neurons ATRX binds certain regions of the cellular genome associated with the constitutive heterochromatin modification H3K9me3 (Noh et al, 2015). Importantly, this distribution of ATRX differs from what is seen in murine dermal fibroblasts (Fig EV1D and E) and other non‐neuronal cells, where there is a high degree of colocalization between ATRX and PML (Alandijany et al, 2018). Following treatment with IFNα, we found a redistribution of ATRX staining and colocalization between ATRX and the formed PML‐NBs, but the majority of ATRX staining remained outside the context of PML‐NBs (Fig EV1D and E). Similar to PML, sympathetic SCG and sensory TG neurons isolated from both postnatal and adult mice were devoid of detectable puncta of Daxx staining (Fig EV1E), and we did not observe extensive Daxx staining in untreated neurons as we did for ATRX. We were unable to directly co‐stain for Daxx and PML; however, treatment of neurons with IFNα did induce punctate Daxx staining that strongly colocalized with puncta of ATRX (Fig EV1E), which given our previous observation of ATRX colocalization with PML following type I IFN treatment we used as a correlate for PML‐NBs. We were also interested in SUMO‐1, which has been shown to be required for formation of PML‐NBs (Zhong et al, 2000a). Similar to ATRX and Daxx, treatment of neurons with IFNα induced punctate SUMO‐1 staining in P6 SCG neurons that colocalized with PML puncta (Fig EV1F). Therefore, PML‐NBs containing their well‐characterized associated proteins are not detected in cultured primary neurons but form in response to type I IFN exposure.
Type I IFN treatment specifically at time of infection restricts reactivation of HSV‐1 from primary sympathetic neurons without affecting initial infectivity or LAT expression
Because we observed that primary SCG neurons are largely devoid of PML‐NBs and that PML‐NBs form upon treatment with type I IFN treatment, we first wanted to clarify that latency was maintained in the absence of IFN and presumably without PML‐NB formation, consistent with our previous data (Cuddy et al, 2020). SCG neurons were infected at a multiplicity of infection (MOI) of 7.5 plaque forming units (PFU)/cell with HSV‐1 Us11‐GFP presence of acyclovir (ACV). The ACV was removed after 6 days, and the neuronal cultures were monitored to ensure the no GFP‐positive neurons were present (Fig 2A). We found that latency could be established and maintained for up to 5 days following removal of ACV (Fig 2B). Reactivation was triggered by PI3K inhibition using LY294002, as previously described (Camarena et al, 2010; Kim et al, 2012; Kobayashi et al, 2012; Cliffe et al, 2015), and quantified based on the number of Us11‐GFP neurons in the presence of WAY‐150138 which blocks packaging of progeny genomes and thus cell‐to‐cell spread (van Zeijl et al, 2000). These data therefore indicate that exogenous IFN is not required to induce a latent state in this model system.
Figure 2. Type I IFN treatment solely at time of infection inhibits LY294002‐mediated reactivation of HSV‐1 in primary sympathetic SCG neurons.
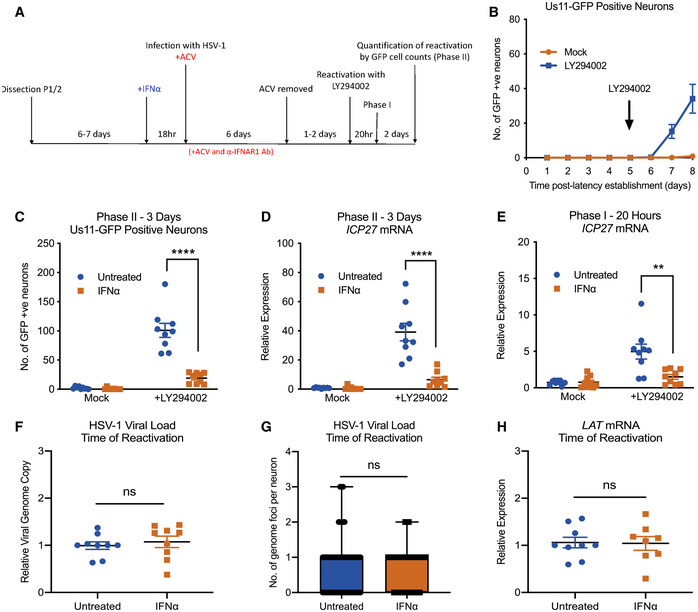
- Schematic of the primary postnatal sympathetic neuron‐derived model of HSV‐1 latency.
- Reactivation from latency is quantified by Us11‐GFP expressing neurons following addition of the PI3K inhibitor LY294002 (20 μM) in the presence of WAY‐150168, which prevents cell‐to‐cell spread. The arrow indicates the time of LY294002 treatment at 5 days post‐establishment of latency. n = 9 biological replicates.
- Number of Us11‐GFP expressing neurons at 3 days post‐LY294002‐induced reactivation in P6 SCG neuronal cultures infected with HSV‐1 in the presence or absence of IFNα (600 IU/ml), then treated with an α‐ΙFNAR1 neutralizing antibody. n = 9 biological replicates.
- RT–qPCR for viral mRNA transcripts at 3 days post‐LY294002‐induced reactivation of SCGs infected with HSV‐1 in the presence or absence of IFNα. n = 9 biological replicates.
- RT–qPCR for viral mRNA transcripts at 20 h post‐LY294002‐induced reactivation in SCGs infected with HSV‐1 in the presence of absence of IFNα. n = 9 biological replicates.
- Relative amount of viral DNA at time of reactivation (8 dpi) in SCG neurons infected with HSV‐1 in the presence or absence of IFNα (600 IU/ml). n = 9 biological replicates.
- Quantification of vDNA foci detected by click chemistry at time of reactivation (8 dpi) in SCG neurons infected with HSV‐1 in the presence or absence of IFNα (600 IU/ml). n = 60 genomes from 3 biological replicates.
- LAT mRNA expression at time of reactivation (8 dpi) in neurons infected with HSV‐1 in the presence or absence of IFNα (600 IU/ml). n = 9 biological replicates.
Data information: Data represent the mean ± SEM. Statistical comparisons were made using a Mann–Whitney test (ns not significant, **P < 0.01, ****P < 0.0001).
Source data are available online for this figure.
We next turned our attention to whether type I IFN treatment at the time of infection impacted the ability of HSV to establish latency or reactivate in this model system. SCG neurons were pretreated with IFNα (600 IU/ml) for 18 h and during the initial 2 h HSV inoculation. Following inoculation, IFNα was washed out and an IFNAR1 blocking antibody was used to prevent subsequent type I IFN signaling through the receptor. To confirm the effectiveness of the IFNAR1 ab to block detectable IFN signaling, we validated it by its ability to block ISG expression (ISG15) in cultured SCG neurons by RT–qPCR (Fig EV2A). Reactivation was induced and initially quantified based on the number of GFP‐positive neurons at 3 days post‐stimuli. We found that full reactivation was restricted in neurons exposed to type I IFN just prior to and during de novo infection (Fig 2C). We further confirmed this IFNα‐mediated restriction of latency by the induction of lytic mRNAs upon reactivation. IFNα treatment at the time of infection significantly decreased the expression of immediate‐early gene (ICP27), early gene (ICP8), and late gene (gC) at 3 days post‐reactivation (Figs 2D and EV2AB and C). There were very few GFP‐positive neurons and little to no viral gene expression in mock reactivated controls, further indicating that latency can be established in the presence and absence of IFN.
Figure EV2. Type I IFN treatment solely at time of infection inhibits LY294002‐mediated reactivation of HSV‐1 in primary sympathetic SCG neurons.
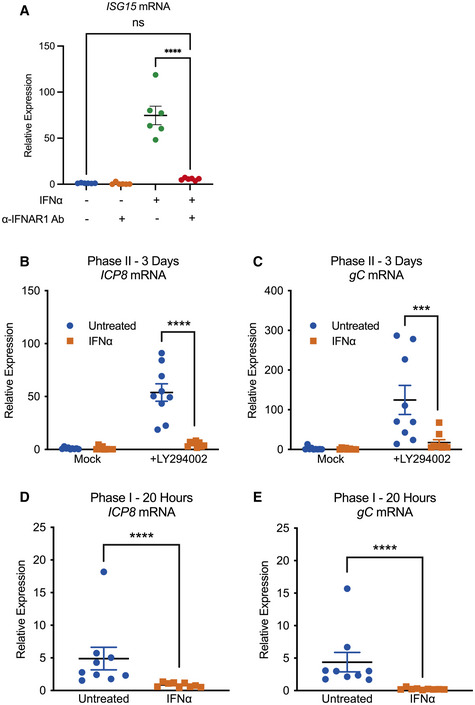
-
ART–qPCR for ISG15 mRNA expression in SCG neurons treated with IFNα (600 IU/ml) in the presence or absence of anti‐mouse IFNAR1 antibody (1:1,000). n = 6 biological replicates.
-
B, CRT–qPCR for viral mRNA transcripts at 3 days post‐LY294002‐induced reactivation of SCGs infected with HSV‐1 in the presence or absence of IFNα (600 IU/ml). n = 9 biological replicates.
-
D, ERT–qPCR for viral mRNA transcripts at 20 h post‐LY294002‐induced reactivation in SCGs infected with HSV‐1 in the presence or absence of IFNα (600 IU/ml). n = 9 biological replicates.
Data information: Data represent the mean ± SEM. Statistical comparisons were made using a one‐way ANOVA with Tukey’s multiple comparison (A) or a Mann–Whitney test (ns not significant, ***P < 0.001, ****P < 0.0001).
Reactivation of HSV in this system proceeds over two phases. GFP‐positive neurons is a readout for full reactivation or Phase II. However, we and others have observed an initial wave of lytic gene expression that occurs prior to and independently of viral DNA replication at around 20 h post‐stimulus, termed Phase I (Du et al, 2011; Kim et al, 2012; Cliffe et al, 2015; Cliffe & Wilson, 2017). Therefore, to determine whether IFNα treatment at the time of infection restricted the Phase I wave of lytic, we carried out RT–qPCR to detect representative immediate‐early (ICP27), early (ICP8), and late (gC) transcripts at 20 h post‐addition of LY294002. We found significantly decreased expression in the IFNα‐treated neurons (Figs 2E and EV2D and E). This is interesting as exogenous type I IFNs have previously been shown to suppress reactivation in murine neurons by preventing Phase I and are rendered ineffective once Phase I viral products accumulate (Linderman et al, 2017). Therefore, type I IFN treatment solely at the time of infection has a long‐term effect on the ability of HSV to initiate lytic gene expression and undergo reactivation.
Because IFN treatment could reduce nuclear trafficking of viral capsids during initial infection or impact infection efficiency, we next determined whether equivalent numbers of viral genomes were present in the neuronal cultures. At 8 dpi, we measured relative viral DNA genome copy numbers in SCG neurons that were treated with IFNα compared to untreated controls and found no significant difference (Fig 2F). To further confirm that equivalent genomes were present in the neuronal nuclei, we infected neurons with HSV‐1‐containing EdC‐incorporated genomes and performed click chemistry to detect vDNA foci. At 8 dpi, we found no significant difference in the average number of vDNA foci per nucleus of neurons treated with IFNα at the time of initial infection compared to untreated controls (Fig 2G). Therefore, the restricted reactivation phenotype mediated by IFNα was not due to a decrease in the number of latent viral genomes.
The decreased reactivation observed with IFNα treatment could be secondary to changes in expression of the LAT and/or directly as a result of decreased viral genome accessibility. The HSV LAT, one of the only highly expressed gene products during latent infection, has been shown to modulate several features of latency, including the viral chromatin structure, lytic gene expression, and neuronal survival, as well as the efficiency of latency establishment and reactivation (Leib et al, 1989; Hill et al, 1990; Trousdale et al, 1991; Gordon et al, 1995; Chen et al, 1997; Garber et al, 1997; Thompson & Sawtell, 1997, 2001; Perng et al, 2000; Knipe & Cliffe, 2008). Therefore, the ability of HSV to undergo reactivation could be due to changes in LAT expression following IFNα treatment. However, when we evaluated LAT expression levels at 8 dpi by RT–qPCR, we found no detectable difference between IFNα‐treated and untreated cultures of neurons. This suggests that the IFNα‐mediated restriction in reactivation does not appear to occur as a result of changes in expression of the LAT (Fig 2H). Therefore, it is possible that the type I IFN‐mediated restriction of HSV latency is due to changes to the latent genome that results in a decreased ability to undergo reactivation following PI3‐kinase inhibition.
