

Abstract
Tuberculosis (TB) is an infectious disease caused by Mycobacterium tuberculosis (Mtb) that places a heavy strain on public health. Host susceptibility to Mtb is modulated by macrophages, which regulate the balance between cell apoptosis and necrosis. However, the role of molecular switches that modulate apoptosis and necrosis during Mtb infection remains unclear. Here, we show that Mtb‐susceptible mice and TB patients have relatively low miR‐342‐3p expression, while mice with miR‐342‐3p overexpression are more resistant to Mtb. We demonstrate that the miR‐342‐3p/SOCS6 axis regulates anti‐Mtb immunity by increasing the production of inflammatory cytokines and chemokines. Most importantly, the miR‐342‐3p/SOCS6 axis participates in the switching between Mtb‐induced apoptosis and necrosis through A20‐mediated K48‐linked ubiquitination and RIPK3 degradation. Our findings reveal several strategies by which the host innate immune system controls intracellular Mtb growth via the miRNA‐mRNA network and pave the way for host‐directed therapies targeting these pathways.
Keywords: apoptosis, inflammation, microRNA, tuberculosis, ubiquitination
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; RNA Biology
The miR‐342/SOCS6 axis promotes anti‐Mtb defense by increasing the production of inflammatory factors, and by switching cell death pathways from necrosis to apoptosis through A20 mediated K48 ubiquitination and degradation of RIPK3.
Introduction
Tuberculosis (TB) is a globally distributed infectious disease caused by Mycobacterium tuberculosis (Mtb). According to recent studies, more than one‐quarter of the global human population has been infected with Mtb (Chakaya et␣al, 2021). However, only about 10% of infected individuals develop active TB, indicating that innate immunity likely plays a critical role in limiting Mtb replication.
MicroRNAs (miRNAs) act by negatively regulating the expression of key genes (Bartel, 2018). Some evidence indicates that host miRNAs may impact the microbial life cycle and pathogenesis (Jopling et␣al, 2005; Huang et␣al, 2007; Liu et␣al, 2016). More commonly, bacteria can regulate the expression of host‐specific miRNAs and weaken the host’s immunity to promote survival and immune evasion (Kumar et␣al, 2015; Liu et␣al, 2018; Fu et␣al, 2020a). Recently, it has also been reported that microbe‐derived miRNAs have a negative effect on host antimicrobial immunity (Sullivan et␣al, 2005; Choy et␣al, 2008). Thus, both bacterial and host miRNAs are likely to influence the relationship between hosts and pathogens. It has been shown that miR‐342‐3p is involved in the immune response in a variety of diseases. For example, overexpression of miR‐342‐3p can greatly suppress the inflammatory response and lipid uptake in THP‐1 cells and can therefore regulate the development of atherosclerosis (Wang et␣al, 2019). In a recent study, miR‐342‐3p was shown to play an anti‐inflammatory role in regulatory T cells and to contribute to glucocorticoid‐mediated treatment of inflammation in murine autoimmune models (Kim et␣al, 2020). However, the role of miR‐342‐3p in anti‐tuberculosis immunity has not yet been reported.
SOCS proteins are negative regulators of the JAK/STAT pathway (Alexander & Hilton, 2004), and accumulating evidence indicates that they are involved in the control of the cytokine networks responsible for adequate and efficient innate immune responses (Yoshimura et␣al, 2007). For example, SOCS1 expression is significantly increased in patients with tuberculosis, where SOCS1 transcript levels are correlated with disease severity (Masood et␣al, 2012; Masood et␣al, 2013; Masood et␣al, 2014). SOCS1, 4, and 5 are highly expressed in mice infected with hypervirulent Mtb (Manca et␣al, 2005). CISH, a founding member of the SOCS family, can mediate control of Mtb in mice shortly after infection (Carow et␣al, 2017). Meanwhile, SOCS6 expression is reportedly enhanced by exosomes from IL‐1β‐primed mesenchymal stem cells; this process is related to osteoarthritic signal regulation (Kim et␣al, 2021). In addition, the homologs of vertebrate SOCS6 in Eriocheir sinensis are critical to the immune response to bacterial and viral infection (Qu et␣al, 2018). These studies suggest that SOCS6 may also play a role in inflammation and infection. However, to our knowledge, there are no currently published studies on the role of SOCS6 in the pathogenesis of tuberculosis.
C3HeB/FeJ (C3H) and C57BL/6J (B6) inbred mice are often used as models in TB susceptibility studies. C3H mice are extremely susceptible to virulent Mtb, have marked lung pathology, and die soon after infection with Mtb. In contrast, B6 mice, a substrain derived from C57BL/6 mice, have been reported to be resistant to Mtb (Kramnik et␣al, 2000; Pan et␣al, 2005; Kramnik, 2008). More specifically, after Mtb stimulation, C3H mouse bone marrow‐derived macrophages (BMDMs) showed characteristic necrosis, whereas B6 BMDMs mainly underwent apoptosis. Previous studies also showed that extreme susceptibility to Mtb was associated with necrosis of Mtb‐susceptible macrophages (Kramnik et␣al, 2000; Pan et␣al, 2005; Wu et␣al, 2015; Leu et␣al, 2017). The precise mechanism for this phenomenon, however, is not well understood. In this study, we used C3H and B6 inbred mice to demonstrate that miR‐342‐3p targets and represses Socs6, a negative regulator of cytokine signaling. We showed that the miR‐342‐3p/SOCS6 axis modulates anti‐Mtb immunity by inducing production of inflammatory cytokines (TNF‐α, IL‐1, IL‐6, and CXCL15) and chemokines (CCL5, CXCL10, and ICAM1). Most importantly, the miR‐342‐3p/SOCS6 axis is involved in the switching between Mtb‐induced apoptosis and necrosis. Collectively, we have demonstrated a new regulatory pathway involved in tissue necrosis during Mtb infection. These findings suggest that new host‐based therapies targeting these pathways might help combat drug‐susceptible and drug‐resistant TB.
Results
MiR‐342‐3p is associated with TB susceptibility
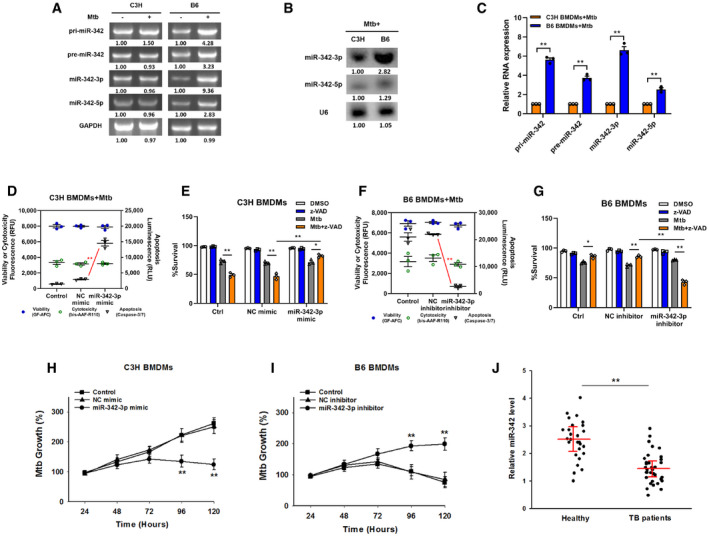
Our group has previously reported that array‐based miRNA profiling can help to shed light on the role of miRNAs in the switching between Mtb‐induced apoptosis and necrosis (Wu et␣al, 2016). We noticed that in macrophages with an Mtb‐resistant phenotype, two of the five highly expressed miRNAs, miR‐342‐5p, and miR‐342‐3p, were spliced from the same precursor (NCBI Accession No. PRJNA279232). Using C3H and B6 BMDMs, we first investigated whether miR‐342 expression was correlated with Mtb infection in mouse macrophages. Semiquantitative RT–PCR analysis showed that the expression levels of primary (pri‐), precursor (pre‐), and mature miR‐342 transcripts were upregulated in Mtb‐stimulated B6 BMDMs but were not found in C3H BMDMs (Fig 1A). The relative levels of mature miR‐342‐5p and miR‐342‐3p in B6 and C3H BMDMs were then examined through northern blotting and quantitative RT–PCR (qRT–PCR). The results showed that miR‐342‐5p and miR‐342‐3p expression was much higher in B6 BMDMs than in C3H BMDMs (Fig 1B and C).
Figure 1. MiR‐342‐3p associates with TB susceptibility.
-
AExpression changes of pri‐, pre‐, and mature miR‐342 transcripts were analyzed by semiquantitative PCR in C3H or B6 BMDMs after Mtb infection. Representative blots from n = 3 biological replicates are shown.
-
B, CThe relative expressions of miR‐342‐3p and miR‐342‐5p were analyzed by northern blotting (B, representative blots from n = 3 biological replicates are shown) or quantitative real‐time PCR (C, data are shown as the mean ± SEM of n = 3 biological replicates) in Mtb‐stimulated C3H and B6 BMDMs.
-
D–GCell death mechanisms of C3H BMDMs transfected with miR‐342‐3p mimic (D), or B6 BMDMs transfected with miR‐342‐3p inhibitor (F), followed by Mtb infection for 36 h. Cell viabilities of C3H BMDMs transfected with miR‐342‐3p mimic (E), or B6 BMDMs transfected with miR‐342‐3p inhibitor (G), followed by stimulation with Mtb or z‐VAD (20 μM) for 24 h. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
H, IMtb growth rates of C3H BMDMs transfected with miR‐342‐3p mimic (H), or B6 BMDMs transfected with miR‐342‐3p inhibitor (I) after Mtb infection. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
JExpression levels of miR‐342‐3p in PBMCs from healthy controls (n = 27) or TB patients (n = 34) were detected by qRT–PCR. Data are shown as the medians ± interquartile ranges.
Data information: ANOVA followed by Bonferroni post hoc test (C‐I) and Mann–Whitney U test (J) were used for data analysis. *P < 0.05, **P < 0.01. Abbreviation: NC, negative control.
Source data are available online for this figure.
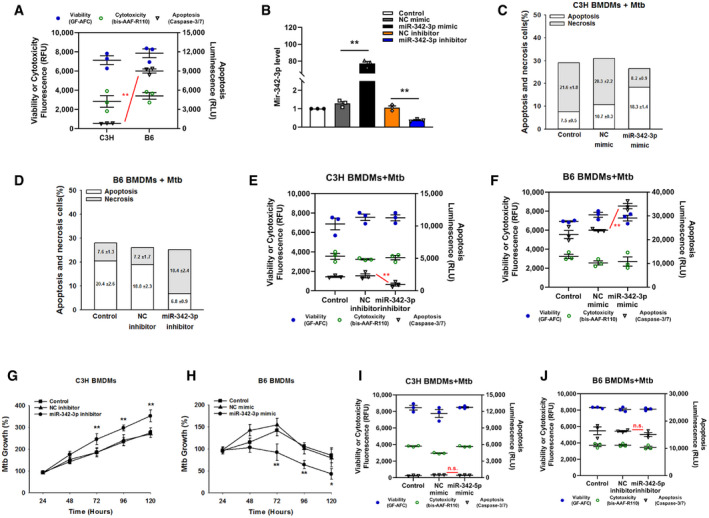
Next, a microRNA mimic and inhibitor were used to investigate whether miR‐342 was related to the Mtb‐resistant phenotype. The mechanism of cell death in Mtb‐infected wild‐type C3H and B6 BMDMs is verified in Fig EV1A, and the effectiveness of the miR‐342‐3p mimic and inhibitor is verified in Fig EV1B. Transfection of C3H BMDMs with the miR‐342‐3p mimic switched Mtb‐induced necrosis to caspase‐dependent cell apoptosis; this switching was inhibited by benzyloxycarbonyl‐Val‐Ala‐Asp‐fluoromethylketone (zVAD, a pan‐caspase inhibitor (Zhang et␣al, 2009)) (Figs 1D and E, and EV1C). On the other hand, inhibition of miR‐342‐3p in B6 BMDMs turned Mtb‐induced apoptosis into caspase‐independent necrosis (Figs 1F and EV1D), and cells treated with both Mtb and zVAD showed decreased cell viability (Fig 1G). Furthermore, transfection of C3H BMDMs with the miR‐342‐3p mimic significantly hindered Mtb growth (Fig 1H), while inhibition of miR‐342‐3p in B6 BMDMs increased Mtb survival and replication (Fig 1I). In addition, treatment of C3H BMDMs with the miR‐342‐3p inhibitor resulted in more severe necrosis, while treatment of B6 BMDMs with the miR‐342‐3p mimic resulted in a greater degree of apoptosis (Fig EV1, EV2, EV3, EV4). Since the other splicing product, miR‐342‐5p, showed no significant effect on the mechanisms of cell death (Fig EV1I and J), we chose to further investigate miR‐342‐3p. As miR‐342‐3p is conserved between mice and humans, we analyzed the production of miR‐342‐3p in peripheral blood mononuclear cells (PBMCs) from 34 TB patients. We observed that TB patients expressed relatively low levels of miR‐342‐3p (Fig 1J). These results suggest that miR‐342‐3p may act as a positive regulator of the immune response to Mtb infection.
