

Abstract
Mfn2 is a mitochondrial fusion protein with bioenergetic functions implicated in the pathophysiology of neuronal and metabolic disorders. Understanding the bioenergetic mechanism of Mfn2 may aid in designing therapeutic approaches for these disorders. Here we show using endoplasmic reticulum (ER) or mitochondria‐targeted Mfn2 that Mfn2 stimulation of the mitochondrial metabolism requires its localization in the ER, which is independent of its fusion function. ER‐located Mfn2 interacts with mitochondrial Mfn1/2 to tether the ER and mitochondria together, allowing Ca2+ transfer from the ER to mitochondria to enhance mitochondrial bioenergetics. The physiological relevance of these findings is shown during neurite outgrowth, when there is an increase in Mfn2‐dependent ER‐mitochondria contact that is necessary for correct neuronal arbor growth. Reduced neuritic growth in Mfn2 KO neurons is recovered by the expression of ER‐targeted Mfn2 or an artificial ER‐mitochondria tether, indicating that manipulation of ER‐mitochondria contacts could be used to treat pathologic conditions involving Mfn2.
Keywords: Ca2+ , ER‐mitochondria tethering, Mfn2, neuritic growth
Subject Categories: Membrane & Intracellular Transport, Metabolism, Neuroscience
The bioenergetic role of Mfn2 relies on its localization in the ER. Mfn2 localization to mitochondria is dispensable in the presence of mitochondrial Mfn1, which tethers ER and mitochondria, and facilitates Ca2+ transfer.
Introduction
Mitochondria are dynamic organelles whose morphology continuously changes through fusion and fission events. This dynamism affects mitochondrial bioenergetic properties, among other functions (Liesa & Shirihai, 2013; Schrepfer & Scorrano, 2016). Mfn1 and Mfn2 are two proteins from the large GTPase family that mediate outer mitochondrial membrane fusion. Despite their high degree of homology, there are functions that Mfn1 and Mfn2 do not share, as evidenced in the phenotypes of Mfn1 and Mfn2 KO mice (Chen et al, 2003; Chen et al, 2007). Mfn1 has been shown to be more efficient at mediating mitochondrial fusion; meanwhile, Mfn2 plays a more prominent role regulating mitochondrial metabolism (Bach et al, 2003; Chen et al, 2003; Eura et al, 2003; Ishihara et al, 2004; Pich et al, 2005). The mechanism through which Mfn2 has a greater impact on mitochondrial energetics than Mfn1 does is not fully understood, although several mechanisms have been proposed. Mfn2 has been reported to regulate the levels of proteins in the OXPHOS system (Pich et al, 2005) and the formation of supercomplexes (Segales et al, 2013). It has also been found to be necessary for coenzyme Q biosynthesis (Mourier et al, 2015). The finding that a small fraction of Mfn2, but not Mfn1, resides in the endoplasmic reticulum (ER) where it interacts, both homo‐ and heterotipically, with mitochondria‐located Mfns and tethers the two organelles together (de Brito & Scorrano, 2008) has also suggested a mechanism by which Mfn2 could regulate mitochondrial metabolism via modulation of Ca2+ transfer from the ER to mitochondria (Chen et al, 2012; Seidlmayer et al, 2019).
The ER and mitochondria establish functional contact in the area called mitochondria‐associated membrane (MAM). The thickness of ER‐mitochondria contact ranges 10–80 nm, with the distance between the organelles being what determines MAM function (Giacomello & Pellegrini, 2016; Herrera‐Cruz & Simmen, 2017). The three most studied functions of the MAM are as follows: (i) lipid synthesis. Most of the enzymes involved in lipid biosynthesis are localized in the ER membrane, but some are located in the mitochondrial membrane. Thus, it is necessary to transfer different lipid intermediates from one organelle to other in order to complete the biosynthetic pathway (Vance, 2014). (ii) Mitochondrial fission. It has been observed that ER tubules mark sites of mitochondrial division (Friedman et al, 2011). (iii) Regulation of Ca2+ homeostasis. The two main organelles responsible for maintaining the Ca2+ homeostasis are the ER and mitochondria. These two organelles exchange Ca2+ which affects their respective functionality. Mitochondrial Ca2+ uptake is produced through the mitochondrial calcium uniporter (Baughman et al, 2011; De Stefani et al, 2011) (MCU); but given its low Ca2+ affinity, this takes place in microdomains with high Ca2+ concentrations in the proximity of the Ca2+‐releasing channels in the ER (Rizzuto et al, 1993; Rizzuto et al, 1998). That is to say, it occurs in the regions of close proximity between the ER and mitochondria. The optimal separation between ER and mitochondria to generate microdomains with high enough Ca2+ concentrations to facilitate Ca2+ uptake by MCU is 12–24 nm (Giacomello & Pellegrini, 2016). Mitochondrial Ca2+ regulates the activity of dehydrogenases of the tricarboxylic acid cycle (TCA) and promotes dephosphorylation, hence activation of pyruvate dehydrogenase (Balaban, 2009). Thus, Ca2+ uptake by mitochondria following its release from ER is necessary to maintain cellular bioenergetics (Cárdenas et al, 2010). However, this Ca2+ transfer must be finely regulated since too little or too much Ca2+ transfer is noxious (Scorrano et al, 2003; Csordás et al, 2006; Cárdenas et al, 2016).
Observations of reduced mitochondrial Ca2+ and increased ER Ca2+ levels in Mfn2 depleted cells (Chen et al, 2012; Seidlmayer et al, 2019) support the view that Mfn2 regulates mitochondrial metabolism by tethering the ER and mitochondria together and thereby regulating mitochondrial Ca2+. However, this has not been studied thoroughly.
By expressing Mfn2 targeted at the ER or mitochondria in Mfn2 KO or Mfn1/2 DKO cells, here, we show that the presence of Mfn2 in the ER is needed in order to tether ER and mitochondria together and enhance mitochondrial energetics. The reestablishment of ER‐mitochondria contact via the expression of ER‐located Mfn2 or the artificial tether ChiMERA (Kornmann et al, 2009) is sufficient to correct the bioenergetic defects in Mfn2 KO fibroblasts. The rescue of neuritic arbor abnormalities in Mfn2 KO neurons by ER‐located Mfn2 or ChiMERA provides a proof of concept that targeting the ER‐mitochondria contacts may be a suitable therapeutic option in pathologic conditions in which Mfn2 is involved.
Results
Defects in mitochondrial bioenergetics in Mfn2 KO cells do not depend on mitochondrial morphology
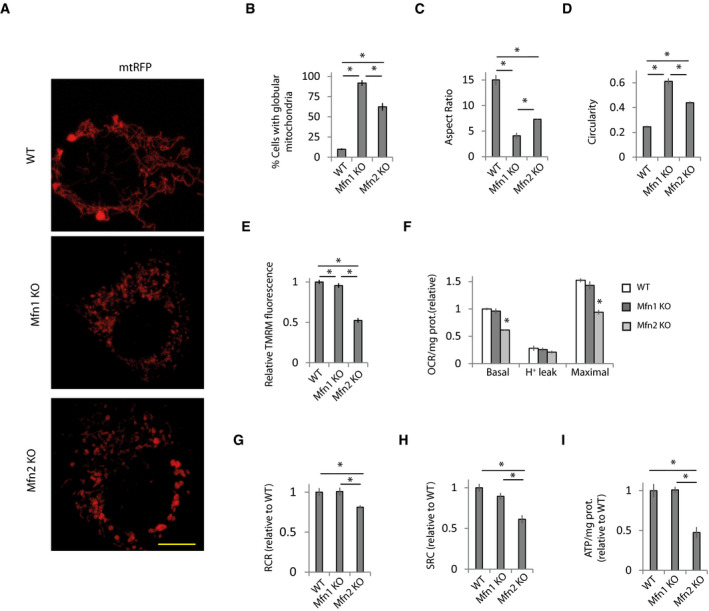
Mfn2 is a potent regulator of mitochondrial bioenergetics, but the mechanism by which Mfn2 maintains mitochondria bioenergetics is not well understood. Elongated mitochondria are associated with increased mitochondrial bioenergetics (Liesa & Shirihai, 2013). However, although Mfn1 KO fibroblasts (Appendix Fig S1A) showed more globular mitochondria than Mfn2 KO fibroblasts (Fig 1A–D), they did not show bioenergetic alterations (Fig 1E–I and Appendix Fig S1B). Meanwhile, Mfn2 KO cells showed reduced mitochondrial membrane potential (MMP) (Fig 1E) and lower basal and maximal oxygen consumption rate (OCR) (Fig 1F) when analyzed using a fluorescent O2 sensor probe, which resulted in reduced respiratory control ratio (RCR) and a reduced spare respiratory capacity (SRC) (Fig 1G and H). Analysis of the OCR using Seahorse XF24 showed a similar pattern (Appendix Fig S1B).
Figure 1. Defects in mitochondrial bioenergetics in Mfn2 KO cells do not depend on mitochondrial morphology.
-
A–D(A) Representative images of WT, Mfn1 KO, and Mfn2 KO MEFs transfected for 48 h with a plasmid encoding mitochondria‐targeted RFP (mt‐RFP). Scale bar: 5 µm. (B) Percentage of cells displaying globular mitochondria (n = 310–668 cells analyzed in 3–6 independent experiments). Data are presented as mean ± SEM. (C) Analysis of aspect ratio (AR) calculated as major axis/minor axis and (D) circularity calculated as 4*π*Area/Perimeter2 (n = 300 mitochondria in three independent experiments). Data are presented as mean ± SEM.
-
EMitochondrial membrane potential (MMP) was determined by measuring TMRM fluorescence (n = 90 cells analyzed in three independent experiments). Data are presented as mean ± SEM.
-
F–H(F) Oxygen consumption rate (OCR) normalized to the amount of protein in WT, Mfn1 KO, and Mfn2 KO fibroblasts. Proton leak was measured after application of oligomycin (1 µM), maximal after CCCP application (10 µM), and oligomycin (n = 3 independent experiments). Data are presented as mean ± SEM. (G) Respiratory control ratio (RCR) was calculated as maximal oxygen consumption rate (OCR)/H+ leak OCR (n = 3 independent experiments). Data are presented as mean ± SEM. (H) Spare respiratory capacity was calculated as maximal OCR‐basal OCR (n = 3 independent experiments). Data are presented as mean ± SEM.
-
IWT MEFs and Mfn1 or Mfn2 KO MEFs were incubated with 2‐DG (10 mM) for 6 h, and ATP levels were measured (n = 3 independent experiments). Data are presented as mean ± SEM.
Data information: *P < 0.05, one‐way ANOVA followed by Tukey’s post hoc test.
Cells adjust metabolic pathways to maintain relatively constant total ATP levels, and therefore, there were not differences in the ATP levels between the different cell lines (Fig EV1A). However, in agreement with the reduced respiratory capacity in Mfn2 KO cells, inhibition of glycolysis with 2‐deoxy‐D‐glucose (2‐DG) caused a drop in ATP levels in Mfn2 KO cells with respect to WT and Mfn1 KO cells (Fig 1I).
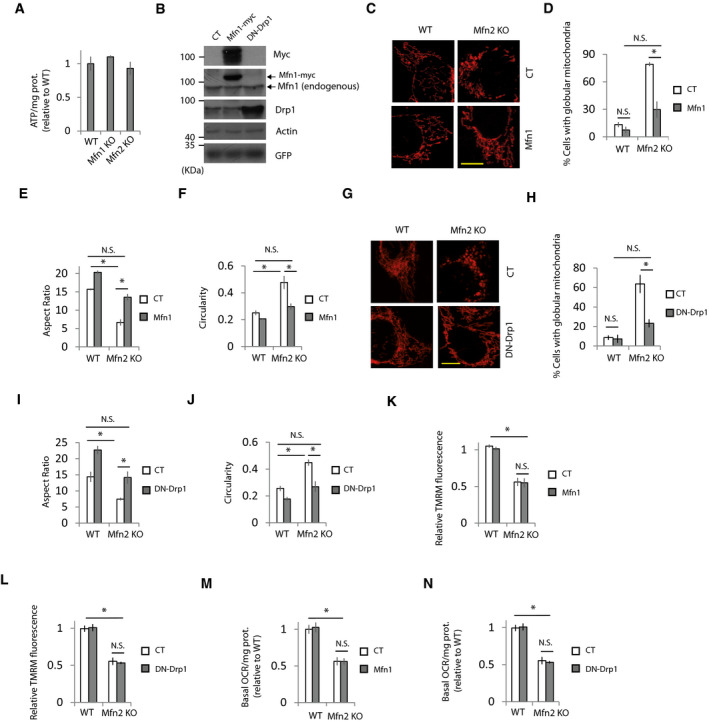
Figure EV1. Bioenergetics defects in Mfn2 KO cells are independent of mitochondrial morphology.
-
AATP levels in the indicated cell lines when metabolic flexibility is allowed (n = 3 independent experiments). Data are presented as mean ± SEM.
-
BRepresentative Western blots of the indicated proteins in Mfn2 KO fibroblasts transfected with plasmids expressing Mfn1, DN‐Drp1, or a control plasmid (CT) (n = 3 independent experiments).
-
C–FExpression of Mfn1 restores mitochondrial morphology in Mfn2 KO MEFs. WT or Mfn2 KO MEFs were co‐transfected with plasmids encoding mt‐RFP and Mfn1 or control plasmids. (C) Representative images. Scale bar = 20 µm. (D) Percentage of cells displaying globular mitochondria (n = 54–414 cells analyzed in three independent experiments), (E) aspect ratio, and (F) circularity (n = 260–300 mitochondria analyzed in three independent experiments). Data are presented as mean ± SEM.
-
G–JExpression of DN‐Drp1 restores mitochondrial morphology in Mfn2 KO MEFs. WT or Mfn2 KO MEFs were co‐transfected with plasmids encoding mt‐RFP and DN‐Drp1 or control plasmids. (G) Representative images. Scale bar = 20 µm. (H) Percentage of cells displaying globular mitochondria (n = 135–470 cells analyzed in three independent experiments), (I) aspect ratio and (J) circularity (n = 300 mitochondria were analyzed in three independent experiments). Data are presented as mean ± SEM.
-
K, LRestoration of mitochondrial morphology by expression of (K) Mfn1 or (L) DN‐Drp1 in Mfn2 KO MEFs has no effect on mitochondrial membrane potential (n = 90 cells analyzed in three independent experiments). Data are presented as mean ± SEM.
-
M, NRestoration of mitochondrial morphology by expression of (M) Mfn1 or (N) DN‐Drp1 in Mfn2 KO MEFs has no effect on basal oxygen consumption rate (n = 3 independent experiments). Data are presented as mean ± SEM.
Data information: *P < 0.05, one‐way ANOVA followed by Tukey’s post hoc test.
Mitochondrial morphology can affect uptake of cationic dyes used to measure MMP (Kowaltowski et al, 2002). However, restoration of mitochondrial morphology in Mfn2 KO cells by expressing exogenous Mfn1 or a dominant‐negative mutant of the pro‐fission protein Drp1 (DN‐Drp1) (Fig EV1, EV2, EV3, EV4, EV5) did not modify the MMP or the basal oxygen consumption in Mfn2 KO cells (Fig EV1, EV2, EV3, EV4, EV5).
