

Abstract
Cilia are thin microtubule‐based protrusions of eukaryotic cells. The swimming of ciliated protists and sperm cells is propelled by the beating of cilia. Cilia propagate the flow of mucus in the trachea and protect the human body from viral infections. The main force generators of ciliary beating are the outer dynein arms (ODAs) which attach to the doublet microtubules. The bending of cilia is driven by the ODAs' conformational changes caused by ATP hydrolysis. Here, we report the native ODA complex structure attaching to the doublet microtubule by cryo‐electron microscopy. The structure reveals how the ODA complex is attached to the doublet microtubule via the docking complex in its native state. Combined with coarse‐grained molecular dynamic simulations, we present a model of how the attachment of the ODA to the doublet microtubule induces remodeling and activation of the ODA complex.
Keywords: cilia, cryo‐electron microscopy, doublet microtubule, outer arm dynein
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Structural Biology
Cryo‐EM structure of the outer dynein arm (ODA) complex attached to the doublet microtubule combined with the coarse‐grained molecular dynamics (MD) simulation reveals the active conformation of the ODA and the activation mechanisms of the ODA complex.

Introduction
Motion is an important aspect of life. In eukaryotes, cilia and flagella are responsible for cell motility. These microscopic hair‐like organelles bend several tens of times per second to generate fluid flows. Cilia in trachea generate the flow of mucus and protect our body from infectious agents such as viruses. There is a canonical axonemal 9 + 2 structure where the central pair microtubules are surrounded by nine doublet microtubules (Fig 1A). The propulsive force generators of cilia and flagella are the axonemal dyneins. As the molecular motors, the axonemal dyneins drive the sliding of doublet microtubules, which is then converted into the bending of the cilium. The axonemal dyneins consist of outer arm dyneins and inner arm dyneins. The outer dynein arm (ODA) regulates the beat frequency while the inner arm dyneins are important for the waveform of the cilium (Brokaw & Kamiya, 1987). Improper assembly of the ODA complex causes ciliopathies in humans (reviewed in Reiter & Leroux, 2017).
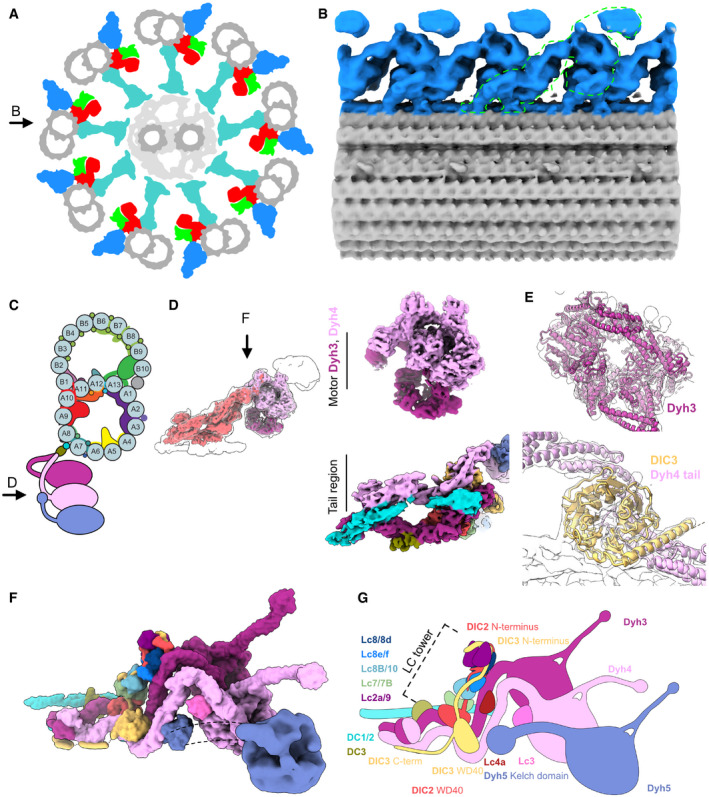
Figure 1. Cryo‐EM structure of the ODA complex on the doublet microtubule.
-
ASchematic diagram of axoneme structure of cilia viewed from the base of the cilia. Doublet microtubule: gray; outer dynein arm: blue; inner dynein arm: red; dynein regulatory complex: green; radial spokes: cyan. Black arrow indicates the view in (B).
-
BThe 24‐nm structure of the doublet microtubule from K40R mutant filtered to 18 Å showing the row of the ODA. The green outline indicates the single ODA complex.
-
CA schematic cartoon of the doublet microtubule and the ODA complex. Arrow indicates the view in (D).
-
DThe focused refined maps of the tail and the heads of the ODA are shown within the map of the entire ODA complex (left). The focused refined maps of Dyh3 and Dyh4 heads and the tail (right).
-
EFitting of models into maps (Dyh3, top; DIC3 and Dyh4 tail, bottom). The α‐helix part of the DIC3 was more structured in our map compared with that of the Shulin–ODA complex.
-
F, GThe surface render of the model (F) and the schematic cartoon (G) of the ODA with the docking complex. The Dyh5 is too flexible to resolve well by cryo‐EM. The Dyh5 head is only shown as the 18‐Å resolution surface render, and the Dyh5 tail is drawn as dotted lines. All the components (ICs, LCs, and DCs) are colored and indicated and will be used consistently in all the figures. C‐terminal side of the DIC3 has more structured region along the Dyh4 HC tail on the doublet.
Unlike cytoplasmic dyneins which walk on microtubules while carrying cargos, axonemal dyneins are anchored firmly on the doublet microtubules (Goodenough & Heuser, 1982; Bui et al, 2008, 2009). The ODA complex is composed of two or three heavy chains (HCs) depending on the species (Diamant & Carter, 2013), two intermediate chains (ICs) and numerous light chains (LCs). For example, human cilia have two‐headed ODA (DNAH5 and DNAH9) while ODA of Tetrahymena contains three heavy chains (Dyh3, Dyh4, and Dyh5). Dynein HCs are the most important force‐producing components, and each HC has a head domain and a tail domain. The three head domains are coupled together at the level of the tail domain. Within the head domain of each ODA HC, there is an AAA+ ring (composed of AAA1 to AAA6 subdomains) where ATP is hydrolyzed to exert force. The ODA complex is stably attached to the A‐tubule of the doublet microtubule and produces force while interacting with the B‐tubule of the neighboring doublet microtubule. The docking of the ODA complex to the A‐tubule of the doublet is mediated by the HC tail domain. The interaction with B‐tubule of the neighboring doublet is mediated by the microtubule‐binding domain (MTBD) at the tip of the stalk which extends from the AAA+ ring. ICs and LCs have various regulatory roles (reviewed in King, 2012). Most of the ICs and LCs interact with the tail domain. However, LC1 is bound to the MTBD of the Dyh3 head (Ichikawa et al, 2015; Toda et al, 2020).
By conventional quick‐frozen deep‐etch replicas and cryo‐electron tomography (cryo‐ET) works, ODAs were shown to form a 24‐nm repeating row on the doublet microtubules (Goodenough & Heuser, 1982; Nicastro et al, 2006; Bui et al, 2012; Lin & Nicastro, 2018). The ODA complex is proposed to be attached via the docking complex (DC) (Owa et al, 2014; Oda et al, 2016a). Within each ODA complex, the three heads are aligned parallel to each other so that all three head domains can interact with the adjacent doublet microtubule in a proper orientation. The architecture of ODA complex has been studied by negative staining EM using purified ODA complexes (Ichikawa et al, 2015) or cryo‐electron tomography (cryo‐ET) of the intact axoneme (Oda et al, 2016b). However, the resolutions were limited in these studies and the modeling of the protein subunits was not possible. Recently, a cryo‐EM structure of the inactive Tetrahymena ODA complex (before its incorporation into cilia) was obtained (Mali et al, 2021). In the inactive form, three head domains are packed together by a regulator protein, Shulin. This conformation is markedly different from the active conformation observed in the axoneme where the three heads are in parallel arrangement. The subunit architecture was also revealed in high resolution for the inactive Shulin–ODA complex (Mali et al, 2021). However, to understand how ODA is incorporated into the axoneme structure and activated, it was crucial to obtain a high‐resolution structure of the ODA complex in the context of the doublet microtubule.
Here, we revealed the Tetrahymena ODA complex on the doublet microtubule at 5.5–7 Å resolutions. Our structure showed in detail how the Tetrahymena ODA complex is attached to the doublet, including its interaction with the DC complex. Very recently, high‐resolution structure of Chlamydomonas ODA complex attached to the doublet microtubule was also reported by cryo‐EM (Walton et al, 2021). This enabled us to compare the ODA structure from different species. Combined with coarse‐grained molecular dynamics (MD) simulations, we have revealed how the ODA complex undergoes an activating rearrangement when it is docked onto the doublet microtubule.