Primary neurons have a memory of prior IFNα exposure characterized by persistence of PML‐NBs
Because we observed a restriction in the ability of HSV to reactivate that occurred 7–8 days following type I IFN exposure, we went on to examine any long‐term changes resulting from IFNα exposure. First, we investigated the kinetics of representative ISG expression. As expected, we saw a robust induction of Isg15 and Irf7 in IFNα‐treated (600 IU/ml) neurons that persisted for at least 42 h post‐treatment post‐addition of IFNα (this represents 1 day post‐infection (dpi)). However, by 8 dpi, the time at which neurons were induced to reactivate, there was no detectable difference in Isg15 or Irf7 expression in IFNα‐treated neurons vs untreated controls (Fig 3A and B), indicating that these representative ISGs were not detectably elevated at the time of reactivation. We also found similar Isg15 and Irf7 expression in HSV‐1‐infected neurons compared to uninfected controls, suggesting that HSV‐1 infection was not impacting IFN signaling pathways at a population level. PML has been previously characterized as an ISG product in non‐neuronal cells (Chelbi‐Alix et al, 1995; Stadler et al, 1995) and is responsive to both IFNβ and IFNγ in latently infected rat sympathetic neurons induced to reactivate (Linderman et al, 2017), and we found an approximate 5‐fold increased expression of Pml in primary sympathetic neurons following IFNα treatment which was less than the increased expression of Irf7 and Isg15 (approximate 250‐fold and 100‐fold increased expression, respectively). Pml expression returned to untreated levels by 1 dpi (Fig 3C).
Figure 3. Type I IFN‐induced PML‐NBs persist in primary sympathetic neurons despite resolution of IFN signaling.
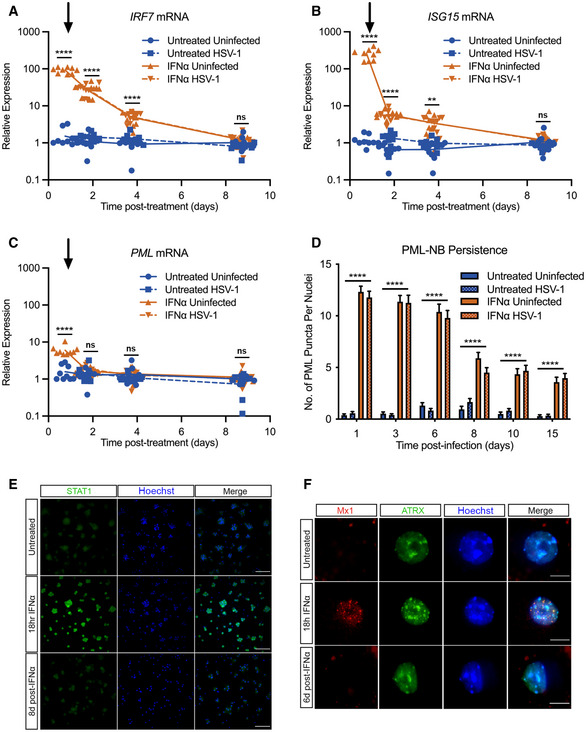
-
A–CKinetics of ISG15, IRF7, and PML mRNA expression at 0.75, 1.75, 3.75, and 8.75 days post‐ΙFNα (600 IU/ml) treatment. Arrow indicates the time of HSV‐1 infection at 18 h post‐interferon treatment. n = 9 biological replicates.
-
DQuantification of PML puncta at 1, 3, 6, 8, 10 and 15 days post‐infection with HSV‐1 in untreated and IFNα (600 IU/ml)‐treated SCG neurons. n ≥ 60 cells from 3 biological replicates.
-
ERepresentative images of P6 SCG neurons treated with IFNα (600 IU/ml) and stained for STAT1 at 18 h and 8 days post‐treatment. Scale bar, 100 μm.
-
FRepresentative images of P6 SCG neurons treated with IFNα (600 IU/ml) and stained for Mx1 at 18 h and 6 days post‐treatment. Scale bar, 20 μm.
Data information: Data represent the mean ± SEM. Statistical comparisons were made using one‐way ANOVA with Tukey’s multiple comparison (A–C) or mixed‐effects analysis with Tukey’s multiple comparison (D). (ns not significant, **P < 0.01, ****P < 0.0001).
Source data are available online for this figure.
Although we did not detect maintained induction of IFN‐stimulated gene expression including Pml, we were intrigued as to whether PML‐NBs persisted throughout the course of infection. To assess this, we first established whether PML‐NBs persist even in the absence of sustained ISG expression. Quantifying the number of PML‐NBs following IFNα (600 IU/ml) treatment, we found that the number of bodies remains elevated through 15 days post‐treatment (Fig 3D). We went on to investigate additional products of ISGs including STAT1 and Mx1 because of the availability of specific antibodies against these proteins. We observed robust STAT1 staining following IFNα exposure for 18 h. However, by 8 days post–infection, we could not detect STAT1 staining in primary neurons indicating that accumulation of this IFNα‐induced protein had returned to baseline (Fig 3E). Similarly, we found induction of punctate Mx1 staining in neurons exposed to IFNα for 18 h that was undetectable by day 6 post‐treatment (Fig 3F). Therefore, exposure of primary neurons to type I IFN led to a modest induction of Pml mRNA but resulted in long‐term persistence of PML‐NBs, even in the absence of continued IFN signaling and when antiviral protein products of other ISGs were undetectable.
PML‐NBs persist and stably entrap latent HSV‐1 genomes only if IFNα is present at the time of initial infection
The persistence of PML‐NBs following IFN exposure raised the possibility that viral genomes are maintained within PML‐NBs only in type I IFN‐treated neurons. This would also suggest that PML‐NB‐associated genomes are less permissive for reactivation and provide us with an experimental system to investigate the contribution of PML‐NBs to the maintenance of HSV latency. To determine whether viral genomes localize with PML‐NBs in type I IFN‐treated neurons, SCG neurons were pretreated with IFNα (600 IU/ml) then infected with HSV‐1EdC at an MOI of 5 PFU/cell in the presence of ACV and IFNα as described above. By co‐staining for PML, we found that a large proportion of vDNA foci colocalized with PML‐NBs in the IFNα‐treated neurons over the course of infection. In untreated neurons that are largely devoid of detectable PML‐NBs, very few genomes were colocalized to PML puncta as expected. Representative images are shown (Fig 4A) and the percent of genome foci colocalized to PML‐NBs is quantified (Fig 4B). Furthermore, high‐resolution Airy scan‐based 3D confocal microscopy of IFNα‐treated neurons revealed that vDNA foci were entrapped within PML‐NBs (Fig 4C and D), as has also been reported upon lytic infection of non‐neuronal cell lines (Alandijany et al, 2018) and in latently infected TG in vivo (Catez et al, 2012), and interestingly, we found that the volume of PML‐NBs associated with vDNA is greater than PML‐NBs not associated with vDNA (Fig 4E). Previous studies have found that colocalization of viral DNA by PML‐NBs during lytic HSV‐1 infection of human fibroblasts occurs independently of type I IFN exposure (Maul et al, 1996; Everett et al, 2004; Everett & Murray, 2005; Alandijany et al, 2018), and we confirmed this was also the case in dermal fibroblasts isolated from postnatal mice (Fig EV3A and B). Therefore, the presence of IFNα during initial infection can impact the long‐term subnuclear localization of latent viral genomes in neurons by inducing PML‐NBs that persist and stably entrap latent viral genomes.
Figure 4. Type I IFN‐induced PML‐NBs stably entrap vDNA throughout a latent HSV‐1 infection of primary sympathetic neurons.
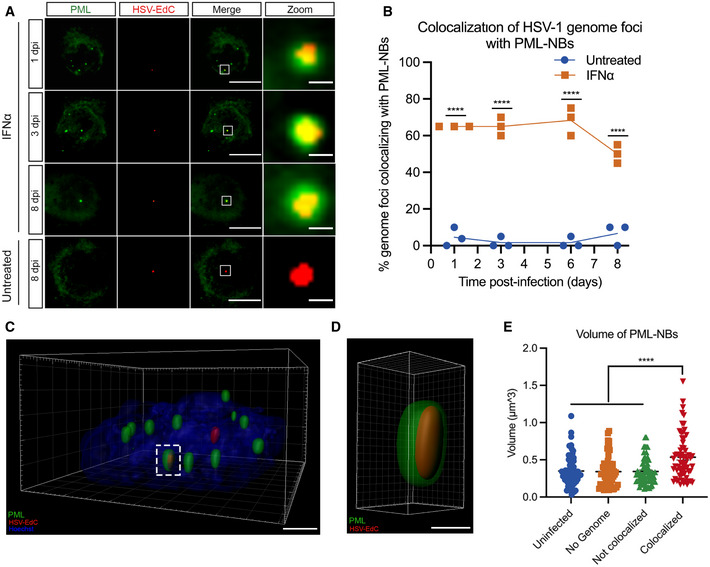
-
ARepresentative images of vDNA foci detected by click chemistry to PML at 1, 3, 6, and 8 dpi in P6 SCG neurons infected with HSV‐1EdC in the presence or absence of IFNα (600 IU/ml). Z‐stack images of individual vDNA foci were acquired. Scale bar, 20 μm. Zoom scale bar, 1 μm.
-
BPercent colocalization of vDNA foci detected by click chemistry to PML at 1, 3, 6, and 8 dpi in SCG neurons infected with HSV‐1EdC in the presence or absence of IFNα (600 IU/ml). Each point represents the percentage of 20 vDNA foci that colocalized to PML from 3 biological replicates.
-
C, D(C) 3D reconstruction of a high‐resolution Z‐series confocal image showing PML entrapment of a HSV‐1EdC vDNA foci. Scale bar, 2 μm. (D) Enlargement of PML entrapped vDNA outlined by white dashed box. Scale bar, 0.5 μm.
-
EQuantification of PML‐NB volume in SCG neurons infected with HSV‐1EdC in the presence or absence of IFNα (600 IU/ml). n = 64 biological replicates.
Data information: Data represent the mean ± SEM. Statistical comparisons were made using a 2‐way ANOVA (B) or a one‐way ANOVA with Tukey’s multiple comparison (E) (****P < 0.0001).
Source data are available online for this figure.
Figure EV3. PML‐NBs entrap vDNA in the absence of type I IFN during lytic HSV‐1 infection of murine dermal fibroblasts.
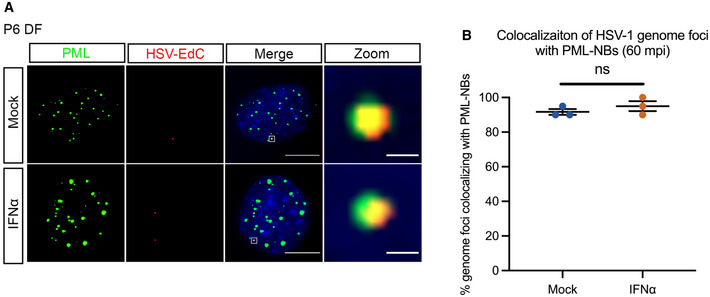
- Representative images of vDNA foci detected by click chemistry to PML at 60 min post‐infection in P6 dermal fibroblasts lytically infected with HSV‐1EdC in the presence or absence of ΙFNα (600 IU/ml). Scale bar, 20 μm. Zoom scale bar, 1 μm.
- Percent colocalization of vDNA foci detected by click chemistry to PML at 60 mpi in P6 dermal fibroblasts infected with HSV‐1EdC in the presence or absence of IFNα (600 IU/ml). Each point represents the percentage of 20 vDNA foci that colocalized to PML from 3 biological replicates.
Data information: Data represent the mean ± SEM. Statistical comparisons were made using a Mann–Whitney test (ns not significant).
Thus far, our data indicate that the presence of IFNα during initial infection determines subnuclear positioning of latent viral genomes and the ability of genomes to reactivate in response to inhibition of PI3‐kinase activity. We considered that type I IFN treatment could have a long‐term effect on cell signaling pathways which could impact the ability of HSV to reactivate. Therefore, to determine the direct versus indirect effects on the viral genome itself, we next investigated whether the timing of IFNα exposure had a differential effect on the ability of viral genomes to reactivate. We treated postnatal SCG neurons with IFNα (600 IU/ml) for 18 h and during the 2 h HSV inoculation (−18 hpi) or exposed neurons to IFNα for 18 h at 3 days prior to infection (−3 dpi). Following pretreatment at −3 dpi or −18 hpi, IFNα was washed out and an IFNAR1 blocking antibody was used. As expected, IFNα during initial infection significantly inhibited HSV reactivation, but surprisingly, IFNα treatment at −3 dpi did not restrict reactivation as shown by the similar number of GFP‐positive neurons at 72 h post‐reactivation when compared to untreated neurons (Fig 5A). Consistent with the reactivation data, we found that vDNA foci did not localize to PML‐NBs in SCG neurons treated with IFNα at −3 dpi (Fig 5B). We confirmed that PML‐NBs were present at the time of infection in neurons treated 3 days prior to infection (Fig 5C), although we did detect slightly fewer PML‐NBs per nucleus in neurons treated −3 dpi compared to −18 hpi (a mean of 17.57 versus 12.47 per nucleus, respectively). We also confirmed comparable recruitment of known PML‐NB‐associated proteins ATRX, Daxx, and SUMO‐1 at 3 days post‐IFNα treatment (Fig EV4A–C). When IFNα treatment of SCG neurons is continued from −3 dpi through infection, or if SCG neurons treated at −3 dpi receive a second treatment of IFNα during infection, then a similar proportion of latent viral genomes colocalize with PML‐NBs as with a single treatment during infection (Fig EV4D). Together with the previous data on genome entrapment in dermal fibroblasts (Fig EV3A and B), these data indicate that type I IFN must be present during infection of neurons, but not necessarily non‐neuronal cells, for vDNA to colocalize with PML‐NBs.