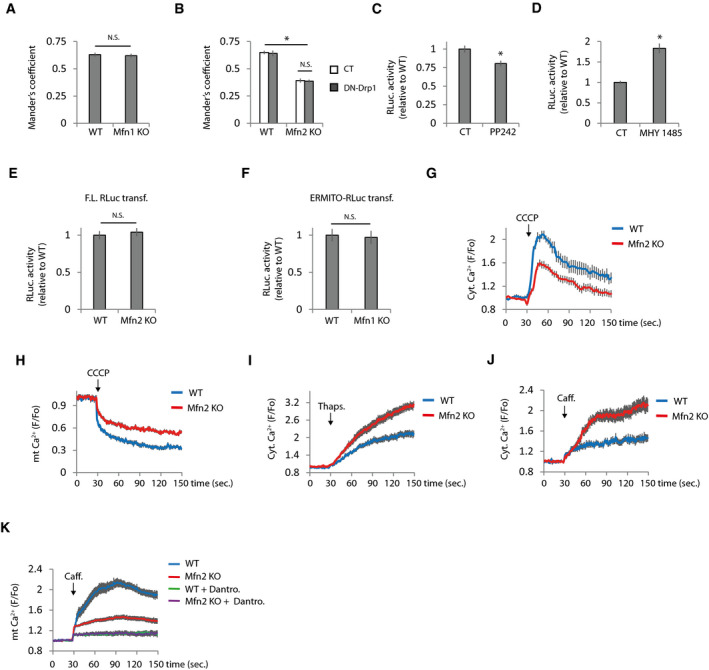
Figure EV2. ER and mitochondrial Ca2+ levels are deregulated in Mfn2 KO cells.
-
AMander’s coefficient of WT and Mfn1 KO cells co‐transfected with plasmids encoding mitochondria‐targeted RFP and ER‐targeted GFP (n = 15 cells analyzed in three independent experiments). Data are presented as mean ± SEM.
-
BRestoration of mitochondrial morphology does not restores ER‐mitochondria colocalization. Mander’s coefficient of WT and Mfn2 KO cells co‐transfected with plasmids encoding mitochondria‐targeted RFP and ER‐targeted GFP and the indicated plasmids (n = 15 cells analyzed in three independent experiments). Data are presented as mean ± SEM.
-
C, DERMITO‐Luc detects changes in ER‐mitochondria contacts induced by manipulation of mTOR pathway. RLuc activity in cells transfected with ERMITO‐Luc and treated with (C) the mTOR inhibitor PP242 (25 µM) (n = 4 independent experiments) or (D) the mTOR activator MHY 1485 (10 µM) (n = 6 independent experiments). Data are presented as mean ± SEM.
-
EThere are not differences in the RLuc activity in the WT and Mfn2 KO cells transfected with plasmid expressing full‐length Renilla Luciferase driven by the CMV promoter (n = 3 independent experiments). Data are presented as mean ± SEM.
-
FRLuc activity in WT and Mfn1 KO cells transfected with ERMITO‐Luc (n = 3 independent experiments). Data are presented as mean ± SEM.
-
G, HDiminished Ca2+ levels in Mfn2 KO mitochondria. (G) Mitochondrial Ca2+ was released from mitochondria by applying CCCP (10 µM), and the rise in cytoplasmic Ca2+ was analyzed with Fluo‐4 (n = 30 cells analyzed from three independent experiments). (H) Analysis of mitochondria Ca2+ levels in WT, and Mfn2 KO cells were transfected with the mitochondria‐targeted Ca2+ sensor mt‐Cepia (n = 30 cells from three independent experiments). Data are presented as mean ± SEM.
-
IIncreased ER Ca2+ levels in Mfn2 KO cells analyzed by measuring cytoplasmic Ca2+ rise with Fluo‐4 after thapsigargin (1 µM) treatment (n = 30 cells from three independent experiments). Data are presented as mean ± SEM.
-
JCaffeine induces higher ER Ca2+ release to the cytoplasm in Mfn2 KO cells. ER Ca2+ release was stimulated with caffeine (20 mM) as indicated, and cytoplasmic Ca2+ levels were analyzed with Fluo‐4 (n = 30 cells from three independent experiments). Data are presented as mean ± SEM.
-
KThe source of Ca2+ after caffeine treatment is from de ER. Inhibition of the RyR with dantrolene (10 µM) prevents caffeine‐induced increase in mitochondrial Ca2+ levels (n = 30 cells from three independent experiments). Data are presented as mean ± SEM.
Data information: *P < 0.05, one‐way ANOVA followed by Tukey’s post hoc test.
Figure EV3. Restoration of bioenergetics defects in Mfn2 KO cells require proper Ca2+ transfer from the ER to mitochondria.
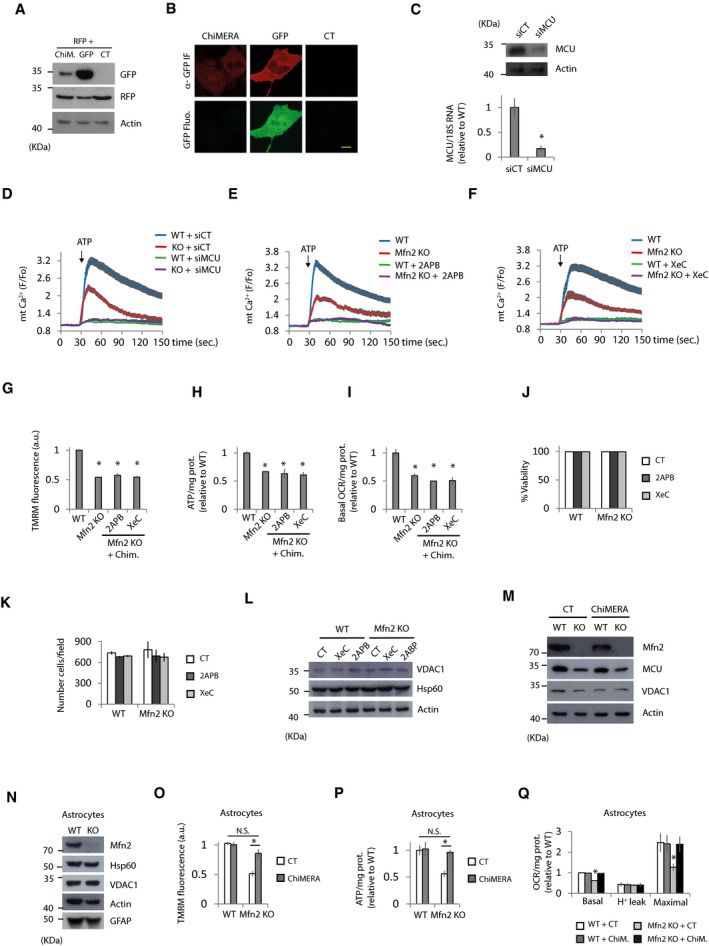
-
A, BChiMERA is weakly expressed in MEFs. (A) Representative Western blots of the indicated proteins in Mfn2 KO fibroblasts transfected with RFP plus ChiMERA, GFP, or control (CT) plasmids as indicated (n = 3 independent experiments). Data are presented as mean ± SEM. (B) Representative confocal images of Mfn2 KO transfected cells with ChIMERA or GFP. Green fluorescent signal cannot be observed in ChIMERA expressing cells, although GFP molecule can be detected by three‐step immunofluorescence using antibodies anti‐GFP. Scale bar = 10 µm.
-
CMCU mRNA and protein levels were analyzed by qPCR and Western blot, respectively, of MEFs transfected with siRNA control (siCT) or a pool of 4 siRNAs targeting MCU (siMCU) (n = 3 independent experiments).
-
DMCU KD (siMCU) abolish mitochondrial Ca2+ uptake when the IP3R is stimulated with ATP (100 µM). WT and Mfn2 KO cells were transfected with siRNA control (siCT) or targeting MCU (siMCU). Mitochondrial Ca2+ uptake was analyzed. (n = 28–30 cells from three independent experiments). Data are presented as mean ± SEM.
-
E, FInhibition of IP3R with (E) 2APB (50 µM) or (F) xestospongin C (XeC) (1.5 µM) prevents mitochondrial Ca2+ uptake caused by ATP treatment (n = 30 cells from three independent experiments). Data are presented as mean ± SEM.
-
G–ITreatment with 2APB or XeC prevents ChiMERA‐mediated rescue of (G) MMP (n = 90 cells analyzed in three independent experiments), (H) ATP levels (n = 3 independent experiments), and (I) basal oxygen consumption rate (n = 3 independent experiments) in Mfn2 KO cells. Data are presented as mean ± SEM.
-
J, K2APB or XeC treatment does not affect cell viability. WT and Mfn2 KO MEFs were treated with XeC (1.5 µM) or 2APB (50 µM) for 16 h, and then cell viability (J) was analyzed by fixing cells, DAPI staining and counting non‐pyknotic nuclei as percentage of the total number of nuclei (n = 3 independent experiments). (K) Total number of nuclei per field was counted (n = 3 independent experiments). Data are presented as mean ± SEM.
-
LMitochondrial mass is not affected by 2APB or XeC treatment. Representative Western blot of the indicated proteins of WT and Mfn2 KO MEFs treated for 16 h with XeC (1.5 µM), 2APB (50 µM) or vehicle (CT) (n = 3 independent experiments).
-
MMfn2 KO show diminished expression levels of MCU but this is not rescued by ChiMERA expression. WT and Mfn2 KO cells were transfected with RFP and ChiMERA or control plasmid. After 24 h, the transfected cells were sorted and plated for another 24 h, when protein extracts were obtained. Representative Western blot of the indicated proteins (n = 3 independent experiments).
-
NWestern blot with the indicated antibodies to characterize cultures of tamoxifen‐inducible Mfn2 KO astrocyte cultures (n = 3 independent experiments).
-
O–QBioenergetic parameters were analyzed in WT and Mfn2 KO astrocytes transfected with ChiMERA or control plasmid. (O) MMP (n = 30 cells from three independent cultures). (P) Analysis of ATP levels after 6 h treatment with 2‐DG (10 mM) (n = 3 independent experiments). (Q) Oxygen consumption rates. Proton leak was measured after application of oligomycin (1 µM), maximal after CCCP application (10 µM), and oligomycin (n = 5 independent experiments). Data are presented as mean ± SEM.
Data information: *P < 0.05, one‐way ANOVA followed by Tukey’s post hoc test except (C) that was used two‐tailed Student’s t‐test.
Figure EV4. ER‐Mfn2 localizes in the ER.
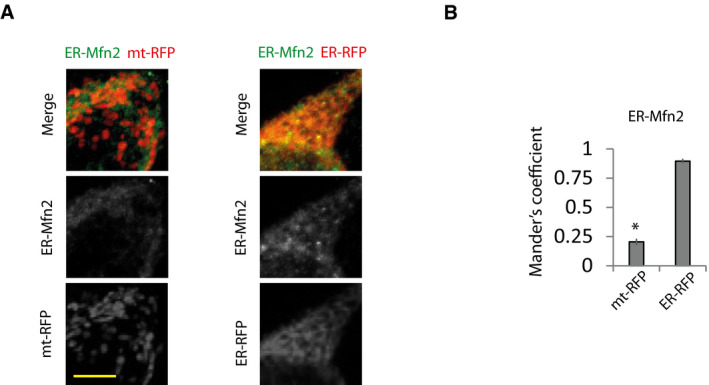
-
A, B(A) Representative immunofluorescence imaging of Mfn1/2 DKO MEFs transfected with ER‐Mfn2 and ER‐RFP or mt‐RFP and (B) colocalization of ER‐Mfn2 with mitochondria or ER analyzed by Mander’s coefficient (n = 10 cells analyzed in three independent experiments). Scale bar = 5 µm. Data are presented as mean ± SEM. *P < 0.05, two‐tailed Student’s t‐test.
Figure EV5. Mitochondria only targeted Mfn2 restores mitochondrial morphology to the same extent as Mfn2 in Mfn1/2 DKO cells.
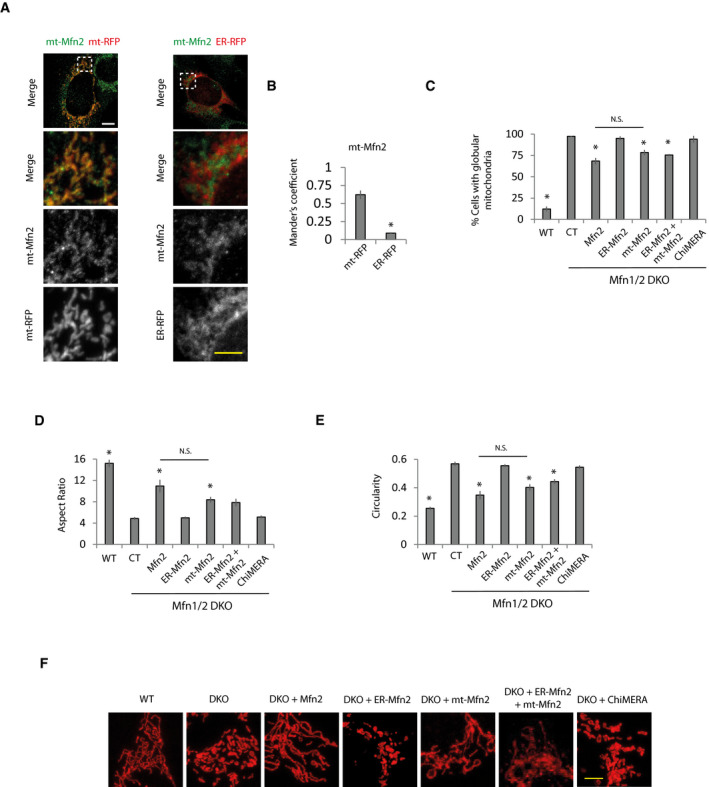
-
ARepresentative immunofluorescence imaging of Mfn1/2 DKO MEFs transfected with mt‐Mfn2 and ER‐RFP or mt‐RFP. Scale bar: 5 µm.
-
BColocalization of mt‐Mfn2 with mitochondria or ER analyzed by Mander’s coefficient (n = 10 cells analyzed in three independent experiments). Data are presented as mean ± SEM. *P < 0.05, two‐tailed Student’s t‐test.
-
C–Fmt‐Mfn2 expression restores mitochondrial morphology to the same extent as Mfn2 expression. Mfn1/2 DKO cells were transfected with control (CT), ChiMERA, or the indicated Mfn2 expression vectors and (C) percentage of cells displaying globular mitochondria (n = 310–668 cells analyzed in 3–6 independent experiments), (D) aspect ratio, and (E) circularity were calculated (n = 300 mitochondria analyzed from three independent experiments). Data are presented as mean ± SEM. (F) Representative images. White scale bar = 10 µm. Yellow scale bar = 5 µm.
Data information: *P < 0.05, one‐way ANOVA followed by Tukey’s post hoc test.
These results indicate that the potential for Mfn2 to maintain mitochondrial bioenergetics is independent of its capacity to modify mitochondrial morphology.
