

Abstract
Epithelial plasticity, or epithelial‐to‐mesenchymal transition (EMT), is a well‐recognized form of cellular plasticity, which endows tumor cells with invasive properties and alters their sensitivity to various agents, thus representing a major challenge to cancer therapy. It is increasingly accepted that carcinoma cells exist along a continuum of hybrid epithelial–mesenchymal (E‐M) states and that cells exhibiting such partial EMT (P‐EMT) states have greater metastatic competence than those characterized by either extreme (E or M). We described recently a P‐EMT program operating in vivo by which carcinoma cells lose their epithelial state through post‐translational programs. Here, we investigate the underlying mechanisms and report that prolonged calcium signaling induces a P‐EMT characterized by the internalization of membrane‐associated E‐cadherin (ECAD) and other epithelial proteins as well as an increase in cellular migration and invasion. Signaling through Gαq‐associated G‐protein‐coupled receptors (GPCRs) recapitulates these effects, which operate through the downstream activation of calmodulin‐Camk2b signaling. These results implicate calcium signaling as a trigger for the acquisition of hybrid/partial epithelial–mesenchymal states in carcinoma cells.
Keywords: calcium, cellular plasticity, E‐cadherin, partial EMT
Subject Categories: Cancer, Membrane & Intracellular Transport, Signal Transduction
Cancer cells exist along a continuum of hybrid or partial EMT states. This is underpinned by a calcium‐dependent molecular mechanism in various carcinomas, resulting in the internalization of epithelial proteins.
Introduction
Cellular plasticity in cancer emerges from an inherent instability of tumor cells and heterogeneous signals in the tumor microenvironment (Yuan et al, 2019). Epithelial plasticity, or epithelial‐to‐mesenchymal transition (EMT), is a well‐recognized form of cellular plasticity whereby cancer cells lose their epithelial features and acquire the more motile characteristics of fibroblasts, leukocytes, and neural crest derivatives (Yang et al, 2020). Epithelial plasticity endows tumor cells with invasive properties, alters their sensitivity to various therapeutic agents, and thus represents a major challenge to effective cancer therapy (Yuan et al, 2019).
EMT is associated classically with changes in gene transcription mediated by “EMT transcription factors” (EMT‐TFs), including members of the Snail, Twist, and Zeb families of DNA‐binding proteins (Nieto et al, 2016). In response to extracellular signals such as TGFβ, EMT‐TFs repress genes associated with epithelial identity (e.g., adherens and tight junction components) and induce genes associated with mesenchymal identity (e.g., intermediate filament proteins and matrix components) (Nieto et al, 2016). At their extremes, epithelial (E) and mesenchymal (M) cells have vastly different phenotypic and functional characteristics. However, it is increasingly accepted that carcinoma cells exist along a continuum of hybrid E‐M states (Grigore et al, 2016; Dongre & Weinberg, 2019; Jolly et al, 2019; Yang et al, 2020). As reversible transitions between E and M phenotypes appear to be important at different stages of metastatic spread (Ocaña et al, 2012; Tsai et al, 2012; Tran et al, 2014; Kröger et al, 2019), carcinoma cells exhibiting such hybrid or partial EMT (P‐EMT) states can more easily navigate barriers to metastatic spread and may thus possess greater metastatic competence (Nieto et al, 2016; Zhang & Weinberg, 2018). Yet, the mechanisms by which cells adopt such intermediate states are unknown.
Using an autochthonous model of pancreatic ductal adenocarcinoma (PDA), we reported that P‐EMT is a common feature of stochastically arising pancreatic tumors (Aiello et al, 2018). P‐EMT in vivo is not associated with transcriptional repression of epithelial genes but rather occurs through a non‐canonical program characterized by the relocalization of epithelial proteins (Aiello et al, 2018). Importantly, this phenomenon is not limited to PDA but also describes the molecular events underlying P‐EMT in a variety of human carcinomas. In this study, we investigated the molecular signals underlying this newly described plasticity program.
Results and Discussion
Ca2+ signaling is enriched in P‐EMT tumor cells
For clues regarding the molecular pathways responsible for P‐EMT, we examined transcriptomic data from our prior in vivo analysis (Aiello et al, 2018). In that study, we used flow cytometry for the epithelial protein E‐cadherin (ECAD) to isolate tumor cells exhibiting an epithelial phenotype (surface ECAD+) or a mesenchymal phenotype (surface ECAD−) and performed RNA‐seq. This analysis stratified tumors into two “EMT subtypes”—those that employed a classical or canonical EMT program (C‐EMT), involving transcriptional repression of epithelial genes, and those that employed an alternative partial EMT program (P‐EMT) characterized by post‐translational internalization of epithelial proteins including ECAD (Aiello et al, 2018 and Fig EV1A and B).
Figure EV1. Enriched pathways in C‐EMT.
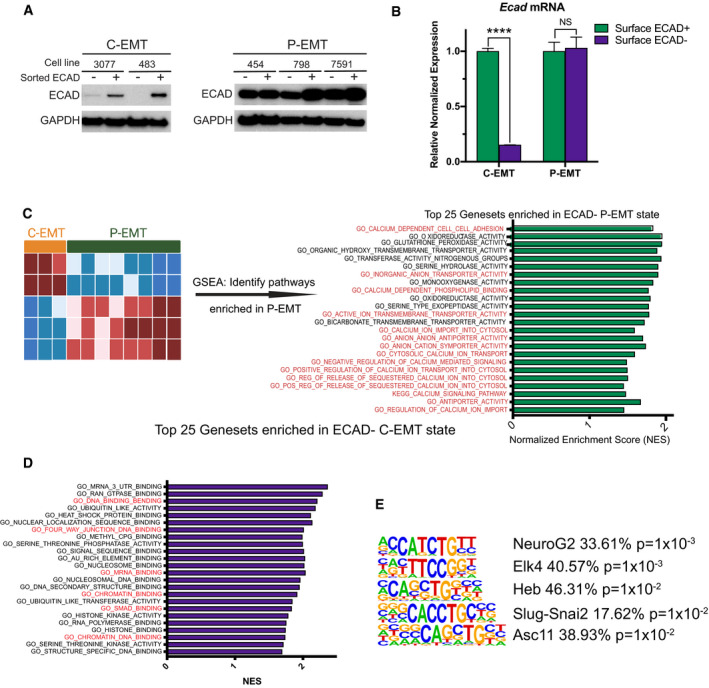
- Western blot comparing ECAD in sorted ECAD+ versus ECAD− cells from two C‐EMT and three P‐EMT murine PDA cell lines.
- qPCR comparing Ecad mRNA in sorted ECAD+ versus ECAD− cells from a P‐EMT (454) and C‐EMT (483) murine PDA cell line. Experiment was performed in triplicate. Statistical analysis by Student’s unpaired t‐test (****P < 0.0001; non‐significant (NS); ± SEM).
- Left: Bioinformatic schematic for GSEA comparing ECAD− versus ECAD+ murine PDA tumor cells in either P‐EMT tumors (n = 8) or C‐EMT (n = 3). Right: GO and KEGG pathways upregulated in P‐EMT tumors. Normalized enrichment score (NES) for the top 25 pathways with a false discovery rate (FDR) < 0.15 are shown, ordered by FDR. Red text indicates pathways of interest.
- Gene ontology (GO) and KEGG pathways upregulated in C‐EMT tumors. NES score for the top 25 pathways with an FDR < 0.15 are shown ordered by FDR value. Red text indicates pathways of interest.
- HOMER Motif analysis comparing C‐EMT ECAD− tumor cells versus C‐EMT ECAD+ tumor cells. The five most enriched pathways shown. Statistical analysis performed in HOMER.
Source data are available online for this figure.
At a global transcriptional level, both subtypes carried robust EMT signatures (Aiello et al, 2018). Hence, our re‐analysis focused on pathways that were exclusively enriched in P‐EMT tumors relative to C‐EMT tumors. To this end, we performed gene set enrichment analysis (GSEA) of the entire dataset—comparing the ECAD+ and ECAD− cell populations of each EMT subtype—and ranked those pathways that were uniquely associated with the P‐EMT program (Fig EV1C). Strikingly, 15 of the top 25 P‐EMT‐associated gene sets were related to ion transport, especially calcium (Figs 1A and EV1C). Likewise, HOMER motif analysis of differentially expressed genes in ECAD− versus ECAD+ cells revealed strong enrichment for nuclear factor of activated T cells (NFAT), a calcium‐regulated transcription factor, only in P‐EMT tumors (Fig 1B). As expected, a complementary analysis of C‐EMT tumors revealed enrichment for pathways related to mRNA binding, DNA/chromatin binding, and SMAD signaling (Fig EV1D), and enrichment for binding sites for the EMT‐TF Slug‐Snai2 (Fig EV1E). Based upon these results, we hypothesized that calcium signaling may contribute to P‐EMT.
Figure 1. Ca2+ signaling is enriched in P‐EMT cells.
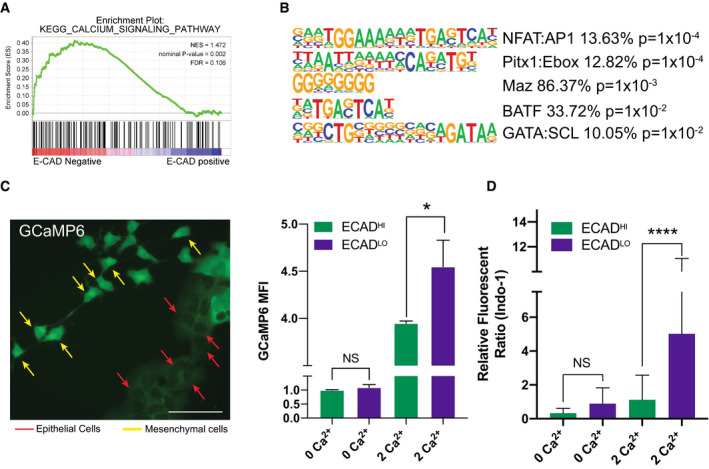
- Representative calcium signature enriched in P‐EMT tumors (comparing ECAD− versus ECAD+ tumor cells). NES, nominal P‐value, and FDR are shown. Statistical analysis performed in GSEA.
- HOMER motif analysis comparing P‐EMT ECAD− versus ECAD+ tumor cells. The five most enriched motifs are shown. Statistical analysis performed in HOMER.
- Representative image of tumor cells transduced with GCaMP6 (Left). Yellow arrows (mesenchymal cells), red arrows (epithelial cells), scale bar = 50 µm. GCaMP6 mean fluorescent intensity (MFI) in ECADHIGH or ECADLOW cells cultured in indicated medium (0 Ca2+ or 2 Ca2+) for 1 h (Right). Representative of three independent murine PDA cell lines run in triplicate. Statistical analysis by Student’s unpaired t‐test (*P < 0.05; non‐significant (NS); ± SD).