Figure 5. HSV‐1 genomes only associate with PML‐NBs when type I IFN is present during initial infection.
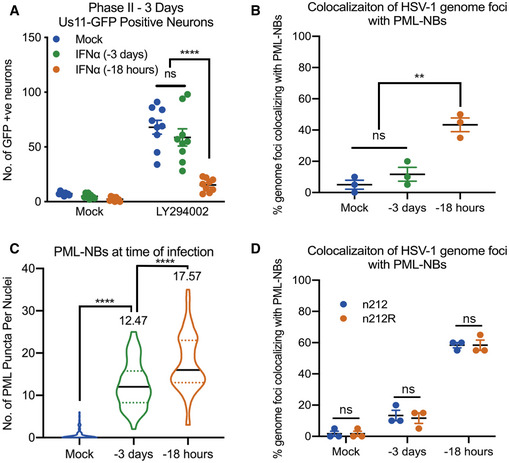
- Number of Us11‐GFP expressing P6 SCG neurons infected with HSV‐1 following IFNα treatment for 18 h prior to infection or for 18 h at 3 days prior to infection. n = 9 biological replicates.
- Percent colocalization of vDNA foci detected by click chemistry to PML at 8 dpi in SCG neurons infected with HSV‐1EdC following IFNα treatment for 18 h prior to infection or for 18 h at 3 days prior to infection. Each point represents the percentage of 20 vDNA foci that colocalized to PML from 3 biological replicates.
- Quantification of PML puncta at time of infection in P6 SCG neurons treated with IFNα (600 IU/ml) for 18 h prior to infection or for 18 h at 3 days prior to infection. n = 60 cells from 3 biological replicates.
- Percent colocalization of vDNA foci detected by click chemistry to PML at 3 dpi in SCG neurons with HSV‐1EdC infected with ICP0‐null mutant HSV‐1, n212, or a rescued HSV‐1 virus, n212R, in P6 SCG neurons treated with IFNα for 18 h prior to infection or for 18 h at 3 days prior to infection. Each point represents the percentage of 20 vDNA foci that colocalized to PML from 3 biological replicates.
Data information: Data represent the mean ± SEM. Statistical comparisons were made using a one‐way ANOVA with Tukey’s multiple comparison (A–C) or a 2‐way ANOVA (D) (ns not significant, **P < 0.01, ****P < 0.0001).
Source data are available online for this figure.
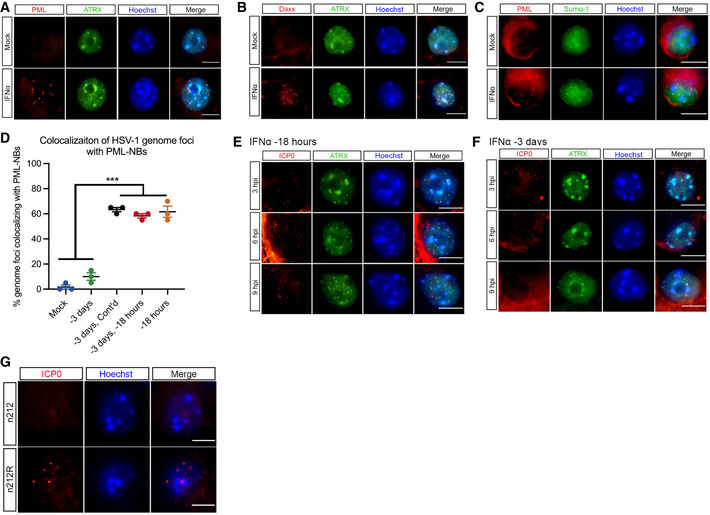
Figure EV4. HSV‐1 genomes only associate with PML‐NBs when type I IFN is present during initial infection.
-
ARepresentative images of untreated or IFNα‐treated (600 IU/ml) P6 SCG neurons stained for PML and ATRX at 3 days post‐treatment.
-
BRepresentative images of untreated or IFNα‐treated (600 IU/ml) P6 SCG neurons stained for Daxx and ATRX at 3 days post‐treatment.
-
CRepresentative images of untreated or IFNα‐treated (600 IU/ml) P6 SCG neurons stained for PML and SUMO‐1 at 3 days post‐treatment.
-
DPercent colocalization of vDNA foci detected by click chemistry to PML at 3 dpi in SCG neurons infected with HSV‐1EdC with or without IFNα (600 IU/ml) present at the time of infection. Each point represents the percentage of 20 vDNA foci that colocalized to PML from 3 biological replicates.
-
E, FRepresentative images of HSV‐1‐infected P6 SCG neurons treated with IFNα (600 IU/ml) for 18 h prior to infection or for 18 h at 3 days prior to infection and stained for ICP0 and ATRX at 3, 6, and 9 h post‐infection.
-
GRepresentative images of P6 SCG neurons infected with n212 or n212R for 8 h and stained for HSV‐1 ICP0.
Data information: Scale bars, 20 μm. Data represent the mean ± SEM. Statistical comparisons were made using a Mann–Whitney test (***P < 0.001).
The HSV‐infected cell protein 0 (ICP0) is a RING‐finger E3 ubiquitin ligase that is synthesized at very early stages of HSV‐1 infection (Boutell et al, 2002). During lytic infection, it localizes to PML‐NBs and disrupts their integrity by targeting PML and other PML‐NB‐associated proteins for degradation (Everett et al, 1998; Muller et al, 1998; Chelbi‐Alix & de The, 1999; Boutell et al, 2002; Boutell et al, 2011; Cuchet‐Lourenco et al, 2012; Alandijany et al, 2018). This activity is required for promoting the efficient onset of HSV‐1 lytic replication, and ICP0‐null mutants exhibit a defect in viral gene expression in certain cell types at low multiplicities of infection (Everett et al, 2008). ICP0 mRNA is also known to be expressed during the establishment of latency (Cliffe et al, 2013). Therefore, the colocalization of latent viral genomes to PML‐NBs and ultimately the ability of HSV to undergo reactivation could be due the presence of IFNα during initial infection and its effect on the localization or amount of ICP0. To investigate the distribution of ICP0 at early time points post‐infection, SCG neurons were treated with IFNα at either −3 dpi or −18 hpi and infected at a MOI of 7.5 PFU/cell with HSV‐1 Us11‐GFP in the presence of acyclovir (ACV). In both treatment groups, ICP0 staining similarly colocalized with puncta of ATRX, a correlate for PML‐NBs, at 3, 6, and 9 h post‐infection (Fig EV4E and F). Interestingly, foci of ATRX still remained even with the presence of ICP0, suggesting that ICP0 is not disrupting the integrity of PML‐NBs in this system. To further investigate the effect of ICP0 on the colocalization of latent viral genomes to PML‐NBs, we generated an EdC‐labeled ICP0‐null mutant strain n212 (Cai & Schaffer, 1989) and rescue (Lee et al, 2016) verified by immunofluorescence (Fig EV4G) and found that the presence or absence of ICP0 had no detectable impact on the ability of vDNA foci to colocalize to PML‐NBs (Fig 5D). Taken together, these data demonstrate that association of latent viral genomes with PML‐NBs in peripheral neurons is dependent on the formation of type I IFN‐induced PML‐NBs and the presence of type I IFN during initial infection and is independent of ICP0 expression.
PML is required for the IFNα‐dependent restriction of HSV‐1 latency
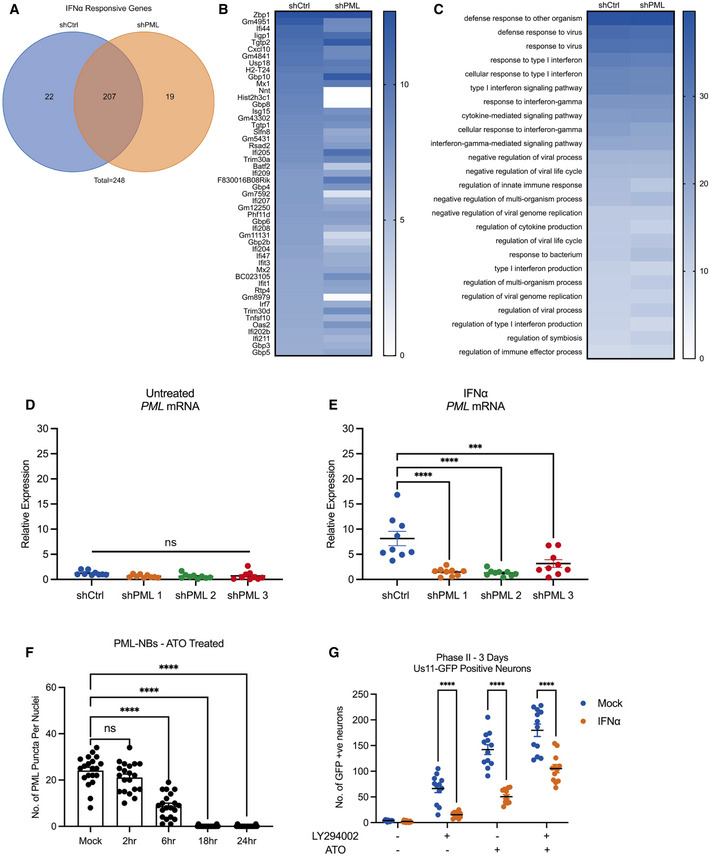
To determine whether the stable association of viral genomes with PML‐NBs directly contributes to the IFNα‐dependent restriction of HSV reactivation, we investigated whether PML depletion was sufficient to restore the ability of the latent viral genomes to reactivate. A previous study has shown that PML‐dependent recruitment of HIRA to ISG promoters contributes to the upregulation of gene expression as a result of cytokine release in response to HSV infection (McFarlane et al, 2019). Although carried out in non‐neuronal cells, this study and others (Ulbricht et al, 2012; Chen et al, 2015; Kim & Ahn, 2015; Scherer et al, 2016) suggest that PML itself may contribute to ISG upregulation, so to determine whether PML was indeed required for ISG stimulation in SCG neurons, we carried out RNA deep sequence analysis in IFNα‐treated neurons depleted of PML. Postnatal SCG neurons were transduced with lentiviral vectors expressing non‐targeting control or PML‐targeting shRNAs (shCtrl and shPML, respectively) and then mock treated or treated with IFNα (600 IU/ml) for 18 h prior to RNA extraction for next‐generation sequencing. High‐confidence reads were used for gene expression and gene ontology (GO) analysis. As expected, treatment of shCtrl transduced neurons with IFNα caused large changes in differentially regulated gene expression, with an enrichment of upregulated genes involved in immune system regulation. Similar to control neurons, PML‐depleted neurons also significantly upregulated the expression of genes involved in the response to IFNα stimulation. We found that of the total of 248 genes upregulated > 1.5‐fold following IFNα treatment, 83.47% of these genes were shared between the shCtrl‐ and shPML‐treated groups (Fig EV5A). Furthermore, we found similar ISG expression (Fig EV5B) and GO pathway enrichment (Fig EV5C). Therefore, in primary SCG neurons, the expression of ISGs in response to exogenous IFNα is largely independent of PML expression.
Figure EV5. PML is not required for ISG induction in primary postnatal sympathetic neurons.
-
AP6 SCG neurons were transduced with either control non‐targeting shRNA or shRNA targeting PML for 3 days, then treated with IFNα (600 IU/ml) for 18 h. Total genes > 1.5‐fold higher in IFNα (600 IU/ml) treated cells than untreated cells were subdivided into 3 groups: shCtrl‐treated neurons only. shPML‐treated neurons only. Both shCtrl and shPML neurons (shared).
-
BGene expression heat map of top 50 most upregulated genes in P6 neurons transduced with control non‐targeting shRNA or shRNA targeting PML for 3 days, then treated with IFNα (600 IU/ml) for 18 h.
-
CHeat map of the top 25 shared upregulated GO terms in P6 neurons transduced with control non‐targeting shRNA or shRNA targeting PML for 3 days, then treated with IFNα for 18 h.
-
D, ERT–qPCR for Pml mRNA expression in SCG neurons transduced with either control non‐targeting shRNA or shRNA targeting PML for 3 days, then treated with IFNα (600 IU/ml) for 18 h. n = 9 biological replicates.
-
FQuantification of detectable, IFNα (600 IU/ml)‐induced PML puncta in P6 SCG neurons treated with ATO (1 μM) for 2, 6, 18, and 24 h. n = 20 cells from 2 biological replicates.