Figure EV1. MiR‐342‐3p is associated with TB susceptibility.
-
ACell death mechanisms of C3H and B6 BMDMs after stimulation with Mtb for 36 h. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
BRelative miRNA expression was detected by qRT–PCR using miR‐342‐3p specific primer. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
C, DCell death mechanisms of C3H BMDMs transfected with miR‐342‐3p mimic (C), or B6 BMDMs transfected with miR‐342‐3p inhibitor (D), followed by Mtb infection for 36 h. Representative data (from n = 3 biological replicates) are shown as the mean ± SEM of technical replicates.
-
E, FCell death mechanisms of C3H BMDMs transfected with miR‐342‐3p inhibitor (E), or B6 BMDMs transfected with miR‐342‐3p mimic (F), followed by Mtb infection for 36 h. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
G, HMtb growth rates of C3H BMDMs transfected with miR‐342‐3p inhibitor (G), or B6 BMDMs transfected with miR‐342‐3p mimic (H) after Mtb infection. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
I, JCell death mechanisms of C3H BMDMs transfected with miR‐342‐5p mimic (I), or B6 BMDMs transfected with miR‐342‐5p inhibitor (J), followed by Mtb infection for 36 h. Data are shown as the mean ± SEM of n = 3 biological replicates.
Data information: ANOVA followed by Bonferroni post hoc test was used for data analysis (A, B, E‐J). *P < 0.05, **P < 0.01. Abbreviation: n.s., not significant. NC, negative control.
Source data are available online for this figure.
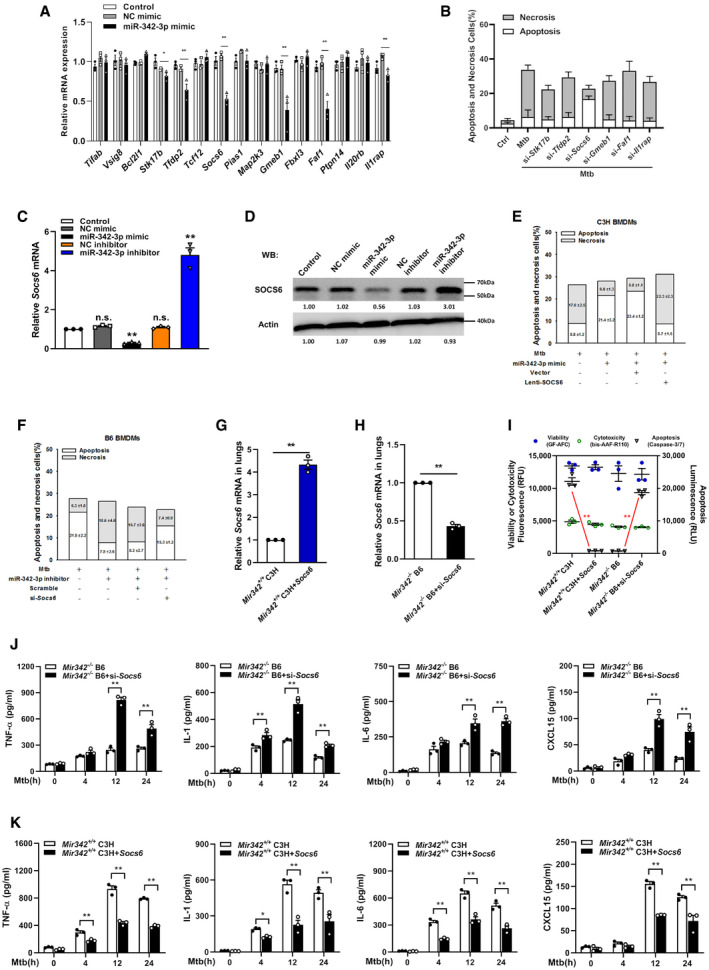
Figure EV2. MiR‐342‐3p directly targets SOCS6 to regulate anti‐Mtb immunity.
-
ARelative expressions of miR‐342‐2p target genes were analyzed by qRT–PCR. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
BCell death mechanisms of RAW264.7 cells transfected with siRNA, followed by Mtb infection for 36 h. Representative data (from n = 3 biological replicates) are shown as the mean ± SEM of technical replicates.
-
C, DMiR‐342‐3p mimic or inhibitor was transfected to RAW264.7 macrophages. After 48 h, cells were collected for qRT–PCR (C, data are shown as the mean ± SEM of n = 3 biological replicates) and Western blotting (D, representative blots from n = 3 biological replicates are shown) to detect the relative levels of SOCS6.
-
EC3H BMDMs were transfected with miR‐342‐3p mimic or SOCS6‐overexpressing lentivirus, followed by Mtb infection. Cell death mechanisms were analyzed, respectively. Representative data (from n = 3 biological replicates) are shown as the mean ± SEM of technical replicates.
-
FB6 BMDMs were transfected with miR‐342‐3p inhibitor or Socs6 siRNA, followed by Mtb infection. Cell death mechanisms were analyzed, respectively. Representative data (from n = 3 biological replicates) are shown as the mean ± SEM of technical replicates.
-
G, HSOCS6‐overexpressing vector (G) or Socs6 siRNA (H) was mixed with polyethylenimine to form a complex, which was used to infect mice by tail vein injection (N/P ratio=8). Lungs were collected for transfection efficiency validation. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
IAlveolar macrophages from mice treated with SOCS6‐overexpressing vector or Socs6 siRNA were collected to analyze cell death mechanisms. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
J, KSecretion of cytokines TNF‐α, IL‐1, IL‐6, and CXCL15 in BMDMs obtained from Mir342 −/− B6 and Mir342 −/− B6 mice supplemented with Socs6 siRNA (J), or from Mir342 +/+ C3H and Mir342 +/+ C3H mice supplemented with SOCS6‐overexpressing vector (K), was detected by ELISA after Mtb stimulation. Data are shown as the mean ± SEM of n = 3 biological replicates.
Data information: ANOVA followed by Bonferroni post hoc test (A, C, I‐K) and two‐tailed Student t test (G, H) were used for data analysis. *P < 0.05, **P < 0.01. Abbreviations: n.s., not significant. NC, negative control.
Source data are available online for this figure.
Figure EV3. SOCS6 has no effects on IFN‐γ, caspase 2, and caspase 9.
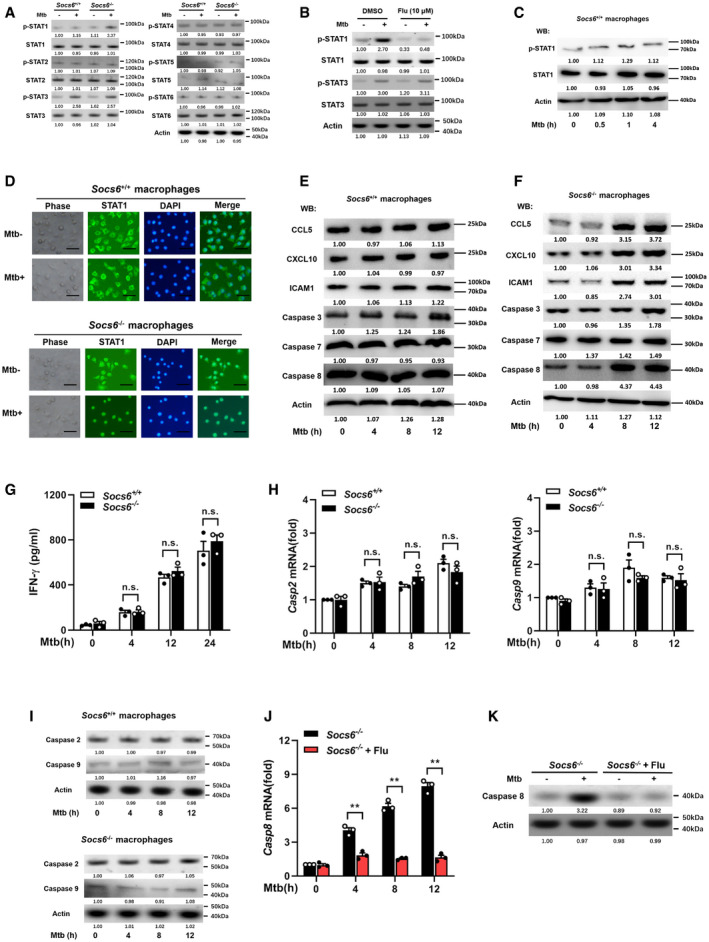
-
APhosphorylation states of STAT family members in response to Mtb stimulation (4 h) in Socs6 +/+ or Socs6 −/− BMDMs were examined by Western blotting. Representative blots from n = 3 biological replicates are shown.
-
BPhosphorylation states of STAT1 and STAT3 in response to Mtb stimulation (4 h) in Socs6 +/+ BMDMs were examined by Western blotting. Fludarabine treatment concentration was 10 μM, and the treatment time was 24 h. Representative blots from n = 3 biological replicates are shown.
-
CPhosphorylation states of STAT1 in response to Mtb stimulation in Socs6 +/+ BMDMs were examined by Western blotting. Representative blots from n = 3 biological replicates are shown.
-
DIntracellular localization of STAT1 in Mtb‐stimulated Socs6 +/+ and Socs6 −/− BMDMs were detected by immunofluorescence. Representative images from n = 3 biological replicates are shown. Scale bar = 100 μm.
-
E, FRelative expressions of chemokines CCL5, CXCL10, ICAM1, and caspase 3, caspase 7, caspase 8 in Mtb‐stimulated Socs6 +/+ (E) or Socs6 −/− (F) BMDMs were detected by Western blotting. Representative blots from n = 3 biological replicates are shown.
-
GELISA was performed to detect the secretion of IFN‐γ in Socs6 +/+ and Socs6 −/− BMDMs during Mtb stimulation. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
H, Icaspase 2, caspase 9 in Mtb‐stimulated Socs6 +/+ or Socs6 −/− BMDMs were detected by qRT–PCR (H, data are shown as the mean ± SEM of n = 3 biological replicates) and Western blotting (I, representative blots from n = 3 biological replicates are shown).
-
J, KCaspase 8 in STAT1‐suppressed Socs6 −/− BMDMs were detected by qRT–PCR (J, data are shown as the mean ± SEM of n = 3 biological replicates) and Western blotting (K, representative blots from n = 3 biological replicates are shown). Fludarabine (10 μM) was used to treat Socs6 −/− BMDMs for 24 h to specifically suppress the activation of STAT1.
Data information: ANOVA followed by Bonferroni post hoc test (G, H, J) was used for data analysis. **P < 0.01. Abbreviation: n.s., not significant.
Source data are available online for this figure.
Figure EV4. RIPK3 is critical for SOCS6‐regulated cell death mechanisms.
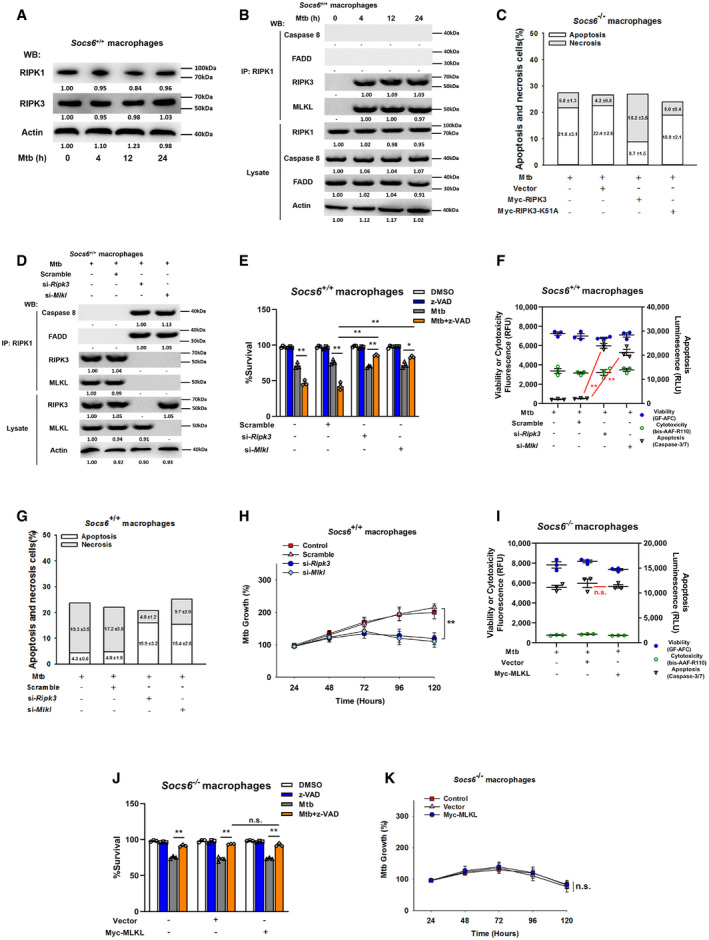
-
ARelative expressions of RIPK1 and RIPK3 were analyzed by Western blotting in Socs6 +/+ BMDMs stimulated with Mtb for 0–24 h. Representative blots from n = 3 biological replicates are shown.