Mfn2 KO cells show alterations in ER and mitochondrial Ca2+ that may mediate impaired bioenergetic function
Mfn2 modulates the ER stress response (Ngoh et al, 2012; Muñoz et al, 2013) and ER stress regulates mitochondrial bioenergetics (Bravo et al, 2011; Balsa et al, 2019; Carreras‐Sureda et al, 2019). We analyzed the three branches of the unfolded protein response (UPR) and found increased activation of the ATF6 and Xbp1 pathways but no changes in the PERK‐eIF2a pathway (Appendix Fig S2A–C). Use of the chemical chaperone 4‐phenylbutyric acid (4PBA) blocked activation of the UPR in Mfn2 KO cells (Appendix Fig S2B and C), but 4PBA had no effect on bioenergetics defects in Mfn2 KO cells (Appendix Fig S2D–F).
Mfn2 has also been reported to modulate mitophagy (Chen & Dorn, 2013; Gong et al, 2015) which could be responsible for reduced mitochondrial bioenergetics in Mfn2 KO cells. However, we observed no changes in the colocalization of LC3 with mitochondria in Mfn2 KO cells or in protein levels of VDAC and Hsp60, two well‐known markers of mitochondrial mass (Appendix Fig S2G and H). Mfn1 and Mfn2 are ubiquitinated by Parkin and degraded upon induction of mitophagy (Gegg et al, 2010; Tanaka et al, 2010), but protein levels of Mfn1 remained unaltered in Mfn2 KO cells (Appendix Fig S2H). Thus, alterations in ER stress or mitophagy do not seem to be the mechanisms responsible for bioenergetics defects in Mfn2 KO cells.
Ca2+ transfer from the ER to mitochondria is essential to maintain mitochondrial energetics (Cárdenas et al, 2010). Ca2+ released from the ER is taken up by mitochondria through the MCU in regions of close contact between the two organelles (Rizzuto et al, 1993; Rizzuto et al, 1998; Baughman et al, 2011; De Stefani et al, 2011). A small fraction of Mfn2 (5–10%) localizes in the ER where it interacts with mitochondrial Mfns and tethers the organelles together (de Brito & Scorrano, 2008). However, whether Mfn2 tethers or promotes separation of the ER and mitochondria has been questioned (Sugiura et al, 2013; Filadi et al, 2015; Leal et al, 2016; Naon et al, 2016). Thus, under our experimental conditions, we tested whether Mfn2 is a tether or a spacer using three different approaches. First, we analyzed Mander’s coefficient, which quantifies the degree of colocalization between fluorophores based on pixel channel overlap. To this end, cells were co‐transfected with plasmids expressing RFP and GFP targeting the mitochondria and the ER, respectively. Using this technique, Mfn2 KO cells showed lower ER‐mitochondria colocalization (Fig 2A and B). This result was not an artifact as consequence of different mitochondrial morphology in WT and Mfn2 KO cells since no changes were observed in Mander’s coefficient in Mfn1 KO cells (Fig EV2A), whose mitochondria are even more globular than those of Mfn2 KO cells (Fig 1A–D). Furthermore, the differences in Mander’s coefficient were maintained when mitochondrial morphology was restored in Mfn2 KO cells by overexpressing DN‐Drp1 (Fig EV2B).
Figure 2. Mfn2 KO cells show alterations in ER and mitochondrial Ca2+ that may mediate impaired bioenergetic function.
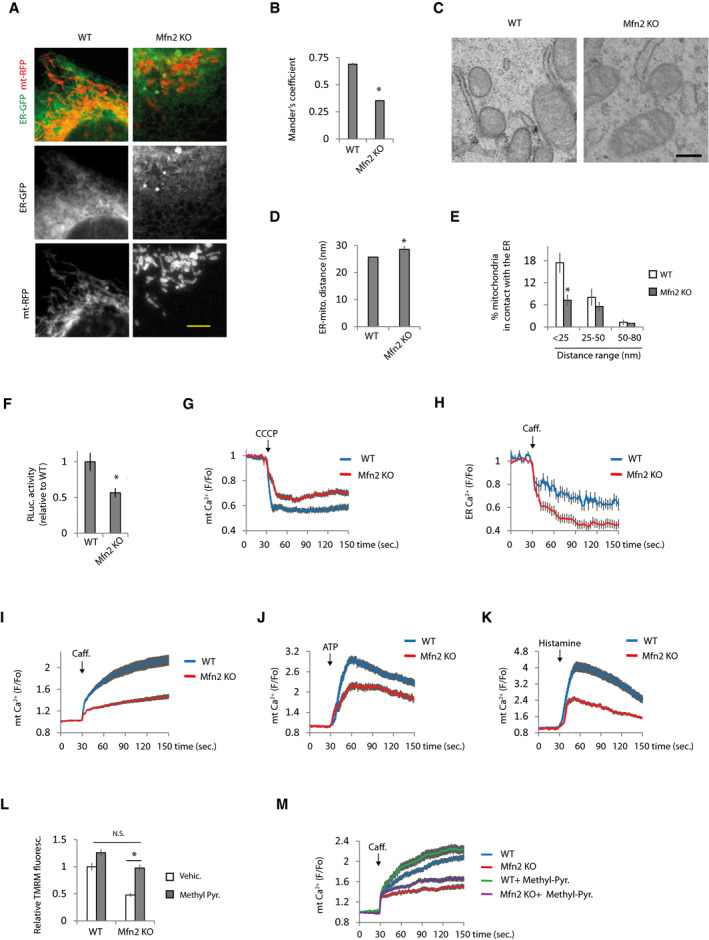
-
A, B(A) Representative images of WT and Mfn2 KO MEFs co‐transfected with plasmids encoding mitochondria‐targeted RFP and ER‐targeted GFP. Scale bar: 5 µm. (B) After 48 h, the cells were fixed and the degree of colocalization between ER and mitochondria based on red and green fluorescent pixel overlap was analyzed using Mander’s coefficient (n = 30 cells analyzed in three independent experiments). Data are presented as mean ± SEM. *P < 0.05, two‐tailed Student’s t‐test.
-
C–E(C) Representative EM micrographs of WT and Mfn2 KO cells. Scale bar: 250 nm. (D) Average of ER‐mitochondria distance (n = 111–168 mitochondria). *P < 0.05, Mann–Whitney test. Data are presented as mean ± SEM. (E) Quantification of the percentage of mitochondria in contact with the ER distributed at the indicated distance range (n = 3 independent experiments in which were analyzed 627–779 mitochondria from 68 to 98 cells). Data are presented as mean ± SEM. *P < 0.05, two‐tailed Student’s t‐test.
-
FWT and Mfn2 KO MEFs were co‐transfected with GFP and ERMITO‐RLuc plasmids. After 24 h, the transfected cells were sorted and plated for another 24 h, after which RLuc was analyzed, the activity of which requires the proper apposition of the ER and mitochondria to reconstitute the split RLuc (n = 3 independent experiments). Data are presented as mean ± SEM. *P < 0.05, two‐tailed Student’s t‐test.
-
Getermination of mitochondrial Ca2+. Fluorescence was recorded every second in WT and Mfn2 KO MEFs that were loaded with Rhod2. Mitochondrial Ca2+ was released with the addition of the uncoupler CCCP (10 µM) as indicated (n = 40 cells from 4 independent experiments).Data are presented as mean ± SEM.
-
HDetermination of ER Ca2+. WT and Mfn2 KO MEFs were transfected with plasmid encoding the ER‐located Ca2+ sensor LAR‐GECO1. After 48 h, fluorescence was recorded every 3 s. ER Ca2+ release was induced with caffeine (20 mM) as indicated (n = 30 cells from 3 independent experiments). Data are presented as mean ± SEM.
-
I–KMitochondrial Ca2+ was determined in WT and Mfn2 KO MEFs loaded with Rhod2. The effect of (I) caffeine‐induced (20 mM), (J) IP3R activation with ATP‐induced (100 µM) and (K) histamine‐induced (100 µM) ER Ca2+ release on mitochondrial Ca2+ was measured (n = 30 cells from 3 independent experiments). Data are presented as mean ± SEM.
-
L, M(L) MMP potential was measured in WT and Mfn2 KO cells treated with vehicle or methyl‐pyruvate (5 mM) for 24 h (n = 3 independent experiments). *P < 0.05, one‐way ANOVA followed by Tukey’s post hoc test. (M) Effect of methyl‐pyruvate in mitochondrial Ca2+ uptake after caffeine stimulation (n = 30 cells from three independent experiments). Data are presented as mean ± SEM.
Data information: Note that experiments in Figs 2D and 3C, and 2E and 3B were performed at the same time, so the values of WT and Mfn2 KO are the same but the figures have been split in two for the sake of linearity.
Second, we used electron microscopy (EM). The ER‐mitochondria contacts showed greater separation between the organelles in Mfn2 KO cells (Fig 2C and D) and the proportion of mitochondria separated from the ER by a distance of < 25 nm was diminished in Mfn2 KO cells (Fig 2E).
Third, we performed a split Renilla luciferase (RLuc) assay. This method is based on the reversible auto‐assembly of two non‐functional portions of RLuc (Stefan et al, 2007); when they are in sufficient proximity, the active enzyme is reconstituted. It was expressed ERMITO‐RLuc, a plasmid encoding a split RLuc in which the N‐terminus of RLuc targets mitochondria and the C‐terminus targets the ER. The N‐ and C‐terminuses are separated by an autocleavable peptide P2A to ensure equal expression of both portions of RLuc. ERMITO‐RLuc also contains a flexible linker to allow reconstitution of RLuc when the two organelles are separated by between 12 and 23.5 nm, in the optimal range of separation for Ca2+ uptake by mitochondria when it is released from the ER (Giacomello & Pellegrini, 2016). Activation of mTORC2 promotes MAM formation (Betz et al, 2013); hence, the mTOR pathway was manipulated to validate the capacity of ERMITO‐Rluc to detect changes in ER‐mitochondria contacts. We observed that mTOR inhibition decreased RLuc activity; meanwhile, mTOR activation increased RLuc activity (Fig EV2C and D).
The RLuc reconstitution approach also revealed a reduction in ER‐mitochondria contacts in Mfn2 KO cells (Fig 2F). This diminished RLuc activity in Mfn2 KO cells was not a consequence of diminished accessibility of the substrate, since there were not differences in RLuc activity between cell lines when full‐length RLuc was transfected (Fig EV2E). As expected, Mfn1 KO cells did not show any alterations in ER‐mitochondria contact using this approach (Fig EV2F). Since the results obtained with all three techniques indicated that ER‐mitochondria contact is reduced in Mfn2 KO cells, we therefore concluded that Mfn2 acts as an ER‐mitochondria tether.
Next, we analyzed whether as a consequence of diminished ER‐mitochondria contacts there are disturbances in mitochondrial and ER Ca2+ levels in WT and Mfn2 KO cells. The Mfn2 KO cells showed reduced mitochondrial Ca2+ levels measured with the mitochondrial Ca2+ sensor Rhod2 (Fig 2G). This was not an effect of diminished Rhod2 accumulation into mitochondria in Mfn2 KO cells because of reduced MMP, since a diminished cytoplasmic Ca2+ rise was also observed in Mfn2 KO cells after depolarizing mitochondria (Fig EV2G), as well as a reduced signal of the mitochondria‐targeted genetic Ca2+ sensor mt‐CEPIA (Fig EV2H). Although Mfn2 KO cells showed increased ER Ca2+ levels (Figs 2H, and EV2I and J), Ca2+ release via ryanodine receptor (RyR) when caffeine was applied resulted in lower Ca2+ transfer from the ER to mitochondria (Fig 2I). The source of caffeine‐induced Ca2+ release was the ER, since it was blocked by the RyR inhibitor dantrolene (Fig EV2K). Although there have been reports of Ca2+ transfer from the ER to mitochondria via RyR (Szalai et al, 2000; Santulli et al, 2015; Guidarelli et al, 2019), the best‐known route of ER‐mitochondria Ca2+ transfer is via the IP3R. Activation of the IP3R with ATP or histamine also showed reduced mitochondrial Ca2+ uptake in Mfn2 KO cells (Fig 2J and K). Disturbances in ER and mitochondrial Ca2+ in Mfn2 KO cells are in agreement with results reported previously (de Brito & Scorrano, 2008). However, in contrast to the results of de Brito and Scorrano, we observed reduced ER to mitochondria Ca2+ transfer using equal amounts of ATP (100 µM). However, de Brito and Scorrano had to stimulate Mfn2 KO cells with 20 times less ATP than WT cells (10 and 200 µM, respectively) to achieve equal ER Ca2+ release between cell lines and observe reduced mitochondrial Ca2+ uptake in Mfn2 KO cells.
MMP is a driving force for mitochondrial Ca2+ uptake. Consequently, reduced Ca2+ transfer from the ER to mitochondria in Mfn2 KO cells could be due to reduced MMP. To test this possibility, cells were treated with methyl‐pyruvate, which freely permeates the inner mitochondrial membrane (Rossi et al, 2020), increasing the MMP of the Mfn2 KO cells to the same levels as those in WT cells (Fig 2L). However, ER to mitochondria Ca2+ transfer in methyl‐pyruvate treated Mfn2 KO cells remained lower than in WT cells (Fig 2M), indicating that reduced MMP in Mfn2 KO cells is not the main cause of the reduced ER‐mitochondria Ca2+ transfer.
These results suggest that defects in mitochondrial bioenergetics in Mfn2 KO cells may be due to deregulation of the ER and mitochondrial Ca2+ levels.
Artificial ER‐mitochondria tethering corrects Ca2+ homeostasis and bioenergetic defects in Mfn2 KO cells
The loss of ER‐mitochondria contacts could be the cause of altered mitochondrial and ER Ca2+ levels. Thus, we wondered whether restoration of ER‐mitochondria apposition would be sufficient to correct disturbed Ca2+ homeostasis in Mfn2 KO cells. To avoid cell death caused by excessive ER‐mitochondria contact (Scorrano et al, 2003; Csordás et al, 2006; Cárdenas et al, 2016), we transfected the MEFs with a yeast expression vector encoding the artificial ER‐mitochondria tether ChiMERA (Kornmann et al, 2009) to obtain low expression of the artificial tether (Fig EV3A and B). ChiMERA consists of GFP flanked by ER and mitochondria localization signals; however although ChiMERA could be detected by three‐step immunofluorescence using anti‐GFP antibodies, no GFP signal was detected, confirming weak expression in MEFs (Fig EV3B). ChiMERA expression restored ER‐mitochondria contact (Fig 3A–D and Appendix Fig S3A and B). Concomitant with the restoration of ER‐mitochondria contact, ChiMERA corrected mitochondrial and ER Ca2+ levels (Fig 3E–H). In agreement with the hypothesis of disturbed mitochondrial Ca2+ being the cause of bioenergetics defects, the restoration of mitochondrial Ca2+ levels by artificially tethering the ER and mitochondria corrected Mfn2 KO bioenergetics defects (Fig 3I–M) without modifying mitochondrial morphology (Fig 3N–R).
Figure 3. Artificial ER‐mitochondria tethering corrects Ca2+ homeostasis and bioenergetics defects in Mfn2 KO cells.