-
GNumber of Us11‐GFP expressing neurons at 3 days post‐treatment with LY294002 (20 μM), ATO (1 μM) or LY294002 (20 μM) + ATO (1 μM) in P6 SCG neuronal cultures infected with HSV‐1 in the presence or absence of IFNα (600 IU/ml). n = 12 biological replicates.
Data information: Data represent the mean ± SEM. Statistical comparisons were made using a one‐way ANOVA with Tukey’s multiple comparison (D–F) or a 2‐way ANOVA (G) (ns not significant, ***P < 0.001, ****P < 0.0001).
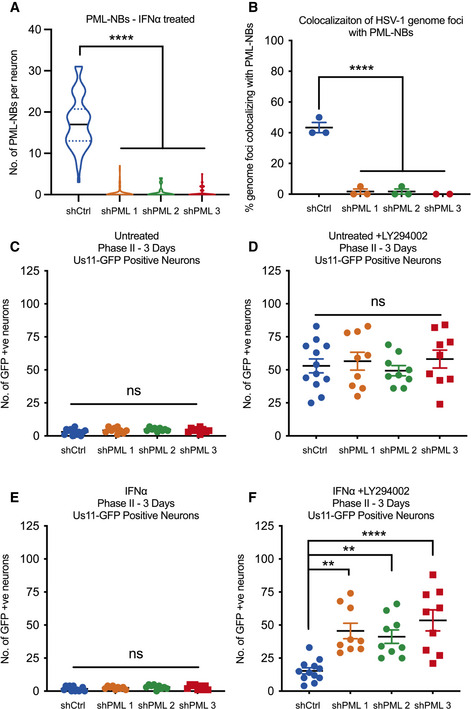
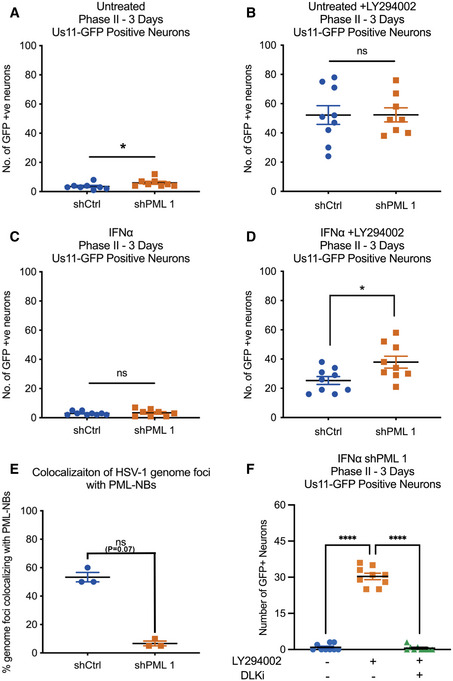
Because PML depletion did not detectably prevent the induction of type I IFN response genes in SCG neurons, we were able to examine the effect of PML depletion prior to infection on the IFNα‐mediated restriction of HSV‐1 reactivation. SCG neurons were transduced with lentiviral vectors expressing different PML‐targeting shRNA or control non‐targeting shRNA. PML depletion was confirmed by average number of PML‐NBs per nucleus (Fig 6A) and Pml mRNA expression level (Fig EV5D and E) in neurons transduced for 3 days then treated with IFNα (600 IU/ml). As expected, we found a significant decrease in the percent of vDNA foci stably colocalizing with PML‐NBs at 8 dpi in the shPML‐treated neurons compared to shCtrl‐treated neurons (Fig 6B). Furthermore, we assessed reactivation in neurons infected with HSV‐1 in the presence or absence of IFNα (150 IU/ml) at 3 days post‐transduction. In these experiments, neurons were infected with a Us11‐GFP gH‐null virus, which is defective in cell‐to‐cell spread and eliminates the need for WAY‐150138 during reactivation. In untreated neurons, we found no difference in reactivation following treatment with LY294002 (Fig 6C and D). In addition, PML depletion had no effect on the number of GFP‐positive neurons in the non‐reactivated samples, indicating that in this system that PML was not required for the establishment of latency. However, in neurons treated with IFNα at the time of initial infection, depletion of PML using either of the three PML shRNAs increased the ability of HSV to reactivate as indicated by a 2.97‐, 2.69‐, and 3.49‐fold increase in GFP‐positive neurons following treatment with LY294002, respectfully (Fig 6E and F). Moreover, there was no significant difference between the PML‐depleted, IFNα‐treated neurons and the non‐IFNα‐treated neurons, indicating that PML depletion fully restored the ability of HSV to reactivate from type Ι IFN‐treated neurons. Taken together, these data demonstrate that type I IFN exposure solely at the time of infection results in entrapment of viral genomes in PML‐NBs and restricts reactivation. This suggests that genome entrapment by PML promotes a more restrictive or deeper form of latency where reactivation is limited.
Figure 6. Depletion of PML with shRNA‐mediated knockdown prior to infection restores HSV‐1 reactivation in type I interferon‐treated primary sympathetic neurons.
-
AQuantification of PML puncta in P6 SCG neurons transduced with either control non‐targeting shRNA or shRNA targeting PML for 3 days, then treated with IFNα (600 IU/ml) for 18 h. n = 60 cells from 3 biological replicates.
-
BQuantification of colocalization of vDNA foci detected by click chemistry to PML in primary SCG neurons transduced with shRNA targeting PML for 3 days prior to being infected with HSV‐1EdC in the presence or absence of IFNα (150 IU/ml). Each point represents the percentage of 20 vDNA foci that colocalized to PML from 3 biological replicates.
-
C–FNumber of Us11‐GFP expressing neurons at 3 days post‐LY294002‐induced reactivation in P6 SCG neuronal cultures transduced with shRNA targeting PML for 3 days prior to infection with HSV‐1 in the presence or absence of IFNα (150 IU/ml). n = 9 biological replicates.
Data information: Data represent the mean ± SEM. Statistical comparisons were made using a one‐way ANOVA with Tukey’s multiple comparison (ns not significant, **P < 0.01, ****P < 0.0001).
Source data are available online for this figure.
Depletion of PML after the establishment of latency enhances reactivation in IFNα‐treated neurons
To explore the long‐term effect of stable PML‐NB‐association on the latent viral genome, we next tested whether PML depletion after the establishment of latency was sufficient to restore the ability of the latent viral genomes to reactivate following treatment with a trigger that may directly disrupt PML‐NBs. Arsenic trioxide (ATO) has been shown to bind directly to PML and disrupt PML‐NBs (Lallemand‐Breitenbach et al, 2008; Zhang et al, 2010; Sides et al, 2011), and we confirmed that ATO (1 μM) fully disrupted IFNα‐induced PML‐NBs in our peripheral neurons by 18 h post‐treatment (Fig EV5F). When we investigated reactivation in neurons that were latently infected in the presence or absence of IFNα, then treated with arsenic trioxide (ATO) at 8 dpi, we found that ATO is a very potent stimulator of reactivation independent of IFNα treatment, indicating that ATO is capable of triggering reactivation of genomes that are either PML‐NB‐associated or not (Fig EV5G). This is likely because ATO is a potent activator of the cell stress response and can result in robust histone phosphorylation (Gehani et al, 2010), which we have previously linked to reactivation (Cliffe et al, 2015). Although ATO could also induce reactivation in the presence of IFNα‐induced PML‐NBs, this reactivation was still less robust than mock‐treated neurons, likely reflecting the time required for disruption of PML‐NBs by ATO.
Therefore, to more specifically determine whether PML depletion restored the ability of neurons to reactivate following treatment with a physiological stimulus of reactivation, neurons were infected with Us11‐GFP gH‐null HSV‐1 virus in the presence or absence of IFNα (150 IU/ml) and subsequently transduced with lentiviral vectors expressing PML‐targeting shRNA or control non‐targeting shRNA at 1 dpi. Under these experimental conditions, PML knockdown post‐infection did not impact LY294002‐induced reactivation in untreated neurons (Fig 7A and C), but did increase the ability of HSV to reactivate from IFNα‐treated neurons in response to treatment with LY294002, as indicated by a 1.3‐fold increase in GFP‐positive neurons, albeit reactivation was not restored to levels seen in untreated neurons (Fig 7B and D). As expected, we found that only a small proportion of vDNA foci stably colocalize with PML‐NBs at 8 dpi in the shPML‐treated neurons compared to vDNA foci in the shCtrl‐treated neurons (Fig 7E). Therefore, PML depletion post‐infection does not result in detectable spontaneous reactivation of PML‐NB‐associated viral genomes, indicating that they are still in a repressed state and/or lack the necessary factors required to initiate gene expression. However, depletion of PML does partially restore the ability of HSV to enter the lytic from IFN‐treated neurons in response to a reactivation stimulus.
Figure 7. Depletion of PML with shRNA‐mediated knockdown post‐infection partially restores LY294002‐mediated HSV‐1 reactivation in type I interferon‐treated primary sympathetic neurons.
-
A–DNumber of Us11‐GFP expressing neurons at 3 days post‐LY294002‐induced reactivation in P6 SCG neuronal cultures transduced with shRNA targeting PML at 1 day post‐infection with HSV‐1 in the presence or absence of IFNα (150 IU/ml). n = 9 biological replicates. Statistical comparisons were made using a Mann–Whitney test (ns not significant, *P < 0.05).
-
EQuantification of colocalization of vDNA foci detected by click chemistry to PML in primary SCG neurons transduced with shRNA targeting PML at 1 day post‐infection with HSV‐1EdC in the presence or absence of IFNα (150 IU/ml). Each point represents the percentage of 20 vDNA foci that colocalized to PML from 3 biological replicates.
-
FNumber of Us11‐GFP expressing neurons at 3 days post‐reactivation in P6 SCG neuronal cultures transduced with shRNA targeting PML at 1 day post‐infection with HSV‐1 in the presence of IFNα (150 IU/ml). Reactivation was induced by LY294002 in the presence of the DLK inhibitor GNE‐3511 (4 μM). n = 9 biological replicates.
Data information: Data represent the mean ± SEM. Statistical comparisons were made using a Mann–Whitney test (A–E) or a one‐way ANOVA with Tukey’s multiple comparison (F) (ns not significant, *P < 0.05, ****P < 0.0001).
Source data are available online for this figure.
Previously, we have shown that reactivation in response to LY294002 is dependent on activation of the neuronal stress pathway involved dual‐leucine zipper kinase (DLK) and JNK activation (Cliffe et al, 2015). To test whether the same cell stress stimuli is required to induce reactivation from genomes released from PML‐NBs upon shRNA‐mediated knockdown of PML, we reactivated in the presence of the DLK inhibitor GNE‐3511 (Patel et al, 2015). GNE‐3511 inhibited LY294002‐mediated reactivation of latent genomes following PML depletion post‐infection (Fig 7F). Therefore, PML‐NBs maintain a restricted form of latency that is more refractory to reactivation, and following PML depletion, viral genomes do not undergo detectable spontaneous reactivation and are still dependent on activation of neuronal cell stress signaling pathways for reactivation.
Discussion
The considerable heterogeneity observed at the neuronal level in the colocalization of viral genomes with different nuclear domains may reflect in different types of latency that are more or less susceptible to reactivation. The determinants of this heterogeneity and a direct link between the subnuclear localization of a latent genome and its ability to reactivate following a given stimulus was not known. Using a primary neuronal model of HSV latency and reactivation, we found that the presence of type I IFN solely at that time of initial infection acts as a key mediator of the subnuclear distribution of latent viral genomes in neurons and promotes a more restricted form of latency that is less capable of reactivation following disruption of NGF signaling. Importantly, we show that activation of the type I IFN signaling pathway in peripheral neurons induces the detectable formation of PML‐NBs, which stably entrap a proportion of latent genomes. Importantly, we show that this IFN‐dependent restriction is mediated by PML, suggesting that PML‐NBs are directly responsible for the observed restriction of reactivation.