-
BSocs6+/+ BMDMs were stimulated with Mtb for 0–24 h, and cell lysates were collected and immunoprecipitated using an anti‐RIPK1 antibody. The recruitment of caspase 8, RIPK3, FADD, and MLKL was analyzed by immunoblots. The lower panel represents the immunoblot analysis of whole cell lysates. Representative blots from n = 3 biological replicates are shown.
-
CCell death mechanisms of Mtb‐infected Socs6 −/− BMDMs that were transfected with plasmids expressing Myc‐RIPK3 or Myc‐RIPK3 K51A mutant as indicated. Representative data (from n = 3 biological replicates) are shown as the mean ± SEM of technical replicates.
-
DSocs6+/+ BMDMs were transfected with Ripk3 siRNA or Mlkl siRNA for 24 h. Afterward, transfected cells were stimulated with Mtb for 12 h, and cell lysates were collected and immunoprecipitated using an anti‐RIPK1 antibody. The recruitment of caspase 8, RIPK3, FADD, and MLKL was analyzed by immunoblot. The lower panel represents the immunoblot analysis of whole cell lysates. Representative blots from n = 3 biological replicates are shown.
-
E–HCell viabilities (E, data are shown as the mean ± SEM of n = 3 biological replicates), cell death mechanisms [F, data are shown as the mean ± SEM of n = 3 biological replicates. G, representative data (from n = 3 biological replicates) are shown as the mean ± SEM of technical replicates], or Mtb growth rates (H, data are shown as the mean ± SEM of n = 3 biological replicates) of Mtb‐infected Socs6 +/+ BMDMs that were transfected with Ripk3 siRNA or Mlkl siRNA as indicated. Z‐VAD treatment concentration was 20 μM, and the treatment time was 24 h.
-
ICell death mechanisms of Socs6 −/− BMDMs transfected with plasmids expressing Myc‐MLKL and stimulated with Mtb for 36 h. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
JCell viabilities of Socs6 −/− BMDMs transfected with plasmids expressing Myc‐MLKL for 24 h and stimulated with Mtb or z‐VAD for 24 h. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
KMtb growth rates of Socs6 −/− BMDMs transfected with plasmids expressing Myc‐MLKL and stimulated with Mtb for 0–120 h. Data are shown as the mean ± SEM of n = 3 biological replicates.
Data information: ANOVA followed by Bonferroni post hoc test (E, F, H‐K) was used for data analysis. *P < 0.05, **P < 0.01. Abbreviation: n.s., not significant.
Source data are available online for this figure.
MiR‐342‐3p enhances the production of inflammatory cytokines
We generated a Mir342 +/+ C3H mouse strain carrying the pBROAD3‐miR‐342 sequence and a Mir342 −/− B6 mouse strain with knockout of the pre‐miR‐342 sequence (Appendix␣Fig S1A and B). Compared to the control group, the survival time of Mir342 +/+ C3H mice infected with 400 CFU Mtb was significantly lengthened (Fig 2A), while the survival time of Mtb‐infected Mir342 −/− B6 mice was remarkably shortened (Fig 2B). We also observed a statistically significant difference in bacterial loads in the lungs, spleens, and livers of Mir342 +/+ C3H mice and their wild‐type littermates (C3H) (Fig 2C) as well as Mir342 −/− B6 mice and their wild‐type littermates (B6) (Fig 2D). Mir342 −/− B6 mice developed massive necrotic lung lesions after intravenous infection (Fig 2E, Appendix Table S1). In contrast, Mtb‐infected Mir342 +/+ C3H mice did not develop any lung lesions and only displayed trace levels of inflammation (Fig 2F, Appendix␣Table S1). Next, we detected the expression of inflammatory cytokines secreted by BMDMs during Mtb infection. Compared with littermate controls, Mir342 +/+ C3H mice secreted increased levels of TNF‐α, IL‐1, IL‐6, and CXCL15 (also known as IL‐8) (Fig 2G). Deletion of miR‐342 in B6 mice resulted in disordered cytokine secretion (Fig 2H). Taken together, these data indicate that miR‐342‐3p plays an important role in the switch between necrosis and apoptosis in Mtb‐infected BMDMs and has a profound effect on anti‐tuberculosis immunity.
Figure 2. MiR‐342‐3p enhances the production of inflammatory cytokines.
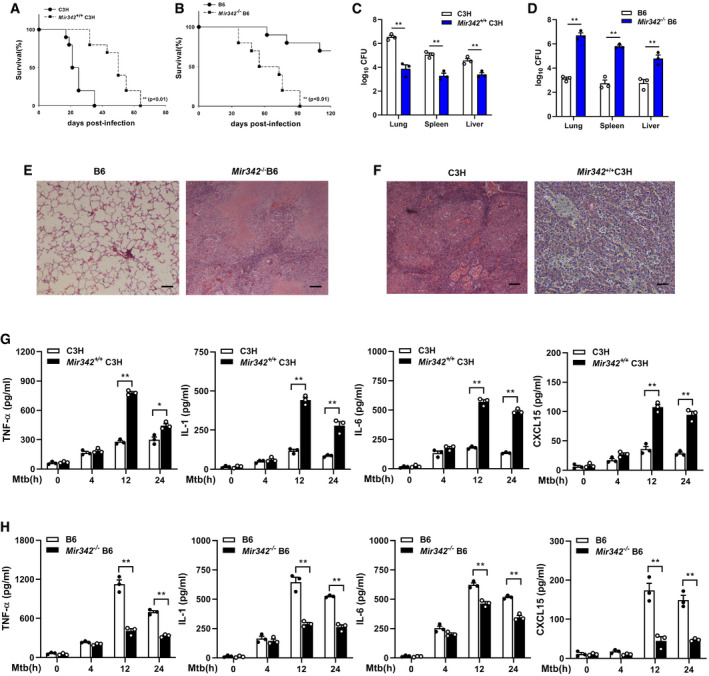
-
A, BSurvival of Mir342 +/+ C3H mice and their wild‐type littermate controls (C3H) (n = 10) (A), or Mir342 −/− B6 mice and their wild‐type littermate controls (B6) (n = 10) (B) after aerosol infection with around 400 CFU Mtb. Data are shown as the Kaplan–Meier curves.
-
C, DMtb bacterial loads in lungs, spleens, and livers of Mir342 +/+ C3H mice and their littermates (C3H) (C), or Mir342 −/− B6 mice and their littermates (B6) at 21 days post‐infection (dpi). Data are shown as the medians ± interquartile ranges of n = 3 biological replicates.
-
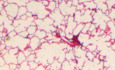
E, FTuberculosis lung lesions of Mir342 −/− B6 mice and their littermates (B6) (E), or Mir342 +/+ C3H mice and their littermates (C3H) (F) were detected by hematoxylin/eosin staining at 21 dpi. Representative images from n = 3 biological replicates are shown. Scale bar = 100 μm.
-
G, HSecretion of cytokines TNF‐α, IL‐1, IL‐6, and CXCL15 in BMDMs obtained from Mir342 +/+ C3H and their littermates (C3H) (G), or from Mir342 −/− B6 and their littermates (B6) (H) after Mtb stimulation, was detected by ELISA. Data are shown as the mean ± SEM of n = 3 biological replicates.
Data information: Log‐rank (Mantel–Cox) test (A, B), Mann–Whitney U test (C, D), and ANOVA followed by Bonferroni post hoc test (G, H) were used for data analysis. *P < 0.05, **P < 0.01.
MiR‐342‐3p directly targets SOCS6 to regulate anti‐Mtb immunity
The targets of miR‐342‐3p were predicted using the PicTar, miRanda, and TargetScan databases. We narrowed the candidate targets to dozens of molecules that may regulate the innate immune response (Fig EV2A). Using an siRNA screening strategy, we chose Socs6 for further investigation based on its effect on the switching between Mtb‐induced apoptosis and necrosis (Fig EV2B). The 3′‐UTR of murine Socs6 contains putative regions that match the miR‐342‐3p seed sequence (Fig 3A). To confirm the predicted regions, these putative matching sequences were amplified and inserted into a psicheck‐2 vector. The vectors were then transfected into HEK‐293T cells for dual‐luciferase reporter (DLR) analysis. Our results showed that the miR‐342‐3p mimic dramatically suppressed the activity of the wild‐type Socs6 3′‐UTR but not that of the double‐mutation type (MUT) group (site 1 + 2 mut) (Fig 3B). qRT–PCR and Western blotting results showed that the miR‐342‐3p mimic considerably decreased SOCS6 mRNA and protein levels (Figs 3C and D, and EV2C and D), indicating that silencing was achieved through a combination of translation repression and mRNA destabilization (Jonas & Izaurralde, 2015). The RNA‐induced silencing complex (RISC) has been reported to mediate the binding of miRNAs and mRNAs and activate Ago (Iwakawa & Tomari, 2015). Therefore, RAW264.7 cells were transfected with a Myc‐tagged Ago2 construct and a miR‐342‐3p mimic. Immunoprecipitation was performed using a Myc antibody, and Socs6 enrichment was determined by qRT–PCR. Cells transfected with the miR‐342‐3p mimic showed significant Socs6 mRNA enrichment compared with cells transfected with the scramble mimic (Fig 3E). These data indicated that Socs6 is a specific degradation target of miR‐342‐3p. Additionally, we discovered that restoration of SOCS6 expression abrogated the effects of miR‐342‐3p on cell death (Figs 3F and EV2E), cell survival (Fig 3G) and Mtb growth (Fig 3H) in C3H BMDMs. Moreover, the anti‐tuberculosis effects of B6 BMDMs, which could be suppressed using the miR‐342‐3p inhibitor, were recovered upon siRNA‐mediated SOCS6 knockdown (Figs 3I–K and EV2F). Meanwhile, we analyzed the relative expression of miR‐342‐3p and Socs6 in 27 healthy individuals and 34 TB patients and found an inverse correlation between miR‐342‐3p and Socs6 (Spearman rank correlation: r = −0.532, P < 0.001, n = 61) (Fig 3L).
Figure 3. MicroRNA‐342‐3p directly targets SOCS6.
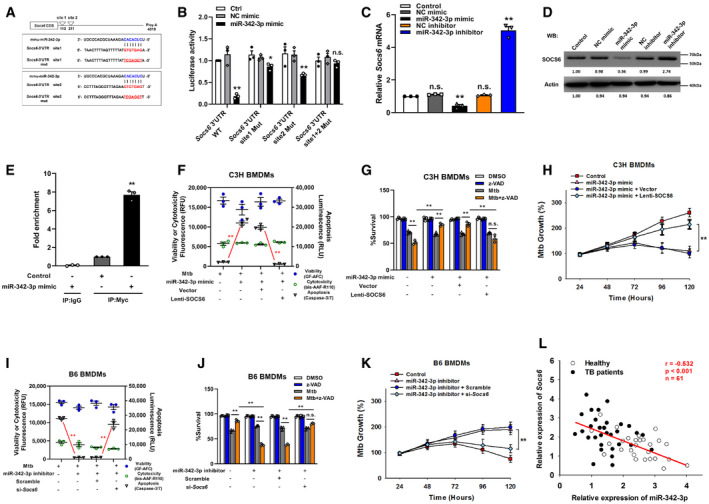
-
APredicted binding sites of miR‐342‐3p and Socs6 3′‐UTR.
-
BHEK‐293T fibroblasts were cotransfected with miR‐342‐3p mimic and luciferase reporter construct containing wild‐type or mutated UTR. After 24 h, cells were collected for luciferase assays. The Y‐axis showed relative luciferase activity. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
C, DMiR‐342‐3p mimic or inhibitor was transfected to NIH‐3T3 fibroblasts. After 48 h, cells were collected for qRT–PCR (C, data are shown as the mean ± SEM of n = 3 biological replicates) and Western blotting (D, representative blots from n = 3 biological replicates are shown) to detect SOCS6 expression.
-
E€ RAW264.7 cells were transfected with Myc‐tagged Ago2 in the presence of miR‐342‐3p mimic or scramble for 24 h. Immunoprecipitation was performed with Myc antibody or IgG, and Socs6 mRNA was detected by qRT–PCR. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
F–HC3H BMDMs were transfected with miR‐342‐3p mimic or SOCS6‐overexpressing lentivirus, followed by stimulation with Mtb or z‐VAD (20 μM) for 24 h. Cell death mechanisms (F), cell viabilities (G), and Mtb growth rates (H) were analyzed, respectively. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
I–KB6 BMDMs were transfected with miR‐342‐3p inhibitor or Socs6 siRNA, followed by stimulation with Mtb or z‐VAD (20 μM) for 24 h. Cell death mechanisms (I), cell viabilities (J), and Mtb growth rates (K) were analyzed, respectively. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
LRelative expressions of miR‐342‐3p and Socs6 in PBMCs from healthy individuals (n = 27) and TB patients (n = 34) were detected by qRT–PCR. The correlation between miR‐342‐3p and Socs6 was analyzed by Spearman test. Spearman rank correlation: r = −0.532, P < 0.001, n = 61.