Results
Cryo‐EM structure of the ODA attached to the doublet microtubule
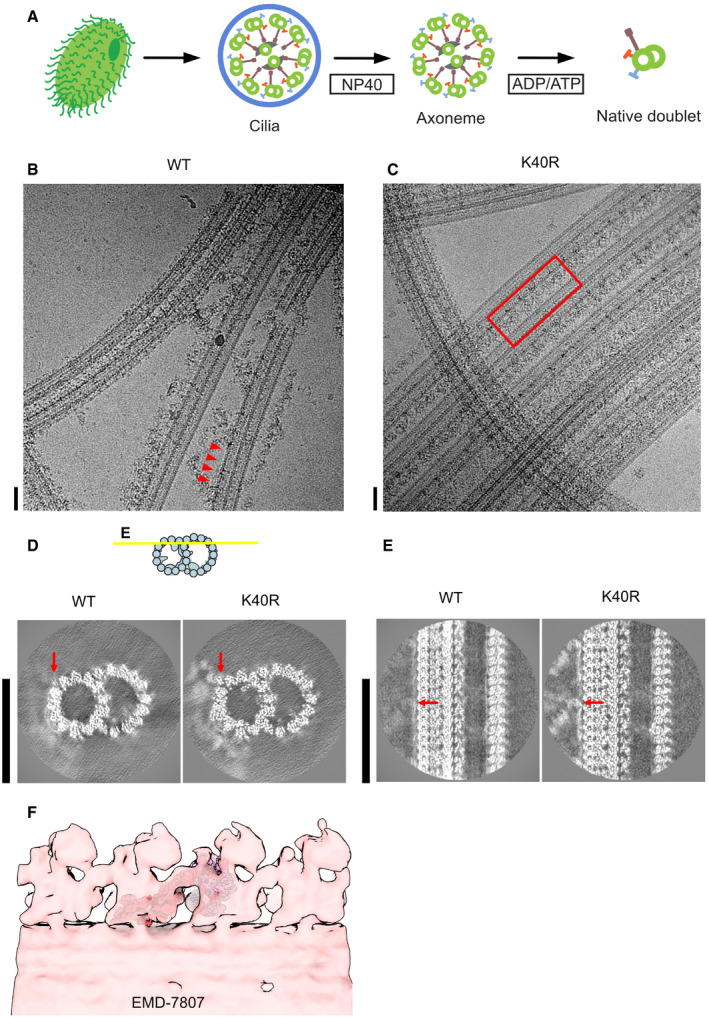
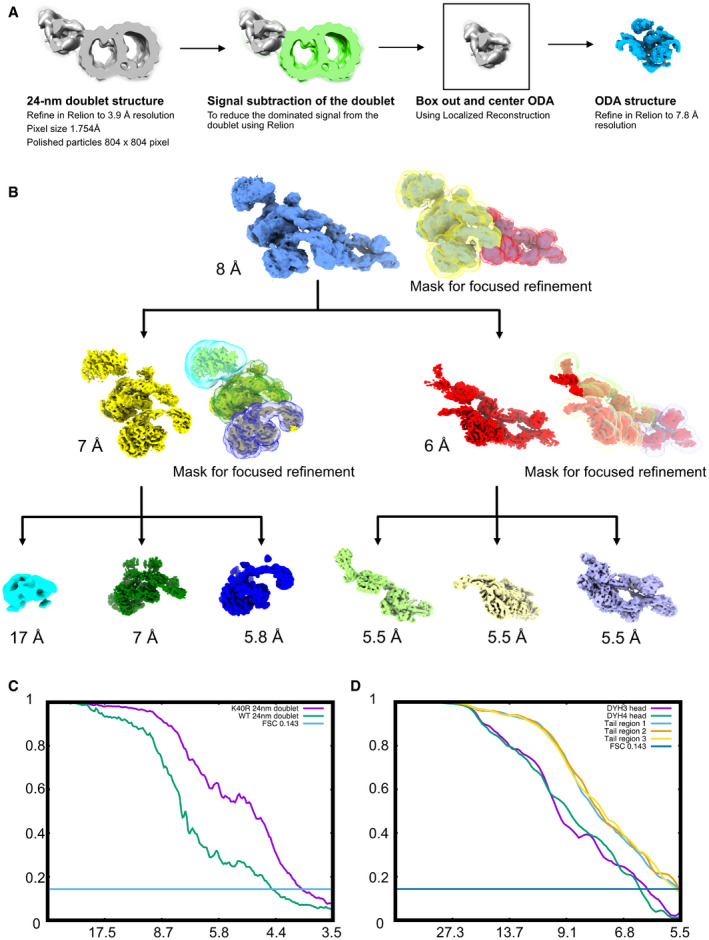
In our previous studies of the doublet microtubule structures, the ODA complexes were removed by high‐salt treatment from doublets (Ichikawa et al, 2017; Ichikawa et al, 2019; Khalifa et al, 2020). Here, to obtain the native structure of the ODA complex on the doublet, we tried to obtain a wild‐type (WT) Tetrahymena doublet microtubule structure without a salt wash (Fig EV1A). The individual doublet microtubules were separated from the rest of the axoneme by induction of microtubule sliding using ATP. However, in the disintegrated WT axonemes, the ODAs tended to detach from the doublet (Fig EV1B) and we were not able to obtain the high‐resolution structure of the ODA by single particle analysis (Fig EV1D and E). We noticed that the sliding of doublets in the ATP‐reactivated K40R (Gaertig et al, 1995) or MEC17‐KO mutant Tetrahymena axonemes (Akella et al, 2010) was less efficient as compared to the WT Tetrahymena axonemes, possibly due to different levels of post‐translational modifications of tubulin. This enabled us to obtain cryo‐EM images of partially split doublet microtubules with attached ODA complexes (Fig EV1C). Using the cryo‐EM images, we first obtained a 24‐nm doublet microtubule unit structure as previously described (Ichikawa et al, 2017; Ichikawa et al, 2019; Khalifa et al, 2020) at a 3.9 Å resolution (Fig EV2A). However, the resolution of the ODA part was not high enough due to its flexibility. Therefore, we performed signal subtraction of the doublet and obtained the ODA structure with a 7.8 Å resolution. From here, different regions of the ODA were further revealed by focused refinement. The tail region's resolution was improved to 5.5 Å, the Dyh3 head part to 5.8 Å, and Dyh4 head part to 7 Å (Fig EV2B). The resolution of the Dyh5 head remained at 17 Å since it was furthest from the doublet and more flexible. The head domains were in a parallel configuration (Fig 1B) as previously shown by cryo‐ET studies (Bui et al, 2008; Lin & Nicastro, 2018). Our structure fits well with previous cryo‐ET structure of the intact axoneme, thereby confirming it is a physiological structure of the ODA complex (Fig EV1F).
Figure EV1. Sample preparation and cryo‐EM of the doublet microtubules.
-
AThe schematic of our isolation strategy for the intact doublet microtubules.
-
B, CMicrographs of doublet microtubules from Tetrahymena WT (B) and K40R mutant (C). The red arrowheads indicate the ODA complex falling off from the doublet. The red rectangle indicates the row of intact ODA in the K40R mutant. Scale bars represent 50 nm.
-
D, E24 nm structure of doublet from WT and K40R showing the DC is intact in both cases while ODA is clearly present only in K40R. Red arrows indicate the docking complex. Scale bars, 50 nm.
-
FFitting of our high‐resolution structure into the tomographic map of Tetrahymena showing it is physiological (ΔRib72B mutant rescued with Rib72B‐GFP) (EMD‐7807, Stoddard et al, 2018).
Figure EV2. Cryo‐EM processing strategy.
-
AAlignment strategy for ODA particles. First, we obtained the 24‐nm doublet structures. After that, we performed signal subtraction of the doublet microtubule. We centered and boxed out the ODA particles and performed refinement of the entire ODA particles.
-
BFocus refinement strategy for different regions of the ODA complex. The Dyh5 seems to be too flexible; therefore, we can only obtain Dyh4 head at 17 Å resolution.
-
CFourier Shell Correlation of the doublet of WT and K40R & MEC17 combined data.
-
DFourier Shell Correlation of the different regions of the ODA complex by focus refinement.
Using the recent Shulin–ODA complex structure from Tetrahymena as a starting model, we were able to build in all the components of the ODA complex except for Dyh5's head (Fig 1C–G). Since Dyh5 is not conserved in vertebrates (Lin et al, 2014; Ueno et al, 2014), an atomic model for conserved part of the ODA complex on the doublet was obtained. The ICs and LCs were also assigned to the density map. The majority of LCs were forming the LC tower as earlier observed in the Shulin–ODA complex (discussed below). Compared with the Shulin–ODA complex, there were some parts of ICs which were structured in our map and modeled (Fig 1E–G). There was a density not observed in the Shulin–ODA complex associated with Dyh4, and LC4 was tentatively assigned to this region (Fig 1F and G). Very recently, Tetrahymena ODA model based on cryo‐EM structure of reconstituted ODA array on the doublet was reported (preprint: Rao et al, 2020). Our model was similar to obtained reconstituted ODA complex validating our model. When we compare our Tetrahymena ODA structure with recent Chlamydomonas ODA structure attached to the doublet (Walton et al, 2021), both structures appeared strikingly similar in general (Fig EV3). In our structure, there was also an additional density of two segments of coiled coil with a globular domain at the end which is running along the tail domain of the ODA (cyan parts in Fig 1D, F, and G) (discussed later).
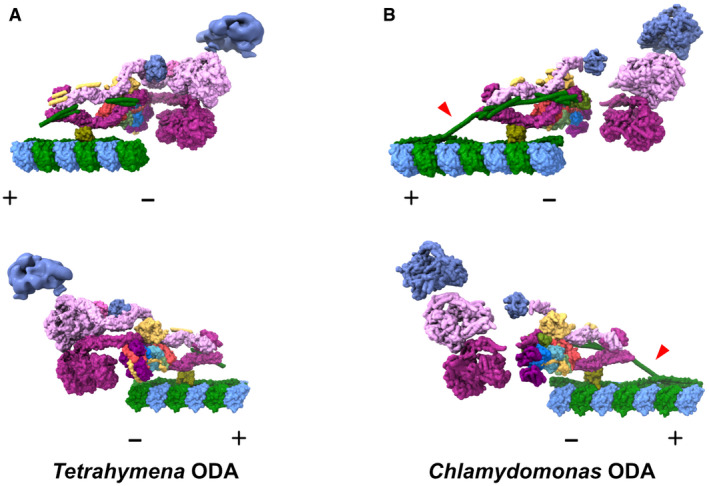
Figure EV3. Comparison of the ODA structures obtained by cryo‐EM.
-
A, BThe model of our Tetrahymena ODA complex (A) and Chlamydomonas ODA complex (PDB ID: 7KZM, Walton et al, 2021) (B) viewed from different sides. The Dyh5 head of Tetrahymena ODA is shown as 18‐Å resolution surface rendering. α‐Tubulin is shown in green and β‐tubulin is shown in blue. The polarities of the doublet microtubules are indicated by + and −. In our Tetrahymena model, there is more modeled region around the linker region. In contrast, there is more modeled region in DC in the Chlamydomonas model from Walton et al (2021), especially the region connecting the DC1/2 on the doublet surface and the extended coiled‐coil region associated with the ODA complex (red arrowheads). These differences could relate to different properties of the Tetrahymena and Chlamydomonas ODA or different ways of sample preparation.
Attachment of the ODA complex to the doublet via the docking complex
The part of DC on the doublet was also visualized in our structure which allowed us to understand how the ODA complex is attached to the doublet (Figs 2 and EV4A). We identified Tetrahymena proteins Q22T00, Q233H6, and I7M2C6 as DC1/2 and 3 following the names of the counterparts of Chlamydomonas (Fig EV4B and C, see Materials & Methods for details). Along the doublet, DC1/2 forms a 24‐nm repeating unit, the same periodicity as the repeating unit of the ODA complex. DC3 is associated with the DC1/2 coiled‐coil region on the doublet serving as a marker for the ODA attachment (Figs 2C and EV4A) as previously proposed (Ma et al, 2019). The α‐helix bundle 3 of Dyh3 HC was interacting with the DC3 (Fig 2C). The tethering of the ODA to the doublet microtubule is mediated by Dyh3 alone, and ICs/LCs are not involved with the microtubule. The DC1/2 also appears to connect to the coiled‐coil density with a gap associated with the neighboring ODA tail domain (dashed line in Figs 2A and EV4H). This model is consistent with the coiled‐coil prediction of DC1/2 with a gap (Fig EV4B) and the previous tagging results of DC2 of Chlamydomonas reinhardtii by cryo‐ET in Oda et al (2016a) (Fig EV4, EV5). Therefore, the density associated with the tail domain corresponds to the C‐terminal side of DC1/2 (Fig EV4I and J). This is also consistent with recent Chlamydomonas ODA structure model (Fig EV3).