- Flow cytometry analysis of ECADHIGH or ECADLOW cells loaded with Indo‐1 and cultured in indicated medium (0 Ca2+ or 2 Ca2+) for 1 h. Representative of two independent cell lines (n indicates the number of cells analyzed in each group, ECADHIGH 0 Ca2+ n = 4,963, ECADLOW 0 Ca2+ n = 14, ECADHIGH 2 Ca2+ n = 6,575, ECADLOW 2 Ca2+ n = 29). Representative of 2 independent murine PDA cell lines run in triplicate. Statistical analysis by Student’s unpaired t‐test (****P < 0.0001; non‐significant (NS); ±SD).
To test this hypothesis, we first asked whether the E‐M state of a cell is correlated with intracellular calcium concentration ([Ca2+]). We previously demonstrated that in the absence of any EMT inducer, carcinoma cells exist in an equilibrium between epithelial and mesenchymal states, whereby EMT and its reverse process mesenchymal‐to‐epithelial transition (MET) occur spontaneously (Aiello et al, 2018; Yuan et al, 2020). We therefore introduced a genetically encoded Ca2+ indicator protein, GCaMP6, into three murine PDA cell lines, designated cells as either E or M based upon morphology (see Materials and Methods), and measured relative levels of fluorescence as a function of cellular state. As shown in Fig 1C, cells with an E morphology (typically part of a colony; red arrows) exhibited low fluorescence levels, while cells with an M morphology (typically spindle‐like and separated from colonies; yellow arrows) exhibited high fluorescence levels. To quantitate this effect, we performed flow cytometry on GCaMP6 expressing tumor cells pre‐incubated with either 0 mM or 2 mM Ca2+ and then determined E‐M status by staining cells for surface ECAD expression. Cells grown in 0 mM Ca2+ exhibited low/background GCaMP6 fluorescence that was equivalent in ECADLow (mesenchymal) and ECADHigh (epithelial) cells. In 2 mM Ca2+, both populations exhibited an increase in GCaMP6 fluorescence; however, ECADLow (mesenchymal) cells exhibited significantly greater fluorescence (Fig 1C). Similar results were obtained with the ratiometric Ca2+ indicator Indo‐1 (Fig 1D). Taken together, these results suggest that carcinoma cells with a mesenchymal phenotype have a higher intracellular [Ca2+] compared to cells with an epithelial phenotype.
Ca2+ influx is sufficient to drive a P‐EMT phenotype and increased migration and invasion
Altered calcium homeostasis has been described in association with several hallmarks of cancer, including proliferation, survival, migration, metastasis, and C‐EMT under certain contexts (Davis et al, 2014; Stewart et al, 2015; Monteith et al, 2017). We therefore sought to determine whether the observed association between intracellular Ca2+ and EMT was correlative or whether Ca2+ influx could directly influence a P‐EMT state. To this end, we treated a panel of murine PDA carcinoma cell lines with either vehicle (DMSO), TGFβ (a potent EMT inducer), or ionomycin, a calcium ionophore that promotes Ca2+ influx (Fig EV2A). Because sustained elevations in intracellular [Ca2+] are toxic to normal cells (Roderick & Cook, 2008), we first confirmed that the treatment did not reduce the viability of our carcinoma cells (Fig EV2B).
Figure EV2. Activation of calcium signaling does not induce cell death and promotes EMT in human carcinoma cells.
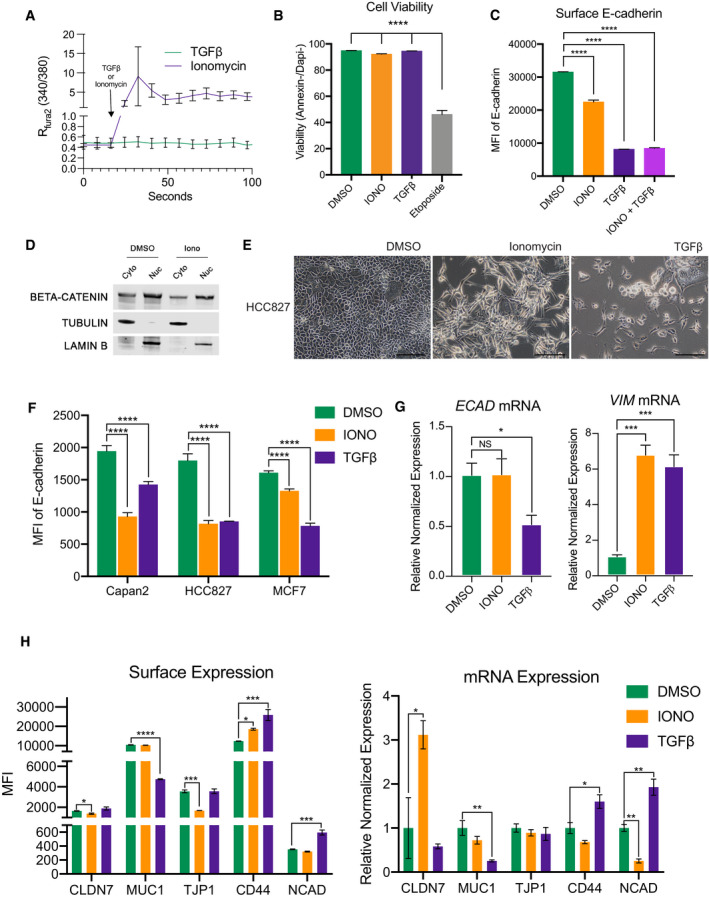
- Representative average Fura‐2 Ca2+ measurements after addition of 2.5 µM ionomycin (purple) or 10 ng/ml TGFβ (green). Colored line indicates average of n = 12 cells. Black error bars represent SD. Two murine PDA cell lines were analyzed.
- Flow cytometry analysis of cellular viability in murine PDA cell lines. Viable cells were defined as Annexin‐/DAPI‐. Cell lines were treated with DMSO, 2.5 µM ionomycin, or 10 ng/ml TGFβ for 72 h or 50 µM etoposide for 24 h. Experiment was repeated in three different murine PDA cell lines in triplicate. Statistical analysis by ANOVA with significance indicated (****P < 0.0001; error bars indicate SD).
- Representative surface E‐cadherin MFI after treatment with DMSO, 2.5 µM ionomycin, 10 ng/ml TGFβ, or 2.5 µM ionomycin with 10 ng/ml TGFβ for 48 h in murine PDA line. Two different murine PDA cell lines analyzed in duplicate. Statistical analysis by ANOVA with significance indicated (****P < 0.0001; error bars indicate SD).
- Western blot showing cytoplasmic/nuclear fractionation of β‐catenin, tubulin (cytoplasmic marker), and Lamin B (nuclear marker) in HCC827 after treatment with ionomycin for 48 h.
- Representative 20× images of the human lung cancer cell line HCC827 after treatment with 7 µM ionomycin for 2 days or 10 ng/ml TGFβ for 10 days. Scale bars = 100 µm.
- Mean fluorescent intensity of surface E‐cadherin of human pancreatic (Capan2), lung (HCC827), or breast (MCF7) carcinoma cell lines following treatment with DMSO, 7 µM ionomycin for 3 days, or 10 ng/ml TGFβ for 10 days. Experiment was performed in triplicate. Statistical analysis by ANOVA with significance indicated (****P < 0.0001; error bars indicate SD).
- Representative relative mRNA expression of ECAD and VIM after treatment with DMSO, 7 µM ionomycin for 3 days, or 10 ng/ml TGFβ for 10 days in human cell lines. Experiment was repeated in three different human cell lines in triplicate. Statistical analysis by ANOVA with significance indicated (*P < 0.05; ***P < 0.001; NS, non‐significant; error bars indicate SD).
- Representative MFI of surface expression of indicated protein (Left) and respective mRNA (Right) in human HCC827 treated with DMSO, 7 µM ionomycin, or 10 ng/ml TGFβ for 3 days. Experiment was repeated in two different human cell lines analyzed in duplicate (*P < 0.05; **P < 0.01; ***P < 0.001, ****P < 0.0001; error bars indicate SEM).
Source data are available online for this figure.
Next, we examined the effects on cell morphology following treatment with these agents. As expected, TGFβ‐treated cells adopted a spindle‐like morphology and complete loss of ECAD staining, consistent with an EMT (Fig 2A, bottom row). Ionomycin treatment also led to significant changes in cell morphology; however, cells retained expression of ECAD (Fig 2A, middle panel). Notably, ECAD was found in the cytoplasm of ionomycin‐treated cells, as compared to the predominant membrane staining observed in vehicle‐treated cells (Fig 2A, compare middle and top rows). This loss of membrane staining was confirmed by flow cytometry, which revealed a significant reduction of surface ECAD (Fig 2B). Others in the field have noted that ionomycin‐induced loss of membranous ECAD can lead to activation of cadherin–catenin complex members such as β‐catenin (Ito et al, 1999), but Western blot analysis in our cells found no difference in nuclear accumulation of β‐catenin (Fig EV2D). This is consistent with our previous study that showed β‐catenin relocalizes from the membrane to endocytic vesicles. Next, we performed a time‐course experiment, collecting protein and mRNA samples at 24 h intervals following treatment with ionomycin or TGFβ. As expected, TGFβ treatment resulted in a rapid decrease of ECAD at both protein and mRNA level (Fig 2C). By contrast, cells treated with ionomycin exhibited no change in ECAD mRNA or protein (whole cell lysate), while vimentin, a mesenchymal marker, was upregulated at the protein level following both treatments (Fig 2C). Interestingly, co‐treatment with both ionomycin and TGFβ reduced surface ECAD to levels similar to TGFβ alone (Fig EV2C), likely due to the rapid induction of classic EMT‐TFs by TGFβ.
Figure 2. Ca2+ influx is sufficient to drive a P‐EMT phenotype and increased migration and invasion.