Data information: ANOVA followed by Bonferroni post hoc test (B, C, E‐K) was used for data analysis. *P < 0.05, **P < 0.01. Abbreviations: n.s., not significant. NC, negative control.
Source data are available online for this figure.
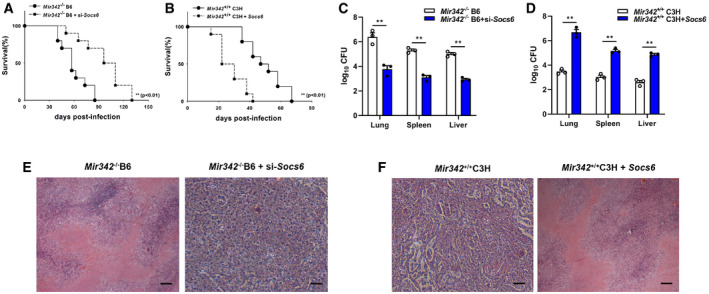
To confirm our findings, mice were intravenously infected with plasmids encoding SOCS6 or Socs6 siRNA (Fig EV2G and H). Survival time was lengthened in Mir342 −/− B6 mice transfected with Socs6 siRNA, with lower bacterial loads in the indicated organs (Fig 4A and C). We also observed that survival time was shortened in Mir342 +/+ C3H mice transfected with the SOCS6‐overexpressing vector, with significantly higher bacterial loads in the indicated organs (Fig 4B and D). Additionally, Mir342 −/− B6 mice with Socs6 knockdown showed pulmonary inflammation rather than extensive TB nodules (Fig 4E, Appendix␣Table S1), while the lungs of Mir342 +/+ C3H mice transfected with SOCS6‐overexpressing vector developed severe necrotic lesions (Fig 4F, Appendix␣Table S1). Furthermore, the alveolar macrophages from Mir342 −/− B6 mice with Socs6 knockdown showed characteristic apoptosis, whereas those from Mir342 +/+ C3H mice with SOCS6 overexpression mainly underwent necrosis (Fig EV2I). We also detected the expression of inflammatory cytokines. As expected, TNF‐α, IL‐1, IL‐6, and CXCL15 secretion was enhanced by SOCS6 knockdown in Mir342 −/− B6 mice and hampered by SOCS6 overexpression in Mir342 +/+ C3H mice (Fig EV2J and K). Taken together, these data suggest that Socs6 is a direct target of miR‐342‐3p and is responsible for anti‐tuberculosis immunity in macrophages.
Figure 4. SOCS6 participates in anti‐Mtb immunity.
-
A, BSurvival of Mir342 −/− B6 and Mir342 −/− B6 mice supplemented with Socs6 siRNA (n = 10) (A), or Mir342 +/+ C3H and Mir342 +/+ C3H mice supplemented with SOCS6‐overexpressing vector (n = 10) (B), after aerosol infection with around 400 CFU Mtb. Data are shown as the Kaplan–Meier curves.
-
C, DMtb bacterial loads in lungs, spleens, and livers of Mir342 −/− B6 and Mir342 −/− B6 mice supplemented with Socs6 siRNA (C), or Mir342 +/+ C3H and Mir342 +/+ C3H mice supplemented with SOCS6‐overexpressing vector (D) at 21 dpi. Data are shown as the medians ± interquartile ranges of n = 3 biological replicates.
-
E, FTuberculosis lung lesions of Mir342 −/− B6 and Mir342 −/− B6 mice supplemented with Socs6 siRNA (E), or Mir342 +/+ C3H and Mir342 +/+ C3H mice supplemented with SOCS6‐overexpressing vector (F) at 21 dpi were detected by hematoxylin/eosin staining. Representative images from n = 3 biological replicates are shown. Scale bar = 100 μm.
Data information: Log‐rank (Mantel–Cox) test (A, B) and Mann–Whitney U test (C, D) were used for data analysis. **P < 0.01.
SOCS6 regulates the expression of chemokines by modulating JAK/STAT signaling
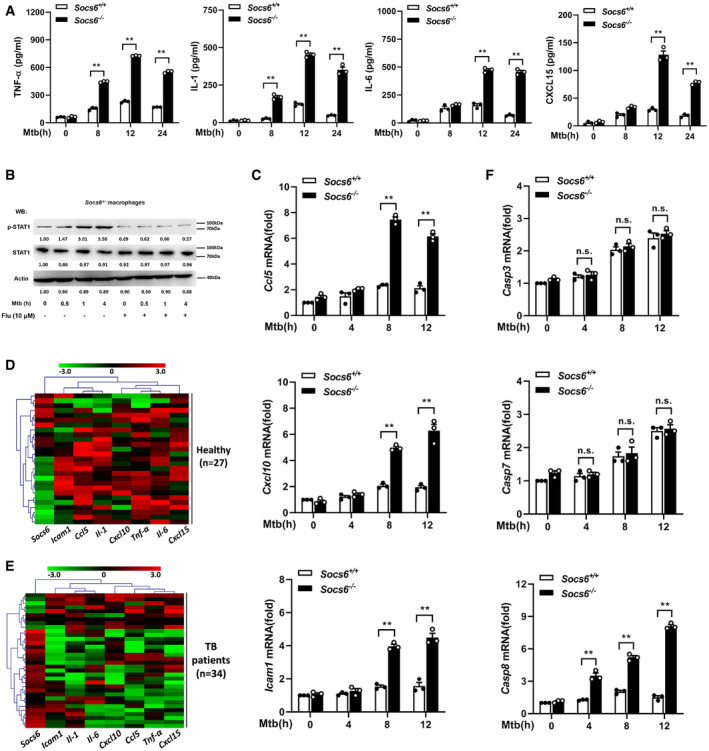
As a suppressor of cytokine signaling, SOCS6 can directly suppress the production of cytokines (Yoshimura et␣al, 2007). To gain insight into the role of Socs6 in the innate immune response, we measured the release of cytokines in BMDMs obtained from Mtb‐infected Socs6 +/+ and Socs6 −/− mice. The release of TNF‐α, IL‐1, IL‐6, and CXCL15 from Mtb‐infected macrophages was enhanced in the absence of SOCS6 (Fig 5A). Since SOCS proteins are known to negatively regulate the JAK/STAT signaling pathway (Alexander & Hilton, 2004), we examined the phosphorylation levels of the STAT family in Mtb‐infected Socs6 +/+ and Socs6 −/− BMDMs. The results suggested that STAT1 might be negatively regulated by SOCS6 signaling (Fig EV3A). Furthermore, we discovered that STAT1 phosphorylation was dramatically upregulated in Mtb‐infected Socs6 −/− BMDMs and could be blocked by a specific STAT1 inhibitor called fludarabine (Figs 5B and EV3B). In contrast, there was no significant fluctuation in STAT1 phosphorylation in Socs6 +/+ BMDMs post‐Mtb infection (Fig EV3C). STAT phosphorylation and nuclear translocation are key steps in the activation of the JAK/STAT signaling pathway. Therefore, we next detected the intracellular localization of STAT1 by immunofluorescence. In Mtb‐infected Socs6 +/+ BMDMs, fluorescence was mainly distributed in the cytoplasm (Fig EV3D). However, in infected Socs6 −/− BMDMs, most of the fluorescence was nuclear (Fig EV3D). These results suggested that SOCS6 might prevent inflammation and promote bacterial growth by repressing STAT1. This was further confirmed by testing the expression of several interferon‐stimulated genes that can be directly regulated by STAT1. Knocking out Socs6 augmented CCL5, CXCL10, and ICAM1 expression in Mtb‐infected cells (Figs 5C and EV3E and F). We also evaluated the expression of Socs6 and inflammatory factors in clinical samples. Cluster analysis showed that, in both healthy and diseased patients, expression levels of Socs6 and inflammatory factors were inversely correlated. Specifically, compared to healthy individuals, most TB patients displayed relatively higher expression of Socs6 and lower expression of inflammatory factors (Fig 5D and E). Since the release of IFN‐γ is closely related to Mtb infection, we tested whether SOCS6 regulated IFN‐γ expression. The results showed that IFNγ expression was increased in both Socs6 +/+ and Socs6 −/− cells following Mtb infection (Fig EV3G). Therefore, we hypothesize that SOCS6 does not play a role in the expression of IFN‐γ. Additionally, since STAT1‐mediated cell death is essential for host immunity against pathogens (O'Shea et␣al, 2015), we tested the expression of apoptosis‐associated caspases, such as caspases 2, 3, 7, 8, and 9 (Fulda & Debatin, 2002; Sironi & Ouchi, 2004; Hong et␣al, 2019; Song et␣al, 2019). The results showed that Mtb‐infected Socs6 +/+ and Socs6 −/− BMDMs expressed significantly different levels of caspase 8 (Figs 5F and EV3E, F, H and I) and that these levels were specifically regulated by STAT1 (Fig EV3J and K). Collectively, these results indicate that the production of STAT1‐mediated inflammatory factors plays an important role in the SOCS6 regulatory network, which controls the immune response to Mtb infection.
Figure 5. SOCS6 regulates the expression of chemokines via modulating JAK/STAT signaling.
-
AELISA was performed to detect the secretion of cytokines TNF‐α, IL‐1, IL‐6, and CXCL15 in Socs6 +/+ and Socs6 −/− BMDMs during Mtb stimulation. Data are shown as the mean ± SEMof n = 3 biological replicates.
-
BPhosphorylation states of STAT1 in response to Mtb stimulation in Socs6 −/− BMDMs were examined by Western blotting. Fludarabine treatment concentration was 10 μM, and the treatment time was 24 h. Representative blots from n = 3 biological replicates are shown.
-
C–FRelative expressions of chemokines CCL5, CXCL10, ICAM1 (C), and caspase 3, caspase 7, caspase 8 (F) in Mtb‐stimulated Socs6 +/+ or Socs6 −/− BMDMs were detected by qRT–PCR. Data are shown as the mean ± SEM of n = 3 biological replicates. Cluster analysis of clinical samples. Heat maps showed relatively high (red) or low (green) expression levels of Socs6 and inflammatory factors in PBMCs from healthy individuals (n = 27) (D) and TB patients (n = 34) (E).
Data information: ANOVA followed by Bonferroni post hoc test (A, C, F) was used for data analysis. **P < 0.01. Abbreviation: n.s., not significant.
Source data are available online for this figure.
SOCS6 regulates cell death mechanisms by modulating the recruitment of RIPK3
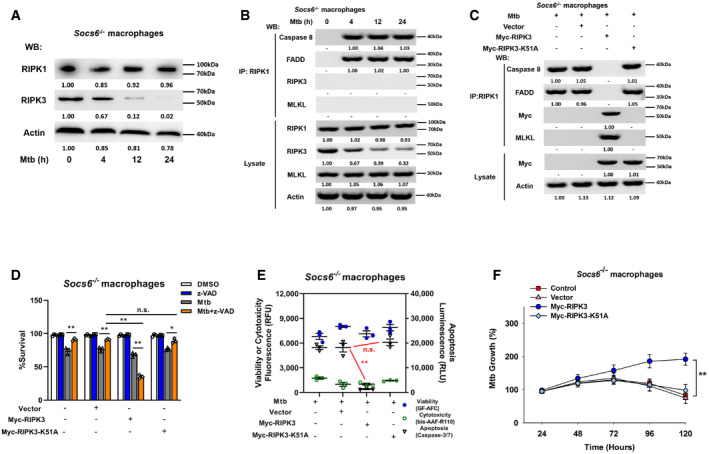
To understand the role of the cell death complex in the SOCS6 regulatory network, we analyzed the expression levels of RIPK1 and RIPK3 in Mtb‐infected Socs6 +/+ and Socs6 −/− BMDMs. Interestingly, RIPK3 was decreased in Mtb‐infected Socs6 −/− BMDMs, while RIPK1 was not affected by SOCS6 expression (Figs 6A and EV4A). To investigate the composition of the cell death complex during Mtb infection, we first infected Socs6 +/+ and Socs6 −/− BMDMs with Mtb for 4–24 h and then performed immunoprecipitation for RIPK1 followed by immunoblotting for caspase 8, RIPK3, FADD, and MLKL. In Socs6 +/+ BMDMs, we detected formation of a necrosis‐related complex, whereas in Socs6 −/− BMDMs, we observed assembly of the RIPK1‐FADD‐Caspase 8 complex, which can induce apoptosis (Figs 6B and EV4B). We then overexpressed either an empty vector or Myc‐tagged RIPK3 in Socs6 −/− BMDMs. Compared with the empty vector control, transfection of RIPK3 led to substantial recruitment of MLKL (Fig 6C). However, K51A‐mutant Myc‐tagged RIPK3, which lacks kinase activity (He et␣al, 2009), does not induce MLKL recruitment (Fig 6C). To further confirm the role of RIPK3 in Mtb‐induced necrosis, we examined cell viability (Fig 6D), cell death mechanisms (Figs 6E and EV4C), and Mtb growth rate (Fig 6F) in RIPK3‐overexpressing Socs6 −/− BMDMs. The results showed that recruitment of RIPK3 to RIPK1 contributed to the switch from apoptosis to necrosis in Mtb‐stimulated BMDMs. We then knocked down RIPK3 or MLKL with siRNAs in Mtb‐stimulated Socs6 +/+ BMDMs and found that RIPK3 and MLKL were both required for complex formation (Fig EV4D). Furthermore, Mtb‐induced necrosis was attenuated upon knockdown of RIPK3 or MLKL in Mtb‐stimulated Socs6 +/+ BMDMs (Fig EV4E–H). However, unlike RIPK3, overexpression of MLKL alone did not alter necrosis in Socs6 −/− BMDMs (Fig EV4I–K). Taken together, these data demonstrate that recruitment of RIPK3 to RIPK1 switches apoptosis to necrosis in Mtb‐stimulated BMDMs.