Figure 2. Interactions between the ODA complex and the DC.
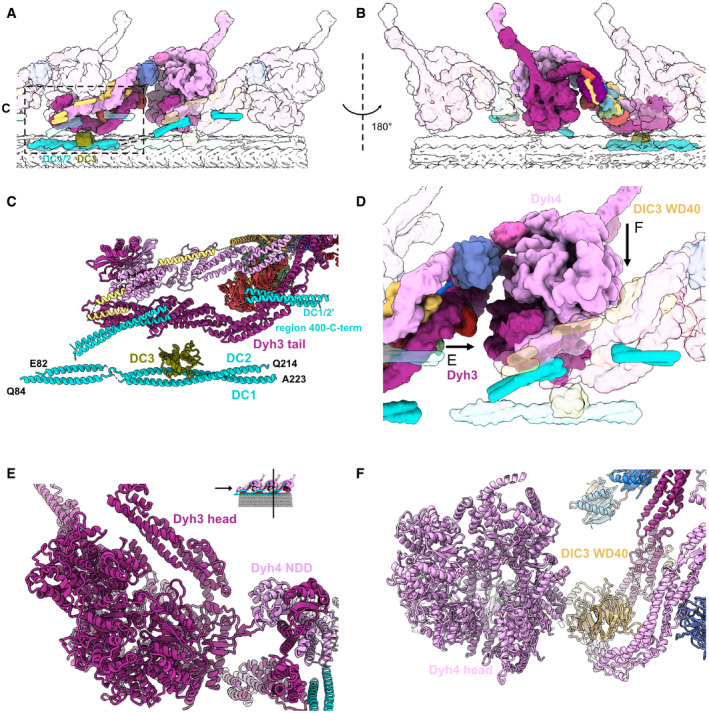
-
A, BThe stacking of the ODA complex in the axoneme viewed from inside (A) and outside (B) the cilia. All the components belonging to the middle ODA complex are shown in color. The proximal and distal ODA complexes are shown in transparent.
-
CModel of the DC and the tail of the ODA.
-
DThe interaction of the head of the ODA complex with the tail of the next ODA complex unit. Dyh3 head interacts with the dimerization domain NDD of the proximal Dyh4. The Dyh4 head interacts with the DIC3 WD40 of the proximal ODA complex. Arrows indicate the views in (E) and (F).
-
EModel view of the interaction between the Dyh3 head with the dimerization domain NDD of the proximal ODA complex.
-
FModel view of the interaction between the Dyh4 head with the DIC3 WD40 of the proximal ODA complex.
Figure EV4. Modeling of the docking complex.
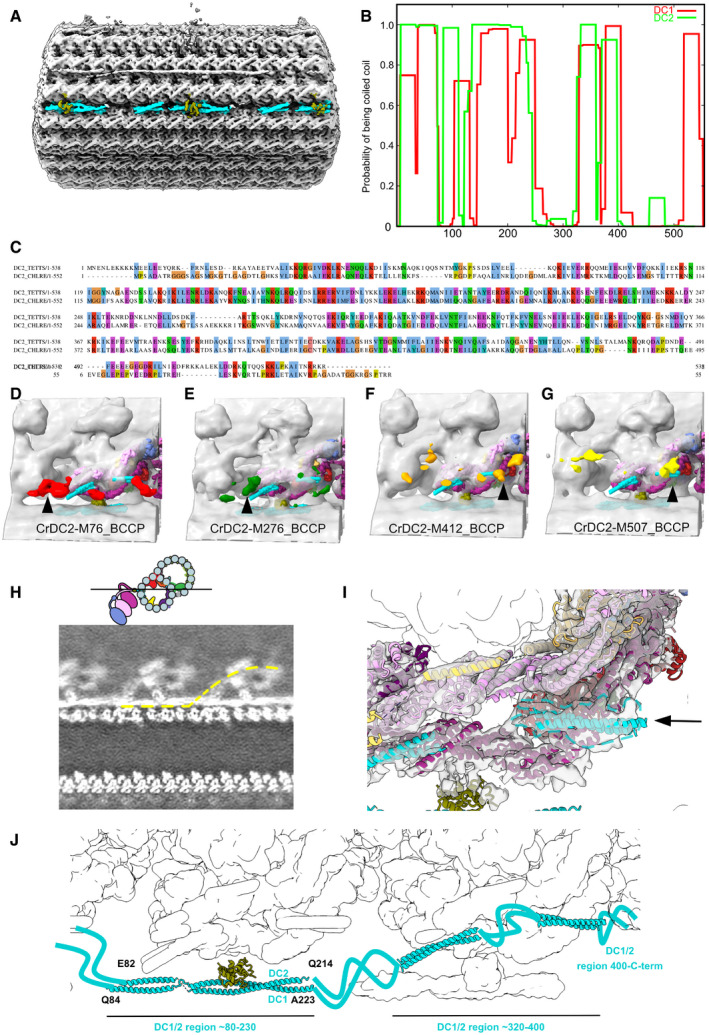
-
AThe DC density on the doublet microtubule between PFs‐A7 and A8.
-
BPrediction of coiled coil for DC1 and DC2 using COILS with a window size of 28 (Lupas et al, 1991).
-
CSequence alignment of Chlamydomonas DC1 and Tetrahymena CCDC151 homolog (Q22T00).
-
D–GStructures of Chlamydomonas ODA reconstituted on microtubules with biotin carboxyl carrier protein (BCCP) tagged in different regions of DC2 (residue 76, 276, 412, and 507) from Oda et al (2016a) (EMD‐6508, 6509, 6510, 6511). The enhanced signals of BCCP‐tag are indicated in colors.
-
HSlice from a density map showing the docking complex (position indicated in the cartoon). The yellow line indicates one continuous DC1/2.
-
IThe globular density at the end of the coiled coil of DC1/2 (black arrow and dotted line).
-
JThe model of the DC based on our analysis.
Figure EV5. Data related to the remodeling of the ODA complex.
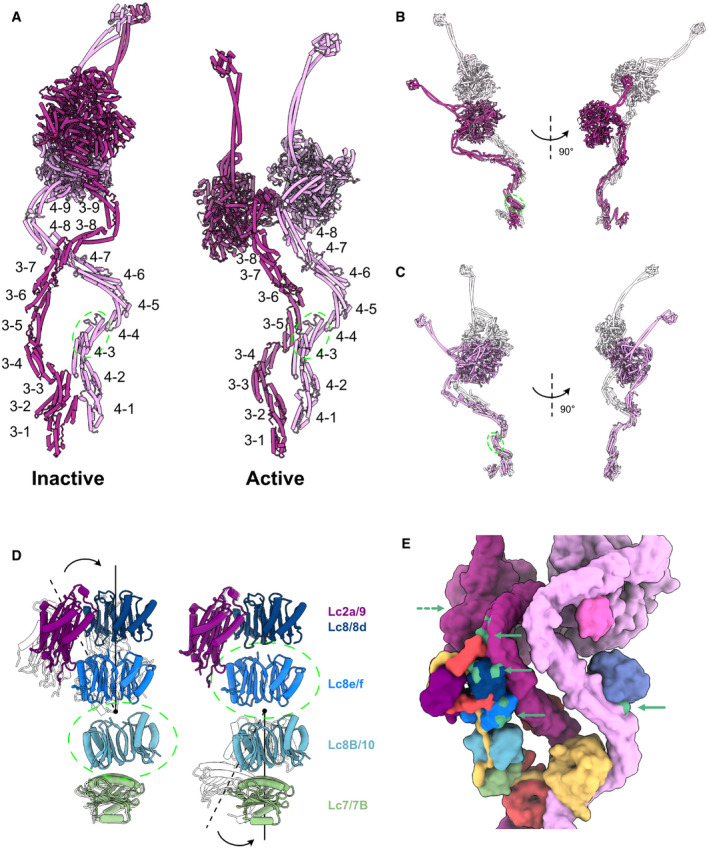
-
AThe labeling of helix bundles in the inactive and active Dyh3 and Dyh4. Inactive and active structures are aligned on helix bundle 4 of Dyh4 (residues 414–513).
-
BAlignment of the inactive and active Dyh3 at helix bundle 3 (residue 448–536) showing ˜ 90‐degree rotation of the head domain (top).
-
CAlignment of inactive and active Dyh4 at helix bundle 3 (residue 414–513) showing compressing conformational changes (bottom).
-
DBending conformational change in the LC tower. LC tower from the active ODA complex is in colors, and the LC tower from the Shulin–ODA is shown in transparent. LC towers are aligned based on either Lc8B/10 (left) or Lc8e/f (right) as indicated by green dashed circles.
-
ERegions of the ODA that interact with Shulin in the inactive conformation (green regions with green arrowhead) are spread out in the active conformation. The dash arrow indicates the region of Dyh3 head interacting with C3 domain of Shulin, now at the back of the view.
The ODA complex is anchored to the doublet by the above‐mentioned two sites: DC1/2 and DC3. Over these docking sites, there is a layer composed of Dyh3 HC, DIC2's WD domain, LC tower, and the C‐terminal side of DC1/2. Another layer of Dyh4 and DIC3 is further built onto this layer (Fig 2A and B). The three head domains are stacked together in a parallel configuration. The stacking of both the tail and the head domains is important for the proper attachment of the ODA onto the doublet.
While the ODA complex is not directly in contact with the doublet microtubule in its native state, ODAs can be reconstituted on preassembled singlet microtubules or doublets lacking DCs with 24‐nm periodicity (Ueno et al, 2008; Owa et al, 2014). Our structure suggests that the ODA can be reconstituted on the microtubule without DCs by the interaction of the ODA heads with the proximal neighboring ODA tail domains (Figs 1B and 2D–F). More specifically, the head of Dyh3 seems to interact with the N‐terminal dimerization domain (NDD) of the proximal ODA Dyh4 tail while the Dyh4 head interacts with DIC3 WD40 domain of the proximal ODA. These interactions probably enable the spontaneous alignment and stacking of the ODA complexes on the doublet microtubule without the help of the DCs. Similar tail‐to‐head interaction was recently observed in reconstituted ODA array on the doublet microtubule (preprint: Rao et al, 2020). Since our structure is the native structure of ODA on the doublet, our structure validated their model of the interaction. The interactions between head and tail of neighboring ODA complexes were also observed in Chlamydomonas ODA (Walton et al, 2021).