PML‐NBs typically number 1–30 bodies per nucleus in non‐neuronal cells (Bernardi & Pandolfi, 2007). In the mouse nervous system, however, Pml mRNA expression levels have previously been found to be low as measured by in situ hybridization (Gray et al, 2004). PML protein is enriched in neural progenitor cells, but the induction of differentiation results in the downregulation of PML both at a transcriptional and protein level, and Pml mRNA expression is undetectable in post‐mitotic neurons in many regions of the developing brain (Regad et al, 2009). Our findings in postnatal peripheral neurons further support these observations. Pml expression in adult mouse neurons varies considerably between brain regions but is generally confined to the gray matter (Hall et al, 2016). Although implicated to play a role in regulating circadian rhythms (Miki et al, 2012), synaptic plasticity (Bloomer et al, 2007) and the response to toxic proteins that cause neurodegenerative disorders (Yamada et al, 2001; Kumada et al, 2002; Mackenzie et al, 2006; Chort et al, 2013), PML regulation, and function in adult nervous system is still largely unknown. In our study, we could not detect PML‐NBs in adult primary neurons isolated from the SCG or the TG. In contrast to our findings, PML‐NBs have previously been shown to be present in adult mouse and human TG neurons by FISH and immunofluorescence (Catez et al, 2012; Maroui et al, 2016). However, no quantification was done in these studies, and Catez et al, (2012) describe subpopulations of adult TG neurons that did not display any PML signal in the nucleus. In addition, characterization of PML distribution in adult TG neurons by IF‐FISH of ganglia isolated in vivo may reflect prior exposure to type I IFNs or other signaling molecules. The functional significance of peripheral neurons lacking PML‐NBs is unclear, but could be linked to the capacity of neurons to undergo dynamic rearrangement of local and global nuclear architecture during maturation or neuronal excitation. An absence of PML‐NBs in neurons could also contribute to their resistance to apoptosis, as PML has also been shown to play a role in cell death through the induction of both p53‐dependent and p53‐independent apoptotic pathways (Quignon et al, 1998; Wang et al, 1998; Guo et al, 2000). Whether PML‐mediated regulation of these pathways occurs in the context of PML‐NBs or by PML itself is unclear, but interestingly, the pro‐apoptotic functions of Daxx, a PML‐NB‐associated protein, may require localization to PML‐NBs in certain cell types (Croxton et al, 2006). Furthermore, our in vitro model using pure populations of intact neurons is devoid of the immune responses and complexities of intact animals, and we cannot rule out the possibility that axotomy or the processing of the neurons ex vivo could lead to PML‐NB disruption or dispersal. However, notwithstanding these caveats, primary neurons provide an excellent model system to understand the impact of extrinsic immune factors and PML‐NBs to the altering the nature of HSV latency.
Peripheral neurons are capable of responding to type I IFN signaling, given the robust induction in ISG expression and formation of PML‐NBs following treatment with IFNα, and this is supported by a number of previous studies (Yordy et al, 2012; Katzenell & Leib, 2016; Song et al, 2016; Linderman et al, 2017; Barragan‐Iglesias et al, 2020). Importantly, however, peripheral neurons produce little to no type I interferons upon HSV infection (Yordy et al, 2012; Rosato & Leib, 2014), indicating that IFN production arises from other surrounding infected cells. Infected fibroblasts at the body surface, as well as professional immune cells, have been shown to produce high levels of IFNα/β after HSV infection (Hochrein et al, 2004; Li et al, 2006; Rasmussen et al, 2007; Rasmussen et al, 2009). In addition, there is evidence of elevated type I IFN in peripheral ganglia during HSV‐1 infection (Carr et al, 1998), suggesting that glial or immune cells located adjacent to peripheral neuron cell bodies are capable of type I IFN production. It will be important to delineate if the inflammatory environment at the initial site of infection acts on neuronal axons to prime the neuron for a more repressed latent infection or if inflammatory cytokines in the ganglia are crucial for promoting a more repressive state. Although responsive to IFN, primary peripheral and cortical mouse neurons have previously been shown to have inefficient type I IFN‐mediated antiviral protection compared to non‐neuronal mitotic cells (Yordy et al, 2012; Kreit et al, 2014). One study showed that DRG neurons are less responsive to type I IFN signaling and used an absence of cell death upon IFN treatment as one of their criteria (Yordy et al, 2012). It should be noted that different cell types display specific responses to type I IFN signaling and peripheral neurons have even been reported to be more protected from cell death stimuli following IFN treatment (Chang et al, 1990). Furthermore, a previous study found that inducible reactivation of HSV‐1 from latently infected neuronal cultures is transiently sensitive to type I IFNs (Linderman et al, 2017). Our model of HSV‐1 latency and reactivation in primary sympathetic neurons highlights a type I IFN response that is PML‐dependent and suggests a role for neuronal IFN signaling in promoting a more restricted latent HSV‐1 infection.
Prior to this study, it was not clear whether viral genomes associated with PML‐NBs were capable of undergoing reactivation. In response to inhibition of NGF signaling, our data demonstrate that PML‐NB‐associated genomes are more restricted for reactivation given that (i) IFN induces PML‐NB formation and increased association with viral genomes with PML‐NBs, (ii) IFN pretreatment promotes restriction of viral reactivation, and (iii) the ability of viral genomes to reactivate from IFN‐treated neurons increases with PML knockdown either prior to or following infection. Previous work by Cohen et al (2018) showed that quiescent genomes associated with PML‐NBs in fibroblasts can be transcriptionally reactivated by induced expression of ICP0. However, this previous study did not address the capability of viral genomes to reactivate in the absence of viral lytic protein (i.e., during reactivation from latency in neurons). In a further study using primary neurons, treatment of quiescently infected neurons with the histone deacetylase inhibitor, trichostatin A (TSA), could lead to disruption of PML‐NBs and induce active viral transcription in a subset of PML‐NB‐associated genomes (Maroui et al, 2016). However, the mechanisms of reactivation following TSA treatment are not known and may be directed via altering the HSV chromatin structure or indirect via increasing the acetylation levels of histones or non‐histone proteins, including PML. How increased acetylation relates to the physiological triggers that induce HSV reactivation is not clear. In contrast, loss of neurotrophic signaling can occur in response to known physiological stimuli that trigger HSV reactivation (Suzich & Cliffe, 2018). Although we cannot rule out the possibility that different stimuli have the potential for PML‐NB‐associated genomes to undergo reactivation, this study clearly demonstrates that at least one well‐characterized trigger of reactivation cannot efficiently induce PML‐NB‐associated genomes to undergo transcription.
Our results identify a persistence of PML‐NBs, an IFN‐mediated innate immune response, that allows for long‐term restriction of latent viral genomes in the absence of continued ISG expression. Interestingly, type I IFN‐induced PML‐NBs persisted for up to 15 days post‐treatment both in the presence and in the absence of viral infection. Given the absence of PML‐NBs in our untreated peripheral neurons, this induction and persistence could represent neuron‐specific innate immune memory. The persistence of PML‐NBs in neurons may alter the subsequent response to IFN and/or viral infection, and it will be interesting to determine whether there is trained immunity in neurons such that subsequent responses differ from the first exposure. What is clear from our results, however, is the role of PML and IFN exposure in sustained repression of the latent HSV genome. Even in the absence of known chromatin changes that occur on the PML‐associated viral genome, this long‐term effect on the ability of the HSV‐1 genome to respond to an exogenous signal and restriction of reactivation is reminiscence of the classical definition of an epigenetic change (of course in the case of post‐mitotic neurons in the absence of inheritance).
Here, we demonstrate that there are different types of HSV latency dependent on the subnuclear positioning of the viral genome and ability to reactivate. Genomes associated with PML‐NBs are one form of restricted latency in our system. PML‐NBs are known to play a role in the restriction of viral gene expression in non‐neuronal cells, but the potential mechanism of PML‐NB‐mediated HSV gene silencing in neurons is unknown. During latency, the viral genome is enriched with histone post‐translational modifications (PTMs) consistent with repressive heterochromatin, including H3K9me2/3 and H3K27me3, and it is possible that PML‐NBs play a role in the association of viral genomes with core histones, repressive PTMs, or heterochromatin‐associated proteins (Wang et al, 2005; Cliffe et al, 2009; Kwiatkowski et al, 2009), or instead via physical compaction of the viral genome in PML‐NBs. Although PML‐NBs promote a more restricted form of latency, we have shown that latency can be established in the absence of IFN treatment and PML‐NBs. Even in IFN‐treated neurons, only a proportion of the latent viral genomes colocalized with PML‐NBs. This indicates that latent genomes associate with other subnuclear regions and proteins that may promote the assembly and/or maintenance of repressive heterochromatic histone modifications. This supports previous observations that HSV‐1 viral genomes also colocalize with centromeric repeats and other, undefined nuclear domains in latently infected TG in vivo (Catez et al, 2012). For example, the viral genome is known to be enriched for H3K27me3 (Wang et al, 2005; Cliffe et al, 2009; Kwiatkowski et al, 2009), which can be bound by Polycomb group proteins. Interestingly, we have found that the multifunctional, chromatin remodeler protein ATRX has abundant nuclear staining in neurons and, in contrast to non‐neuronal cells, is localized outside of PML‐NBs. ATRX staining overlapped with Hoechst DNA staining in our primary neurons, suggesting its localization with AT‐rich heterochromatin regions (Bucevicius et al, 2019). Ultimately, the latent viral genomes are likely bound by ATRX, Polycomb group proteins, or other repressive cellular proteins independently of PML‐NBs. Investigating the identity, mechanism of targeting, and role of these proteins in the induction and maintenance of latency will ultimately facilitate the development of antiviral therapeutics that target the latent stage of infection to prevent reactivation.
Materials and Methods
Reagents
Compounds used in the study are as follows: Acycloguanosine, FUDR, LY 294002, Nerve Growth Factor 2.5S (Alomone Labs), Primocin (Invivogen), Aphidicolin (AG Scientific), IFN‐α (EMD Millipore IF009), IFN‐β (EMD Millipore IF011), ΙFN‐γ (EMD Millipore IF005), and IFN‐λ2 (PeproTech 250‐33); WAY‐150138 was kindly provided by Pfizer, Dr. Jay Brown and Dr. Dan Engel at the University of Virginia, and Dr. Lynn Enquist at Princeton University. Compound information and concentrations used can be found below in Table 1.
Table 1.
Compounds used and concentrations.
| Compound | Supplier | Identifier | Concentration |
|---|---|---|---|
| Acycloguanosine | Millipore Sigma | A4669 | 10 µM, 50 µM |
| FUDR | Millipore Sigma | F‐0503 | 20 µM |
| L‐Glutamic Acid | Millipore Sigma | G5638 | 3.7 µg/ml |
| LY 294002 | Tocris | 1130 | 20 µM |
| IFNα | EMD Millipore | IF009 | 150 IU/ml, 600 IU/ml |
| IFNβ | EMD Millipore | IF011 | 150 IU/ml |
| IFNγ | EMD Millipore | IF005 | 150 IU/ml, 500 IU/ml |
| IFNλ2 | PeproTech | 250‐33 | 100 ng/ml, 500 ng/ml |
| NGF 2.5S | Alomone Labs | N‐100 | 50 ng/ml |
| Primocin | Invivogen | ant‐pm‐1 | 100 µg/ml |
| Aphidicolin | AG Scientific | A‐1026 | 3.3 µg/ml |
| WAY‐150138 | Pfizer | N/A | 10 µg/ml |
| AFDye 555 Azide Plus | Click Chemistry Tools | 1479‐1 | 10 μM |
Preparation of HSV‐1 virus stocks
HSV‐1 stocks of eGFP‐Us11 Patton were grown and titrated on Vero cells obtained from the American Type Culture Collection (Manassas, VA). Cells were maintained in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% FetalPlex (Gemini Bio‐Products) and 2 mM l‐Glutamine. eGFP‐Us11 Patton (HSV‐1 Patton strain with eGFP reporter protein fused to true late protein Us11 (Benboudjema et al, 2003)) was kindly provided by Dr. Ian Mohr at New York University.
StayPut Us11‐GFP was created by inserting an eUs11‐GFP tag into the previously created gH‐deficient HSV‐1 SCgHZ virus (strain SC16) through co‐transfection of SCgHZ viral DNA and pSXZY‐eGFP‐Us11 plasmid (Forrester et al, 1992). StayPut Us11‐GFP is propagated and titrated on previously constructed Vero F6 cells, which contain copies of the gH gene under the control of an HSV‐1 gD promoter, as described in Forrester et al, (1992). Vero F6s are maintained in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% FetalPlex (Gemini Bio‐Products). They are selected with the supplementation of 250 ug/mL of G418/Geneticin (Gibco).
Primary neuronal cultures
Sympathetic neurons from the Superior Cervical Ganglia (SCG) of post‐natal day 0‐2 (P0‐P2) or adult (P21‐P24) CD1 Mice (Charles River Laboratories) were dissected as previously described (Cliffe et al, 2015). Sensory neurons from trigeminal ganglia (TG) of post‐natal day 0‐2 (P0‐P2) CD1 mice (Charles River Laboratories) were dissected using the same protocol. Sensory neurons from TG of adult were dissected as previously described (Bertke et al, 2011) with a modified purification protocol using Percoll from the protocol published by Malin et al (2007). Rodent handling and husbandry were carried out under animal protocols approved by the Animal Care and Use Committee of the University of Virginia (UVA). Ganglia were briefly kept in Leibovitz’s L‐15 media with 2.05 mM l‐glutamine before dissociation in collagenase type IV (1 mg/ml) followed by trypsin (2.5 mg/ml) for 20 min each at 37°C. Dissociated ganglia were triturated, and approximately 10,000 neurons per well were plated onto rat tail collagen in a 24‐well plate. Sympathetic neurons were maintained in CM1 (Neurobasal® Medium supplemented with PRIME‐XV IS21 Neuronal Supplement (Irvine Scientific), 50 ng/ml Mouse NGF 2.5S, 2 mM l‐Glutamine, and Primocin). Aphidicolin (3.3 µg/ml) was added to the CM1 for the first five days post‐dissection to select against proliferating cells. Sensory neurons were maintained in the same media supplemented with GDNF (50 ng/ml; PeproTech 450‐44).