Figure 6. SOCS6 determines cell death mechanisms via modulating recruitment of RIPK3.
-
ARelative expressions of RIPK1 and RIPK3 were analyzed by Western blotting in Socs6 −/− BMDMs stimulated with Mtb for 0–24 h. Representative blots from n = 3 biological replicates are shown.
-
BSocs6−/− BMDMs were stimulated with Mtb for 0–24 h, cell lysates were collected and immunoprecipitated using an anti‐RIPK1 antibody. The recruitment of caspase 8, RIPK3, FADD, and MLKL was analyzed by immunoblots. The lower panel represents the immunoblot analysis of whole cell lysates. Representative blots from n = 3 biological replicates are shown.
-
CSocs6−/− BMDMs were transfected with plasmids expressing Myc‐RIPK3 or Myc‐RIPK3 K51A mutant for 24 h. Afterward, transfected cells were stimulated with Mtb for 12 h, and cell lysates were collected and immunoprecipitated using an anti‐RIPK1 antibody. The recruitment of caspase 8, RIPK3, FADD, and MLKL was analyzed by immunoblot. The lower panel represents the immunoblot analysis of whole cell lysates. Representative blots from n = 3 biological replicates are shown.
-
D–FCell viabilities (D), cell death mechanisms (E), or Mtb growth rates (F) of Mtb‐infected Socs6 −/− BMDMs that were transfected with plasmids expressing Myc‐RIPK3 or Myc‐RIPK3 K51A mutant as indicated. Z‐VAD treatment concentration was 20 μM and the treatment time was 24 h. Data are shown as the mean ± SEM of n = 3 biological replicates.
Data information: ANOVA followed by Bonferroni post hoc test (D‐F) was used for data analysis. *P < 0.05, **P < 0.01. Abbreviation: n.s., not significant.
Source data are available online for this figure.
A20 controls the degradation of RIPK3 through K48‐linked ubiquitination
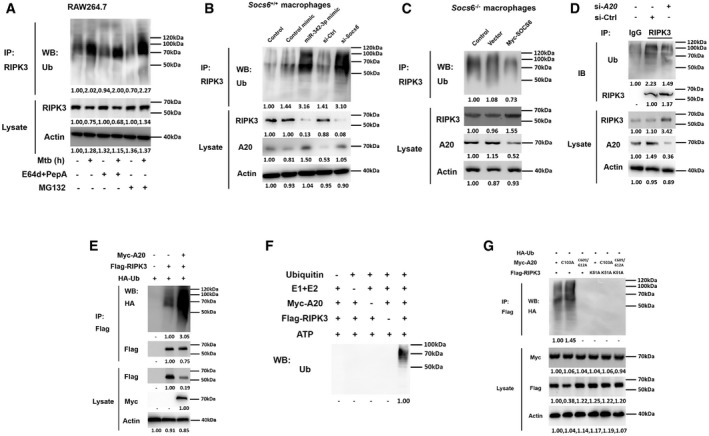
Since RIPK3 overexpression caused Socs6 −/− BMDMs to switch from apoptosis to necrosis, we further investigated the RIPK3 degradation pathway. The degradation of RIPK3 was blocked by the proteasome inhibitor MG132 but not by lysosome protease inhibitors, such as E64d and pepstatin A (E64d/PepA) (Fig 7A). This indicated that RIPK3 undergoes proteasome‐dependent rather than lysosome‐dependent degradation. Both the miR‐342‐3p mimic and Socs6 siRNA facilitated RIPK3 ubiquitination and subsequent degradation in Mtb‐stimulated Socs6 +/+ BMDMs (Fig 7B). Additionally, SOCS6 overexpression suppressed the ubiquitination of RIPK3 in Mtb‐infected Socs6 −/− BMDMs (Fig 7C).
Figure 7. A20 controls degradation of RIPK3 through ubiquitin chains.
-
ARAW264.7 cells were pretreated with or without E64d/PepA or MG132 for 4 h and then stimulated with or without Mtb for 12 h as indicated. Cell lysates were collected and immunoprecipitated using an anti‐RIPK3 antibody, and the ubiquitination of endogenous RIPK3 was detected using an anti‐Ub antibody. Representative blots from n = 3 biological replicates are shown.
-
B, CSocs6+/+ BMDMs were transfected with miR‐342‐3p mimic or Socs6 siRNA (B), and Socs6 −/− BMDMs were transfected with plasmid expressing Myc‐SOCS6 (C) for 24 h as indicated. Transfected cells were then stimulated with Mtb for another 12 h, cell lysates were collected for immunoprecipitation, and ubiquitination of endogenous RIPK3 was detected by immunoblot. Representative blots from n = 3 biological replicates are shown.
-
DRAW264.7 cells were transfected with A20 siRNA, and cell lysates were collected for immunoprecipitation and immunoblot. Representative blots from n = 3 biological replicates are shown.
-
E–GRAW264.7 cells were transfected with plasmids expressing A20 and RIPK3 (E), or A20 and RIPK3 mutants (G) as indicated, cell lysates were collected for immunoprecipitation and immunoblot. Myc‐A20 or Flag‐RIPK3 purified from transfected HEK‐293T cells was incubated with ATP, E1, E2, and ubiquitin as indicated. The in␣vitro ubiquitination of RIPK3 was analyzed by immunoblot using an anti‐Ub antibody (F). Representative blots from n = 3 biological replicates are shown.
Source data are available online for this figure.
It has been reported that RIPK3 can be modified by several E3 ligases, such as cIAP1/2, A20 (also known as TNFAIP3), and CHIP (Tenev et␣al, 2011; Onizawa et␣al, 2015; Seo et␣al, 2016). In this study, we found that A20 expression was correlated with RIPK3 ubiquitination levels (Fig 7B and C). More specifically, the ubiquitination of endogenous RIPK3 was suppressed by A20 siRNA (Fig 7D), while the ubiquitination of Flag‐tagged RIPK3 was induced by A20 in RAW264.7 cells (Fig 7E). A20 directly targets RIPK3 for degradation through the in␣vitro ubiquitination system (Fig 7F). These data suggest that A20 is a direct E3 ligase of RIPK3.
The ovarian tumor (OTU) domain of A20 functions as a deubiquitinating (DUB) enzyme that hydrolyzes ubiquitin chains (Onizawa et␣al, 2015). In addition to its DUB activity, A20 displayed E3 ubiquitin ligase activity that is mediated by seven Cys2‐Cys2 zinc‐finger (ZF) domains (Polykratis et␣al, 2019). This gives A20 dual ubiquitin editing capabilities. It is known that the C103A substitution in the OTU domain of A20 blocks deubiquitinating activity, whereas C609A and C612A substitutions in the zinc finger‐4 (ZnF4) domain disrupt E3 ligase activity (Ma & Malynn, 2012; Lork & Beyaert, 2017). As shown in Fig 7G, introduction of C609A/C612A, but not C103A, blocked the ubiquitination of RIPK3, indicating that ZnF4 was responsible for catalyzing ubiquitination. In addition, a K51A kinase mutant of RIPK3 was unable to be ubiquitinated (Fig 7G), supporting the view that the kinase activity of RIPK3 is required for the formation of necrotic complexes. We also conducted RIPK3 binding studies with various A20 mutants to determine whether the ZnF4 domain contributes to the recognition of RIPK3. The results indicated that the A20 ZnF4 region was essential for RIPK3 binding (Appendix␣Fig S2A and B). To precisely define the ubiquitination of RIPK3, we cotransfected RIPK3 with A20 and ubiquitin mutants, where only K48 and/or K63 were available for polymerization (Appendix␣Fig S2C). The results showed that both K48‐ and K63‐linked ubiquitin chains contributed to A20‐mediated RIPK3 ubiquitination (Appendix Fig S2D). Notably, unlike K48‐linked ubiquitination, K63‐linked ubiquitination is independent of RIPK3 proteasomal degradation (Appendix␣Fig S2E).
The A20‐mediated cell death mechanism is independent of inflammatory responses
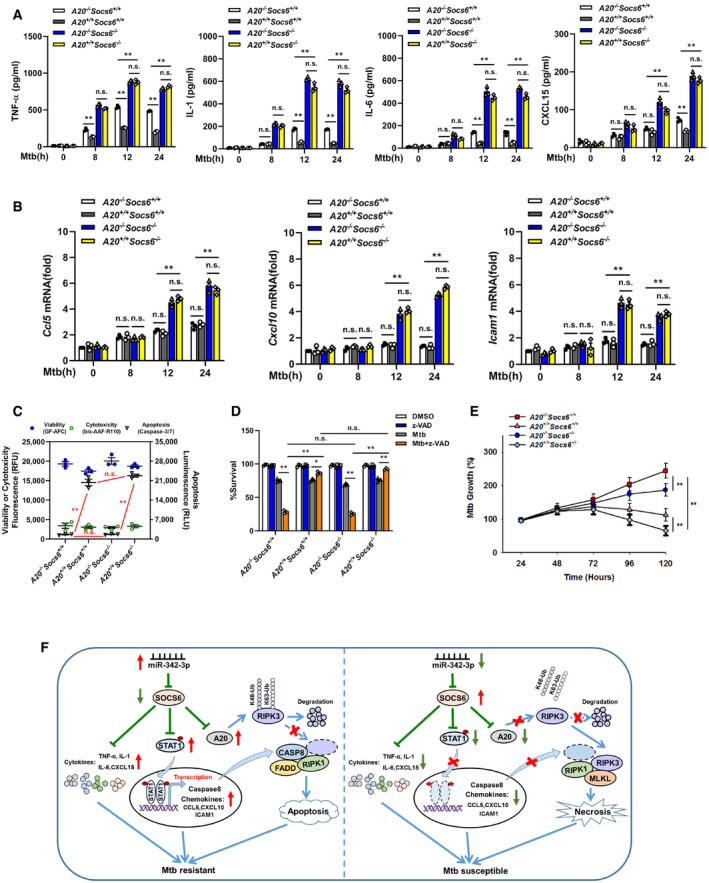
To understand the role of A20 in the miR‐342/SOCS6‐mediated immune response to Mtb infection, BMDMs from mice with different genotypes were infected with Mtb and measured the release of cytokines (TNF‐α, IL‐1, IL‐6, CXCL15) and chemokines (Ccl5, Cxcl10, Icam1). The results showed that the release of cytokines and chemokines was significantly higher in Socs6 −/− BMDMs than in Socs6 +/+ BMDMs (Fig 8A and B). There was no difference in the release of cytokines and chemokines in Socs6 −/− BMDMs with and without A20 (Fig 8A and B). In addition, assays measuring cell death mechanisms (Fig 8C, Appendix␣Fig S3) and cell viability (Fig 8D) showed that, in A20‐overexpressing or A20‐knockout cells, SOCS6 expression had no impact on the mechanism of cell death. Taken together, these data indicated that A20‐regulated RIPK3 ubiquitination and the formation of a death complex were independent of SOCS6‐mediated cytokine and chemokine production in response to Mtb stimulation. Mtb growth results confirmed that the A20‐mediated cell death mechanism switch and the SOCS6‐mediated inflammatory response played synergistic roles in controlling Mtb replication: Cells with high expression of A20 and low expression of SOCS6 had the strongest resistance to Mtb (Fig 8E).
Figure 8. A20‐mediated cell death mechanism is independent of inflammatory responses.
-
A, BProduction of cytokines TNF‐α, IL‐1, IL‐6, CXCL15 (A), or chemokines Ccl5, Cxcl10, Icam1 (B) in the A20 −/− Socs6 +/+, A20 +/+ Socs6 +/+, A20 −/− Socs6 −/−, and A20 +/+ Socs6 −/− BMDMs after Mtb stimulation. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
C–ECell death mechanisms (C), cell viabilities (D), or Mtb growth rates (E) of A20 −/− Socs6 +/+, A20 +/+ Socs6 +/+, A20 −/− Socs6 −/−, and A20 +/+ Socs6 −/− BMDMs stimulated with Mtb. Z‐VAD treatment concentration was 20 μM, and the treatment time was 24 h. Data are shown as the mean ± SEM of n = 3 biological replicates.