Remodeling of the tail and the head
It has been recently proposed that the ODA complex, before its assembly on the doublet, is in an inactive form with the regulatory protein Shulin (Mali et al, 2021). To understand how ODA rearranges into an active form on the doublet, we compared the structures of the Shulin–ODA complex with our ODA complex on the doublet (Figs 3 and EV5). The overall appearance of the active ODA complex on the doublet was quite different from that of the inactive Shulin–ODA complex, which has a more elongated conformation (Fig 3A). The elongated conformation of the inactive Shulin–ODA complex stems from the positions of the motor domains which are fixed in pre‐powerstroke conformations (discussed later). The most unchanged region was the base region of the Dyh4 HC up to the bundle 4 region where the DIC3's WD40 domain resides (Figs 3B and EV5A). Dyh3 HC, DIC2, and LC tower were rotating together about ˜ 90 degrees relative to the Dyh4's base region (Fig 3B). With this configuration change, Dyh3 and Dyh4 HCs are no longer crossed to each other and freed. In the active conformation, DIC2's WD40 domain interacts with Dyh4 HC, and the WD40 domain of DIC3 is docked onto Dyh3 HC. Within the HCs, there were also conformational changes. The Dyh3 showed more change in configuration with ˜ 90‐degree rotation of the head compared with the inactive form whereas Dyh4 HC showed rather a compressing movement of the head side compared with Shulin–ODA structure (Fig EV5A–C). These conformational changes are mediated by hinge‐like motions between the α‐helical bundles (Fig 3C). The center region (bundles 4–8) of Dyh3 showed a milder conformational change compared with other regions of the Dyh3 (Fig 3C) since the LC tower is associated with this region. In detail, there is a slight change in conformation of both Dyh3 and LC tower (Figs 3D and EV5D). The conformational change was due to the association of α‐helix bundle 8 of the Dyh3 with LC8/8d. This interaction was hindered in the Shulin–ODA complex since Shulin is keeping Dyh3's bundle 8 away from LC8/8d. These tail rearrangements align the head domains to parallel configuration on the doublet (discussed later).
Figure 3. Comparison of the active ODA complex structure on the doublet and the inactive Shulin–ODA complex structure.
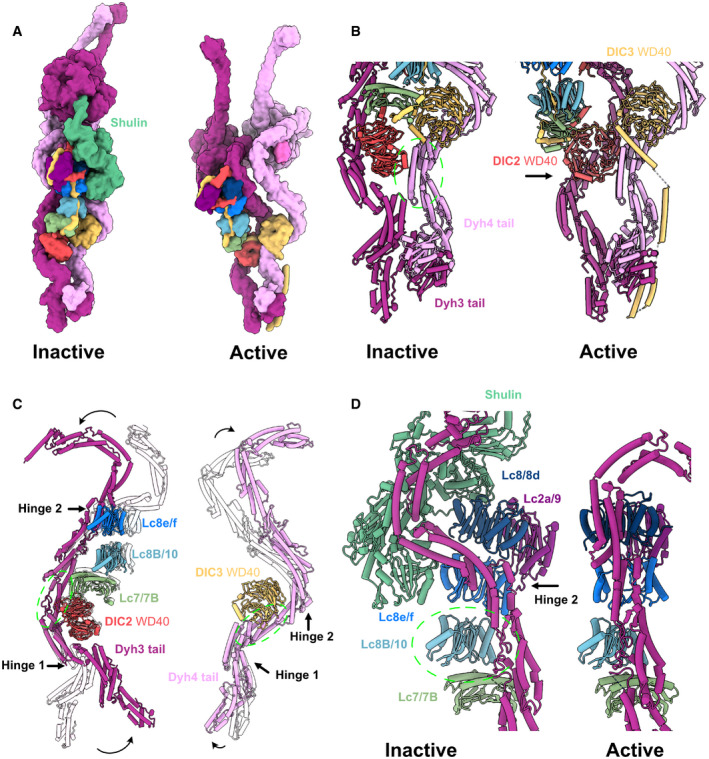
-
AThe surface rendering of the inactive ODA model (with bound Shulin in green) and the active ODA model. Dyh5 is not shown.
-
BRemodeling of Dyh3 and Dyh4 tails and associated IC/LCs. The green circle indicates the region of alignment (Dyh4 residues 414–513, helix bundle 3). Dyh4 tail does not exhibit large conformational change while Dyh3 rotates almost 90 degrees, evident by the rotation of DIC2 WD40 domain.
-
CSuperimposition of Dyh3 and Dyh4 tails between the inactive (transparent) and the active ODA (Dyh3 is aligned based on res. 535–646, helix bundle 5 while Dyh4 is aligned on res. 414–513, helix bundle 4). The Dyh3 tail exhibits two hinges with larger rotations while the Dyh4 tail also exhibits two hinges with smaller rotations.
-
DThe changes in interaction between Dyh3 tail and the LC tower due to the remodeling of inactive (with Shulin in green) to active conformations (two structures are aligned based on Lc8b/10). The conformations of Dyh3 bundle 8, Lc8/8d, and Lc2a/9 are significantly different. The release of the Shulin leads to the rotation of the Dyh3 tail, which causes the different interaction pattern between Dyh3 and LC8/8d.
Head domain structure of the ODA on the doublet
In the Shulin–ODA complex, the heads are fixed in the pre‐powerstroke positions (Fig 4A). In our active ODA complex structure, the linkers are in the post‐powerstroke configuration both in the Dyh3 and Dyh4 heads. Therefore, the heads are ready to go through transition to next ATPase cycle with Shulin detached. Linker of the Dyh3 head was in a canonical post‐powerstroke conformation (Fig 4A). As for the Dyh4 head, LC3 was docked onto the AAA+ ring in the post‐powerstroke configuration of the linker. LC3 was interacting with AAA4 of the binding position of the Dyh4 AAA+ ring. However, the binding scheme was different from that of Lis1 which also binds to the AAA3 of cytoplasmic dynein (Toropova et al, 2014) (Fig 4B). LC3 still interacts with AAA4 but it was closer to AAA3. LC1 was previously shown to bind to the MTBD of Dyh3 head (Ichikawa et al, 2015; Toda et al, 2020). LC3 is the second component found to be associated with the head domain of the ODA complex. Since LC3 was interacting with both linker and the AAA+ ring, we carefully examined the linker position of Dyh4. The linker was going at the middle of the apostate and the ADP state and slightly leaning toward AAA4 (Fig 4C). This is a novel configuration of dynein linker. To perform powerstroke, LC3 is thought to be detached from the AAA+ ring (Fig 4D).
Figure 4. Structure of the head domain in ODA on the doublet.
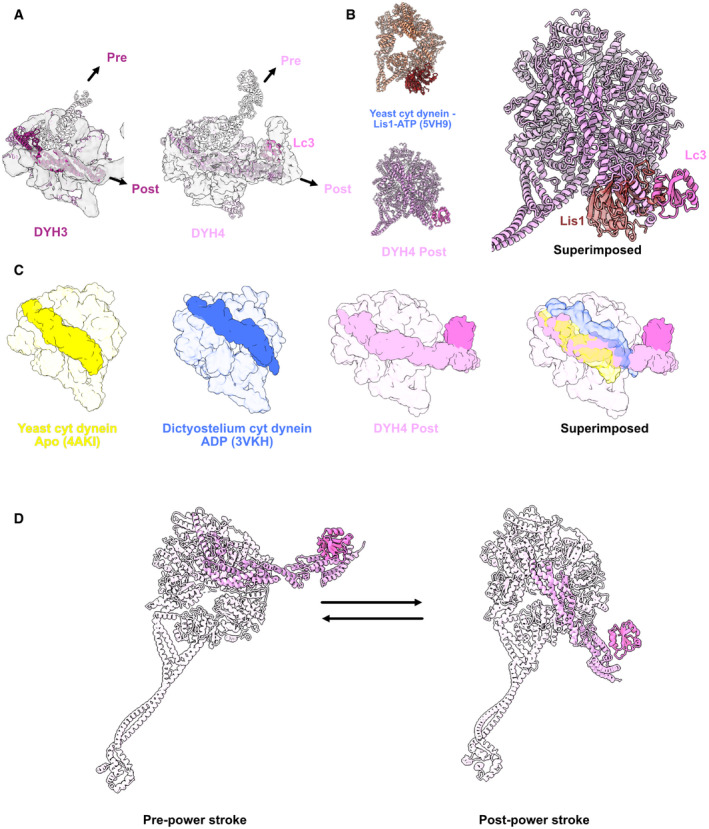
-
AModels of the Dyh3 and Dyh4 heads in post‐powerstroke conformations are fitted in the cryo‐EM maps of the ODA on the doublet. The Dyh3 and Dyh4 linkers in pre‐powerstroke conformations in the inactive Shulin–ODA model are also superimposed (shown in transparent).
-
BComparison of yeast cytoplasmic dynein bound to Lis1 (PDB: 5VH9) with the Dyh4 bound to LC3. LC3 also binds to the AAA+ ring but in an opposite site from Lis1.
-
CComparison of the linker of Dyh4 in post‐powerstroke conformation with the yeast cytoplasmic dynein in apostate and dictyostelium cytoplasmic dynein in ADP state reveals that the Dyh4 linker is positioned in between of the linker positions in apo and ADP states. The structures are aligned based on the AAA+ ring without linker.
-
DModel for the conformation changes from pre‐powerstroke to post‐powerstrokes in Dyh4.
Docking of the ODA complex to the DC induces the remodeling
How the regulatory protein Shulin detaches from the ODA complex? When Shulin–ODA structure was docked onto the doublet microtubule, the Shulin region did not show a steric clash (Fig 5A). Therefore, we then examined the Shulin‐binding sites in the ODA complex on the doublet. Even though there was no other structure covering the Shulin‐binding sites, the regions involved in the interaction with Shulin were dispersed due to the rearrangement of the tail domain as well as the head domains (Fig EV5E). From these results, the remodeling of the ODA itself likely causes the detachment of Shulin. To understand the cause of the remodeling of the ODA complex, we focused on the region where ODA binds to the doublet. As mentioned above, the base region of the Dyh4 tail did not change significantly (Fig 5A). In contrast, the tail region of Dyh3 remodeled and was located closer to the doublet (Fig 5A and B). This remodeled region coincides with the part which interacts with DC3. In addition, the remodeled Dyh3 interacted with extended coiled‐coil region of DC1/2 (Fig 5C). The interaction between the head and the neighboring tail also thought to pull the Dyh3 HC.