Establishment and reactivation of latent HSV‐1 infection in primary neurons
Latent HSV‐1 infection was established in P6‐8 sympathetic neurons from SCGs. Neurons were cultured for at least 24 h without antimitotic agents prior to infection. The cultures were infected with eGFP‐Us11 (Patton recombinant strain of HSV‐1 expressing an eGFP reporter fused to true late protein Us11) or StayPut. Neurons were infected at a multiplicity of infection (MOI) of 7.5 PFU/cell with eGFP‐Us11 and at an MOI of 5 PFU/cell with StayPut (assuming 1.0 × 104 neurons/well/24‐well plate) in DPBS + CaCl2 + MgCl2 supplemented with 1% fetal bovine serum, 4.5 g/l glucose, and 10 µM Acyclovir (ACV) for 2–3 h at 37°C. Post‐infection, inoculum was replaced with CM1 containing 50 µM ACV and an anti‐mouse IFNAR1 antibody (Leinco Tech I‐1188, 1:1,000) for 5–6 days, followed by CM1 without ACV. Reactivation was carried out in DMEM/F12 (Gibco) supplemented with 10% fetal bovine serum, Mouse NGF 2.5S (50 ng/ml), and Primocin. WAY‐150138 (10 µg/ml) was added to reactivation cocktail to limit cell‐to‐cell spread. Reactivation was quantified by counting number of GFP‐positive neurons or performing reverse transcription–quantitative PCR (RT–qPCR) of HSV‐1 lytic mRNAs isolated from the cells in culture.
Analysis of mRNA expression by reverse transcription–quantitative PCR (RT–qPCR)
To assess relative expression of HSV‐1 lytic mRNA, total RNA was extracted from approximately 1.0 × 104 neurons using the Quick‐RNA™ Miniprep Kit (Zymo Research) with an on‐column DNase I digestion. mRNA was converted to cDNA using the SuperScript IV First‐Strand Synthesis system (Invitrogen) using random hexamers for first‐strand synthesis and equal amounts of RNA (20–30 ng/reaction). To assess viral DNA load, total DNA was extracted from approximately 1.0 × 104 neurons using the Quick‐DNA™ Miniprep Plus Kit (Zymo Research). qPCR was carried out using Power SYBR™ Green PCR Master Mix (Applied Biosystems). The relative mRNA or DNA copy number was determined using the comparative C T (ΔΔC T) method normalized to mRNA or DNA levels in latently infected samples. Viral RNAs were normalized to mouse reference gene GAPDH. All samples were run in duplicate on an Applied Biosystems™ QuantStudio™ 6 Flex Real‐Time PCR System and the mean fold change compared to the reference gene calculated. Primers used are described in Table 2.
Table 2.
Primers used for RT–qPCR.
| Primer | Sequence 5’–3’ |
|---|---|
| mGAP 1SF | CAT GGC CTT CCG TGT GTT CCT A |
| mGAP 1SR | GCG GCA CGT CAG ATC CA |
| ICP27 F | GCA TCC TTC GTG TTT GTC ATT CTG |
| ICP27 R | GCA TCT TCT CTC CGA CCC CG |
| ICP8 1SF | GGA GGT GCA CCG CAT ACC |
| ICP8 1SR | GGC TAA AAT CCG GCA TGA AC |
| gC #1 F | GAG TTT GTC TGG TTC GAG GAC |
| gC #1R | ACG GTA GAG ACT GTG GTG AA |
| PML F | GGG AAA CAG AGG AGC GAG TT |
| PML R | AAG GCC TTG AGG GAA TTG GG |
| ISG15 F | CAA GCA GCC AGA AGC AGA CT |
| ISG15 R | CCC AGC ATC TTC ACC TTT AGG |
| IRF7 F | CCA GTT GAT CCG CAT AAG GT |
| IRF7 R | GAG GCT CAC TTC TTC CCT ATT T |
| LAT F | TGT GTG GTG CCC GTG TCT T |
| LAT R | CCA GCC AAT CCG TGT CGG |
Immunofluorescence
Neurons were fixed for 15 min in 4% formaldehyde and blocked in 5% bovine serum albumin and 0.3% Triton X‐100 and incubated overnight in primary antibody. Antibodies used are described in Table 3. Following primary antibody treatment, neurons were incubated for one hour in Alexa Fluor® 488‐, 555‐, and 647‐conjugated secondary antibodies for multi‐color imaging (Invitrogen). Nuclei were stained with Hoechst 33258 (Life Technologies). Unless indicated otherwise, z‐stack images of entire nuclei were acquired using an sCMOS charge‐coupled device camera (pco.edge) mounted on a Nikon Eclipse Ti Inverted Epifluorescent microscope and processed into 2D projection images using the NIS‐Elements software (Nikon) Extended Depth of Focus (EDF) plug‐in. Images were further analyzed and processed using ImageJ.
Table 3.
Antibodies used for immunofluorescence and concentrations
| Antibody | Supplier | Identifier/RRID | Concentration |
|---|---|---|---|
|
Anti‐PML Mouse monoclonal |
EMD Millipore |
MAB3738 AB_2166836 |
1:200 |
|
Anti‐Beta‐III Tubulin Chicken polyclonal |
Millipore sigma |
AB9354 AB_570918 |
1:500 |
|
Anti‐ATRX Rabbit polyclonal |
Santa Cruz Bio |
sc‐15408 AB_2061023 |
1:250 |
|
Anti‐Daxx Mouse monoclonal |
Santa Cruz Bio |
sc‐8043 AB_627405 |
1:250 |
|
Anti‐STAT1 Rabbit monoclonal |
Cell Signaling Technologies |
14994 AB_2737027 |
1:400 |
|
Anti‐Mx1/2/3 Mouse monoclonal |
Santa Cruz Bio |
sc‐166412 AB_2147714 |
1:250 |
|
Anti‐HSV‐1 ICP0 Mouse monoclonal |
East Coast Bio | H1A027 | 1:200 |
|
Anti‐SUMO‐1 Rabbit monoclonal |
Abcam |
Ab32058 AB_778173 |
1:250 |
|
Anti‐IFNAR1 Mouse monoclonal |
Leinco Tech |
I‐1188 AB_2830518 |
1:1,000 |
|
F(ab’)2 Anti‐Mouse IgG Alexa Fluor® 555 Goat polyclonal |
Thermo Fisher |
A21425 AB_2535846 |
1:1,000 |
|
F(ab’)2 Anti‐Rabbit IgG Alexa Fluor® 488 Goat polyclonal |
Thermo Fisher |
A11070 AB_2534114 |
1:1,000 |
|
F(ab’)2 Anti‐Rabbit IgG Alexa Fluor® 488 Goat polyclonal |
Thermo Fisher |
A11017 AB_2534084 |
1:1,000 |
|
Anti‐Chicken IgY Alexa Fluor® 647 Goat polyclonal |
Abcam |
ab150175 AB_2732800 |
1:1,000 |
Click chemistry
For EdC‐labeled HSV‐1 virus infections, an MOI of 5 was used. EdC‐labeled virus was prepared using a previously described method (McFarlane et al, 2019). Click chemistry was carried out as described previously (Alandijany et al, 2018) with some modifications. Neurons were washed with CSK buffer (10 mM HEPES, 100 mM NaCl, 300 mM Sucrose, 3 mM MgCl2, 5 mM EGTA) and simultaneously fixed and permeabilized for 10 min in 1.8% methanol‐free formaldehyde (0.5% Triton X‐100, 1% phenylmethylsulfonyl fluoride (PMSF)) in CSK buffer, then washed twice with PBS before continuing to the click chemistry reaction and immunostaining. Samples were blocked with 3% BSA for 30 min, followed by click chemistry using EdC‐labeled HSV‐1 DNA and the Click‐iT EdU Alexa Flour 555 Imaging Kit (Thermo Fisher Scientific, C10638) according to the manufacturer’s instructions with AFDye 555 Picolyl Azide (Click Chemistry Tools, 1288). For immunostaining, samples were incubated overnight with primary antibodies in 3% BSA. Following primary antibody treatment, neurons were incubated for one hour in Alexa Fluor® 488‐ and 647‐conjugated secondary antibodies for multi‐color imaging (Invitrogen). Nuclei were stained with Hoechst 33258 (Life Technologies). Epifluorescence microscopy images were acquired at 60× using an sCMOS charge‐coupled device camera (pco.edge) mounted on a Nikon Eclipse Ti Inverted Epifluorescent microscope using NIS‐Elements software (Nikon). Images were analyzed and processed using ImageJ. Confocal microscopy images were acquired using a Zeiss LSM 880 confocal microscope using the 63× Plan‐Apochromat oil immersion lens (numerical aperture 1.4) using 405, 488, 543, and 633 nm laser lines. Zen black software (Zeiss) was used for image capture, generating cut mask channels, and calculating weighted colocalization coefficients. Exported images were processed with minimal adjustment using Adobe Photoshop and assembled for presentation using Adobe Illustrator.
Preparation of lentiviral vectors
Lentiviruses expressing shRNA against PML (PML‐1 TRCN0000229547, PML‐2 TRCN0000229549, PML‐3 TRCN0000314605), or a control lentivirus shRNA (Everett et al, 2006) were prepared by co‐transfection with psPAX2 and pCMV‐VSV‐G (Stewart et al, 2003) using the 293LTV packaging cell line (Cell Biolabs). Supernatant was harvested at 40‐ and 64‐h post‐transfection. Sympathetic neurons were transduced overnight in neuronal media containing 8μg/ml protamine and 50 μM ACV.
RNA sequence analysis
Reads were checked for quality using FASTQC (v0.11.8), trimmed using BBMAP (v3.8.16b), and aligned to the mouse genome with GENCODE (vM22) annotations using STAR (v2.7.1a). Transcripts per million calculations were performed by RSEM (v1.3.1), the results of which were imported into R (v4.0.2) and Bioconductor (v3.12) using tximport (v1.18.0). Significant genes were called using DESeq2, using fold change cutoffs and p‐value cutoffs of 0.5 and 0.05, respectively. Results were visualized using Heatplus (v2.36.0), PCAtools (v2.2.0), and UpSetR (v1.4.0). Functional enrichment was performed using GSEA and Metascape.
Statistical analysis
Power analysis was used to determine the appropriate sample sizes for statistical analysis. All statistical analysis was performed using Prism V8.4. A Mann–Whitney test was used for all experiments where the group size was 2. All other experiments were analyzed using a one‐way ANOVA with Tukey’s multiple comparison. Specific analyses are included in the figure legends. For all reactivation experiments measuring GFP expression, viral DNA, gene expression, or DNA load, individual biological replicates were plotted (an individual well of primary neurons) and all experiments were repeated from pools of neurons from at least 3 litters.
Author contributions
JBS: Conceptualization, formal analysis, investigation, methodology, validation, visualization, writing—original draft, writing—review and editing. SRC: Investigation, validation. HB: Investigation. SD: Investigation, validation. EK: Data curation, formal analysis, methodology, software. ARS: Investigation. AB: Investigation. CB: Resources, writing—review and editing. ARC: Conceptualization, formal analysis, funding acquisition, investigation, validation, methodology, project administration, supervision, validation, visualization, writing—original draft, writing—review and editing.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Expanded View Figures PDF
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Dr. Ian Mohr at New York University for the gift of the Us11‐GFP virus and Gary Cohen at the University of Pennsylvania for SCgHZ. This work was supported by R21AI151340 (ARC), R01NS105630 (ARC), The Owens Family Foundation (ARC), NIH/NEI F30EY030397 (JBS), NIH/NIAID T32AI007046 (JBS and SRC), T32GM007267 (JBS), NIH/NIGMS T32GM008136 (SD) and MRC (https://mrc.ukri.org) MC_UU_12014/5 (CB).
EMBO reports (2021) 22: e52547.
See also: SK Weller & NA Deluca (September 2021)
Data availability
The RNA‐seq dataset generated and analyzed in the current study is available at the NCBI Gene Expression omnibus (accession number GSE166738; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166738).