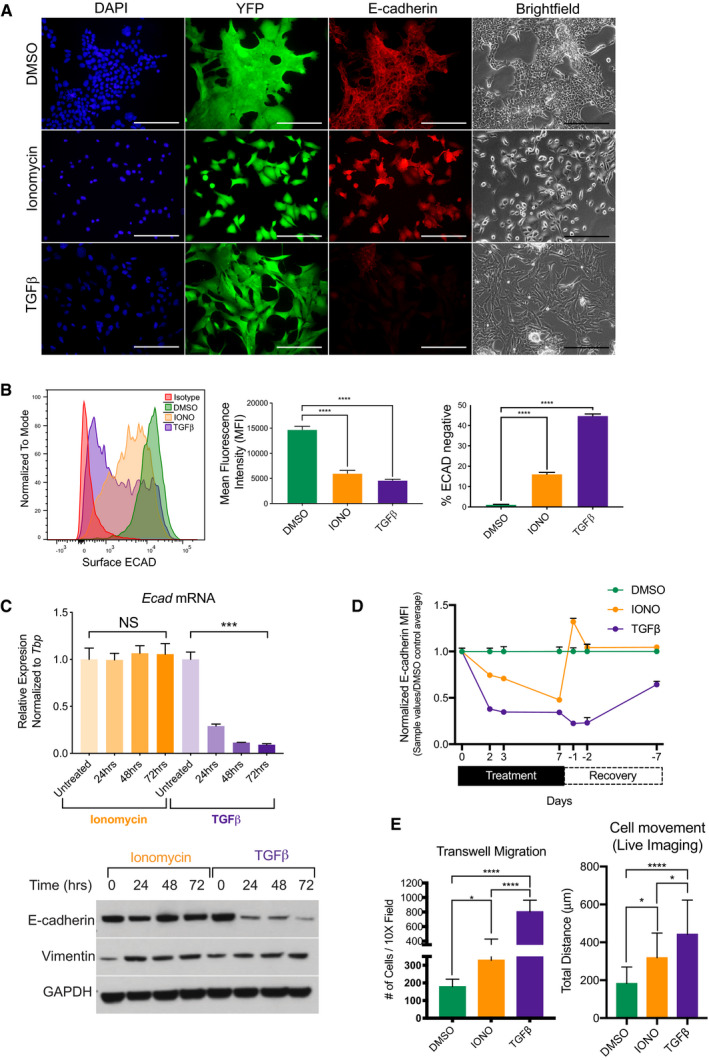
- Representative 20× brightfield and fluorescent images of murine YFP+ tumor cells treated for 72 h with vehicle control (DMSO), 2.5 µM ionomycin, or 10 ng/ml TGFβ and co‐stained for E‐cadherin (red) and DAPI (blue). Scale bar is 100 µm.
- Representative flow cytometry analysis of surface ECAD (left), MFI of surface ECAD (middle), and percentage of surface ECAD‐negative cells (right) in murine PDA cells, after treatment with DMSO, 2.5 µM ionomycin (IONO), and 10 ng/ml TGFβ for 72 h. Representative of three independent murine PDA cell lines run in triplicate. Statistical analysis by ANOVA (****P < 0.0001; non‐significant (NS); ±SD).
- Top: Relative mRNA expression of Ecad after treatment with 2.5 µM ionomycin or 10 ng/ml TGFβ for denoted time. Bottom: Western blot analysis of total E‐cadherin and Vimentin protein for denoted time. Experiment was run in duplicate with three different murine PDA cell lines. Statistical analysis by ANOVA (***P < 0.001; non‐significant (NS); ± SEM).
- Normalized MFI of surface E‐cadherin in PDA cells treated with DMSO, 2.5 µM ionomycin (IONO), and 10 ng/ml TGFβ for 7 days, followed by withdrawal of ionomycin or TGFβ for 7 days as indicated on the x‐axis. Treated cells were analyzed at days 2, 3, and 7. Withdrawn cells were analyzed on days 1, 2, and 7. Experiment was performed in duplicate in two different murine PDA cell lines. Error bars indicate SD.
- Quantification of live cellular migration over 4 h after treatment with DMSO, 2.5 µM ionomycin (IONO), or 10 ng/ml TGFβ for 48 h. (n = 16 cells per condition. Experiment was run in triplicate in two murine PDA different cell lines) (Right). Quantification of transwell migration after treatment with DMSO, 2.5 µM ionomycin (IONO), or 10 ng/ml TGFβ for 72 h (n = 2 cell lines, three replicates per cell line with 3 20× images taken per transwell) (left). Statistical analysis by ANOVA (*P < 0.05; ****P < 0.0001; non‐significant (NS); ± SD).
Source data are available online for this figure.
To further understand the kinetics between ionomycin and TGFβ, we treated cells for 7 days and then withdrew treatment for 7 days and monitored surface expression of ECAD at set intervals. By day 7, both treatments exhibited equal amounts of loss of surface ECAD. Upon withdrawal, TGFβ–treated cells slowly returned expression of surface ECAD, as seen in a similar study (Yuan et al, 2020). Contrastingly, after only 1 day of withdrawal of ionomycin, surface expression of ECAD overshot DMSO control and by 2 days completely rebounded (Fig 2D).
To examine whether Ca2+‐induced P‐EMT is generalizable to human cancer cells, we treated human lung (HCC827), breast (MCF7), and PDA (Capan2) carcinoma lines with either ionomycin or TGFβ. While both agents prompted striking morphological changes and a decrease in surface ECAD levels (Fig EV2E and F), ionomycin‐treated cells exhibited no change in ECAD mRNA and an increase in VIM mRNA (Fig EV2G), consistent with our results using murine lines. We further investigated other epithelial genes in HCC827 (Claudin‐7, Mucin‐1, and Tight Junction Protein 1) and mesenchymal genes (CD44 and N‐cadherin) by flow cytometry and mRNA. While TGFβ elicited an increase in NCAD, both agents led to increases in CD44 at the protein and mRNA level (Fig EV2H). Ionomycin treatment led to a significant decrease in surface expression of CLDN7 and TJP1, while TGFβ treatment led to a decrease in Mucin‐1 (Fig EV2H, left). Notably, these decreases in surface expression of CLDN7 and TJP1 occurred irrespectively of mRNA (Fig EV2H, right). Of note, P‐EMT in human cells occurred more rapidly with ionomycin than TGFβ. Specifically, robust changes in cellular phenotype and surface ECAD were observed after only 48 h of ionomycin treatment, while comparable changes following TGFβ treatment were not observed before day 10 (Fig EV2E).
Increases in migration and invasion are hallmarks of EMT. To determine whether increased intracellular [Ca2+] prompts cells to move, we performed live‐cell imaging following treatment with TGFβ, ionomycin, or vehicle. Cells treated with ionomycin exhibited a 1.7‐fold increase in movement, as compared to a 2.4‐fold increase following TGFβ treatment (Fig 2E; Movies [Link], [Link], [Link]). To study invasion, we performed a Boyden chamber transwell migration assay and measured cellular mobility through Matrigel. Ionomycin treatment led to a significant increase in cellular invasion (Fig 2E). Taken together, these results indicate that carcinoma cells treated with ionomycin exhibit multiple phenotypic characteristics of P‐EMT: (i) adoption of a mesenchymal morphology, (ii) internalization of ECAD protein without change in corresponding mRNA levels, (iii) acquisition of mesenchymal protein expression, and (iv) an increase in migratory and invasive properties.
Ionomycin treatment induces mesenchymal gene transcription without repressing epithelial gene transcription
Classically, EMT has been associated with the simultaneous repression of epithelial genes and induction of mesenchymal genes. We reported previously that while loss of the epithelial program occurs post‐transcriptionally in P‐EMT, the induction of mesenchymal genes at the mRNA level is comparable in P‐EMT and C‐EMT (Aiello et al, 2018). To obtain a global picture of changes in gene expression associated with increased intracellular [Ca2+], we treated murine PDA cell lines with vehicle, ionomycin, or TGFβ for 48 h and performed RNA‐sequencing (Fig 3A). By principal component analysis (PCA), TGFβ‐treated samples were widely separated from vehicle‐ and ionomycin‐treated samples, suggesting that ionomycin treatment brought about comparatively fewer transcriptional differences (Fig EV3A). Next, we examined the abundance of specific epithelial and mesenchymal transcripts previously implicated in EMT. As expected, TGFβ treatment led to a robust downregulation of mRNAs for epithelial genes such as ECAD, EpCAM, and epithelial cytokeratins, while ionomycin treatment had little effect on the abundance of these epithelial transcripts (Figs 3A and EV3B). Consistent with our previous in vivo findings regarding P‐EMT and C‐EMT, both ionomycin and TGFβ treatment resulted in an upregulation of mesenchymal‐related transcripts (Fig 3A), and GSEA confirmed that the transcriptomes of ionomycin‐treated cells were highly enriched for EMT signatures (Fig 3B). As expected, calcium‐related signatures were strongly enriched following ionomycin treatment, whereas these signatures were not enriched following TGFβ treatment (Fig 3C). We then compiled lists of genes that were differentially expressed between ECAD+ and ECAD− cells from our in vivo analysis and compared them to the genes whose mRNAs changed following TGFβ or ionomycin treatment in vitro. TGFβ treated samples were strongly enriched for the C‐EMT signature, whereas ionomycin‐treated samples were strongly enriched for the P‐EMT signature (Fig 3D). Finally, a multinomial logistic regression (MLR)‐based quantitative EMT metric (Chakraborty et al, 2020) revealed that TGFβ‐treated cells exhibited a more mesenchymal state relative to those treated with DMSO or ionomycin (Fig EV3C). Thus, TGFβ treatment recapitulates the transcriptional changes associated with C‐EMT programs in vivo, while ionomycin treatment recapitulates the transcriptional changes associated with P‐EMT programs in vivo.
Figure 3. Ionomycin treatment induces mesenchymal gene transcription without repressing epithelial gene transcription.
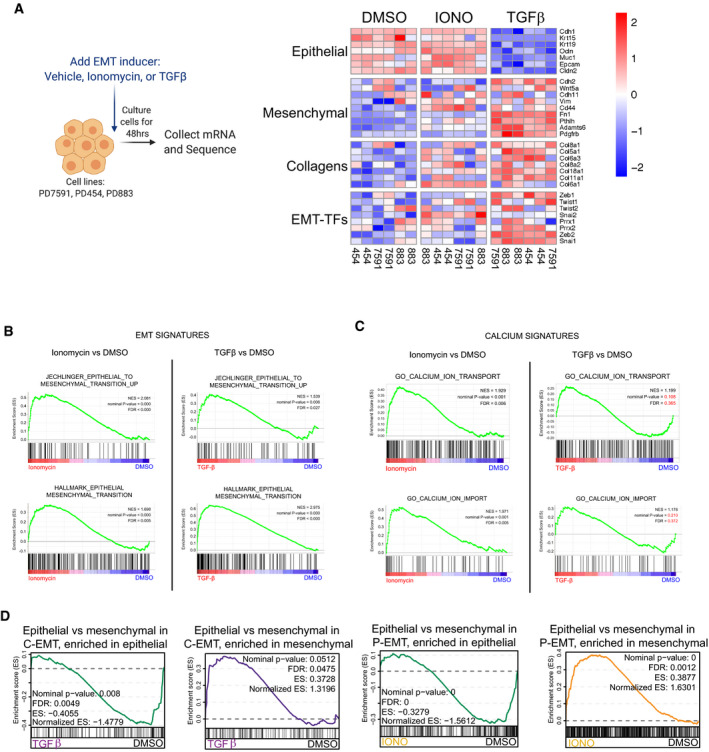
- Schematic for RNA‐sequencing after EMT induction for 48 h. Heatmap illustration of EMT‐related genes in three independent murine PDA cell lines, sequenced in duplicate, and treated with DMSO, 2.5 µM ionomycin, or 10 ng/ml TGFβ. Scale indicates Z‐score.