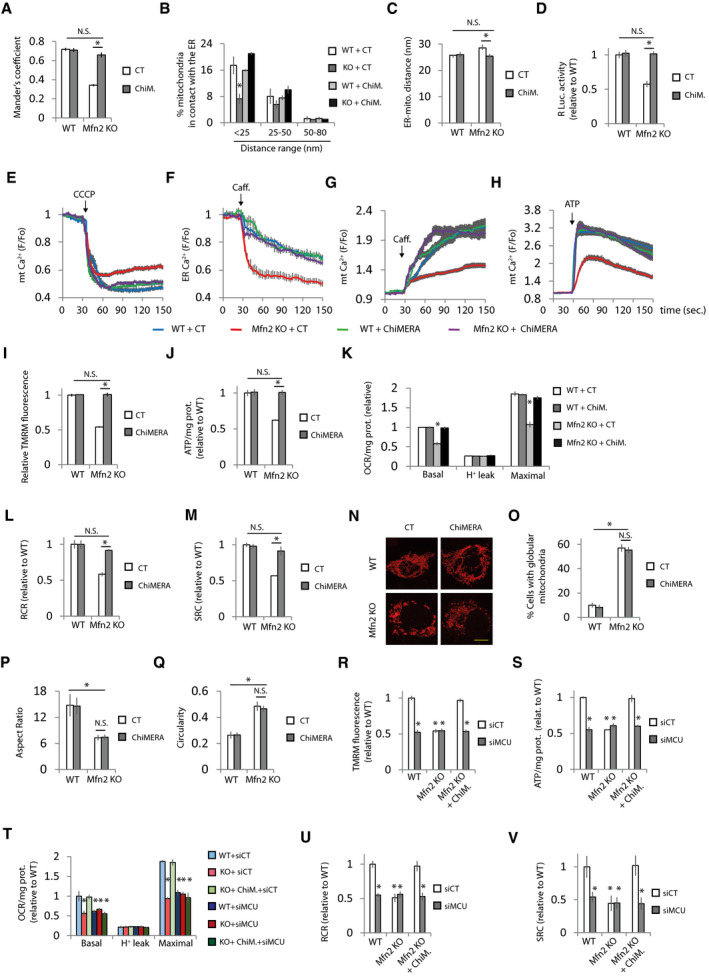
-
Amt‐RFP, ER‐GFP, and ChIMERA or control (CT). ER‐mitochondria colocalization was analyzed using Mander’s coefficient (n = 15 cells analyzed in three independent experiments). Data are presented as mean ± SEM.
-
B, CGFP and ChiMERA or control (CT). After 24 h, the transfected cells were sorted and plated for another 24 h, after which were processed for EM. (B) Quantification of the percentage of mitochondria in contact with the ER distributed at the indicated distance range (n = 3 independent experiments in which were analyzed 598–779 mitochondria from 68 to 98 cells). *P < 0.05, one‐way ANOVA followed by Tukey’s post hoc test. (C) Average of ER‐mitochondria distance (n = 111–193 mitochondria). Data are presented as mean ± SEM. *P < 0.05, Kruskal‐Wallis test followed by Dunn’s post hoc test.
-
DGFP, ERMITO‐Rluc, and ChiMERA or control plasmid. After 24 h, the transfected cells were sorted and plated for another 24 h, after which RLuc was analyzed (n = 3 independent experiments). Data are presented as mean ± SEM.
-
EGFP and ChiMERA or control (CT) plasmid. Mitochondrial Ca2+ was determined. Mitochondria were uncoupled with CCCP (10 µM) as indicated (n = 30 cells from 4 independent experiments). Data are presented as mean ± SEM.
-
FLAR‐GECO1 and ChiMERA or control (CT) plasmid. ER Ca2+ was determined. ER Ca2+ release was induced with caffeine (20 mM) as indicated (n = 30 cells from 3 independent experiments). Data are presented as mean ± SEM.
-
GGFP and ChiMERA or control (CT) plasmid. The effect of caffeine‐induced (20 mM) ER Ca2+ release on mitochondrial Ca2+ was measured (n = 30 cells from three independent experiments). Data are presented as mean ± SEM.
-
HGFP and ChiMERA or control (CT) plasmids. The effect of ATP‐induced (100 µM) ER Ca2+ release on mitochondrial Ca2+ was measured (n = 30 cells from three independent experiments). Data are presented as mean ± SEM.
-
IGFP and ChiMERA or control plasmid. MMP was determined. The values were normalized to surrounding untransfected cells (n = 90 cells analyzed in three independent experiments). Data are presented as mean ± SEM.
-
JGFP and ChiMERA or control plasmid. After 24 h, the transfected cells were sorted and plated for another 24 h, after which cells were treated with 2‐DG (10 mM) for 6 h, and ATP levels were measured (n = 3 independent experiments). Data are presented as mean ± SEM.
-
K–MGFP and ChiMERA or control plasmid. After 24 h, the transfected cells were sorted and plated for another 24 h, when (K) oxygen consumption was measured and (L) RCR and (M) SRC were calculated (n = 4 independent experiments). Data are presented as mean ± SEM.
-
N–Qmt‐RFP and ChiMERA or control plasmid, and after 48 h, they were fixed. N) Representative images. Scale bar = 5 µm. (O) The percentage of globular mitochondria (n = 175–408 cells analyzed in three independent experiments), (P) aspect ratio, and (Q) circularity were calculated (n = 300 mitochondria analyzed in three independent experiments). Data are presented as mean ± SEM.
-
R–VGFP and the indicated combination of non‐targeting siRNA (siCT), siRNA targeting MCU (siMCU), ChiMERA, or control (indicated as WT or Mfn2 alone) plasmids. (R) MMP was analyzed (n = 90 cells analyzed in three independent experiments). (S) ATP levels of GFP+ cells was analyzed after 24 h treatment with 2‐DG (10 mM) (n = 3 independent experiments). (T) OCR was analyzed and (U) cell respiratory control ration and (V) spare respiratory capacity were calculated (n = 3 independent experiments). Data are presented as mean ± SEM.
Data information: *P < 0.05, one‐way ANOVA followed by Tukey’s post hoc test. Note that experiments in Figs 2D and 3C, 2E and 3B, 3G and 4I, 3J and 4K, 3K and 4L, 3L and 4M, and 3M and 4N were performed at the same time, so the values of WT and Mfn2 KO transfected with control plasmid are the same but the figures have been split in two for the sake of linearity.
ER‐located IP3R releases Ca2+ that is taken up by mitochondria through the MCU. To verify that ChiMERA reestablishes mitochondrial energetics in Mfn2 KO cells by allowing Ca2+ transfer from ER to mitochondria, we knocked down MCU (Fig EV3C), which abolished the transfer of Ca2+ from the ER to mitochondria (Fig EV3D). In agreement with the need for the transfer of ER Ca2+ to mitochondria for the reestablishment of mitochondrial energetics by ChiMERA, MCU KD completely abolished the ChiMERA‐mediated bioenergetics recovery of bioenergetics and caused bioenergetic defects in WT cells (Fig 3R–V).
Inhibition of ER to mitochondria Ca2+ transfer through the IP3R with two unrelated inhibitors, 2‐aminoethoxydiphenylborane (2APB) and xestospongin C (XeC) (Fig EV3E and F), also blocked the bioenergetics rescue by ChiMERA (Fig EV3, EV4, EV5). The effect of the IP3R inhibitors was not due to loss of cell viability (Fig EV3J and K) or to a decrease in mitochondrial mass (Fig EV3L).
There are conflicting reports regarding whether MCU expression levels are diminished in Mfn2 KO MEFs (Filadi et al, 2015; Naon et al, 2016), probably because its expression changes with cell density (Naon et al, 2016). In our experimental conditions, we observed reduced MCU levels in Mfn2 KO cells; however, these were not recovered by ChiMERA expression (Fig EV3M). Therefore, although MCU levels may contribute to reduced mitochondrial Ca2+ uptake in Mfn2 KO cells, our results suggest that MCU expression levels are sufficient for Ca2+ uptake and that the limiting factor is the ER‐mitochondria apposition.
The role of Mfn2 as regulator of mitochondrial metabolism in different cell types is generally accepted. We tested whether the restoration of ER‐mitochondria contacts could also rescue the bioenergetics phenotype in a cell type other than MEFs. Mfn2 was deleted in primary astrocyte cultures of tamoxifen‐inducible Mfn2 KO mice (Fig EV3N). Mfn2 KO astrocytes showed bioenergetics defects that were also rescued by ChiMERA expression (Fig EV3, EV4, EV5).
These results indicate that Mfn2 regulates mitochondrial bioenergetics by allowing ER‐mitochondria apposition and proper Ca2+ exchange between these organelles.
ER‐located Mfn2 and mitochondrial Mfns are necessary to maintain mitochondrial bioenergetics
As expected, expression of Mfn2 rescued the ER‐mitochondria contacts, and consequently, Ca2+ disturbances and bioenergetics in Mfn2 KO cells (Appendix Fig S4A–I). The ER‐mitochondria tethering role of Mfn2 requires the presence of Mfn2 in the ER to form homo‐ and heterotypic interactions with Mfns located in mitochondria (de Brito & Scorrano, 2008). Given that the expression of Mfn1 is unaffected in Mfn2 KO fibroblasts (Appendix Figs S1A and S2H), we reasoned that expression of ER‐targeted Mfn2 could reestablish mitochondrial bioenergetics in Mfn2 KO cells through its interaction with endogenous Mfn1. As expected, expression of an Mfn2 construct in which the C‐terminus, after the transmembrane domain, had been replaced by the stretch of hydrophobic amino acids (IYFFT) that targets Mfn2 at the ER (ER‐Mfn2; (Rojo et al, 2002; de Brito & Scorrano, 2008) in Mfn2 KO cells, showed ER localization and could not be detected in mitochondria (Fig EV4A and B). ER‐Mfn2 increased ER‐mitochondrial contact in Mfn2 KO cells (Fig 4A and B, Appendix Fig S4A) without affecting mitochondrial morphology (Fig 4C–F). In agreement with the importance of ER‐mitochondria apposition for mitochondrial bioenergetics, ER‐Mfn2 partially normalized mitochondrial and ER Ca2+ levels (Fig 4G–I) as well as mitochondrial bioenergetics in Mfn2 KO cells (Fig 4J–N). The bioenergetic effect of ER‐Mfn2 in Mfn2 KO cells depended on contact with mitochondrial Mfn1, since expression of ER‐Mfn2 in double Mfn1/2 KO cells had no effect on ER‐mitochondria contacts (Fig 5A and B, Appendix Fig S5A) and consequently did not restore ER and mitochondrial Ca2+ levels (Fig 5C–E) or mitochondrial bioenergetics (Fig 5F–J). However, co‐expression with the ER‐located Mfn2 of an Mfn2 in which the Ct had been switched with the ActA mitochondrial localization peptide (Pistor et al, 1994), i.e., a mitochondrial only Mfn2 (mt‐Mfn2; Fig EV5A and B) (de Brito & Scorrano, 2008), completely restored ER‐mitochondria contact (Fig 5A and B) and reversed the ER and mitochondrial Ca2+ disturbances and the bioenergetics defects (Fig 5C–J), as did expression of full‐length Mfn2 did (Appendix Fig S5A–I). mt‐Mfn2 restored mitochondrial morphology to the same extent as Mfn2 in Mfn1/2 DKO cells (Fig EV5C–F), but had no effect on mitochondrial bioenergetics on its own (Fig 5F–J). This provides further evidence that Mfn2 bioenergetic properties rely on its ER‐mitochondria tethering rather than on its mitochondrial fusion function. The lack of effect of the expression of Mfn2 targeting a single organelle suggests that the rescue observed when mt‐ and ER‐targeted Mfn2 are expressed together is not consequence of the overexpression.
Figure 4. ER‐located Mfn2 is necessary to maintain mitochondrial bioenergetics.
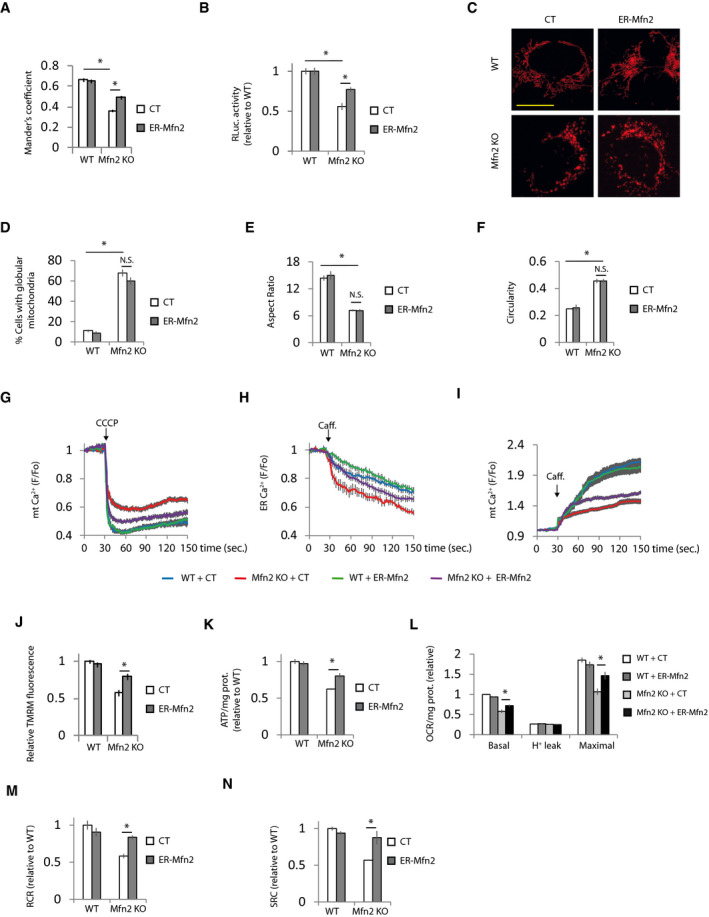
-
Amt‐RFP and ER‐GFP and ER‐targeted Mfn2 (ER‐Mfn2) or a control plasmid (CT). Mitochondria‐ER colocalization was analyzed using Mander’s coefficient (n = 15 cells analyzed in three independent experiments). Data are presented as mean ± SEM.
-
BGFP and ERMITO‐RLuc and ER‐Mfn2 or control plasmid. After 24 h, the transfected cells were sorted and plated for another 24 h, after which RLuc was analyzed (n = 4 independent experiments). Data are presented as mean ± SEM.
-
C–Fmt‐RFP and ER‐Mfn2 or control (CT) plasmid. After 48 h, the cells were fixed. (C) Representative images. Scale bar = 5 µm. (D) Mitochondrial morphology was analyzed (n = 113–422 cells analyzed in three independent experiments) and (E) aspect ratio and (F) circularity calculated (n = 360 mitochondria analyzed in three independent experiments). Data are presented as mean ± SEM.
-
GGFP and ER‐Mfn2 or control (CT) plasmid. Mitochondrial Ca2+ was determined. Mitochondria were uncoupled with CCCP (10 µM) as indicated (n = 30 cells from three independent experiments). Data are presented as mean ± SEM.
-
HLAR‐GECO1 and ER‐Mfn2 or control (CT) plasmid. ER Ca2+ levels were determined. ER Ca2+ release was induced with caffeine (20 mM) as indicated (n = 35 cells from four independent experiments). Data are presented as mean ± SEM.
-
IGFP and ER‐Mfn2 or control (CT) plasmid. The effect of caffeine‐induced (20 mM) ER Ca2+ release on mitochondrial Ca2+ was measured (n = 30 cells from three independent experiments). Data are presented as mean ± SEM.