References
- Alandijany T, Roberts APE, Conn KL, Loney C, McFarlane S, Orr A, Boutell C (2018) Distinct temporal roles for the promyelocytic leukaemia (PML) protein in the sequential regulation of intracellular host immunity to HSV‐1 infection. PLoS Pathog 14: e1006769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baringer JR, Pisani P (1994) Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann Neurol 36: 823–829 [DOI] [PubMed] [Google Scholar]
- Baringer JR, Swoveland P (1973) Recovery of herpes‐simplex virus from human trigeminal ganglions. N Engl J Med 288: 648–650 [DOI] [PubMed] [Google Scholar]
- Barragan‐Iglesias P, Franco‐Enzastiga U, Jeevakumar V, Shiers S, Wangzhou A, Granados‐Soto V, Campbell ZT, Dussor G, Price TJ (2020) Type I interferons act directly on nociceptors to produce pain sensitization: implications for viral infection‐induced pain. J Neurosci 40: 3517–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benboudjema L, Mulvey M, Gao Y, Pimplikar SW, Mohr I (2003) Association of the herpes simplex virus type 1 Us11 gene product with the cellular kinesin light‐chain‐related protein PAT1 results in the redistribution of both polypeptides. J Virol 77: 9192–9203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi R, Pandolfi PP (2007) Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8: 1006–1016 [DOI] [PubMed] [Google Scholar]
- Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, Margolis TP (2011) A5‐positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J Virol 85: 6669–6677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop CL, Ramalho M, Nadkarni N, May Kong W, Higgins CF, Krauzewicz N (2006) Role for centromeric heterochromatin and PML nuclear bodies in the cellular response to foreign DNA. Mol Cell Biol 26: 2583–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomer WA, VanDongen HM, VanDongen AM (2007) Activity‐regulated cytoskeleton‐associated protein Arc/Arg3.1 binds to spectrin and associates with nuclear promyelocytic leukemia (PML) bodies. Brain Res 1153: 20–33 [DOI] [PubMed] [Google Scholar]
- Boutell C, Cuchet‐Lourenco D, Vanni E, Orr A, Glass M, McFarlane S, Everett RD (2011) A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog 7: e1002245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutell C, Sadis S, Everett RD (2002) Herpes simplex virus type 1 immediate‐early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J Virol 76: 841–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucevicius J, Keller‐Findeisen J, Gilat T, Hell SW, Lukinavicius G (2019) Rhodamine‐Hoechst positional isomers for highly efficient staining of heterochromatin. Chem Sci 10: 1962–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral JM, Oh HS, Knipe DM (2018) ATRX promotes maintenance of herpes simplex virus heterochromatin during chromatin stress. Elife 7: e40228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai WZ, Schaffer PA (1989) Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J Virol 63: 4579–4589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV (2010) Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV‐1 latency in neurons. Cell Host Microbe 8: 320–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DJ, Veress LA, Noisakran S, Campbell IL (1998) Astrocyte‐targeted expression of IFN‐alpha1 protects mice from acute ocular herpes simplex virus type 1 infection. J Immunol 161: 4859–4865 [PubMed] [Google Scholar]
- Catez F, Picard C, Held K, Gross S, Rousseau A, Theil D, Sawtell N, Labetoulle M, Lomonte P (2012) HSV‐1 genome subnuclear positioning and associations with host‐cell PML‐NBs and centromeres regulate LAT locus transcription during latency in neurons. PLoS Pathog 8: e1002852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Martin DP, Johnson EM Jr (1990) Interferon suppresses sympathetic neuronal cell death caused by nerve growth factor deprivation. J Neurochem 55: 436–445 [DOI] [PubMed] [Google Scholar]
- Chelbi‐Alix MK, Pelicano L, Quignon F, Koken MH, Venturini L, Stadler M, Pavlovic J, Degos L, de The H (1995) Induction of the PML protein by interferons in normal and APL cells. Leukemia 9: 2027–2033 [PubMed] [Google Scholar]
- Chelbi‐Alix MK, de The H (1999) Herpes virus induced proteasome‐dependent degradation of the nuclear bodies‐associated PML and Sp100 proteins. Oncogene 18: 935–941 [DOI] [PubMed] [Google Scholar]
- Chen SH, Kramer MF, Schaffer PA, Coen DM (1997) A viral function represses accumulation of transcripts from productive‐cycle genes in mouse ganglia latently infected with herpes simplex virus. J Virol 71: 5878–5884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wright J, Meng X, Leppard KN (2015) Promyelocytic Leukemia Protein Isoform II Promotes Transcription Factor Recruitment To Activate Interferon Beta and Interferon‐Responsive Gene Expression. Mol Cell Biol 35: 1660–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chort A, Alves S, Marinello M, Dufresnois B, Dornbierer J‐G, Tesson C, Latouche M, Baker DP, Barkats M, El Hachimi KHet al (2013) Interferon beta induces clearance of mutant ataxin 7 and improves locomotion in SCA7 knock‐in mice. Brain 136: 1732–1745 [DOI] [PubMed] [Google Scholar]
- Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M (2015) Neuronal stress pathway mediating a histone Methyl/Phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18: 649–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Coen DM, Knipe DM (2013) Kinetics of facultative heterochromatin and polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. MBio 4: e00590‐12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Garber DA, Knipe DM (2009) Transcription of the herpes simplex virus latency‐associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83: 8182–8190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Knipe DM (2008) Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J Virol 82: 12030–12038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Wilson AC (2017) Restarting lytic gene transcription at the onset of herpes simplex virus reactivation. J Virol 91: e01419‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clynes D, Higgs DR, Gibbons RJ (2013) The chromatin remodeller ATRX: a repeat offender in human disease. Trends Biochem Sci 38: 461–466 [DOI] [PubMed] [Google Scholar]
- Cohen C, Corpet A, Roubille S, Maroui MA, Poccardi N, Rousseau A, Kleijwegt C, Binda O, Texier P, Sawtell Net al (2018) Promyelocytic leukemia (PML) nuclear bodies (NBs) induce latent/quiescent HSV‐1 genomes chromatinization through a PML NB/Histone H3.3/H3.3 Chaperone Axis. PLoS Pathog 14: e1007313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croxton R, Puto LA, de Belle I , Thomas M, Torii S, Hanaii F, Cuddy M, Reed JC (2006) Daxx represses expression of a subset of antiapoptotic genes regulated by nuclear factor‐kappaB. Cancer Res 66: 9026–9035 [DOI] [PubMed] [Google Scholar]
- Cuchet‐Lourenco D, Vanni E, Glass M, Orr A, Everett RD (2012) Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO‐independent degradation. J Virol 86: 11209–11222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddy SR, Schinlever AR, Dochnal S, Seegren PV, Suzich J, Kundu P, Downs TK, Farah M, Desai BN, Boutell Cet al (2020) Neuronal hyperexcitability is a DLK‐dependent trigger of herpes simplex virus reactivation that can be induced by IL‐1. eLife 9: e58037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T, Zhou G, Roizman B (2011) HSV‐1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency‐associated transcript and miRNAs. Proc Natl Acad Sci 108: 18820–18824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efstathiou S, Preston CM (2005) Towards an understanding of the molecular basis of herpes simplex virus latency. Virus Res 111: 108–119 [DOI] [PubMed] [Google Scholar]
- Everett RD, Chelbi‐Alix MK (2007) PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89: 819–830 [DOI] [PubMed] [Google Scholar]
- Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J (1998) The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110‐ and proteasome‐dependent loss of several PML isoforms. J Virol 72: 6581–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Maul GG (1994) HSV‐1 IE protein Vmw110 causes redistribution of PML. EMBO J 13: 5062–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Murray J (2005) ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J Virol 79: 5078–5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Murray J, Orr A, Preston CM (2007) Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J Virol 81: 10991–11004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Parada C, Gripon P, Sirma H, Orr A (2008) Replication of ICP0‐null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J Virol 82: 2661–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A (2006) PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol 80: 7995–8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Sourvinos G, Leiper C, Clements JB, Orr A (2004) Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10‐like complexes. J Virol 78: 1903–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester A, Farrell H, Wilkinson G, Kaye J, Davis‐Poynter N, Minson T (1992) Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J Virol 66: 341–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber DA, Schaffer PA, Knipe DM (1997) A LAT‐associated function reduces productive‐cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J Virol 71: 5885–5893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrick D, Samara V, McDowell TL, Smith AJ, Dobbie L, Higgs DR, Gibbons RJ (2004) A conserved truncated isoform of the ATR‐X syndrome protein lacking the SWI/SNF‐homology domain. Gene 326: 23–34 [DOI] [PubMed] [Google Scholar]
- Gehani SS, Agrawal‐Singh S, Dietrich N, Christophersen NS, Helin K, Hansen K (2010) Polycomb group protein displacement and gene activation through MSK‐dependent H3K27me3S28 phosphorylation. Mol Cell 39: 886–900 [DOI] [PubMed] [Google Scholar]
- Gordon YJ, Romanowski EG, Araullo‐Cruz T, Kinchington PR (1995) The proportion of trigeminal ganglionic neurons expressing herpes simplex virus type 1 latency‐associated transcripts correlates to reactivation in the New Zealand rabbit ocular model. Graefes Arch Clin Exp Ophthalmol 233: 649–654 [DOI] [PubMed] [Google Scholar]
- Gray PA, Fu H, Luo P, Zhao Q, Yu J, Ferrari A, Tenzen T, Yuk DI, Tsung EF, Cai Zet al (2004) Mouse brain organization revealed through direct genome‐scale TF expression analysis. Science 306: 2255–2257 [DOI] [PubMed] [Google Scholar]
- Greger JG, Katz RA, Ishov AM, Maul GG, Skalka AM (2005) The cellular protein daxx interacts with avian sarcoma virus integrase and viral DNA to repress viral transcription. J Virol 79: 4610–4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotzinger T, Jensen K, Will H (1996) The interferon (IFN)‐stimulated gene Sp100 promoter contains an IFN‐gamma activation site and an imperfect IFN‐stimulated response element which mediate type I IFN inducibility. J Biol Chem 271: 25253–25260 [DOI] [PubMed] [Google Scholar]
- Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, Pandolfi PP (2000) The function of PML in p53‐dependent apoptosis. Nat Cell Biol 2: 730–736 [DOI] [PubMed] [Google Scholar]
- Hall MH, Magalska A, Malinowska M, Ruszczycki B, Czaban I, Patel S, Ambrożek‐Latecka M, Zołocińska E, Broszkiewicz H, Parobczak Ket al (2016) Localization and regulation of PML bodies in the adult mouse brain. Brain Struct Funct 221: 2511–2525 [DOI] [PubMed] [Google Scholar]
- Hendricks RL, Weber PC, Taylor JL, Koumbis A, Tumpey TM, Glorioso JC (1991) Endogenously produced interferon alpha protects mice from herpes simplex virus type 1 corneal disease. J Gen Virol 72(Pt. 7): 1601–1610 [DOI] [PubMed] [Google Scholar]
- Hill JM, Sedarati F, Javier RT, Wagner EK, Stevens JG (1990) Herpes simplex virus latent phase transcription facilitates in vivo reactivation. Virology 174: 117–125 [DOI] [PubMed] [Google Scholar]
- Hochrein H, Schlatter B, O'Keeffe M, Wagner C, Schmitz F, Schiemann M, Bauer S, Suter M, Wagner H (2004) Herpes simplex virus type‐1 induces IFN‐alpha production via Toll‐like receptor 9‐dependent and ‐independent pathways. Proc Natl Acad Sci USA 101: 11416–11421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ives AM, Bertke AS (2017) Stress hormones epinephrine and corticosterone selectively modulate herpes simplex virus 1 (HSV‐1) and HSV‐2 productive infections in adult sympathetic, but not sensory, neurons. J Virol 91: e00582‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CA, Fernandez M, Herc K, Bosnjak L, Miranda‐Saksena M, Boadle RA, Cunningham A (2003) Herpes simplex virus type 2 induces rapid cell death and functional impairment of murine dendritic cells in vitro. J Virol 77: 11139–11149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada R, Yang W, Zhang Y, Patel MC, Yang Y, Ouda R, Dey A, Wakabayashi Y, Sakaguchi K, Fujita Tet al (2018) Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc Natl Acad Sci 115: E9162–E9171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenell S, Leib DA (2016) Herpes simplex virus and interferon signaling induce novel autophagic clusters in sensory neurons. J Virol 90: 4706–4719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC (2012) Transient reversal of episome silencing precedes VP16‐dependent transcription during reactivation of latent HSV‐1 in neurons. PLoS Pathog 8: e1002540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YE, Ahn JH (2015) Positive role of promyelocytic leukemia protein in type I interferon response and its regulation by human cytomegalovirus. PLoS Pathog 11: e1004785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Cliffe A (2008) Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6: 211–221 [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Wilson AC, Chao MV, Mohr I (2012) Control of viral latency in neurons by axonal mTOR signaling and the 4E‐BP translation repressor. Genes Dev 26: 1527–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MF, Coen DM (1995) Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J Virol 69: 1389–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreit M, Paul S, Knoops L, De Cock A, Sorgeloos F, Michiels T (2014) Inefficient type I interferon‐mediated antiviral protection of primary mouse neurons is associated with the lack of apolipoprotein l9 expression. J Virol 88: 3874–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumada S, Uchihara T, Hayashi M, Nakamura A, Kikuchi E, Mizutani T, Oda M (2002) Promyelocytic leukemia protein is redistributed during the formation of intranuclear inclusions independent of polyglutamine expansion: an immunohistochemical study on Marinesco bodies. J Neuropathol Exp Neurol 61: 984–991 [DOI] [PubMed] [Google Scholar]