- GSEA of ionomycin versus DMSO (left) or TGFβ versus DMSO (right) for EMT signatures. NES, nominal P‐value, and FDR are shown. Statistical analysis performed in GSEA.
- GSEA of ionomycin versus DMSO (left) or TGFβ versus DMSO (right) for calcium signatures. NES, nominal P‐value, and FDR are shown. Statistical analysis performed in GSEA.
- Leading edge plots showing enrichment of C‐EMT gene signatures in TGFβ‐treated cells (left) or P‐EMT gene signatures in ionomycin (IONO)‐treated cells (right), based on GSEA. Enrichment scores (ES), NES, nominal P‐value, and FDR are shown. Statistical analysis performed in GSEA.
Figure EV3. Epithelial proteins are conserved in exosomes.
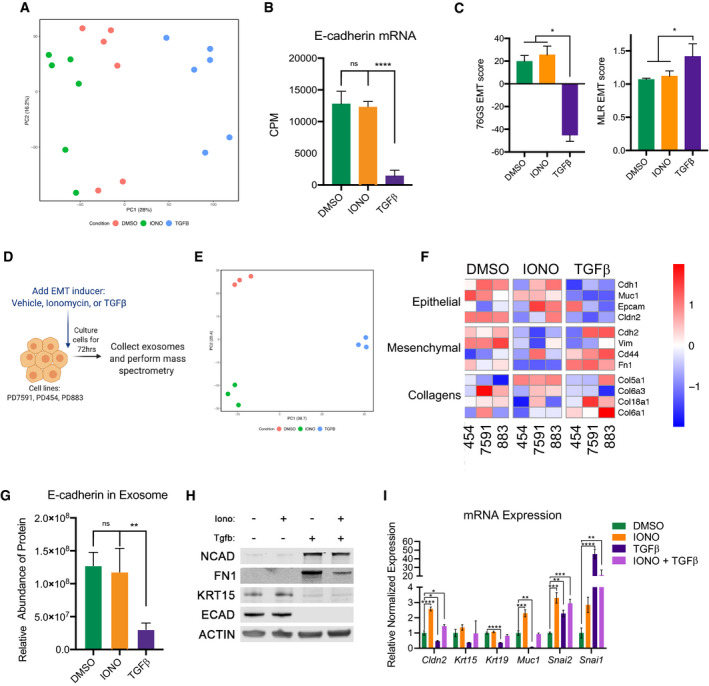
- Principal component analysis of differentially expressed genes of murine PDA cell lines treated for 48 h with DMSO, 2.5 µM ionomycin, or 10 ng/ml TGFβ. Each cell line per condition was sequenced in duplicate.
- Counts per million reads (CPM) of three cell lines pooled together after treatment with DMSO, ionomycin (IONO), or TGFβ as described in (A). Statistical analysis by ANOVA with significance indicated (****P < 0.0001; NS, non‐significant; error bars indicate SD).
- EMT scores calculated via 76GS (top) or MLR (bottom) methods of murine PDA cell lines after treatment with DMSO, ionomycin (IONO), or TGFβ for 48 h. Error bars represent SD. *P < 0.05 for Students’ two‐tailed t‐test. N = 3.
- Schematic for exosome collection and mass spectrometry after EMT induction for 72 h.
- Principal component analysis of proteins contained within exosomes in murine PDA cell lines treated for 72 h with DMSO, 2.5 µM ionomycin, or 10 ng/ml TGFβ. Three different cells line analyzed per condition.
- Heatmap illustrating the representation of EMT‐related proteins found within exosomes in murine PDA cell lines treated for 72 h with DMSO, 2.5 µM ionomycin, or 10 ng/ml TGFβ. Scale indicates Z‐score.
- Relative abundance of E‐cadherin protein within exosomes of three cell lines pooled together in duplicate treated with DMSO, ionomycin (IONO), or TGFβ. Statistical analysis by ANOVA with significance indicated (**P < 0.01; NS, non‐significant; error bars indicate SD).
- Western blot analysis showing expression levels of NCAD, FN1, KRT15, ECAD, and ACTIN after treatment with DMSO, 2.5 µM ionomycin, 10 ng/ml TGFβ, or 2.5 µM ionomycin with 10 ng/ml TGFβ, for 3 days in murine cell lines.
- Representative relative mRNA expression of Cldn2, Krt15, Krt19, Snai2, Snai1 after treatment with DMSO, 2.5 µM ionomycin, 10 ng/ml TGFβ, or 2.5 µM ionomycin with 10 ng/ml TGFβ, for 3 days in murine cell lines. Statistical analysis by ANOVA with significance indicated (*P < 0.05; **P < 0.01, ***P < 0.001, ****P < 0.0001; error bars indicate SD).
Source data are available online for this figure.
To investigate changes in protein abundance accompanying these divergent EMT programs, we examined tumor‐derived exosomes as a window into the proteome. We collected exosomes from three cell lines after 72 h of treatment with vehicle, ionomycin, or TGFβ and performed mass spectrometry, capturing approximately 2,700 proteins (Fig EV3D). The three treatments resulted in highly distinctive proteomes as visualized by PCA (Fig EV3E). Consistent with our mRNA results, TGFβtreatment depleted epithelial proteins from tumor‐derived exosomes, whereas ionomycin had little effect on the abundance of these proteins (Fig EV3F and G). Western blot analysis of whole‐cell extracts confirmed that ionomycin had no effect on epithelial protein levels (Fig EV3H). Co‐treatment with ionomycin and TGFβ decreased epithelial and increased mesenchymal proteins (Fig EV3H), in agreement with reduced surface ECAD by flow cytometry (Fig EV2C). As expected, co‐treatment resulted in an intermediate state at the RNA level (Fig EV3I). These results confirm that the epithelial program is preserved at the protein level following ionomycin‐induced P‐EMT.
GPCR signaling through Gαq induces P‐EMT
Many signals could potentially drive a P‐EMT by mobilizing intracellular Ca2+. We reasoned that G‐protein‐coupled receptors (GPCRs)—particularly those that utilize a Gαq subunit, which mobilizes Ca2+ following receptor engagement—might play such a role, as GPCRs are frequently overexpressed in cancer (Dorsam & Gutkind, 2007; Bar‐Shavit et al, 2016). Furthermore, the Gαq subunit (Gnaq) was upregulated in P‐EMT tumors in vivo (Fig EV4A). To test this hypothesis, we employed a synthetic ligand system in which GPCR‐Gαq signaling can be induced with the experimentally derived drug clozapine‐N‐oxide (CNO) (Armbruster et al, 2007; Conklin et al, 2008; Vaqué et al, 2013). We generated two murine PDA lines overexpressing Gαq‐GPCR Designer Receptors Exclusively Activated by Designer Drugs (DREADD), an engineered receptor that mobilizes Ca2+ following addition of CNO (Fig 4A, top). Treatment with CNO resulted in a change in cell morphology and loss of surface ECAD, recapitulating the phenotypes observed with ionomycin (Fig 4B). Of note, a transient calcium spike, as elicited by ATP, was not sufficient to cause this phenotype (Fig 4A, bottom), suggesting that sustained but not transient Ca2+ mobilization is required for induction of P‐EMT.
Figure EV4. Calcineurin activity is dispensable for calcium‐induced EMT.
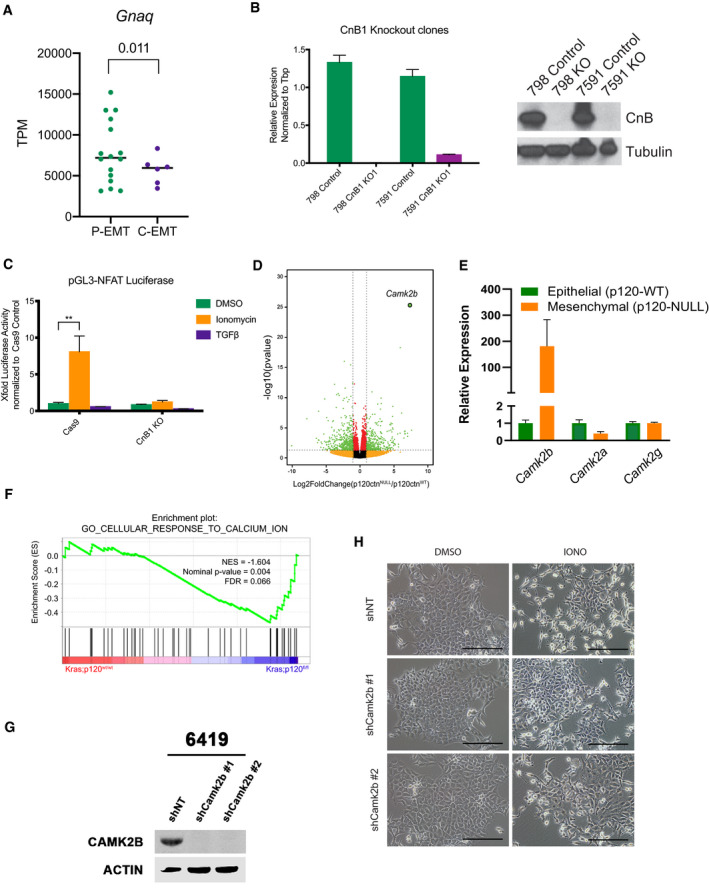
- TPM of Gnaq comparing murine P‐EMT and C‐EMT tumors in vivo. P‐EMT (n = 16, ECAD+ and ECAD− sorted fractions combined), C‐EMT (n = 6, ECAD+ and ECAD− sorted fractions combined), statistical analysis by Student’s unpaired t‐test, black line indicates the median.
- Left: Relative mRNA expression of CnB in two murine PDA cell lines (PD798 and PD7591) comparing Cas9 control cells to CnB knockout (KO). Right: Western blot analysis showing loss of CnB in KO cells. Experiment performed in triplicate. Error bars indicate SD.
- Representative NFAT luciferase activity in Cas9 control and CnB knockout murine PDA cells after treatment with DMSO, 2.5 µM ionomycin (IONO), or 10 ng/ml TGFβ for 24 h. Experiment performed in two murine PDA cell lines in duplicate. Statistical analysis by ANOVA with significance indicated (**P < 0.01; all other comparisons to DMSO are non‐significant; error bars indicate SD).