-
J–NGFP and ER‐Mfn2 or control (CT) plasmid. (J) MMP was determined. The values were normalized to surrounding untransfected cells. (n = 90 cells analyzed in three independent experiments). (K) After 24 h, the transfected cells were sorted and plated for another 24 h, after which cells were treated with 2‐DG (10 mM) for 6 h, and ATP levels were measured (n = 3 independent experiments). (L) Oxygen consumption was measured, and (M) RCR and (N) SRC were calculated (n = 4 independent experiments). Data are presented as mean ± SEM.
Data information: *P < 0.05, one‐way ANOVA followed by Tukey’s post hoc test. Note that experiments in Fig 4A and Appendix Fig S4E, Fig 4D and Appendix Fig S4B, Figs 4K and 3I, 4L and 3J, 4M and 3K, 4N and 3L, 4G and Appendix Fig S4G, Fig 4H and Appendix Fig S4H, and Fig 4J and Appendix Fig S4J were performed at the same time, so the values of WT and Mfn2 KO transfected with control plasmid are the same but the figures have been split in two for the sake of linearity.
Figure 5. ER‐located Mfn2 and mitochondrial Mfns are necessary to maintain mitochondrial bioenergetics.
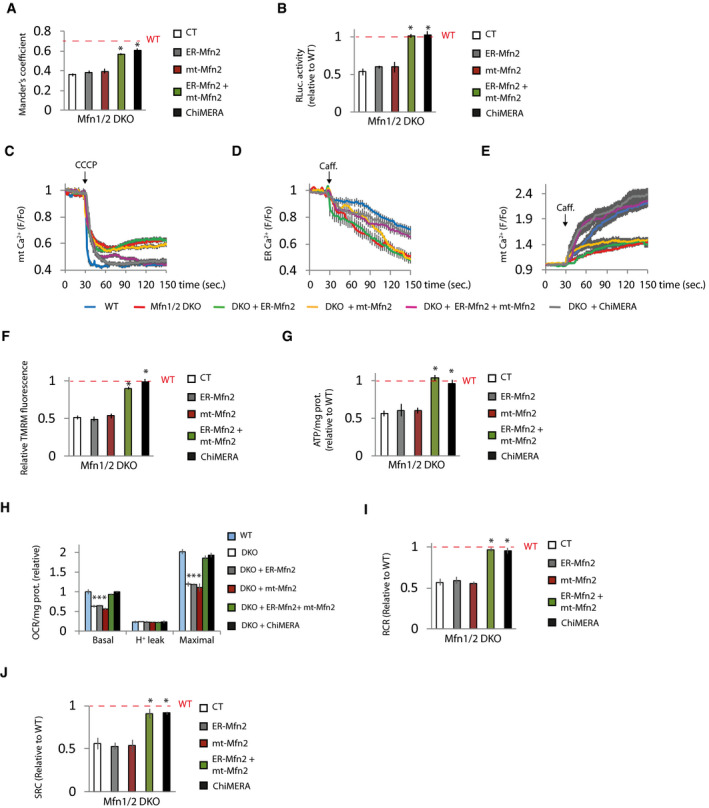
-
Amt‐RFP, ER‐GFP, and plasmids expressing the indicated proteins. Mitochondria‐ER colocalization was analyzed using Mander’s coefficient (n = 15 cells analyzed in three independent experiments). Data are presented as mean ± SEM.
-
BGFP and ERMITO‐RLuc and indicated expression vectors. After 24 h, the transfected cells were sorted and plated for another 24 h, after which RLuc was analyzed (n = 3 independent experiments). Data are presented as mean ± SEM.
-
CGFP and the indicated plasmids. After 48 h, mitochondrial Ca2+ was determined (n = 30 cells from 3 independent experiments). Data are presented as mean ± SEM.
-
DLAR‐GECO1 and the indicated plasmids. After 48 h ER Ca2+ was released with caffeine (20 mM) as indicated (n = 25–27 cells from three independent experiments). Data are presented as mean ± SEM.
-
EGFP and the indicated plasmid. After 48 h, the effect of caffeine‐induced (20 mM) ER Ca2+ release on mitochondrial Ca2+ was measured (n = 25 cells from three independent experiments). Data are presented as mean ± SEM.
-
F–JGFP and the indicated plasmids. (F) After 48 h, mitochondrial membrane potential was determined by measuring TMRM fluorescence. The values were normalized to surrounding untransfected cells. (n = 90 cells analyzed in three independent experiments). (G–I) After 24 h, the transfected cells were sorted and plated for another 24 h. (G) Cells were treated with 2‐DG (10 mM) for 6 h, and ATP levels were measured (n = 3 independent experiments). (H) Oxygen consumption was measured and (I) RCR and (J) SRC were calculated (n = 3 independent experiments). Data are presented as mean ± SEM.
Data information: The red dashed line indicates the WT cells results. *P < 0.05 versus Mfn1/2 DKO cells, one‐way ANOVA followed by Tukey’s post hoc test. Note that experiments in Fig 5A and Appendix Fig S5B, Fig 5C and Appendix Fig S5G, and Fig 5D and Appendix Fig S5H were performed at the same time, so the values of WT and Mfn2 KO transfected with control plasmid are the same but the figures have been split in two for the sake of linearity.
Ca2+ homeostasis and mitochondrial bioenergetics in Mfn1/2 DKO cells were also restored by ChiMERA, further supporting the notion that Mfn2 primarily controls mitochondrial bioenergetics by regulating ER‐mitochondria contact (Fig 5C–J). Taken together, these results indicate that the presence of Mfn2 in the ER is necessary to establish ER‐mitochondria communication through homo‐ and heterotypic interactions with mitochondrial Mfns, which are required for the Mfn2 bioenergetics function.
Mfn2‐mediated ER‐mitochondria tethering is necessary for neurite outgrowth
In order to strengthen these results obtained in fibroblasts, we sought a physiological process in which the ER‐mitochondria tethering role of Mfn2 is necessary. Neurite outgrowth requires increasing amounts of energy to fuel the anabolic pathways needed to enlarge neuronal processes (Mattson et al, 2008; Oruganty‐Das et al, 2012). Primary cortical neurons show extensive neurite growth between days 3 and 7 (Dotti et al, 1988). Throughout this period of neurite outgrowth, neurons increase ER‐mitochondria colocalization (Fig 6A and Appendix Fig S6A), which correlates with increased mitochondrial mass and Mfn2 expression (Fig 6B). Deletion of the Mfn2 gene from primary cortical cultures of tamoxifen‐inducible Mfn2 KO neurons (Fig 6C) resulted in less ER‐mitochondria colocalization (Fig 6D) with no change in mitochondrial mass (Fig 6C), which suggests that increased levels of Mfn2 in more mature neurons may play a key role in increased ER‐mitochondria colocalization during neuronal maturation. The co‐expression of ER‐Mfn2 and mt‐Mfn2, or ChiMERA, led to the complete rescue of ER‐mitochondria colocalization in Mfn2 KO neurons (Fig 6D and Appendix Fig S6B).
Figure 6. Mfn2‐mediated ER‐mitochondria tethering is necessary for neurite outgrowth.
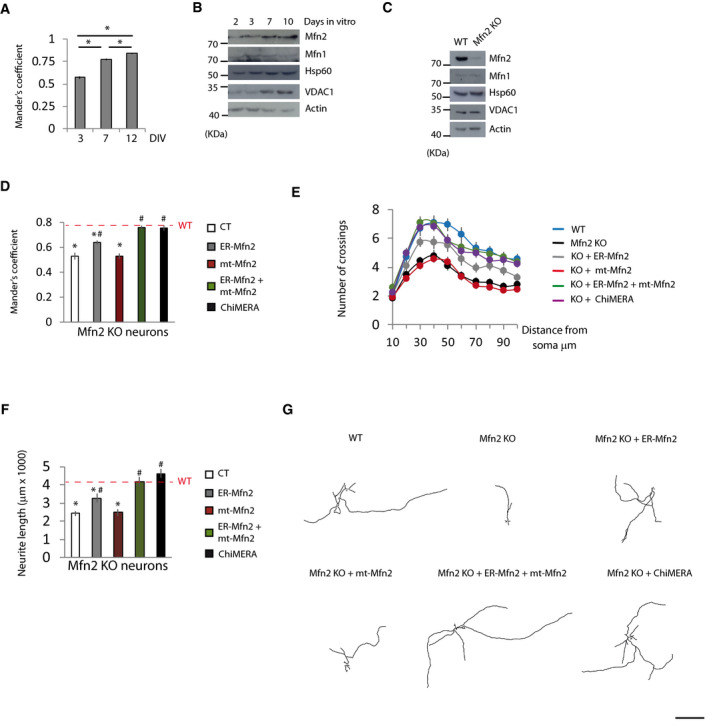
-
APrimary cortical neurons were transfected 48 h before the indicated day in vitro (DIV) with mt‐RFP and ER‐GFP, fixed at the indicated DIV, and mitochondria‐ER colocalization was analyzed using Mander’s coefficient (n = 21 neurons were analyzed in three independent experiments). Data are presented as mean ± SEM.
-
BRepresentative Western blots of the indicated proteins during the neuronal maturation in vitro (n = 3 independent experiments).
-
CRepresentative Western blots of the indicated proteins of WT and Mfn2 KO neurons at DIV7 (n = 3 independent experiments).
-
DPrimary neurons of tamoxifen‐inducible Mfn2 KO mice were transfected at DIV3 with ER‐GFP, mt‐RFP plus the indicated plasmids. Tamoxifen‐treated (for 72 h) and vehicle‐treated neurons (WT; red dashed line) were fixed, and mitochondria‐ER colocalization was analyzed using Mander’s coefficient (n = 15 neurons analyzed in three independent experiments). Data are presented as mean ± SEM.
-
E–G(E) Sholl analysis and (F) neurite length measurement and representative tracings (G) of primary cortical neurons of WT and Mfn2 KO mice co‐transfected at DIV4 with GFP and the indicated plasmids. After 72 h, the neurons were fixed, immunofluorescence with anti‐GFP antibodies was performed and neurite length was measured (n = 30 neurons from three independent experiments). Data are presented as mean ± SEM.
Data information: *P < 0.05 versus WT; # P < 0.05 versus Mfn2 KO neurons, one‐way ANOVA followed by Tukey’s post hoc test. Scale bar: 500 µm.
We have previously shown that Mfn2 KD causes reduced MMP in neurons (Martorell‐Riera et al, 2014). In agreement with the results shown here in fibroblasts, co‐expression of ER‐Mfn2 plus mt‐Mfn2, or ChiMERA, reversed the reduction in MMP in Mfn2 KO neurons (Appendix Fig S6C). These results indicate that, as in fibroblasts, Mfn2 localization in the ER is necessary to maintain mitochondrial metabolism in neurons, although additional bioenergetic parameters should be analyzed to confirm this conclusion.
The neuritic growth defects observed in vivo in Mfn2 KO mice (Chen et al, 2007) were confirmed in in vitro primary culture of Mfn2 KO neurons (Fig 6E–G). Mfn2 KO cortical neurons did not exhibit mitochondrial motility defects (Appendix Fig S6D); thus, it is highly unlikely that neuritic growth defects in Mfn2 KO neurons are due to alterations in mitochondrial trafficking. Reduced neurite complexity and length in Mfn2 KO neurons were completely reversed by overexpression of ER‐Mfn2 plus mt‐Mfn2, or by ChiMERA expression (Fig 6E–G). Interestingly, mt‐Mfn2 expression alone had no effect on neuronal architecture or length, whereas ER‐Mfn2 expression led to a partial reversal of the neurite defects. Together, these results indicate that Mfn2‐dependent ER‐mitochondria tethering is necessary for neuronal maturation in vitro.
Discussion
Mfn2 mutations are the most common cause of the neuropathy Charcot‐Marie‐Tooth disease type 2A (CMT2A) (Zuchner et al, 2004). Alterations in Mfn2 expression have also been observed in chronic neurodegenerative disorders, acute traumatic episodes such as stroke, and cardiometabolic diseases (Bach et al, 2003; Bach et al, 2005; Wang et al, 2009; Shirendeb et al, 2011; Marsboom et al, 2012; Martorell‐Riera et al, 2014). Given the well‐established role of Mfn2 as a regulator of mitochondrial metabolism (Bach et al, 2003; Sebastián et al, 2012; Schneeberger et al, 2013; Segales et al, 2013; Martorell‐Riera et al, 2014; Boutant et al, 2017; Tur et al, 2020; Xu et al, 2020) and the importance of mitochondrial metabolism in the physiopathology of these disorders, understanding the bioenergetic mechanism of Mfn2 may aid in the design of therapeutic approaches for disorders in which Mfn2 is involved.
In this study, we show that Mfn2 regulation of mitochondrial metabolism requires its location in the ER, where it regulates ER‐mitochondria tethering. This allows Ca2+ transfer from the ER to mitochondria, which enhances mitochondrial metabolism.
Mfn2 is primarily a mitochondrial protein. However, around 5–10% of Mfn2 is located in the ER, where it establishes homo‐ and heterotypic contact with Mfns located in the mitochondria (de Brito & Scorrano, 2008). Nonetheless, controversial remains over whether Mfn2 tethers or separates ER and mitochondria (Sugiura et al, 2013; Filadi et al, 2015; Leal et al, 2016; Naon et al, 2016). Under our experimental conditions, Mfn2 acts to tether them, as indicated by the results obtained using three different techniques. Mfn2 must be located in the ER for it to play its bioenergetic role, but the localization of Mfns, either Mfn1 or Mfn2, in mitochondria is also necessary to establish contacts between the ER and mitochondria. This is supported by our findings that ER‐targeted Mfn2 restores mitochondrial energetics in Mfn2 KO cells but not in Mfn1/2 DKO cells, unless an Mfn2 targeting only mitochondria is also expressed. In the absence of ER‐located Mfn2, this latter mitochondrial only Mfn2 has no effect on mitochondrial energetics. Interestingly, it has previously been shown that a cytosolic Mfn2 lacking the transmembrane domain, which is defective in terms of fusion, enhances mitochondrial respiration but the effect is completely lost in Mfn2 KO cells (Segales et al, 2013). This observation could be explained if cytosolic Mfn2 stabilizes contact between the ER and mitochondrial Mfns. A redox‐dependent oligomerization of Mfn2 has been reported that induces mitochondrial fusion (Shutt et al, 2012), but there are no data indicating whether cytosolic Mfn2 can stabilize transorganelle Mfn contact or its oligomerization.