Figure 5. Remodeling of the tail domain and detachment of Shulin.
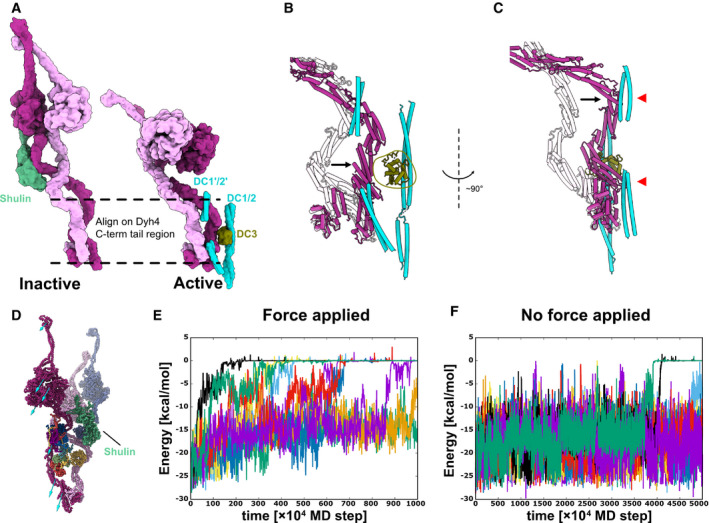
-
AModels of the inactive and the active ODA. Dyh3 and Dyh4 parts from the models are shown with Shulin and docking complex, respectively. The two structures are aligned based on the Dyh4 residues 414–513 (helix bundle 3).
-
B, CDetailed comparison of the Dyh3 before and after the remodeling. With the two structures aligned as in (A), Dyh3 tail in the active conformation indicates significant shift toward DC3 (B). Similarly, the Dyh3 tail in the active conformation indicates a significant shift toward the extended coiled‐coil region of DC1/2 (˜ 320–400 aa). Red arrowheads indicate the interaction sites of Dyh3 with the extended coiled‐coil region of DC1/2.
-
DCoarse grain molecular dynamics of the inactive Shulin–ODA complex. External force was applied in the direction of the arrow to simulate the remodeling of Dyh3 upon binding to the doublet microtubule. Shulin is shown in green.
-
E, FEnergy trajectories between Shulin and the ODA complex with (E) and without (F) external force applied to Dyh3. 8 out of 10 setups showed detachment of Shulin (energy = 0) with external force in 1,000 × 104 MD steps, which roughly corresponds to 10 μs. (E). In contrast, only two out of 10 trajectories showed Shulin detachment without applied force even with 5,000 × 104 MD steps, which approximately corresponds to 50 μs. (F). It should be noted that our coarse‐grained MD simulation is performed in the condition where Shulin binding to the ODA complex is weakened, but it still showed the difference in detachment rate of Shulin in response to the global conformational change in the ODA complex.
To test whether the remodeling of the Dyh3 HC causes the detachment of Shulin, we performed a coarse‐grained MD simulation. Here, coarse‐grained MD simulation was chosen over all‐atom simulation for below reasons. First, the dynein is a huge protein complex, and therefore, the all‐atom simulation was unfavorable. Previous all‐atom simulation has shown dynein molecule's stability and flexibility (Kamiya et al, 2016; Can et al, 2019); however, it is difficult to track conformational change in dynein complex by all‐atom simulation. Secondly, previous coarse‐grained MD simulation has successfully captured the conformational change in cytoplasmic dynein, and it clarified the allosteric conformational change cascade of dynein’s motor domain (Kubo et al, 2017; Goldtzvik et al, 2018). Lastly, we intended to understand the relationship between the global conformational change in the ODA and detachment of Shulin rather than the details of the residues. Thus, we have performed a coarse‐grained MD simulation in the presence of external force. External force was applied to Dyh3 of the Shulin–ODA complex to mimic the remodeling of Dyh3 (Fig 5D, see Materials & Methods for the details). The coarse‐grained MD simulation was performed from 1,000 × 104 to 5,000 × 104 MD steps. This time scale was chosen since the allosteric conformational change in dynein was captured with 1,000 × 104 MD steps in the previous MD simulation using AICG2+ (Kubo et al, 2017). Though the correspondence of MD step with the real time is weak, one MD step roughly corresponds to ˜ 1 ps. Thus, each trajectory corresponds to approximately 10–50 μs. To see the detachment of Shulin in these time scales, the interaction between Shulin and the ODA complex was weakened (see Materials and Methods for the details). In the presence of applied force, in eight out of ten MD trajectories, Shulin detached within 1,000 × 104 MD steps (Fig 5E, Movie EV1). In contrast, without external force applied, only two out of ten trajectories showed detachment within 5,000 × 104 MD steps, which is a five times longer time frame than the case with external force (Fig 5F). Therefore, the attraction by DCs and the adjacent ODA is likely to be important for Shulin detachment.
Discussion
Our structure presents a physiological binding state of the Tetrahymena ODA to the doublet via the DC complex in an unprecedented resolution. The Tetrahymena ODA complex is docked onto the doublet by two sites via DCs. In addition, the ODA interacts with the neighboring ODA complex. These interactions are crucial for the stable attachment of the ODA to the doublet and therefore force generation by the ODA complex within cilia. The ODA is bottomed up by DC “heals” so that it can associate with the neighboring doublet properly. In different species, DCs might tune the ODA interaction schemes. DC3 in Tetrahymena and Chlamydomonas contains several EF‐hand motifs. We speculate that the interaction of DC3 to the ODA can change based on the intracellular calcium ion concentration, leading to the regulation of ODA activity. In Chlamydomonas, high concentration of calcium ions is proposed to induce a symmetric waveform instead of the normal asymmetric breaststroke waveform (Bessen et al, 1980). The docking sites of the Tetrahymena ODA to the DC1/2 and 3 were similar to those of Chlamydomonas ODA (Walton et al, 2021). In contrast, the density connecting the DC1/2 regions on the doublet and the ODA complex was more prominent in Chlamydomonas ODA (Walton et al, 2021) (red arrowheads in Fig EV3B). The coiled‐coil density of this region in Tetrahymena ODA was weaker, and we could not model this region (Fig EV3A). This could reflect the different properties of the DCs from different species.
DC3 is not conserved in vertebrates. In humans, the DC assembly affects CCDC114, CCDC151, and ARMC4 (Hjeij et al, 2014). Therefore, ARMC4 might act as DC3 in humans. However, ARMC4 (1,044 aa) is a significantly larger protein than DC3 (309 aa in Tetrahymena). Comparing the tomography maps of human cilia and Tetrahymena, the human ODA also docks on the doublet at two sites (Appendix Fig S1B) (Lin et al, 2014a; Stoddard et al, 2018). However, both docking sites are different from the Tetrahymena ODA. The docking site close to Tetrahymena DC3 docking site is shifted (Appendix Fig S1B), while the other docking site attaches to PF‐A7, instead of attaching to the DC1/2 between PFs‐A7 and A8 as is the case in Tetrahymena. In humans, mutations leading to early termination or frameshift of CCDC151 (p.Glu109X, p.Glu309X, and p.Ser419X) and CCDC114 (p.Ala248Serfs52 and p.Ser432Argfs*7) cause primary ciliary dyskinesia (Knowles et al, 2013; Hjeij et al, 2014; Zhang et al, 2019) (Appendix Fig S1A). These mutations map to both the regions of the DC1/2 of Tetrahymena‐binding site to the doublet and also binding to the Dyh3 tails as proposed by our structures. This implies the importance of the extended coiled‐coil region of DC1/2 for the assembly of the ODA and for the remodeling of the ODA.
By comparing the active ODA complex structure with the recently obtained inactive form of the ODA complex, we were able to reveal the details of the remodeling of the ODA complex. The observed docking of the LC3 to the AAA+ ring of Dyh4 in the active ODA complex was surprising. Previous cryo‐ET studies revealed that the βHC of the sea urchin sperm (equivalent to the Dyh4 of Tetrahymena) can undergo an ATPase cycle (Lin et al, 2014b). Therefore, in the ATP cycle of the Dyh4, the linker detaches from and re‐attaches to the AAA+ ring (Fig 4D). LC3 is a thioredoxin component which was proposed to regulate the ODA activity in response to redox state of the cilia (Wakabayashi & King, 2006). Since LC3 was shown to interact with other proteins within the cilia depending on the redox state, LC3 might recruit other regulatory proteins to the Dyh4 AAA+ ring to modulate its motor activity. In the recent Chlamydomonas ODA structure, the linker of the β‐HC, which corresponds to Tetrahymena Dyh4, is in a canonical post‐powerstroke configuration (Walton et al, 2021). Furthermore, the Chlamydomonas γ‐HC (equivalent to Tetrahymena Dyh3) showed the linker configuration similar to that of Tetrahymena Dyh4. This suggests that there is a complex regulatory mechanism of the ATPase cycle in the ODA complex.
Though our coarse‐grained MD simulation was performed with weakened Shulin interaction, it captured the effects of conformational change in ODA complex with Shulin detachment. Based on our structure and coarse‐grained MD simulation, we present a model of how the ODA attachment induces remodeling and activation of the ODA complex (Fig 6). First, the Shulin–ODA complex docks onto the doublet using DC3 as a marker (Fig 6A). Next, the Dyh3 tail region starts to remodel. The extended coiled‐coil region of DC1/2 catches the Dyh3 tail domain and facilitates the remodeling of the Dyh3 tail region (Fig 6B). Due to the remodeling in the Dyh3 tail region, the binding of Shulin becomes destabilized. After Shulin detaches, the remodeling of the ODA complex is propagated toward the heads (Fig 6C). Without Shulin, the head domains would be released to be in a parallel configuration. Furthermore, the linker would be in post‐powerstroke configurations and ready to exert force in cilia. Interaction of the head domain and the neighboring ODA complex would enhance this process. Our model illustrates a simple yet robust activation mechanism of the ODA complex which does not require other external factors.
Figure 6. Model of the activation mechanism of the ODA.

-
AShulin–ODA docks on the doublet through interaction between the tail region of Dyh3 and the DC3.