- Volcano plot showing significantly upregulated and downregulated genes in p120ctnNull/p120ctnWT murine PDA cells. Statistical analysis performed in DEseq. Vertical dashed lines represent Log2 fold change equal to 1, horizontal dashed line represents −log10 equal to 1, red dots are non‐significant genes, and green dots are significant genes with P‐value β 0.1 and |Log2FC ≥ 2|.
- mRNA expression of Camk2 genes in p120ctnWT and p120ctnNull cells.
- Bioinformatic schematic for GSEA comparing p120wt/wt and p120fl/fl tumor cells. Statistical analysis performed in GSEA.
- Western blot analysis showing expression levels of CAMK2B after shRNA knockdown in murine PDA cells (6419).
- Representative 20× brightfield images of shNT, shCAMK2B #1, and shCAMK2B #2, murine PDA cells treated for 72 h with DMSO or 2.5 µM Ionomycin. Scale bar is 100 µm.
Source data are available online for this figure.
Figure 4. Gαq signaling acts through calmodulin to induce P‐EMT.
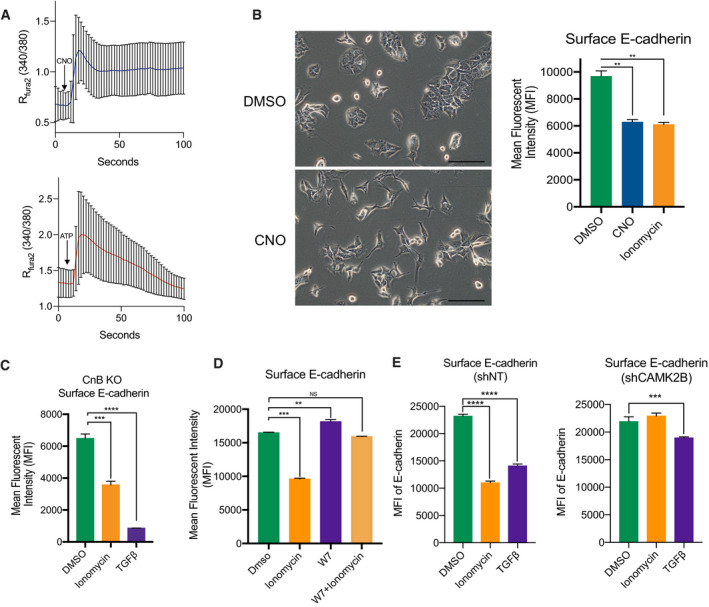
- Fura‐2 Ca2+ measurements after addition of 5 µM CNO (Top) or 10 µM ATP (Bottom) in murine PDA cells expressing a Gαq‐DREADD. Blue or red line indicates average of n = 22 cells. ± SD. Tracings are representative of experiments performed twice in 2 independent cell lines.
- Representative 20× brightfield images of Gαq‐DREADD‐expressing cells 48 h after addition of CNO (Left). Scale bar = 100 µm. MFI of surface E‐cadherin in cells expressing a Gαq‐DREADD after treatment with DMSO, 5 µM CNO, or 2.5 µM ionomycin (Right). Both experiments performed in triplicate in 2 independent cell lines. Statistical analysis by ANOVA (**P < 0.01; ± SD).
- MFI of surface E‐cadherin in CnB knockout lines after 48‐h treatment with DMSO, 2.5 µM ionomycin, or 10 ng/ml TGFβ. Experiment was repeated in two different murine PDA cell lines in triplicate. Statistical analysis by ANOVA (***P < 0.001; ****P < 0.00001; ± SD).
- MFI of surface E‐cadherin of murine PDA cells pretreated with 20 µM W7 for 24 h and then DMSO, 2.5 µM ionomycin, or 20 µM W7 with 2.5 µM ionomycin for 24 h. Experiment was repeated in two different murine PDA cell lines in triplicate. Statistical analysis by ANOVA (**P < 0.01; ***P < 0.001; non‐significant (NS); ±SD).
- MFI of surface E‐cadherin of shRNA non‐targeting (shNT) (left) or shCAMK2B (right) after 48 h treatment with DMSO, 2.5 µM ionomycin, or 10 ng/ml TGFβ. Experiment was repeated in two different murine PDA cell lines. Statistical analysis by ANOVA (***P < 0.001; ****P < 0.00001; ±SD).
Calcineurin (CaN) is dispensable for calcium‐induced P‐EMT
An increase in intracellular [Ca2+] results in the activation of several downstream mediators, including the transcription factor NFAT, the adaptor protein calmodulin, and calcium‐activated protein kinases. Based on our HOMER motif analysis (Fig 1B), we initially hypothesized that the calcium responsive transcription factor NFAT might be responsible for the P‐EMT phenotype. In support of such a hypothesis, several in vitro and in vivo studies have reported a role for NFAT in tumor development, migration, and EMT (Mancini & Toker, 2009; Baumgart et al, 2014; Singh et al, 2015; preprint: Subbalakshmi et al, 2020). To test this hypothesis, we inactivated the NFAT regulator calcineurin (CaN), which is required for the activity of nearly all NFAT family members (NFAT1‐4) (Crabtree & Olson, 2002; Heit et al, 2006). To this end, we deleted the calcineurin B (CnB) regulatory subunit in two PDA cell lines (Fig EV4B) and used the pGL3‐NFAT luciferase reporter, which contains 3× NFAT binding sequences upstream of a luciferase reporter, to confirm that CnB KO cells lacked detectable NFAT activity following ionomycin treatment (Fig EV4C). Nevertheless, CnB KO cells treated with ionomycin continued to exhibit a P‐EMT phenotype (Fig 4C), indicating that CaN and NFAT activity is dispensable for Ca2+‐induced P‐EMT.
Calmodulin (CaM) and Camk2b act downstream of Ca2+ mobilization to induce P‐EMT
We next considered calmodulin (CaM), which acts as a multifunctional calcium‐binding protein upstream of calcineurin and NFAT. When calcium binds to the EF‐hands of calmodulin, a conformational shift allows calmodulin to interact with and regulate a variety of kinases, phosphatases, and other signaling proteins (Villalobo & Berchtold, 2020). Because the mammalian genome contains three dispersed CaM genes, whose functions are essential for cell viability, genetic targeting of CaM is challenging. Consequently, we tested the ability of the CaM antagonist W7 to inhibit P‐EMT (see Methods). Pretreatment of cells with W7 for 24 h blocked ionomycin‐induced P‐EMT, as measured by loss of surface ECAD (Fig 4D). Moreover, W7 treatment alone resulted in increased surface ECAD expression (Fig 4D). These results indicate that CaM is necessary for Ca2+‐mediated P‐EMT.
CaM binds to and potentially activates several other proteins that may mediate our P‐EMT phenotype. To determine the downstream mechanisms at play, we utilized RNA‐seq from a recent study in our laboratory that compared EMT induced by deletion of p120catenin (p120), a regulator of ECAD stability (Reichert et al, 2018); GSE96729). Importantly, loss of p120 destabilizes membranous ECAD protein leading to its internalization and subsequent degradation (reviewed in Kourtidis et al, 2013) and is likely modeling a P‐EMT state. Differential gene expression analysis of p120‐null (mesenchymal) tumor cells versus p120‐intact (epithelial) tumor cells revealed that the most upregulated gene in cells lacking p120 was calcium/calmodulin‐dependent protein kinase II Beta (Camk2b) (Fig EV4D). Only Camk2b expression, but not Camk2a or Camk2g was upregulated in the mesenchymal state (Fig EV4E). GSEA also revealed an enrichment of calcium signaling in p120‐null tumor cells (Fig EV4F), likely indicating a P‐EMT state. To test whether Camk2b was regulating P‐EMT, we used shRNA to knockdown Camk2b in several cell lines (Fig EV4G). While non‐targeting controls continued to respond to ionomycin, knockdown of Camk2b blocked the decrease of surface E‐cadherin (Figs 4E and EV4H). Of note, TGFβ induced EMT may also be blunted in cells lacking Camk2b. Thus, a CaM‐Camk2b signaling axis regulates the P‐EMT phenotype.
Concluding remarks
While EMT has long been associated with tumor cell migration and chemoresistance, recent studies have pointed to the importance of a hybrid E‐M state—P‐EMT—as a state exhibiting greater plasticity and metastatic potential (Nieto et al, 2016; Zhang & Weinberg, 2018). Ca2+ signaling is known to play a role in cellular migration and cancer progression (Monteith et al, 2017) and a requirement for Ca2+ mobilization has previously been reported in hypoxia‐ or EGF‐induced EMT in breast cancer (Davis et al, 2014). Additionally, we have recently demonstrated that the parathyroid hormone‐related protein (PTHrP) governs EMT and metastasis in PDAC (Pitarresi et al, 2021), and others have shown that PTHrP stimulates release of intracellular Ca2+ stores (reviewed in (John Martin, 2016)). Here, using an unbiased approach based on in vivo observations, we discovered that Ca2+ mobilization induces a stable P‐EMT characterized by the relocalization of epithelial proteins and increased invasion and migration. Given that our in vitro findings recapitulate the cellular phenotypes observed in autochthonous tumors (Aiello et al, 2018), we conclude that extracellular signals prompting an increase in Ca2+ flux, potentially through Gαq‐coupled GPCRs, are at least one mechanism by which tumor cells achieve a partial EMT state.
These findings raise several questions. First, which GPCR(s) may be responsible for EMT in native tumors, and what are their cognate ligands? The mammalian genome contains hundreds of GPCRs, many of which are orphan receptors. We identified ˜90 GPCRs that are differentially expressed between P‐EMT and C‐EMT tumors in vivo (Fig EV5A), a number that is impractical to study sequentially. This issue highlights a broader problem in the EMT field, which is that the physiologic inducers of EMT in vivo remain ill‐defined. Consequently, it is possible (or even likely) that multiple signals act in combination within the tumor microenvironment to achieve critical thresholds of intracellular calcium, TGFβ pathway activation, and/or other EMT mediators.
Figure EV5. GPCRs expressed in C‐EMT and P‐EMT pancreatic tumors and a GPCR‐calcium‐calmodulin‐Camk2b signaling axis.