-
FSchematic representation of miR‐342 regulated anti‐Mtb immunity.
Data information: ANOVA followed by Bonferroni post hoc test (A‐E) was used for data analysis. *P < 0.05, **P < 0.01. Abbreviation: n.s., not significant.
In summary, our data show that the miR‐342‐3p/SOCS6 axis strengthens the anti‐Mtb immune system by increasing the production of inflammatory cytokines and chemokines and by switching cell death from necrosis to apoptosis through A20‐mediated K48 ubiquitination and RIPK3 degradation (Fig 8F).
Discussion
In this study, we found that SOCS6 is regulated by miR‐342‐3p and thus mediates multiple downstream immune response pathways. Considering that other SOCS family members have similar structures to SOCS6, we examined whether they could also be regulated by miR‐342‐3p. The results showed that miR‐342‐3p had no significant effect on the 3’UTR activity of other SOCS family members (Appendix␣Fig S4A), indicating that miR‐342‐3p has a relatively specific regulatory effect on SOCS6. We also examined the effects of SOCS6 on the regulation of other SOCS and STAT family members (Fig EV3A, Appendix␣Fig S4B–D). The results showed that SOCS6 negatively regulates the expression of SOCS7 (Appendix␣Fig S4B). However, siRNA knockdown experiments indicated that SOCS7 is dispensable for Mtb‐induced cell death in C3H and B6 mice (Appendix␣Fig S4C and D). STAT3 activation was also observed in Mtb‐infected Socs6 +/+ macrophages (Fig EV3A). A similar STAT3 activation pattern was also detected in Socs6 −/− cells (Fig EV3A). Thus, we speculate that STAT3 activation has no effect on SOCS6 and that the pathway by which SOCS6 mediates downstream STAT1 is relatively independent of other STAT family members. However, this needs to be validated with further experiments.
Apoptosis and necrosis are two common forms of cell death that play essential roles in the development and maintenance of homeostasis in multicellular organisms (Green & Llambi, 2015). In Mtb‐infected macrophages, necrosis is associated with uncontrollable cellular damage, providing a niche for Mtb replication before it escapes into the extracellular milieu (Lerner et␣al, 2017). Apoptosis, on the other hand, results in programmed cell suicide, thus limiting the replication of Mtb (Behar et␣al, 2011; Zhao et␣al, 2017). It has been reported that in␣vitro modulation of apoptosis affects mycobacterial cell‐to‐cell spread, suggesting an unambiguous relationship between apoptosis and the propagation of Mtb (Aguilo et␣al, 2013). We found that the recruitment of RIPK1 and FADD to caspase 8 resulted in apoptotic cell death in Mtb‐infected macrophages. On the other hand, when the complex was formed with RIPK1, RIPK3, and MLKL, necrotic cell death was observed. This result is consistent with previous reports showing that RIPK3 acts as a molecular switch between TNF‐induced apoptosis and necrosis in mouse embryonic fibroblasts and that high levels of RIPK3 can switch apoptosis into necrosis in certain cell types. Interestingly, Stutz et␣al (2018) argued that RIPK3 does not play a fundamental role in regulating inflammatory responses or necrosis in␣vivo. In our current work, we found that RIPK3 did not affect the production of several inflammatory cytokines, including TNF‐α and IL‐1, which is consistent with Stutz’s findings. However, we also found that recruitment of RIPK3 to the cytoplasmic complex contributed to the necrosis of Mtb‐infected macrophages. We hypothesize that SOCS6 may play as‐yet undescribed roles in various aspects of macrophage activities, including activation, differentiation, and Mtb‐induced cell death mechanisms.
Many bacterial pathogens exploit the host ubiquitination system to promote pathogenesis. For example, the host E3 ubiquitin ligase ANAPC2 interacts with mycobacterial protein Rv0222 to suppress the expression of pro‐inflammatory cytokines (Wang et␣al, 2020). A20, an inhibitor of NF‐κB signaling, has dual ubiquitination and deubiquitination roles within the proteasomal degradation pathway and is highly connected to the immune response network (Vereecke et␣al, 2009; Sokhi et␣al, 2018; Priem et␣al, 2020). A20 also contains seven ZF domains, which mediate its E3 ubiquitin ligase and ubiquitin‐binding activities (Lork & Beyaert, 2017). We found that upregulation of A20 facilitates RIPK3 ubiquitination, resulting in apoptotic cell death in Mtb‐stimulated macrophages. We also showed that the ZF4 domain is responsible for A20‐mediated K48‐ and K63‐linked ubiquitination of RIPK3. K48‐linked ubiquitin chains tag substrates for proteasomal degradation, whereas K63‐linked ubiquitin chains fulfill diverse proteasome‐independent cellular roles, including endocytosis, DNA damage responses, and immune responses (Yau & Rape, 2016). Consistent with previous reports, we found that the proteasomal degradation of RIPK3 depended on K48 ubiquitination alone. However, the functional role of K63‐linked ubiquitination of RIPK3 has not been fully elucidated. It would be interesting to further investigate the specific role of K63‐ubiquitinated RIPK3 in determining the fate of macrophages.
Cytokines such as IL‐1 mediate susceptibility to Mtb. Mir342 −/−B6 mice, which were susceptible to Mtb, showed low levels of inflammatory factors (TNF‐α, IL‐1, IL‐6, and CXCL15) along with severe lung lesions. Ji et␣al (2019) reported that IL‐1 plays a protective role during Mtb infection, which is consistent with our results. Interestingly, they also found that elevated levels of IL‐1α/β proteins in B6J.C3‐Sst1 C3HeB/FeJKrmn mice (which are susceptible to Mtb) failed to control Mtb (Ji et␣al, 2019). This difference might be due to the different genotypes of the mice used in these studies or the “double‐edged sword” character of cytokines in the immune system.
The release of cytokines and chemokines was dramatically higher in Socs6 −/− macrophages than in Socs6 +/+ macrophages. This suggested that A20 regulates the degradation of RIPK3 and that the induction of apoptosis is independent of SOCS6‐mediated inflammatory responses. Interestingly, the presence or absence of A20 exerted different effects on cytokine release in Socs6 +/+ macrophages but not in Socs6 −/− macrophages. We speculate that this may be because SOCS6 has a stronger inhibitory effect on cytokines than A20. Although A20 can be used as a negative feedback regulator of NF‐κB to inhibit the production of cytokines, its effect is far smaller than that of SOCS6. Therefore, the absence of SOCS6 will result in a large increase in cytokine expression; in this case, the regulatory effect of A20 appears to be minimal. However, this hypothesis needs to be verified by further experiments.
C3H (TB susceptible) and B6 (TB resistant) inbred mice were used as models in this study. We found that the deletion of miR‐342 in B6 mice resulted in susceptibility to Mtb infection, as evidenced by acceleration of bacterial load within organs and development of necrotic lung lesions. Additionally, we observed an inverse correlation between the expression of miR‐342‐3p and tuberculosis. These data indicate that miR‐342‐3p participates in a new regulatory pathway involved in tissue necrosis. Despite the substantial amount of work presented, there are still some limitations and remaining questions. For example, we performed miR‐342‐3p knockout in B6 mice and miR‐342‐3p overexpression in C3H mice. Since the expression of miR‐342‐3p is not the only difference between these mouse strains, interventions (knockout and overexpression) performed in both strains would be better for direct comparisons. Next, we used macrophages derived from SOCS6‐knockout C3H mice (Socs6 −/−) and their wild‐type littermates (Socs6 +/+) as in␣vitro models to study the effects of SOCS6 on downstream signaling pathways and cell death mechanisms. Although the amount of SOCS6 protein was exquisitely manipulated by the interventions as expected (Appendix Fig S4E), these manipulations are not a perfect substitute for alterations in miR‐342‐3p levels. We also observed an inverse correlation between miR‐342‐3p expression and tuberculosis in clinical PBMC samples. Because other cell types may also serve as a source of miR‐324‐3p, we believe that using pure macrophages rather than PBMCs would provide a more convincing argument. In addition, PBMCs extracted from whole blood might not have even been in contact with Mtb. In the future, it would be worth investigating how the upregulation of miR‐342‐3p is initiated and whether it depends on direct contact with Mtb. We speculate that infection signals could be transmitted through cellular communication, such as endocrine communication, which means that the signals could be transmitted throughout the organism via the circulatory system. However, more clinical data and experiments are required to test this hypothesis. In this study, z‐VAD, a pan‐caspase inhibitor, was used to distinguish cell death mechanisms. A miR‐342‐3p inhibitor induced a switch from apoptosis to necrosis in Mtb‐infected B6 BMDMs. Interestingly, cell survival was further decreased upon the addition of z‐VAD. This observation is consistent with previous reports (He et␣al, 2009; Zhang et␣al, 2009). We speculate that, in addition to caspase inhibition, z‐VAD might be involved in the regulation of cell death patterns via additional mechanisms. The potential roles of z‐VAD in cell death require further investigation.
Materials and Methods
Ethic statements
This study was carried out in strict accordance with the Guidelines for the Care and Use of Animals of Chongqing University. All animal experimental procedures were approved by the Animal Ethics Committees of the School of Life Sciences, Chongqing University. Blood samples were obtained from Chongqing Public Health Medical Center in accordance with the guidelines of the local ethics committee. Patients providing blood samples were given informed consent. The ethics committee approved this consent procedure.
Mice
All animal study protocols were reviewed and approved by Chongqing University School of Life Sciences review boards for animal studies. C3HeB/FeJ and C57BL/6J mice were purchased from Jackson Laboratory. Mir342 +/+ C3H mice (C3HeB/FeJ‐Tg (mROSA‐Mir342)1cyagen) and Mir342 −/− B6 mice (C57BL/6J‐Mir342 tm1cyagen) were constructed and identified by Cyagen Biosciences (Guangzhou, China). Littermate wild‐type mice were used as controls. Briefly, in order to generate Mir342 +/+ C3H mice, pre‐miR‐342 sequence was cloned into pBROAD3 vector and transgenic mice were obtained by pronuclear microinjection. Mir342 −/− B6 mice were generated as previously described (Dooley et␣al, 2017). The Socs6 −/− mice (C3HeB/FeJ‐Socs6 em1cyagen) were purchased from Cyagen Biosciences (Guangzhou, China). Littermate wild‐type mice were used as controls. The pBROAD3‐A20 vector was microinjected into the zygotes from Socs6 +/+ (wild‐type) and Socs6 −/− mice to generate A20‐overexpressing mice (A20 +/+ Socs6 +/+, A20 +/+ Socs6 −/−). A20‐knockout mice (A20 −/− Socs6 +/+, A20 −/− Socs6 −/−) were generated from Socs6 +/+ and Socs6 −/− mice using CRISPR/Cas9.
Blood samples
TB patients (n = 34) were diagnosed based on clinical presentation and radiological signs and were confirmed by sputum culture positivity. Subjects were excluded if they had autoimmune disorders or other infectious diseases. The average age was 45, and 55.9% of patients were male. The control individuals (n = 27) came from healthy donors. The inclusion criteria were no previous history of TB, and no evidence of TB‐related infiltration in chest X‐rays. The average age was 42, and 51.9% of the controls were male. All protocols were approved by the local ethics committee of the Public Health Medical Center, Chongqing, China, and signed informed consent was obtained from subjects.
Plasmids construction
The 3′‐UTR of Socs6 (Genebank Accession number: NM_018821.5) was amplified and inserted the psicheck‐2 vector (Promega) through standard molecular cloning methods and confirmed by sequencing. Full‐length coding sequence of Socs6 was inserted into pCDH‐MCS‐T2A‐Puro‐MSCV (System Biosciences, Palo Alto, CA, USA); the recombinant construct was named pCDH‐SOCS6. The lentiviral vector pCDH‐SOCS6 and packaging plasmids such as pMD2.G and psPAX2 were cotransfected into HEK‐293T cells to generate lentivirus. The RIPK3 (Genebank Accession number: NM_019955.2) and A20 (Genebank Accession number: NM_009397.3) mutants were generated by overlapped extension PCR method based on wild‐type plasmids. The K48R, K63R, and K48/63R mutants were kindly provided by Dr. Yonghui Zheng (Michigan State University). The wild‐type Ubiquitin and other mutants were purchased from Addgene. All the primers used for plasmids construction are listed in Appendix␣Table S2.
Bacterial strains and infection
The Mycobacterial strain H37Rv (ATCC, Manassas, VA, USA) was grown in Middlebrook 7H9 (BD Biosciences, San Jose, CA, USA) medium supplemented with 10% OADC (BD Biosciences) and 0.05% Tween‐80 (Sigma‐Aldrich, St. Louis, MO, USA), or on Middlebrook 7H10 agar (BD Biosciences) supplemented with 10% OADC. For mice infection, 6‐week‐old mice were randomly allocated into groups and aerosol infected with approximately 400 CFU Mtb. The mice were killed at the indicated time. The organs were collected for CFU assay or histopathology analysis. All mice were age‐ and sex‐matched in each experiment. Every effort was made to minimize animal pain, suffering, and distress and reduce the number of animals used.