-
BExtended coiled‐coil region of the DC1/2 catches the ODA complex at the tail domain and facilitates the remodeling of the tail. Head domain of Dyh3 also interacts with the neighboring ODA complex's tail. Therefore, the Dyh3 is “pulled” toward the doublet and binding of the Shulin destabilizes.
-
CShulin detaches from the ODA complex and the head domains are remodeled and aligned parallel to each other. Head domains are also in post‐powerstroke conformations and ready to exert force.
The remodeling of the tail involves an interaction with the extended coiled‐coil part of DC1/2. This is the reminiscence of the cytoplasmic dynein's case. Cytoplasmic dynein‐1 takes inactive conformation with two heads stacked together (Torisawa et al, 2014), when it binds to dynactin and adaptor proteins, such as BICD2, BICDR1, and HOOK3, the tail domain remodels, and the two heads becomes separated (Zhang et al, 2017; Urnavicius et al, 2018). These adaptor proteins are also coiled‐coil proteins and interact with the cytoplasmic dynein heavy chain by two sites to stabilize the remodeled conformation. In the cytoplasmic dynein, one of the ICs' WD domain interacts with the other remodeled HC similarly observed in the ODA's ICs. Thus, similar mechanisms are exploited for cytoplasmic dynein‐1 and the ODA for the activation. The stacking of the head is also conserved with cytoplasmic dynein‐2 (IFT dynein) (Toropova et al, 2017; Toropova et al, 2019). Thus, it would be interesting to clarify whether there are similar adaptor proteins to remodel the tail when dynein‐2 is working in the retrograde intraflagellar transport. The heterodimeric inner arm dynein‐f has different subunit components and head arrangement compared with the ODA (Heuser et al, 2012). Therefore, future study is needed to show whether the same mechanism is exploited with two‐headed axonemal dynein‐f when incorporated to the doublet.
In conclusion, we have shown the ODA complex structure on the doublet by cryo‐EM and obtained insights into how the ODA is activated inside the cilia. Our findings would be the important foundation to understand the generation of ciliary structure.
Materials and Methods
Growth of Tetrahymena cells for isolation
1 ml of the saturated Tetrahymena cells in SPP media (Gorovsky et al, 1975) was transferred into 50 ml liquid SPP and grown overnight with shaking at 150 rpm at 30°C in the shaker incubator (Thermo Fisher MAXQ8000). The overnight 50 ml culture was added to 900 ml of SPP and grown for approximately two days with shaking at 150 rpm at 30°C (MAXQ8000) until the cell density reached an OD at 600 nm of 0.7.
Cilium isolation by dibucaine treatment
The Tetrahymena culture was centrifuged at 700 g for 10 min at 22°C with slow deceleration (Avanti, Rotor JLA‐8.1000). The cell pellet was resuspended with 10 ml of SPP, and the volume was adjusted to 24 ml. The cell suspension was transferred to a 250‐ml Erlenmeyer flask on ice. 1 ml dibucaine in SPP at 25 mg/ml was added to the flask to make a final concentration of 1 mg/ml, and the cell suspension was gently swirled over ice for exactly 60 s. 75 ml of ice‐cold liquid SPP media was immediately added, and the 100 ml cilium suspension was then split into two 50‐ml conical tubes. The cell suspension was centrifuged at 2,000 g for 10 min at 4°C with slow deceleration (Sorvall ST 16R, Rotor 75003181). The cilium‐containing supernatant was transferred to four centrifuge tubes for the Beckman Coulter JA 25.50 rotor, approximately 20 ml per tube. The tubes containing the cilium‐containing supernatant were centrifuged at 17,000 g for 40 min at 4°C with slow deceleration (Avanti, Rotor JA25.50). The supernatants were discarded, and the pellets were carefully washed with the ice‐cold Cilia Wash Buffer (50 mM HEPES at pH 7.4, 3 mM MgSO4, 0.1 mM EGTA, 1 mM DTT, 250 mM sucrose), before resuspension in 250 μl of the ice‐cold Cilia Wash Buffer. The cilia were pelleted by centrifugation with 16,000 g for 10 min at 4°C in a microfuge, flash‐frozen with liquid nitrogen, and stored in the −80°C.
Purification of intact doublet microtubule
The thawed cilium pellet was resuspended in 250 μl ice‐cold Cilia Final Buffer (50 mM HEPES at pH 7.4, 3 mM MgSO4, 0.1 mM EGTA, 1 mM DTT, 0.5% trehalose, 1 mM PMSF). The concentration of total protein was measured using the Bradford Assay. The resuspended cilium sample was centrifuged at 16,000g for 10 min at 4°C in a microfuge (Eppendorf, Centrifuge 5415D). The supernatant was discarded, and the pellet was resuspended once more with 250 μl Cilia Final Buffer. NP‐40 alternative (Millipore Sigma, 492016) was added to the final concentration of 1.5%, and the total solution was resuspended. The samples were incubated on ice for 30 min before the demembraned cilia were centrifuged at 16,000 g for 10 min at 4°C in a microfuge (Eppendorf, Centrifuge 5415D). The supernatant was discarded, and the axoneme‐containing pellet was resuspended to 245 μl of Cilia Final Buffer. Final concentration of 1 mM ADP was added to the axonemes and incubated at room temperature for 10 min. Then, final concentration of 0.1 mM ATP was added to the solution and incubated at the room temperature for 10 min.
Cryo‐EM sample preparation
Wild‐type, K40R, and MEC17‐KO doublet concentrations were adjusted to 3 mg/ml. Quantifoil R2/2 (Electron Microscopy Sciences, #Q2100CR2) were treated with chloroform overnight and then glow discharged (30 s at 25 mAh). 3.5 μl of axoneme was applied to the pretreated grids and blotted by the Vitrobot Mark IV using blot force 3, blot time of 5 s, and drain time of 0.5 s.
Cryo‐EM data acquisition
Movies of the doublet microtubules were acquired at 64kx nominal magnification (calculated pixel size of 1.370 Å/pixel) with counting mode using Titan Krios 300 kV FEG electron microscope (Thermo Fisher Scientific) with the direct electron detector K3 Summit (Gatan, Inc.) and the BioQuantum energy filter (Gatan, Inc.). The data were acquired using SerialEM (Mastronarde, 2005). The WT and MEC17 datasets were collected using a total dose of 45 electrons per Å2 over 40 frames. The K40R dataset was with a total dose of 73 electrons per Å2 per frame over 30 frames. The defocus range was between −1.0 and −3.0 μm.
Cryo‐EM image processing
The movies were motion corrected and dose‐weighted using MotionCor2 (Zheng et al, 2017) implemented in Relion 3 (Zivanov et al, 2018), and the contrast transfer function parameters were estimated using Gctf (Zhang, 2016). After discarding micrographs with apparent drift and ice contamination, bad contrast transfers function estimation (3,074, 2,410, 4,283 micrographs for WT, K40R and MEC17 data, respectively). K40R and MEC17 data were merged since the ODA structures were the same in both mutants (S. K. Yang, S. Kubo, K. Peri, C. S. Black, M. Valente‐Paterno, J. Gaertig, M. Ichikawa, K. H. Bui, unpublished data). The filaments were picked using e2helixboxer (Tang et al, 2007).
The particles of 512 × 512 pixels were initially picked with 8‐nm periodicity, binned twice, and pre‐aligned using a modified version of the Iterative Helical Real Space Reconstruction script (Egelman, 2007) in SPIDER (Frank et al, 1996) to work with non‐helical symmetry. After that, the alignment parameters were transferred to Frealign and the particles were aligned by Frealign for 6 iterations (Grigorieff, 2007). The alignment parameters were then converted into Relion 3.0. The particles were scaled to 1.744 Å/pixel (box size 402 pixel) for image processing to reduce memory usage. The 8‐nm particles then underwent iterative per‐particles‐defocus refinement and Bayesian polishing in Relion 3.
The signal of the tubulin lattice was subtracted from each particle. The subtracted particles were subjected to 3D classification into three classes to obtain the 24‐nm repeat particles with the ODA in the center. The 24‐nm particles were then refined, resulting in resolution of 4.3 and 3.9 Å for WT, and K40R & MEC17 combined data from 71,462 and 139,548 particles, respectively. At this point, it is clear that the WT doublet did not retain a lot of ODA, only the docking complex (Fig EV1D and E). Therefore, we continued the next step with the K40R and MEC17 combined dataset.
The 24‐nm particles were boxed out at a box size of 804 pixel at 1.744 Å/pixel and underwent Bayesian polishing. The ODA is then centered and boxed out of the imaging using the Localized Reconstruction in Scipion (De la Rosa‐Trevín et al, 2016; Abrishami et al, 2020). The particles containing the ODA were then refined using Relion 3 and cryoSPARC (Punjani et al, 2017; Zivanov et al, 2018). We first refined a single entire ODA region (Fig EV2A). Then, we refined the ODA tail and head regions separately using focus refinement with masks for different regions (Fig EV2B). The resolutions for different regions of the ODA are in the range of 5.5–7 Å (Fig EV2, EV3, EV4, EV5).
Model building of the ODA complex on the doublet
First, the Shulin–ODA complex model was separated into two regions: (i) Dyh3 & IC/LC tower region, and (ii) Dyh4, Dyh5, and DIC3's WD domain. These two regions were rigid body fitted into the active ODA map based on the tail region. Then, ICs and LCs were rigid body fitted one by one to our cryo‐EM map. Dyh3 and Dyh4 were segmented into several regions and further fitted to the density. Since the IC and LC regions were fitted quite well to our map at this point, the density corresponding to ICs and LCs was subtracted from the whole tail map, and then, the HC regions were fitted to the map more precisely. For the head part, the whole head domains were fitted to our cryo‐EM map. We noticed that the linker parts of the Shulin–ODA complex were sticking out since our maps are in post‐powerstroke conformations. Thus, the linker part was segmented and fitted separately. For Dyh4, the linker region and LC3 were fitted together to our map and then fitted further separately. Finally, the model was integrated and refined by Coot (Emsley et al, 2010) in our map and real space refined using Phenix (Adams et al, 2010).
Docking complex modeling
DC2 (Q233H6) and DC3 (I7M2C6) of Tetrahymena were found by blasting UniProt database using the Chlamydomonas DC2 (A8JF70) and DC3 (Q7Y0H2). Their initial models were constructed by MODELLER 9.19 using the AutoModel class with GA341 and DOPE model assessment (Šali & Blundell, 1993) from the DC2 and DC3 structures (PDB 6U42). Q233H6 and I7M2C6 were then modified in Coot (Emsley et al, 2010) in the density of the 24‐nm average doublet microtubule structure to make them consistent with the density map (Fig EV4A), in which the docking complex density is clearer. In our map, the density of helices of the docking complex is reliable but side chain densities are not very clear.