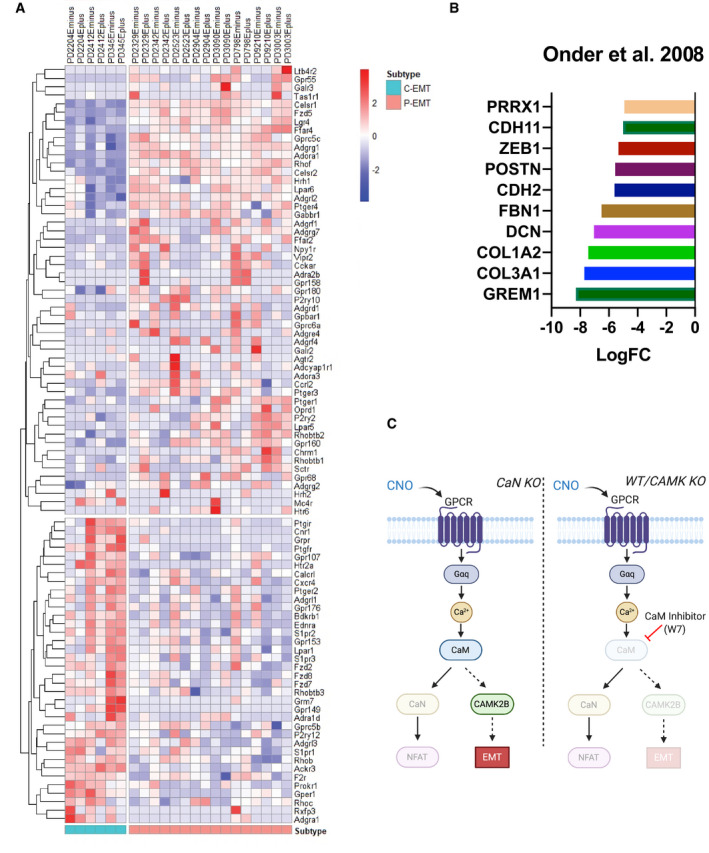
- Heatmap illustrating GPCRs differentially expressed between P‐EMT and C‐EMT murine PDA tumors in vivo. Scale bar indicates Z‐score.
- Re‐analyzed EMT‐related genes upregulated during adherens junction loss from Onder et al, 2008.
- Graphical model illustrating GPCR‐calcium‐calmodulin‐Camk2b signaling axis resulting in P‐EMT.
Secondly, the conduits through which intracellular [Ca2+] reach levels sufficient for P‐EMT remain unknown. Tumors are known to upregulate a number of calcium channels to promote progression and invasion (Azimi & Monteith, 2016; Monteith et al, 2017). These channels can mobilize Ca2+ from several different stores, including the endoplasmic reticulum, the extracellular space, or mitochondria (Clapham, 2007). While we found that tumor cells within a mesenchymal state indeed have more intracellular calcium, the source of this calcium—and whether it derives from an influx of extracellular calcium or a release from intracellular stores—remains unknown and warrants further investigation.
Another question concerns the mechanism by which elevated intracellular [Ca2+] dampens the epithelial program while simultaneously upregulating a mesenchymal one, thus bringing about a hybrid state. Previous work has suggested that loss of the adherens junctions is sufficient to induce many mesenchymal genes (Onder et al, 2008), but the initial stimulus resulting in adherens junction loss remains unknown (Fig EV5B). Our data indicate that increased intracellular [Ca2+] activates a calmodulin‐Camk2b signaling axis that results in an internalization of adherens junctions and other epithelial proteins. This internalization could also upregulate mesenchymal genes, as suggested by other studies, or Camk2b could play a direct role in this upregulation. In support of a direct role for CAMK2 in regulating EMT, a recent study in squamous cell carcinoma and lung carcinoma found that a CAMK2‐CD44 axis promotes a hybrid EMT state (Pastushenko et al, 2020). Our work revealed that the beta isoform of CAMK2 plays a significant role in mediating calcium‐induced EMT. In addition, our loss of function studies also implicates CAMK2b in TGFβ‐induced EMT. How this kinase regulates epithelial plasticity, and the possibility that it represents a point of convergence of the P‐EMT and C‐EMT programs, remains to be elucidated.
Several studies have suggested that tumor cells expressing a hybrid/partial EMT state exhibit increased plasticity and metastatic potential relative to those that have undergone a classical or canonical EMT. We therefore studied the kinetics of P‐EMT and C‐EMT by comparing the effects of withdrawing ionomycin versus TGFβ, respectively. Upon removal of ionomycin, surface E‐cadherin levels quickly rebounded to pre‐treatment (even surpassing the DMSO control on the first day). By contrast, TGFβ withdrawal led to a more gradual return of surface E‐cadherin to pre‐treatment levels. These findings are not surprising given the greater dependence of C‐EMT on transcriptional repression of epithelial genes, and they have implications for tumor biology. Given the widely held view that MET is important for metastatic colonization (Tsai et al, 2012), it is possible that tumor cells that have undergone a Ca2+‐induced P‐EMT may have an enhanced ability to rapidly revert to an epithelial state and thereby more efficiently colonize a metastatic site. Finally, our findings raise the possibility that the Ca2+‐driven EMT programs described herein play a role in cellular plasticity in other contexts, such as embryonic development, as is the case for classical EMT programs driven by epithelial gene repression.
In summary, our data suggest a model wherein activation of a Gαq‐associated GPCR mobilizes calcium, which in turn acts through Ca2+‐calmodulin‐Camk2b axis to prompt changes in gene expression, the cytoskeleton, and protein trafficking, resulting in a stable, partial/hybrid E‐M state (Fig EV5C). As the Ca2+‐mediated effects described here are observed across multiple murine and human cell lines, we propose that this P‐EMT program is likely to operate widely and have therapeutic implications.
Materials and Methods
Cell lines
Murine PDA cell lines PD7591 (female), PD798 (male), PD483 (female), PD3077 (male), PD454 (male), and PD883 (male) were derived from primary KPCY tumors of mixed genetic background. All human cell lines (Capan2, MCF7, HCC827) were obtained originally from ATCC. PD6419 was derived from C57BL/6 KPCY tumors. Cell lines were regularly tested for mycoplasma using MycoAlert Mycoplasma Detection Kit (Lonza). Murine cell lines were cultured in Dulbecco’s modified Eagle medium/F12 medium supplemented with 5 mg/ml d‐glucose (Invitrogen), 0.1 mg/ml soybean trypsin inhibitor type I (Invitrogen), 5 ml/l insulin‐transferrin‐selenium (ITS Premix; BD Biosciences), 25 µg/ml bovine pituitary extract (Gemini Bio‐Products), 5 nM 3,3′,5‐triiodo‐L‐thyronine (Sigma), 1 µM dexamethasone (Sigma), 100 ng/ml cholera toxin (Sigma), 10 mM nicotinamide (Sigma), 5% Nu‐serum IV culture supplement (Thermo Fisher Scientific), and antibiotics (gentamicin 150 µg/ml, Gibco; amphotericin B 0.25 µg/ml, Invitrogen). PD6419 were cultured as previously described (Li et al, 2018). Human PDA and breast cell lines were cultured in Dulbecco’s modified Eagle medium supplemented to 10% decomplemented fetal bovine serum and 1% penicillin/streptomycin. Human lung cell lines were cultured in RPMI 1640 supplemented to 10% decomplemented fetal bovine serum with 1% penicillin/streptomycin. All lines were cultured at 37°C, 5% CO2, 21% O2, and 100% humidity. Cell lines were maintained and passaged according to ATCC‐recommended procedures.
Drugs and ligands
Mouse (Cell Signaling Technology (CST) 5231LC) or human (CST 8915LC) TGFβ was resuspended in 20 mM citrate pH 3.0 per instructions. Ionomycin calcium salt (CST 9995S) was resuspended in DMSO per instructions. W7‐HCl (Santa Cruz sc‐201501) was resuspended in DMSO per instructions. All solutions were used as indicated in text or figure legends.
Flow cytometry and fluorescence‐activated cell sorting (FACS)
For detailed protocol, see references (Aiello et al, 2018; Norgard & Stanger, 2020). Briefly, for ECAD flow cytometry, cells were dissociated to single cells with Hank’s enzyme‐free cell dissociation solution (EMD Millipore). Cells were washed in staining solution (HBSS with 5% FBS and DNase I (Sigma) and stained with anti‐ECAD (BioLegend 147308 or 147319), anti‐human CD325 (N‐cadherin) (BioLegend 350808), anti‐mouse/human CD44 (BioLegend 103012), anti‐human CD227 (MUC‐1) (BioLegend 355607), anti‐Claudin‐7 (ab27487), anti‐ZO‐1/TJP1 (Invitrogen 61‐7300), or isotype control (BioLegend 400418 or 400430) at 1:100 in staining solution on ice for 30 min. For unconjugated antibodies (Claudin‐7 and TJP1), cells were stained for an additional 30 min with APC donkey anti‐rabbit (Jackson ImmunoResearch). Cells were washed 3× in staining solution, filtered through a 70‐µM strainer, and counterstained with DAPI prior to flow cytometry on a LSR II or FACSJazz, BD. FlowJo software was used for analysis.
Lentivirus production and transduction
Transfection of 293Ts for lentivirus production was performed using Opti‐MEM I (GIBCO), DNA (expression plasmid, psPAX2, and pVSVg mixed in a 4:2:1 ratio), and polyethylenimine (PEI, Polysciences) at a 3:1 ratio with total DNA. Recipient cells were transduced with filtered, unconcentrated viral supernatant in the presence of 8 μg/ml polybrene. Puromycin antibiotic selection at 5–10 μg/ml was applied for at least 72 h.
shRNA
For shRNA experiments, cells were transduced with Mission pLKO.1‐puro Lentiviral particles with non‐target (shNT; 5′ CCGGGATACCTAACTCAGGAAACCACTCGAGTGGTTTCCTGAGTTAGGTATCTTTTTG 3′), shCamk2b #1 (5′ CCGGCCTGCTGAAGCATTCCAACATCTCGAGATGTTGGAATGCTTCAGCAGGTTTTT 3′), or shCamk2b #2 (5′ CCGGGACTGTGGAATGTCTGAAGAACTCGAGTTCTTCAGACATTCCACAGTCTTTTTG 3′) and stable colonies selected with puromycin (Sigma).
RNA Isolation, real‐time–quantitative PCR (RT–qPCR)
RNA was prepared from cultured tumor cells using RNeasy Mini Kit (Qiagen) or NucleoSpin RNA (Takara). For RT–qPCR, cDNA was generated using High‐capacity cDNA Reverse Transcription Kit (Life Technologies). RT–qPCR analysis was performed with SsoAdvanced SYBR (Bio‐Rad) using a CFX384 Real‐Time System (Bio‐Rad). Transcript quantities were determined using the difference of Ct method, and values were normalized to the expression of GAPDH or TBP. Primer sequences are listed in Table EV1.