Soon after the discovery of mitofusins, it was observed that expression of Mfn2 maintains mitochondrial bioenergetics more efficiently than expression of Mfn1, despite Mfn1 being more efficient at mediating mitochondrial fusion (Bach et al, 2003; Eura et al, 2003; Ishihara et al, 2004). Diminishment of Mfn2 expression as well as Mfn2 deletion have been shown to reduce mitochondrial respiration in different cell types (Bach et al, 2003; Sebastián et al, 2012; Schneeberger et al, 2013; Martorell‐Riera et al, 2014; Boutant et al, 2017; Li et al, 2019; Tur et al, 2020; Xu et al, 2020), including fibroblasts (Yao et al, 2019; Hu et al, 2020). However, Kawalec et al (2015) have reported increased oxygen consumption in Mfn2 KO MEFs, although they did not observe changes in RCR and found reduced MMP and mitochondrial ATP production, concluding that Mfn2 KO MEFs have impaired bioenergetics. The reason for this discrepancy in oxygen consumption remains unclear. One explanation could be that Kawalec et al measured the oxygen consumption in suspension cells after trypsinization. Interestingly, another two studies measuring oxygen consumption in suspension cells also found increased OCR in Mfn2 KO MEFs (Thaher et al, 2018; Wolf et al, 2019). Loss of cell‐matrix contact modifies mitochondrial metabolism (Werner & Werb, 2002; Irwin et al, 2003; Gough et al, 2009; Wegrzyn et al, 2009). One proposed mechanism regulating cell‐matrix‐dependent mitochondrial metabolism is through non‐canonical Stat3 functions, in which Stat3 localizes in mitochondria, the ER and MAM via integrin signaling (Gough et al, 2009; Wegrzyn et al, 2009; Visavadiya et al, 2016; Avalle et al, 2019). Consequently, it is possible that loss of cell anchorage specifically reduces respiration in WT cells but not in Mfn2 KO cells. However, further research is required to determine whether Mfn2 KO cells are insensitive to respiratory changes due to loss of outside‐in signaling and the mechanisms involved. Discrepancies have also been reported on how Mfn2 deletion affects mitochondrial respiration in brown adipocytes (Boutant et al, 2017; Mahdaviani et al, 2017). The reason for this discrepancy is unclear, but appears to be caused by systemic effects involving feeding, sex, and the temperature at which the mice were housed.
Several mechanisms have been proposed to explain how Mfn2 regulates mitochondrial metabolism. These include Mfn2 expression increasing the levels of different proteins in the electron transport chain (Pich et al, 2005); that Mfn2 stabilizes the formation of supercomplexes (Segales et al, 2013); and that it promotes the synthesis of coenzyme Q (Mourier et al, 2015). With the discovery that Mfn2 acts as an ER‐mitochondrial tether, it was shown that Mfn2 regulates Ca2+ transfer from the ER to mitochondria with the subsequent bioenergetic effect (de Brito & Scorrano, 2008; Chen et al, 2012; Seidlmayer et al, 2019). The increase in mitochondrial Ca2+ activates isocitrate dehydrogenase and α‐ketoglutarate dehydrogenase of TCA and PDH, which catalyze the conversion of pyruvate to acetyl‐CoA, which in turn fuels the TCA (Balaban, 2009). Mitochondria uptakes Ca2+ through the MCU, which has a low affinity for Ca2+. Thus, to be functional, microdomains with high Ca2+ concentrations are required. These are achieved via close apposition of the ER releasing channels with the MCU (Rizzuto et al, 1993; Rizzuto et al, 1998). The data reported here using organelle‐specific forms of Mfn2 and the artificial ER‐mitochondria tether ChiMERA strongly suggest that the main factor regulating mitochondrial energetics via Mfn2 is the promotion of ER‐mitochondria tethering, and therefore, the presence of Mfn2 in the ER is necessary. These results do not rule out the participation of the other mechanisms; in fact, they could be downstream effects of alterations in ER‐mitochondria contact. For instance, mitochondrial cristae remodeling in postprandial liver is produced through a mechanism linked to Mfn2‐mediated ER‐mitochondria tethering (Sood et al, 2014), and coenzyme Q biosynthetic proteins assemble into domains at ER‐mitochondria contact (Subramanian et al, 2019).
Our results are in contrast with the MCU KO mice grown in a mixed DC1 background being viable and with no remarkable bioenergetics deficit (Pan et al, 2013). However, inbreed MCU KO mice with a C57BL/6 background dies at E10.5‐13.5 (Pan et al, 2013). The different background could explain the metabolic phenotype in Mfn2 KO MEFs, which come from a C57BL/6 background (Chen et al, 2003). In fact, liver‐specific MCU KO in the C57BL/6 background showed reduced respiratory capacity (Tomar et al, 2019). The fact that MCU KO mice from a DC1 background is not born at Mendelian rate (Pan et al, 2013) also suggests a significant amount of embryonic lethality and adaptations in the surviving embryos during development. Indeed, extramitochondrial adaptations, including transcriptional reprogramming, have been described in transgenic mice expressing dominant‐negative MCU in myocardium from a CD1 background (Rasmussen et al, 2015).
Here, we show that the bioenergetic function of Mfn2 is independent of its pro‐fusion activity, in agreement with other results (Pich et al, 2005; Segales et al, 2013). This is supported by data showing that Mfn2 targeting the ER restores mitochondrial bioenergetics in Mfn2 KO fibroblasts without affecting mitochondrial morphology or transient expression of a dominant‐negative mutant of Drp1, and overexpression of Mfn1 restores mitochondrial morphology in Mfn2 KO fibroblasts without affecting the mitochondrial bioenergetics. Moreover, ER‐mitochondria tethering with the artificial tether ChiMERA restores mitochondrial bioenergetics in Mfn2 KO and Mfn1/2 DKO cells without affecting mitochondrial morphology. These results shown here refer to the mechanism by which Mfn2 regulates mitochondrial bioenergetics and explains why Mfn2, with a lower degree of fusion activity than Mfn1, is more efficient at enhancing mitochondrial bioenergetics.
ER‐mitochondria contact is an important regulator of Ca2+ homeostasis in non‐neuronal and neuronal cells (Rizzuto et al, 2012). Therefore, disturbances in ER‐mitochondria contacts have profound effects on neuronal physiology. Knocking down the ER‐resident tether PDZD8 shows dysregulated Ca2+ dynamics in dendrites (Hirabayashi et al, 2017) whose morphogenesis and maturation constitute an important process affecting the cognitive abilities of an organism (Mauceri et al, 2011). The scaffold protein disrupted‐in‐schizophrenia 1 (DISC1) which is linked to cognitive and emotional deficits is located in the MAM, where it interacts with IP3R and modulates ER‐mitochondria Ca2+ transfer (Park et al, 2017). Mitochondria and ER dysfunction has been linked to the progression of neurodegenerative disorders. In recent years, several reports have associated disturbances in ER‐mitochondria contact with neurodegenerative disorders such as Alzheimer’s disease and ALS (Stoica et al, 2014; Krols et al, 2016; Paillusson et al, 2016; De Mario et al, 2017; Area‐Gomez et al, 2018). The involvement of Mfn2 in ER‐mitochondria disturbances in these neurodegenerative disorders has not been studied, but interestingly, reductions in Mfn2 levels have been reported in these diseases (Wang et al, 2009; Manczak et al, 2011; Russell et al, 2013). Future research should study the role of Mfn2 in establishing ER‐mitochondria contact in these neurodegenerative disorders.
Axon and dendritic outgrowth requires high energy provision to support the considerable anabolic effort involved in generating new biomass to extend neurites’ supposes. Here we show that, in agreement with the reduction in dendritic branching in Purkinje cell‐specific Mfn2 KO mice (Chen et al, 2007), deletion of Mfn2 in the period a maximal neurite outgrowth results in diminished neurite growth and complexity in cortical neurons. Restoration of the Mfn2‐dependent ER‐mitochondria contact established during neuronal maturation in Mfn2 KO neurons by expression of ER‐targeted Mfn2 or the artificial tether ChiMERA completely rescued the neurite growth defects. These results indicate that restoration of ER‐mitochondria contact could be used as a therapeutic strategy when Mfn2 expression is impaired. For these purposes, it is critical to continue to advance our knowledge of how ER‐mitochondria tethering is regulated.
Materials and Methods
Cell culture and transfections
WT, Mfn1 KO, Mfn2 KO, and Mfn1/2 DKO mouse embryonic fibroblasts (MEFs) (Chen et al, 2003) and HeLa cells were maintained in DMEM, 1 g/l glucose supplemented with 10% fetal bovine serum (FBS) in subconfluent cultures. Transfections were performed with Lipofectamine 2000 (Thermo Scientific) in DMEM without FBS. Four hours after transfections, medium was replaced for fresh DMEM with 10% FBS. The experiments were performed 48 h after transfection. For knockdown experiments, 25 ng of mouse MCU (M‐062849‐01) or a non‐targeting control (D‐001810‐01‐05) siRNA‐SMART pool (Dharmacon) containing a pool of 4 siRNAs was used.
Cortical mouse neurons were cultured as described previously (Papadia et al, 2008) from E17.5 Mfn2‐floxed UBC‐CRE‐ERT2 mice with neurobasal growth medium (NBA) supplemented with B27 (Invitrogen) and 1% FBS. These mice ubiquitously express a tamoxifen‐inducible CRE recombinase (cre‐ERT2) that allows global inactivation of the Mfn2 locus, flanked by two LoxP sites upon (Z)‐4OH‐tamoxifen treatment. 4OH‐Tamoxifen (1 µM) was added to the neuronal cultures at DIV3; 72 h later was analyzed neuritic length and complexity. Transfections were performed with Lipofectamine 2000 (Thermo Scientific). Prior to transfection, the neurons were transferred from growth medium to a medium composed of 10% MEM (Invitrogen) and 90% salt‐glucose‐glycine (SGG) medium (SGG: 114 mM NaCl, 0.219% NaHCO3, 5.292 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 1 mM glycine, 30 mM glucose, 1 mM glutamine, 0.5 mM sodium pyruvate, 0.1% phenol red; osmolarity 325 mosm/l). After 2.5 h, the transfection medium was replaced with NBA. Experiments were performed 72 h after transfection.
Cortical mouse astrocytes were cultured from brain homogenates of P0‐P1 Mfn2‐floxed UBC‐CRE‐ERT2 mice. Astrocytes were grown in DMEM, 4.5 g/l glucose supplemented with 1% glutamine, and 10% FBS. After approximately 10 days, astrocytic cultures were agitated on a mechanical shaker at 200 rpm for 3 h to eliminate superficial microglia, washed twice with PBS, detached with Trypsin/EDTA, and re‐seeded. Transfections were performed with Lipofectamine 2000 (Thermo Scientific) 3 days before trypsinization. After transfection, tamoxifen (1 µM) was added to the medium.
Plasmids and cloning
DN‐Drp1 (Addgene plasmid 26049; Drp1‐K38A‐myc) and Mfn1‐Myc (Addgene plasmid 23212) were a gift from Dr. David Chan (Chen et al, 2003). ER‐LAR‐GECO1 (Addgene plasmid 61244) was a gift from Dr. Robert Campbell (Wu et al, 2014). ER‐GFP (Addgene plasmid 15108; pAC‐GFPC1‐Sec16β) was a gift from Dr. Tom Rapoport. ATF6‐Luc (Addgene plasmid #11976; p5xATF6‐GL3) was a gift from Dr. Ron Prywes (Wang et al, 2000). The ChiMERA was a gift from Dr. Benoît Kornmann (Kornmann et al, 2009). pCMV‐CEPIA3mt was a gift from Masamitsu Iono (Suzuki et al, 2014). ER‐Mfn2, Mfn2, and mt‐RFP were gifts from Dr. Manuel Rojo (Rojo et al, 2002). GFP‐LC3 was a gift from Dr. Antonio Zorzano (Sancho et al, 2012).
For construction of mt‐Mfn2 nucleotides from 404 to 1,842 of human Mfn2, cDNA was amplified with the primers: forward 5′‐ggg tag agg gca cag atg gcc atg‐3′ and reverse 5′‐ aaa gga tcc tgc aag ctt ggt cct gga tgt caa gga ggc cag −3′. The amplified product contains sequences that produce BamHI (italics) and HindIII (bold) restriction sites at the 3′ end. Next, it was substituted from nucleotide 430 to the 3′ end from Mfn2 plasmid with the PCR product digested with the same enzymes. To this construct, lacking transmembrane and C‐terminus of Mfn2, it was ligated an oligonucleotide with BamHI and HindIII sticky ends codifying for the mitochondrial targeting signal of ActA (amino acids 613–639).
Split Renilla luciferase plasmid (ERMITO‐RLuc) was synthesized and cloned in pCDN3.1(+) (Thermo Fisher Scientific) based in the split RLuc targeting the ER and mitochondria designed by Lim et al (2015), but both portions of the split RLuc are expressed in the same plasmid separated by an autocleavable peptide P2A (ATNFSLLKQAGDVEENPGP). The construct contains the N‐terminus of the RLuc (amino acids 1–91) fused at the N‐terminus to a flexible linker (GGGGS)6 and the mitochondrial targeting sequence of AKAP1 (amino acids 34–63) as well as the RLuc C‐terminus (amino acids 92–311) fused to the ER localization sequence of UBC6 (amino acids 233–250).
All newly generated constructs were confirmed by sequencing.
Oxygen consumption assay
Extracellular oxygen consumption was measured with the MitoXpres Xtra Oxygen Consumption Assay kit (Agilent) following the manufacturer’s instructions. Briefly, cells were grown in 96‐well plates to confluence. In the case of transfected cells, they were co‐transfected with GFP in 6–well plates, and 48 h later, they were cell sorted with transfected cells seeded in 96‐well plates and assayed 24 h later. Before the assay, the culture medium was replaced by fresh DMEM 10% FBS plus MitoXpress Xtra reagent (diluted 1:10) and two drops of mineral oil were promptly added. Plate was read at 380 nm excitation and 650 nm emission, every minute for 3 h. Proton leak and maximal OCR was assayed after the addition of oligomycin (1 µM) and CCCP (10 µM) and oligomycin, respectively. For analysis, after 20–40’ initial decrease in fluorescence due to O2 back diffusion through the body of the microplate reader (Will et al, 2006), the fluorescence signal starts to increase in a linear way for at least the following 2 h. Fluorescence values were plotted against time, and the slope of the linear portion was determined. The values were normalized by total protein levels, quantified using a Pierce BCA Protein Assay Kit (Thermo Scientific).
For OCR using the Agilent Seahorse XF24, 30,000 MEFs were plated in Seahorse XF24 Cell Culture Microplate. After 24 h, oxygen consumption was measured. Injection ports on the sensor cartridge were loaded with 5 μM oligomycin (oli; complex V inhibitor) to distinguish the percentage of oxygen consumption devoted to ATP synthesis and the percentage of oxygen consumption needed to overcome the natural proton leak across the inner mitochondrial membrane, 2 μM FCCP to calculate the maximal respiratory capacity of cells, 5 μM rotenone (complex I inhibitor), and 15 μM antimycin A (complex III inhibitor) to calculate the remaining respiration due to residual oxygen consumption. During the sensor calibration, cells were kept in a 37°C incubator without CO2 in 450 μl of respiration buffer (Seahorse XF Base Medium supplemented with 5 mM glucose, 2 mM glutamine, and 0.5 mM pyruvate). Plates were immediately placed into the calibrated Seahorse XF24 flux analyzer for mitochondrial bioenergetic analysis. OCR was normalized for total protein/well.
ATP measurement
ATP levels were measured using the ATPlite Luminescence Assay System (Perkin‐Elmer) on the Infinite 200 PRO multimode reader (TECAN) following the manufacturer's instructions. Cells were previously treated with 10 mM 2‐deoxy‐D‐glucose (2‐DG) for 6 h to inhibit glycolytic ATP production. ATP levels were normalized by total protein levels, quantified using Pierce BCA Protein Assay Kit (Thermo Scientific).
Luciferase reporter assay
Cells were transfected with ATF6‐driven firefly luciferase‐based reporter plasmid along with a Renilla expressing vector (CMV‐RL; Promega). Forty‐eight hours after transfection, luciferase assays were performed using the Dual Glo Luciferase Assay system (Promega) with firefly luciferase‐based reporter gene activity normalized to the Renilla control.