DC1 from Chlamydomonas (A8IPZ5) did not yield a homolog in Tetrahymena by blasting UniProt database. Since we could clearly see a density at the place of DC1 in the density map and DC1 from Chlamydomonas is not conserved in humans, we reasoned that there is another coiled‐coil protein in place of DC1. In humans, CCDC151 is known to be important for the docking complex and causes ciliopathy similar to CCDC114 (human DC2 homolog) (Hjeij et al, 2014; Zhang et al, 2019). The human CCDC151 localizes to respiratory ciliary axonemes and interacts with CCDC114 (DC2 homolog). The proposed homolog of CCDC151 in Chlamydomonas is ODA10. While this protein is important for the ODA assembly in Chlamydomonas (Dean & Mitchell, 2013), it does not seem to localize in the axoneme‐like DC1/2 from the MS pattern (Pazour et al, 2005).
Using BLAST on UniProt database, Q22T00 was identified as a homolog for CCDC151. To see the MS profile of Q22T00, MS analyses were performed for untreated and salt‐treated doublet samples as described in Dai et al (2020). The MS profiles of Q22T00 were similar to DC2 and DC3 (Appendix Table S1). The coiled‐coil prediction of Q22T00 using COILS program (Lupas et al, 1991) also coincided with Chlamydomonas DC1 (Fig EV4B). Therefore, we concluded that despite the weak conservation, Q22T00 is the DC1 in Tetrahymena. We constructed the initial model of Q22T00 to the DC1 using MODELLER. Q22T00 was then modeled by Coot in the density of the 24‐nm average doublet microtubule structure (Fig EV4A). The secondary prediction of Q22T00 fitted well with helical density observed in the density map. For the extended coiled‐coil part of DC1/2, poly‐alanine model was generated based on two regions of the coiled‐coil of BICDR1 model (105–161 and 202–245 aa from PDB: 6F1T) and fitted to our cryo‐EM map.
Visualization
The maps and models are visualized using ChimeraX (Goddard et al, 2018).
MD simulation
The inactive Shulin–ODA structure (PDB ID: 6ZYW) had some missing residues. For MD simulation, we modeled loops for missing regions by MODELLER (Šali & Blundell, 1993). At the same time, two intermediated parts of Dyh5 (531–636, 3,270–3,382) and both ends of the Dic2 and Dic3 sequences (1–74, 636–667, and 1–17, 511–670, respectively) were removed. Then, we did a coarse‐grained MD simulation using this model. In the model, 1 amino acid was treated as 1 bead and each bead located at the Cα position. The force field for observing dynamics used is AICG2+ (Li et al, 2012; Li et al, 2014). By using AICG2+, the coarse‐grained MD approaches have been successfully employed to study large‐scale protein dynamics, such as the bending motion of microtubules (Ichikawa et al, 2019), the rotational movement of F0‐motor of the yeast ATP synthase (Kubo et al, 2020), and the scheme of conformational change in cytoplasmic dynein (Kubo et al, 2017). Here, we aimed to gain insights into the relationship between global conformational change and detachment of Shulin. In the AICG2+, the reference structure was assumed as the most stable conformation, and their parameters are defined from the reference of all‐atom structures. First, by comparing the inactive Shulin–ODA model with the active ODA structure on the doublet, chains that keep their binding and chains which change their binding schemes were determined by visual inspection. Based on this information, we adjusted the parameters that determine the attraction between the chains (see Supplementary text for the equation and the parameters in Appendix). First, since Dyh3, Dyh4, and Dyh5 clearly have a different contact style, we set the attraction force between these chains to be 0.5 times of the default. Next, since the contacts between Dyh3, Dic2, and the LC tower were not significantly changed, we increased the attraction by a factor of 10 in order to treat this chain as a rigid body. Also, since there was no significant difference between Dyh4 and Dic3 contacts, we increased the force of attraction by a factor of 10. Lastly, the attractive force between Shulin and others was decreased by 0.3 times the default in order to observe the Shulin detachment in a reasonable simulation time. After that, we run the simulations on several systems. When we applied the force, the force strength and direction was as follows: force direction, Dyh4 370th residue to Dyh3 480th residue; force power, f(x) = 0.0001184, f(y) = 0.0002144, f(z) = 0.0003164. (CafeMol Unit); force type, PULL_UNIT_CF (see CafeMol Manual, Kenzaki et al, 2011). We performed 10 simulations for all systems using CafeMol version 2.1 (Kenzaki et al, 2011). Each MD simulation took 1,000 × 104 MD steps with external force and 5,000 × 104 MD steps without external force. The simulations were conducted by the underdamped Langevin dynamics at 300 K temperature. We set the friction coefficient to 0.02 (CafeMol unit), and default values were used for others.
Author contributions
KHB and MI conceived the project and designed the experiments. JG generated mutant Tetrahymena strains. SKY and CSB performed culture of the cells and purification of microtubule fractions from cilia with the help of DD and MV‐P. SKY and CSB performed vitrification of the grids and collected data. SKY, SK, and KHB performed the cryo‐EM data analysis of the doublet. SK, KHB, and MI generated a model of the ODA complex on the doublet. SK performed the MD simulation. SK, MI, and KHB interpreted the structure. All authors were involved in the manuscript writing process.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Expanded View Figures PDF
Supplementary Movie S1
Movie EV1
Alignment
Acknowledgements
We thank Drs. Kaustuv Basu, Kelly Sears, Mike Strauss (Facility for Electron Microscopy Research), Ferdos Abid Ali, and Andrew P. Carter for helping with data collection, processing, and modeling. This work was supported by JST, PRESTO Grant Number JPMJPR20E1, JSPS KAKENHI Grant Numbers JP19K23726 and JP20K15733, and the Foundation for Nara Institute of Science and Technology (R2290001) to MI. SK is supported by JSPS Overseas Research Fellowships. KHB is supported by the grants from Canadian Institutes of Health Research (PJT‐156354), Natural Sciences and Engineering Research Council of Canada (RGPIN‐2016‐04954), and Canadian Institute for Advanced Research (Azieli Global Scholars Program).
EMBO reports (2021) 22: e52911.
Contributor Information
Muneyoshi Ichikawa, Email: michikawa@bs.naist.jp.
Khanh Huy Bui, Email: huy.bui@mcgill.ca.
Data availability
The data produced in this study are available in the following databases:
Cryo‐EM map: EMDB EMD‐23926 (https://www.emdataresource.org/EMD‐23926)
Model coordinates: PDB 7MOQ (https://www.rcsb.org/structure/7MOQ)
Mass spectrometry data: DataDryad: https://doi.org/10.5061/dryad.p2ngf1vqv (https://doi.org/10.5061/dryad.p2ngf1vqv)
References
- Abrishami V, Ilca SL, Gomez‐Blanco J, Rissanen I, de la Rosa‐Trevín JM, Reddy VS, Carazo J‐M, Huiskonen JT (2020) Localized reconstruction in scipion expedites the analysis of symmetry mismatches in Cryo‐EM data. Prog Biophys Mol Biol 160: 43–52 [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L‐W, Kapral GJ, Grosse‐Kunstleve RWet al (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr Sect D: Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akella JS, Wloga D, Kim J, Starostina NG, Lyons‐Abbott S, Morrissette NS, Dougan ST, Kipreos ET, Gaertig J (2010) MEC‐17 is an α‐tubulin acetyltransferase. Nature 467: 218–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessen M, Fay RB, Witman GB (1980) Calcium control of waveform in isolated flagellar axonemes of Chlamydomonas. J Cell Biol 86: 446–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brokaw CJ, Kamiya R (1987) Bending patterns of Chlamydomonas flagella: IV. Mutants with defects in inner and outer dynein arms indicate differences in dynein arm function. Cell Motil Cytoskeleton 8: 68–75 [DOI] [PubMed] [Google Scholar]
- Bui KH, Sakakibara H, Movassagh T, Oiwa K, Ishikawa T (2008) Molecular architecture of inner dynein arms in situ in Chlamydomonas reinhardtii flagella. J Cell Biol 183: 923–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui KH, Sakakibara H, Movassagh T, Oiwa K, Ishikawa T (2009) Asymmetry of inner dynein arms and inter‐doublet links in Chlamydomonas flagella. J Cell Biol 186: 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui KH, Yagi T, Yamamoto R, Kamiya R, Ishikawa T (2012) Polarity and asymmetry in the arrangement of dynein and related structures in the Chlamydomonas axoneme. J Cell Biol 198: 913–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Can S, Lacey S, Gur M, Carter AP, Yildiz A (2019) Directionality of dynein is controlled by the angle and length of its stalk. Nature 566: 407–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai D, Ichikawa M, Peri K, Rebinsky R, Bui KH (2020) Identification and mapping of central pair proteins by proteomic analysis. Biophys Physicobiol 17: 71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa‐Trevín JM, Quintana A, del Cano L, Zaldívar A, Foche I, Gutiérrez J, Gómez‐Blanco J, Burguet‐Castell J, Cuenca‐Alba J, Abrishami Vet al (2016) Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J Struct Biol 195: 93–99 [DOI] [PubMed] [Google Scholar]
- Dean AB, Mitchell DR (2013) Chlamydomonas ODA10 is a conserved axonemal protein that plays a unique role in outer dynein arm assembly. Mol Biol Cell 24: 3689–3696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamant AG, Carter AP (2013) Dynein family classification. In Encyclopedia of Biophysics, Roberts GCK (ed.), pp 552–572. Berlin, Heidelberg: Springer; [Google Scholar]
- Egelman EH (2007) The iterative helical real space reconstruction method: surmounting the problems posed by real polymers. J Struct Biol 157: 83–94 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr Sect D: Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A (1996) SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol 116: 190–199 [DOI] [PubMed] [Google Scholar]
- Gaertig J, Cruz MA, Bowen J, Gu L, Pennock DG, Gorovsky MA (1995) Acetylation of lysine 40 in alpha‐tubulin is not essential in Tetrahymena thermophila. J Cell Biol 129: 1301–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE (2018) UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci 27: 14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldtzvik Y, Mugnai ML, Thirumalai D (2018) Dynamics of allosteric transitions in dynein. Structure 26: 1664–1677 [DOI] [PubMed] [Google Scholar]