In vivo transfection
The in␣vivo transfection of mice was performed by tail vein injection (N/P ratio = 8) using the jetPEI in␣vivo transfection reagent (Polyplus Transfection Inc, New York, NY, USA) according to the manufacturer’s instruction as previously described (Sutherland et␣al, 2014; Li et␣al, 2016). Injections were repeated 3 times per week for the living mice.
Cell culture, transfection, and treatment
NIH‐3T3, HEK‐293T, and RAW264.7 cells were obtained from American Type Culture Collection (ATCC) and grown adherently to plastic plates (Corning Costar, Cambridge, MA, USA). The culture medium was composed of Dulbecco's modified Eagle's medium (DMEM, Gibco, USA) and 10% fetal bovine serum (Gibco). Double‐stranded mmu‐miR‐342‐3p (miRBase accession number MIMAT0000590, UCUCACACAGAAAUCGCACCCGU) mimic, single‐stranded mmu‐miR‐342‐3p inhibitor and their corresponding negative controls (NC mimic or inhibitor) were purchased from GenePharma (GenePharma, Shanghai, China). BMDMs were obtained by culturing bone marrow cells as previously described (Sutavani et␣al, 2018). In brief, bone marrow cells were cultured in RPMI‐1640 (Gibco) supplemented with 10% heat‐inactivated FBS, 2 mM l‐glutamine, 1% nonessential amino acids, 100 unit penicillin, 100 µg streptomycin, and 22 ng M‐CSF (PeproTech, Rocky Hill, NJ, USA)/ml for 6 d at 37°C, 5% CO2. After 6 d of culture, adherent macrophages were switched into antibiotic‐free media and seeded with 105 cells per well. BMDMs were infected with Mtb at a MOI of 5 for in␣vitro research. Human PBMCs were obtained by density gradient centrifugation (Ficoll, Sigma‐Aldrich) from fresh heparinized whole blood. All cells were maintained in a humidified, 37°C incubator with 5% CO2.
Identification of cell death mechanism
ApoTox‐Glo Triplex Assay (Promega, Madison, WI, USA) was used to identify the cell death mechanism following manufactory instructions. In brief, GF‐AFC and bis‐AAF‐R110 substrates were added to the cell culture medium. Cells were incubated for 30 min at 37°C, and fluorescence data were collected to assess viability and cytotoxicity. Then, 100 μl of Caspase‐Glo 3/7 reagent was added to the mixture. Cells were incubated for 30 min at room temperature. Luminescence data were collected to assess the caspase activation. Apoptosis or necrosis result was determined according to the manual: Cell apoptotic death was with a high level of caspase activation, and cell necrotic death was with a low level of caspase activation. In addition, Annexin V/PI staining was also used to identify apoptotic and necrotic cells as previously described (Pan et␣al, 2005). Briefly, at indicated time points, cells were stained with Annexin V/PI (Sigma‐Aldrich), fixed with 1% paraformaldehyde, and then analyzed using a BD LSR II flow cytometer (BD Biosciences). Data were analyzed with FlowJo data analysis software.
In vitro CFU assay
CFU assay was performed by plating tenfold serial dilutions of each tissue homogenate on Middlebrook 7H10 agar plates. The colonies were counted after 4–6 weeks of incubation at 37°C in 5% CO2.
Histopathology analysis
Pathology scoring system was used in histopathology analysis as previously described with partial modification (Wu et␣al, 2015). Pathological changes of lung lobes were examined and evaluated by semiquantitative gross pathology scoring system: 0 = no visible lesions; 1 = no external gross lesions, but lesions seen upon slicing; 2 = less than five gross lesions with a diameter < 4 mm; 3 = more than five gross lesions with a diameter < 4 mm; 4 = more than one distinct gross lesion with a diameter > 4 mm; 5 = gross coalescing lesions. The scores of the individual lobes were added to calculate the lung score. Typical lesions were fixed with 10% buffered formaldehyde for more than 24 h, embedded in paraffin, sectioned, and stained with H&E according to the standard procedure. Photographs were obtained by microscopy (Carl Zeiss, Jena, Germany).
Immunoprecipitation and immunoblot analysis
Immunoblot analysis was performed as previously described (Fu et␣al, 2020b). Cell lysates were fractionated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). Blots were probed with 1/1,000 anti‐SOCS6 (PA5‐50263), 1/1,000 anti‐STAT1 (AHO0832), 1/500 anti‐phospho‐STAT1 (44‐376G) (Thermo Fisher Scientific, San Jose, CA, USA), 1/500 anti‐Ubiquitin (sc‐271289), 1/500 anti‐HA (sc‐7392), 1/500 anti‐Myc (sc‐56634), 1/500 anti‐Caspase 8 (sc‐5263, 1/30 for IP), 1/500 anti‐CXCL10 (sc‐374092), 1/500 anti‐ICAM1 (sc‐18853), 1/500 anti‐CCL5 (sc‐373984), 1/500 anti‐STAT3 (sc‐8019), 1/500 anti‐phospho‐STAT3 (sc‐8059), 1/500 anti‐STAT4 (sc‐398228), 1/500 anti‐phospho‐STAT4 (sc‐28296), 1/500 anti‐STAT6 (sc‐374021), 1/500 anti‐phospho‐STAT6 (sc‐136019), 1/500 anti‐SOCS1 (sc‐518028), 1/500 anti‐SOCS3 (sc‐73045), 1/500 anti‐SOCS4 (sc‐135566), 1/500 anti‐SOCS5 (sc‐100858), 1/500 anti‐SOCS7 (sc‐137241), 1/200 anti‐Caspase 2 (sc‐53928), 1/200 anti‐Caspase 9 (sc‐56076) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), 1/1,000 anti‐MLKL (SAB1302339), 1/1,000 anti‐RIPK1 (SAB3500420) (Sigma‐Aldrich), 1/1,000 anti‐RIPK3 (NBP1‐77299, 1/200 for IP) (Novus Biologicals, Englewood, CO, USA), 1/500 anti‐STAT2 (72604), 1/500 anti‐phospho‐STAT2 (88410), 1/500 anti‐STAT5 (25653), 1/500 anti‐phospho‐STAT5 (9359), 1/500 anti‐SOCS2 (2779) (Cell Signaling Technology, Inc., Danvers, MA, USA), 1/1,000 anti‐Actin (AA128), and 1/1,000 anti‐Flag (AF519, 1/100 for IP) (Beyotime, Jiangsu, China) antibodies. Immunoblots were revealed using SuperSignal west pico substrate (Thermo Fisher Scientific). The relative intensity of protein bands was quantified using ImageJ software and normalized to controls. For IP, cells were collected and lysed with 1× lysis buffer (Thermo Fisher Scientific), and afterward, the lysates were incubated with appropriate antibodies and Protein A/G beads (Thermo Fisher Scientific) at 4°C overnight. The beads were washed three times with IP buffer (Thermo Fisher Scientific), followed by immunoblot analysis.
Luciferase assays
Luciferase assays were performed using the Dual‐Luciferase Reporter (DLR) Assay System (Promega) as previously described (Wu et␣al, 2015). In brief, cells were cotransfected with luciferase reporter plasmid and internal control plasmid pRL‐SV40. Cells were lysed for DLR assays 24 h after treatment. Data were collected with a VICTOR X5 Multilabel Plate␣Reader (PerkinElmer, Waltham, MA, USA) and relative luciferase activities were measured by firefly luciferase luminescence divided by renilla luciferase luminescence.
Cell viability assays
Cell viability was performed using a Cell Titer‐Glo Luminescent Cell Viability Assay kit (Promega) following manufactory instructions. In brief, 100 μl of Cell Titer‐glo reagent was added to the cell culture medium. Cells were then incubated at room temperature for 10 min. Luminescent data were collected with a VICTOR X5 Multilabel Plate␣Reader (PerkinElmer).
Real‐time qRT–PCR
We used qRT–PCR to analyze gene expression. Total RNA samples were isolated using TRIzol reagent (Invitrogen, Life Technologies, CA, USA). Purified RNA was reverse‐transcribed using a SYBR PrimeScript RT–PCR Kit (Takara, Otsu, Shiga, Japan). The expression of mRNAs was quantified using a SYBR Premix ExTaq II Kit (Takara). qRT–PCR was performed on Bio‐Rad CFX‐96 system (Bio‐Rad, Hercules, CA, USA), and results were normalized to β‐actin mRNA levels. We also used qRT–PCR to analyze miRNA expression. Total RNA samples were isolated using mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA). Purified RNA was reverse‐transcribed using a miScript II RT Kit (Qiagen, Hilden, Germany). The expressions of mature miRNAs were quantified using a miScript SYBR Green PCR Kit, which contained 10× miScript Universal Primer (Qiagen), and were performed according to the manufacturer’s instructions. Quantization of U6 was performed to normalize miRNA expression levels. Data were analyzed using the 2−ΔΔCt method. All primer sequences used for qRT–PCR are listed in Appendix␣Table S3.
Enzyme‐linked immunosorbent assays
RAW264.7 and BMDMs were infected with Mtb for the indicated periods of time, and equal volumes of control and infected culture supernatants cleared by centrifugation were assayed by ELISA. TNF‐α, IL‐1, IL‐6, and CXCL15 ELISA kits were purchased from e‐Biosciences (Thermo Fisher Scientific). The concentration of each cytokine was calculated against a standard curve.
Statistical analysis
Sample size was based on empirical data from pilot experiments. The investigators were blinded during data collection and analysis. Data are presented as mean ± SEM and analyzed by ANOVA with Bonferroni post hoc test unless otherwise indicated. A value of P < 0.05 was considered significant.
Author contributions
HW, XW, and ZL conceived and designed the study. HW, BF, XL, ST, RZ, WX, HZ, Shanfu. Z, QZ, YW, KF, LS, Shaolin. Z, WN, and KCP performed the experiments. HW, XW, and ZL analyzed the data. HW and BF wrote the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Source Data for Figure␣1D
Source Data for Figure␣3
Source Data for Figure␣5
Source Data for Figure␣6
Source Data for Figure␣7
Acknowledgements
The authors would like to thank Dr. Yonghui Zheng (Michigan State University) for providing Ubiquitin mutants plasmids, Dr. Tao Li (Wuhan University) for his assistance in mice feeding and histopathology analysis, and Dr. Rui Wang (Sun Yat‐sen University) for technical support. This work was supported by the National Natural Science Foundation of China (No. 81970008, 82000020, 31702205, and 81700015), the Fundamental Research Funds for the Central Universities (No. 2019CDYGZD009 and 2020CDJYGRH‐1005), and the Natural Science Foundation of Chongqing, China (No. cstc2020jcyj‐msxmX0460, cstc2020jcyj‐bshX0105, and cstc2018jcyjAX0372). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
EMBO reports (2021) 22: e52252.
Contributor Information
Zhifeng Li, Email: qc_zhifengli@126.com.
Xingsheng Wang, Email: xingswang@163.com.
Haibo Wu, Email: hbwu023@cqu.edu.cn.
Data availability
No primary data sets were generated and deposited.