Cell viability
Cell viability was determined by counting the number of 4′,6′‐diamidino‐2‐phenylindole (DAPI)‐stained non‐pyknotic/necrotic nuclei as a percentage of the total. For each treatment, 550–1,000 cells were scored across several random fields within three independent experiments.
Imaging studies
Cells were visualized using a Zeiss LSM 880 confocal microscope. For MMP analysis, cells were visualized using Leica DMIRB microscope equipped with Leica DFC550 camera (Leica Lasertechnik GmbH, Mannheim, Germany). Images were taken and analyzed with the same settings. The analysis was performed using ImageJ software (Schneider et al, 2012).
Mitochondrial membrane potential
To quantify MMP, cells were loaded for 30 min at 37°C with tetramethylrhodamine methylester (TMRM; Sigma) at non‐quenching concentration (6 nM) in phenol red‐free HBSS with 10 mM HEPES and 5.5 mM glucose (Gibco, 14025‐050). Transfected cells were identified by co‐transfecting with GFP expression plasmid. Single cells were monitored, TMRM was excited at 540 nm and emission was measured using a 570‐nm filter. For analysis, ROIs of the same surface were drawn in areas completely occupied by mitochondria and fluorescence intensity was analyzed using ImageJ (Schneider et al, 2012). Background values obtained after mitochondrial depolarization with CCCP (10 µM, concentration that in titration experiments completely depolarized mitochondria in all the cell lines used) were subtracted from the TMRM fluorescence values. The MMP was compared to that observed in surrounding untransfected cells and subsequently expressed as a percentage of that observed in untransfected cells.
Mitochondrial morphology
Cells were transfected with mitochondria‐targeted RFP. After 48 h, cells were fixed, and nuclei were stained with DAPI. The cells with tubular or globular mitochondria were counted. Aspect ratio (AR) was calculated as major axis/minor axis, and circularity was calculated as 4*π*Area/Perimeter2.
Mitophagy
WT and Mfn2 KO MEFs were transfected with plasmids expressing GFP‐LC3 and mt‐RFP. After 48 h, cells were fixed in 4% paraformaldehyde in phosphate‐buffered saline. Images were taken at 63× magnification and the number of GFP+/RFP+ (yellow) puncta per cell were counted.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde, permeabilized, blocked, and incubated overnight at 4°C with primary antibodies anti‐Myc (1:500, M4439, Sigma) o anti‐GFP (1:750, A11122, Life technologies). For anti‐myc, binding was visualized using Alexa 488 secondary antibody; anti‐GFP binding was visualized using biotinylated secondary antibody/Cy3‐conjugated streptavidin.
ER‐mitochondria co‐localization
Cells were co‐transfected with plasmids expressing mitochondria‐targeted RFP and ER‐targeted GFP. Cross‐sectional images from fixed cells were taken at 63× magnification using a Zeiss LSM 880 confocal microscope (Life Imaging Services) and analyzed using JaCoP software (Bolte & Cordelières, 2006) and Manders’ algorithm (Manders et al, 1993).
Mitochondrial mobility
Cortical neurons were transfected with a plasmid expressing mt‐RFP at DIV3. After 72 h, series of confocal images using 63× magnification of axonal mitochondria from neurons kept in medium with 5 mM glucose were taken every 2 s for 5 min. Images were taken in an area approximately 50 µm away from the soma. Kymographs showing the temporal positioning of mitochondria through the axon were generated. Axons were straightened using the Straighten plugin in ImageJ, resliced, and then z‐projected (sum intensities). Immobile mitochondria were identified as straight lines and moving mitochondria were identified as diagonal lines.
Neuronal morphometric analysis
Cortical neurons were transfected with a plasmid expressing GFP at DIV3; the neurons were then fixed 72 h later with 4% paraformaldehyde, permeabilized, blocked, and incubated over‐night at 4°C with anti‐GFP antibody (1:750, A11122, Life Technologies). Antibody binding was visualized using a Alexa 488‐conjugated secondary antibody (1:500, Thermo). Preparations were mounted in VECTASHIELD Mounting Medium with DAPI (Vector Laboratories).
Images were taken blindly at 4× magnification using an Olympus BX61 microscope equipped with an Olympus DP70 camera. Neurites were manually traced and analyzed using the Simple Neurite Tracer software (Longair et al, 2011). For Sholl analysis, the shell interval was set at 10 µm.
Ca2+ measurement
Cells were cultured in 14‐mm round glass bottom microwell dishes (MatTek Corporation). Images were taken at 40× magnification using a Zeiss LSM 880 confocal microscope (Life Imaging Services) and analyzed using Fiji software.
For mitochondrial Ca2+ analysis, cells were loaded with 5 µM Rhod2 (Life Technologies) for 30 min at 4°C followed by extensive washout of the dye for 20 additional minutes with phenol red‐free HBSS medium at 37°C. Rhod2 was excited at 540 nm, and emission was measured using a 570‐nm filter. Images were taken every second. Mitochondria were uncoupled with CCCP (10 µM) to release matrix Ca2+ which allows an estimation of mitochondrial Ca2+ content.
For ER Ca2+ analysis, the cells were transfected with the ER‐targeted red fluorescent genetically encoded calcium indicator protein LAR‐GECO1. Caffeine (20 mM), ATP (100 µM), or histamine (100 µM) was applied to induce ER Ca2+ release.
Cytoplasmic Ca2+ was monitored with Fluo‐4 (Life Technologies). Cells were loaded 2 μM Fluo‐4 for 30 min at 37°C, after 2 washes with HBSS were incubated for additional 45 min at room temperature and excited at 488 nm and emission captured with a 516‐nm filter.
Electron microscopy
Cells were fixed in 2.5% glutaraldehyde in 0.1 M Sörensen’s phosphate buffer (0.1 M NaH2PO4/Na2HPO4, pH 7.2) for 1h at room temperature. A pellet was obtained followed by washing three times in Sörensen’s phosphate buffer. Cell pellets were postfixed in 1% osmium tetroxide, 0.8% potassium ferricyanide in Sörensen’s phosphate buffer for 2 h at 4°C, washed four times with Milli Q water, and placed overnight in Sörensen’s phosphate buffer at 4°C. Samples were then dehydrated with graded series of acetone solutions: 50, 70, 90, 98%, and 100%. Samples were gradually infiltrated with series of epon resin:acetone (1:3, 1:1, 3:1, and pure epon resin) and polymerized in the oven at 60°C for 48 h. The blocks were sectioned (60 nm) using an ultramicrotome. Stained sections with uranyl acetate and lead citrate were observed using a Jeol 1010 Transmission Electron Microscope, and pictures were taken using a 1Kx1K Megaview CCD camera. Mitochondria separated from the ER by < 80 nm were analyzed in blind fashion.
Renilla complementation
Cells were co‐transfected with ERMITO‐RLuc and GFP. After 48 h, the transfected cells were cell sorted and seeded in 96‐well plates. 24 h after plating, ViviRENTM Life Cell Substrate (Promega) was added, following the manufacturer’s instructions, and luciferase activity was read after 5 min. Luciferase values were normalized according to total protein levels, quantified using the Pierce BCA Protein Assay Kit (Thermo Scientific).
Western blot
Total cell lysates were boiled at 100°Cfor 5 min in 1.5× sample buffer (1.5 M Tris pH 6.8; 15% Glycerol; 3% SDS; 7.5% β‐mercaptoethanol; 0.0375% bromophenol blue). Gel electrophoresis was performed using 9% polyacrylamide gels. The gels were blotted onto PVDF membranes, which were then blocked for 1 h at room temperature with 5% (w/v) non‐fat dried milk in PBS with 0.05% Tween 20. The membranes were then incubated overnight at 4°Cwith the primary antibodies diluted in blocking solution as follows: Mfn2 (1:2,000; Ab56889; Abcam), Mfn1 (1:250; sc‐50330; Santa Cruz Biotechnology), Drp1 (1:1,000, 611112, BD Biosciences) VDAC1 (1:1,000; Ab14734; Abcam), HSP60 (1:2,000; Ab190828; Abcam), MCU (1:1,000; #14997; Cell Signaling Technology), p‐eIF2alpha (1:200; sc‐101670; Santa Cruz Biotechnology), eIF2alpha (1:1,000; ab242148; Abcam), myc (1:500, M4439, Sigma), GFP (1:1,000, A11122, Life Technologies), RFP (1:500, 600‐401‐379, Rockland), GFAP (1:500, Z0334, Dako), and actin (1:10,000, A4700, Sigma). To visualize Western blots, HRP‐based secondary antibodies were used followed by chemiluminescent detection on Kodak X‐Omat film.
RNA isolation, RT–PCR, and qPCR
RNA was isolated using a PureLinkTM RNA mini kit (Life Technologies). For qPCR, cDNA was synthesized from RNA using the SuperScript® III First‐Strand Synthesis SuperMix (Thermo Scientific) following the manufacturer’s instructions. qPCR was performed in a StepOne Real‐Time PCR System (Applied Biosystem) using the GoTaq qPCR Master Mix (Promega) according to the manufacturer’s instructions. The primers used were: MCU‐F: 5′‐AGA GGA GGA TCG GGG AAT CG −3′, MCU‐R: 5′‐ TCT TCA CGT CGT TCA GCG TC −3′, Xbp1‐F: 5′‐ GAG TCC GCA GCA GGT G‐3′, Xbp1‐R: 5′‐ GTG TCA GAG TCC ATG GGA‐3′ and 18S‐F: 5′‐ GTG GAG CGA TTT GTC TGG TT −3′, 18S‐R: 5′‐ CAA GCT TAT GAC CCG CAC TT −3′.
Statistical analysis
Statistical testing involved two‐tailed Student’s t‐tests. For any multiple comparisons within data sets, we used one‐way ANOVA followed by Tukey’s post hoc test. For nonparametric test, Mann–Whitney test was used for two comparisons, and for multiple comparisons, we used Kruskal–Wallis followed by Dunn’s post hoc test. All data are presented as mean ± SEM of at least three independent experiments (n).
Author contributions
SC‐D designed and performed most of the experiments and analyzed data; RL‐A, GR‐P, PT‐M, CM‐S, MS‐M designed and performed experiments and analyzed data; AG‐N and FV designed and performed Seahorse XF24 experiments, and analyzed data; MR and OMM‐E analyzed data and had critical input into the manuscript preparation; FXS conceived, designed and performed experiments, analyzed data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Appendix
Expanded View Figures PDF
Acknowledgements
This work was supported by research grants from the Spanish Ministerio de Economía y Competitividad/FEDER funds (SAF2014‐59872‐P and SAF2017‐86622‐C2‐1‐R to FXS), Velux Stiftung (Project 1262 to FXS) and La Caixa Foundation (LCF/PR/HR17/52150009 to OMM‐S). María de Maeztu Unit of Excellence (Institute of Neurosciences, University of Barcelona) MDM‐2017‐0729, Ministry of Science, Innovation and Universities. SC‐D is the recipient of a FI predoctoral fellowship from AGAUR/Generalitat de Catalunya and MS‐M was the recipient of a predoctoral fellowship from Fundación Tatiana Pérez de Guzmán el Bueno.
EMBO reports (2021) 22: e51954.
Data availability
This study includes no data deposited in external repositories.