- Goodenough UW, Heuser JE (1982) Substructure of the outer dynein arm. J Cell Biol 95: 798–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorovsky MA, Yao MC, Keevert JB, Pleger GL (1975) Isolation of micro‐and macronuclei of Tetrahymena pyriformis. In Methods in Cell Biology, Vol. 9, Prescott DM (ed.), pp 311–327. Cambridge, MA: Academic Press; [DOI] [PubMed] [Google Scholar]
- Grigorieff N (2007) FREALIGN: high‐resolution refinement of single particle structures. J Struct Biol 157: 117–125 [DOI] [PubMed] [Google Scholar]
- Heuser T, Barber CF, Lin J, Krell J, Rebesco M, Porter ME, Nicastro D (2012) Cryoelectron tomography reveals doublet‐specific structures and unique interactions in the I1 dynein. Proc Natl Acad Sci USA 109: E2067–E2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjeij R, Onoufriadis A, Watson C, Slagle C, Klena N, Dougherty G, Kurkowiak M, Loges N, Diggle C, Morante Net al (2014) CCDC151 mutations cause primary ciliary dyskinesia by disruption of the outer dynein arm docking complex formation. Am J Hum Genet 95: 257–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa M, Khalifa AAZ, Kubo S, Dai D, Basu K, Maghrebi MAF, Vargas J, Bui KH (2019) Tubulin lattice in cilia is in a stressed form regulated by microtubule inner proteins. Proc Natl Acad Sci USA 116: 19930–19938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa M, Liu D, Kastritis PL, Basu K, Hsu TC, Yang S, Bui KH (2017) Subnanometre‐resolution structure of the doublet microtubule reveals new classes of microtubule‐associated proteins. Nat Commun 8: 15035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa M, Saito K, Yanagisawa H‐A, Yagi T, Kamiya R, Yamaguchi S, Yajima J, Kushida Y, Nakano K, Numata Oet al (2015) Axonemal dynein light chain‐1 locates at the microtubule‐binding domain of the γ heavy chain. Mol Biol Cell 26: 4236–4247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya N, Mashimo T, Takano Y, Kon T, Kurisu G, Nakamura H (2016) Elastic properties of dynein motor domain obtained from all‐atom molecular dynamics simulations. Protein Eng Des Sel 29: 317–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenzaki H, Koga N, Hori N, Kanada R, Li W, Okazaki KI, Yao X‐Q, Takada S (2011) CafeMol: a coarse‐grained biomolecular simulator for simulating proteins at work. J Chem Theory Comput 7: 1979–1989 [DOI] [PubMed] [Google Scholar]
- Khalifa AAZ, Ichikawa M, Dai D, Kubo S, Black CS, Peri K, McAlear TS, Veyron S, Yang SK, Vargas Jet al (2020) The inner junction complex of the cilia is an interaction hub that involves tubulin post‐translational modifications. eLife 9: e52760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King SM (2012) Integrated control of axonemal dynein AAA(+) motors. J Struct Biol 179: 222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles M, Leigh M, Ostrowski L, Huang L, Carson J, Hazucha M, Yin W, Berg J, Davis S, Dell Set al (2013) Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia. Am J Hum Genet 92: 99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo S, Li W, Takada S (2017) Allosteric conformational change cascade in cytoplasmic dynein revealed by structure‐based molecular simulations. PLoS Comput Biol 13: e1005748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo S, Niina T, Takada S (2020) Molecular dynamics simulation of proton‐transfer coupled rotations in ATP synthase FO motor. Sci Rep 10: 8225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Terakawa T, Wang W, Takada S (2012) Energy landscape and multiroute folding of topologically complex proteins adenylate kinase and 2ouf‐knot. Proc Natl Acad Sci USA 109: 17789–17794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wang W, Takada S (2014) Energy landscape views for interplays among folding, binding, and allostery of calmodulin domains. Proc Natl Acad Sci USA 111: 10550–10555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Nicastro D (2018) Asymmetric distribution and spatial switching of dynein activity generates ciliary motility. Science 360: eaar1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Okada K, Raytchev M, Smith MC, Nicastro D (2014) Structural mechanism of the dynein power stroke. Nat Cell Biol 16: 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Yin W, Smith MC, Song K, Leigh MW, Zariwala MA, Knowles MR, Ostrowski LE, Nicastro D (2014) Cryo‐electron tomography reveals ciliary defects underlying human RSPH1 primary ciliary dyskinesia. Nat Commun 5: 5727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J (1991) Predicting coiled coils from protein sequences. Science 252: 1162–1164 [DOI] [PubMed] [Google Scholar]
- Ma M, Stoyanova M, Rademacher G, Dutcher SK, Brown A, Zhang R (2019) Structure of the decorated ciliary doublet microtubule. Cell 179: 909–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali GR, Ali FA, Lau CK, Begum F, Boulanger J, Howe JD, Chen ZA, Rappsilber J, Skehel M, Carter AP (2021) Shulin packages axonemal outer dynein arms for ciliary targeting. Science 371: 910–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005) Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152: 36–51 [DOI] [PubMed] [Google Scholar]
- Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, McIntosh JR (2006) The molecular architecture of axonemes revealed by cryoelectron tomography. Science 313: 944–948 [DOI] [PubMed] [Google Scholar]
- Oda T, Abe T, Yanagisawa H, Kikkawa M (2016a) Docking‐complex‐independent alignment of Chlamydomonas outer dynein arms with 24‐nm periodicity in vitro. J Cell Sci 129: 1547–1551 [DOI] [PubMed] [Google Scholar]
- Oda T, Abe T, Yanagisawa H, Kikkawa M (2016b) Structure and function of outer dynein arm intermediate and light chain complex. Mol Biol Cell 27: 1051–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owa M, Furuta A, Usukura J, Arisaka F, King SM, Witman GB, Kamiya R, Wakabayashi K (2014) Cooperative binding of the outer arm‐docking complex underlies the regular arrangement of outer arm dynein in the axoneme. Proc Natl Acad Sci USA 111: 9461–9466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Agrin N, Leszyk J, Witman GB (2005) Proteomic analysis of a eukaryotic cilium. J Cell Biol 170: 103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA (2017) cryoSPARC: algorithms for rapid unsupervised cryo‐EM structure determination. Nat Methods 14: 290–296 [DOI] [PubMed] [Google Scholar]
- Rao Q, Wang Y, Chai P, Kuo YW, Han L, Yang R, Yang Y, Howard J, Zhang K (2020) Cryo‐EM structures of outer‐arm dynein array bound to microtubule doublet reveal a mechanism for motor coordination. bioRxiv 10.1101/2020.12.08.415687 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter JF, Leroux MR (2017) Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol 18: 533–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815 [DOI] [PubMed] [Google Scholar]
- Stoddard D, Zhao Y, Bayless BA, Gui L, Louka P, Dave D, Suryawanshi S, Tomasi RFX, Dupuis‐Williams P, Baroud CNet al (2018) Tetrahymena RIB72A and RIB72B are microtubule inner proteins in the ciliary doublet microtubules. Mol Biol Cell 29: 2566–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ (2007) EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157: 38–46 [DOI] [PubMed] [Google Scholar]
- Toda A, Nishikawa Y, Tanaka H, Yagi T, Kurisu G (2020) The complex of outer‐arm dynein light chain‐1 and the microtubule‐binding domain of the γ heavy chain shows how axonemal dynein tunes ciliary beating. J Biol Chem 295: 3982–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torisawa T, Ichikawa M, Furuta A, Saito K, Oiwa K, Kojima H, Furuta K, Toyoshima YY (2014) Autoinhibition and cooperative activation mechanisms of cytoplasmic dynein. Nat Cell Biol 16: 1118–1124 [DOI] [PubMed] [Google Scholar]
- Toropova K, Mladenov M, Roberts AJ (2017) Intraflagellar transport dynein is autoinhibited by trapping of its mechanical and track‐binding elements. Nat Struct Mol Biol 24: 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toropova K, Zalyte R, Mukhopadhyay AG, Mladenov M, Carter AP, Roberts AJ (2019) Structure of the dynein‐2 complex and its assembly with intraflagellar transport trains. Nat Struct Mol Biol 26: 823–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toropova K, Zou S, Roberts AJ, Redwine WB, Goodman BS, Reck‐Peterson SL, Leschziner AE (2014) Lis1 regulates dynein by sterically blocking its mechanochemical cycle. eLife 3: e03372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno H, Bui KH, Ishikawa T, Imai Y, Yamaguchi T, Ishikawa T (2014) Structure of dimeric axonemal dynein in cilia suggests an alternative mechanism of force generation. Cytoskeleton 71: 412–422 [DOI] [PubMed] [Google Scholar]
- Ueno H, Yasunaga T, Shingyoji C, Hirose K (2008) Dynein pulls microtubules without rotating its stalk. Proc Natl Acad Sci USA 105: 19702–19707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnavicius L, Lau CK, Elshenawy MM, Morales‐Rios E, Motz C, Yildiz A, Carter AP (2018) Cryo‐EM shows how dynactin recruits two dyneins for faster movement. Nature 554: 202–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi K, King SM (2006) Modulation of Chlamydomonas reinhardtii flagellar motility by redox poise. J Cell Biol 173: 743–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton T, Wu H, Brown A (2021) Structure of a microtubule‐bound axonemal dynein. Nat Commun 12: 477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016) Gctf: Real‐time CTF determination and correction. J Struct Biol 193: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Foster HE, Rondelet A, Lacey SE, Bahi‐Buisson N, Bird AW, Carter AP (2017) Cryo‐EM reveals how human cytoplasmic dynein is auto‐inhibited and activated. Cell 169: 1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WZ, Li DP, Wei SJ, Guo T, Wang J, Luo H, Yang YF, Tan ZP (2019) Whole‐exome sequencing identifies a novel CCDC151 mutation, c. 325G> T (p. E109X), in a patient with primary ciliary dyskinesia and Situs inversus. J Hum Genet 64: 249–252 [DOI] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH (2018) New tools for automated high‐resolution cryo‐EM structure determination in RELION‐3. eLife 7: e42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Expanded View Figures PDF
Supplementary Movie S1
Movie EV1
Alignment
Data Availability Statement
The data produced in this study are available in the following databases:
Cryo‐EM map: EMDB EMD‐23926 (https://www.emdataresource.org/EMD‐23926)
Model coordinates: PDB 7MOQ (https://www.rcsb.org/structure/7MOQ)
Mass spectrometry data: DataDryad: https://doi.org/10.5061/dryad.p2ngf1vqv (https://doi.org/10.5061/dryad.p2ngf1vqv)