Plasmid construction and cloning
GCaMP6 (a gift from Bruce Freedman) was cloned into pCDH‐FHC‐EF1 using BamH1 and Swa1. GCaMP6 was transduced in YFP− tumor cells that were generated by inserting sgRNA toward YFP into lentiCRISPR v2, generously provided by Feng Zhang (Addgene plasmid # 52961) using BsmBI. Primers used are listed in Table EV2.
Measurement of cytosolic Ca2+ flow cytometry by GCaMP6
Tumor cells expressing GCaMP6 were dissociated with Hank’s enzyme‐free cell dissociation solution (EMD Millipore). Cells were then stained with anti‐ECAD (BioLegend 147308 or 147319) or isotype control (BioLegend 400418 or 400430) at 1:100 in staining solution on ice for 30 min. Cells were washed three times in staining solution and then placed in either 0 or 2 mM Tyrodes solution on ice. After equilibrium for 1 h, cells were stained with DAPI prior to flow cytometric analysis on BD LSR II flow cytometer. FlowJo software was used for analysis.
Measurement of cytosolic Ca2+ flow cytometry by Indo‐1 calcium indicator
Cells were loaded with Indo‐1 at a final concentration of 1.5 μM for 45 min at 37°C in an incubator. Cells were washed twice with PBS and dissociated with Hank’s enzyme‐free cell dissociation solution (EMD Millipore). Cells were then stained with anti‐ECAD (BioLegend 147308 or 147319) or isotype control (BioLegend 400418 or 400430) at 1:100 in staining solution on ice for 30 min. Cells were washed three times in staining solution and then placed in either 0 or 2 mM Tyrodes solution on ice. After equilibrium for 1 h, cells were stained with DAPI prior to flow cytometric analysis on BD LSR II flow cytometer. Single‐cell ratios of bound to unbound were performed using FlowJo software.
Measurement of cytosolic Ca2+ changes
Cytosolic Ca2+ was assessed by plating cells on glass coverslips. Cells were loaded with Fura‐2 AM (Invitrogen F1221) at 1 μM for 30 min at 37°C. Time‐lapse images were recorded (2s interval) using a NikonTi system using a 20×/0.75 NA objective for fluorescence at 340 nm excitation/515 nm emission (Ca2+‐bound Fura‐2) and 380nm excitation/515 nm emission (Ca2+‐free Fura‐2). ATP, TGFβ, CNO, or ionomycin were spiked in after equilibration. Data were analyzed with ImageJ and presented as traces of mean (bold line) and SEM (error bars) for all data points (time versus fluorescent ratio).
Matrigel invasion assay
Transwell inserts (Corning 3464) were coated with Matrigel and placed above complete DMEM. Cells were serum‐starved in 0.2% serum DMEM for 24 h. Cells were seeded in serum‐free DMEM on top of the Matrigel layer and incubated for 24 h. Transwell inserts were removed, washed three times, and fixed in 4% PFA for 15 min. Fixed Matrigel inserts were washed three times, stained with DAPI, and imaged.
Live cell migration
Cells were seeded on glass coverslips and treated for 24 h prior to imaging with DMSO, Ionomycin (2.5 μM), or TGFβ (5 ng/ml). Images were recorded every 8 min using an ImageXpress Micro 4 High Content Imaging Device. IMARIS (Bitplane, Oxford Instruments) was used to quantify migration and create movies.
Cell viability assay
Cells were plated in a six‐well dish and treated with DMSO, Ionomycin (2.5 μM), or TGFβ (5 ng/ml) for 48 h. Etoposide (50 μM) was added for 24 h as a positive control. Cells were stained with eBioscience Annexin V Apoptosis Detection Kit APC (88‐8007‐72) according to manufacture instructions. DAPI (1 mg/ml) was added prior to analysis by flow cytometry.
RNA‐seq, GSEA, and HOMER
RNA samples were extracted using the Qiagen RNeasy or Takara NucleoSpin RNA kit following manufacturer’s instructions. RNA was sent out to Novogene for library preparation and high‐throughput sequencing using Illumina sequencers to generated paired‐end results. Raw counts of gene transcripts were obtained using an alignment‐independent tool, Salmon (https://combine‐lab.github.io/salmon/), using standard settings. The raw count matrix was subsequently imported into R‐studio (R version 3.5) and used as input file for DESeq2 analysis (https://bioconductor.org/packages/release/bioc/html/DESeq2.html) with default settings from online software instruction for normalization and differential gene expression analysis. Salmon was used to normalize and quantitate gene expression in transcripts‐per‐million (tpm) through quasi‐alignment. Differentially expressed genes were used as input for principal component analysis (PCA), gene set enrichment analysis (GSEA) (https://www.gsea‐msigdb.org/gsea/index.jsp), and motif analysis using HOMER (http://homer.ucsd.edu/homer/ngs/index.html). Detailed scripts and parameters used for each steps of analysis could be provided by reasonable request to the authors.
Western blot analysis
Cells were washed with ice‐cold PBS and lysed in an appropriate amount of RIPA buffer. Equal amounts of protein were run in reducing conditions on SDS–PAGE gels and transferred to PVDF membrane, blocked in 5% non‐fat milk in PBS plus 0.1% Tween‐20 for 1 h at room temperature. After blocking, membranes were incubated in primary antibody diluted in 5% non‐fat milk in PBS plus 0.1% Tween‐20 overnight at 4°C. After PBS‐T washes, membranes were incubated with horseradish peroxidase‐conjugated secondary antibodies (Jackson Immunoresearch) diluted in 5% non‐fat milk in PBS plus 0.1% Tween‐20 for 1 h at room temperature. Primary antibodies used are located in Table EV3.
Immunofluorescent staining (IF)
Cells were seeded into 8‐well Nunc Lab‐Tek II chamber slides (Thermo Scientific) and fixed in 4% paraformaldehyde for 15 min. Fixed cells were blocked and permeabilized in PBS with 0.3% Triton‐X and 5% donkey serum for 1 h. After blocking, cells were incubated in primary antibody diluted in 5% donkey serum overnight at 4°C. After PBS‐T washes, cells were incubated in fluorescently conjugated secondary antibodies and mounted with Aqua Poly/Mount (Polysciences, Inc). Slides were visualized using an Olympus IX71 inverted multicolor fluorescent microscope equipped with a DP71 camera. Primary antibodies used are located in Table EV3.
NFAT reporter assay
pGL3‐NFAT luciferase (Addgene #17870) and pRL‐SV40 (Promega E2231) were transfected into tumor cells using Lipofectamine 2000 at a ratio of 3:1 for PDAC cells. Media was changed after 6 h. Cells were treated with DMSO, Ionomycin (2.5 μM), or TGFβ (5 ng/ml) for 24 h. Luciferase activity was analyzed using dual‐luciferase reporter assay system (Promega E1910) and read on a luminometer.
Exosome purification, characterization, and analyses
Exosomes were purified by sequential ultracentrifugation, as described previously (Hoshino et al, 2015). In brief, cell contamination was removed from 3‐ to 4‐day cell culture supernatant by centrifugation at 500 g for 10 min. To remove apoptotic bodies and large cell debris, the supernatants were then spun at 3,000 g for 20 min, followed by centrifugation at 12,000 g for 20 min to remove large microvesicles. Finally, exosomes were collected by spinning at 100,000 g for 70 min. Exosomes were washed in PBS and pelleted again at 100,000 g for 70 min by ultracentrifugation in a Beckman Coulter Optima XE or XPE ultracentrifuge. The final exosomes pellet was resuspended in PBS, and protein concentration was measured by BCA (Pierce, Thermo Fisher Scientific). Exosome size and particle number were analyzed using the LM10 or DS500 nanoparticle characterization system (NanoSight, Malvern Instruments) equipped with a violet laser (405 nm).
Data‐dependent analysis of exosomes samples
Exosome samples (5 µg—adjusted based on BCA measurements) were dried by vacuum centrifugation and re‐dissolved in 30–50 µl 8 M Urea/50 mM ammonium bicarbonate/10 mm DTT. Following lysis and reduction, proteins were alkylated using 20 or 30 mM iodoacetamide (Sigma). Proteins were digested with Endopeptidase Lys C (Wako) in < 4 M urea followed by trypsinization (Promega) in < 2 M Urea. Peptides were desalted and concentrated using Empore C18‐based solid‐phase extraction prior to analysis by high‐resolution/high‐mass accuracy reversed phase (C18) nano‐LC‐MS/MS. Typically, 30% of samples were injected. Peptides were separated on a C18 column (12 cm / 75 µm, 3 µm beads, Nikkyo Technologies) at 200 or 300 nl/min with a gradient increasing from 1% buffer B/95% buffer A to 40% buffer B/60% buffer A in typically 90 or 120 min (buffer A: 0.1% formic acid, buffer B: 0.1% formic acid in 80% acetonitrile). Mass spectrometers (Q‐Exactive, Q‐Exactive Plus, Q‐Exactive‐HF, or Fusion Lumos, Thermo Scientific) were operated in data‐dependent (DDA) positive ion mode.
Proteomic database search
High‐resolution/high‐mass accuracy nano‐LC‐MS/MS data were processed using Proteome Discoverer 1.4.1.14/Mascot 2.5. Mouse data were queried against UniProt’s Complete MOUSE proteome (March, 2020; 55,412 sequences) using the following parameters: enzyme: trypsin/P, maximum allowed missed cleavage sites: 2, monoisotopic precursor mass tolerance: 10 ppm, monoisotopic fragment mass tolerance: 0.02 Da, dynamic modifications: oxidation (M), acetyl (protein N‐term), static modification: carbamidomethyl (C). Percolator was used to calculate peptide false discovery rates (FDR), which was calculated per file. 1% FDR was applied to each separate LC‐MS/MS file. For exosome samples that had been in contact with fetal bovine serum (FBS, exemplified by samples that originated from cell culture), an FBS‐specific database was concatenated to the mouse databases when querying the data.
EMT score analysis
EMT scores for RNA‐seq data were calculated using MLR and 76GS EMT scoring methods (Chakraborty et al, 2020). The higher the MLR score, the more mesenchymal the sample is. The higher the 76GS score, the more epithelial the sample is.
Software
PRISM software was used for the statistical analysis and data visualization (http://www.graphpad.com). ImageJ software (U.S. National Institutes of Health) was used for data analysis. The R language and environment for statistical computing and graphics (https://www.r‐project.org) were utilized in this study for the statistical and bioinformatics analysis of RNA‐seq. The R packages used for the analysis described in the method section were obtained from the Bioconductor (https://www.bioconductor.org) and CRAN (https://cran.r‐project.org/web/packages/). BioRender (https://biorender.com) was used to create illustrations.
Quantification and statistical analysis
Statistical analysis of multiple comparisons was performed using ANOVA with Tukey’s multiple comparison test, and comparisons between two groups were performed using Students’ unpaired t‐test. All statistical analyses were performed with GraphPad Prism 8. Error bars show standard deviation (SD) or standard error of the mean (SEM) as indicated.