References
- Area‐Gomez E, de Groof A, Bonilla E, Montesinos J, Tanji K, Boldogh I, Pon L, Schon EA (2018) A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis 9: 335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalle L, Camporeale A, Morciano G, Caroccia N, Ghetti E, Orecchia V, Viavattene D, Giorgi C, Pinton P, Poli V (2019) STAT3 localizes to the ER, acting as a gatekeeper for ER‐mitochondrion Ca2+ fluxes and apoptotic responses. Cell Death Differ 26: 932–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, Laville M, Guillet C, Boirie Y, Wallberg‐Henriksson Het al (2005) Expression of Mfn2, the charcot‐marie‐tooth neuropathy type 2A gene, in human skeletal muscle : effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor α and interleukin‐6. Diabetes 54: 2685–2693 [DOI] [PubMed] [Google Scholar]
- Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JRet al (2003) Mitofusin‐2 determines mitochondrial network architecture and mitochondrial metabolism. J Biol Chem 278: 17190–17197 [DOI] [PubMed] [Google Scholar]
- Balaban RS (2009) The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta Bioenerget 1787: 1334–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsa E, Soustek MS, Thomas A, Cogliati S, Garcia‐Poyatos C, Martin‐Garcia E, Jedrychowski M, Gygi SP, Enriquez JA, Puigserver P (2019) ER and nutrient stress promote assembly of respiratory chain supercomplexes through the PERK‐eIF2alpha axis. Mol Cell 74: 877–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher‐Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RLet al (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476: 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz C, Stracka D, Prescianotto‐Baschong C, Frieden M, Demaurex N, Hall MN (2013) mTOR complex 2‐Akt signaling at mitochondria‐associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci 110: 12526–12534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelières FP (2006) A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224: 213–232 [DOI] [PubMed] [Google Scholar]
- Boutant M, Kulkarni SS, Joffraud M, Ratajczak J, Valera‐Alberni M, Combe R, Zorzano A, Cantó C (2017) Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J 36: 1543–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, Quiroga C, Rodriguez AE, Verdejo HE, Ferreira Jet al (2011) Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 124: 2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610 [DOI] [PubMed] [Google Scholar]
- Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung K‐H, Yang J, Parker Iet al (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142: 270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárdenas C, Müller M, McNeal A, Lovy A, Jaňa F, Bustos G, Urra F, Smith N, Molgó J, Diehl Jet al (2016) Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep 14: 2313–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreras‐Sureda A, Jana F, Urra H, Durand S, Mortenson DE, Sagredo A, Bustos G, Hazari Y, Ramos‐Fernandez E, Sassano MLet al (2019) Non‐canonical function of IRE1alpha determines mitochondria‐associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat Cell Biol 21: 755–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, McCaffery JM, Chan DC (2007) Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130: 548–562 [DOI] [PubMed] [Google Scholar]
- Chen Y, Csordás G, Jowdy C, Schneider TG, Csordás N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem Set al (2012) Mitofusin 2‐containing mitochondrial‐reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca2+ crosstalk. Circ Res 111: 863–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW (2013) PINK1‐phosphorylated mitofusin 2 Is a parkin receptor for culling damaged mitochondria. Science 340: 471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnóczky G (2006) Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol 174: 915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mario A, Quintana‐Cabrera R, Martinvalet D, Giacomello M (2017) (Neuro)degenerated mitochondria‐ER contacts. Biochem Biophys Res Comm 483: 1096–1109 [DOI] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R (2011) A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476: 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotti C, Sullivan C, Banker G (1988) The establishment of polarity by hippocampal neurons in culture. J Neurosci 8: 1454–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eura Y, Ishihara N, Yokota S, Mihara K (2003) Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem 134: 333–344 [DOI] [PubMed] [Google Scholar]
- Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2015) Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc Natl Acad Sci USA 112: E2174–E2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334: 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Chau K‐Y, Rojo M, Schapira AHV, Taanman J‐W (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin‐dependent manner upon induction of mitophagy. Hum Mol Genet 19: 4861–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomello M, Pellegrini L (2016) The coming of age of the mitochondria‐ER contact: a matter of thickness. Cell Death Differ 23: 1417–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW (2015) Parkin‐mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350: aad2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE (2009) Mitochondrial STAT3 supports ras‐dependent oncogenic transformation. Science 324: 1713–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidarelli A, Fiorani M, Cerioni L, Cantoni O (2019) Calcium signals between the ryanodine receptor‐ and mitochondria critically regulate the effects of arsenite on mitochondrial superoxide formation and on the ensuing survival vs apoptotic signaling. Redox Biol 20: 285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera‐Cruz MS, Simmen T (2017) Over Six decades of discovery and characterization of the architecture at mitochondria‐associated membranes (MAMs). In organelle contact sites: from molecular mechanism to disease, Tagaya M, Simmen T (eds), pp 13–31. Singapore: Singapore; [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Kwon S‐K, Paek H, Pernice WM, Paul MA, Lee J, Erfani P, Raczkowski A, Petrey DS, Pon LAet al (2017) ER‐mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 358: 623–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Chen H, Zhang L, Lin X, Li X, Zhuang H, Fan H, Meng T, He Z, Huang Het al (2020) The AMPK‐MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 17: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin WA, Bergamin N, Sabatelli P, Reggiani C, Megighian A, Merlini L, Braghetta P, Columbaro M, Volpin D, Bressan GMet al (2003) Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat Genet 35: 367–371 [DOI] [PubMed] [Google Scholar]
- Ishihara N, Eura Y, Mihara K (2004) Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci 117: 6535–6546 [DOI] [PubMed] [Google Scholar]
- Kawalec M, Boratyńska‐Jasińska A, Beręsewicz M, Dymkowska D, Zabłocki K, Zabłocka B (2015) Mitofusin 2 deficiency affects energy metabolism and mitochondrial biogenesis in MEF cells. PLoS One 10: e0134162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P (2009) An ER‐mitochondria tethering complex revealed by a synthetic biology screen. Science 325: 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaltowski AJ, Cosso RG, Campos CB, Fiskum G (2002) Effect of Bcl‐2 overexpression on mitochondrial structure and function. J Biol Chem 277: 42802–42807 [DOI] [PubMed] [Google Scholar]
- Krols M, van Isterdael G, Asselbergh B, Kremer A, Lippens S, Timmerman V, Janssens S (2016) Mitochondria‐associated membranes as hubs for neurodegeneration. Acta Neuropathol 131: 505–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal NS, Schreiner B, Pinho CM, Filadi R, Wiehager B, Karlström H, Pizzo P, Ankarcrona M (2016) Mitofusin‐2 knockdown increases ER–mitochondria contact and decreases amyloid β‐peptide production. J Cell Mol Med 20: 1686–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Han J, Jia L, Hu X, Chen L, Wang Y (2019) PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell 10: 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesa M, Shirihai OS (2013) Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17: 491–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim Y, Cho I‐T, Schoel LJ, Cho G, Golden JA (2015) Hereditary spastic paraplegia‐linked REEP1 modulates endoplasmic reticulum/mitochondria contacts. Ann Neurol 78: 679–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longair MH, Baker DA, Armstrong JD (2011) Simple neurite tracer: open source software for reconstruction, visualization and analysis of neuronal processes. Bioinformatics 27: 2453–2454 [DOI] [PubMed] [Google Scholar]
- Mahdaviani K, Benador IY, Su S, Gharakhanian RA, Stiles L, Trudeau KM, Cardamone M, Enríquez‐Zarralanga V, Ritou E, Aprahamian Tet al (2017) Mfn2 deletion in brown adipose tissue protects from insulin resistance and impairs thermogenesis. EMBO Rep 18: 1123–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Calkins MJ, Reddy PH (2011) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet 20: 2495–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA (1993) Measurement of co‐localization of objects in dual‐colour confocal images. J Microsc 169: 375–382 [DOI] [PubMed] [Google Scholar]
- Marsboom G, Toth PT, Ryan JJ, Hong Z, Wu X, Fang Y‐H, Thenappan T, Piao L, Zhang HJ, Pogoriler Jet al (2012) Dynamin‐related protein 1–mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ Res 110: 1484–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martorell‐Riera A, Segarra‐Mondejar M, Muñoz JP, Ginet V, Olloquequi J, Pérez‐Clausell J, Palacín M, Reina M, Puyal J, Zorzano Aet al (2014) Mfn2 downregulation in excitotoxicity causes mitochondrial dysfunction and delayed neuronal death. EMBO J 33: 2388–2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60: 748–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauceri D, Freitag HE, Oliveira Ana MM, Bengtson CP, Bading H (2011) Nuclear calcium‐VEGFD signaling controls maintenance of dendrite arborization necessary for memory formation. Neuron 71: 117–130 [DOI] [PubMed] [Google Scholar]
- Mourier A, Motori E, Brandt T, Lagouge M, Atanassov I, Galinier A, Rappl G, Brodesser S, Hultenby K, Dieterich Cet al (2015) Mitofusin 2 is required to maintain mitochondrial coenzyme Q levels. J Cell Biol 208: 429–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz JP, Ivanova S, Sánchez‐Wandelmer J, Martínez‐Cristóbal P, Noguera E, Sancho A, Díaz‐Ramos A, Hernández‐Alvarez MI, Sebastián D, Mauvezin Cet al (2013) Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J 32: 2348–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernández‐Alvarez MIet al (2016) Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc Natl Acad Sci USA 113: 11249–11254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngoh GA, Papanicolaou KN, Walsh K (2012) Loss of mitofusin 2 promotes endoplasmic reticulum stress. J Biol Chem 287: 20321–20332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oruganty‐Das A, Ng T, Udagawa T, Goh Eyleen LK, Richter Joel D (2012) Translational control of mitochondrial energy production mediates neuron morphogenesis. Cell Metab 16: 789–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillusson S, Stoica R, Gomez‐Suaga P, Lau DHW, Mueller S, Miller T, Miller CCJ (2016) There's something wrong with my MAM; the ER–mitochondria axis and neurodegenerative diseases. Trends Neurosci 39: 146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DAet al (2013) The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15: 1464–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadia S, Soriano FX, Léveillé F, Martel M‐A, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska Vet al (2008) Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat Neurosci 11: 476–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Lee SB, Suh Y, Kim S‐J, Lee N, Hong J‐H, Park C, Woo Y, Ishizuka K, Kim J‐Het al (2017) DISC1 modulates neuronal stress responses by gate‐keeping ER‐mitochondria Ca2+ transfer through the MAM. Cell Rep 21: 2748–2759 [DOI] [PubMed] [Google Scholar]
- Pich S, Bach D, Briones P, Liesa M, Camps M, Testar X, Palacín M, Zorzano A (2005) The Charcot–Marie–Tooth type 2A gene product, Mfn2, up‐regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet 14: 1405–1415 [DOI] [PubMed] [Google Scholar]
- Pistor S, Chakraborty T, Niebuhr K, Domann E, Wehland J (1994) The ActA protein of Listeria monocytogenes acts as a nucleator inducing reorganization of the actin cytoskeleton. EMBO J 13: 758–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen TP, Wu Y, Joiner M‐L, Koval OM, Wilson NR, Luczak ED, Wang Q, Chen B, Gao Z, Zhu Zet al (2015) Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci USA 112: 9129–9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T (1993) Microdomains with high Ca2+ close to IP3‐sensitive channels that are sensed by neighboring mitochondria. Science 262: 744–747 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, De Stefani D, Raffaello A, Mammucari C (2012) Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol 13: 566–578 [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766 [DOI] [PubMed] [Google Scholar]
- Rojo M, Legros F, Chateau D, Lombès A (2002) Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci 115: 1663–1674 [DOI] [PubMed] [Google Scholar]
- Rossi A, Rigotto G, Valente G, Giorgio V, Basso E, Filadi R, Pizzo P (2020) Defective mitochondrial pyruvate flux affects cell bioenergetics in Alzheimer’s disease‐related models. Cell Rep 30: 2332–2348.e2310 [DOI] [PubMed] [Google Scholar]
- Russell AP, Wada S, Vergani L, Hock MB, Lamon S, Léger B, Ushida T, Cartoni R, Wadley GD, Hespel Pet al (2013) Disruption of skeletal muscle mitochondrial network genes and miRNAs in amyotrophic lateral sclerosis. Neurobiol Dis 49: 107–117 [DOI] [PubMed] [Google Scholar]
- Sancho A, Duran J, García‐España A, Mauvezin C, Alemu EA, Lamark T, Macias MJ, DeSalle R, Royo M, Sala Det al (2012) DOR/Tp53inp2 and Tp53inp1 constitute a metazoan gene family encoding dual regulators of autophagy and transcription. PLoS One 7: e34034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Xie W, Reiken SR, Marks AR (2015) Mitochondrial calcium overload is a key determinant in heart failure. Proc Natl Acad Sci USA 112: 11389–11394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger M, Dietrich M, Sebastián D, Imbernón M, Castaño C, Garcia A, Esteban Y, Gonzalez‐Franquesa A, Rodríguez I, Bortolozzi Aet al (2013) Mitofusin 2 in POMC Neurons Connects ER Stress with Leptin Resistance and Energy Imbalance. Cell 155: 172–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrepfer E, Scorrano L (2016) Mitofusins, from mitochondria to metabolism. Mol Cell 61: 683–694 [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139 [DOI] [PubMed] [Google Scholar]
- Sebastian D, Hernandez‐Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu Pet al (2012) Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA 109: 5523–5528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segales J, Paz JC, Hernandez‐Alvarez MI, Sala D, Munoz JP, Noguera E, Pich S, Palacin M, Enriquez JA, Zorzano A (2013) A form of Mitofusin 2 (Mfn2) lacking the transmembrane domains and the C‐terminal end stimulates metabolism in muscle and liver cells. Am J Physiol Endocrinol Metab 305: E1208–E1221 [DOI] [PubMed] [Google Scholar]
- Seidlmayer LK, Mages C, Berbner A, Eder‐Negrin P, Arias‐Loza PA, Kaspar M, Song M, Dorn GW, Kohlhaas M, Frantz Set al (2019) Mitofusin 2 is essential for IP3‐mediated sr/mitochondria metabolic feedback in ventricular myocytes. Front Physiol 10: 733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, Hemachandra Reddy P (2011) Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington's disease: implications for selective neuronal damage. Hum Mol Genet 20: 1438–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutt T, Geoffrion M, Milne R, McBride HM (2012) The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep 13: 909–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood A, Jeyaraju DV, Prudent J, Caron A, Lemieux P, McBride HM, Laplante M, Tóth K, Pellegrini L (2014) A Mitofusin‐2–dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc Natl Acad Sci USA 111: 16017–16022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan E, Aquin S, Berger N, Landry CR, Nyfeler B, Bouvier M, Michnick SW (2007) Quantification of dynamic protein complexes using Renilla luciferase fragment complementation applied to protein kinase A activities in vivo. Proc Natl Acad Sci USA 104: 16916–16921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau K‐F, Vizcay‐Barrena G, Lin W‐L, Xu Y‐F, Lewis Jet al (2014) ER‐mitochondria associations are regulated by the VAPB‐PTPIP51 interaction and are disrupted by ALS/FTD‐associated TDP‐43. Nat Commun 5: 3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian K, Jochem A, Le Vasseur M, Lewis S, Paulson BR, Reddy TR, Russell JD, Coon JJ, Pagliarini DJ, Nunnari J (2019) Coenzyme Q biosynthetic proteins assemble in a substrate‐dependent manner into domains at ER–mitochondria contacts. J Cell Biol 218: 1353–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride H, Fukuda T, Matsushita Net al (2013) MITOL regulates endoplasmic reticulum‐mitochondria contacts via mitofusin2. Mol Cell 51: 20–34 [DOI] [PubMed] [Google Scholar]
- Suzuki J, Kanemaru K, Ishii K, Ohkura M, Okubo Y, Iino M (2014) Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat Commun 5: 4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai G, Csordás G, Hantash BM, Thomas AP, Hajnóczky G (2000) Calcium signal transmission between ryanodine receptors and mitochondria*. J Biol Chem 275: 15305–15313 [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen D‐F, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaher O, Wolf C, Dey PN, Pouya A, Wüllner V, Tenzer S, Methner A (2018) The thiol switch C684 in Mitofusin‐2 mediates redox‐induced alterations of mitochondrial shape and respiration. Neurochem Int 117: 167–173 [DOI] [PubMed] [Google Scholar]
- Tomar D, Jaña F, Dong Z, Quinn WJ III, Jadiya P, Breves SL, Daw CC, Srikantan S, Shanmughapriya S, Nemani Net al (2019) Blockade of MCU‐mediated Ca2+ uptake perturbs lipid metabolism via PP4‐dependent AMPK dephosphorylation. Cell Rep 26: 3709–3725.e3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tur J, Pereira‐Lopes S, Vico T, Marín EA, Muñoz JP, Hernández‐Alvarez M, Cardona P‐J, Zorzano A, Lloberas J, Celada A (2020) Mitofusin 2 in macrophages links mitochondrial ROS production, cytokine release, phagocytosis, autophagy, and bactericidal activity. Cell Rep 32: 108079 [DOI] [PubMed] [Google Scholar]
- Vance JE (2014) MAM (mitochondria‐associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta 1841: 595–609 [DOI] [PubMed] [Google Scholar]
- Visavadiya NP, Keasey MP, Razskazovskiy V, Banerjee K, Jia C, Lovins C, Wright GL, Hagg T (2016) Integrin‐FAK signaling rapidly and potently promotes mitochondrial function through STAT3. Cell Commun Signal 14: 32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Lee H‐g, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci 29: 9090–9103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R (2000) Activation of ATF6 and an ATF6 DNA binding site by the ER stress response. J Biol Chem 275: 27013–27020 [DOI] [PubMed] [Google Scholar]
- Wegrzyn J, Potla R, Chwae Y‐J, Sepuri N, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka Aet al (2009) Function of mitochondrial Stat3 in cellular respiration. Science 323: 793–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner E, Werb Z (2002) Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J Cell Biol 158: 357–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will Y, Hynes J, Ogurtsov VI, Papkovsky DB (2006) Analysis of mitochondrial function using phosphorescent oxygen‐sensitive probes. Nat Protoc 1: 2563–2572 [DOI] [PubMed] [Google Scholar]
- Wolf C, Zimmermann R, Thaher O, Bueno D, Wüllner V, Schäfer MKE, Albrecht P, Methner A (2019) The charcot–marie tooth disease mutation R94Q in MFN2 decreases ATP production but increases mitochondrial respiration under conditions of mild oxidative stress. Cells 8: 1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Prole D, Shen Yi, Lin Z, Gnanasekaran A, Liu Y, Chen L, Zhou H, Chen SR, Usachev Yet al (2014) Red fluorescent genetically encoded Ca2+ indicators for use in mitochondria and endoplasmic reticulum. Biochem J 464: 13–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Wu Z, He Y, Chen Z, Xu K, Yu W, Fang W, Ma C, Moqbel S, Ran Jet al (2020) MFN2 contributes to metabolic disorders and inflammation in the aging of rat chondrocytes and osteoarthritis. Osteoarthritis Cartilage 28: 1079–1091 [DOI] [PubMed] [Google Scholar]
- Yao CH, Wang R, Wang Y, Kung CP, Weber JD, Patti GJ (2019) Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife 8: e41351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züchner S, Mersiyanova IV, Muglia M, Bissar‐Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek Jet al (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐Tooth neuropathy type 2A. Nat Genet 36: 449–451 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Appendix
Expanded View Figures PDF
Data Availability Statement
This study includes no data deposited in external repositories.