- Kwiatkowski DL, Thompson HW, Bloom DC (2009) The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol 83: 8173–8181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand‐Breitenbach V, Jeanne M, Benhenda S, Nasr R, Lei M, Peres L, Zhou J, Zhu J, Raught B, de The H (2008) Arsenic degrades PML or PML‐RARalpha through a SUMO‐triggered RNF4/ubiquitin‐mediated pathway. Nat Cell Biol 10: 547–555 [DOI] [PubMed] [Google Scholar]
- Lallemand‐Breitenbach V, de The H (2010) PML nuclear bodies. Cold Spring Harb Perspect Biol 2: a000661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Raja P, Knipe DM (2016) Herpesviral ICP0 protein promotes two waves of heterochromatin removal on an early viral promoter during lytic Infection. MBio 7(1): e02007‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib DA, Bogard CL, Kosz‐Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA (1989) A deletion mutant of the latency‐associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63: 2893–2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Elsaesser SJ, Noh K‐M, Stadler SC, Allis CD (2010) Daxx is an H3.3‐specific histone chaperone and cooperates with ATRX in replication‐independent chromatin assembly at telomeres. Proc Natl Acad Sci USA 107: 14075–14080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Zhang J, Kumar A, Zheng M, Atherton SS, Yu FS (2006) Herpes simplex virus 1 infection induces the expression of proinflammatory cytokines, interferons and TLR7 in human corneal epithelial cells. Immunology 117: 167–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linderman JA, Kobayashi M, Rayannavar V, Fak JJ, Darnell RB, Chao MV, Wilson AC, Mohr I (2017) Immune escape via a transient gene expression program enables productive replication of a latent pathogen. Cell Rep 18: 1312–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashchuk V, Everett RD (2010) Regulation of ICP0‐null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J Virol 84: 4026–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JZ, Russell TA, Spelman T, Carbone FR, Tscharke DC (2014) Lytic gene expression is frequent in HSV‐1 latent infection and correlates with the engagement of a cell‐intrinsic transcriptional response. PLoS Pathog 10: e1004237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Baker M, West G, Woulfe J, Qadi N, Gass J, Cannon A, Adamson J, Feldman H, Lindholm Cet al (2006) A family with tau‐negative frontotemporal dementia and neuronal intranuclear inclusions linked to chromosome 17. Brain 129: 853–867 [DOI] [PubMed] [Google Scholar]
- Malin SA, Davis BM, Molliver DC (2007) Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc 2: 152–160 [DOI] [PubMed] [Google Scholar]
- Maroui MA, Callé A, Cohen C, Streichenberger N, Texier P, Takissian J, Rousseau A, Poccardi N, Welsch J, Corpet Aet al (2016) Latency entry of herpes simplex virus 1 is determined by the interaction of its genome with the nuclear environment. PLoS Pathog 12: e1005834–e1005828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul GG (1998) Nuclear domain 10, the site of DNA virus transcription and replication. BioEssays 20: 660–667 [DOI] [PubMed] [Google Scholar]
- Maul GG, Ishov AM, Everett RD (1996) Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type‐1. Virology 217: 67–75 [DOI] [PubMed] [Google Scholar]
- McFarlane S, Orr A, Roberts APE, Conn KL, Iliev V, Loney C, da Silva Filipe A, Smollett K, Gu Q, Robertson Net al (2019) The histone chaperone HIRA promotes the induction of host innate immune defences in response to HSV‐1 infection. PLoS Pathog 15: e1007667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Xu Z, Chen‐Goodspeed M, Liu M, Van Oort‐Jansen A, Rea MA, Zhao Z, Lee CC, Chang KS (2012) PML regulates PER2 nuclear localization and circadian function. EMBO J 31: 1427–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikloska Z, Cunningham AL (2001) Alpha and gamma interferons inhibit herpes simplex virus type 1 infection and spread in epidermal cells after axonal transmission. J Virol 75: 11821–11826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikloska Z, Danis VA, Adams S, Lloyd AR, Adrian DL, Cunningham AL (1998) In vivo production of cytokines and beta (C‐C) chemokines in human recurrent herpes simplex lesions–do herpes simplex virus‐infected keratinocytes contribute to their production? J Infect Dis 177: 827–838 [DOI] [PubMed] [Google Scholar]
- Moorlag S, Roring RJ, Joosten LAB, Netea MG (2018) The role of the interleukin‐1 family in trained immunity. Immunol Rev 281: 28–39 [DOI] [PubMed] [Google Scholar]
- Muller S, Matunis MJ, Dejean A (1998) Conjugation with the ubiquitin‐related modifier SUMO‐1 regulates the partitioning of PML within the nucleus. EMBO J 17: 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll MP, Hann W, Shivkumar M, Harman LE, Connor V, Coleman HM, Proenca JT, Efstathiou S (2016) The HSV‐1 latency‐associated transcript functions to repress latent phase lytic gene expression and suppress virus reactivation from latently infected neurons. PLoS Pathog 12: e1005539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh KM, Maze I, Zhao D, Xiang B, Wenderski W, Lewis PW, Shen L, Li H, Allis CD (2015) ATRX tolerates activity‐dependent histone H3 methyl/phos switching to maintain repetitive element silencing in neurons. Proc Natl Acad Sci USA 112: 6820–6827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Cohen F, Dean BJ, De La Torre K, Deshmukh G, Estrada AA, Ghosh AS, Gibbons P, Gustafson A, Huestis MPet al (2015) Discovery of dual leucine zipper kinase (DLK, MAP3K12) inhibitors with activity in neurodegeneration models. J Med Chem 58: 401–418 [DOI] [PubMed] [Google Scholar]
- Perng GC, Jones C, Ciacci‐Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn ABet al (2000) Virus‐induced neuronal apoptosis blocked by the herpes simplex virus latency‐associated transcript. Science 287: 1500–1503 [DOI] [PubMed] [Google Scholar]
- Proenca JT, Coleman HM, Connor V, Winton DJ, Efstathiou S (2008) A historical analysis of herpes simplex virus promoter activation in vivo reveals distinct populations of latently infected neurones. J Gen Virol 89: 2965–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quignon F, De Bels F, Koken M, Feunteun J, Ameisen JC, de The H (1998) PML induces a novel caspase‐independent death process. Nat Genet 20: 259–265 [DOI] [PubMed] [Google Scholar]
- Rasmussen SB, Jensen SB, Nielsen C, Quartin E, Kato H, Chen ZJ, Silverman RH, Akira S, Paludan SR (2009) Herpes simplex virus infection is sensed by both Toll‐like receptors and retinoic acid‐inducible gene‐ like receptors, which synergize to induce type I interferon production. J Gen Virol 90: 74–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SB, Sorensen LN, Malmgaard L, Ank N, Baines JD, Chen ZJ, Paludan SR (2007) Type I interferon production during herpes simplex virus infection is controlled by cell‐type‐specific viral recognition through Toll‐like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J Virol 81: 13315–13324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regad T, Bellodi C, Nicotera P, Salomoni P (2009) The tumor suppressor Pml regulates cell fate in the developing neocortex. Nat Neurosci 12: 132–140 [DOI] [PubMed] [Google Scholar]
- Richter ER, Dias JK, Gilbert JE II, Atherton SS (2009) Distribution of herpes simplex virus type 1 and varicella zoster virus in ganglia of the human head and neck. J Infect Dis 200: 1901–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato PC, Leib DA (2014) Intrinsic innate immunity fails to control herpes simplex virus and vesicular stomatitis virus replication in sensory neurons and fibroblasts. J Virol 88: 9991–10001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainz B Jr, Halford WP (2002) Alpha/Beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1. J Virol 76: 11541–11550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM (1997) Comprehensive quantification of herpes simplex virus latency at the single‐cell level. J Virol 71: 5423–5431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer M, Otto V, Stump JD, Klingl S, Muller R, Reuter N, Muller YA, Sticht H, Stamminger T (2016) Characterization of recombinant human cytomegaloviruses encoding IE1 mutants L174P and 1–382 reveals that viral targeting of PML bodies perturbs both intrinsic and innate immune responses. J Virol 90: 1190–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalginskikh N, Poleshko A, Skalka AM, Katz RA (2013) Retroviral DNA methylation and epigenetic repression are mediated by the antiviral host protein Daxx. J Virol 87: 2137–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sides MD, Block GJ, Shan B, Esteves KC, Lin Z, Flemington EK, Lasky JA (2011) Arsenic mediated disruption of promyelocytic leukemia protein nuclear bodies induces ganciclovir susceptibility in Epstein‐Barr positive epithelial cells. Virology 416: 86–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R, Koyuncu OO, Greco TM, Diner BA, Cristea IM, Enquist LW (2016) Two modes of the axonal interferon response limit alphaherpesvirus neuroinvasion. MBio 7: e02145–e2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M, Chelbi‐Alix MK, Koken MH, Venturini L, Lee C, Saib A, Quignon F, Pelicano L, Guillemin MC, Schindler Cet al (1995) Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene 11: 2565–2573 [PubMed] [Google Scholar]
- Stevens JG, Wagner EK, Devi‐Rao GB, Cook ML, Feldman LT (1987) RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 235: 1056–1059 [DOI] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PAet al (2003) Lentivirus‐delivered stable gene silencing by RNAi in primary cells. RNA 9: 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzich JB, Cliffe AR (2018) Strength in diversity: Understanding the pathways to herpes simplex virus reactivation. Virology 522: 81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM (1997) The herpes simplex virus type 1 latency‐associated transcript gene regulates the establishment of latency. J Virol 71: 5432–5440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM (2001) Herpes simplex virus type 1 latency‐associated transcript gene promotes neuronal survival. J Virol 75: 6660–6675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trousdale MD, Steiner I, Spivack JG, Deshmane SL, Brown SM, MacLean AR, Subak‐Sharpe JH, Fraser NW (1991) In vivo and in vitro reactivation impairment of a herpes simplex virus type 1 latency‐associated transcript variant in a rabbit eye model. J Virol 65: 6989–6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbricht T, Alzrigat M, Horch A, Reuter N, von Mikecz A , Steimle V, Schmitt E, Kramer OH, Stamminger T, Hemmerich P (2012) PML promotes MHC class II gene expression by stabilizing the class II transactivator. J Cell Biol 199: 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Shiels C, Sasieni P, Wu PJ, Islam SA, Freemont PS, Sheer D (2004) Promyelocytic leukemia nuclear bodies associate with transcriptionally active genomic regions. J Cell Biol 164: 515–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q‐Y, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM (2005) Herpesviral latency‐associated transcript gene promotes assembly of heterochromatin on viral lytic‐gene promoters in latent infection. Proc Natl Acad Sci 102: 16055–16059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R, Pandolfi PP (1998) PML is essential for multiple apoptotic pathways. Nat Genet 20: 266–272 [DOI] [PubMed] [Google Scholar]
- Warren KG, Brown SM, Wroblewska Z, Gilden D, Koprowski H, Subak‐Sharpe J (1978) Isolation of latent herpes simplex virus from the superior cervical and vagus ganglions of human beings. N Engl J Med 298: 1068–1069 [DOI] [PubMed] [Google Scholar]
- Wilcox CL, Johnson EM (1987) Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J Virol 61: 2311–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CL, Smith RL, Freed CR, Johnson EM (1990) Nerve growth factor‐dependence of herpes simplex virus latency in peripheral sympathetic and sensory neurons in vitro. J Neurosci 10: 1268–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Roizman B (2017) The SP100 component of ND10 enhances accumulation of PML and suppresses replication and the assembly of HSV replication compartments. Proc Natl Acad Sci USA 114: E3823–E3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Sato T, Shimohata T, Hayashi S, Igarashi S, Tsuji S, Takahashi H (2001) Interaction between neuronal intranuclear inclusions and promyelocytic leukemia protein nuclear and coiled bodies in CAG repeat diseases. Am J Pathol 159: 1785–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yordy B, Iijima N, Huttner A, Leib D, Iwasaki A (2012) A neuron‐specific role for autophagy in antiviral defense against herpes simplex virus. Cell Host Microbe 12: 334–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zeijl M , Fairhurst J, Jones TR, Vernon SK, Morin J, LaRocque J, Feld B, O'Hara B, Bloom JD, Johann SV (2000) Novel class of thiourea compounds that inhibit herpes simplex virus type 1 DNA cleavage and encapsidation: resistance maps to the UL6 gene. J Virol 74: 9054–9061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, Liang WX, Song AX, Lallemand‐Breitenbach V, Jeanne Met al (2010) Arsenic trioxide controls the fate of the PML‐RARalpha oncoprotein by directly binding PML. Science 328: 240–243 [DOI] [PubMed] [Google Scholar]
- Zhong S, Muller S, Ronchetti S, Freemont PS, Dejean A, Pandolfi PP (2000a) Role of SUMO‐1‐modified PML in nuclear body formation. Blood 95: 2748–2752 [PubMed] [Google Scholar]
- Zhong S, Salomoni P, Pandolfi PP (2000b) The transcriptional role of PML and the nuclear body. Nat Cell Biol 2: E85–E90 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Expanded View Figures PDF
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The RNA‐seq dataset generated and analyzed in the current study is available at the NCBI Gene Expression omnibus (accession number GSE166738; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE166738).