Author contributions
Conceptualization: RJN, JRP, BZS. Development of methodology: RJN, JRP, MKJ, JSG, DL, BDF, JKF, AKR, BZS. Data curation (provided reagents, analyzed data, provided facilities): RJN, JRP, RM, NMA‐C, DB, JL, TY, MDW, IDM, IWF, DNR‐B, I‐KK, JBB, RP, CTB, XF, MC, PC, MKJ, JSG, DL, BDF, JKF. Formal Analysis and interpretation of data (statistical and computational analysis): RJN, JRP, KS, PC, MKJ. Writing—review and editing: RJN, JRP, NMA‐C, AKR, BZS. Study supervision: BZS.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Review Process File
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View
Source Data for Figure 2
Acknowledgements
This work was supported by NIH CA236269, CA229803, DK083355, CA01369645, CA221094, 1K08DK109292, and DK050306 and by CPRIT RR190029. JRP was supported by the American Gastroenterology Association Bern Schwartz Research Scholar Award in Pancreatic Cancer, and Hopper‐Belmont Foundation Inspiration Award. MKJ was supported by Ramanujan Fellowship awarded by Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Government of India (SB/S2/RJN‐049/2018). We thank the Stanger laboratory for input and technical help, the Penn Flow Cytometry Core for help with cell sorting, and Gordon Ruthel of the Penn Vet Imaging Core for help with live‐cell imaging.
EMBO reports (2021) 22: e51872.
Data availability
All sequencing data have been deposited in the Gene Expression Omnibus (GEO): GSE157892. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157892
References
- Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, Yamazoe T, Black T, Sahmoud A, Furth EEet al (2018) EMT subtype influences epithelial plasticity and mode of cell migration. Dev Cell 45: 681–695.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL (2007) Evolving the lock to fit the key to create a family of G protein‐coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci USA 104: 5163–5168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimi I, Monteith GR (2016) Plasma membrane ion channels and epithelial to mesenchymal transition in cancer cells. Endocr Relat Cancer 23: R517–R525 [DOI] [PubMed] [Google Scholar]
- Bar‐Shavit R, Maoz M, Kancharla A, Nag JK, Agranovich D, Grisaru‐Granovsky S, Uziely B (2016) G protein‐coupled receptors in cancer. Int J Mol Sci 17: 1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart S, Chen NM, Siveke JT, König A, Zhang JS, Singh SK, Wolf E, Bartkuhn M, Esposito I, Heßmann Eet al (2014) Inflammation‐Induced NFATc1‐STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D. Cancer Discov 4: 688–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty P, George JT, Tripathi S, Levine H, Jolly MK (2020) Comparative study of transcriptomics‐based scoring metrics for the epithelial‐hybrid‐mesenchymal spectrum Fronti Bioeng Biotechnol 8: 220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE (2007) Calcium signaling. Cell 131: 1047–1058 [DOI] [PubMed] [Google Scholar]
- Conklin BR, Hsiao EC, Claeysen S, Dumuis A, Srinivasan S, Forsayeth JR, Guettier J‐M, Chang Wc, Pei Y, McCarthy KDet al (2008) Engineering GPCR signaling pathways with RASSLs. Nat Methods 5: 673–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN (2002) NFAT signaling: choreographing the social lives of cells. Cell 109: S67–S79 [DOI] [PubMed] [Google Scholar]
- Davis FM, Azimi I, Faville RA, Peters AA, Jalink K, Putney JW, Goodhill GJ, Thompson EW, Roberts‐Thomson SJ, Monteith GR (2014) Induction of epithelial‐mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 33: 2307–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongre A, Weinberg RA (2019) New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol 20: 69–84 [DOI] [PubMed] [Google Scholar]
- Dorsam RT, Gutkind JS (2007) G‐protein‐coupled receptors and cancer. Nat Rev Cancer 7: 79–94 [DOI] [PubMed] [Google Scholar]
- Grigore A, Jolly M, Jia D, Farach‐Carson M, Levine H (2016) Tumor Budding: the name is EMT. Partial EMT. J Clin Med 5: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit JJ, Apelqvist ÅA, Gu X, Winslow MM, Neilson JR, Crabtree GR, Kim SK (2006) Calcineurin/NFAT signalling regulates pancreatic β‐cell growth and function. Nature 443: 345–349 [DOI] [PubMed] [Google Scholar]
- Hoshino A, Costa‐Silva B, Shen T‐L, Rodrigues G, Hashimoto A, Tesic Mark M, Molina H, Kohsaka S, Di Giannatale A, Ceder Set al (2015) Tumour exosome integrins determine organotropic metastasis. Nature 527: 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Okamoto I, Araki N, Kawano Y, Nakao M, Fujiyama S, Tomita K, Mimori T, Saya H (1999) Calcium influx triggers the sequential proteolysis of extracellular and cytoplasmic domains of E‐cadherin, leading to loss of β‐catenin from cell‐cell contacts. Oncogene 18: 7080–7090 [DOI] [PubMed] [Google Scholar]
- John Martin T (2016) Parathyroid hormone‐related protein, its regulation of cartilage and bone development, and role in treating bone diseases. Physiol Revi 96: 831–871 [DOI] [PubMed] [Google Scholar]
- Jolly MK, Somarelli JA, Sheth M, Biddle A, Tripathi SC, Armstrong AJ, Hanash SM, Bapat SA, Rangarajan A, Levine H (2019) Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol Ther 194: 161–184 [DOI] [PubMed] [Google Scholar]
- Kourtidis A, Ngok SP, Anastasiadis PZ (2013) P120 catenin: an essential regulator of cadherin stability, adhesion‐induced signaling, and cancer progression. Prog Mol Biol Transl Sci 116: 409–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kröger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL, Thiru P, Bierie B, Ye X, Burge CBet al (2019) Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci USA 116: 7353–7362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, Richman LP, Lin JH, Sun YH, Rech AJet al (2018) Tumor cell‐intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 49: 178–193.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini M, Toker A (2009) NFAT proteins: emerging roles in cancer progression. Nat Rev Cancer 9: 810–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteith GR, Prevarskaya N, Roberts‐Thomson SJ (2017) The calcium–cancer signalling nexus. Nat Rev Cancer 17: 373–380 [DOI] [PubMed] [Google Scholar]
- Nieto MA, Huang RY‐J, Jackson RA, Thiery JP (2016) EMT: 2016. Cell 166: 21–45 [DOI] [PubMed] [Google Scholar]
- Norgard RJ, Stanger BZ (2020) Isolation and identification of EMT subtypes. In Methods in molecular biology, Campbell K, Theveneau E (eds) pp 315–326. New York, NY: Springer US; [DOI] [PubMed] [Google Scholar]
- Ocaña OH, Córcoles R, Fabra Á, Moreno‐Bueno G, Acloque H, Vega S, Barrallo‐Gimeno A, Cano A, Nieto MA (2012) Metastatic colonization requires the repression of the epithelial‐mesenchymal transition inducer Prrx1. Cancer Cell 22: 709–724 [DOI] [PubMed] [Google Scholar]
- Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA (2008) Loss of E‐cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 68: 3645–3654 [DOI] [PubMed] [Google Scholar]
- Pastushenko I, Mauri F, Song Y, de Cock F, Meeusen B, Swedlund B, Impens F, Van Haver D, Opitz M, Thery Met al (2020) Fat1 deletion promotes hybrid EMT state, tumour stemness and metastasis. Nature 589: 448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitarresi JR, Norgard RJ, Chiarella AM, Suzuki K, Bakir B, Sahu V, Li J, Zhao J, Marchand B, Wengyn MDet al (2021) PTHrP drives pancreatic cancer growth and metastasis and reveals a new therapeutic vulnerability. Cancer Discov 11: 1774–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert M, Bakir B, Moreira L, Pitarresi JR, Feldmann K, Simon L, Suzuki K, Maddipati R, Rhim AD, Schlitter AMet al (2018) Regulation of epithelial plasticity determines metastatic organotropism in pancreatic cancer. Dev Cell 45: 696–711.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roderick HL, Cook SJ (2008) Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer 8: 361–375 [DOI] [PubMed] [Google Scholar]
- Singh SK, Chen N‐M, Hessmann E, Siveke J, Lahmann M, Singh G, Voelker N, Vogt S, Esposito I, Schmidt Aet al (2015) Antithetical NFATc1‐Sox2 and p53‐miR200 signaling networks govern pancreatic cancer cell plasticity. EMBO J 34: 517–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart TA, Yapa KTDS, Monteith GR (2015) Altered calcium signaling in cancer cells. Biochim Biophys Acta ‐ Biomembr 1848: 2502–2511 [DOI] [PubMed] [Google Scholar]
- Subbalakshmi AR, Kundnani D, Biswas K, Ghosh A, Hanash SM, Tripathi SC, Jolly MK (2020) NFATc acts as a non‐canonical phenotypic stability factor for a hybrid epithelial/mesenchymal phenotype. bioRxiv 10.1101/2020.04.18.047803 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HD, Luitel K, Kim M, Zhang K, Longmore GD, Tran DD (2014) Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res 74: 6330–6340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J (2012) Spatiotemporal regulation of epithelial‐mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22: 725–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaqué J, Dorsam R, Feng X, Iglesias‐Bartolome R, Forsthoefel D, Chen Q, Debant A, Seeger M, Ksander B, Teramoto Het al (2013) A Genome‐wide RNAi screen reveals a trio‐regulated Rho GTPase Circuitry transducing mitogenic signals initiated by G protein‐coupled receptors. Mol Cell 49: 94–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalobo A, Berchtold MW (2020) The role of calmodulin in tumor cell migration, invasiveness, and metastasis. Int J Mol Sci 21: 765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, Campbell K, Cano A, Casanova J, Christofori Get al (2020) Guidelines and definitions for research on epithelial–mesenchymal transition. Nat Rev Mol Cell Biol 21: 341–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Natesan R, Sanchez‐Rivera FJ, Li J, Bhanu NV, Yamazoe T, Lin JH, Merrell AJ, Sela Y, Thomas SKet al (2020) Global regulation of the histone mark H3K36ME2 underlies epithelial plasticity and metastatic progression. Cancer Discov 10: 854–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Norgard RJ, Stanger BZ (2019) Cellular plasticity in cancer. Cancer Discov 9: 837–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Weinberg RA (2018) Epithelial‐to‐mesenchymal transition in cancer: complexity and opportunities. Front Med 12: 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Review Process File
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View
Source Data for Figure 2
Data Availability Statement
All sequencing data have been deposited in the Gene Expression Omnibus (GEO): GSE157892. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE157892