References
- Aguilo JI, Alonso H, Uranga S, Marinova D, Arbues A, de Martino A , Anel A, Monzon M, Badiola J, Pardo Jet␣al (2013) ESX‐1‐induced apoptosis is involved in cell‐to‐cell spread of Mycobacterium tuberculosis . Cell Microbiol 15: 1994–2005 [DOI] [PubMed] [Google Scholar]
- Alexander WS, Hilton DJ (2004) The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol 22: 503–529 [DOI] [PubMed] [Google Scholar]
- Bartel DP (2018) Metazoan microRNAs. Cell 173: 20–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan H, Divangahi M, Remold HG (2011) Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis . Mucosal Immunol 4: 279–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carow B, Gao Y, Teran G, Yang XO, Dong C, Yoshimura A, Rottenberg ME (2017) CISH controls bacterial burden early after infection with Mycobacterium tuberculosis in mice. Tuberculosis 107: 175–180 [DOI] [PubMed] [Google Scholar]
- Chakaya J, Khan M, Ntoumi F, Aklillu E, Razia F, Mwaba P, Kapata N, Mfinanga S, Hasnain SE, Katoto PDet␣al (2021) Global Tuberculosis Report 2020 ‐ Reflections on the Global TB burden, treatment and prevention efforts. Int J Infect Dis 10.1016/j.ijid.2021.02.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy EY, Siu KL, Kok KH, Lung RW, Tsang CM, To KF, Kwong DL, Tsao SW, Jin DY (2008) An Epstein‐Barr virus‐encoded microRNA targets PUMA to promote host cell survival. J Exp Med 205: 2551–2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley J, Lagou V, Pasciuto E, Linterman MA, Prosser HM, Himmelreich U, Liston A (2017) No functional role for microRNA‐342 in a mouse model of pancreatic acinar carcinoma. Front Oncol 7: 101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B, Xue W, Zhang H, Zhang R, Feldman K, Zhao Q, Zhang S, Shi L, Pavani KC, Nian Wet␣al (2020a) MicroRNA‐325‐3p facilitates immune escape of Mycobacterium tuberculosis through targeting LNX1 via NEK6 accumulation to promote anti‐apoptotic STAT3 signaling. MBio 11: e00557–e1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B, Yin S, Lin X, Shi L, Wang Y, Zhang S, Zhao Q, Li Z, Yang Y, Wu H (2020b) PTPN14 aggravates inflammation through promoting proteasomal degradation of SOCS7 in acute liver failure. Cell Death Dis 11: 803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, Debatin KM (2002) IFNgamma sensitizes for apoptosis by upregulating caspase‐8 expression through the Stat1 pathway. Oncogene 21: 2295–2308 [DOI] [PubMed] [Google Scholar]
- Green DR, Llambi F (2015) Cell Death Signaling. Cold Spring Harb Perspect Biol 7: a006080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He SD, Wang L, Miao L, Wang T, Du FH, Zhao LP, Wang XD (2009) Receptor interacting protein kinase‐3 determines cellular necrotic response to TNF‐alpha. Cell 137: 1100–1111 [DOI] [PubMed] [Google Scholar]
- Hong JY, Chung K‐S, Shin J‐S, Lee J‐H, Gil H‐S, Lee H‐H, Choi E, Choi J‐H, Hassan AHE, Lee YSet␣al (2019) The Anti‐proliferative activity of the hybrid TMS‐TMF‐4f compound against human cervical cancer involves apoptosis mediated by STAT3 inactivation. Cancers 11: 1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H (2007) Cellular microRNAs contribute to HIV‐1 latency in resting primary CD4+ T lymphocytes. Nat Med 13: 1241–1247 [DOI] [PubMed] [Google Scholar]
- Iwakawa HO, Tomari Y (2015) The functions of microRNAs: mRNA decay and translational repression. Trends Cell Biol 25: 651–665 [DOI] [PubMed] [Google Scholar]
- Ji DX, Yamashiro LH, Chen KJ, Mukaida N, Kramnik I, Darwin KH, Vance RE (2019) Type I interferon‐driven susceptibility to Mycobacterium tuberculosis is mediated by IL‐1Ra. Nat Microbiol 4: 2128–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas S, Izaurralde E (2015) Towards a molecular understanding of microRNA‐mediated gene silencing. Nat Rev Genet 16: 421–433 [DOI] [PubMed] [Google Scholar]
- Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P (2005) Modulation of hepatitis C virus RNA abundance by a liver‐specific microRNA. Science 309: 1577–1581 [DOI] [PubMed] [Google Scholar]
- Kim D, Nguyen QT, Lee J, Lee SH, Janocha A, Kim S, Le HT, Dvorina N, Weiss K, Cameron MJet␣al (2020) Anti‐inflammatory roles of glucocorticoids are mediated by Foxp3(+) regulatory T cells via a miR‐342‐dependent mechanism. Immunity 53: 581–596.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Shin DI, Choi BH, Min BH (2021) Exosomes from IL‐1beta‐primed mesenchymal stem cells inhibited IL‐1beta‐ and TNF‐alpha‐mediated inflammatory responses in osteoarthritic SW982 cells. Tissue Eng Regen Med 10.1007/s13770-020-00324-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramnik I (2008) Genetic dissection of host resistance to Mycobacterium tuberculosis: the sst1 locus and the Ipr1 gene. Curr Top Microbiol Immunol 321: 123–148 [DOI] [PubMed] [Google Scholar]
- Kramnik I, Dietrich WF, Demant P, Bloom BR (2000) Genetic control of resistance to experimental infection with virulent Mycobacterium tuberculosis . Proc Natl Acad Sci USA 97: 8560–8565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Sahu SK, Kumar R, Subuddhi A, Maji RK, Jana K, Gupta P, Raffetseder J, Lerm M, Ghosh Zet␣al (2015) MicroRNA let‐7 modulates the immune response to Mycobacterium tuberculosis infection via control of A20, an inhibitor of the NF‐kappaB pathway. Cell Host Microbe 17: 345–356 [DOI] [PubMed] [Google Scholar]
- Lerner TR, Borel S, Greenwood DJ, Repnik U, Russell MR, Herbst S, Jones ML, Collinson LM, Griffiths G, Gutierrez MG (2017) Mycobacterium tuberculosis replicates within necrotic human macrophages. J Cell Biol 216: 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JS, Chen ML, Chang SY, Yu SL, Lin CW, Wang H, Chen WC, Chang CH, Wang JY, Lee LNet␣al (2017) SP110b controls host immunity and susceptibility to tuberculosis. Am J Respir Crit Care Med 195: 369–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, He S, Li R, Zhou X, Zhang S, Yu M, Ye Y, Wang Y, Huang C, Wu M (2016) Pseudomonas aeruginosa infection augments inflammation through miR‐301b repression of c‐Myb‐mediated immune activation and infiltration. Nat Microbiol 1: 16132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Chen J, Wang P, Li H, Zhou Y, Liu H, Liu Z, Zheng R, Wang L, Yang Het␣al (2018) MicroRNA‐27a controls the intracellular survival of Mycobacterium tuberculosis by regulating calcium‐associated autophagy. Nat Commun 9: 4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, da Cunha AP , Rezende RM, Cialic R, Wei Z, Bry L, Comstock LE, Gandhi R, Weiner HL (2016) The host shapes the gut microbiota via fecal microRNA. Cell Host Microbe 19: 32–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lork MVK, Beyaert R (2017) CYLD, A20 and OTULIN deubiquitinases in NF‐kappa B signaling and cell death: so similar, yet so different. Cell Death Differ 24: 1172–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A, Malynn BA (2012) A20: linking a complex regulator of ubiquitylation to immunity and human disease. Nat Rev Immunol 12: 774–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca C, Tsenova L, Freeman S, Barczak AK, Tovey M, Murray PJ, Barry C, Kaplan G (2005) Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak‐Stat pathway. J Interferon Cytokine Res 25: 694–701 [DOI] [PubMed] [Google Scholar]
- Masood KI, Hussain R, Rao N, Rottenberg ME, Salahuddin N, Irfan M, Hasan Z (2014) Differential early secreted antigen target (ESAT) 6 kDa‐induced IFN‐gamma and SOCS1 expression distinguishes latent and active tuberculosis. J Infect Dev Ctries 8: 59–66 [DOI] [PubMed] [Google Scholar]
- Masood KI, Rottenberg ME, Carow B, Rao N, Ashraf M, Hussain R, Hasan Z (2012) SOCS1 gene expression is increased in severe pulmonary tuberculosis. Scand J Immunol 76: 398–404 [DOI] [PubMed] [Google Scholar]
- Masood KI, Rottenberg ME, Salahuddin N, Irfan M, Rao N, Carow B, Islam M, Hussain R, Hasan Z (2013) Expression of M. tuberculosis‐induced suppressor of cytokine signaling (SOCS) 1, SOCS3, FoxP3 and secretion of IL‐6 associates with differing clinical severity of tuberculosis. BMC Infect Dis 13: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onizawa M, Oshima S, Schulze‐Topphoff U, Oses‐Prieto JA, Lu T, Tavares R, Prodhomme T, Duong B, Whang MI, Advincula Ret␣al (2015) The ubiquitin‐modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol 16: 618–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A (2015) The JAK‐STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med 66: 311–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Yan BS, Rojas M, Shebzukhov YV, Zhou H, Kobzik L, Higgins DE, Daly MJ, Bloom BR, Kramnik I (2005) Ipr1 gene mediates innate immunity to tuberculosis. Nature 434: 767–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polykratis A, Martens A, Eren RO, Shirasaki Y, Yamagishi M, Yamaguchi Y, Uemura S, Miura M, Holzmann B, Kollias Get␣al (2019) A20 prevents inflammasome‐dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin‐binding domain. Nat Cell Biol : 731–742 [DOI] [PubMed] [Google Scholar]
- Priem D, van Loo G , Bertrand MJM (2020) A20 and cell death‐driven inflammation. Trends Immunol 41: 421–435 [DOI] [PubMed] [Google Scholar]
- Qu C, Xu QS, Lu MM, Wang FF, Liu ZQ, Liu DY, Yang W, Yi QL, Wang LL, Song LS (2018) The involvement of suppressor of cytokine signaling 6 (SOCS6) in immune response of Chinese mitten crab Eriocheir sinensis . Fish Shellfish Immunol 72: 502–509 [DOI] [PubMed] [Google Scholar]
- Seo J, Lee E‐W, Sung H, Seong D, Dondelinger Y, Shin J, Jeong M, Lee H‐K, Kim J‐H, Han SYet␣al (2016) CHIP controls necroptosis through ubiquitylation‐ and lysosome‐dependent degradation of RIPK3. Nat Cell Biol 18: 291–302 [DOI] [PubMed] [Google Scholar]
- Sironi JJ, Ouchi T (2004) STAT1‐induced apoptosis is mediated by caspases 2, 3, and 7. J Biol Chem 279: 4066–4074 [DOI] [PubMed] [Google Scholar]
- Sokhi UK, Liber MP, Frye L, Park S, Kang K, Pannellini T, Zhao B, Norinsky R, Ivashkiv LB, Gong S (2018) Dissection and function of autoimmunity‐associated TNFAIP3 (A20) gene enhancers in humanized mouse models. Nat Commun 9: 658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Ping Y, Zhang K, Yang L, Li F, Zhang C, Cheng S, Yue D, Maimela NR, Qu Jet␣al (2019) Low‐dose IFNgamma induces tumor cell stemness in tumor microenvironment of non‐small cell lung cancer. Cancer Res 79: 3737–3748 [DOI] [PubMed] [Google Scholar]
- Stutz MD, Ojaimi S, Ebert G, Pellegrini M (2018) Is receptor‐Interacting protein kinase 3 a viable therapeutic target for Mycobacterium tuberculosis infection? Front Immunol 9: 1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D (2005) SV40‐encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 435: 682–686 [DOI] [PubMed] [Google Scholar]
- Sutavani RV, Phair IR, Barker R, McFarlane A, Shpiro N, Lang S, Woodland A, Arthur JSC (2018) Differential control of Toll‐like receptor 4‐induced interleukin‐10 induction in macrophages and B cells reveals a role for p90 ribosomal S6 kinases. J Biol Chem 293: 2302–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland TE, Logan N, Ruckerl D, Humbles AA, Allan SM, Papayannopoulos V, Stockinger B, Maizels RM, Allen JE (2014) Chitinase‐like proteins promote IL‐17‐mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat Immunol 15: 1116–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain Ket␣al (2011) The ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell 43: 432–448 [DOI] [PubMed] [Google Scholar]
- Vereecke L, Beyaert R, van Loo G (2009) The ubiquitin‐editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 30: 383–391 [DOI] [PubMed] [Google Scholar]
- Wang L, Wu J, Li J, Yang H, Tang T, Liang H, Zuo M, Wang J, Liu H, Liu Fet␣al (2020) Host‐mediated ubiquitination of a mycobacterial protein suppresses immunity. Nature 577: 682–688 [DOI] [PubMed] [Google Scholar]
- Wang L, Xia JW, Ke ZP, Zhang BH (2019) Blockade of NEAT1 represses inflammation response and lipid uptake via modulating miR‐342‐3p in human macrophages THP‐1 cells. J Cell Physiol 234: 5319–5326 [DOI] [PubMed] [Google Scholar]
- Wu H, Wang Y, Zhang Y, Yang M, Lv J, Liu J, Zhang Y (2015) TALE nickase‐mediated SP110 knockin endows cattle with increased resistance to tuberculosis. Proc Natl Acad Sci USA 112: E1530–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Guo Z, Yao K, Miao Y, Liang S, Liu F, Wang Y, Zhang Y (2016) The transcriptional foundations of Sp110‐mediated macrophage (RAW264.7) Resistance to Mycobacterium tuberculosis H37Ra. Sci Rep 6: 22041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau R, Rape M (2016) The increasing complexity of the ubiquitin code. Nat Cell Biol 18: 579–586 [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Naka T, Kubo M (2007) SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 7: 454–465 [DOI] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han JH (2009) RIP3, an energy metabolism regulator that switches TNF‐induced cell death from apoptosis to necrosis. Science 325: 332–336 [DOI] [PubMed] [Google Scholar]
- Zhao X, Khan N, Gan H, Tzelepis F, Nishimura T, Park SY, Divangahi M, Remold HG (2017) Bcl‐xL mediates RIPK3‐dependent necrosis in M. tuberculosis‐infected macrophages. Mucosal Immunol 10: 1553–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Source Data for Figure␣1D
Source Data for Figure␣3
Source Data for Figure␣5
Source Data for Figure␣6
Source Data for Figure␣7
Data Availability Statement
No primary data sets were generated and deposited.